

Summary
With hundreds of copies of rDNA, it is unknown whether they possess sequence variations that form different types of ribosomes. Here, we developed an algorithm for long-read variant calling, termed RGA, which revealed that variations in human rDNA loci are predominantly insertion-deletion (indel) variants. We developed full-length rRNA sequencing (RIBO-RT) and in situ sequencing (SWITCH-seq), which showed that translating ribosomes possess variation in rRNA. Over 1,000 variants are lowly expressed. However, tens of variants are abundant and form distinct rRNA subtypes with different structures near indels as revealed by long-read rRNA structure probing coupled to dimethyl sulfate sequencing. rRNA subtypes show differential expression in endoderm/ectoderm-derived tissues, and in cancer, low-abundance rRNA variants can become highly expressed. Together, this study identifies the diversity of ribosomes at the level of rRNA variants, their chromosomal location, and unique structure as well as the association of ribosome variation with tissue-specific biology and cancer.
Keywords: ribosome, repetitive genomic elements, ribosome heterogeneity, rDNA, rRNA, rRNA sequence variations, rDNA sequence variations, rRNA structure, rRNA association with development and cancer, in situ sequencing
Graphical abstract
Highlights
-
•
Indels are main variants of human rRNA as shown by a new computational pipeline
-
•
A long-read rRNA variation atlas and DMS sequencing identifies different ribosomes
-
•
rRNA subtypes are chromosome specific and visualized by novel single-cell technologies
-
•
rRNA subtypes change expression in human tissues and rRNA variants in cancer
Rothschild et al. discover rRNA sequence variations in translating ribosomes, which form novel rRNA subtypes encoded on different chromosomes. These ribosomes have different structures and can be visualized in single cells. They show that while rRNA sequences are usually discarded from sequencing analyses, they associate with human physiology and disease.
Introduction
The ribosome is a complex, ancient machine responsible for all protein synthesis, with a core ribosomal RNA (rRNA) structure that is conserved across all kingdoms of life. The primary transcript of the rRNA genes is the large 45S pre-rRNA, which contains the 18S, 5.8S, and 28S rRNAs as well as transcribed spacer regions. In humans, rRNA genes are present in hundreds of ribosomal DNA (rDNA) copies in tandem repeats that are spread across multiple chromosomal loci.1 These high rDNA copy numbers are thought to be necessary to produce millions of ribosomes in each cell. Nevertheless, these hundreds of rDNA copies allow for sequence variation between copies, as was first noted in mice and humans almost 50 years ago.2
It remains an outstanding challenge to understand whether ribosomes are different at the level of rRNA and how many ribosome subtypes may exist. For the last several decades we have therefore had a limited knowledge of fundamental differences in the translational machinery and limited insight beyond the textbook view of ribosome composition. A very significant study showed that rRNA sequence variations between different rDNA loci lead to novel rRNA functions in E. coli, where sequence variations between the bacterial rDNA operons support growth under stress conditions.3 In particular, they found that naturally occurring rRNA sequence variation can modulate bacterial ribosome function, central aspects of gene expression regulation, and cellular physiology. This inspiring work raises the possibility that this is general and could be the case in humans. In humans, by examining short-read sequencing data of the 1000 Genomes Project (1KGP),4 two previous studies discovered hundreds of positions in rDNA bearing sequence variants.5,6 A major challenge faced by these former studies arises from limitations in short-read sequencing where there is depletion of GC-rich sequences. Specifically, certain regions within rDNA possess >80% GC content on average.6,7,8 This results in two types of problems in variant calling: (1) false negatives, caused by the inability to identify variants in regions with poor sequence coverage; and (2) false positives, caused by PCR errors in low-coverage regions. Moreover, these GC-rich regions are highly repetitive, which makes short-read variant discovery tools inaccurate.9 As a result, the latter two previous studies5,6 reported contradictory results likely because of these major caveats. Nevertheless, these studies did raise interest in investigating rRNA sequence variation, inspiring future studies. Interestingly, the copy number of the rDNA loci was also shown to be important for gene expression and cellular homeostasis.10,11,12,13 Moreover, rDNA copy number was associated with age and cancer.14,15,16 In cancer, both rDNA copy-number gain and loss have been reported in multiple studies.17,18,19,20,21 This calls for further investigation into whether rDNA copy-number changes are coupled with sequence variations and whether such changes affect human health.
Recently, long-read sequencing enabled the successful complete assembly of a human genome by the Telomere-2-Telomere (T2T) consortium, including positioning of rDNA copies in the five acrocentric chromosomes.22 Moreover, in the mouse and Arabidopsis plant genomes, rDNA variants were grouped into haplotypes, and a few rRNA variants were found to be expressed in tissues using short reads.5,23,24 However, in order to find low-frequency variations between full-length rDNA paralog copies, development of new computational methods is necessary. Long reads offer the possibility for distinguishing between paralog genes. However, existing common methods for long-read variant calling, such as DeepVariant25 and Clair,26 are primarily designed for detecting variants in single-copy regions. For paralog genes where low-frequency variants exist between copies, accurate variant calling is lacking.9 Moreover, it remains an open question whether rRNA haplotypes are expressed from the human genome, what their abundances are, and whether such variability is linked to human physiology. There, too, long-read sequencing and analysis of full-length rRNA is necessary This highlights the need for new approaches to comprehensively characterize human ribosome diversity.
