

Abstract
In multiple myeloma, as in B‐cell malignancies, mono‐ and especially bi‐allelic TP53 gene inactivation is a high‐risk factor for treatment resistance, and there are currently no therapies specifically targeting p53 deficiency. In this study, we evaluated if the loss of cell cycle control in p53‐deficient myeloma cells would confer a metabolically actionable vulnerability. We show that CTP synthase 1 (CTPS1), which encodes a CTP synthesis rate‐limiting enzyme essential for DNA and RNA synthesis in lymphoid cells, is overexpressed in samples from myeloma patients displaying a high proliferation rate (high MKI67 expression) or a low p53 score (synonymous with TP53 deletion and/or mutation). This overexpression of CTPS1 was associated with reduced survival in two cohorts. Using scRNA‐seq analysis in 24 patient samples, we further demonstrate that myeloma cells in the S or G2/M phase display high CTPS1 expression. Pharmacological inhibition of CTPS1 by STP‐B induced cell cycle arrest in early S phase in isogenic NCI‐H929 or XG7 TP53 +/+, TP53 −/−, and TP53 R175H/R175H cells and in a TP53 −/R123STOP patient sample. The functional annotation of transcriptional changes in 10 STP‐B‐treated myeloma cell lines revealed a decrease in protein translation and confirmed the blockade of cells into the S phase. The pharmacological inhibition of ATR, which governs the intrinsic S/G2 checkpoint, in STP‐B‐induced S‐phase arrested cells synergistically induced cell death in TP53 +/+, TP53 −/−, and TP53 R175H/R175H isogenic cell lines (Bliss score >15). This combination induced replicative stress and caspase‐mediated cell death and was highly effective in resistant/refractory patient samples with TP53 deletion and/or mutation and in TP53 −/− NCI‐H929 xenografted NOD‐scid IL2Rgamma mice. Our in vitro, ex vivo, and in vivo data provide the rationale for combined CTPS1 and ATR inhibition for the treatment of p53‐deficient patients.
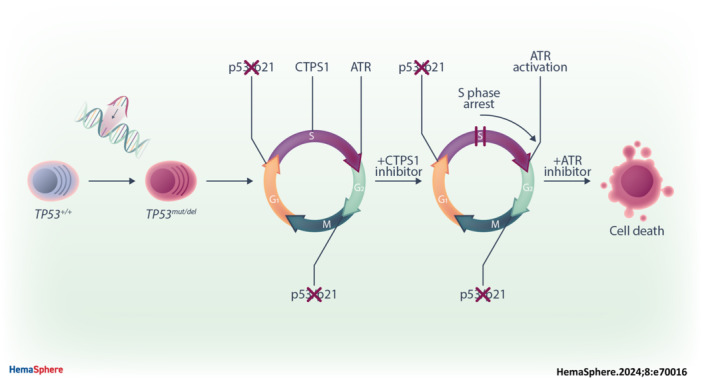
INTRODUCTION
Over the past decade, impressive therapeutic progress has been made in the treatment of patients with multiple myeloma (MM), thanks to the introduction of antibodies (such as anti‐CD38, drug conjugate, and bispecific) or the development of cell therapies. 1 , 2 , 3 , 4 , 5 These treatments have increased response rates and the duration of response, even in refractory/resistant patients. However, it remains unclear whether these new treatments will effectively overcome resistance in patients with TP53 defects, characterized by the deletion of the 17p locus (del17p) and/or TP53 mutation. 6 In the meantime, treatment better adapted to these patients is still needed, as serial relapses, after initially efficient treatment, ultimately lead to the selection of cells with a greater number of TP53 hits. 7 , 8
Abnormalities in TP53 lead to the loss of p53 function, which is crucial for cellular response to stress. 9 While the loss of p53 response is an advantage for cancer cells to escape apoptosis, it also reveals other vulnerabilities that can be targeted: cells with TP53 hits indeed fail to survive stress such as viral infection, or massive exposure to ROS. 9 , 10 , 11 p53‐deficient cells also lose p53‐mediated cell cycle checkpoints, but retain the S/G2 checkpoints, which was recently shown to be controlled by ATR. ATR stops cells from entering the G2 phase until their replication is complete, by preventing FOXM1 phosphorylation, and its inhibition precipitates replication‐defective cells into mitotic catastrophe. 12 , 13 By abrogating control of the cell cycle checkpoint, loss of p53 may offer a vulnerability to the inhibition of ATR, which plays a central role in orchestrating the molecular response to replication stress. 14 Targeting replication stress in cancer using ATR, CHK1, or WEE1 inhibitors is under clinical evaluation, with or without chemotherapy, or targeted therapies interfering with nucleotide and DNA synthesis. 15 Among them, CTP synthase 1 (CTPS1) inhibitors appear to be of particular interest in cancers of lymphoid origin. Indeed, CTPS1, which is a rate‐limiting step enzyme in de novo CTP synthesis, is mainly expressed in lymphoid cells, is involved in activation‐induced lymphoid cell proliferation, and is overexpressed in tumor cells of lymphoid origin. 16 , 17 , 18 Of note, in mantle cell lymphoma, CTPS1 expression was shown to inversely correlate to patient survival. 19 Although CTPS1 and its homolog CTPS2 catalyze the CTP de novo synthesis, only CTPS1 is necessary for the maintenance and growth of lymphoid T and B cells. 18 , 20 Recently, STP‐B, a molecule that selectively targets CTPS1 and which was initially developed to inhibit lymphocyte proliferation in immune disorders, has recently shown significant efficacy in vitro in both lymphoma and myeloma cell lines. 18 , 21 , 22 A compound from the same chemical series is currently being evaluated in a clinical trial (NCT05463263).
In this article, we have evaluated the therapeutic potential of STP‐B‐mediated CTPS1 targeting in TP53‐deficient myeloma cells in combination with ATR inhibition, in vitro in TP53 +/+/TP53 −/−/TP53 mut CRISPR/Cas9 isogenic myeloma cell lines, ex vivo in del17p patient samples, and in vivo in TP53 −/− myeloma xenografted mice.
MATERIALS AND METHODS
Human myeloma cell lines and patient samples
Samples from MM patients were obtained at the University Hospital (MYRACLE cohort, NTC03807128). 23 The isolation of mononuclear cells, purification of myeloma cells, and assessment of del17p status by fluorescence in situ hybridization, as well as the characterization of human myeloma cell lines (HMCLs), have all been reported previously. 24 , 25 Routine authentication of HMCLs was carried out using a flow cytometry process that has been previously reported. 26 Interleukin‐6‐dependent and ‐independent myeloma cells were cultured in RPMI‐1640 with 5% foetal calf serum supplemented or not with 3 ng/mL IL‐6.
Generation of TP53 −/− and TP53 R175H/R175H HMCLs
For NCI‐H929 and XG7 cell lines, TP53 −/− clones were obtained by CRISPR/Cas9 as previously described using two lentiviruses that contain Cas9 or an inducible single‐guide RNA (sgRNA) directed against the TP53 codons 157 and 158, after transient culture with 10 μM Nutlin3a to kill unmodified TP53 +/+ cells, prior to cloning by limiting dilution. 27 Control TP53 +/+ clones (n = 3) were obtained from noninduced infected cells after limiting dilution. TP53 R175H/R175H XG7 clones (n = 3) were obtained through homologous recombination following the manufacturer's instructions (Integrated DNA Technologies [IDT]): 500,000 cells were transiently electroporated (Nucleofector II) in the presence of Cas9 electroporation enhancer (IDT), with an RNP complex, that is, 18 pmol Cas9 nuclease preincubated with 22 pmol AATO550‐conjugated tracerRNA and 22 pmol TP53 specifically methylated sgRNA (5′‐mAmGmCACAATGACGGAGGTTGTG‐3′), and a DNA donor containing the R175H (A) and silent mutations (CCC) in codons 173 and 174 in order to prevent additional cleavage (5′‐CATGGCCATCTACAAGCAGTCACAGCACATGACGGAGGTTGTCCGCCACTGCCCCCACCATGAGCGCTGCTCAGATAGCGATGGTGAG‐3′). After electroporation, cells were cultured in complete medium in the presence of 1 μM HDR enhancer V2 to prevent NHEJ recombination during 24 h (IDT). After 3 days, cells were transiently cultured for 2 days with 10 μM Nutlin3a to kill TP53 +/+ unmodified XG7 cells prior to cloning by limiting dilution. Clones were characterized for TP53 sequence and overexpression of p53 protein to select three independent clones harboring the R175H mutation.
Reagents and antibodies
STP‐B was provided by Step Pharma. 28 , 29 The pan‐caspase inhibitor Q‐VD‐OPh, NAC (N‐acetyl cysteine), and the ATR inhibitors VE‐821 and AZD6738 were purchased from Sigma‐Aldrich or MedChemExpress. Anti‐CD138‐PE monoclonal antibody (mAb) came from Beckman Coulter, Annexin‐V‐APC from Immunotools, anti‐BrdU‐AF647 mAb from BD Pharmingen, and γ‐H2AX‐AF647 and antipuromycin mAb from Millipore.
