

Abstract
Persistent human papillomavirus (HPV) infection is necessary but insufficient for viral oncogenesis. Additional contributing co-factors, such as immune evasion and viral integration have been implicated in HPV-induced cancer progression. It is widely accepted that HPV+ keratinocytes require co-culture with fibroblasts to maintain viral episome expression, yet the exact mechanisms for this have yet to be elucidated. Here we present comprehensive RNA sequencing and proteomic analysis demonstrating that fibroblasts not only support the viral life cycle, but reduce HPV+ keratinocyte transformation. Our co-culture models offer novel insights into HPV-related transformation mechanisms.
Keywords: stroma, HPV, human papillomavirus, oropharyngeal cancer, microenvironment, fibroblasts, transformation
1. Introduction
Human papillomaviruses (HPVs) infect the basal keratinocytes of differentiating squamous epithelia [1]. Some current estimates suggest there may be more than 400 types of HPV, however, there are approximately 12 high-risk HPV types with the capacity to cause cancer in the general population [2–4]. HPV-related cancers (HPV+ cancers) continue to contribute to approximately 5% of the worldwide cancer burden [5–14]. HPV 16 is responsible for the majority of HPV+ cancers, contributing to 54% of cervical cancers and ~90% of HPV+ oropharyngeal squamous cell carcinoma (HPV+OPC) [3,5,8–10,15–17]. While these HPV+ cancers remain prevalent, the majority of total infections are asymptomatic, self-limiting, and clear before cancer progression [3,18–23]. Persistent HPV infection is a necessary component of cancer development but is not considered sufficient without additional co-factors [15,24]. One key factor in maintaining viral persistence is the ability of HPV to evade host immunity [22,23,25–29]. Numerous studies have demonstrated that HPV suppresses innate immune-related signaling in both infected epithelia and neighboring stromal fibroblasts [22,23,25–36]. Suppression of immune-related genes allows for immune evasion, which is critical for viral persistence and may play a role in cancer development [37,38].
The stroma is a complex connective tissue comprised of numerous cell types; the main component of the dermal stroma is fibroblasts [15,39–41]. Fibroblasts support tissue homeostasis via the secretion of all components of the extracellular matrix (ECM) and facilitate stromal extracellular signaling; factors produced by fibroblasts are key for angiogenesis, inflammation, wound healing, and are necessary for the proper differentiation of keratinocytes [23,41,42]. Keratinocyte differentiation is critical for the HPV lifecycle [43,44]. While HPV exclusively infects basal keratinocytes, viral gene products alter the secretion of host factors, indirectly affecting neighboring keratinocytes, fibroblasts and immune cells in the local microenvironment [22,23,45]. Given the complexity of the tissue infected and the transformation process, the relationship between HPV and epithelial-stromal communication remains at a nascent phase and further investigations are warranted [15,23].
The importance of stromal support in the microenvironment is now an emerging field in the context of overall cancer progression, as well as HPV-induced transformation and carcinogenesis [15,23,26,39,40,45–56]. Precise mechanisms for viral transformation and progression mechanisms remain unclear; however, persistent viral oncogene expression contributes to clear epithelial growth advantages [27,57–59,59–64]. HPV E6 and E7 are considered the major viral oncoproteins that contribute to carcinogenesis via altering cellular tumor suppressor pathways; E6 targets and degrades p53, while E7 targets and degrades retinoblastoma protein (pRb) [18,37,57,65,66]. The lesser characterized minor oncoprotein, HPV E5, appears to regulate cellular transformation, immune modulation, and response to cell signaling events [23,57,67]. While the expression of E6 and E7 extends the proliferative capacity of epithelial cells, fibroblasts have demonstrated a cooperative role in the induction of cell immortalization [15,68–71]. E5 has also demonstrated regulatory interactions as an innate immune suppressor in the adjacent stroma, thus contributing to viral persistence [22,23]. Of note, the viral DNA binding protein, E2, is not proposed to be oncogenic but has also been reported to be involved in the suppression of the innate immune response and is crucial for viral episome persistence [28,29,72–75].
Oncogene expression alone is considered insufficient for carcinogenesis, and other indeterminate events have been implicated in transformation [76]. During the HPV lifecycle, the viral genome exists in an episomal form in basal keratinocytes. Conversely, when aberrant HPV genome integration events occur, they have been noted as contributing factors in transformation; viral integration correlates with increased viral oncogene expression, loss of functional E2, cellular growth advantages, enhanced tumor progressiveness, cervical cancer progression, and poor clinical prognostics of HPV+OPC [25,27,59–61,77–85]. It is generally accepted that HPV+ keratinocyte cell lines must be grown in co-culture with fibroblasts to support viral episome maintenance [80,86,87]. HPV+ keratinocytes maintained in the absence of fibroblasts are noted to quickly integrate or lose viral genome expression [87,88]. From these observations, fibroblasts are influential on the HPV episomal status of adjacent keratinocytes, suggesting their role in regulating this transforming factor. The mechanisms of episomal regulatory control via fibroblasts have yet to be elucidated.
We previously reported the value of fibroblast co-culture both in the context of HPV episomal maintenance and as a model for better predicting in vitro to in vivo translational treatment paradigms [88]. In our previous analysis, we demonstrated that mitomycin C (MMC) growth-arrested murine 3T3-J2 fibroblasts (referred to as J2s moving forward) supported HPV16 long control region (LCR) transcriptional regulation [88]. We further investigated HPV protein expression and host protein signaling observed in the presence or absence of J2s [88]. N/Tert-1 cells (telomerase immortalized foreskin keratinocytes, HPV negative), HFK+E6E7 (foreskin keratinocytes immortalized by the viral oncogenes only), and HFK+HPV16 (foreskin keratinocytes immortalized by the entire HPV16 genome, replicating as an episome), were cultured in the presence or absence of J2s. We demonstrated that HFK+HPV16 maintained in J2 had measurable E7 protein levels; however, when J2s were removed for one week, E7 protein expression was lost [88]. Conversely, there were no significant alterations in E7 protein levels in HFK+E6E7 in the presence or absence of J2s, suggesting a partial reliance on the expression of the LCR or the full genome for the ability of fibroblasts to regulate viral protein expression [88]. Alterations in the protein levels of p53, pRb, and γH2AX were also demonstrated to be altered in the presence of J2 and further suggested fibroblasts may alter host protein expression that is supportive of HPV viral genome regulation [88].
In this report, we utilized RNA sequencing (RNA-seq) and proteomic analysis for a global and comprehensive approach to investigate keratinocyte signaling impacted by fibroblasts. Our investigation confirmed the prior observation, that HPV downregulates portions of innate immune signaling [23,28,29,89–91]. Further separation of keratinocytes grown in the presence or absence of J2s revealed the novel observation that fibroblasts impact the transformation potential of keratinocytes. N/Tert-1+HPV16 cells grown with J2s showed a gene regulation pattern similar to that of a suprabasal layer. Gene ontology (GO) analysis indicated that fibroblasts supported the viral life cycle, and that keratinocytes were less transformed compared to those grown without J2s. In contrast, N/Tert-1+E6E7 cells grown with J2s showed a greater tendency toward transformation than those grown without J2s, especially in relation to altered cell cycle regulation, and oncogenic cytokine expression. Proteomic analysis further supported these observations. Our results confirm that the expression of episomal HPV is necessary to regulate optimal viral-host interactions. Integration would mimic results observed in N/Tert-1+E6E7 cells, and the presence of fibroblasts promote a much more transformed genotype. Overall, our findings suggest that both monoculture and fibroblast co-culture approaches are useful for future studies on HPV-related transformation.
2. Materials and methods
2.1. Cell Culture
N/Tert-1 cells and all derived cell lines have been described previously and were maintained in keratinocyte-serum free medium (K-SFM; Invitrogen), and supplemented with previously described antibiotics [27–29,88,92–95].
2.2. Culture and mitomycin C (MMC) inactivation of 3T3-J2 mouse embryonic fibroblast feeder cells, and co-culture with keratinocytes
As previously described, 3T3-J2 immortalized mouse embryonic fibroblasts (J2) were grown in DMEM and supplemented with 10% FBS [88]. 80–90% confluent plates were supplemented with 4μg/ml of MMC in DMSO (Cell Signaling Technology) for 4–6 hours at 37°C. MMC-supplemented medium was removed and cells were washed with 1xPBS.
Cells were trypsinized, centrifuged at 800 rcf for 5 mins, washed once with 1xPBS, centrifuged again, and resuspended at 2 million cells per mL. Quality control of inactivation (lack of proliferation) was monitored for each new batch of mitomycin-C. Unless otherwise stated, 100-mm plate conditions were continually supplemented with 1×106 J2 every 2–3 days. Before trypsinization or harvesting, plates were washed to remove residual J2.
2.3. RNA isolation
The SV total RNA isolation system kit (Promega) was utilized to isolate RNA from cells, as per the manufacturer’s protocol.
2.4. Human Sequences RNA-seq Bioinformatics Pipeline
Library preparation, sequencing, and pre-processing of samples was performed by Novogene. Novogene uses in-house scripts to clean raw reads, filtering out low-quality reads, and reads containing adapter sequences. The genome index was built and cleaned sequences were aligned to the reference human genome using Hisat2 v2.05 [96,97]. Raw gene expression levels were quantified with featureCounts v1.5.0-p3 and then normalized to fragments per kilobase per million (FPKM) [98]. Differential expression analysis was performed using DESeq2 R package v1.20.0 between three experimental groups N/Tert-1, N/Tert-1+E6/E7, and N/Tert-1+HPV16 treated with J2 fibroblasts (n=3 in each group) and their paired controls respectively (untreated). P-values were adjusted using the Benjamini and Hochberg’s approach for controlling the false discovery rate (FDR), where significance for a differentially expressed gene was determined at FDR < 0.05 [99].
2.4. Gene Ontology Enrichment Analysis
GO enrichment analysis of differentially expressed genes was implemented by the clusterProfiler R package, in which gene length bias was corrected [100,101]. GO terms with corrected P-value < 0.05 were considered significantly enriched by differential expressed genes. Heatmaps were generated with the `pheatmap` R package using z-score normalized FPKM gene expression averages for each sample condition.
2.5. HPV16 sequences RNA-seq Bioinformatics Pipeline
Fastq files from Novogene were examined for quality using FastQC and quality control reports were collated by multiQC [102,103]. Reads were filtered to remove low quality sequences and adapter sequences were trimmed using trimmomatic v 0.39 [104]. A genome index was built and all sequences were aligned to the GRCh38.d1.vd1 Reference Sequence, part of the Genomic Data Commons GDC data harmonization pipeline, using STAR aligner v 2.7.9.a [105]. Samtools v1.16.1 was used to index and filter the bam file for reads aligned to HPV16 [106]. The HPV16 filtered bam files were converted back to fastq files using bedtools [107]. The HPV16 fastq sequences were re-aligned to an HPV16 reference genome from NCBI and raw gene expression levels were counted using featureCounts. Raw counts were then normalized using EdgeR’s calcNormFactors scaling factor of trimmed mean of M-values (TMM) normalization. EdgeR’s quasi-likelihood F-test (QLF) method was then used for differential expression analysis of each gene between three experimental groups N/Tert-1, N/Tert-1+E6/E7, and N/Tert-1+HPV16 treated with J2 fibroblasts (n=3 in each group) and their paired controls respectively (untreated) [108–110]. The p-value of each QLF test was adjusted using a Benjamini-Hochberg False Discovery Rate (FDR) multiple testing correction using the basic R stats package p.adjust function. Genes passing the FDR cut-off threshold of ≤ 0.05 for significance were considered statistically significantly different.
2.6. Real-time PCR (qPCR)
A high-capacity cDNA reverse-transcription kit from Invitrogen was used to synthesize cDNA from RNA and processed for qPCR. qPCR was performed on 10 ng of the cDNA isolated. cDNA and relevant primers were mixed with PowerUp SYBR green master mix (Applied Biosystems), and real-time PCR was performed using the 7500 Fast real-time PCR system, using SYBR green reagent. Expression was quantified as relative quantity over GAPDH using the 2−ΔΔCT method. Primer used are as follows. HPV16 E2 F, 5′-ATGGAGACTCTTTGCCAACG-3′; HPV16 E2 R, 5′-TCATATAGACATAAATCCAG-3′; HPV16 E6 F, 5′-TTGAACCGAAACCGGTTAGT-3′; HPV16 E6 R, 5′-GCATAAATCCCGAAAAGCAA-3′; MX1 F, 5′-GGTGGTCCCCAGTAATGTGG-3′; MX1 R, 5′-CGTCAAGATTCCGATGGTCCT-3′; STAT1 F, 5′-CAGCTTGACTCAAAATTCCTGGA-3′; STAT1 R, 5′-TGAAGATTACGCTTGCTTTTCCT-3′; STAT2 F, 5′-CCAGCTTTACTCGCACAGC-3′; STAT2 R, 5′-AGCCTTGGAATCATCACTCCC-3′; STAT3 F, 5′-CAGCAGCTTGACACACGGTA-3′; STAT3 R, 5′-AAACACCAAAGTGGCATGTGA-3′; p53 F, 5′-GAGGTTGGCTCTGACTGTACC-3′; p53 R, 5′-TCCGTCCCAGTAGATTACCAC-3′; Glyceraldehyde-3-phosphate dehydrogenase (GAPDH) F, 5′-GGAGCGAGATCCCTCCAAAAT-3′; GAPDH R, 5′-GGCTGTTGTCATACTTCTCATGG-3′.
