

Abstract
Though the fragile histidine triad gene product, Fhit, was discovered and characterized as a tumor suppressor 13 years ago, its sequence, structure, and cellular location did not provide clues to aid discovery of its mechanisms of suppression. Recently, using chemical cross‐linkers and immunoprecipitation, a Fhit protein complex was identified that includes Hsp60 and Hsp10 which may mediate Fhit stability and mitochondrial localization, where Fhit binds and stabilizes ferredoxin reductase (Fdxr); when Fdxr is overexpressed, it can lead to production of reactive oxygen species (ROS) that induce apoptosis. Cancer cells expressing endogenous or exogenous Fhit, when exposed to H2O2, an oxidative stress, produce higher levels of apoptosis‐inducing ROS than matched, Fhit‐negative cells; the Fhit‐negative cancer cells survive, carrying DNA damage. In addition to this mitochondrial function, Fhit‐overexpression in cancer cells exposed to replicative stress‐inducing agents leads to enhanced caspase 3 activation and apoptosis, due to defective Chk1 activation. Thus, damage to the fragile FHIT locus leads to reduced expression of Fhit protein, and makes a two‐pronged contribution to development of preneoplastic clonal expansion: (1) absence or reduction of Fhit leads to reduced expression of Fdxr and reduced ROS‐induced apoptosis; (2) cells that escape ROS‐ or replicative stress‐induced apoptosis can carry misrepaired DNA damage. The aberrant DNA damage response checkpoint in Fhit‐deficient preneoplasias and cancers may make these lesions targets for inhibitors of proteins such as Parp1 and Chk1 with important roles in checkpoint responses, as observed for BRCA1‐deficient cancer cells that also exhibit DNA damage repair deficiencies. (Cancer Sci 2009; 100: 1145–1150)
Abbreviations:
- DDR
DNA damage response
- Fdxr
ferredoxin reductase
- Fhit
fragile histidine triad protein
- ROS
reactive oxygen species
More than 760 published reports on the fragile FHIT gene and its gene product Fhit have established its role in protecting against tumor development. Fhit protein expression is lost in many cancers and its loss in some types of cancer has prognostic significance. Fhit can be lost in the early steps of preneoplasia and its loss contributes to further genetic alterations in the preneoplastic cells. Fhit knockout mice show increased susceptibility to development of induced and spontaneous tumors, and Fhit gene therapy contributes to tumor prevention and regression in animal models.( 1 , 2 ) Though it was shown early after Fhit discovery that Fhit overexpression can initiate the apoptotic response in cancer cells,( 3 , 4 , 5 ) only recently have mechanisms through which Fhit participates in signal pathways been elucidated.
The FHIT gene encompasses the most active common human fragile site and is thus exquisitely sensitive to intragenic alterations by DNA damaging agents that can lead to allele loss early in the preneoplasia,( 6 , 7 , 8 ) coincident with activation of a DNA damage checkpoint.( 9 , 10 )
Defining how Fhit induces apoptosis and suppresses tumors has been a challenge because interacting proteins, effectors of Fhit signals, were difficult to identify. Nevertheless, the study of Fhit‐deficient tissue‐ and cancer‐derived cells in vitro has led to important conclusions: repair protein‐deficient cancers are more likely to be Fhit‐deficient;( 11 , 12 ) Fhit‐deficient cells show enhanced resistance to DNA damage–induced cell killing;( 13 , 14 , 15 , 16 , 17 , 18 ) Fhit indirectly affects S‐phase checkpoint and DNA repair;( 14 , 19 ) and oxidative stress, coupled with error‐prone DNA damage repair, allows long‐term survival of genotoxin‐exposed Fhit‐deficient hematopoietic stem cells.( 16 ) Results of a recent study have suggested that Fhit is necessary for protecting cells from accumulation of DNA damage, through modulation of checkpoint proteins Hus1 and phosphoChk1,( 14 ) contributing to accumulation of abnormal checkpoint phenotypes in cancer development. To determine mechanisms employed by Fhit in modulating responses to oxidative and replicative stress and to define consequences of Fhit loss, we have isolated Fhit‐interacting proteins and examined biologic effects of activation of the Fhit complex.( 15 , 20 )
This review of recent studies of Fhit function draws connections among the results of various studies, suggesting that Fhit is involved in a self‐perpetuating loop of damage to fragile genomic regions, followed by inactivation of expression of Fhit protein; Fhit protein is necessary for the correct execution of the DNA damage checkpoint and associated repair, so its loss leads to further unrepaired DNA damage and mutation, as illustrated in summary form in Figure 1.
Figure 1.