Cell death assays
Myeloma cells were treated with STP‐B (1–10,000 nM) alone or in combination with VE‐821 (1.25–5 μM) for 24–72 h. Cell viability was assessed by CellTiter‐Glo assay. Cell death was assessed using Annexin‐V staining (HMCLs) or loss of CD138 staining (patient samples) with flow cytometry. 10 , 24
Transcriptomic analysis
Genomic profiling of HMCLs, clones, or patient samples (MYRACLE) was determined by 3′ Seq RNA profiling (SRP), as described previously. 27 Single‐cell RNA‐sequencing (scRNA‐seq) analysis of patient samples has been previously described (EGA50000000044). 27 Expression profiling analysis was carried out on 684 patient samples from the MMRF‐CoMMpass cohort (https://themmrf.org) and 139 patients in the CASSIOPEA clinical trial. 30
Functional p53 score
A functional 8‐gene p53 score, based on the ranked expression of eight p53‐target genes, that is, APOBEC3H, BAX, CDKN1A, DDB2, EDA2R, MDM2, PHLDA3, and RPS27L, was calculated using the rank‐based gene set scoring method with the singscore package in R, as previously described, with minor adaptations (the 8‐gene score was adapted from the 13‐gene score). 27 , 31
MM xenograft model
NCI‐H929 TP53 −/− cells were engrafted subcutaneously in NOD‐scid IL2Rgamma (NSG) mice (1 × 106 cells/mouse). After 1 week, mice with a tumor volume of 100 mm3 were randomly distributed into four groups of seven mice that were treated, or not treated, with STP‐B (30 mg/kg) injected subcutaneously and/or with AZD6738 (25 mg/kg) delivered by oral gavage. STP‐B was formulated in 10% benzyl alcohol and 90% castor oil, while AZD6738 was prepared in 10% DMSO, 40% PEG300, 5% Tween‐80, and 45% saline solution.
Statistical analysis
Statistical analyses were carried out using the nonparametric Mann–Whitney, Kruskal–Wallis, Friedman, and the parametric two‐way analysis of variance tests, as indicated in the legends of the figures. Survival analyses were carried out using the log‐rank (Mantel–Cox) test. Statistical significance is indicated in the figures with symbols *p < 0.05, **p < 0.01, ***p < 0.001, and ****p < 0.0001.
RESULTS
Increased CTPS1 expression is associated with proliferation and TP53 deficiency in myeloma cells
CTPS1 and CTPS2 expression was analyzed in 684 MM patients from the MMRF‐CoMMpass cohort, characterized for both del17p and TP53 mutation, and in 139 patients from the CASSIOPEA clinical trial including 137 characterized for del17p. 27 In MMRF‐CoMMpass patients, CTPS1, and not CTPS2, expression was significantly higher in 58 of the samples with TP53 hits, that is, del17p (n = 34), del17p and TP53 mutation (n = 19), or TP53 mutation (n = 5), compared to 626 samples without (p = 0.0007 and p = 0.3, respectively; Figure 1A). Similarly, in CASSIOPEA patients, CTPS1, and not CTPS2, expression was significantly higher in 12 samples with del17p versus 125 without (p = 0.0039 and p = 0.83, respectively; Figure 1A). To determine whether high CTPS1 expression in the TP53 abnormal cells was related to p53 or to proliferation, we analyzed the expression of MKI67 and CCNB1 and used a functional score to reflect p53 transcriptomic activity (p53 score). In both cohorts, MKI67 (p < 0.0001 and p = 0.0006 in MMRF‐CoMMpass and CASSIOPEA, respectively) and CCNB1 (p < 0.0001 and p = 0.0006 in MMRF‐CoMMpass and CASSIOPEA, respectively) expression was higher in TP53 abn samples compared to TP53 wt samples (Figure 1A). CTPS1 and MKI67 expression correlated in both cohorts independently of TP53 status (Figure 1B, upper panels). Using a functional 8‐gene p53 score derived from the 13‐gene score we previously reported (Figure S1), 27 we further evaluated whether CTPS1 expression could reflect p53 activity: indeed, CTPS1 expression and the p53 score showed significant correlation in samples from patients in MMRF‐CoMMpass and CASSIOPEA (p < 0.0001 and p = 0.007, respectively), and the correlation was restricted to samples with a normal TP53 status (Figure 1B, middle panels). Interestingly, the p53 score and MKI67 expression positively correlated in the TP53 wt samples in both cohorts and negatively correlated in the TP53 abn samples in MMRF‐CoMMpass (the weak del17p number in CASSIOPEA excluded definitive analysis; Figure 1B, lower panel). These results might suggest that a high p53 score in TP53 wt samples reflects a high proliferation rate, while a low p53 score in TP53 abn samples reflects low p53 transcriptomic activity despite the high proliferation rate. Taken together, these results showed that samples with high proliferation rates and/or abnormal TP53 status displayed high CTPS1 expression.
Figure 1.
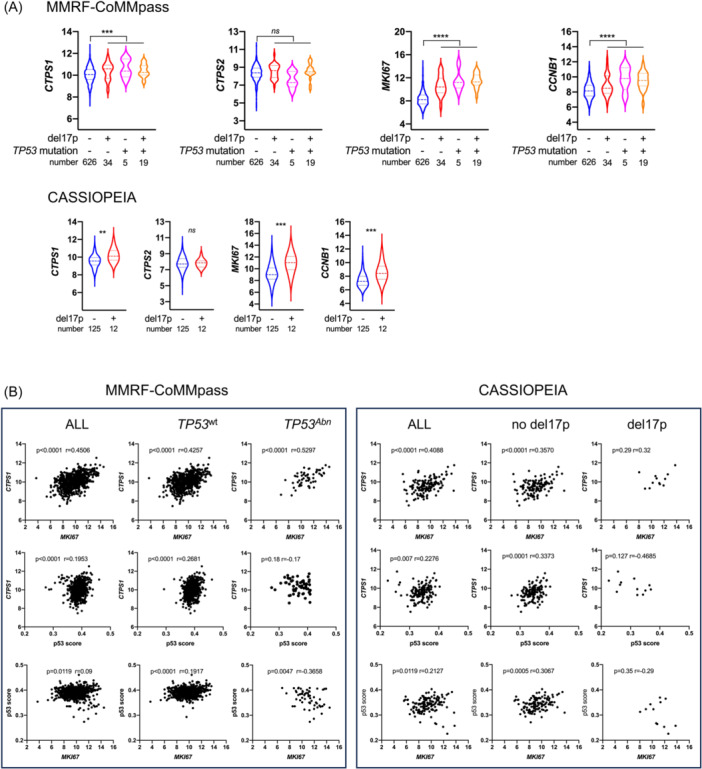
CTP synthase 1 (CTPS1) expression is associated with proliferation and is increased in TP53 abnormal myeloma cells. (A) TP53 abnormal myeloma cells overexpress CTPS1, MKI67, and CCNB1. Upper panel: Expression of CTPS1, CTPS2, MKI67, and CCNB1 was assessed by RNA‐seq in myeloma cells from 684 patients (MMRF‐CoMMpass) annotated for TP53 status (TP53 deletion and/or TP53 mutation). TP53 status was considered abnormal when a VAF was inferior to −0.5 (deletion) or superior to 0.3 (mutation). Lower panel: Expression of CTPS1, CTPS2, MKI67, and CCNB1 was assessed by RNA‐seq in 137 myeloma samples (CASSIOPEIA) annotated for del17p. Del17p was considered positive when 50% of myeloma cells displayed 17p deletion. Statistical significance was determined using the Mann–Whitney test. (B) CTPS1 expression correlates with MKI67 expression or p53 score in patient samples. The expression of CTPS1 in the MMRF‐coMMpass or CASSIOPEIA cohort was analyzed according to MKI67 expression (upper panels), p53 score (middle panels) in all, TP53 wt , TP53 abn MMRF‐coMMpass samples or in all, del17p, and no del17p samples in CASSIOPEIA, respectively. Correlation between p53 score and MKI67 expression was assessed in all TP53 wt , TP53 abn MMRF‐CoMMpass samples or in all del17p, no del17p samples in CASSIOPEIA, respectively (lower panels). Statistical significance was determined using Spearman's test. Statistical significance is indicated in the figures with symbols **p < 0.01, ***p < 0.001 and ****p < 0.0001. ns, Not significant.
Lastly, to confirm this direct association between CTPS1 expression and proliferation in myeloma samples, we used scRNA‐seq technology. Myeloma cells (n = 55,114) were identified within 23 mononuclear bone marrow samples, and one pleural effusion sample, as reported previously (Figure S2). 27 Expression of CTPS1, MKI67, and CTPS2 was analyzed in 40,266, 11,936, and 2912 myeloma cells in G1, S, and G2/M phase, respectively (Figure 2A): CTPS1, but not CTPS2, was expressed throughout the cell cycle, with maximum expression in the S and G2/M phases (Figure 2B).
Figure 2.
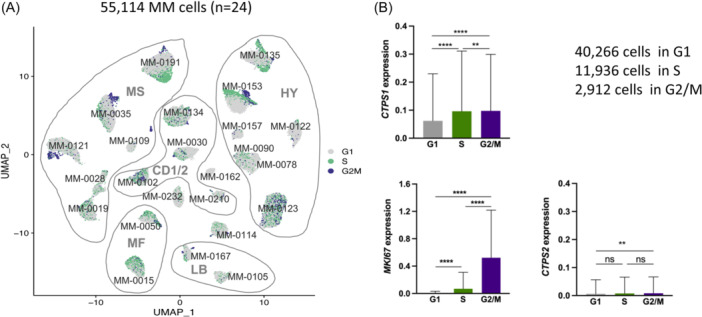
Myeloma cells in S and G2M phases overexpress CTP synthase 1 (CTPS1). (A) Myeloma cells in S and G2/M phases overexpress CTPS1 and not CTPS2. UMAP (uniform manifold approximation and projection) representation of myeloma cells from 24 samples (23 bone marrow and one pleural effusion). 27 The myeloma classification of samples is indicated (MS, CD‐1/2, MF, LB, and HY). Mononuclear cells from the bone marrow or pleural effusion (MM‐0191) of 24 multiple myeloma patients were cultured overnight in RPMI‐1640 with 10% fetal calf serum and 3 ng/mL interleukin‐6 before undergoing cell processing and single‐cell RNA‐sequencing analysis. Myeloma cells were identified as described previously. 27 Cell cycle signature was calculated using the AddModuleScore function of the Seurat package in R (gray: G1 phase; green: S phase; purple: G2/M phase). (B) The graphs represent the mean ± SD of CTPS1, MKI67, and CTPS2 expression in 55,114 myeloma cells according to the cell cycle phase. Statistical significance was determined using the Kruskal–Wallis test with Dunn's multiple comparison test. Statistical significance is indicated in the figures with symbols **p < 0.01 and ****p < 0.0001. ns, Not significant.