2.7. Exo V
PCR based analysis of viral genome status was performed using methods described by Myers et al. [111]. 20 ng of genomic DNA was either treated with exonuclease V (RecBCD, NEB), in a total volume of 30 ul, or left untreated for 1 hour at 37°C followed by heat inactivation at 95°C for 10 minutes. 2 ng of digested/undigested DNA was then quantified by real time PCR, as noted above, using and 100 nM of primer in a 20 μl reaction. Nuclease free water was used in place of the template for a negative control. The following cycling conditions were used: 50°C for 2 minutes, 95°C for 10 minutes, 40 cycles at 95°C for 15 seconds, and a dissociation stage of 95°C for 15 seconds, 60°C for 1 minute, 95°C for 15 seconds, and 60°C for 15 seconds. Separate PCR reactions were performed to amplify HPV16 E6 F: 5’- TTGCTTTTCGGGATTTATGC-3’ R: 5’-CAGGACACAGTGGCTTTTGA-3’, HPV16 E2 F:5’-TGGAAGTGCAGTTTGATGGA-3’ R: 5’- CCGCATGAACTTCCCATACT-3’, human mitochondrial DNA F: 5’-CAGGAGTAGGAGAGAGGGAGGTAAG-3’ R: 5’-TACCCATCATAATCGGAGGCTTTGG −3’, and human GAPDH DNA F: 5’-GGAGCGAGATCCCTCCAAAAT-3’ R: 5’- GGCTGTTGTCATACTTCTCATGG-3’
2.8. Proteomic sample preparation
The samples were digested using commercially available PreOmics iST sample clean up protocol. To the sample containing approximately 100ug of protein, 70ul of lysis buffer was added and mixed, followed by an incubation for 10 minutes at 950C; 1000rpm. 50ul of DIGEST solution was added to the mixture, which was then incubated at 370C for 3hrs at 500 rpm. After the digestion, 100ul of STOP solution was added and mixed properly. The digest was then centrifuged at 3800rcf; 3min to ensure complete flow through and washed with 200ul of WASH 1 and 200ul of WASH 2 solution followed by centrifugation after each wash. The cartridge was then placed to the fresh collection tube and 100ul of ELUTE solution was added and centrifuged at 3800rcf; 3min to ensure complete flow through. This step was repeated one more time to ensure maximum recovery. The elutes were then placed in a vacuum evaporator at 450C until completely dried.
2.9. LC-MS/MS.
LC-MS/MS analysis were performed using a Q-Exactive HF-X (Thermo) tandem mass spectrometer coupled to an Easy nLC 1200 (Thermo) nanoflow UPLC system. The LC-MS/MS system was fitted with an Easy spray ion source and an Acclaim PepMap 75μm x 2cm nanoviper C18 3μm x 100Å pre-column in series with an Acclaim PepMap RSLC 75μm x 50cm C18 2μm bead size (Thermo). The mobile phase consists of Buffer A (0.1% formic acid in water) and Buffer B (80% acetonitrile in water,0.1% formic acid). 500ng of peptides were injected onto the above column assembly and eluted with an acetonitrile/0.1% formic acid gradient at a flow rate of 300 nL/min over 2 hours. The nano-spray ion source was operated at 1.9 kV. The digests were analyzed using a data dependent acquisition (DDA) method acquiring a full scan mass spectrum (MS) followed by 40 tandem mass spectra (MS/MS) in the high energy C-trap Dissociation HCD spectra). This mode of analysis produces approximately 50,000 MS/MS spectra of ions ranging in abundance over several orders of magnitude. Not all MS/MS spectra are derived from peptides.
2.10. Proteomic Data Analysis
The data were analyzed in Proteome Discoverer (ver 3.0) using the Sequest HT search algorithm and the Human database. Proteins were identified at an FDR < 0.01 and quantification used the peptide intensities. Raw protein abundances were normalized in Proteome Discoverer using the “Total Peptide Abundance” method. Differential Enrichment of protein abundance was performed using the `DEP` package v. 1.26 [112]. First, we filtered for proteins detected in two of three replicates of at least one of the experimental conditions. Variance stabilizing transformation of remaining protein intensity observations was performed using the `vsn` package v 3.72 via the ǹormalize_vsn` function [113]. The quantile regression-based left-censored (QRILC) method was used as the missing value imputation approach. The differential enrichment test was conducted pairwise on each protein using limma v 3.60.4 between three experimental groups N/Tert-1, N/Tert-1+E6/E7, and N/Tert-1+HPV16 treated with J2 fibroblasts (n=3 in each group) and their paired controls (untreated), respectively [114]. Proteins were identified as significantly differentially expressed between the control and experimental groups with a Benjamini-Hochberg adjusted p-value of < 0.05, and a |log2-fold change| > 0.58.
2.11. Immunoblotting
Cells were trypsinized, washed with PBS and resuspended in 2x pellet volume NP40 protein lysis buffer (0.5% Nonidet P-40, 50 mM Tris [pH 7.8], 150 mM NaCl) supplemented with protease inhibitor (Roche Molecular Biochemicals) and phosphatase inhibitor cocktail (MilliporeSigma). Cell suspension was incubated on ice for 20 min and then centrifuged for 20 min at 184,000 rcf at 4 °C. Protein concentration was determined using the Bio-Rad protein estimation assay according to manufacturer’s instructions. 50 μg protein was mixed with 2x Laemmli sample buffer (Bio-Rad) and heated at 95 °C for 5 min. Protein samples were separated on Novex 4–12% Tris-glycine gel (Invitrogen) and transferred onto a nitrocellulose membrane (Bio-Rad) at 30V overnight using the wet-blot transfer method. Membranes were then blocked with Odyssey (PBS) blocking buffer (diluted 1:1 with PBS) at room temperature for 1 hr. and probed with indicated primary antibody diluted in Odyssey blocking buffer, overnight. Membranes were washed with PBS supplemented with 0.1% Tween (PBS-Tween) and probed with the Odyssey secondary antibody (goat anti-mouse IRdye 800CW or goat anti-rabbit IRdye 680CW) (Licor) diluted in Odyssey blocking buffer at 1:10,000. Membranes were washed twice with PBS-Tween and an additional wash with 1X PBS. After the washes, the membrane was imaged using the Odyssey® CLx Imaging System and ImageJ was used for quantification, utilizing GAPDH as internal loading control. Primary antibodies used for western blotting studies are as follows: pRb 1:1000 (Santa Cruz, sc-102), p53 1:1000 (Cell Signaling Technology, CST-2527, and CST-1C12), γH2AX 1:500 (Cell Signaling Technology, CST-80312 and CST-20E3).
2.12. Reproducibility, research integrity, and statistical analysis
All experiments were carried out at least in triplicate in all of the cell lines indicated. Keratinocytes were typed via cell line authentication services. All images shown are representatives from triplicate experiments. Student’s t-test or analysis of variance was used to determine significance as appropriate: *P < 0.05, **P < 0.01, ***P < 0.001.
3. Results
3.1. Differential Genomic Landscapes altered by fibroblasts in keratinocytes
The utility of a supportive fibroblast feeder layer is broadly accepted as essential for maintaining an episomal HPV genome in primary keratinocyte models, and is a necessary component of 3D models for HPV lifecycle analysis where it is chiefly responsible for proper keratinocyte differentiation [57,68,77,87,88,115–122]. While the coculture of keratinocytes with fibroblast feeders is accepted, the full mechanism of how fibroblasts aid in HPV episomal maintenance has yet to be deciphered. It is worth noting that 2D coculture may represent interactions that occur in the basal layer, while far more complex spatial and temporal regulatory mechanisms are likely involved in 3D models and in vivo. This analysis focuses on short-term 2D interactions, with the aim of investigating 3D models in the future.
We previously demonstrated that fibroblast co-culture was important for maintaining HPV episomes, influenced HPV16 LCR transcriptional regulation, and supported the expression of HPV16 E7 protein in human foreskin keratinocytes immortalized with HPV16 (HFK+HPV16) [88]. We also observed that fibroblasts altered host protein levels which could affect viral genome regulation [88]. Taking a more global approach to investigate signaling impacted by fibroblasts, N/Tert-1, N/Tert-1+E6/E7, and N/Tert-1+HPV16 cells were cultured in the presence or absence of J2s for one week. These matched samples were then subjected to bulk RNA-seq analysis, and label-free liquid chromatography-mass spectrometry-based proteomic analysis (LC-MS/MS).
For RNA-seq, triplicate sample data were combined to assess differential gene expression analysis. Initial comparisons were made in large batched sets; cell lines were either not separated based on the presence or absence of J2, or grouped as all mono-culture vs all co-culture. They were compared in the following large sets: N/Tert-1 vs N/Tert-1+HPV16, N/Tert-1+E6E7 vs N/Tert-1+HPV16, N/Tert-1 vs N/Tert-1+E6E7, and monoculture control vs co-culture “+J2”. Evaluations of datasets were then further compared based on the presence or absence of J2 in each individual N/Tert-1, N/Tert-1+E6E7, or N/Tert-1+HPV16 cell line and cross-compared. Our data revealed numerous genes significantly differentially expressed 1.5 fold or greater when cross-comparing our samples (DEG gene counts presented in Figure 1A, Quantitative correlation presented in Figure 1B). A full list of these genes can be found in Supplementary Material S1. The expression level of the HPV16 genes used to generate the gene expression data is given in Supplementary Table S2. Novogene and further bioinformatic analysis identified the most affected canonical pathways, upstream regulators, diseases, and functions predicted to be altered in this data set; significant observations are given in Supplementary Tables S3. The most notable HPV differential expression and GO enrichment observations were alterations in innate immune signaling, including altered cytokine and chemokine activity; additional alterations in cellular communication potential, tight junction regulation, and growth factor signaling events were also differentially regulated (GO enrichment plots summarized in Figures 2A-C). When grouped as a whole, fibroblasts significantly altered GO enrichment associated with angiogenesis, differentiation, extracellular matrix organization, and both cytokine and growth factor-related activity (Figure 2D).
Figure 1. Global comparison of RNA-seq.
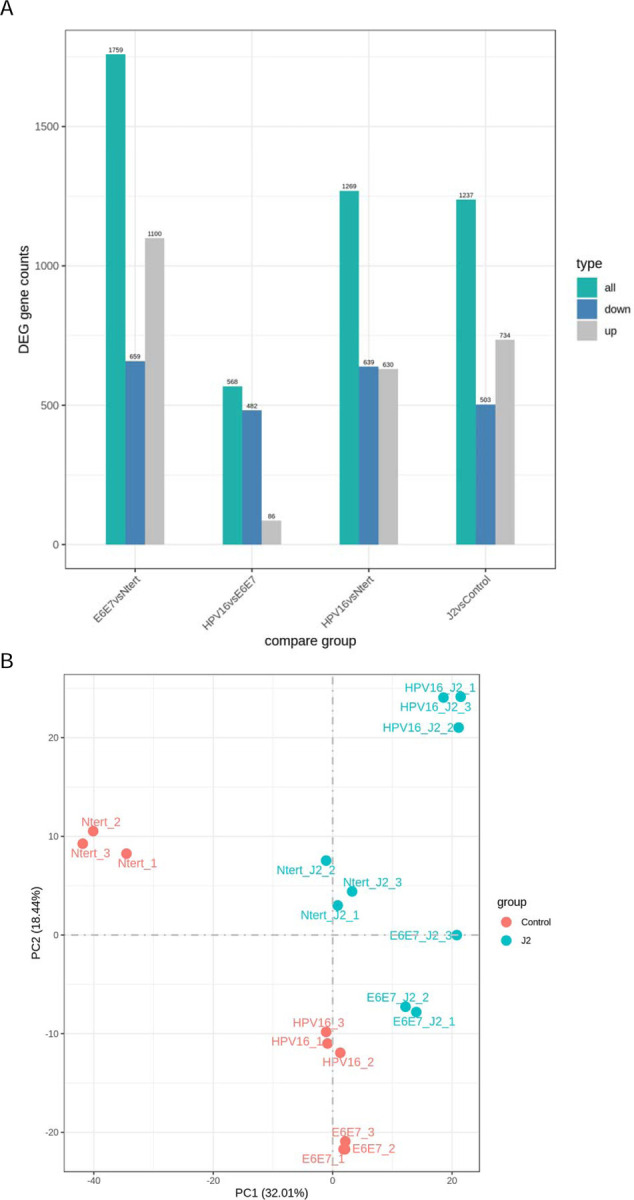
1A. RNA-seq differential expression (DEG) analysis histogram comparison of the number of significant differential genes (including up-regulation and down-regulation) for each combination. 1B. Principal component analysis (PCA) analysis on the gene expression value (FPKM) of all samples.
Figure 2. Gene ontology (GO) enrichment analysis histograms demonstrate differential regulation between N/Tert-1 cell lines and between mono vs co-culture.


The 30 most significantly GO terms are displayed. All Terms are separated according to major categories of biological processes (BP), cell components (CC), molecular functions (MF) and categories of upregulated and downregulated expression of noted GO. 2A. Grouped N/Tert-1+HPV16 are compared to Grouped N/Tert-1. 2B. Grouped N/Tert-1+HPV16 are compared to Grouped N/Tert-1+E6E7. 2C. Grouped N/Tert-1+E6E7 are compared to Grouped N/Tert-1. 2D. Grouped fibroblast co-culture cell line sets (J2) are compared to Grouped mono-culture cell line sets (Control).
As previously reported, numerous gene sets related to interferon (IFN) response were significantly reduced in the N/Tert-1+HPV16 group, over that of both N/Tert-1 and N/Tert-1+E6E7 groups (Figures 2A-B) [28,123]. Of note, fibroblasts were not utilized when preparing our N/Tert-1-related cultures in previous RNAseq analysis [28,29]. Various interleukins and CXCL family members were also significantly downregulated in grouped N/Tert-1+HPV16 when compared to grouped N/Tert-1 and N/Tert-1+E6E7 (Figures 2A-B). Reactome enrichment further highlighted the following genes concerning the aforementioned significantly downregulated networks: BST2, CREB5, CSF1, CX3CL1, CXCL1, CXCL2, CXCL3, IFI27, IFI35, IFI6, IFIT1, IFITM1, IFITM3, IL18R1, IL6, IRF7, ISG15, HLA-B, LIF, MMP9, MX1, MX2, OAS1, OAS2, OAS3, PIK3R3, PTAFR, RIPK3, RSAD2, SAMHD1, STAT1, TRIM22, UBE2L6, USP18, XAF1 (Supplemental Tables S3). The observation that HPV downregulates innate immune functions is not novel, but highlights the consistency of our observations with others [23,28,29,89–91].