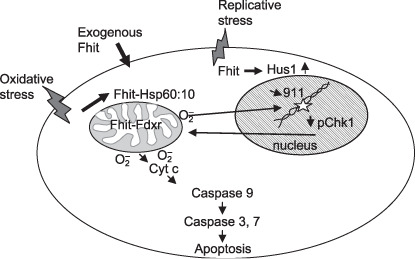
Roles of fragile histidine triad gene product (Fhit) in response to cellular stress. Depiction of replicative and oxidative stress pathways affected by Fhit expression. The lines between the nucleus and mitochondrion indicate that ROS produced in mitochondria influence the cell cycle and cause DNA damage, while nuclear DNA damage can lead to apoptosis pathways that involve the mitochondrial pathway.
Fhit and oxidative stress
The Fhit protein complex. To identify proteins that interact with Fhit to affect downstream apoptotic pathways, we used chemical protein cross‐linking and proteomics methods to isolate and characterize a Fhit protein complex involved in triggering Fhit‐mediated apoptosis.( 15 )
Using liquid‐chromatography tandem mass spectrometry, six proteins, Hsp60, Hsp10, malate dehydrogenase, electron transfer flavoprotein b, mitochondrial aldehyde dehydrogenase, and ferredoxin reductase (Fdxr), were identified, all with mitochondrial subcellular localization. The finding that candidate partners for Fhit protein were mitochondrial proteins suggested that Fhit must also be partially distributed in the mitochondria. Fhit mitochondrial localization was confirmed by subcellular fractionation and immunofluorescence localization studies in cancer‐derived cell lines expressing endogenous or exogenous Fhit. We then focused on Hsp60 and Hsp10 as possible chaperonins and on Fdxr, a mitochondrial respiratory chain protein,( 21 ) transactivated by p53 and involved in responses to therapeutic drugs.( 22 , 23 ) Hsp60 and 10 are molecular chaperones found in complex and are important for the folding and import of proteins into mitochondria.( 24 ) Experiments in which Hsp60 and 10 expression was down‐modulated by specific antisense oligonucleotides provided evidence that the Hsp60/10 complex was responsible for Fhit mitochondrial localization. To examine protein–protein direct interactions in the Fhit complex, purified Fhit was mixed with purified Hsp10, Hsp60, or Fdxr and experiments were performed to detect direct interactions among the protein pairs. Fhit interacted directly with Hsp60 and Fdxr, but not with Hsp10.( 20 )
Fhit–Fdxr interaction generates ROS and induces apoptosis. We were especially interested in the Fhit–Fdxr interaction because of previous studies of the p53 tumor suppressor( 22 , 23 ) showing that p53 is a transcriptional activator of Fdxr, through which ROS are produced and apoptosis is induced. Thus, we studied the biologic consequences of the Fhit–Fdxr interaction. Fdxr, the 54 kDa mammalian mitochondrial cytochrome P‐450 NADPH reductase, is located on the matrix side of the inner mitochondrial membrane, and is responsible for transferring electrons from NADPH, via the single electron shuttle ferredoxin‐cytochrome P‐450, to substrates during steroidogenesis.( 21 ) Under substrate‐limiting conditions, electrons can leak from this shuttle system and generate ROS.( 24 ) Fdxr mediates p53‐dependent, 5‐fluorouracil‐induced apoptosis in colorectal cancer cells, through the generation of ROS,( 22 , 23 ) critical regulators of apoptosis.( 25 ) Overexpression of Fdxr increases sensitivity of tumor cells to apoptosis on H2O2 treatment, through ROS production. Fhit prevents destabilization of Fdxr protein by protecting it from proteasome degradation,( 15 ) and the Fhit–Fdxr interaction generates ROS production and is involved in Fhit‐mediated apoptosis (see Fig. 2).
Figure 2.
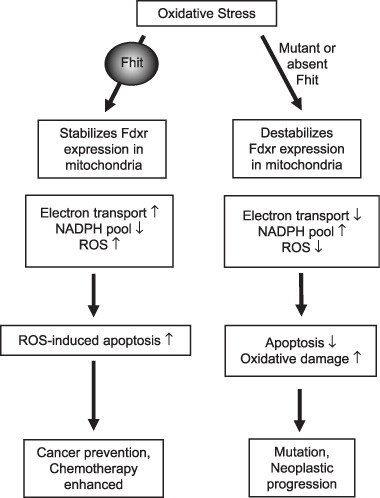
Fragile histidine triad gene product (Fhit) and oxidative stress. When cancer cells are exposed to extreme oxidative stress, the outcome depends on whether Fhit protein is expressed. In Fhit positive cells exposed to peroxide, cytosolic Fhit enters mitochondria through interaction with stress proteins Hsp60 and Hsp10, where Fhit binds to and stabilizes Fdxr. An excess of Fdxr relative to its substrate can lead to ROS production followed by apoptosis. In the absence of Fhit, Fdxr is less stable, leading to less ROS production and reduced apoptosis.