As expected, the patients with a high expression of MKI67 or CTPS1, and not of CTPS2, had significantly reduced survival in both cohorts, and multivariate Cox analysis showed that CTPS1 expression retained significance, independently from proliferation (Figure S3). Collectively, these results show that CTPS1 was highly expressed in proliferating MM cells, particularly in cells with TP53 hits, and that high CTPS1 levels were associated with poor outcomes, justifying its selective targeting in p53‐deficient cells.
TP53 status had no impact on CTPS1 expression in cell lines
To confirm that CTPS1 expression is not under the direct control of p53, in contrast to BAX expression, we analyzed CTPS1 and CTPS2 expression in 16 HMCLs and in 15 isogenic TP53 +/+, TP53 −/–, and TP53 R175H/R175H clones (Figure S4), using differential gene expression sequencing (DGE‐seq), as previously reported. 27 As shown in Figure 3A,B, CTPS1 was significantly expressed at higher levels than CTPS2 in both HMCLs and isogenic clones (p ≤ 0.001), and TP53 status did not significantly impact the constitutive CTPS1 expression (the under expression of CTPS2 in two TP53 −/− and eight TP53 −/mut HMCLs was not confirmed in the NCI‐H929 or XG7 TP53 isogenic clones, excluding the direct impact of p53 on CTPS2 expression). To assess the direct regulation by p53, we used Nutlin3a, an MDM2 inhibitor that prevents p53 degradation and, therefore, increases p53 expression. We showed that CTPS1 or CTPS2 expression was not regulated upon the activation of the p53 pathway, in contrast to BAX, a well‐known target of p53, whose expression was increased by Nutlin3a in a p53‐dependent manner (Figure 3B, lower panels). These results clearly confirmed that p53 did not directly control the expression of CTPS1 and CTPS2.
Figure 3.
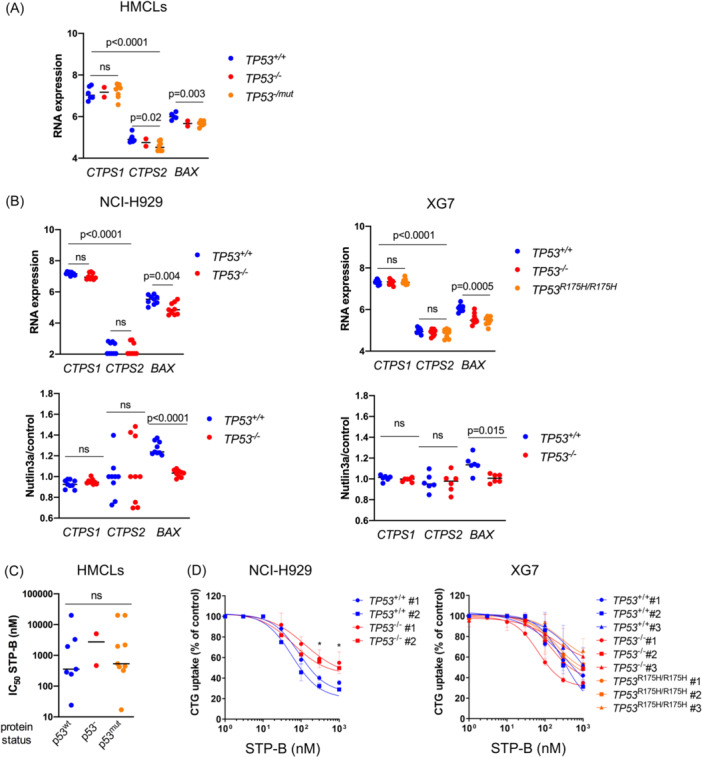
CTP synthase 1 (CTPS1) expression and response to CTPS1 inhibitor are not regulated by p53. (A) CTPS1, and not CTPS2, is highly expressed in TP53 abnormal myeloma cells. CTPS1, CTPS2, and BAX expression was assessed by SRP in 16 human myeloma cell lines (HMCLs). Statistical significance was determined using the Kruskal–Wallis and Mann–Whitney tests. (B) The expression of CTPS1 and CTPS2 is not regulated by p53. Upper panel: CTPS1, CTPS2, and BAX expression was assessed using SRP in the six isogenic NCI‐H929 clones (three TP53 +/+ and three TP53 −/− clones), as well as in the nine XG7 clones (three TP53 +/+, three TP53 −/−, and three TP53 R175H/R175H). Statistical significance was determined using the Kruskal–Wallis test. Lower panel: CTPS1, CTPS2, and BAX expression was assessed using SRP in the NCI‐H929 and XG7 clones treated, or not, with Nutlin3a (10 μM in NCI‐H929 and 2 μM in XG7) for 18 h. Results are expressed as the fold expression in Nutlin3a‐treated cells over untreated cells. Statistical significance was determined using the Kruskal–Wallis and Mann–Whitney tests. (C, D) TP53 abnormal myeloma cells are sensitive to CTPS1 inhibitor STP‐B. Response to STP‐B was assessed by CellTiter‐Glo assay after 72 h of culture in (C) 18 HMCLs and in (D) TP53 isogenic clones from NCI‐H929 or XG7, as indicated. STP‐B half‐maximal inhibitory concentration (IC50) values were compared in HMCLs according to their p53 protein status. STP‐B IC50 values of HMCLs and clones are detailed in Table 1. Results represent the mean of two to three independent experiments performed in triplicate. Statistical significance was determined using the Kruskal–Wallis and Mann–Whitney tests. ns, Not significant.
To assess the efficacy of targeting CTPS1 in TP53‐deficient myeloma cells, we used STP‐B, a selective inhibitor of CTPS1. 28 Treatment with STP‐B for 72 h resulted in a loss of CTG incorporation in all HMCLs tested (median IC50 = 500 nM), irrespective of p53 status (wild type, deleted, and mutated) (Figure 3C and Table 1). There was no significant difference in STP‐B IC50 values between TP53 +/+ and TP53 −/− or TP53 R175H/R175H clones from NCI‐H929 or XG7 (p = 0.21; Figure 3D and Table 1), although TP53 −/− clones from NCI‐H929 appeared less sensitive to a high STP‐B concentration (≥300 nM) than the control clones. These results showed that CTPS1 expression is independent of TP53 status in cell lines and that CTPS1 inhibition efficiently reduces the viability of p53‐deficient myeloma cells.
Table 1.
STP‐B IC50 values in HMCLs and isogenic clones.
| HMCLs | TP53 sequence | p53 WB | Cytogenetic group | STP‐B IC50 (nM) |
|---|---|---|---|---|
| TP53 wt | ||||
| AMO1 | +/+ | + (53 kDa) | OTHER | 285 |
| MDN | +/+ | + (53 kDa) | CCND1 | 24 |
| MM1S | +/+ | + (53 kDa) | MAF | 3287 |
| NAN11 | +/− | + (53 kDa) | MAF | >10,000 |
| NCI‐H929 | +/+ | + (53 kDa) | MS | 245 |
| XG6 | +/+ | + (53 kDa) | MAF | 1937 |
| XG7 | +/+ | + (53 kDa) | MS | 356 |
| TP53Abn | ||||
| JIM3 | R273C/− | + (53 kDa) | MS | 2181 |
| JJN3 | −/− | ‐ | MAF | ND |
| KMM1 | C135F/S241F | + (53 kDa) | OTHER | 424 |
| KMS12PE | R337L/− | + (53 kDa) | CCND1 | >10,000 |
| L363 | S261T/− | +/− (53 kDa) | MAF | 440 |
| LP1 | E286K/− | + (53 kDa) | MS | ND |
| NAN1 | E180STOP/− | ‐ | MAF | 5031 |
| NAN3 | R248Q/− | + (53 kDa) | MS | 1966 |
| NAN6 | Del exons 7–9/− | + (35 kDa) | MAF | 322 |
| NAN8 | D21Y/− | ‐ | MS | 466 |
| OPM2 | R175H/− | + (53 kDa) | MS | ND |
| U266 | A161T/− | + (53 kDa) | CCND1 | ND |
| XG2 | C176Y/R213STOP | + (53 kDa) | OTHER | 533 |
| XG5 | R282W/− | + (53 kDa) | CCND1 | >10,000 |
| XG11 | C135Y/− | + (53 kDa) | CCND1 | 17 |
| TP53 clones | ||||
| NCI‐H929 TP53 +/+ #1 | +/+ | + (53 kDa) | MS | 165 |
| NCI‐H929 TP53 +/+ #2 | +/+ | + (53 kDa) | MS | 93 |
| NCI‐H929 TP53 −/− #1 | −/− | ‐ | MS | 1043 |
| NCI‐H929 TP53 −/− #2 | −/− | ‐ | MS | 486 |
| XG7 TP53 +/+ #1 | +/+ | + (53 kDa) | MS | 552 |
| XG7 TP53 +/+ #2 | +/+ | + (53 kDa) | MS | 379 |
| XG7 TP53 +/+ #3 | +/+ | + (53 kDa) | MS | 1108 |
| XG7 TP53 −/− #1 | −/− | ‐ | MS | 129 |
| XG7 TP53 −/− #2 | −/− | ‐ | MS | 453 |
| XG7 TP53 −/− #3 | −/− | ‐ | MS | >10,000 |
| XG7 TP53 R175H/R175H #1 | R175H/R175H | + (53 kDa) | MS | 661 |
| XG7 TP53 R175H/R175H #2 | R175H/R175H | + (53 kDa) | MS | 991 |
| XG7 TP53 R175H/R175H #3 | R175H/R175H | + (53 kDa) | MS | >10,000 |
Note: STP‐B IC50 value in 18 HMCLs and 13 TP53 isogenic clones from NCI‐H929 and XG7 was determined using a 72‐h CellTiter‐Glo assay. TP53 status (mutation and/or deletion), cytogenetic group, and p53 status are indicated.