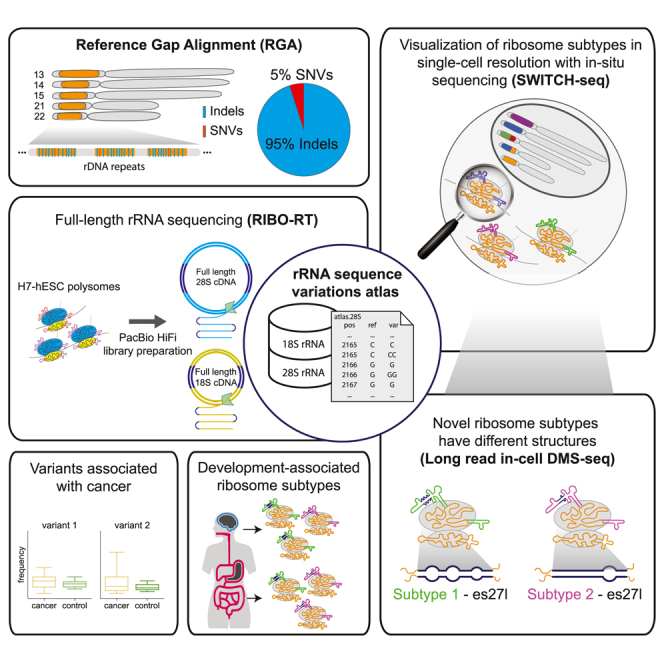
To address this need in the field, we devised an efficient novel computational algorithm to detect all variations between paralogs, termed RGA (reference gap alignment). Applied to the long-read 1KGP dataset, we discovered hundreds of rDNA sequence variations enriched with previously undiscovered insertion-deletions (indels). We further developed a novel methodology to perform long-read sequencing on rRNA in actively translating ribosomes to identify variants (RIBO-RT). Using this method, we discovered that ribosomes have different subtypes with rRNA variants that are genomically encoded by rDNA clustered on distinct chromosomes. Additionally, using an in situ rRNA sequencing platform that we developed (SWITCH-seq), we discovered that variants belonging to different rRNA subtypes are co-expressed in single cells. We then used structure probing coupled with long-read sequencing to find that 28S subtypes have different rRNA structures. Lastly, we found that these subtypes are differentially expressed in human tissues and that low-abundance variants are elevated in certain cancers. Together, these results suggest that ribosomes with unique sequence variation may be used to modulate different cellular programs underlying human physiology and disease.
Results
Indels are the main variants of the human rDNA loci
How rDNA variation shapes the presence of unique ribosomes in the cell remains an important open question. Previous studies that analyzed the 1KGP dataset for discovery of rDNA variants reported discordant results. Parks et al.5 reported hundreds of variants in both the 18S and 28S, yet 75% of variants were not made publicly available, making a comparison to this dataset problematic. Nonetheless, Fan et al.6 reported notable differences from Parks et al. by suggesting that the 18S has low variation and also reporting many fewer variant positions in the 28S. Moreover, Parks et al. reported only 2.7% of variants being indels, while Fan et al. reported 19.2% indels. Here, considering the limitations of short reads in rDNA variant discovery and their inability to distinguish between rDNA paralogs, we decided to re-evaluate the variants in the human rDNA genes.
Until recently, the 1KGP dataset included only short-read genome sequencing.27 Yet as of 2022, the 1KGP includes PacBio’s HiFi long-read sequencing for 30 individuals from diverse ancestral origins, which could serve as a better method for accurately calling variants. Here, we compared the rDNA variants captured by short and long reads from the same individuals and addressed the discrepancies between previous studies (Figures 1A and 1B; Table S1).
Figure 1.

1000 Genomes Project and H7 human embryonic stem cell rRNA and rDNA variant extraction pipeline with high correlation between 28S rDNA and rRNA high-abundance variant frequencies
(A) Graphical illustration of the dataset analyzed consisting of 30 individuals from the 1000 genomes project (1KGP) with both short- and long-read sequencing.
(B) Comparison of single-nucleotide variants (SNVs) and insertion-deletions (indels) across studies.
(C) Graphical illustration of the reference gap alignment (RGA) method used for variant discovery in 18S and 28S sequences.
(D) 18S and 28S rDNA/rRNA sequence extraction pipeline from H7 human embryonic stem cell (H7-hESC).
(E) Scatterplot of 28S rRNA frequency (x axis) and rDNA frequency (y axis) for reference and alternate alleles. Alternate alleles are marked in red if their frequencies in rDNA and rRNA agree or in orange if they differ significantly. Reference alleles are marked in gray. A dashed black line indicates rRNA frequency equal to 10%. Spearman and Pearson correlations for rRNA frequency and rDNA frequency between alternate alleles alone are presented (calculated on variants, red dots alone).
(F) Stacked bar plots of allele frequencies at positions with variants with frequency >10% in both rRNA and rDNA. The nucleotide sequence matching the alleles are indicated inside the bar plots for variants with >10% allele frequency (“–” indicates deletion). The reference allele is indicated in gray, and alternate alleles are indicated in color.
When analyzing the short-read data, we followed the pre-processing steps as performed in previous studies of marking duplicate reads suspected to be PCR artifacts. This step discarded 97% of reads. However, it is unknown whether duplicate reads are PCR biases given the high rDNA paralog copy numbers, which highlights the limitation of short-read sequencing for rDNA variant discovery. Next, to call variants including rare variants, which are not expected to follow germline variant frequencies in high paralog rDNA copy numbers, we tested two common somatic variant calling methods for short reads: LoFreq∗,28 which was used by Parks et al., and Mutect2.29 Specifically, Mutect2 was chosen instead of the germline variant caller used by Fan et al. because germline variant callers will not detect rare variants found between paralogs. Using the LoFreq∗ method, which is known to be sensitive,28 we found 1,582 positions with variants compared to 861 positions with variants with Mutect2 (Tables S2 and S3). Notably, both methods detected 23% indels, which is on par with the indel percentage reported by Fan et al. Given the difference in the proportion of indel frequencies between the previous two studies, we compared variant quality scores of single-nucleotide variants (SNVs) and indels (Table S3, Mutect2 false-discovery-rate [FDR]-corrected log10 likelihood ratio score of variant existence). Here, we found that indel variants were enriched with high-confidence p values (Figure S1, p value <10−15 on comparing SNVs and indel likelihood ratio scores using Kolmogorov-Smirnov test for goodness of fit). Notably, tandem repeats and GC-rich sequences in the human genome were shown to be prone to chromosomal breakage and were found to be enriched in indels.30 Therefore, the 23% indel frequency derived solely from short-read data may under-represent the true indel frequency in these samples. To test this, we next examined the HiFi long-read data from the same samples.