Generation of intracellular ROS is an early event in the apoptosis of lung cancer cells induced by treatment with paclitaxel,( 26 ) and Fhit expressing cells are more sensitive to paclitaxel and cisplatin.( 27 , 28 ) Cisplatin induces Fdxr expression and the cisplatin‐induced apoptotic pathway is associated with ROS generation.( 29 ) Fhit expression increases sensitivity to oxidative injury through participation with Fdxr in ROS generation and enhances ROS‐related apoptotic effects of chemotherapeutic agents. The finding that ROS generation is involved in Fhit‐mediated apoptosis emphasizes the importance of Fhit loss as a negative prognostic factor in various clinical settings.
Fhit mutants in the mitochondrial pathway. To define Fhit structural features that affect interactions, downstream signaling, and biological outcomes, cancer cells expressing Fhit mutants with amino acid substitutions that alter enzymatic activity, enzyme substrate binding, or phosphorylation were used. Cancer cell clones stably expressing mutants that do not bind substrate or cannot be phosphorylated, showed decreased binding to Hsp60 and Fdxr, and reduced mitochondrial localization. Expression of Fhit or mutants that bind interactor proteins results in oxidative damage and accumulation of cells in subG1 fractions after peroxide treatment; non‐interacting mutants were defective in these effects (see Fig. 3). Cancer clones expressing non‐complexing Fhit mutants showed reduction of Fhit tumor suppressor activity, confirming that substrate binding, interaction with Hsps, mitochondrial localization, and interaction with Fdxr are important for Fhit tumor suppressor function.( 20 )
Figure 3.
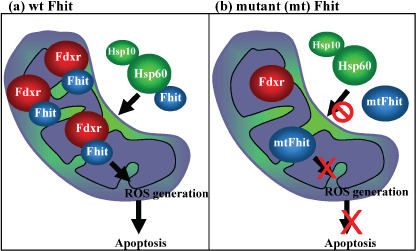
Fragile histidine triad gene product (Fhit) and Fhit mutants in mitochondria. (a) WT Fhit binds Fdxr in the mitochondrion, where stabilized Fdxr participates in electron transport pathways that, in absence of sufficient substrate, can lead to ROS production. (b) The right panel shows that Fhit proteins carrying point mutations known to abrogate Fhit apoptosis‐inducing ability may be defective in binding to Hsp60, may show reduced mitochondrial localization, or may not interact with Fdxr, and are thus unable to induce apoptosis after application of oxidative stress.
Response of Fhit‐deficient hematopoietic stem cells to genotoxic stress. To explore roles for Fhit in hematopoietic cells, Ishii et al.( 16 ) used Fhit‐deficient bone marrow cells from Fhit knockout mice,( 30 , 31 ) and examined the response to a benzene metabolite, hydroquinone. Treatment with hydroquinone leads to increased intracellular production of ROS, which can produce DNA double‐strand breaks; when imperfectly repaired,( 32 ) double‐strand breaks can lead to deleterious genetic changes. We found that Fhit‐deficient hematopoietic cells exposed to hydroquinone were resistant to the suppression of stem cell colony formation observed with wild‐type (WT) hematopoietic cells. In vivo – transplanted, hydroquinone‐exposed, Fhit‐deficient bone marrow cells also escaped the bone marrow suppression. Immunohistochemical analyses of bone marrow transplants showed reduced expression of Bax in Fhit‐deficient bone marrow, suggesting insensitivity to apoptosis. Also, the assessment of DNA damage showed that presence of the oxidized base 8‐hydroxyguanosine, a marker of DNA damage, was reduced in Fhit‐deficient bone marrow, as was production of intracellular ROS. Antioxidant treatment reduced the hydroquinone‐induced suppression of colony formation by WT hematopoietic cells, suggesting that decreased oxidative damage to Fhit‐deficient bone marrow cells allowed the survival advantage. Homologous recombination repair predominated in Fhit‐deficient cells but was not error‐free, as shown by a higher incidence of 6‐thioguanine‐resistant colonies. Tissues of the hydroquinone‐exposed, Fhit‐deficient bone marrow–transplanted mice showed evidence of preneoplastic alterations, including accumulation of histone H2AX‐positive DNA damage. Thus, reduced oxidative stress, coupled with efficient but not error‐free DNA damage repair, allowed unscheduled long‐term survival of genotoxin‐exposed Fhit‐deficient hematopoietic stem cells carrying deleterious mutations.
If overexpression of Fhit in cells exposed to extreme extrinsic stressful agents leads to excess ROS production and apoptosis, then what is happening in the Fhit‐negative cells exposed to extreme stress? They produce less ROS, so are they not, in fact, better off than Fhit‐expressing cells? And how does this scenario relate to what is happening in normal cells experiencing intrinsic, physiologic levels of ROS due to normal metabolism? It may be that answers can be found by performing experiments similar to those first done in p53 WT‐expressing and deficient cells. Sablina et al.,( 33 ) examined the differences between the effects of p53 down‐modulation by siRNA methods in several cell types under physiological conditions, i.e. without exposure to exogenous stress. Under these conditions, it was found that absence of p53 had a deleterious effect, while presence of p53 had a survival effect, due to transcriptional activation of pro‐survival genes, such as members of the Sestrin family. On the other hand, under conditions of extreme extrinsic oxidative stress, p53, like Fhit, causes enhancement of expression of Fdxr and increased production of ROS, followed by apoptosis.