Abbreviations: HMCL, human myeloma cell line; IC50, half‐maximal inhibitory concentration; ND, not determined; WB, western blot.
CTPS1 inhibitor STP‐B induced early S‐phase cell cycle arrest in p53‐proficient and p53‐deficient cells
To characterize the functional consequences of selective CTPS1 inhibition in myeloma cells, transcriptomic analysis was carried out using SRP in 10 HMCLs selected to represent myeloma genomic heterogeneity and different TP53 statuses (Table 1). Following STP‐B treatment (STP‐B IC50 for 24 h), 11 genes were upregulated in STP‐B‐treated cells, and 31 were downregulated (false discovery rate <0.05; Figure 4A). Using the Reactome database, the functional annotation of the transcriptomic response to STP‐B in 10 HMCLs identified a significant decrease in the G2/M‐phase signature (PLK1, CCNB1, CDC20, and CEP63) and in the protein translation signature (RPL22, RPL23, RPL27, and RPS3A; Figure 4A,B). To further analyze the functional impact of CTPS1 inhibition on the cell cycle, NCI‐H929 and XG7 clones, as well as TP53 /−R213STOP myeloma cells from a patient with plasma cell leukemia (PCL#1), were incubated for 24 h with STP‐B, before bromodeoxyuridine (BrdU)/propidium iodide staining. As illustrated in Figure 4C and summarized in Figure 4D, STP‐B induced a significant accumulation of cells blocked in the early S phase in TP53 +/+, TP53 −/−, and TP53 R175H/R175H clones, as well as in TP53 −/R213STOP PCL#1 (mean increase 2.3‐fold, p = 0.001). The accumulation in the early S phase was associated with a consequent decrease in G1, late S, and G2/M phases (mean decrease 1.7‐, 1.3‐, and 3.1‐fold, respectively, p = 0.001). Taken together, these results suggest that CTP depletion resulting from CTPS1 inhibition induced early S‐phase arrest, irrespective of TP53 status.
Figure 4.
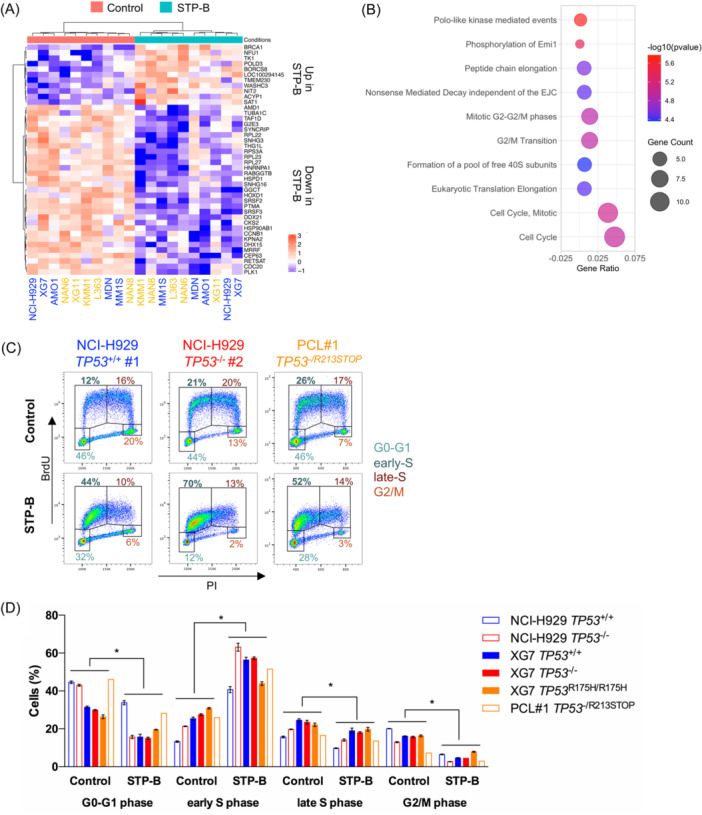
STP‐B induces early S‐phase cell cycle arrest in myeloma cells regardless of their TP53 status. (A) STP‐B modulates the expression of 42 genes in myeloma cell lines Transcriptomic profiling was performed using SRP in 10 human myeloma cell lines (HMCLs) (five TP53 wt HMCLs annotated in blue and five TP53 abnormal HMCLs in yellow; see Table 1). HMCLs were treated for 24 h with STP‐B IC50 before RNA extraction and sequencing. Genes with significant differential expression (log 2 fold change <−0.5 or >0.5 and adjusted p < 0.05) were selected and shown in the supervised hierarchical clustering (control vs. STP‐B). (B) CTPS1 inhibition affects transcriptional programs related to cell cycle and translation. Pathway enrichment analysis was conducted using Reactome. The graph represents the 10 pathways most significantly affected by STP‐B treatment. (C, D) STP‐B induces early S‐phase cell cycle arrest in TP53 abnormal clones and in a patient sample. Cells were treated for 24 h with 200 nM STP‐B before incubation with bromodeoxyuridine (BrdU) from 30 (clones) to 90 min (patient sample). Cells were then fixed, permeabilized, stained with anti‐BrdU antibody and propidium iodide (PI), and analyzed using flow cytometry. (C) Representative BrdU/PI profiles of two NCI‐H929 clones (TP53 +/+ #1 and TP53 −/− #2) and of myeloma cells derived from a patient with plasma cell leukemia (PCL) with an abnormal TP53 status (PCL#1 TP53 −/R213STOP). (D) The bar graph summarizes the percentage of cells in G1, early S, late S, and G2/M phases in TP53 isogenic clones from NCI‐H929 and XG7 and in the PCL#1 patient sample treated or not with STP‐B. Statistical significance was determined using the Wilcoxon test. Statistical significance is indicated in the figures with symbol *p < 0.05.
Simultaneous CTPS1 and ATR targeting showed synergistic efficacy in TP53‐deficient myeloma cells
As STP‐B efficiently induced S‐phase arrest in p53‐deficient cells, we thought about inhibiting ATR, the S/G2 cell cycle checkpoint, so as to induce mitotic catastrophe and cell death. 12 ATR is of particular interest in MM as it is rarely deleted or mutated, and myeloma cells have been shown to be addicted to ATR. 32 , 33 Transcriptomic analysis showed that TP53 status did not impact ATR expression in HMCLs, isogenic myeloma clones, or patient samples (Figure 5A–C). Single‐cell analysis further showed that ATR was well‐expressed irrespective of cell cycle phase cells (Figure 5D). We next used VE‐821, a selective ATP‐competitive inhibitor of ATR, which has minimal activity against ATM, to assess the efficacy in combination with STP‐B. 33 Indeed, 200 nM STP‐B with 2.5 µM VE‐821 induced 37.5% and 53.5% cell death in four TP53 wt and five TP53 abnormal HMCLs, respectively (p = 0.9; Figure 6A, left panel). The efficacy of the combination was similar in NCI‐H929 and XG7 clones, irrespective of TP53 status, although it was slightly less effective in TP53 R175H/R175H compared to TP53 +/+ XG7 clones (mean death was 55% versus 64%, respectively; Figure 6A, middle and right panels).
Figure 5.
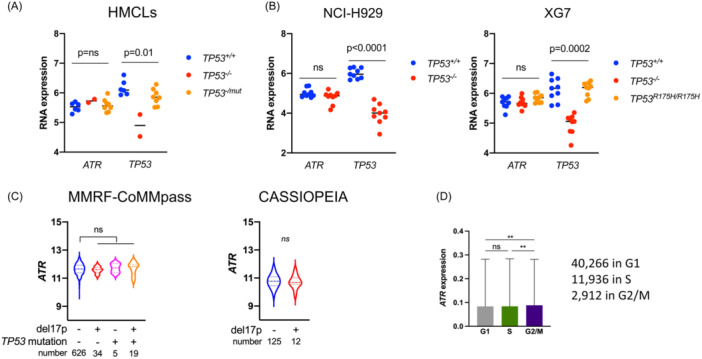
ATR is expressed in multiple myeloma (MM) cells irrespective of TP53 status and cell cycle. (A) ATR expression was analyzed in TP53 +/+, TP53 −/−, or TP53 −/mut human myeloma cell lines using SRP. (B) ATR expression was analyzed in TP53 +/+, TP53 −/−, or TP53 R175H/R175H XG7 and/or NCI‐H929 clones using SRP 8B. (C) ATR expression was analyzed according to the presence of del17p and/or TP53 mutation in the MMRF‐CoMMpass cohort and according to the presence of del17p in the CASSIOPEIA cohort. (D) ATR expression in the 55,114 MM cells (see Figure 2) according to the cell cycle phase (mean ± SD). Statistical significance was determined using the Kruskal–Wallis test with Dunn's multiple comparison test. Statistical significance is indicated in the figures with symbol **p < 0.01. ns, Not significant.