Several interesting significant alterations in GO enrichment were observed when N/Tert-1 cell lines were further separated based on the presence or absence of J2. N/Tert-1+HPV16 continuously maintained in J2 co-culture demonstrated significant upregulation of interleukin antagonist genes and genes related to inflammation and cell motility, while expression of IFN-induced genes remained downregulated (Figures 3A-J). Genes related to B-cell recruitment and the compliment pathway, also were enriched in N/Tert-1+HPV16 maintained in J2 (Figures 3A,C). The GO enrichment of N/Tert-1+E6E7 in the presence or absence of J2, in comparison to N/Tert-1+HPV16 in the presence or absence of J2, was markedly different. N/Tert-1+E6E7 grown in the presence of J2 exhibited the most significant increase in GO enrichment of genes related to IFN, indicating that the expression of the full viral genome is necessary for their repression (Figures 3A-J). This would correspond to observations that both E2 and E5 have been tied to the regulation of innate immunity [23,28,29]. While IFN is known to regulate viral infections, IFN-mediated activation of the Janus kinase (JAK)-signal transducer activator of transcription (STAT) has also been associated with cancer progression, including HPV+ cervical cancer [124,125]. Specifically, HPV oncoproteins have previously been shown to activate JAK/STAT [125]. GO enrichment, and qPCR validation demonstrate that N/Tert-1+E6E7 cells cocultured with fibroblasts, markedly upregulate STAT1,2,3 expression; in comparison, N/tert-1+HPV16 keratinocytes cocultured with fibroblasts have significantly lower expression of these genes (Figures 3E-H).
Figure 3. Fibroblasts differentially regulate GO enrichment in relation to innate immune function.

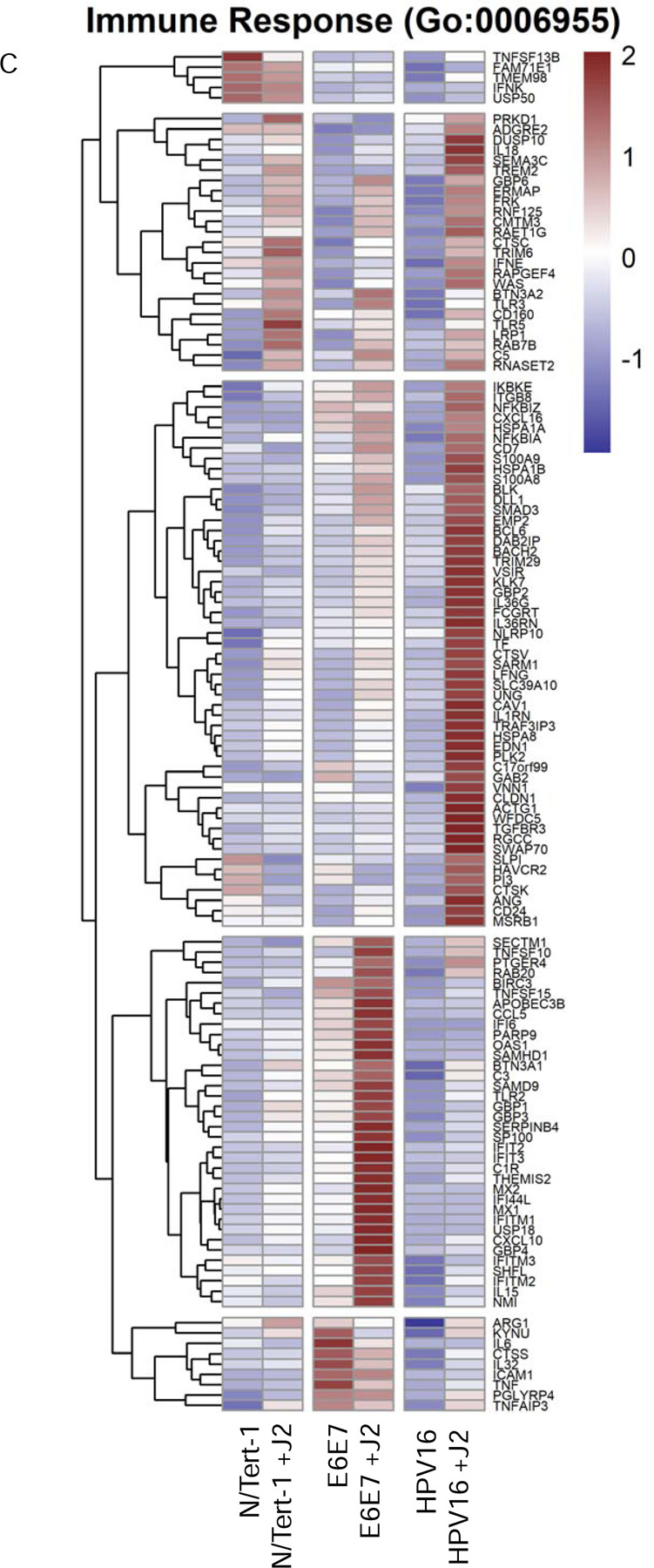
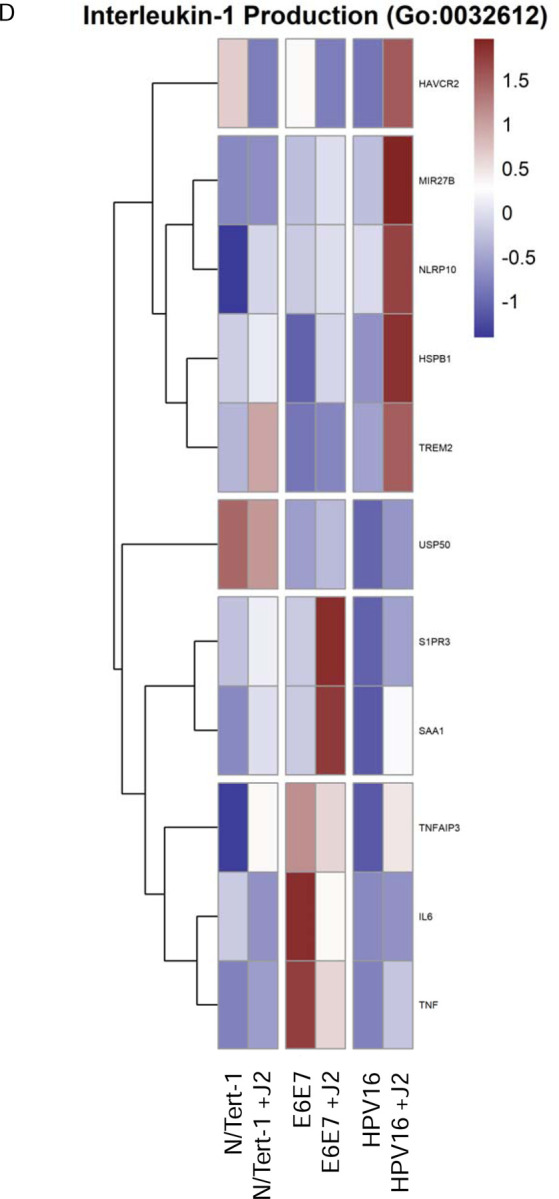


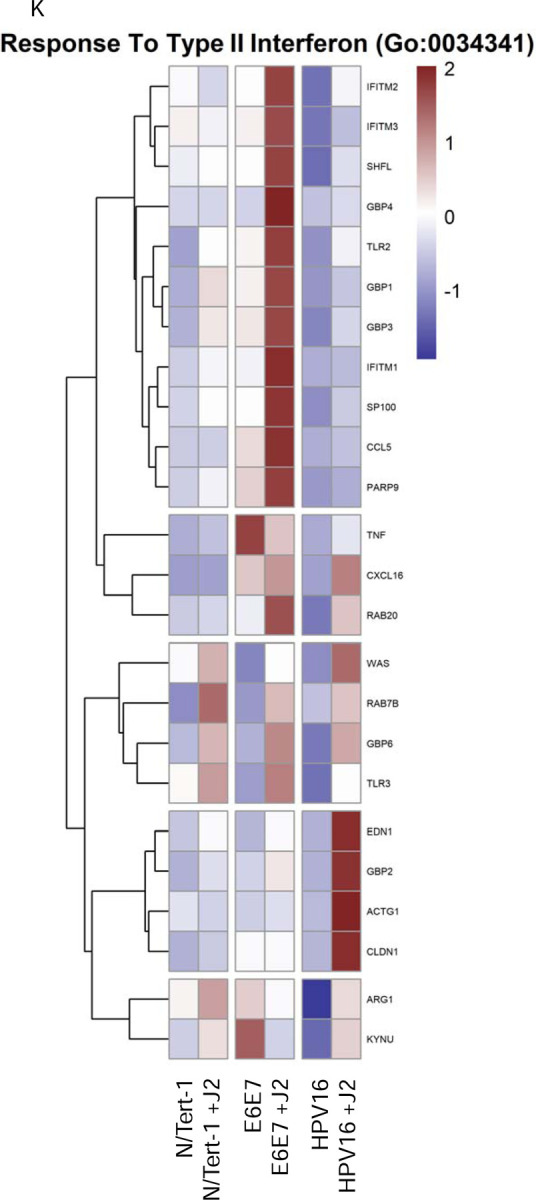
3A. Heat map demonstrating significant GO:0045087 innate immune regulation across all groups. 3B. qPCR validation of MX1 RNA expression, presented in log scale. 3C. Heat map demonstrating significant GO:0006955 innate immune response across all groups. 3D. Heat map demonstrating significant GO:0032612 interleukin-1 production across all groups. 3E. Heatmap demonstrating significant STAT RNA expression across all groups. 3F. qPCR validation of STAT1 RNA expression, presented in log scale. 3G. qPCR validation of STAT2 RNA expression, presented in log scale. 3H. qPCR validation of STAT3 RNA expression. 3I. Heat map demonstrating significant GO:0035456 response to interferon beta across all groups. 3J. Heat map demonstrating significant GO:0034340 response to type I interferon across all groups. 3K. Heat map demonstrating significant GO:0034341 response to type II interferon across all groups.
Another noteworthy observation in our GO enrichment cross-comparison, was the alterations observed in genes related to cell junctions, particularly with tight junctions (TJs) and cell-cell signaling control (Figure 4). TJs are comprised of a complex group of molecules, and are associated with the suprabasal and intermediate layers of epithelia. While numerous TJ proteins are downregulated in the transformation process, others are overexpressed and mislocalized [126,127]. Such dysregulation of TJ proteins is associated with epithelial-to-mesenchymal transition (EMT) and invasive phenotypes, including in HPV+ cervical cancer and HPV16 E7 has been shown to alter the expression and localization of TJ-associated claudins [127–129]. Twist1 is also associated with EMT; its transcriptional activation of Claudin-4 has been shown to promote cervical cancer migration and invasion [130–132]. Our analysis shows partial upregulation of TJ components in E6E7+ cells by coculture with fibroblasts, and a significant upregulation in HPV16+ keratinocytes (Figures 4A,C). In particular, there was a marked increase in TJ assembly proteins in both cell lines, including claudins, which are crucial to tight junction integrity (Figure 4A,C). Here, we suggest that this is a model for stages of transformation. The decreased expression of junctional proteins seen in N/Tert-1+E6E7 is more analogous to later, neoplastic stages of transformation; when the viral genome is integrated, E6E7 is overexpressed and there is a progression towards EMT. Meanwhile, the increased expression of TJ components in HPV16+ keratinocytes cultured with fibroblasts is analogous to early viral lifecycle stages. Furthermore, by inducing increased levels of TJ components in infected keratinocytes, the virus induces an environment that mimics a suprabasal phenotype, which is important for the amplification stage of the viral lifecycle [82,118,133]. As large complexes, TJs facilitate signal transduction and are involved in cell proliferation, migration, differentiation, and survival, all of which are beneficial to the viral lifecycle [134]. The comparison to E6E7+ keratinocytes indicates that the upregulation of junctional proteins seen in HPV16+ cells is likely driven by other viral factors, possibly E2, although this warrants further investigation. It would be interesting to further dissect the impact of keratinocyte-fibroblast co-culture upon the subcellular localization of these TJ components and any resulting downstream effects on cell invasive capacity in both E6E7+ and full-genome containing cell lines.
Figure 4. Fibroblasts differentially regulate GO enrichment in relation to cell signaling and epithelial-to-mesenchymal (EMT) progression.