LoFreq∗ and Mutect2 did not work on the long-read sequencing data. To identify all positions with sequence variants, we developed a new computational method for accurate variant calling between paralogs, which we term RGA (see STAR Methods). We align all reads against a common reference and report all variants at a given position with respect to this reference (Figures 1C and S2; STAR Methods). Our method reports at every position with respect to the 18S/28S reference if that position has a variant and calls its identity. The only parameters in our method are the standard pairwise alignment parameters: a mismatch penalty, a gap-opening penalty, and a gap-extension penalty (see STAR Methods for more details). When benchmarking the global sequence alignment parameters, these resulted in similar indel proportions (Table S4). In agreement with previous studies,5,6 we found that rDNA is highly variable, yet using our method we discovered that the vast majority of variants are short indels and not SNVs in all 30 samples. Specifically, when examining each reference position individually we found that, on average, 95% of variants are GC-rich indels (Table S5).
Since this indel proportion found with long reads is markedly different from the results obtained with short reads (Figure 1B), we cross-validated our reported variants in three ways. As first indel validation, we decided to compare our results from our primary long-read sequencing technology, namely HiFi, with an alternative, Oxford Nanopore (ONT). Importantly, ONT is much more error prone compared to HiFi, with an estimated 13% error rate in ONT compared to 0.1% error rate in HiFi.31,32,33 Since 1KGP long-read sequencing was only performed on HiFi, we tested this using the Genome In A Bottle (GIAB) dataset, which consists of two trio families, where both HiFi and ONT were performed on the same samples. Here, when examining the HiFi dataset of GIAB, in agreement with the 1KGP HiFi results, we discovered that 96% of variants are indels (Tables S1 and S6; STAR Methods). When using the ONT dataset as a validation dataset, 81% of variants found in HiFi were replicated in the ONT dataset (Table S7 and STAR Methods). Notably, the variants that were not identified by ONT consisted of insertions and SNVs but not deletions (Table S8). Additionally, after retaining variants found at frequencies above the ONT error rate, 87% of found variants were indels (Table S7, filtering variants with allele frequency smaller than 0.13). As another method to cross-validate our RGA variant caller, we examined variant frequencies in the family members of the GIAB trio dataset. We compared variant frequencies for variants that appeared only in the child, in a single parent, or in both parents. We expected here that high-abundance variants would be inherited and, indeed, all variants with frequency greater than 2% were found in at least one of the parents. This held true for both for SNVs and indels (Figure S3).
Finally, we tested whether short reads contain reads with indel variants that are not identified by short-read variant callers. To this end, we used the indels identified with long reads by the RGA method as a reference of existing sequence variations and mapped the short reads from the 1KGP to this reference (STAR Methods). This directly tests whether the variants found in the long reads are also detected in the short reads. Notably, mapping variants is different from de novo variant discovery, since here we count short reads that perfectly match the indels as opposed to variant-calling tools, which are reference free and need to appear in sufficiently high frequency to be discovered. Surprisingly, despite estimated low coverage of the rDNA loci, we were able to detect 928 nucleotide variants, which included 896 indels and 32 SNVs (STAR Methods and Table S9). Previous variant callers that have analyzed the 1KGP were not able to identify most indels, likely because they are too low in abundance (LoFreq∗, Mutect2, and DNA-sequencing pipeline Sentieon, Release 201911). Our results show that short reads can be mapped to indels given a reference of variants identified with long-read sequencing.
Our results highlight that previous studies that used short-read sequencing missed the majority of the variants found in the rDNA loci. Most of our identified rDNA variants were found at GC-rich regions depleted from short-read sequencing. We conclude that indels are the main variants in rDNA both between and within individuals. These findings also highlight the need to curate a reference of rDNA variants and the importance of our accurate variant-calling method together with long-read sequencing in confidently assigning rDNA variants.
An atlas of 18S and 28S human rDNA variants validated in rRNA of translating ribosomes and single-cell microscopy
It is unknown whether the rDNA copies with sequence variants found in the human genome are transcribed and are found in functionally translating ribosomes. With no human reference of different rRNA subtypes, studies performing RNA sequencing (RNA-seq) completely ignore rRNA variants, thus limiting our understanding of the contribution of rRNA to human physiology and disease.
Sequencing of rRNA has been historically technically challenging.6,34 Here, in addition to our computational RGA variant discovery method, by optimizing long-read sequencing we have successfully developed an experimental method for full-length rRNA sequencing of 18S and 28S from translating ribosomes, which we named RIBO-RT. To extract rRNA from translationally active ribosomes, we first employed sucrose gradient fractionation whereby ribosomes can be separated into free ribosomal subunits and translationally active ribosomes, which contain one or more ribosomes bound to the same mRNA. We extracted RNA from translating ribosome-containing fractions (Figure S4), performed reverse transcription in denaturing conditions, and sequenced complete 18S and 28S rRNA by HiFi long-read sequencing (Figure 1D and STAR Methods). We selected a human embryonic stem cell line (H7-hESC) and, using long-read sequencing, sequenced the 18S and 28S from both its rDNA and rRNA (Figure 1D). We obtained 58,495 sequences of the 18S and 14,430 sequences of the 28S rRNAs from translating ribosomes (STAR Methods). With this approach and our variant discovery method, we were able to coherently characterize the 18S and 28S H7-hESC rRNA variants. Most importantly, since rRNA is known to be heavily modified,35 our strategy of matching rRNA to genomic rDNA from the same cell enables us to distinguish modifications or sequencing errors from true sequence variants belonging to different rDNA alleles.