In future experiments it will be important to determine if down‐modulation of Fhit expression in normal or cancer cells under physiological conditions, with no applied stress, will lead to accumulation of cells in apoptosis, as observed by Sablina et al.( 33 ) in cells with down‐modulated p53 (see projected outcome of such experiments in Fig. 4).
Figure 4.
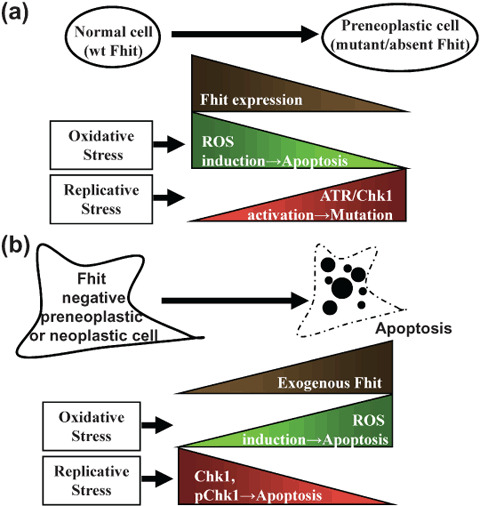
Fragile histidine triad gene product (Fhit) and replicative stress. (a) In non‐cancer cells exposed to replication stress and DNA damage, activation of the checkpoint results in the PCNA‐like Hus1/Rad1/Rad9 clamp recognition of damage foci and ATR and Chk1 activation, leading to cell cycle arrest, repair, and survival. In some cells the damage will cause deletion at the FRA3B/FHIT locus and loss of Fhit. In this case, the checkpoint is likely to be aberrant, and cells will accumulate mutations. (b) In Fhit‐deficient cells expressing exogenous Fhit and exposed to severe replication stress, the Chk1 pathway is not appropriately activated, resulting in uncoupling of recognition of DNA damage and activation of Chk1, leading to apoptosis.
Fhit and replicative stress
Role of Fhit in cells exposed to replicative stress. Chk1, the downstream phosphorylation target of ATR, protects cells from apoptosis induced by DNA replication inhibitors. Myers et al.( 34 ) determined the role of ATR and Chk1 protein kinases in control of apoptosis following replication stress.
ATR/Chk1 signaling pathways were manipulated using siRNA‐mediated depletion or specific inhibitors in tumor cell lines and fibroblasts exposed to thymidine or hydroxyurea. ATR or Chk1 depletion strongly enhanced cell death in the cells tested. The replication stress‐triggered apoptotic pathway was characterized by activation of caspase 3 in both p53‐proficient and ‐deficient cells. It was concluded that the ATR–Chk1 signaling pathway plays a major role in the regulation of death in response to DNA replication stress. This elegant study may explain results we observed( 14 , 19 ) in UV treated esophageal cancer cells. We assessed effects of expression of exogenous WT and mutant Fhit in the checkpoint response to UV‐induced DNA damage.( 15 , 19 ) The introduction of exogenous WT Fhit in Fhit‐deficient UV‐treated esophageal cancer cells caused up‐modulation of Hus1 expression but Chk1 was poorly activated and rapidly down‐modulated, triggering cell death. Expression of the Fhit mutant proteins known to be defective in suppressor function resulted in Chk1 activation and phosphoChk1 expression, indicating activation of the checkpoint. The results suggested that in the cancer cells, WT Fhit overexpression caused abortive checkpoint activation in response to UV stress. Absence of Fhit in the Fhit‐deficient cancer cells allowed survival of DNA‐damaged cancer cells, leading to a cycle of damage and survival of cells carrying mutations.
Impaired homologous recombination repair in Fhit‐negative sebaceous gland carcinomas. Sebaceous gland carcinomas are rare malignancies of the skin; >50% exhibit microsatellite instability due to defective mismatch repair. But a significant fraction shows microsatellite stability associated with loss of Fhit expression.( 30 , 35 ) Becker et al.( 35 ) hypothesized that in sebaceous gland carcinomas with microsatellite stability and loss of Fhit, effector molecules participating in homologous recombination repair, such as BRCA1/2, might be somatically inactivated, and showed that Fhit‐negative sebaceous gland carcinomas displayed loss of heterozygosity and biallelic deletions of the BRCA1 gene in five of 10 cases. Tumor‐specific genomic losses close to BRCA2 were also observed. A homozygous p53 R248W gain‐of‐function mutation was identified in one of seven sebaceous gland carcinomas and it is known that p53 R248W mutants inactivate ATM‐directed homologous recombination repair. This sebaceous gland carcinoma also exhibited genomic deletions at the BRCA1 and BRCA2 loci, and at the constitutively fragile sites FRA3B/FHIT and FRA16D/WWOX. The study demonstrated that microsatellite‐stable Fhit‐negative sebaceous gland carcinomas accumulate mutations that target components of the repair network.