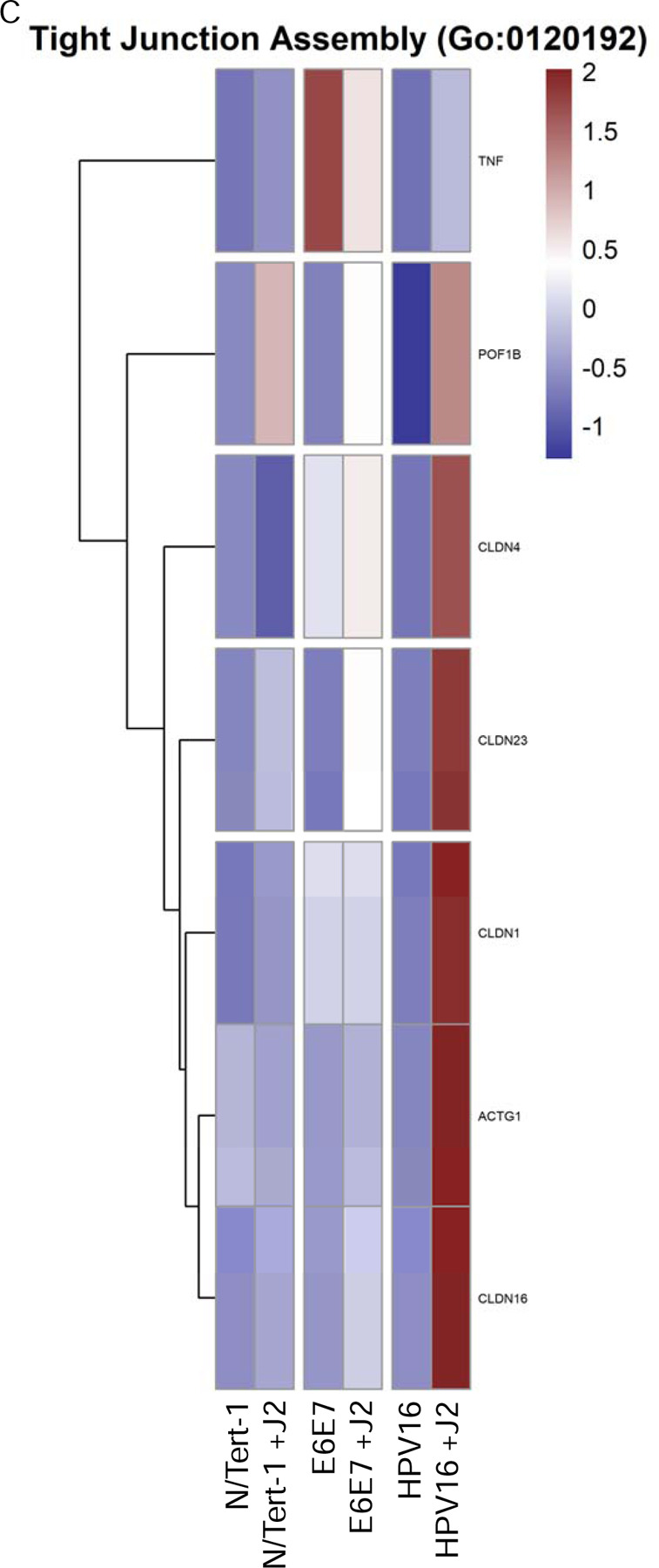
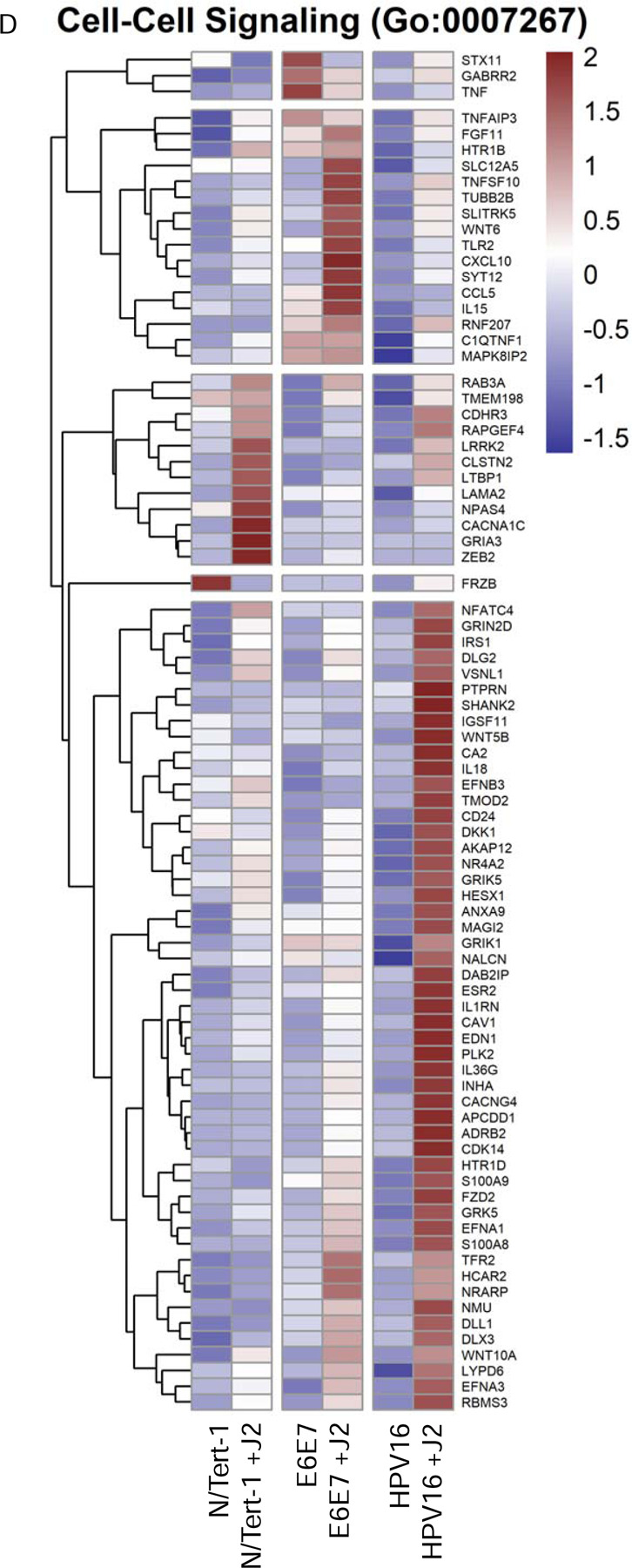

4A. Heat map demonstrating significant GO:0098609 cell-cell adhesion across all groups. 4B. Heatmap demonstrating significant TWIST RNA expression across all groups. 4C. Heat map demonstrating significant GO:0120192 tight junction assembly across all groups. 4D. Heat map demonstrating significant GO:0007267 cell-cell signaling across all groups. 4E. Heat map demonstrating significant GO:0033209 TNF across all groups. 4F. Heat map demonstrating significant CXC chemokines across all groups.
Chemokines are small molecules and secretory peptides are associated with cellular signaling and are broadly divided into subfamilies based on their amino acid motifs: XC, CC, CXC, and CXXXC [135,136]. Chemokine ligands, work jointly with specific chemokine receptors, to control a broad range of biological processes [135,136]. CXC family members are further divided into ELR+ and ELR- members, based on the presence or absence of a Glu-Leu-Arg (ELR) motif in their N-terminus [135]. ELR+ CXC chemokines are associated with the progression of cancer, conversely downregulation of these has been found to suppress the motility of cancer [135]. On the other hand, ELR- CXC chemokines are associated with tumor-suppressive effects [135]. Chemokine-related GO enrichment observed in N/Tert-1+HPV16 grown in the presence of J2 was highly indicative of a less tumorigenic genotype (Figure 4F). This suggests that fibroblasts are likely playing a role in preventing the transformation of HPV+ keratinocytes. Moreover, GO enrichment of TWIST expression (Figure 4B) demonstrated that N/Tert-1+HPV16 grown in the presence of J2 is indicative of a less transformed genotype [132,137,138]. CXC-related signaling is known to impact EMT and cancer progression via interactions with β-catenin, TNF, and Notch/Wnt signaling [135,136,139–142]. While these signaling pathways can have both tumor-promoting and suppressive roles that are cancer-dependent, it is clear that fibroblasts are altering the GO enrichment of N/Tert-1+HPV16 grown in the presence of J2, and this has implications in the mechanism of HPV16-driven carcinogenesis (Figure 4).
As we previously observed protein alterations in p53, pRb, and γH2AX in our human foreskin (HFK) cell lines, we also confirmed this trend via western blotting in the N/Tert-1 lines used for this analysis, and assessed GO enrichment in relation to these [88]. Again, fibroblasts enhanced p53 and γH2AX protein expression in all N/Tert-1 lines, while pRb was enhanced in N/Tert-1 and N/Tert-1+E6E7 (Figure 5A). GO enrichment revealed that TP53 was not enhanced at the RNA expression level, indicating that fibroblast enhancement of p53 protein expression, is likely mediated at the level of translation, post-translation, or protein stability, however, some p53 inducible proteins did appear to be regulated at the level of RNA (GO enrichment Figure 5B, p53 qPCR time course validation 5C-E) [88]. TP53I13, TP53TG1, and TP53TG5 overexpression have been linked to the inhibition of cell proliferation and tumor suppression [143–145]. Enhancement of these tumor suppressors in N/Tert-1+HPV16 grown in the presence of J2, again suggests that fibroblasts promote a less transformed genotype (Figure 5B). GO enrichment related to Rb signaling is less clear. However, the observed RB1, RBL1, RB1CC1, RBBP4P1 RNA upregulation (Figure 5F) in N/Tert-1+HPV16 grown in the presence of J2, is suggestive of a less transformed genotype [146–149]. H2AX RNA upregulation was demonstrated in both N/Tert-1+E6E7 and N/Tert-1+HPV16 grown in the presence of fibroblasts (Figure 5G), indicating a partial role in the previously observed J2 enhancement of γH2AX protein (the phosphorylated form of the H2AX variant) [88].
Figure 5. Fibroblasts differentially regulate p53, pRb, and histone related expression.


5A. N/Tert-1 (lanes 1,2) N/Tert-1+E6E7 (lanes 3,4), N/Tert-1+HPV16 (lanes 5,6) cells were seeded on day 0 and grown in the presence or absence of J2s for 1 week. Cells were washed to remove J2s in noted conditions, trypsinized, lysed, and analyzed via western blotting for pRb, p53, and γH2AX. GAPDH was utilized as a loading control. 5B. Heat map demonstrating significant p53 GO enrichment all groups. 5C. N/Tert-1, 5D. N/Tert-1+E6E7, and 5E. N/Tert-1+HPV16 were grown in the presence or absence of J2s for 3 weeks. Time course of p53 RNA is presented at fold control of day 1. 5F. Heat map demonstrating significant pRb RNA enrichment all groups. 5G. Heat map demonstrating significant histone RNA enrichment in all groups.
Another significant observation from our GO enrichment cross-comparisons were alterations in genes associated with cell cycle regulation and progression (Figure 6). Cell cycle regulation and progression are notably altered during oncogenic transformation and HPV-related transformation [1,150–152]. N/Tert-1+E6E7 cells cocultured with fibroblasts, markedly upregulated GO enrichment related to cell cycle regulation, cell cycle progression, cell division, and mitotic progression; these alterations were highly suggestive of significant transformation (Figures 6A-G)[153–155]. Conversely, N/Tert-1+HPV16 grown in the presence of J2 upregulated GO enrichment in tissue development that was highly suggestive of a less transformed genotype (Figure 6H). In particular, the expression of KRT4 and KRT13 decreases in transformed epithelial cells; N/Tert-1+HPV16 grown in the presence of J2 instead showed enhanced KRT13 and KRT4 levels [156]. Likewise, HPV16+ keratinocytes maintained in J2 exhibited enhanced stress response GO enrichment, including the upregulation of a number of genes related to tumor suppression (Figure 6I). Again, highlighting the ability of fibroblasts to differentially regulated transformative genotypes.
Figure 6. Fibroblasts differentially regulate cell cycle, tissue development, and stress response related GO enrichment.

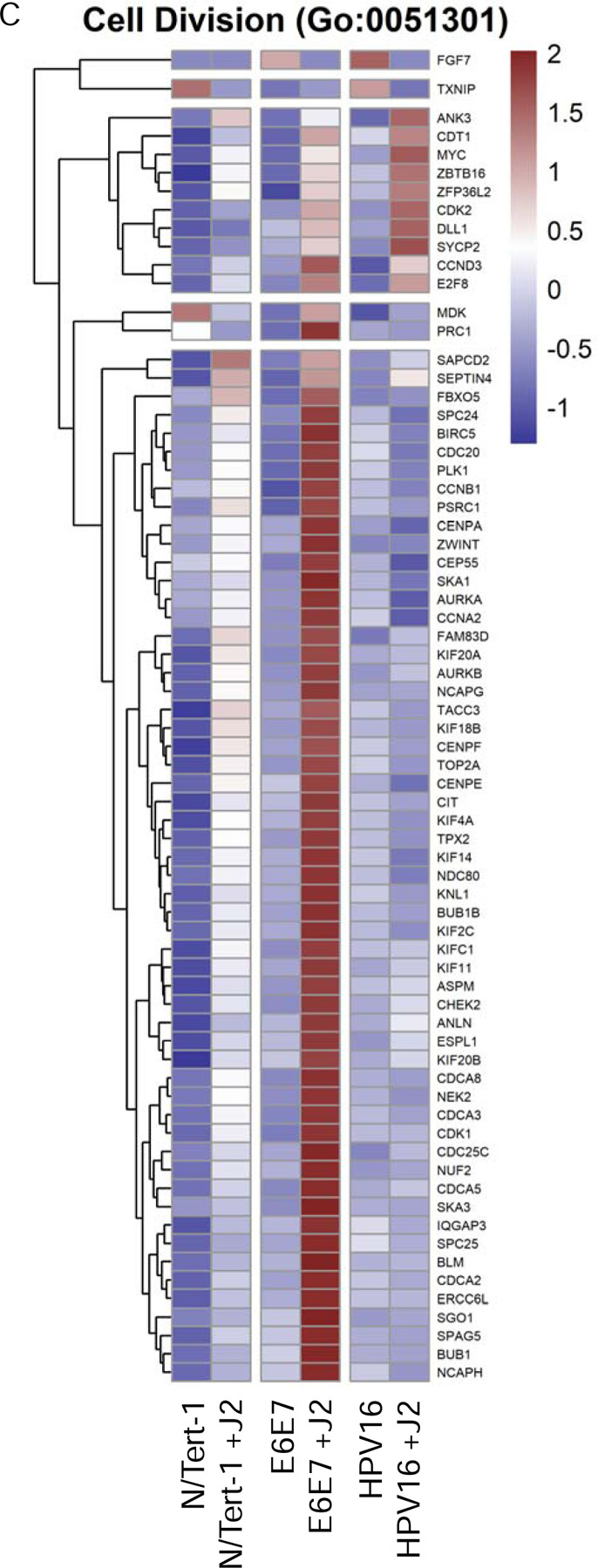



6A. Heat map demonstrating significant GO:0022402 cell cycle progression across all groups. 6B. Heat map demonstrating significant GO:0007049 cell cycle across all groups. 6C. Heat map demonstrating significant GO:0051301 cell division across all groups. 6D. Heat map demonstrating significant GO:1903047 mitotic cell cycle progress across all groups. 6E. Heat map demonstrating significant GO:0000278 mitotic cell cycle across all groups. 6F. Heat map demonstrating significant GO:0010564 regulation of cell cycle process across all groups. 6G. Heat map demonstrating significant GO:0051726 regulation of cell cycle across all groups. 6H. Heat map demonstrating significant GO:0009888 tissue development across all groups. 6I. Heat map demonstrating significant GO:0006950 response to stress across all groups.
3.2. Differential HPV RNA Expression Altered by Fibroblasts in Keratinocytes
We and others have demonstrated the importance of fibroblast co-culture for viral episome maintenance in HPV+ keratinocytes [87,88,122]. As previously demonstrated in HFK+HPV16, N/Tert-1+HPV16 grown in the presence of fibroblasts for one week demonstrated significantly enhanced integration events in the absence of J2 (Figure 7A) [88]. Mining of viral reads from RNA-seq data was performed and interpreted utilizing a technique previously developed [17,157,158]. RNA differential expression analysis demonstrated that N/Tert-1+HPV16 grown in the presence of J2 had significantly higher levels of E2, E5, E6, and E7 transcripts than cells grown in the absence of J2 (RNA-seq reads in Figure 7B, E2, and E6 qPCR time course validation in Figures 7C and 7D, respectively). Alternatively, N/Tert-1+E6E7 grown in the presence of J2 expressed lower RNA transcripts of E7, and similar E6 transcripts in comparison to cells grown in the absence of J2 (RNA-seq reads in Figure 7B and E6 qPCR time course validation in Figure 7E).