Figure 6.
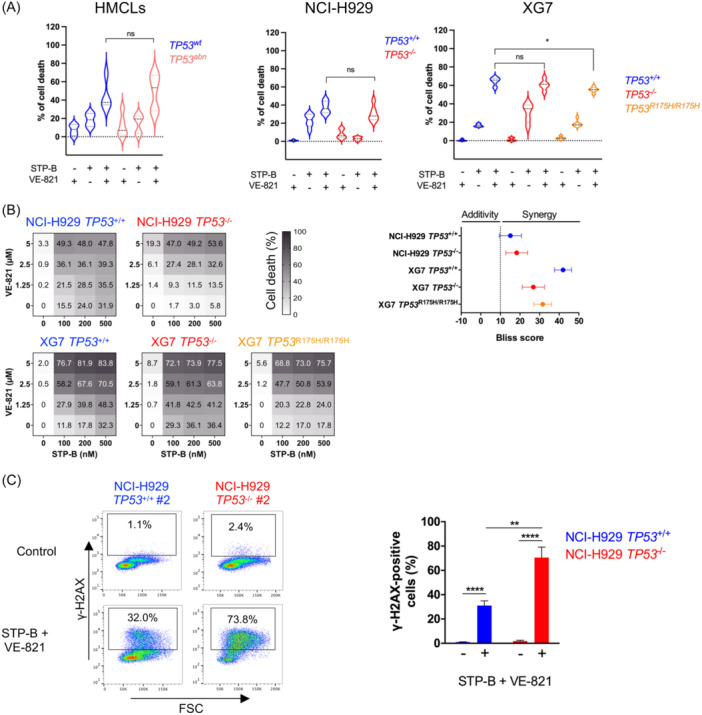
The inhibition of CTPS1 and ATR is highly synergistic in p53‐deficient myeloma cells in vitro. (A) The STP‐B and VE‐821 combination is efficient in TP53 abnormal human myeloma cell lines (HMCLs) and in isogenic clones. Left panel: Four TP53 wt HMCLs (AMO1, BCN, XG6, and XG7) and five TP53 abnormal HMCLs (Karpas620, KMS12PE, NAN8, U266, and XG11) were treated for 72 h with 200 nM STP‐B, 2.5 μM VE‐821 or both (except XG11, which was treated with 10 nM STP‐B and 1 μM VE‐821). Cell death was assessed by Annexin‐V staining using flow cytometry. Statistical analysis was performed using the Mann–Whitney test. Middle and right panels: TP53 +/+ (n = 2) and TP53 −/− (n = 2) clones from NCI‐H929, as well as TP53 +/+ (n = 2), TP53 −/− (n = 2) and TP53 R175H/R175H (n = 3) clones from XG7, were treated for 72 h with or without 200 nM STP‐B and/or 2.5 μM VE‐821, and cell death was assessed with Annexin‐V staining using flow cytometry. The data represents the results of two independent experiments. Statistical analyses were performed using the Mann–Whitney test. (B) STP‐B and VE‐821 combination highly synergizes in TP53 abnormal isogenic clones. Left panel: TP53 +/+ (n = 2) and TP53 −/− (n = 2) clones from NCI‐H929, as well as TP53 +/+ (n = 2), TP53 −/− (n = 2), and TP53 R175H/R175H (n = 3) clones from XG7, were treated for 72 h with increasing concentrations of STP‐B (100, 200, and 500 nM) and/or VE‐821 (1.25, 2.5, or 5 μM). Cell death was assessed using Annexin‐V staining (left panels). Right panel: Bliss synergy scores were calculated using SynergyFinder. Data represent the mean ± SD of two to three experiments. (C) STP‐B and VE‐821 combination induces DNA damage. Cells were treated for 48 h with 200 nM STP‐B and 2.5 µM VE‐821, permeabilized and stained with AF647‐conjugated anti‐γ‐H2AX antibody. Fluorescence was assessed by FACs analysis. Left panel: Data are representative of one experiment. Right panel. The percentage of γ‐H2AX‐positive cells was determined in two TP53 +/+ (#1, #2) and two TP53 −/− (#1, #2) NCI‐H929 clones. The graph represents the mean ± SD of three independent experiments. Statistical significance is indicated in the figures with symbols *p < 0.05, **p < 0.01 and ****p < 0.0001. ns, Not significant.
The STP‐B/VE‐821 combination proved highly synergistic, as shown by the Bliss scores, which were above 15 for all the clones (Figure 6B). The strong cytotoxicity of the combination was associated with γ‐H2AX staining, indicating an increase in DNA strand breaks, a hallmark of mitotic catastrophe (Figure 6C).
Simultaneous targeting of CTPS1 and ATR induced a caspase‐dependent dell death and disrupted protein translation
To further characterize cell death induced by STP‐B and VE‐821, we first assessed whether cell death was strictly related to CTPS activity by complementing the culture medium with CTP to counteract inhibition of the de novo CTP synthesis induced by STP‐B. 21 Indeed, daily addition of 200 μM of CTP highly prevented cell death induced by STP‐B alone or in combination with ATR (Figure 7A): taking all TP53 +/+, TP53 −/− NCI‐H929 and XG7 clones together, the mean inhibition of cell death induced by STP‐B alone or in combination with VE‐821 was 102.4% ± 7.5% and 92.6% ± 14.4%, respectively.
Figure 7.
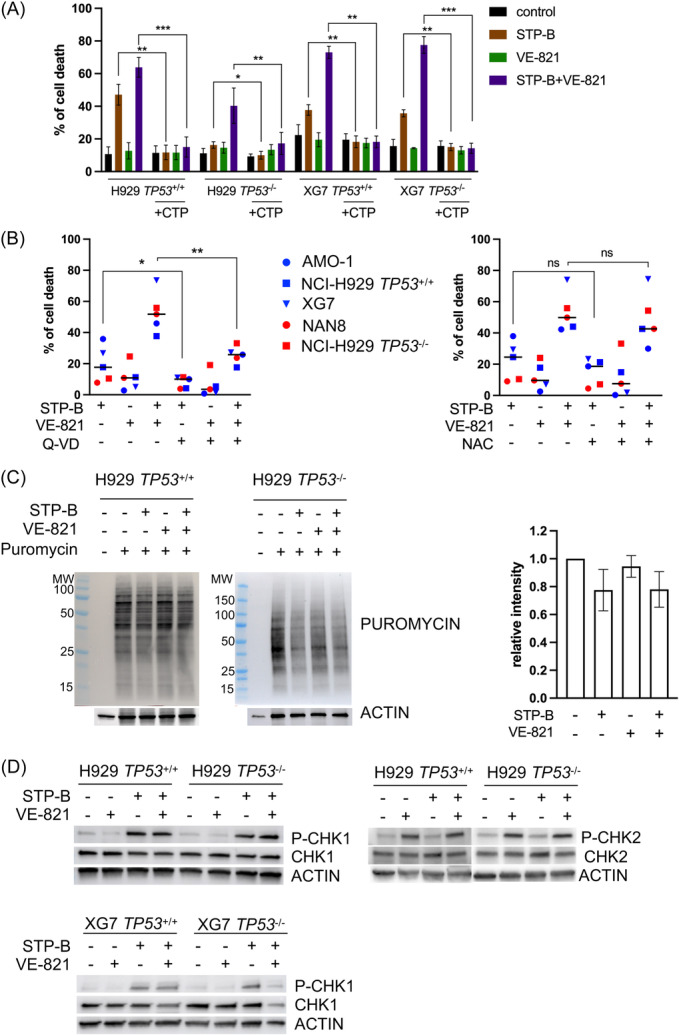
Cell death induced by STP‐B and VE‐821 is strictly dependent on CTP and involves caspase activation. (A) The addition of CTP inhibits cell death induced by STP‐B alone or in combination with VE‐821. Cells were treated for 72 h with 200 nM STP‐B, 2.5 μM VE‐821, or both, and CTP (200 μM) was added daily to the culture medium. Cell death was assessed by Annexin‐V staining using flow cytometry. The graph represents the mean ± SD of three to four independent experiments. Statistical analyses were performed using the paired t‐test. (B) Cell death induced by STP‐B alone or in combination with VE‐821 is partly inhibited by the pan‐caspase inhibitor Q‐VD and not impaired by N‐acetyl cysteine (NAC). Cells were treated for 72 h with 200 nM STP‐B and 2.5 µM VE‐821 alone or in combination with or without 50 µM Q‐VD (left panel) or 5 mM NAC (right panel). The graph represents the mean of cell death induced in AMO1, NAN8, NCI‐H929 TP53 −/−, and NCI‐H929 TP53 +/+ and XG7 (n = 2 experiments for each cell line and clone). Cell death was assessed by Annexin‐V staining using flow cytometry. Statistical analysis was performed using the paired t‐test. (C) STP‐B alone or in combination with VE‐821 inhibits protein synthesis. Cells were treated overnight with 200 nM STP‐B and 2.5 µM VE‐821 alone or in combination, and 1 μM puromycin was added 30 min prior to lysis, protein extraction, and western blotting, as previously described. 22 (D) STP‐B and VE‐821 induce CHK1 and CHK2 phosphorylation. Cells were treated for 24 h with 200 nM STP‐B and 2.5 µM VE‐821 alone or in combination, prior to lysis, protein extraction, and western blotting. Statistical significance is indicated in the figures with symbols *p < 0.05, **p < 0.01 and ***p < 0.001. ns, Not significant.