Fhit loss and DNA damage response in breast cancer subtypes. Breast cancer subtypes, identified through gene expression signatures, define cancers with different prognostic features, including luminal, HER2 overexpressing, and basal‐like.( 36 , 37 ) Basal‐like breast cancers are triple negative tumors that can be identified by immunohistochemistry positivity for CK5/6 and/or EGFR and negativity for ER, PR, and HER2.( 38 , 39 ) This subtype, for which there is no targeted therapy, account for ~15% of all breast cancers but >50% of breast cancers of some cohorts of patients studied in Africa.( 40 ) Mutant BRCA1‐associated breast cancers show basal‐like features and BRCA1 inactivation is a common event in sporadic basal‐like breast tumors.( 41 , 42 ) In various studies of sporadic breast cancers, 40% or more were positive for Fhit, whereas only 9% of BRCA1 mutant tumors were Fhit positive, suggesting that the BRCA1 pathway is important in protecting the FRA3B/FHIT locus from damage;( 12 ) inactivation of the BRCA1 pathway in basal/triple negative cancers may contribute to loss of expression of Fhit in these cancers or vice versa. When >800 breast cancers on tissue micro arrays were divided into subtypes, triple negative tumors showed loss of Fhit in ~90%;( 43 ) see Figure 5 for example. The results suggested that reduced Fhit expression could have a role in pathogenesis of basal‐like differentiation in breast cancer. In preliminary experiments, three DDR proteins (γH2AX, pChk2, p53) were expressed significantly more frequently in basal‐like tumors than in other subtypes (Huebner and colleagues, unpublished data).
Figure 5.
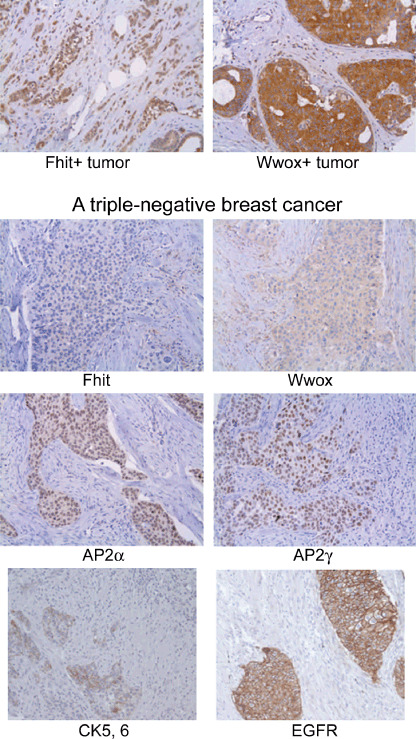
Absence of fragile histidine triad gene product (Fhit) in a triple negative breast cancer. In the top panels strong expression of Fhit and Wwox is illustrated, in contrast to the negative and very low expression of Fhit and Wwox, respectively, in a triple negative tumor (lower panels). This tumor also shows moderate expression of CK5/6, moderate nuclear expression of AP2α and ‐γ, and strong membranous expression of EGFR (×200 for each photo). This figure was adapted from Figure 1 in reference 43; in this reference, Fhit and Wwox, encoded by the two most active chromosome fragile sites, were investigated for expression in specific subtypes of breast cancer and absence of expression of both of these suppressors was significantly associated with the basal‐like/triple negative subtype.
DDR checkpoint proteins are activated in preneoplastic and neoplastic lesions of skin, lung, and breast,( 9 , 10 ) and it was proposed that checkpoint activation acts as a barrier to cancer progression until mutations in checkpoint genes allow growth of cells through the checkpoint, leading to genome instability and cancer progression. Loss of heterozygosity at the FHIT locus is concomitant with DDR checkpoint activation,( 9 , 10 ) so perhaps DNA breakage at the fragile gene loci may trigger the DDR checkpoint; then loss of expression of the FHIT gene due to this breakage alters the DDR checkpoint. Since basal‐like breast cancers show loss or reduction of Fhit protein, these tumors may have defective, activated DDR checkpoints and may be especially sensitive to inhibitors of checkpoint proteins, as are BRCA1‐deficient cancers.( 43 , 44 , 45 , 46 , 47 ) Indeed, it has been reported that Chk1 protein is highly expressed in triple negative breast cancers.( 48 )
Perspective
Because Fhit is not a transcription factor, resides in the cytoplasm, and does not exhibit known protein‐interacting domains or motifs that give clues to function, it has been a challenge to determine its functions. Nevertheless, progress has been made in discovering its involvement in cellular functions of major importance for keeping cells normal. We know that it binds Hsp60, enters mitochondria, binds Fdxr, and contributes to generation of ROS; we also know that Fhit has a role in the cellular response to replication fork stress, though the mechanism through which it affects this response is not well defined. We believe that Fhit signals, at least partially, through effects on stability of proteins such as Fdxr, Hus1, possibly Chk1, and Cyclin D; of these we have demonstrated interaction only with Fdxr thus far.