Figure 7. Fibroblasts support viral RNA expression and episomal maintenance in HPV+keratinocytes.

7A. N/Tert-1+HPV16 cells were grown in the presence or absence of J2s for 1 week. Cells were washed to removed J2, then lysed and analyzed for DNA expression of E2 and E6 via the exonuclease V assay, in comparison to GAPDH and mitochondrial DNA controls. Results are presented as percent integration as calculated from the cut ratio of matched GAPDH. **P < 0.01. 7B. Differential expression data from RNAseq from average normalized reads of E6, E7, E2, and E5 matched to HPV reference genome. Exact significance is presented for each (student’s t-test), NS represents no significance. 7C-E. qPCR time course validation of E2 and E6 RNA expression in N/Tert-1+E6E7 and N/Tert-1+HPV16 in the presence or absence of J2 for 3 weeks, 7D is presented in log scale. *P < 0.05. **P < 0.01.
3.3. Differential Proteomic Landscapes Altered by Fibroblasts in Keratinocytes
For label-free LC-MS/MS proteomic comparison, matched triplicate samples were harvested at the same time as RNA-seq; differential protein expression and bioinformatic analysis was performed, cross-matched to RNA-seq, and further assessed by bioinformatics. Processed datasets are available in Supplementary Data S4. Exact comparative analysis is presented as Venn diagrams in Figure 8 and comparative heatmaps in Figure 9. While mRNA expression precedes protein translation, the exact correlation between transcript levels and protein abundance is often poor; correlative assessments can instead be utilized for biomarker trends [159–162]. The Human Protein Atlas was first consulted to assess if comparative analysis supported our RNAseq observations that fibroblasts regulate the transformation potential in HPV+ keratinocytes [163–165]. Many oncogenic proteins were significantly downregulated in N/Tert-1+HPV16 cells grown in the presence of J2; clinical pathology observations have proven that high expression of these proteins correlates with poor prognostics in either cervical cancer and/or head and neck cancer [163–165]. Fibroblast downregulation of these markers in N/Tert-1+HPV16 is suggestive of less transformation, which is in agreement with the observed changes in EMT markers in the RNA analysis. Global profiling of trends confirmed differentially regulated subgroups in relation to transformation events. Our overall observations suggest that fibroblasts influence genotypic profiles that support the viral lifecycle while inhibiting oncogenic progression in HPV+ keratinocytes. This fibroblast regulation pattern is inversed in E6E7+ keratinocytes, where oncogene expression is outside the control of E2.
Figure 8. Differential expression Venn diagrams comparing significant up or down regulation via fibroblasts in RNA-seq and proteomic analysis.

The sum of all the numbers in the circle represents the total number in the compared groups, and the overlapping area indicates the number of differential genes shared between the groups, as shown in the following figures. 8A,B. Cross comparison of N/Tert-1 downregulation, and upregulation, respectively via fibroblasts. 8C,D. Cross comparison of N/Tert-1+E6E7 downregulation, and upregulation, respectively via fibroblasts. 8E,F. Cross comparison of N/Tert-1+HPV16 downregulation, and upregulation, respectively via fibroblasts.
Figure 9. RNA-seq and proteomic cross comparisons demonstrate fibroblasts differentially regulate GO enrichment in relation to innate immune function and cell-cell adhesion.


9A. Heat map demonstrating significant GO:0006955 immune response across all groups. 9B. Matched heat map analysis of significant proteome alterations of GO:0006955 across all groups. 9C. Heat map demonstrating significant GO:0098609 cell-cell adhesion across all groups. 9D. Matched heat map analysis of significant proteome alterations of GO:0098609 across all groups. Dotted lines are added to help visually compare similar matched sets.
4. Discussion
Decades of research have continued to improve the model systems utilized to mimic HPV infection and progression. Despite the increasing availability of improved models, a current challenge in the field is that these disease models still do not fully replicate the tissue complexity of the various epithelial sites where severe diseases develop [24,166]. The addition of fibroblast feeder cells for the generation of epithelial cell lines has improved both the efficiency of immortalization attempts, as well as contributing to tissue complexity in 2D growth settings [69,70]. Primary keratinocyte lines are easily generated for many epithelial sites of HPV infections, however primary cell lines do not allow for longitudinal studies [167]. Primary cultures can be immortalized with HPV; however, “control” cell lines are limited due to the nature of primary cell culture. Immortalized primary human keratinocytes using the catalytic subunit of telomerase (hTERT) have been generated for use as longitudinal “control” cell lines, however expression of hTERT alone is often insufficient for the immortalization of human keratinocytes [168]. Successfully immortalized keratinocyte lines like telomerase (hTERT) immortalized primary foreskin keratinocytes (N/Tert-1), the spontaneously immortalized normal immortal keratinocytes (NIKS), or the adult epidermis cell line generated from the periphery of a malignant melanoma (HaCaT) are thus utilized as surrogates for long term “control” comparisons [168,169]. HPV E6 and E7 can likewise be exploited to immortalize keratinocytes with improved efficiency, however, they are no longer completely null of HPV [57,71,170]. To assess how fibroblasts modulate viral-keratinocyte interactions, we carefully evaluated the most effective approach to control for all relevant factors. For this reason, we chose to utilize our well-characterized and matched N/Tert-1 keratinocyte lines [28,29,74,88,95,171].
Genomic and proteomic assessments in short-term 2D cultures revealed that fibroblasts promoted a less transformed state in N/Tert-1+HPV, whereas N/Tert-1+E6E7 may be more transformed in the presence of fibroblasts. The exact nature of oncogenic transformation remains largely speculative, although a number of biomarkers are well characterized in this progression [46,57,60,66,77,122,129,131,170,172]. Our studies confirmed that N/Tert-1+HPV maintained in fibroblasts sustained HPV episomes, consistent with a less progressed HPV genotypic state (Figure 7A) [77,79,80,88,158]. Likewise, host expression of host signaling regulation, was also suggestive of a less transformed state; specifically tight junction regulation, CXC chemokine expression, TNF-related signaling, and TWIST expression were most compelling (Figure 4). Conversely, when comparing the signaling regulation of N/Tert-1+E6E7 maintained in fibroblasts, the genotypic regulation presented the biological antithesis of the aforementioned observations (Figure 4). Additionally, N/Tert-1+E6E7 maintained in fibroblasts exhibited significant enhancement of cell cycle regulation that was suggestive of transformation (Figure 6). True longitudinal HPV transformation has yet to be demonstrated in traditional cell culture; our observations suggest that alterations in cell culture maintenance conditions are worth consideration for future analysis.
Organotypic raft cultures have also been used for the broad examination of how high-risk HPVs may drive neoplasia and cancer [166]. It is well noted that fibroblasts serve a fundamental role in epithelial differentiation and the viral lifecycle in this 3D model [23,43,44,173–175]. While 3D cultures present a model for reconstructing the viral lifecycle, these cultures are not useful for traditional cell maintenance. Likewise, 2D culture models can also be utilized to examine the viral lifecycle employing a calcium gradient medium, but differentiation also presents finite time points [166]. Future studies in our lab will extrapolate the transformation-related alterations presented, and assess how fibroblasts continue to regulate viral-host interactions temporally, spatially, and in the context of differentiation. These alterations will be considered at various stages of transformation, in 2D and 3D models, and in the context of both normal and cancer-associated fibroblasts.
5. Conclusion
Both our research and that of others have shown that interactions between fibroblasts and keratinocytes in HPV models are critical for maintaining episomal HPV genomes, influencing keratinocyte differentiation, and regulating viral transcription [23,43,44,52,88,121,173–175]. Here we present RNAseq analysis revealing that fibroblasts may regulate the transformation potential in HPV+ keratinocytes by regulating cytokine activity, cell junction proteins, and innate immune signaling. Proteomic analysis further supported these findings, highlighting fibroblasts’ ability to modulate protein expression linked to oncogenic transformation. Overall, fibroblasts were found to influence both viral and host cell signaling, promoting HPV lifecycle maintenance while potentially limiting cancer progression in HPV+ keratinocytes; conversely, E6E7+ keratinocytes were more transformed in the presence of fibroblasts and may present a more neoplastic model.
Supplementary Material
Highlights.
Fibroblasts support HPV RNA expression and episomal maintenance in HPV+ keratinocytes
Fibroblasts reduce EMT related expression in HPV+ keratinocytes
Fibroblasts promote EMT related expression in E6E7+ keratinocytes
Acknowledgments
This work was supported by the VCU Philips Institute for Oral Health Research, the VCU Quest Fund, the National Institute of Dental and Craniofacial Research/NIH/DHHS R03 DE029548, and the National Cancer Institute-designated Massey Cancer Center grant P30 CA016059. Services in support of the research project were provided by the VCU Massey Comprehensive Cancer Center Bioinformatics Shared Resource. Massey is supported, in part, with funding from NIH-NCI Cancer Center Support Grant P30 CA016059. Services and products in support of the research project were generated by the VCU Massey Comprehensive Cancer Center Proteomics Shared Resource, supported, in part, with funding from NIH-NCI Cancer Center Support Grant P30 CA016059.
Footnotes
Declaration of competing interest
The authors declare that they have no known competing financial interests or personal relationships that might have appeared to influence the work reported in this article.
Appendix A. Supplementary data
Data availability statement
Following the 2023 NIH data management and sharing policy, all data resulting from the development of projects will be available in scientific communications presented at conferences and in manuscripts that will be published in peer-reviewed scientific journals. Data will be deposited in the Open Science Framework (OSF) platform. OSF can be accessed at https://osf.io. VCU is an OSF institutional member, and OSF is an approved generalist repository for the 2023 NIH data management and sharing policy.