In agreement with our 1KGP and GIAB rDNA results, we found that the H7-hESC rDNA is highly variable and is enriched with indels. Moreover, 96% and 84% of the hESC variants are also found in the 1KGP and GIAB datasets, respectively. This is generally concordant with expected rates of replication based on these small sample sizes of the 1KGP and GIAB datasets. Moreover, we find high agreement in the frequency of variants between the H7-hESC and other datasets (Figure S5). Additionally, rDNA variants are transcribed into functionally translating ribosomes, as they are present in polysome fractions (Figure S6). Specifically, we found 270 positions with variants in the 18S and 858 positions with variants in the 28S, corresponding to about one variant for every six rRNA positions (Figures S6 and S7). When comparing monosome to polysome fractions, we observed high correlation in variant frequencies between fractions (Figure S8 and Table S10). Additionally, we long-read sequenced the 18S and 28S from translating ribosomes from an additional commonly used cell line, K562, using the same extraction protocol as described in STAR Methods (Figure S9). We found that 95% of the H7-hESC variants are also found in the tested human cell line (Figure S10 and Table S11). Most variants (59%) are found in expansion segment (ES) regions (Figure S11, ES/non-ES regions are annotated). These regions vary in sequence both within and among different species, nearly doubling the eukaryotic rRNA sequence relative to that of prokaryotes.36 ESs have recently been shown to bind ribosome-associated proteins and transcripts, yet their functions remain poorly understood.36,37,38,39,40
Most importantly, with accurate variant calling and full coverage of the underlying rDNA and transcribed rRNA, we can measure the frequency of each variant between the rDNA copies and rRNA expression levels. We distinguish possible modifications or sequencing errors from certain sequence variants belonging to different rDNA alleles by calling variants with similar frequencies measured in rDNA and rRNA as high-confidence alternate allele variants, while those significantly deviating between their rDNA and rRNA frequencies are called as low-confidence alternate alleles (Figure 1E and Table S12; for Fisher’s exact test measuring the association between rDNA and rRNA alternate allele frequencies, high-confidence alternate alleles are marked in red and low-confidence alternate alleles in orange). Surprisingly, we did not identify any abundant high-confidence variants within the 18S. This suggests that rRNA variation is not tolerated within the small ribosome subunit. While most variants have low abundance, in the 28S we found 23 variants at 17 positions with frequencies above 10% in both rRNA and rDNA (Figures 1E and 1F; Table S13; Figure S7C, annotated positions, minor allele frequency with dashed line marking 0% frequency). Notably, for the 28S high-abundance variants, there is very good agreement between variant frequencies in rRNA and rDNA (Figure 1E, Pearson correlation r = 0.93, correlating all variants colored in red and orange).
Next, we focused on the 28S high-abundance variants. For most positions, the RNA45S5 reference allele is the major allele found in the sequenced H7-hESC line (Figure 1F, reference allele in gray; Data S1, nucleotide atlas). Yet some alternate alleles were more abundant than the 28S RNA45S5 reference alleles (Figure 1F, gray for the reference allele and red for variants). Notably, high-abundance variants are only located in four ES regions (es7l, es15l, es27l, and es39l) and one non-ES region, helix 28S:h11 (Figure 1F). Moreover, in the es7l, es15l, and es27l regions, we observed that variants can be grouped and characterized by indels of GGC in tandem repeats. GGX tandem repeats were recently suggested capable of forming G-quadruplex structures,41 while other works suggested that such repeats can form other higher-order structures.42,43 While the function of these ESs is largely unknown, a growing body of research supports various roles in translation regulation. For example, es27l has been shown to be important for control of translation fidelity and binding of ribosome-associated proteins for several processes, such as initiator methionine cleavage from the nascent polypeptide44 or acetylation of nascent polypeptides.45 Moreover, es39l interacts with the signal recognition particle, which identifies the signal sequence on nascent polypeptides emerging from the translating ribosome.46 Interestingly, the most abundant variant in the non-ES helices, a G-to-A substitution at position 60 in 28S:h11, is considered unique to humans. The alternate allele, A, is the reference allele for other mammals including chimpanzees.47,48
For these aforementioned highly abundant variants, a strong correlation between the frequency of a variant’s occurrence among rDNA copies and its expression levels in rRNA indicates both the authenticity of these sequence variants and their likely co-expression within individual cells. To explore this hypothesis, we developed a template-switching-based in situ sequencing method, SWITCH-seq, to visualize variant ribosomes in individual HeLa cells (Figure 2A). This approach involved designing a reverse transcription primer to target constant non-variable regions downstream of the selected rRNA variant regions. Specifically, we selected regions for which we could design a primer for each of the ES regions with abundant variants (STAR Methods and Table S14). The process of SWITCH-seq begins with performing reverse transcription on fixed HeLa cells, wherein a known sequence of choice is attached to the 3′ end of cDNA (template switching) (Figure 2A and STAR Methods). This step incorporates the variant of interest into the cDNA, which is subsequently amplified into in situ cDNA amplicons through enzymatic circularization and rolling circle amplification. These amplicons encapsulating the rRNA variants are then anchored into a hydrogel network for sequential in situ imaging using a confocal microscope (STAR Methods). We conducted multiple rounds of imaging that capture both the site immediately upstream of the variant, where no variants are expected, and the variant site itself, where the presence of variants is anticipated (Figures 2B and S12). As predicted, we successfully observed both the reference and alternate alleles (Figures 2B, 2C, and S12). Furthermore, the frequencies of the reference and variant alleles corresponded with their frequencies in the H7-hESC samples (Table S15). We conclude that rRNA variants observed at high frequency in the H7-hESC rRNA and rDNA and in the rDNA across the 1KGP and GIAB samples form ribosomes that are co-expressed in individual cells that can be visualized at single-cell resolution.
Figure 2.

rRNA variants are co-expressed in individual cells as visualized by in situ sequencing
(A) Graphical illustration of SWITCH-seq pipeline.
(B) Two rounds of representative fluorescent in situ sequencing images of HeLa cells (DAPI staining in blue) are presented for the es39l-probed region. We identified a non-variable base C (magenta) at position 4912. At position 4913, two alternative sequences were revealed: the known reference sequence C (cyan) and the alternate variant U (yellow). Data shown as mean percentage ± SD. n = 4 images.
(C) Representative fluorescent images of HeLa cells (DAPI staining in blue) showcase three highly abundant rRNA variants. The positions of the variants are indicated at the bottom of the images, while the reference (magenta) and alternate (green) alleles are indicated at the top, along with their respective rRNA frequencies. Data shown as mean percentage ± SD. n = 4 images per allele.
As an important resource for studying human rRNA variations, we create the first comprehensive atlas of all H7-ESC rRNA 18S and 28S rRNA variants at different resolutions from nucleotide variants to gene 28S haplotype groups that we later describe as separate subtypes (Figure S11; Data S1, S2, S3, S4, and S5 and Tables S16 and S17 for region annotation and atlas building as described in STAR Methods).