We next determined the involvement of caspase or ROS in cell death by using a caspase inhibitor or an ROS scavenger, the impact on protein translation by measuring puromycin incorporation, and the induction of replicative stress by assessing CHK1 and CHK2 phosphorylation.
The addition of the pan‐caspase inhibitor Q‐VD inhibited cell death induced by STP‐B alone or in combination with VE‐821 by about 50% in five HMCLs or clones tested: STP‐B‐induced cell death was 8% ± 2% versus 20% ± 5% in Q‐VD versus control, p = 0.06, and STP‐B+VE‐821‐induced cell death was 25.5% ± 2.5% versus 53% ± 6% in Q‐VD versus control, p = 0.0051, respectively, indicating the involvement of caspase in cell death in the five HMCLs or clones tested (Figure 7B, left panel). In contrast, the addition of 5 mM NAC did not inhibit cell death, excluding the involvement of ROS (Figure 7B, middle panel).
As expected, puromycin incorporation in TP53 +/+ or TP53 −/− NCI‐H929 cells was decreased by STP‐B, confirming a decrease in protein translation (23% ± 10%) that was not further modified by VE‐821 (22% ± 0.09%; Figure 7C). 22 In both TP53 +/+ and TP53 −/− NCI‐H929 and XG7 cells, STP‐B induced a strong CHK1 phosphorylation that was not inhibited by VE‐821 at 24 h. Of note, VE‐821 alone or with STP‐B induced P‐CHK2, suggesting an activation of ATM pathway (Figure 7D). 33
Combined inhibition of CTPS1 and ATR was efficient in p53‐deficient cells ex vivo and in vivo
The efficacy of the combination was then assessed, ex vivo, in bone marrow and peripheral blood samples from 17 newly diagnosed (D, n = 5), relapsed (R, n = 6), and resistant/refractory (RR, n = 6) patients. After 72 h of culture with 200 nM STP‐B, 2.5 μM VE‐821, or a combination of them, median cell death was 2.2% (0–24.9), 12.6% (0–28.7), and 18.3% (0–59.9), respectively (Figure 8A). Cell death induced by the combination was additive to synergistic in 17 samples and highly effective in RR samples (median of cell death was 38.6%; Table 2). In the 15 samples with known del17p status (five with del17p and 10 without), the median death was 42.9% versus 7.45% in samples with del17p compared to those without del17p (p = 0.008) and the combination was synergistic in the five del17p samples (p = 0.02) (Figure 8B). The efficacy of the combination in del17p samples prompted us to assess the efficacy of STP‐B and AZD6738 in vivo, with a clinical grade, orally available inhibitor of ATR, using xenografted TP53 −/− NCI‐H929 NSG mice. 34 The mice were treated over 2 weeks, 5 days a week, and then left untreated for 2 days (Figure 8C), middle panel). Each drug induced a significant delay in tumor growth as single agents, and their combination was highly effective: between Days 1 and 12, the increase in tumor size was 10.0‐, 6.4‐, 3.5‐fold, and 1.6‐fold in control, STP‐B, AZD6738, and combo groups, respectively (Figure 8C, middle and right panel). Both single and combined compounds were well‐tolerated, with body weight variations of less than 10% (Figure S5).
Figure 8.
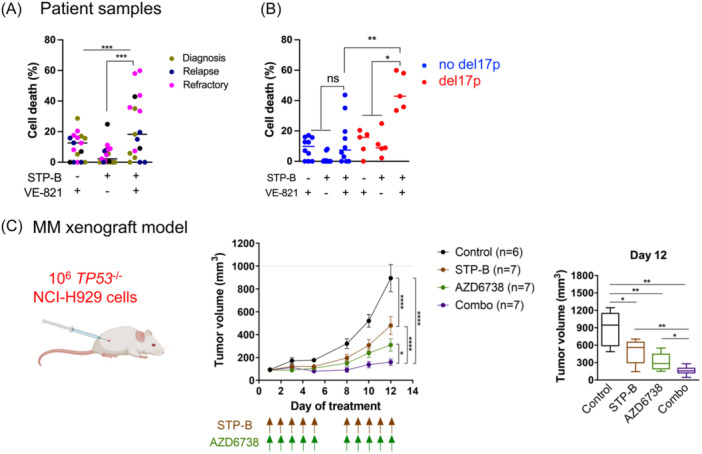
The inhibition of CTP synthase 1 (CTPS1) and ATR is synergistic in p53‐deficient myeloma cells ex vivo and in vivo. (A) The STP‐B and VE‐821 combination is efficient in del17p patient samples. Bone marrow (n = 15) or peripheral blood (n = 2) samples from patients with multiple myeloma at different stages (n = 5 diagnosis, n = 6 relapse, or n = 6 resistant/refractory) were treated for 72 h with 200 nM STP‐B, 2.5 μM VE‐821, or both. Cell death was assessed by the loss of CD138 staining using flow cytometry. Left panel: The graph represents the results in the 17 samples. (B) The graph represents the results in 15 samples according to their del17p status. Statistical analyses were performed using the Friedman test with Dunn's multiple comparisons test (left panel) or the Mann–Whitney test and paired t‐test (right panel). (C) STP‐B and ATR inhibitor AZD6738 prevent NCI‐H929 TP53 −/− cell growth in NSG mice. NSG mice were subcutaneously injected with 106 NCI‐H929 TP53 −/− cells (left panel). After 1 week, mice with detectable tumors were randomly distributed into four groups of seven mice and treated with STP‐B (30 mg/kg), AZD6738 (25 mg/kg), or both, as indicated by the arrows (middle panel). The graphs represent the tumor volume kinetics (mean ± SD, middle panel) and the tumor volume on Day 12 (right panel). Statistical analyses were performed using the two‐way analysis of variance with Tukey's multiple comparisons (middle graph) or the Mann–Whitney test (right graph). Statistical significance is indicated in the figures with symbols *p < 0.05, **p < 0.01 and ***p < 0.001. ns, Not significant; NSG, NOD‐scid IL2Rgamma.
Table 2.
Characteristics of patient samples.
| Characteristics of patient samples | Myeloma cell death (%) | |||||||
|---|---|---|---|---|---|---|---|---|
| Samples | Disease | Stade | 17p | Cytogenetic | CD138 (%) | VE‐821 | STP‐B | Combo |
| MM#1 | MM | D | t(4;14) | 43.0 | 17.1 | 0 | 3 | |
| MM#2 | MM | D | ND | 9.0 | 0 | 0 | 0 | |
| MM#3 | MM | D | ND | 4.5 | 5.2 | 0 | 5.8 | |
| MM#4 | MM | D | ND | t(4;14)− | 28.0 | 28.7 | 5.5 | 18.3 |
| MM#5 | MM | D | t(11;14)− t(4;14)− | 38.0 | 15.7 | 0 | 35.1 | |
| MM#6 | MM | R | t(4;14) | 3.5 | 0 | 0 | 0 | |
| MM#7 | MM | R | t(11;14) | 5.5 | 12.7 | 8.1 | 15.0 | |
| MM#8 | MM | R | del17p | t(11;14)− t(4;14)− | 30.0 | 15.8 | 11.2 | 35.8 |
| MM#9 | MM | R | t(11;14) | 35.0 | 0 | 0 | 0 | |
| MM#10 | MM | R | t(11;14) | 16.0 | 7 | 0.5 | 9.1 | |
| MM#11 | MM | R | del17p | t(11;14)− t(4;14)− | 6.0 | 0 | 24.9 | 42.9 |
| MM#12 | MM | RR | ND | 22.0 | 16.0 | 7.3 | 19.6 | |
| MM#13 | MM | RR | ND | ND | 35.0 | 0 | 4.9 | 7.4 |
| MM#14 | MM | RR | t(11;14)− | 20.0 | 12.6 | 0.6 | 43.7 | |
| MM#15 | MM | RR | del17p | ND | 43.0 | 17.6 | 2.2 | 33.5 |
| PCL#1 | PCL | RR | del17p | t(14;20) | 95.0 | 8.2 | 8.4 | 59.9 |
| PCL#2 | PCL | RR | del17p | ND | 10.0 | 20.4 | 8.9 | 58 |
Note: Bone marrow or peripheral blood samples were collected from patients with MM or PCL, respectively. Chromosomal abnormalities (del17p and translocation) were assessed by fluorescence in situ hybridization. Myeloma cells were identified by CD138 staining with flow cytometry. Cell death was determined by the loss of CD138+ cells after 72 h of culture with or without STP‐B (200 nM) and/or VE‐821 (2.5 μM).
Abbreviations: D, diagnosis; MM, multiple myeloma; ND, not determined; PCL, plasma cell leukemia; R, relapse; RR, refractory/resistant.