References
- [1].Doorbar J, Quint W, Banks L, Bravo IG, Stoler M, Broker TR, et al. The biology and life-cycle of human papillomaviruses. Vaccine 2012;30 Suppl 5:F55–70. 10.1016/j.vaccine.2012.06.083. [DOI] [PubMed] [Google Scholar]
- [2].Bzhalava D, Eklund C, Dillner J. International standardization and classification of human papillomavirus types. Virology 2015;476:341–4. 10.1016/j.virol.2014.12.028. [DOI] [PubMed] [Google Scholar]
- [3].Parkin DM, Bray F. Chapter 2: The burden of HPV-related cancers. Vaccine 2006;24 Suppl 3:S3/11–25. 10.1016/j.vaccine.2006.05.111. [DOI] [PubMed] [Google Scholar]
- [4].Burd EM. Human papillomavirus and cervical cancer. Clin Microbiol Rev 2003;16:1–17. 10.1128/CMR.16.1.1-17.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Gribb JP, Wheelock JH, Park ES. Human Papilloma Virus (HPV) and the Current State of Oropharyngeal Cancer Prevention and Treatment. Del J Public Health 2023;9:26–8. 10.32481/djph.2023.04.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Cogliano V, Baan R, Straif K, Grosse Y, Secretan B, Ghissassi FE. Carcinogenicity of human papillomaviruses. Lancet Oncol 2005;6:204. 10.1016/S1470-2045(05)70086-3. [DOI] [PubMed] [Google Scholar]
- [7].Saraiya M, Unger ER, Thompson TD, Lynch CF, Hernandez BY, Lyu CW, et al. US Assessment of HPV Types in Cancers: Implications for Current and 9-Valent HPV Vaccines. JNCI J Natl Cancer Inst 2015;107:djv086. 10.1093/jnci/djv086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Brianti P, De Flammineis E, Mercuri SR. Review of HPV-related diseases and cancers. New Microbiol 2017;40:80–5. [PubMed] [Google Scholar]
- [9].Marur S, D’Souza G, Westra WH, Forastiere AA. HPV-associated head and neck cancer: a virus-related cancer epidemic. Lancet Oncol 2010;11:781–9. 10.1016/S1470-2045(10)70017-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].HPV and Cancer - NCI 2019. https://www.cancer.gov/about-cancer/causes-prevention/risk/infectious-agents/hpv-and-cancer (accessed August 14, 2024). [Google Scholar]
- [11].Liao C-I, Francoeur AA, Kapp DS, Caesar MAP, Huh WK, Chan JK. Trends in Human Papillomavirus–Associated Cancers, Demographic Characteristics, and Vaccinations in the US, 2001–2017. JAMA Netw Open 2022;5:e222530. 10.1001/jamanetworkopen.2022.2530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Huang J, Deng Y, Boakye D, Tin MS, Lok V, Zhang L, et al. Global distribution, risk factors, and recent trends for cervical cancer: A worldwide country-level analysis. Gynecol Oncol 2022;164:85–92. 10.1016/j.ygyno.2021.11.005. [DOI] [PubMed] [Google Scholar]
- [13].Siegel RL, Miller KD, Wagle NS, Jemal A. Cancer statistics, 2023. CA Cancer J Clin 2023;73:17–48. 10.3322/caac.21763. [DOI] [PubMed] [Google Scholar]
- [14].Malik S, Sah R, Muhammad K, Waheed Y. Tracking HPV Infection, Associated Cancer Development, and Recent Treatment Efforts—A Comprehensive Review. Vaccines 2023;11:102. 10.3390/vaccines11010102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Spurgeon ME, Lambert PF. Human Papillomavirus and the Stroma: Bidirectional Crosstalk during the Virus Life Cycle and Carcinogenesis. Viruses 2017;9:219. 10.3390/v9080219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Jenkins D. A review of cross-protection against oncogenic HPV by an HPV-16/18 AS04-adjuvanted cervical cancer vaccine: Importance of virological and clinical endpoints and implications for mass vaccination in cervical cancer prevention. Gynecol Oncol 2008;110:S18–25. 10.1016/j.ygyno.2008.06.027. [DOI] [PubMed] [Google Scholar]
- [17].James CD, Otoa R, Youssef AH, Fontan CT, Sannigrahi MK, Windle B, et al. HPV16 genome structure analysis in oropharyngeal cancer PDXs identifies tumors with integrated and episomal genomes. Press 2024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].zur Hausen H. Papillomaviruses in the causation of human cancers - a brief historical account. Virology 2009;384:260–5. 10.1016/j.virol.2008.11.046. [DOI] [PubMed] [Google Scholar]
- [19].Moscicki A-B, Schiffman M, Burchell A, Albero G, Giuliano AR, Goodman MT, et al. Updating the natural history of human papillomavirus and anogenital cancers. Vaccine 2012;30 Suppl 5:F24–33. 10.1016/j.vaccine.2012.05.089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Moscicki A-B, Ma Y, Farhat S, Jay J, Hanson E, Benningfield S, et al. Natural history of anal human papillomavirus infection in heterosexual women and risks associated with persistence. Clin Infect Dis Off Publ Infect Dis Soc Am 2014;58:804–11. 10.1093/cid/cit947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Wei F, Goodman MT, Xia N, Zhang J, Giuliano AR, D’Souza G, et al. Incidence and Clearance of Anal Human Papillomavirus Infection in 16 164 Individuals, According to Human Immunodeficiency Virus Status, Sex, and Male Sexuality: An International Pooled Analysis of 34 Longitudinal Studies. Clin Infect Dis Off Publ Infect Dis Soc Am 2023;76:e692–701. 10.1093/cid/ciac581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Bodily J, Laimins LA. Persistence of human papillomavirus infection: keys to malignant progression. Trends Microbiol 2011;19:33–9. 10.1016/j.tim.2010.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Raikhy G, Woodby BL, Scott ML, Shin G, Myers JE, Scott RS, et al. Suppression of Stromal Interferon Signaling by Human Papillomavirus 16. J Virol 2019;93:10.1128/jvi.00458–19. 10.1128/jvi.00458-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].De Gregorio V, Urciuolo F, Netti PA, Imparato G. In Vitro Organotypic Systems to Model Tumor Microenvironment in Human Papillomavirus (HPV)-Related Cancers. Cancers 2020;12:1150. 10.3390/cancers12051150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Koneva LA, Zhang Y, Virani S, Hall PB, McHugh JB, Chepeha DB, et al. HPV Integration in HNSCC Correlates with Survival Outcomes, Immune Response Signatures, and Candidate Drivers. Mol Cancer Res MCR 2018;16:90–102. 10.1158/1541-7786.MCR-17-0153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Barros MR, de Melo CML, Barros MLCMGR, de Cássia Pereira de Lima R, de Freitas AC, Venuti A. Activities of stromal and immune cells in HPV-related cancers. J Exp Clin Cancer Res 2018;37:137. 10.1186/s13046-018-0802-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].James CD, Fontan CT, Otoa R, Das D, Prabhakar AT, Wang X, et al. Human Papillomavirus 16 E6 and E7 Synergistically Repress Innate Immune Gene Transcription. mSphere 2020;5:e00828–19. 10.1128/mSphere.00828-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Evans MR, James CD, Loughran O, Nulton TJ, Wang X, Bristol ML, et al. An oral keratinocyte life cycle model identifies novel host genome regulation by human papillomavirus 16 relevant to HPV positive head and neck cancer. Oncotarget 2017;8:81892–909. 10.18632/oncotarget.18328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Evans MR, James CD, Bristol ML, Nulton TJ, Wang X, Kaur N, et al. Human Papillomavirus 16 E2 Regulates Keratinocyte Gene Expression Relevant to Cancer and the Viral Life Cycle. J Virol 2019;93:e01941–18. 10.1128/JVI.01941-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Bienkowska-Haba M, Luszczek W, Keiffer TR, Guion LGM, DiGiuseppe S, Scott RS, et al. Incoming human papillomavirus 16 genome is lost in PML protein-deficient HaCaT keratinocytes. Cell Microbiol 2017;19. 10.1111/cmi.12708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Chang YE, Laimins LA. Microarray analysis identifies interferon-inducible genes and Stat-1 as major transcriptional targets of human papillomavirus type 31. J Virol 2000;74:4174–82. 10.1128/jvi.74.9.4174-4182.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Arany I, Tyring SK. Status of local cellular immunity in interferon-responsive and - nonresponsive human papillomavirus-associated lesions. Sex Transm Dis 1996;23:475–80. 10.1097/00007435-199611000-00007. [DOI] [PubMed] [Google Scholar]
- [33].Alcocer-González JM, Berumen J, Taméz-Guerra R, Bermúdez-Morales V, Peralta-Zaragoza O, Hernández-Pando R, et al. In vivo expression of immunosuppressive cytokines in human papillomavirus-transformed cervical cancer cells. Viral Immunol 2006;19:481–91. 10.1089/vim.2006.19.481. [DOI] [PubMed] [Google Scholar]
- [34].Fichorova RN, Anderson DJ. Differential expression of immunobiological mediators by immortalized human cervical and vaginal epithelial cells. Biol Reprod 1999;60:508–14. 10.1095/biolreprod60.2.508. [DOI] [PubMed] [Google Scholar]
- [35].Uhlorn BL, Jackson R, Li S, Bratton SM, Van Doorslaer K, Campos SK. Vesicular trafficking permits evasion of cGAS/STING surveillance during initial human papillomavirus infection. PLoS Pathog 2020;16:e1009028. 10.1371/journal.ppat.1009028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Samuel CE. Antiviral actions of interferons. Clin Microbiol Rev 2001;14:778–809, table of contents. 10.1128/CMR.14.4.778-809.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Gaglia MM, Munger K. More than just oncogenes: mechanisms of tumorigenesis by human viruses. Curr Opin Virol 2018;32:48–59. 10.1016/j.coviro.2018.09.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Senba M, Mori N. Mechanisms of virus immune evasion lead to development from chronic inflammation to cancer formation associated with human papillomavirus infection. Oncol Rev 2012;6:e17. 10.4081/oncol.2012.e17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Tripathi M, Billet S, Bhowmick NA. Understanding the role of stromal fibroblasts in cancer progression. Cell Adhes Migr 2012;6:231–5. 10.4161/cam.20419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Bhowmick NA, Neilson EG, Moses HL. Stromal fibroblasts in cancer initiation and progression. Nature 2004;432:332–7. 10.1038/nature03096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Barcellos-Hoff MH. Stroma. In: Dubitzky W, Wolkenhauer O, Cho K-H, Yokota H, editors. Encycl. Syst. Biol., New York, NY: Springer; 2013, p. 2017–9. 10.1007/978-1-4419-9863-7_1384. [DOI] [Google Scholar]
- [42].Kendall RT, Feghali-Bostwick CA. Fibroblasts in fibrosis: novel roles and mediators. Front Pharmacol 2014;5:123. 10.3389/fphar.2014.00123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Lambert PF, Ozbun MA, Collins A, Holmgren S, Lee D, Nakahara T. Using an immortalized cell line to study the HPV life cycle in organotypic “raft” cultures. Methods Mol Med 2005;119:141–55. 10.1385/1-59259-982-6:141. [DOI] [PubMed] [Google Scholar]
- [44].Meyers C. Organotypic (raft) epithelial tissue culture system for the differentiation-dependent replication of papillomavirus. Methods Cell Sci 1996;18:201–10. 10.1007/BF00132885. [DOI] [Google Scholar]
- [45].Sahebali S, Van den Eynden G, Murta EF, Michelin MA, Cusumano P, Petignat P, et al. Stromal issues in cervical cancer: a review of the role and function of basement membrane, stroma, immune response and angiogenesis in cervical cancer development. Eur J Cancer Prev 2010;19:204–15. [DOI] [PubMed] [Google Scholar]
- [46].Chung S-H, Shin MK, Korach KS, Lambert PF. Requirement for Stromal Estrogen Receptor Alpha in Cervical Neoplasia. Horm Cancer 2013;4:50–9. 10.1007/s12672-012-0125-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Epithelial–Stromal Interactions Modulating Penetration of Matrigel Membranes by HPV 16-Immortalized Keratinocytes. J Invest Dermatol 1997;109:619–25. 10.1111/1523-1747.ep12337594. [DOI] [PubMed] [Google Scholar]
- [48].Alkasalias T, Moyano-Galceran L, Arsenian-Henriksson M, Lehti K. Fibroblasts in the Tumor Microenvironment: Shield or Spear? Int J Mol Sci 2018;19:1532. 10.3390/ijms19051532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Mao X, Xu J, Wang W, Liang C, Hua J, Liu J, et al. Crosstalk between cancer-associated fibroblasts and immune cells in the tumor microenvironment: new findings and future perspectives. Mol Cancer 2021;20:131. 10.1186/s12943-021-01428-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Monteran L, Erez N. The Dark Side of Fibroblasts: Cancer-Associated Fibroblasts as Mediators of Immunosuppression in the Tumor Microenvironment. Front Immunol 2019;10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Rahrotaban S, Mahdavi N, Abdollahi A, Yazdani F, Kaghazloo A, Derakhshan S. Carcinoma-associated Fibroblasts are a Common Finding in the Microenvironment of HPV-positive Oropharyngeal Squamous Cell Carcinoma. Appl Immunohistochem Mol Morphol AIMM 2019;27:683–8. 10.1097/PAI.0000000000000687. [DOI] [PubMed] [Google Scholar]
- [52].Smola H, Stark H-J, Thiekötter G, Mirancea N, Krieg T, Fusenig NE. Dynamics of Basement Membrane Formation by Keratinocyte–Fibroblast Interactions in Organotypic Skin Culture. Exp Cell Res 1998;239:399–410. 10.1006/excr.1997.3910. [DOI] [PubMed] [Google Scholar]
- [53].Truffi M, Sorrentino L, Corsi F. Fibroblasts in the Tumor Microenvironment. Adv Exp Med Biol 2020;1234:15–29. 10.1007/978-3-030-37184-5_2. [DOI] [PubMed] [Google Scholar]
- [54].Almangush A, Jouhi L, Haglund C, Hagström J, Mäkitie AA, Leivo I. Tumor-stroma ratio is a promising prognostic classifier in oropharyngeal cancer. Hum Pathol 2023;136:16–24. 10.1016/j.humpath.2023.03.010. [DOI] [PubMed] [Google Scholar]
- [55].Sharma V, Letson J, Furuta S. Fibrous stroma: Driver and passenger in cancer development. Sci Signal 2022;15:eabg3449. 10.1126/scisignal.abg3449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [56].Bremnes RM, Dønnem T, Al-Saad S, Al-Shibli K, Andersen S, Sirera R, et al. The role of tumor stroma in cancer progression and prognosis: emphasis on carcinoma-associated fibroblasts and non-small cell lung cancer. J Thorac Oncol Off Publ Int Assoc Study Lung Cancer 2011;6:209–17. 10.1097/JTO.0b013e3181f8a1bd. [DOI] [PubMed] [Google Scholar]
- [57].Basukala O, Banks L. The Not-So-Good, the Bad and the Ugly: HPV E5, E6 and E7 Oncoproteins in the Orchestration of Carcinogenesis. Viruses 2021;13:1892. 10.3390/v13101892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [58].DeFilippis RA, Goodwin EC, Wu L, DiMaio D. Endogenous human papillomavirus E6 and E7 proteins differentially regulate proliferation, senescence, and apoptosis in HeLa cervical carcinoma cells. J Virol 2003;77:1551–63. 10.1128/jvi.77.2.1551-1563.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [59].Francis DA, Schmid SI, Howley PM. Repression of the integrated papillomavirus E6/E7 promoter is required for growth suppression of cervical cancer cells. J Virol 2000;74:2679–86. 10.1128/jvi.74.6.2679-2686.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [60].Hoppe-Seyler K, Bossler F, Braun JA, Herrmann AL, Hoppe-Seyler F. The HPV E6/E7 Oncogenes: Key Factors for Viral Carcinogenesis and Therapeutic Targets. Trends Microbiol 2018;26:158–68. 10.1016/j.tim.2017.07.007. [DOI] [PubMed] [Google Scholar]