28S variants assemble to distinct ribosome subtypes
Since we successfully obtained full-length 18S and 28S rRNA with variations, we address the outstanding question of whether variations lead to the formation of different ribosome subtypes. We focused here on the 28S, since 18S variants appeared at low frequency. For the 28S, we found high agreement between rRNA and rDNA variant frequency, so we first asked which rDNA variants are co-located on the same 28S rDNA copy. To this end, we calculated the correlation coefficient (Pearson’s r2) between positions across all 28S H7-hESC rDNA sequences. This is analogous to measuring the linkage disequilibrium (LD) coefficient in population genetics, albeit across paralogous copies within a single genome rather than across individuals in a population. Notably, we found low global LD structure between highly abundant rDNA variants (Figure 3A, showing LD for rDNA positions with found rRNA frequency >10%), supporting recent findings indicating high rates of non-allelic gene conversion across the acrocentric chromosomes.49 The highest LD (r2 > 0.2) was found between the es27l and all other regions. Comparing different regions, we found LD between four regions, 28S:h11, es15l, es27l, and es39l, where in each region we identified a position with higher linkage to the other three regions (Figure 3A; four positions are annotated, position 60 being 28S:h11, with higher linkage to other positions). By considering the variants at this subset of positions, we found a total of 21 different haplotypes in both rDNA and rRNA (Figure S13). For testing of whether haplotypes can be considered as different 28S subtype variants, we further analyzed two independent long-read DNA datasets: (1) the fully assembled genome from the T2T with 219 rDNA copies with their chromosome location22; and (2) the GIAB HiFi dataset.50 In agreement with the H7-hESC results, three out of the four positions with higher linkage to other positions in the H7-hESC had higher LD in the GIAB dataset (Figures 3B and S14). Since these three variants are linked to variants at other positions, we define the haplotypes formed by positions 60, 3513, and 4913, belonging to regions 28S:h11, es27l, and es39l, respectively, as different 28S haplotypes (Figure 3C).
Figure 3.

28S subtypes found by haplotype analysis
(A) Correlation coefficient (Pearson’s r2) heatmap between positions across H7-hESC 28S rDNA with variant frequency >10%. x axis and y axis are annotated by regions. Helix regions are annotated by light blue, and ES regions are annotated by yellow. Individual positions with higher r2 between regions are also indicated.
(B) Same as (A) for the Genome In A Bottle (GIAB) dataset.
(C) Haplotype digit code to variant sequence conversion at the three positions with higher r2 in (A) and (B).
(D) Bray-Curtis principal coordinate analysis (PCoA) of 386 H7-hESC 28S rDNA sequences (upper panel) and 386 28S rDNA sequences from each GIAB sample (lower panel). Each dot is a complete 28S rDNA sequence with similarity between sequences measured on 6-mers. The colors correspond to coloring an rDNA sequence by its 3-position haplotype described in (C). Numbers in the x and y labels represent the PCoA explained variance.
(E) Telomere-to-telomere (T2T) haplotype distribution across the five acrocentric chromosomes. The rDNA acrocentric arms are presented in a schematic cartoon with proportions of rDNA haplotypes in different colors as found in the matching table below. Haplotypes match the 3-position haplotypes in (C). We indicate the rDNA copy number of each haplotype in every chromosome.
Previously it was shown that the rDNA array is composed of highly homogenized tandem clusters.51 We therefore next asked whether different 28S haplotypes are spatially separated in the genome as different subtypes. For the H7-hESC, we have 386 complete 28S rDNA sequences and in the GIAB dataset, we randomly subsampled each GIAB sample to 386 complete 28S rDNA sequences. For these datasets, we do not know rDNA-chromosome positioning. Notably, by comparison of 28S rDNA sequence similarities, we detected distinct 28S sequence groups in both hESC and GIAB (Figure 3D, principal coordinate analysis [PCoA] of Bray-Curtis dissimilarities between 28S sequences52; STAR Methods). Here, the different clusters in PCoA space match different 28S haplotypes. Specifically, we observed that 28S sequences of a given haplotype are more similar to one another in their entire sequence compared to 28S rRNAs of other haplotypes (Figure 3D). When plotting individual haplotypes, there is less observed structure in individual haplotypes as compared to the combined data (Figure S15). In the T2T assembly, rDNA copies have chromosome coordinates, which enables us to measure 28S subtype presence at the five acrocentric chromosomes. Remarkably, we discovered that 28S haplotypes are largely chromosome specific (Figure 3E). When analyzing the 1KGP dataset, we find that all of the haplotypes found in high frequency in the H7-hESC, GIAB, and the T2T CHM13 genomes are also present in the 1KGP genomes. However, when examining haplotype frequency changes, we find high standard deviation (SD) between their frequencies across individuals (Table S18). This variability limits our understanding of the 28S haplotypes, which may be caused by high rates of non-allelic gene conversion across rDNA copies. Taken together, our results support the view that 28S haplotypes are genomically separated and belong to different subtypes.
Ribosomes of different 28S subtypes have different structures
We next asked whether different 28S subtypes have different ribosome structures. Notably, the abundant variants in the hESC were found in four different ES regions that were never previously resolved by cryoelectron microscopy. Here, we treated the hESC sample with dimethyl sulfate (DMS), which covalently modifies the RNA at regions where the rRNA is accessible to allow for structure probing of the RNA (STAR Methods). Using our RIBO-RT method for sequencing full-length 28S with our RGA variant calling on DMS-treated hESCs, we obtained an accessibility map of the 28S. Importantly, we are able to predict the structure of the full-length ES regions, which was not previously possible.
We compared the two most abundant 28S subtypes and their linked variants and found that they have different structures (Figures 4A–4C, with 22% and 30% frequency of subtypes 1 and 2, respectively; STAR Methods). While our method with DMS results in a full-length accessibility map of the 28S, secondary structure prediction becomes less accurate for long RNA sequences. Given that ESs have tentacle-like extensions that protrude from the ribosome, we assumed that the core non-ES rRNA is not affected by changes in the ES regions, which allowed us to focus on the structures of individual ESs. Most interestingly, we discovered that the ESs es7l, es15l, and es27l have major DMS accessibility and structure differences when comparing the two subtypes observed at the GGC sites in es7l, es15l, and es27l (Figure 4A, ES region box annotations; Figures S16–S20 for es7l, es15l, and es27l). When focusing on es27l, the second-longest ES, we noticed that the largest accessibility difference between the subtypes was at the site where es27l subtypes differ, at the GGC indel. Specifically, the subtype with one fewer tandem-repeat GGC insertion before the AG at position 3513 of the 28S showed greater DMS accessibility at position 3513 and its vicinity (Figures 4D and 4E). This GGC expands a six-tandem-repeat GGC, i.e., (GGC)6, which changes the region’s structure. Moreover, for the es27l region we found local structure changes near the sequence variants, which opens the possibility that there are proteins or transcripts that may interact with the subtype with the GGCAG variant but not with the AG variant (Figures 4D and 4E, region marked in red). Taken together, our DMS results provide evidence of structural differences for different ribosome subtypes.