Earlier searches for Fhit‐interacting proteins pointed to several candidate proteins, none of which we could confirm as interactors by coimmunoprecipitaton experiments, including Ubc9, tubulin, Mdm2, and beta‐catenin.( 49 , 50 , 51 , 52 ) Weiske et al.( 52 ) recently reported that Fhit binds directly to beta‐catenin, an effector in the Wnt pathway that is dysregulated in various cancers, and that Fhit binding to beta‐catenin repressed transcription of target genes such as Cyclin D1 and Survivin; we have, in fact, observed repression of Cyclin D1 and Survivin by Fhit( 53 ) (Semba and Huebner, unpublished); but we have not been able to observe coimmunoprecipitation of Fhit and beta‐catenin. We do not have an explanation for the difference between the Weiske et al. study and our attempted coimmunoprecipitation of Fhit and beta‐catenin, but we have investigated only endogenous proteins in MCF7 cells and we were also unable to find evidence of endogenous Fhit in the nuclear fraction of these cells. Identification and confirmation of other Fhit protein interactors by interested investigators has the potential to advance understanding of the mechanisms of Fhit involvement in centrally important signal pathways.
In future studies it will also be important to confirm interactions of Fhit with the other potential interactors identified in our previous protein cross‐linking study:( 15 ) malate dehydrogenase, mitochondrial aldehyde dehydrogenase 2, and electron transfer protein b; if the interactions are confirmed and the signal pathways characterized, as for Fdxr, additional Fhit functions may be revealed. Important next steps will involve examination of effects of endogenous Fhit level on responses to intrinsic stress in normal cells and cells in early stages of preneoplasia.
Acknowledgment
We are grateful to our past and present laboratory members and collaborators who have contributed to the work summarized here.
References
- 1. Pichiorri F, Ishii H, Okumura H et al . Molecular parameters of genome instability: roles of fragile genes at common fragile sites. J Cell Biochem 2008; 104: 1525–33. [DOI] [PubMed] [Google Scholar]
- 2. Ishii H, Dumon KR, Vecchione A et al . Potential cancer therapy with the fragile histidine triad gene: review of the preclinical studies. JAMA 2001; 286: 2441–9. [DOI] [PubMed] [Google Scholar]
- 3. Ji L, Fang B, Yen N et al . Induction of apoptosis and inhibition of tumorigenicity and tumor growth by adenovirus vector‐mediated fragile histidine triad (FHIT) gene overexpression. Cancer Res 1999; 59: 3333–9. [PubMed] [Google Scholar]
- 4. Sard L, Accornero P, Tornielli S et al . The tumor‐suppressor gene FHIT is involved in the regulation of apoptosis and in cell cycle control. Proc Natl Acad Sci USA 1999; 96: 8489–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Trapasso F, Krakowiak A, Cesari R et al . Designed FHIT alleles establish that Fhit‐induced apoptosis in cancer cells is limited by substrate‐binding. Proc Natl Acad Sci USA 2003; 100: 1592–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Huebner K, Croce CM. Cancer and the FRA3B/FHIT fragile locus: it's a HIT. Br J Cancer 2003; 88: 1501–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Tsantoulis PK, Kotsinas A, Sfikakis PP et al . Oncogene‐induced replication stress preferentially targets common fragile sites in preneoplastic lesions. A genome‐wide study. Oncogene 2008; 27: 3256–64. [DOI] [PubMed] [Google Scholar]
- 8. Durkin SG, Ragland RL, Arlt MF et al . Replication stress induces tumor‐like microdeletions in FHIT/FRA3B. Proc Natl Acad Sci USA 2008; 105: 246–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Bartkova J, Horejsi Z, Koed K et al . DNA damage response as a candidate anti‐cancer barrier in early human tumorigenesis. Nature 2005; 434: 864–70. [DOI] [PubMed] [Google Scholar]
- 10. Gorgoulis VG, Vassiliou LV, Karakaidos P et al . Activation of the DNA damage checkpoint and genomic instability in human precancerous lesions. Nature 2005; 434: 907–13. [DOI] [PubMed] [Google Scholar]
- 11. Ingvarsson S, Agnarsson BA, Sigbjornsdottir BI et al . Reduced Fhit expression in familial and sporadic breast carcinomas. Cancer Res 1999; 59: 2682–9. [PubMed] [Google Scholar]