- [61].Jeon S, Lambert PF. Integration of human papillomavirus type 16 DNA into the human genome leads to increased stability of E6 and E7 mRNAs: implications for cervical carcinogenesis. Proc Natl Acad Sci U S A 1995;92:1654–8. 10.1073/pnas.92.5.1654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [62].Morrison MA, Morreale RJ, Akunuru S, Kofron M, Zheng Y, Wells SI. Targeting the human papillomavirus E6 and E7 oncogenes through expression of the bovine papillomavirus type 1 E2 protein stimulates cellular motility. J Virol 2011;85:10487–98. 10.1128/JVI.05126-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [63].Riley RR, Duensing S, Brake T, Münger K, Lambert PF, Arbeit JM. Dissection of human papillomavirus E6 and E7 function in transgenic mouse models of cervical carcinogenesis. Cancer Res 2003;63:4862–71. [PubMed] [Google Scholar]
- [64].Nees M, Geoghegan JM, Munson P, Prabhu V, Liu Y, Androphy E, et al. Human Papillomavirus Type 16 E6 and E7 Proteins Inhibit Differentiation-dependent Expression of Transforming Growth Factor-β2 in Cervical Keratinocytes. Cancer Res 2000;60:4289–98. [PubMed] [Google Scholar]
- [65].McLaughlin-Drubin ME, Meyers J, Munger K. Cancer associated human papillomaviruses. Curr Opin Virol 2012;2:459–66. 10.1016/j.coviro.2012.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [66].Mirabello L, Yeager M, Yu K, Clifford GM, Xiao Y, Zhu B, et al. HPV16 E7 Genetic Conservation Is Critical to Carcinogenesis. Cell 2017;170:1164–1174.e6. 10.1016/j.cell.2017.08.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [67].Venuti A, Paolini F, Nasir L, Corteggio A, Roperto S, Campo MS, et al. Papillomavirus E5: the smallest oncoprotein with many functions. Mol Cancer 2011;10:140. 10.1186/1476-4598-10-140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [68].Chapman S, Liu X, Meyers C, Schlegel R, McBride AA. Human keratinocytes are efficiently immortalized by a Rho kinase inhibitor. J Clin Invest 2010;120:2619–26. 10.1172/JCI42297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [69].Liu X, Ory V, Chapman S, Yuan H, Albanese C, Kallakury B, et al. ROCK inhibitor and feeder cells induce the conditional reprogramming of epithelial cells. Am J Pathol 2012;180:599–607. 10.1016/j.ajpath.2011.10.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [70].Dakic A, DiVito K, Fang S, Suprynowicz F, Gaur A, Li X, et al. ROCK inhibitor reduces Myc-induced apoptosis and mediates immortalization of human keratinocytes. Oncotarget 2016;7:66740–53. 10.18632/oncotarget.11458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [71].Fu B, Quintero J, Baker CC. Keratinocyte growth conditions modulate telomerase expression, senescence, and immortalization by human papillomavirus type 16 E6 and E7 oncogenes. Cancer Res 2003;63:7815–24. [PubMed] [Google Scholar]
- [72].Morgan IM, DiNardo LJ, Windle B. Integration of Human Papillomavirus Genomes in Head and Neck Cancer: Is It Time to Consider a Paradigm Shift? Viruses 2017;9:208. 10.3390/v9080208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [73].Fontan CT, James CD, Prabhakar AT, Bristol ML, Otoa R, Wang X, et al. A Critical Role for p53 during the HPV16 Life Cycle. Microbiol Spectr 2022;10:e0068122. 10.1128/spectrum.00681-22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [74].Prabhakar AT, James CD, Fontan CT, Otoa R, Wang X, Bristol ML, et al. Human Papillomavirus 16 E2 Interaction with TopBP1 Is Required for E2 and Viral Genome Stability during the Viral Life Cycle. J Virol 2023;97:e00063–23. 10.1128/jvi.00063-23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [75].Prabhakar AT, James CD, Das D, Fontan CT, Otoa R, Wang X, et al. Interaction with TopBP1 Is Required for Human Papillomavirus 16 E2 Plasmid Segregation/Retention Function during Mitosis. J Virol 2022;96:e0083022. 10.1128/jvi.00830-22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [76].McBride AA, Münger K. Expert Views on HPV Infection. Viruses 2018;10:94. 10.3390/v10020094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [77].McBride AA, Warburton A. The role of integration in oncogenic progression of HPV-associated cancers. PLoS Pathog 2017;13:e1006211. 10.1371/journal.ppat.1006211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [78].Akagi K, Li J, Broutian TR, Padilla-Nash H, Xiao W, Jiang B, et al. Genome-wide analysis of HPV integration in human cancers reveals recurrent, focal genomic instability. Genome Res 2014;24:185–99. 10.1101/gr.164806.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [79].Balaji H, Demers I, Wuerdemann N, Schrijnder J, Kremer B, Klussmann JP, et al. Causes and Consequences of HPV Integration in Head and Neck Squamous Cell Carcinomas: State of the Art. Cancers 2021;13:4089. 10.3390/cancers13164089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [80].Dall KL, Scarpini CG, Roberts I, Winder DM, Stanley MA, Muralidhar B, et al. Characterization of naturally occurring HPV16 integration sites isolated from cervical keratinocytes under noncompetitive conditions. Cancer Res 2008;68:8249–59. 10.1158/0008-5472.CAN-08-1741. [DOI] [PubMed] [Google Scholar]
- [81].Kamal M, Lameiras S, Deloger M, Morel A, Vacher S, Lecerf C, et al. Human papilloma virus (HPV) integration signature in Cervical Cancer: identification of MACROD2 gene as HPV hot spot integration site. Br J Cancer 2021;124:777–85. 10.1038/s41416-020-01153-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [82].Jeon S, Allen-Hoffmann BL, Lambert PF. Integration of human papillomavirus type 16 into the human genome correlates with a selective growth advantage of cells. J Virol 1995;69:2989–97. 10.1128/JVI.69.5.2989-2997.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [83].Yu L, Majerciak V, Lobanov A, Mirza S, Band V, Liu H, et al. HPV oncogenes expressed from only one of multiple integrated HPV DNA copies drive clonal cell expansion in cervical cancer. mBio 2024;15:e0072924. 10.1128/mbio.00729-24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [84].Fan J, Fu Y, Peng W, Li X, Shen Y, Guo E, et al. Multi-omics characterization of silent and productive HPV integration in cervical cancer. Cell Genomics 2023;3:100211. 10.1016/j.xgen.2022.100211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [85].Mainguené J, Vacher S, Kamal M, Hamza A, Masliah Planchon J, Baulande S, et al. Human papilloma virus integration sites and genomic signatures in head and neck squamous cell carcinoma. Mol Oncol 2022;16:3001–16. 10.1002/1878-0261.13219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [86].Rheinwald JG, Green H. Epidermal growth factor and the multiplication of cultured human epidermal keratinocytes. Nature 1977;265:421–4. 10.1038/265421a0. [DOI] [PubMed] [Google Scholar]
- [87].Coursey TL, McBride AA. Development of Keratinocyte Cell Lines containing Extrachromosomal Human Papillomavirus Genomes. Curr Protoc 2021;1:e235. 10.1002/cpz1.235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [88].James CD, Lewis RL, Fakunmoju AL, Witt AJ, Youssef AH, Wang X, et al. Fibroblast Stromal Support Model for Predicting Human Papillomavirus-Associated Cancer Drug Responses 2024:2024.04.09.588680. 10.1101/2024.04.09.588680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [89].SONG D, LI H, LI H, DAI J. Effect of human papillomavirus infection on the immune system and its role in the course of cervical cancer. Oncol Lett 2015;10:600–6. 10.3892/ol.2015.3295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [90].Westrich JA, Warren CJ, Pyeon D. Evasion of host immune defenses by human papillomavirus. Virus Res 2017;231:21–33. 10.1016/j.virusres.2016.11.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [91].Nunes RAL, Morale MG, Silva GÁF, Villa LL, Termini L. Innate immunity and HPV: friends or foes. Clin Sao Paulo Braz 2018;73:e549s. 10.6061/clinics/2018/e549s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [92].Nulton TJ, Olex AL, Dozmorov M, Morgan IM, Windle B. Analysis of The Cancer Genome Atlas sequencing data reveals novel properties of the human papillomavirus 16 genome in head and neck squamous cell carcinoma. Oncotarget 2017;8:17684–99. 10.18632/oncotarget.15179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [93].Bristol ML, Wang X, Smith NW, Son MP, Evans MR, Morgan IM. DNA Damage Reduces the Quality, but Not the Quantity of Human Papillomavirus 16 E1 and E2 DNA Replication. Viruses 2016;8:175. 10.3390/v8060175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [94].James CD, Saini S, Sesay F, Ko K, Felthousen-Rusbasan J, Iness AN, et al. Restoring the DREAM Complex Inhibits the Proliferation of High-Risk HPV Positive Human Cells. Cancers 2021;13:489. 10.3390/cancers13030489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [95].James CD, Prabhakar AT, Otoa R, Evans MR, Wang X, Bristol ML, et al. SAMHD1 Regulates Human Papillomavirus 16-Induced Cell Proliferation and Viral Replication during Differentiation of Keratinocytes. mSphere 2019;4:e00448–19. 10.1128/mSphere.00448-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [96].Kim D, Paggi JM, Park C, Bennett C, Salzberg SL. Graph-based genome alignment and genotyping with HISAT2 and HISAT-genotype. Nat Biotechnol 2019;37:907–15. 10.1038/s41587-019-0201-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [97].Mortazavi A, Williams BA, McCue K, Schaeffer L, Wold B. Mapping and quantifying mammalian transcriptomes by RNA-Seq. Nat Methods 2008;5:621–8. 10.1038/nmeth.1226. [DOI] [PubMed] [Google Scholar]
- [98].Liao Y, Smyth GK, Shi W. featureCounts: an efficient general purpose program for assigning sequence reads to genomic features. Bioinforma Oxf Engl 2014;30:923–30. 10.1093/bioinformatics/btt656. [DOI] [PubMed] [Google Scholar]
- [99].Love MI, Huber W, Anders S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol 2014;15:550. 10.1186/s13059-014-0550-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [100].Young MD, Wakefield MJ, Smyth GK, Oshlack A. Gene ontology analysis for RNA-seq: accounting for selection bias. Genome Biol 2010;11:R14. 10.1186/gb-2010-11-2-r14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [101].Wu T, Hu E, Xu S, Chen M, Guo P, Dai Z, et al. clusterProfiler 4.0: A universal enrichment tool for interpreting omics data. The Innovation 2021;2:100141. 10.1016/j.xinn.2021.100141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [102].Babraham Bioinformatics - FastQC A Quality Control tool for High Throughput Sequence Data n.d. http://www.bioinformatics.babraham.ac.uk/projects/fastqc/ (accessed August 28, 2024). [Google Scholar]
- [103].Ewels P, Magnusson M, Lundin S, Käller M. MultiQC: summarize analysis results for multiple tools and samples in a single report. Bioinforma Oxf Engl 2016;32:3047–8. 10.1093/bioinformatics/btw354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [104].Bolger AM, Lohse M, Usadel B. Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinforma Oxf Engl 2014;30:2114–20. 10.1093/bioinformatics/btu170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [105].Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, Jha S, et al. STAR: ultrafast universal RNA-seq aligner. Bioinforma Oxf Engl 2013;29:15–21. 10.1093/bioinformatics/bts635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [106].Danecek P, Bonfield JK, Liddle J, Marshall J, Ohan V, Pollard MO, et al. Twelve years of SAMtools and BCFtools. GigaScience 2021;10:giab008. 10.1093/gigascience/giab008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [107].Quinlan AR, Hall IM. BEDTools: a flexible suite of utilities for comparing genomic features. Bioinforma Oxf Engl 2010;26:841–2. 10.1093/bioinformatics/btq033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [108].Robinson MD, McCarthy DJ, Smyth GK. edgeR: a Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 2010;26:139–40. 10.1093/bioinformatics/btp616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [109].McCarthy DJ, Chen Y, Smyth GK. Differential expression analysis of multifactor RNA-Seq experiments with respect to biological variation. Nucleic Acids Res 2012;40:4288–97. 10.1093/nar/gks042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [110].Chen Y, Lun ATL, Smyth GK. From reads to genes to pathways: differential expression analysis of RNA-Seq experiments using Rsubread and the edgeR quasi-likelihood pipeline. F1000Research 2016;5:1438. 10.12688/f1000research.8987.2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [111].Myers JE, Zwolinska K, Sapp MJ, Scott RS. An Exonuclease V–qPCR Assay to Analyze the State of the Human Papillomavirus 16 Genome in Cell Lines and Tissues. Curr Protoc Microbiol 2020;59:e119. 10.1002/cpmc.119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [112].Zhang X, Smits AH, van Tilburg GB, Ovaa H, Huber W, Vermeulen M. Proteome-wide identification of ubiquitin interactions using UbIA-MS. Nat Protoc 2018;13:530–50. 10.1038/nprot.2017.147. [DOI] [PubMed] [Google Scholar]
- [113].Huber W, von Heydebreck A, Sültmann H, Poustka A, Vingron M. Variance stabilization applied to microarray data calibration and to the quantification of differential expression. Bioinforma Oxf Engl 2002;18 Suppl 1:S96–104. 10.1093/bioinformatics/18.suppl_1.s96. [DOI] [PubMed] [Google Scholar]
- [114].Ritchie ME, Phipson B, Wu D, Hu Y, Law CW, Shi W, et al. limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res 2015;43:e47. 10.1093/nar/gkv007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [115].Psyrri A, DiMaio D. Human papillomavirus in cervical and head-and-neck cancer. Nat Clin Pract Oncol 2008;5:24–31. 10.1038/ncponc0984. [DOI] [PubMed] [Google Scholar]
- [116].Huh WK, Joura EA, Giuliano AR, Iversen O-E, de Andrade RP, Ault KA, et al. Final efficacy, immunogenicity, and safety analyses of a nine-valent human papillomavirus vaccine in women aged 16–26 years: a randomised, double-blind trial. The Lancet 2017;390:2143–59. 10.1016/S0140-6736(17)31821-4. [DOI] [PubMed] [Google Scholar]
- [117].Kavanagh K, Pollock KG, Cuschieri K, Palmer T, Cameron RL, Watt C, et al. Changes in the prevalence of human papillomavirus following a national bivalent human papillomavirus vaccination programme in Scotland: a 7-year cross-sectional study. Lancet Infect Dis 2017;17:1293–302. 10.1016/S1473-3099(17)30468-1. [DOI] [PubMed] [Google Scholar]
- [118].Meeting IWG on the E of CR to H, Cancer IA for R on. Human Papillomaviruses. World Health Organization; 2007. [Google Scholar]