Figure 4.

In-cell dimethyl sulfate with long-read sequencing shows that 28S subtypes have different RNA 2D structure
(A) Changes in dimethyl sulfate (DMS) accessibility between two most abundant 28S subtypes across the complete 28S molecule. Subtypes are defined by the sequence variants observed at positions 60 (28S:h11), 3513 (es27l), and 4913 (es39l), according to the numbering in NR_146117.1. Above is an illustration of the two subtypes, together with the annotations for the aforementioned regions and other regions with large differences in accessibility. x axis is the nucleotide position along the 28S, and y axis is the absolute percentage of DMS accessibility differences at a given position for a window size of 10 nucleotides.
(B) Illustration of es27l predicted secondary structure for subtype 1 (A,GGCAG,T). Detailed RNA 2D structure of the whole subtype 1 es27l is shown in Figure S19.
(C) Illustration of es27l predicted secondary structure for subtype 2 (G,AG,C). Detailed RNA 2D structure of the whole subtype 2 es27l is shown in Figure S20.
(D) Zoomed-in predicted RNA secondary structure of subtype 1 es27l between positions 3310 and 3552(+3). RNA secondary structures are colored by DMS reactivity, and helix confidence estimates are depicted as green percentages. Regions with major differences are annotated by the red box. Nucleotides with differing accessibility between the two subtypes are highlighted by blue stars.
(E) Zoomed-in predicted RNA secondary structure of subtype 2 es27l between positions 3310 and 3552. Detailed description of annotation is the same as (D).
Quantifying the relative abundance of rRNA variants in expression data
Previously, ten rRNA variants were annotated and showed changed expression between mouse tissues.5 Here, we found that one of these rRNA variants replicates in our atlas. This prompted us to check rRNA variations across human tissues by analyzing the publicly available Genotype-Tissue Expression (GTEx) short-read RNA-seq dataset to test whether rRNA variant frequencies are associated with human tissue biology (see STAR Methods for atlas usage instructions). Previous studies comparing mRNA across tissues in the GTEx dataset found tissue-specific, including brain-specific, gene expression.53,54 Here, we analyzed 2,618 samples from 332 individuals and 44 tissues from GTEx and asked whether rRNA subtypes differ in their expression in these tissues (Figure 5A). We hypothesized that the rRNA subtypes that we identified as highly expressed in the hESC may be important for tissue development. Strikingly, the most abundant subtypes in GTEx significantly differed in expression in many tissues (Figures 5B–5D, upper panels; Figures S21–S25; Tables S19 and S20, p < 0.05, FDR-corrected Mann-Whitney U rank-sum test). Notably, when comparing subtype expression levels across tissues, we observed significant differences between tissues derived from the ectoderm and endoderm germ layers (Figures 5B–5D [lower panels] and Table S21, FDR-corrected rank-sum test comparing subtype relative abundances of ectoderm-derived tissues in blue and endoderm-derived tissues in red). Most of the ectoderm-derived tissues belong to brain tissues, and most endoderm-derived tissues are digestive-system tissues (Figure 5A, endoderm- and ectoderm-derived tissues are labeled). Taken together, our results support major changes in the expression of rRNA subtypes across tissues.
Figure 5.

rRNA subtype expression is tissue specific and differs between tissues derived from the ectoderm and endoderm lineages
(A) Schematic representation of the GTEx analysis focusing on cortex versus stomach comparison. We map rRNA reads from different samples to rRNA subtypes. Per rRNA subtype, we illustrate variant expression comparison between tissues. Upper panel: boxplot comparing rRNA subtype expression in cortex and stomach samples. Bottom panel: median rRNA subtype expression across all tissues. Cortex and stomach are annotated, and all ectoderm and endoderm tissues are highlighted in blue/red.
(B) Upper panel: boxplot comparing the expression levels of the rRNA subtype with the haplotype G,AG,C (positions 60, 3513, 4913) in cortex and testis samples. Bottom panel: scatterplot showing the median frequency of the rRNA subtype from the upper panel across all tissues. Tissues derived from ectoderm are marked in blue, tissues derived from endoderm in red, and other tissues in gray. The cortex and testis that were shown in the top panel are annotated with a line.
(C) Same as (B) for the rRNA subtype with the haplotype A,GGCAG,U highlighting gastroesophageal junction and cerebellum samples.
(D) Same as (B) for the rRNA subtype with the haplotype G,GGCAG,C highlighting smooth muscle and spinal cord samples.
Lastly, we asked whether changes in the expression of rRNA variants are associated with cancer. For this, we used 10,030 samples of short-read RNA-seq with clinical phenotypes from The Cancer Genome Atlas (TCGA).55 When comparing cancer types, we found distinct expression patterns of rRNA regional variants across cancers (see STAR Methods for atlas usage instructions and Figures S26–S31 and Table S22 for region annotations). To test whether rRNA variants are cancer specific, we compared cancer biopsies to control biopsies from the same tissues. Surprisingly, we identified specific rRNA regional variants with significantly different expression levels in control and cancer biopsies for 11 cancer types (Figure 6 and Table S22 for alternate allele regional variant abundances; Table S23, p < 0.05 after FDR correction, bootstrapping 10,000 times, Mann-Whitney U rank-sum test with subsampling controls to match the number of cancer samples). These include rRNA variants that, while they appeared in low abundance in both the H7-hESC and control biopsies, are found to be elevated in cancer biopsies. Thus, even low-abundance variants hold immense importance as disease biomarkers.
Figure 6.