DISCUSSION
The aim of this study was to determine whether the inhibition of CTPS1, an enzyme critical for CTP synthesis and essential for sustaining DNA synthesis in normal and malignant lymphoid cells, could constitute an actionable vulnerability in TP53‐deficient myeloma cells. 16 , 17 , 18 CTPS1 was indeed overexpressed in patient samples with del17p and/or TP53 mutations, compared to samples without TP53 hits in two different patient cohorts (MMRF‐CoMMpass and CASSIOPEIA). CTPS1 expression correlated with MKI67 and CCNB1 expression, both of which were increased in patient samples with del17p and/or TP53 mutation. The analysis of CTPS1 and MKI67 expression and the p53 score values showed that high CTPS1 expression was synonymous with high MKI67 expression, which correlated to a p53 score positively in TP53 wt myeloma cells and negatively in TP53 Abnormal myeloma cells. The association between CTPS1 expression and proliferation was further confirmed at a single‐cell level in 24 patient samples, with CTPS1 expression being significantly higher in cells in the S and G2/M phases than in the G1 phase. We showed that patients with high CTPS1 or MKI67 expression had a shortened survival period in univariate and multivariate analyses. The high CTPS1 expression in TP53 abnnormal samples not only reflects their high proliferation rate but also constitutes a selective vulnerability. CTPS1 appears, therefore, to be a very interesting target for patients with a high‐risk presentation, that is, proliferation index with or without TP53 hits.
In vitro, the CTPS1 expression was similar in TP53 abnormal and TP53 wt HMCLs, and in isogenic TP53 +/+, TP53 −/−, and TP53 R175H/R175H clones as well, confirming that CTPS1 expression was not under p53 control. By using the CTPS1 pharmacological inhibitor STP‐B, we showed that STP‐B induced cell cycle arrest in the early S phase and synergized with the ATR inhibitor VE‐821 in TP53 wt and TP53 abnormal myeloma cell lines: this combination was efficient irrespective of p53 expression and function, whereas a single hit in TP53 induced resistance to chemotherapeutical agents, with this resistance being increased by biallelic hit. 35 Of note, we had previously found that isogenic TP53 −/− myeloma cells were 5–10‐fold more resistant to BH3 mimetics than TP53 wt, the resistance being caused by the decrease in BAX expression and subsequently in the amount of MCL1/BAX complexes in TP53 −/− cells. 27 Our current results show that cell death induced by the combined inhibition of CTPS1 and ATR overcomes the p53‐mediated apoptosis resistance and was independent of BAX expression (data not shown). These results clearly show that the CTPS1/ATR inhibition‐induced cell death is independent of p53‐BAX deficiency, which mediates treatment resistance. Such a combination might be of interest to p53‐deficient patients for whom there is still an unmet medical need.
We confirmed the high STP‐B specificity to inhibit CTP synthesis by showing that CTP complementation totally inhibited cell death by STP‐B with or without VE‐821. The effectiveness of STP‐B in preventing tumor growth in vivo suggests that tumor dependency on CTP synthesis is not overcome by the tumor microenvironment.
The STP‐B and VE‐821 combination induced replicative stress with strong phosphorylation of CHK1 and CHK2, indicating activation of the ATR and ATM pathways, which has recently been reported in glioma upon CTP deprivation. 36 It will be interesting to further decipher the signaling cascades induced by CTP deprivation in combination with inhibition of ATR, ATM, or DNA‐PK.
As regards, the mechanism of cell death, the lack of impact of NAC addition on cell death rules out a major role for ROS and ferroptosis, while the 50% inhibition of cell death induced by Q‐VD indicates an involvement of caspases and suggests activation of apoptosis. Moreover, STP‐B decreases protein translation, which impairs the synthesis of short half‐life proteins such as MCL1, which is a major survival protein in myeloma. 22 , 37
Ex vivo, combination was particularly efficient in samples from refractory/resistant patients and/or from patients with high‐risk features such as del17p and/or leukemic stage. Although the number of samples from high‐risk patients in this study was limited (n = 5), our results argue for further evaluation of the combination in this patient group. In vivo, in mice, we showed that the combination of STP‐B and AZD6738 was well‐tolerated, and it strongly inhibited the growth of NCI‐H929 TP53 −/− cells. The efficacy of STP‐B, in vivo, is under evaluation in several clinical trials. Although ATR is a well‐known, not‐new target for cancer therapy, the efficacy of its inhibition in vivo has been reported quite recently. 38 Several recent clinical trials assessing the tolerance of ATR inhibitors as single agents have reported good tolerance and even some antitumor efficacy. 39 , 40 However, the efficacy of ATR inhibition is clearly increased in combination with drugs that interfere with DNA replication or repair. We, and others, have previously reported that ATR inhibitors synergized with chemotherapies such as melphalan, to induce mitotic catastrophe in p53‐deficient myeloma cells. 32 , 41 While melphalan has wide‐ranging side effects, STP‐B is unlikely to be toxic to normal nonlymphoid cells since they do not express CTPS1, but CTPS2, against which STP‐B is not effective. The combination of ATR inhibitors with the dual inhibition of CTPS1 and CTPS2 was reported to induce synthetic lethality in MYC‐overexpressing cell lines, confirming the interest in targeting CTPS1 and ATR in cancer. 42 Inhibitors targeting other kinases involved in the replication stress (CHK1 and WEE1) were also shown to synergistically induce cell death with STP‐B in myeloma cell lines. 21 Inhibiting ATR appears particularly attractive in myeloma because ATR is well‐expressed, rarely deleted or mutated in patient samples at diagnosis or in HMCLs (which are derived from patients with the most aggressive presentation, i.e., extramedullary disease), and is expressed in patient samples with high‐risk features (high proliferation and/or low p53 score). Although we cannot rule out that the combined efficacy of STP‐B and ATR inhibitors ex vivo in patient samples may be, at least partially, related to the inhibition of STP‐B‐induced protein synthesis, which decreases the expression of critical proteins with short half‐life, such as MCL1 or CCND1, it seems unlikely that the combination with ATR inhibitors involved such mechanisms to induce cell death. 22 Nevertheless, multivariate analysis showed that the CTPS1 expression was predictive of reduced overall survival independently of MKI67 expression, suggesting that CTPS1 has other impacts than DNA synthesis on myeloma escape to therapies.
To summarize, our results show that the dual‐targeting of CTPS1 and ATR induces mitotic catastrophe in p53‐deficient patient myeloma cells and cell lines. This provides a rationale for developing therapies for refractory/resistant myeloma patients with TP53 deficiency, who can be identified using an 8‐gene p53 score or CTPS1 expression, and for whom there is still an unmet clinical need.
AUTHOR CONTRIBUTIONS
Romane Durand: Conceptualization; formal analysis; investigation; writing—original draft preparation. Céline Bellanger: Software; formal analysis. Géraldine Descamps: Investigation. Christelle Dousset: investigation. Sophie Maïga: Investigation. Jennifer Derrien: Software; formal analysis. Laura Thirrourad: Investigation. Louise Bouard: Conceptualization. Patricia Gomez‐Bougie: Writing—review and editing. Marie‐Claire Devilder: Methodology. Hélène Asnagli: Resources. Andrew Parker: Resources. Philip Beer: Resources; conceptualization. Philippe Moreau: Funding acquisition; resources. Cyrille Touzeau: Funding acquisition; resources. Agnès Moreau‐Aubry: Methodology. David Chiron: Conceptualization; writing—review and editing. Catherine Pellat‐Deceunynck: Supervision; conceptualization; formal analysis; investigation; writing—original draft preparation.
CONFLICT OF INTEREST STATEMENT
Hélène Asnagli, Andrew Parker, and Philip Beer are employees of Step Pharma. The remaining authors declare no conflict of interest.
FUNDING
This work was supported by grants from FFRMG, Ligue contre le Cancer du Grand‐Ouest (35, 44), INCA (INCa‐DGOS‐INSERM_12558, INCa‐DGOS‐INSERM‐ITMO Cancer_18011), and Action Cancer 44. R. D. was supported by SIRIC ILIAD and CALYM.
Supporting information
Supporting information.
Supporting information.
Supporting information.
Supporting information.
Supporting information.
ACKNOWLEDGMENTS
The authors acknowledge the Therassay core facility for its technical expertise and help. The authors acknowledge the Cytocell‐Flow Cytometry and FACS core facility (SFR Bonamy, BioCore, Inserm UMS 016, CNRS UAR 3556, Nantes, France) for its technical expertise and help, member of the Scientific Interest Group (GIS) Biogenouest and the Labex IGO program supported by the French National Research Agency (no. ANR‐11‐LABX‐0016‐01). The authors are grateful to the Genomics Core Facility GenoA, a member of Biogenouest and France Genomique, and to the Bioinformatics Core Facility BiRD, a member of Biogenouest and Institut Français de Bioinformatique (IFB) (ANR‐11‐INBS‐0013), for the use of their resources and their technical support. The coMMpass data were generated as part of the Multiple Myeloma Research Foundation (MMRF) Personalized Medicine Initiative (https://themmrf.org).
DATA AVAILABILITY STATEMENT
Data that support the findings of this study are available from the corresponding author upon reasonable request.