- 12. Turner B, Ottey M, Potoczek M et al . The FHIT/FRA3B locus and repair deficient cancers. Cancer Res 2002; 62: 4054–60. [PubMed] [Google Scholar]
- 13. Ottey M, Han SY, Druck T et al . Fhit deficient normal and cancer cells are mitomycin C and UVC resistant. Br J Cancer 2004; 91: 1669–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Ishii H, Mimori K, Inoue H et al . Fhit modulates the DNA damage checkpoint response. Cancer Res 2006; 66: 11287–92. [DOI] [PubMed] [Google Scholar]
- 15. Trapasso F, Pichiorri F, Gasparo M et al . Fhit interaction with ferredoxin reductase triggers generation of reactive oxygen species and apoptosis. J Biol Chem 2008; 283: 13736–44. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 16. Ishii H, Mimori K, Ishikawa K et al . FHIT‐deficient hematopoietic stem cells survive hydroquinone treatment long‐term, carrying precancerous alterations. Cancer Res 2008; 68: 3662–70. [DOI] [PubMed] [Google Scholar]
- 17. Hu B, Han S, Wang X et al . Involvement of the Fhit gene in the ionizing radiation‐activated ATR/CHK1 pathway. J Cell Physiol 2005; 202: 518–23. [DOI] [PubMed] [Google Scholar]
- 18. Hu B, Wang H, Wang X et al . Fhit and CHK1 have opposing effects on homologous recombination repair. Cancer Res 2005; 65: 8613–16. [DOI] [PubMed] [Google Scholar]
- 19. Ishii H, Wang Y, Huebner K. A Fhit‐ing role in the DNA damage checkpoint response. Cell Cycle 2007; 6: 1044–8. [DOI] [PubMed] [Google Scholar]
- 20. Pichiorri F, Okumura H, Nakamura T et al . Correlation of Fhit structural features with effector interactions and biological functions. J Biol Chem 2009; 284: 1040–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Kimura T, Suzuki K. Components of the electron transport system in adrenal steroid hydroxylase. Isolation and properties of non‐heme iron protein (adrenodoxin). J Biol Chem 1967; 242: 485–91. [PubMed] [Google Scholar]
- 22. Hwang PM, Bunz F, Yu J et al . Ferredoxin reductase affects p53‐dependent, 5‐fluorouracil‐induced apoptosis in colorectal cancer cells. Nat Med 2001; 7: 1111–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Liu G, Chen X. The ferredoxin reductase gene is regulated by the p53 family and sensitizes cells to oxidative stress‐induced apoptosis. Oncogene 2001; 21: 7195–204. [DOI] [PubMed] [Google Scholar]
- 24. Hanukoglu I, Rapoport R, Weiner L et al . Electron leakage from the mitochondrial NADPH‐adrenodoxin reductase‐adrenodoxin‐P450scc (cholesterol side chain cleavage) system. Arch Biochem Biophys 1993; 305: 489–98. [DOI] [PubMed] [Google Scholar]
- 25. Danial NN, Korsmeyer SJ. Cell death: critical control points. Cell 2004; 116: 205–19. [DOI] [PubMed] [Google Scholar]
- 26. Alexandre J, Batteux F, Nicco C et al . Accumulation of hydrogen peroxide is an early and crucial step for paclitaxel‐induced cancer cell death both in vitro and in vivo. Int J Cancer 2006; 119: 41–8. [DOI] [PubMed] [Google Scholar]
- 27. Andriani F, Perego P, Carenini N et al . Increased sensitivity to cisplatin in non‐small cell lung cancer cell lines after FHIT gene transfer. Neoplasia 2006; 8: 9–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Kim CH, Yoo JS, Lee CT et al . FHIT protein enhances paclitaxel‐induced apoptosis in lung cancer cells. Int J Cancer 2006; 118: 1692–8. [DOI] [PubMed] [Google Scholar]
- 29. Kerley‐Hamilton JS, Pike AM, Li N et al . A p53‐dominant transcriptional response to cisplatin in testicular germ cell tumor‐derived human embryonal carcinoma. Oncogene 2005; 24: 6090–100. [DOI] [PubMed] [Google Scholar]
- 30. Fong LY, Fidanza V, Zanesi N et al . Muir‐Torre‐like syndrome in Fhit deficient mice. Proc Natl Acad Sci USA 2000; 97: 4742–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Zanesi N, Fidanza V, Fong LY et al . The tumor spectrum in FHIT‐deficient mice. Proc Natl Acad Sci USA 2001; 98: 10250–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Winn LM. Homologous recombination initiated by benzene metabolites: a potential role of oxidative stress. Toxicol Sci 2003; 72: 143–9. [DOI] [PubMed] [Google Scholar]