Cancer-specific rRNA variant expression
(A) Boxplot showing the distribution of rRNA frequencies of the top expressed, alternate allele regional variant, of es9s, ES:es9s:6_d14_r115, across TCGA cancer and control samples for liver hepatocellular carcinoma. The boxplot is overlaid with a categorical scatterplot which saturates at values with over 20 samples, and not all points are displayed. The sample sizes of cancer and controls is indicated in parentheses, and the p value is indicated at the top (Table S23).
(B) Same as (A) for region es5l, ES:es5l:12_d1_r1, in kidney renal clear cell carcinoma.
(C) Same as (A) for region es9l, ES:es9l:21_d2_r8, in uterine corpus endometrial carcinoma.
(D) Same as (A) for region es10l, ES:es9l:21_d2_r8, in colon carcinoma.
(E) Same as (A) for region es13l, ES:es13l:2_d3_r141, in prostate adenocarcinoma.
(F) Same as (A) for region es45l, ES:es45l:7_d2_r45, in breast invasive carcinoma.
We conclude that our atlas enables direct measurement of rRNA variant changes in expression data. Moreover, we showed that atlas variants are present in translating ribosomes and that they are differentially expressed across tissues and cancer types.
Discussion
Here, by developing a pipeline for long-read sequencing and analysis of rDNA and rRNA from actively translating ribosomes, we measured for the first time variant frequencies in rDNA and rRNA and used in situ sequencing microscopy to validate co-variant expression in individual cells. With this atlas we have enabled greater understanding of the often neglected yet ubiquitous rRNA-seq data and have built an atlas of functional human 18S and 28S rRNA variants at different resolutions, from nucleotide position variants to 28S gene-level subtypes, as a useful resource for studying rRNA variations and composition across biological conditions (Data S1, S2, S3, S4, and S5).
In our study we have discovered chromosome-associated rDNA subtypes, revealing that different ribosome subtypes based on rRNA sequence variation exist. It may be possible that spatial separation of rDNA subtypes enable regulation of their expression at the chromosome level through allelic inactivation of rDNA loci or inactivation of nucleolar organizer regions (NORs) in the distal junction.56,57,58 This may enable global remodeling of rDNA transcription and promote specific ribosome subtypes to be expressed within individual cells. Additionally, using DMS structure probing of full-length 28S, we discovered that different rRNA subtypes have different structures at ES regions, including different DMS accessibility profiles. Since these ES regions are solvent exposed and highly flexible, these ES variations may fine-tune regulation of mRNA translation based on differential association with ribosome-associated proteins, mRNA transcripts, or other factors. Moreover, by analyzing the GTEx dataset, we observed differential expression of rRNA subtypes between tissues belonging to ectoderm and endoderm lineages. This pattern might hint at specialized functions of different ribosome subtypes. Long-lived cells associated with the nervous system might require ribosome subtypes that emphasize translation fidelity over speed as compared to rapidly dividing cells in the digestive tract that require constant replacement given harsh local environments. Indeed, our lab and others have previously shown that es27l plays a role in translation fidelity through association with ribosome-associated proteins.44,59,60 Such interactions that trade speed over fidelity might be fine-tuned by the expression of different rRNA subtypes.
Finally, by analyzing the TCGA dataset, we discovered that some low-abundance rRNA variants in control biopsies were elevated in cancer biopsies. However, the mechanism of elevated expression of such variations remains unknown. One possible mechanism may be enhanced transcription of specific rDNA copies bearing coding sequence variants, and, interestingly, it was shown that lung adenocarcinoma samples were enriched with somatic and germline mutations at rDNA promoter regions.61 Alternatively, de novo somatic mutations may increase certain rDNA variant frequencies. Future work is needed to understand whether they promote oncogenic ribosome activity and how they are regulated. Therefore, our results provide another layer of ribosome specificity wherein cancer cells might deploy a particular rRNA variant that is more compatible with their cellular fitness. Importantly, we found that specific rRNA variants may be used as biomarkers for disease. Notably, 5-fluorouracil, a common chemotherapy drug, was recently shown to incorporate into rRNA and promote drug resistance by changing mRNA translation.62 It may be that drugs directly target specific rRNA variants, and further examination would be needed to test whether they should be used for cancer-specific therapies. Together, our results reveal the presence of structurally different ribosomes at the level of rRNA and provide the first atlas to distinguish different types of ribosomes and link them to different cellular programs, including those underlying human health and disease.
Limitations of the study
In this paper we created an atlas of human rRNA sequence variations in translating ribosomes, which we correlate with development as well as cancer. In this study we do not demonstrate that expression differences of rRNA variants have functional implications on human development and disease. In the TCGA dataset, control samples do not belong to the same matched cancer biopsy, and some cancer types have low control sample sizes. In our haplotype analysis we found high SD between haplotype frequencies across individuals, which limits our understanding of their functional significance. The expressed rRNA variants belong to the H7-hESC and K562 cell lines. It is likely that there are rRNA variants that are expressed in other human cells or samples not found in these cell lines. The RGA method is not limited to variant discovery between paralog genes; it can be applied for variant discovery between any related sequences, for example in detecting variants between amplicon sequences.