- [119].Vats A, Trejo-Cerro O, Thomas M, Banks L. Human papillomavirus E6 and E7: What remains? Tumour Virus Res 2021;11:200213. 10.1016/j.tvr.2021.200213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [120].The Expression of HPV E6/E7 mRNA In Situ Hybridization in HP... : International Journal of Gynecological Pathology n.d. https://journals.lww.com/intjgynpathology/Fulltext/2023/01000/The_Expression_of_HPV_E6_E7_mRNA_In_Situ.2.aspx (accessed March 17, 2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [121].Coursey TL, Van Doorslaer K, McBride AA. Regulation of Human Papillomavirus 18 Genome Replication, Establishment, and Persistence by Sequences in the Viral Upstream Regulatory Region. J Virol 2021;95:e0068621. 10.1128/JVI.00686-21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [122].Rossi NM, Dai J, Xie Y, Lou H, Boland JF, Yeager M, et al. Extrachromosomal Amplification of Human Papillomavirus Episomes as a Mechanism of Cervical Carcinogenesis 2021:2021.10.22.465367. 10.1101/2021.10.22.465367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [123].Evans MR, James CD, Loughran O, Nulton TJ, Wang X, Bristol ML, et al. Correction: An oral keratinocyte life cycle model identifies novel host genome regulation by human papillomavirus 16 relevant to HPV positive head and neck cancer. Oncotarget 2019;10:3312. 10.18632/oncotarget.26962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [124].Liu S, Imani S, Deng Y, Pathak JL, Wen Q, Chen Y, et al. Targeting IFN/STAT1 Pathway as a Promising Strategy to Overcome Radioresistance. OncoTargets Ther 2020;13:6037–50. 10.2147/OTT.S256708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [125].Valle-Mendiola A, Gutiérrez-Hoya A, Soto-Cruz I. JAK/STAT Signaling and Cervical Cancer: From the Cell Surface to the Nucleus. Genes 2023;14. 10.3390/genes14061141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [126].Hernández-Monge J, Garay E, Raya-Sandino A, Vargas-Sierra O, Díaz-Chávez J, Popoca-Cuaya M, et al. Papillomavirus E6 oncoprotein up-regulates occludin and ZO-2 expression in ovariectomized mice epidermis. Exp Cell Res 2013;319:2588–603. 10.1016/j.yexcr.2013.07.028. [DOI] [PubMed] [Google Scholar]
- [127].Xu Y-N, Deng M-S, Liu Y-F, Yao J, Xiao Z-Y. Tight junction protein CLDN17 serves as a tumor suppressor to reduce the invasion and migration of oral cancer cells by inhibiting epithelial-mesenchymal transition. Arch Oral Biol 2022;133:105301. 10.1016/j.archoralbio.2021.105301. [DOI] [PubMed] [Google Scholar]
- [128].Zhang W-N, Li W, Wang X-L, Hu Z, Zhu D, Ding W-C, et al. CLDN1 expression in cervical cancer cells is related to tumor invasion and metastasis. Oncotarget 2016;7:87449–61. 10.18632/oncotarget.13871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [129].Uc PY, Miranda J, Raya-Sandino A, Alarcón L, Roldán ML, Ocadiz-Delgado R, et al. E7 oncoprotein from human papillomavirus 16 alters claudins expression and the sealing of epithelial tight junctions. Int J Oncol 2020;57:905–24. 10.3892/ijo.2020.5105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [130].Zhu J, Jiang Q. Twist1 mediated transcriptional activation of Claudin 4 promotes cervical cancer cell migration and invasion. Oncol Lett 2023;26:335. 10.3892/ol.2023.13921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [131].Yu X, He T, Tong Z, Liao L, Huang S, Fakhouri WD, et al. Molecular mechanisms of TWIST1-regulated transcription in EMT and cancer metastasis. EMBO Rep 2023;24:e56902. 10.15252/embr.202356902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [132].de Freitas Silva BS, Yamamoto-Silva FP, Pontes HAR, Pinto Júnior D dos S. E-cadherin downregulation and Twist overexpression since early stages of oral carcinogenesis. J Oral Pathol Med Off Publ Int Assoc Oral Pathol Am Acad Oral Pathol 2014;43:125–31. 10.1111/jop.12096. [DOI] [PubMed] [Google Scholar]
- [133].Peh WL, Middleton K, Christensen N, Nicholls P, Egawa K, Sotlar K, et al. Life cycle heterogeneity in animal models of human papillomavirus-associated disease. J Virol 2002;76:10401–16. 10.1128/jvi.76.20.10401-10416.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [134].Zihni C, Mills C, Matter K, Balda MS. Tight junctions: from simple barriers to multifunctional molecular gates. Nat Rev Mol Cell Biol 2016;17:564–80. 10.1038/nrm.2016.80. [DOI] [PubMed] [Google Scholar]
- [135].Wu T, Yang W, Sun A, Wei Z, Lin Q. The Role of CXC Chemokines in Cancer Progression. Cancers 2022;15:167. 10.3390/cancers15010167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [136].Hughes CE, Nibbs RJB. A guide to chemokines and their receptors. Febs J 2018;285:2944–71. 10.1111/febs.14466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [137].Franco HL, Casasnovas J, Rodríguez-Medina JR, Cadilla CL. Redundant or separate entities?—roles of Twist1 and Twist2 as molecular switches during gene transcription. Nucleic Acids Res 2011;39:1177–86. 10.1093/nar/gkq890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [138].Tang H, Massi D, Hemmings BA, Mandalà M, Hu Z, Wicki A, et al. AKT-ions with a TWIST between EMT and MET. Oncotarget 2016;7:62767–77. 10.18632/oncotarget.11232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [139].Meng P, Liu C, Li J, Fang P, Yang B, Sun W, et al. CXC chemokine receptor 7 ameliorates renal fibrosis by inhibiting β-catenin signaling and epithelial-to-mesenchymal transition in tubular epithelial cells. Ren Fail 2024;46:2300727. 10.1080/0886022X.2023.2300727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [140].Xiu M-X, Liu Y-M, Kuang B. The Role of DLLs in Cancer: A Novel Therapeutic Target. OncoTargets Ther 2020;13:3881–901. 10.2147/OTT.S244860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [141].Kumar V, Vashishta M, Kong L, Wu X, Lu JJ, Guha C, et al. The Role of Notch, Hedgehog, and Wnt Signaling Pathways in the Resistance of Tumors to Anticancer Therapies. Front Cell Dev Biol 2021;9:650772. 10.3389/fcell.2021.650772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [142].Sales-Dias J, Silva G, Lamy M, Ferreira A, Barbas A. The Notch ligand DLL1 exerts carcinogenic features in human breast cancer cells. PLoS ONE 2019;14:e0217002. 10.1371/journal.pone.0217002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [143].Cheng Y, Huang N, Yin Q, Cheng C, Chen D, Gong C, et al. LncRNA TP53TG1 plays an anti-oncogenic role in cervical cancer by synthetically regulating transcriptome profile in HeLa cells. Front Genet 2022;13. 10.3389/fgene.2022.981030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [144].Isaka S, Takei Y, Tokino T, Koyama K, Miyoshi Y, Suzuki M, et al. Isolation and characterization of a novel TP53-inducible gene, TP53TG5, which suppresses growth and shows cell cycle-dependent transition of expression. Genes Chromosomes Cancer 2000;27:345–52. [DOI] [PubMed] [Google Scholar]
- [145].Ge X, Xu M, Cheng T, Hu N, Sun P, Lu B, et al. TP53I13 promotes metastasis in glioma via macrophages, neutrophils, and fibroblasts and is a potential prognostic biomarker. Front Immunol 2022;13:974346. 10.3389/fimmu.2022.974346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [146].RB1 RB transcriptional corepressor 1 [Homo sapiens (human)] - Gene - NCBI n.d. https://www.ncbi.nlm.nih.gov/gene/5925 (accessed August 30, 2024). [Google Scholar]
- [147].RBL1 RB transcriptional corepressor like 1 [Homo sapiens (human)] - Gene - NCBI n.d. https://www.ncbi.nlm.nih.gov/gene/5933 (accessed August 30, 2024). [Google Scholar]
- [148].RB1CC1 RB1 inducible coiled-coil 1 [Homo sapiens (human)] - Gene – NCBI n.d. https://www.ncbi.nlm.nih.gov/gene/9821 (accessed August 30, 2024). [Google Scholar]
- [149].Vélez-Cruz R, Johnson DG. The Retinoblastoma (RB) Tumor Suppressor: Pushing Back against Genome Instability on Multiple Fronts. Int J Mol Sci 2017;18:1776. 10.3390/ijms18081776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [150].Caglar HO, Biray Avci C. Alterations of cell cycle genes in cancer: unmasking the role of cancer stem cells. Mol Biol Rep 2020;47:3065–76. 10.1007/s11033-020-05341-6. [DOI] [PubMed] [Google Scholar]
- [151].Jamasbi E, Hamelian M, Hossain MA, Varmira K. The cell cycle, cancer development and therapy. Mol Biol Rep 2022;49:10875–83. 10.1007/s11033-022-07788-1. [DOI] [PubMed] [Google Scholar]
- [152].Doorbar J, Egawa N, Griffin H, Kranjec C, Murakami I. Human papillomavirus molecular biology and disease association. Rev Med Virol 2015;25 Suppl 1:2–23. 10.1002/rmv.1822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [153].Liu P-F, Chen C-F, Shu C-W, Chang H-M, Lee C-H, Liou H-H, et al. UBE2C is a Potential Biomarker for Tumorigenesis and Prognosis in Tongue Squamous Cell Carcinoma. Diagnostics 2020;10:674. 10.3390/diagnostics10090674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [154].Du R, Huang C, Liu K, Li X, Dong Z. Targeting AURKA in Cancer: molecular mechanisms and opportunities for Cancer therapy. Mol Cancer 2021;20:15. 10.1186/s12943-020-01305-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [155].Jiang A, Zhou Y, Gong W, Pan X, Gan X, Wu Z, et al. CCNA2 as an Immunological Biomarker Encompassing Tumor Microenvironment and Therapeutic Response in Multiple Cancer Types. Oxid Med Cell Longev 2022;2022:5910575. 10.1155/2022/5910575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [156].Soboleva A, Arutyunyan I, Jumaniyazova E, Vishnyakova P, Zarubina D, Nimatov E, et al. Gene-Expression Patterns of Tumor and Peritumor Tissues of Smoking and Non-Smoking HPV-Negative Patients with Head and Neck Squamous Cell Carcinoma. Biomedicines 2024;12:696. 10.3390/biomedicines12030696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [157].Facompre ND, Rajagopalan P, Sahu V, Pearson AT, Montone KT, James CD, et al. Identifying predictors of HPV-related head and neck squamous cell carcinoma progression and survival through patient-derived models. Int J Cancer 2020;147:3236–49. 10.1002/ijc.33125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [158].Nulton TJ, Kim N-K, DiNardo LJ, Morgan IM, Windle B. Patients with integrated HPV16 in head and neck cancer show poor survival. Oral Oncol 2018;80:52–5. 10.1016/j.oraloncology.2018.03.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [159].Day RS, McDade KK, Chandran UR, Lisovich A, Conrads TP, Hood BL, et al. Identifier mapping performance for integrating transcriptomics and proteomics experimental results. BMC Bioinformatics 2011;12:213. 10.1186/1471-2105-12-213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [160].Yang KC, Gorski SM. Protocol for analysis of RNA-sequencing and proteome profiling data for subgroup identification and comparison. STAR Protoc 2022;3:101283. 10.1016/j.xpro.2022.101283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [161].Geiger T, Wehner A, Schaab C, Cox J, Mann M. Comparative Proteomic Analysis of Eleven Common Cell Lines Reveals Ubiquitous but Varying Expression of Most Proteins*. Mol Cell Proteomics 2012;11:M111.014050. 10.1074/mcp.M111.014050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [162].Kosti I, Jain N, Aran D, Butte AJ, Sirota M. Cross-tissue Analysis of Gene and Protein Expression in Normal and Cancer Tissues. Sci Rep 2016;6:24799. 10.1038/srep24799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [163].Uhlén M, Fagerberg L, Hallström BM, Lindskog C, Oksvold P, Mardinoglu A, et al. Proteomics. Tissue-based map of the human proteome. Science 2015;347:1260419. 10.1126/science.1260419. [DOI] [PubMed] [Google Scholar]
- [164].Uhlen M, Zhang C, Lee S, Sjöstedt E, Fagerberg L, Bidkhori G, et al. A pathology atlas of the human cancer transcriptome. Science 2017;357:eaan2507. 10.1126/science.aan2507. [DOI] [PubMed] [Google Scholar]
- [165].Uhlén M, Björling E, Agaton C, Szigyarto CA- K, Amini B, Andersen E, et al. A human protein atlas for normal and cancer tissues based on antibody proteomics. Mol Cell Proteomics MCP 2005;4:1920–32. 10.1074/mcp.M500279-MCP200. [DOI] [PubMed] [Google Scholar]
- [166].Doorbar J. Model systems of human papillomavirus-associated disease. J Pathol 2016;238:166–79. 10.1002/path.4656. [DOI] [PubMed] [Google Scholar]
- [167].Phipps SMO, Berletch JB, Andrews LG, Tollefsbol TO. Aging cell culture: methods and observations. Methods Mol Biol Clifton NJ 2007;371:9–19. 10.1007/978-1-59745-361-5_2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [168].Allen-Hoffmann BL, Schlosser SJ, Ivarie CA, Sattler CA, Meisner LF, O’Connor SL. Normal growth and differentiation in a spontaneously immortalized near-diploid human keratinocyte cell line, NIKS. J Invest Dermatol 2000;114:444–55. 10.1046/j.1523-1747.2000.00869.x. [DOI] [PubMed] [Google Scholar]
- [169].Beilin AK, Gurskaya NG, Evtushenko NA, Alpeeva EV, Kosykh AV, Terskikh VV, et al. Immortalization of Human Keratinocytes Using the Catalytic Subunit of Telomerase. Dokl Biochem Biophys 2021;496:5–9. 10.1134/S1607672921010014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [170].HPV16 E6 and E7 proteins cooperate to immortalize human foreskin keratinocytes. - PMC n.d. https://www.ncbi.nlm.nih.gov/pmc/articles/PMC402081/ (accessed September 10, 2024). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [171].Bristol ML, James CD, Wang X, Fontan CT, Morgan IM. Estrogen Attenuates the Growth of Human Papillomavirus-Positive Epithelial Cells. mSphere 2020;5:e00049–20. 10.1128/mSphere.00049-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [172].Costello LC, Franklin RB. The genetic/metabolic transformation concept of carcinogenesis. Cancer Metastasis Rev 2012;31:123–30. 10.1007/s10555-011-9334-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [173].Anacker DC, Moody CA. Modulation of the DNA damage response during the life cycle of human papillomaviruses. Virus Res 2017;231:41–9. 10.1016/j.virusres.2016.11.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [174].Bienkowska-Haba M, Luszczek W, Myers JE, Keiffer TR, DiGiuseppe S, Polk P, et al. A new cell culture model to genetically dissect the complete human papillomavirus life cycle. PLOS Pathog 2018;14:e1006846. 10.1371/journal.ppat.1006846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [175].Woodby B, Scott M, Bodily J. Chapter Five - The Interaction Between Human Papillomaviruses and the Stromal Microenvironment. In: Pruitt K, editor. Prog. Mol. Biol. Transl. Sci., vol. 144, Academic Press; 2016, p. 169–238. 10.1016/bs.pmbts.2016.09.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Following the 2023 NIH data management and sharing policy, all data resulting from the development of projects will be available in scientific communications presented at conferences and in manuscripts that will be published in peer-reviewed scientific journals. Data will be deposited in the Open Science Framework (OSF) platform. OSF can be accessed at https://osf.io. VCU is an OSF institutional member, and OSF is an approved generalist repository for the 2023 NIH data management and sharing policy.