- 33. Sablina AA, Budanov AV, Ilyinskaya GV et al . The antioxidant function of the p53 tumor suppressor. Nat Med 2005; 11: 1306–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Myers K, Gagou ME, Zuazua‐Villar P et al . ATR and Chk1 suppress a caspase‐3‐dependent apoptotic response following DNA replication stress. PLoS Genet 2009; 5: e1000324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Becker K, Goldberg M, Helmbold P et al . Deletions of BRCA1/2 and p53 R248W gain‐of‐function mutation suggest impaired homologous recombination repair in fragile histidine triad‐negative sebaceous gland carcinomas. Br J Dermatol 2008; 159: 1282–9. [DOI] [PubMed] [Google Scholar]
- 36. Perou CM, Sørlie T, Eisen MB et al . Molecular portraits of human breast tumours. Nature 2000; 406: 747–52. [DOI] [PubMed] [Google Scholar]
- 37. Sørlie T, Perou CM, Tibshirani R et al . Gene expression patterns of breast carcinomas distinguish tumor subclasses with clinical implications. Proc Natl Acad Sci USA 2001; 98: 10869–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Nielsen TO, Hsu FD, Jensen K et al . Immunohistochemical and clinical characterization of the basal‐like subtype of invasive breast carcinoma. Clin Cancer Res 2004; 10: 5367–74. [DOI] [PubMed] [Google Scholar]
- 39. Cheang MC, Voduc D, Bajdik C et al . Basal‐like breast cancer defined by five biomarkers has superior prognostic value than triple‐negative phenotype. Clin Cancer Res 2008; 14: 1368–76. [DOI] [PubMed] [Google Scholar]
- 40. Cleator S, Heller W, Coombs RC. Triple‐negative breast cancer: therapeutic options. Lancet Oncol 2007; 8: 235–44. [DOI] [PubMed] [Google Scholar]
- 41. Turner NC, Reis‐Filho JS, Russell AM et al . BRCA1 dysfunction in sporadic basal‐like breast cancer. Oncogene 2006; 26: 2126–32. [DOI] [PubMed] [Google Scholar]
- 42. Turner NC, Reis‐Filho JS. Basal‐like breast cancer and the BRCA1 phenotype. Oncogene 2006; 25: 5846–53. [DOI] [PubMed] [Google Scholar]
- 43. Guler G, Huebner K, Himmetoglu C et al . Fhit, Wwox and AP2γ expression levels correlate with basal phenotype in breast cancer. Cancer 2009; 115: 899–908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Yehiely F, Moyano JV, Evans JR et al . Deconstructing the molecular portrait of basal‐like breast cancer. Trends Mol Med 2006; 12: 537–44. [DOI] [PubMed] [Google Scholar]
- 45. Bryant HE, Schultz N, Thomas HD et al . Specific killing of BRCA2‐deficient tumours with inhibitors of poly (ADP‐ribose) polymerase. Nature 2005; 434: 913–17. [DOI] [PubMed] [Google Scholar]
- 46. Farmer H, McCabe N, Lord CJ et al . Targeting the DNA repair defect in BRCA mutant cells as a therapeutic strategy. Nature 2005; 434: 917–21. [DOI] [PubMed] [Google Scholar]
- 47. O’Connor MJ, Martin NMB, Smith GCM. Targeted cancer therapies based on the inhibition of DNA strand break repair. Oncogene 2007; 26: 7816–24. [DOI] [PubMed] [Google Scholar]
- 48. Verlinden L, Vandem Bempt I, Eelen G et al . The E2F‐regulated gene Chk1 is highly expressed in triple‐negative estrogen receptor‐/progesterone receptor‐/her1‐breast carcinomas. Cancer Res 2007; 67: 6574–81. [DOI] [PubMed] [Google Scholar]
- 49. Shi Y, Zou M, Farid NR et al . Association of FHIT (fragile histidine triad), a candidate tumour suppressor gene, with the ubiquitin‐conjugating enzyme hUBC9. Biochem J 2000; 352: 443–8. [PMC free article] [PubMed] [Google Scholar]
- 50. Chaudhuri AR, Khan IA, Prasad V et al . The tumor suppressor protein Fhit. A novel interaction with tubulin. J Biol Chem 1999; 274: 24378–82. [DOI] [PubMed] [Google Scholar]
- 51. Nishizaki M, Sasaki J, Fang B et al . Synergistic tumor suppression by coexpression of FHIT and p53 coincides with FHIT‐mediated MDM2 inactivation and p53 stabilization in human non‐small cell lung cancer cells. Cancer Res 2004; 64: 5745–52. [DOI] [PubMed] [Google Scholar]
- 52. Weiske J, Albring KF, Huber O. The tumor suppressor Fhit acts as a repressor of beta‐catenin transcriptional activity. Proc Natl Acad Sci USA 2007; 104: 20344–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Semba S, Trapasso F, Fabbri M et al . Fhit modulation of the Akt‐survivin pathway in lung cancer cells: Fhit‐tyrosine 114 (Y114) is essential. Oncogene 2006; 25: 2860–72. [DOI] [PubMed] [Google Scholar]