REFERENCES
- 1. Moreau P, Attal M, Hulin C, et al. Bortezomib, thalidomide, and dexamethasone with or without daratumumab before and after autologous stem‐cell transplantation for newly diagnosed multiple myeloma (CASSIOPEIA): a randomised, open‐label, phase 3 study. Lancet. 2019;394(10192):29‐38. [DOI] [PubMed] [Google Scholar]
- 2. Moreau P, Garfall AL, van de Donk NWCJ, et al. Teclistamab in relapsed or refractory multiple myeloma. N Engl J Med. 2022;387(6):495‐505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Berdeja JG, Madduri D, Usmani SZ, et al. Ciltacabtagene autoleucel, a B‐cell maturation antigen‐directed chimeric antigen receptor T‐cell therapy in patients with relapsed or refractory multiple myeloma (CARTITUDE‐1): a phase 1b/2 open‐label study. Lancet. 2021;398(10297):314‐324. [DOI] [PubMed] [Google Scholar]
- 4. Munshi NC, Anderson LD, Shah N, et al. Idecabtagene vicleucel in relapsed and refractory multiple myeloma. N Engl J Med. 2021;384(8):705‐716. [DOI] [PubMed] [Google Scholar]
- 5. Manier S, Ingegnere T, Escure G, et al. Current state and next‐generation CAR‐T cells in multiple myeloma. Blood Rev. 2022;54:100929. [DOI] [PubMed] [Google Scholar]
- 6. Corre J, Perrot A, Caillot D, et al. del(17p) without TP53 mutation confers a poor prognosis in intensively treated newly diagnosed patients with multiple myeloma. Blood. 2021;137(9):1192‐1195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Ansari‐Pour N, Samur M, Flynt E, et al. Whole‐genome analysis identifies novel drivers and high‐risk double‐hit events in relapsed/refractory myeloma. Blood. 2023;141(6):620‐633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Tessoulin B, Eveillard M, Lok A, et al. p53 dysregulation in B‐cell malignancies: more than a single gene in the pathway to hell. Blood Rev. 2017;31(4):251‐259. [DOI] [PubMed] [Google Scholar]
- 9. Kruiswijk F, Labuschagne CF, Vousden KH. p53 in survival, death and metabolic health: a lifeguard with a licence to kill. Nat Rev Mol Cell Biol. 2015;16(7):393‐405. [DOI] [PubMed] [Google Scholar]
- 10. Lok A, Descamps G, Tessoulin B, et al. p53 regulates CD46 expression and measles virus infection in myeloma cells. Blood Adv. 2018;2(23):3492‐3505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Tessoulin B, Descamps G, Dousset C, Amiot M, Pellat‐Deceunynck C. Targeting oxidative stress with auranofin or Prima‐1Met to circumvent p53 or Bax/Bak deficiency in myeloma cells. Front Oncol. 2019;9:128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Saldivar JC, Hamperl S, Bocek MJ, et al. An intrinsic S/G2 checkpoint enforced by ATR. Science. 2018;361(6404):806‐810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Galluzzi L, Vitale I, Aaronson SA, et al. Molecular mechanisms of cell death: recommendations of the Nomenclature Committee on Cell Death 2018. Cell Death Differ. 2018;25(3):486‐541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Toledo LI, Altmeyer M, Rask M‐B, et al. ATR prohibits replication catastrophe by preventing global exhaustion of RPA. Cell. 2013;155(5):1088‐1103. [DOI] [PubMed] [Google Scholar]
- 15. Da Costa AABA, Chowdhury D, Shapiro GI, D'Andrea AD, Konstantinopoulos PA. Targeting replication stress in cancer therapy. Nat Rev Drug Discov. 2023;22(1):38‐58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Martin E, Palmic N, Sanquer S, et al. CTP synthase 1 deficiency in humans reveals its central role in lymphocyte proliferation. Nature. 2014;510(7504):288‐292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Martin E, Minet N, Boschat A‐C, et al. Impaired lymphocyte function and differentiation in CTPS1‐deficient patients result from a hypomorphic homozygous mutation. JCI Insight. 2020;5(5):e133880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Minet N, Boschat A‐C, Lane R, et al. Differential roles of CTP synthetases CTPS1 and CTPS2 in cell proliferation. Life Sci Alliance. 2023;6(9):e202302066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Liang JH, Ren YM, Du KX, et al. MYC‐induced cytidine metabolism regulates survival and drug resistance via cGas‐STING pathway in mantle cell lymphoma. Br J Haematol. 2023;202(3):550‐565. [DOI] [PubMed] [Google Scholar]
- 20. Soudais C, Schaus R, Bachelet C, et al. Inactivation of cytidine triphosphate synthase 1 prevents fatal auto‐immunity in mice. Nat Commun. 2024;15(1):1982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Pfeiffer C, Grandits AM, Asnagli H, et al. CTPS1 is a novel therapeutic target in multiple myeloma which synergizes with inhibition of CHEK1, ATR or WEE1. Leukemia. 2024;38(1):181‐192. [DOI] [PubMed] [Google Scholar]
- 22. Durand R, Bellanger C, Kervoëlen C, et al. Selective pharmacologic targeting of CTPS1 shows single‐agent activity and synergizes with BCL2 inhibition in aggressive mantle cell lymphoma. Haematologica. 2024;109(8):2574‐2584. 10.3324/haematol.2023.284345 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Benaniba L, Tessoulin B, Trudel S, et al. The MYRACLE protocol study: a multicentric observational prospective cohort study of patients with multiple myeloma. BMC Cancer. 2019;19(1):855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Gomez‐Bougie P, Maiga S, Tessoulin B, et al. BH3‐mimetic toolkit guides the respective use of BCL2 and MCL1 BH3‐mimetics in myeloma treatment. Blood. 2018;132(25):2656‐2669. [DOI] [PubMed] [Google Scholar]
- 25. Tessoulin B, Moreau‐Aubry A, Descamps G, et al. Whole‐exon sequencing of human myeloma cell lines shows mutations related to myeloma patients at relapse with major hits in the DNA regulation and repair pathways. J Hematol Oncol. 2018;11(1):137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Maïga S, Brosseau C, Descamps G, et al. A simple flow cytometry‐based barcode for routine authentication of multiple myeloma and mantle cell lymphoma cell lines. Cytometry A. 2015;87(4):285‐288. [DOI] [PubMed] [Google Scholar]
- 27. Durand R, Descamps G, Bellanger C, et al. A p53 score derived from TP53 CRISPR/Cas9 HMCLs predicts survival and reveals a major role of BAX in the response to BH3 mimetics. Blood. 2024;143(13):1242‐1258. [DOI] [PubMed] [Google Scholar]
- 28. Asnagli H, Minet N, Pfeiffer C, et al. CTP synthase 1 is a novel therapeutic target in lymphoma. HemaSphere. 2023;7(4):e864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Novak A, Laughton D, Lane R, et al. Discovery and optimization of potent and orally available CTP synthetase inhibitors for use in treatment of diseases driven by aberrant immune cell proliferation. J Med Chem. 2022;65(24):16640‐16650. [DOI] [PubMed] [Google Scholar]
- 30. Alberge J‐B, Kraeber‐Bodéré F, Jamet B, et al. Molecular signature of 18F‐FDG PET biomarkers in newly diagnosed multiple myeloma patients: a genome‐wide transcriptome analysis from the CASSIOPET study. J Nucl Med. 2022;63(7):1008‐1013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Foroutan M, Bhuva DD, Lyu R, Horan K, Cursons J, Davis MJ. Single sample scoring of molecular phenotypes. BMC Bioinformatics. 2018;19(1):404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Botrugno OA, Bianchessi S, Zambroni D, et al. ATR addiction in multiple myeloma: synthetic lethal approaches exploiting established therapies. Haematologica. 2019;105(10):2440‐2447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Reaper PM, Griffiths MR, Long JM, et al. Selective killing of ATM‐ or p53‐deficient cancer cells through inhibition of ATR. Nat Chem Biol. 2011;7(7):428‐430. [DOI] [PubMed] [Google Scholar]
- 34. Foote KM, Lau A, Nissink JWM. Drugging ATR: progress in the development of specific inhibitors for the treatment of cancer. Future Med Chem. 2015;7(7):873‐891. [DOI] [PubMed] [Google Scholar]
- 35. Munawar U, Roth M, Barrio S, et al. Assessment of TP53 lesions for p53 system functionality and drug resistance in multiple myeloma using an isogenic cell line model. Sci Rep. 2019;9(1):18062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Hathaway MR, Gadek KE, Jagana HL, et al. CTPS1 inhibition synergizes with replication stress signaling inhibition in MYC ‐amplified Group 3 medulloblastoma. bioRxiv. 2024. 10.1101/2024.06.03.597242 [DOI] [Google Scholar]
- 37. Gomez‐Bougie P, Ménoret E, Juin P, Dousset C, Pellat‐Deceunynck C, Amiot M. Noxa controls Mule‐dependent Mcl‐1 ubiquitination through the regulation of the Mcl‐1/USP9X interaction. Biochem Biophys Res Commun. 2011;413(3):460‐464. [DOI] [PubMed] [Google Scholar]
- 38. Karnitz LM, Zou L. Molecular pathways: targeting ATR in cancer therapy. Clin Cancer Res. 2015;21(21):4780‐4785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Yap TA, O'Carrigan B, Penney MS, et al. Phase I trial of first‐in‐class ATR inhibitor M6620 (VX‐970) as monotherapy or in combination with carboplatin in patients with advanced solid tumors. J Clin Oncol. 2020;38(27):3195‐3204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Terranova N, Jansen M, Falk M, Hendriks BS. Population pharmacokinetics of ATR inhibitor berzosertib in phase I studies for different cancer types. Cancer Chemother Pharmacol. 2021;87(2):185‐196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Bouard L, Tessoulin B, Descamps G, et al. Inhibition of ATR overcomes chemotherapy resistance in p53 deficient myeloma cells. Blood. 2019;134(Suppl_1):3109. [Google Scholar]
- 42. Sun Z, Zhang Z, Wang Q‐Q, Liu J‐L. Combined inactivation of CTPS1 and ATR is synthetically lethal to MYC‐overexpressing cancer cells. Cancer Res. 2022;82(6):1013‐1024. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting information.
Supporting information.
Supporting information.
Supporting information.
Supporting information.
Data Availability Statement
Data that support the findings of this study are available from the corresponding author upon reasonable request.