STAR★Methods
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Glass-bottom 12-well plates | Mattek | Cat# P12G-1.5-14-F |
| Bind-silane | GE Healthcare | Cat# 17-1330-01 |
| Ethanol | VWR | Cat# 89125-170 |
| Acetic acid | Sigma-Aldrich | Cat# A6283-100ML |
| Poly-D-lysine | Thermo Fisher Scientific | Cat# A3890401 |
| Circular cover glass (12mm) | Electron Microscope Sciences | Cat# 72226-01 |
| Gel slick solution | Lonza | Cat# 50640 |
| 1x PBS | Thermo Fisher Scientific | Cat# 10010049 |
| Paraformaldehyde (PFA) | Electron Microscope Sciences | Cat# 15710-S |
| Methanol | Sigma-Aldrich | Cat# 34860-1L-R |
| Tween 20 | Calbiochem | Cat# 655206 |
| RNaseOUT | Thermo Fisher Scientific | Cat# 10777019 |
| Ultrapure DNA/RNase-free water | Thermo Fisher Scientific | Cat# 10977023 |
| Template switching RT enzyme mix | New England Biolabs | Cat# M0466L |
| dNTP mix | Invitrogen | Cat# 100004893 |
| 5-(3-aminoallyl)-dUTP | Invitrogen | Cat# AM8439 |
| BS(PEG)9 | Thermo Fisher Scientific | Cat# 21582 |
| Glycine | Sigma-Aldrich | Cat# 50046-250G |
| RNase H | New England Biolabs | Cat# M0297L |
| RNase A | Thermo Fisher Scientific | Cat# EN0531 |
| RNase T1 | Thermo Fisher Scientific | Cat# EN0541 |
| BSA, molecular biology grade | New England Biolabs | Cat# B9000S |
| T4 DNA ligase | Thermo Fisher Scientific | Cat# EL0011 |
| Phi29 DNA polymerase | Thermo Fisher Scientific | Cat# EP0094 |
| Methacrylic acid N-hydroxysuccinimide ester | Sigma-Aldrich | Cat# 730300-1G |
| Sodium bicarbonate | Sigma-Aldrich | Cat# S5761-500G |
| Acrylamide solution | Bio-Rad | Cat# 161-0140 |
| Bis-acrylamide solution | Bio-Rad | Cat# 161-0142 |
| 20x SSC | Sigma-Aldrich | Cat# S6639 |
| Ammonium persulfate | Sigma-Aldrich | Cat# A3678 |
| N,N,N,N -Tetramethylethylenediamine | Sigma-Aldrich | Cat# T9281 |
| Formamide | Calbiochem | Cat# 75-12-7 |
| Triton X-100 | Sigma-Aldrich | Cat# 93443 |
| 4′,6-diamidino-2-phenylindole (DAPI) | Sigma-Aldrich | Cat# D9542 |
| Fetal Bovine Serum | Gibco | Cat# 26140079 |
| Trypsin-EDTA (0.5%), no phenol red | Gibco | Cat# 15400-054 |
| DMEM/F12 | Gibco | Cat# 11320033 |
| Accutase | Gibco | Cat# A1110501 |
| mTeSR1 | StemCell Technologies | Cat# 85850 |
| Thiazovivin | Tocris | Cat# 3845 |
| Bicine | Fisher Scientific | Cat# ICN10100580 |
| DMS | Sigma-Aldrich | Cat# D186309 |
| 100% Ethanol | Gold Shield Distributors | Cat# 0412804-PINT |
| 2-Mercaptoethanol | Sigma-Aldrich | Cat# 2-Mercaptoethanol |
| TRIzol™ reagent | Invitrogen | Cat# 15596026 |
| Chloroform | Fisher Scientific | Cat# C298-500 |
| Zymo RNA Clean and Concentrator Kit-5 | Zymo Research | Cat# R1016 |
| TURBO™ DNase | Ambion | Cat# AM2238 |
| SUPERase In RNase Inhibitor | Ambion | Cat# AM2696 |
| Halt™ Protease and Phosphatase Inhibitor Single-Use Cocktail, EDTA-Free (100X) | Thermo Fisher Scientific | Cat# 78443 |
| Sucrose | Millipore | Cat# 8510-OP |
| 10% SDS | Invitrogen | Cat# AM9820 |
| Sodium acetate | Invitrogen | Cat# AM9740 |
| Acid-phenol:chloroform, pH 4.5 with IAA (125:24:1) | Invitrogen | Cat# AM9722 |
| TGIRT-III | InGex | Cat# TGIRT50 |
| Buffer Kit, RNase-free | Invitrogen | Cat# AM9010 |
| PEG 8000 | Promega | Cat# V3011 |
| DTT (Dithiothreitol) | Thermo Fisher Scientific | Cat# A39255 |
| Betaine, 5M Solution | Fisher Scientific | Cat# AAJ77507UCR |
| Hydrochloric acid | Fisher Scientific | Cat# AA33257P6 |
| Sodium hydroxide | Fisher Scientific | Cat# SS255-1 |
| SPRIselect beads | Beckman Coulter | Cat# B23319 |
| Protein LoBind tubes | Eppendorf | Cat# 0030108442 |
| Buffer EB | Qiagen | Cat# 19086 |
| Exo-Resistant Random Primer | Thermo Fisher Scientific | Cat# SO181 |
| NEBNext® Ultra™ II Non-Directional RNA Second Strand Synthesis Module | New England Biolabs | Cat# E6111S |
| SMRTbell prep kit 3.0 | PacBio | Cat# 102-182-700 |
| Experimental models: cell lines | ||
| H7-hESC | Thomson et al.63 | N/A |
| K-562 | ATCC | Cat# CCL-243 |
| HeLa | ATCC | Cat# CCL-2 |
| Oligonucleotides | ||
| 28S RT primer bc11 for K-562 heavy polysome: CTATACGTATATCTATgacaaacccttgtgtcgagg | This paper | N/A |
| 28S RT primer bc12 for K-562 medium polysome: ACACTAGATCGCGTGTgacaaacccttgtgtcgagg | This paper | N/A |
| 28S RT primer bc13 for K-562 light polysome: CTCTCGCATACGCGAGgacaaacccttgtgtcgagg | This paper | N/A |
| 28S RT primer bc14 for K-562 monosome: CTCACTACGCGCGCGTgacaaacccttgtgtcgagg | This paper | N/A |
| 18S RT primer bc11 for K-562 heavy polysome: CTATACGTATATCTATtaatgatccttccgcaggttc | This paper | N/A |
| 18S RT primer bc12 for K-562 medium polysome: ACACTAGATCGCGTGTtaatgatccttccgcaggttc | This paper | N/A |
| 18S RT primer bc13 for K-562 light polysome: CTCTCGCATACGCGAGtaatgatccttccgcaggttc | This paper | N/A |
| 18S RT primer bc14 for K-562 monosome: CTCACTACGCGCGCGTtaatgatccttccgcaggttc | This paper | N/A |
| 28S RT primer bc1 for H7-hESC heavy polysome: CACATATCAGAGTGCGgacaaacccttgtgtcgagg | This paper | N/A |
| 28S RT primer bc2 for H7-hESC medium polysome: ACACACAGACTGTGAGgacaaacccttgtgtcgagg | This paper | N/A |
| 28S RT primer bc3 for H7-hESC light polysome: ACACATCTCGTGAGAGgacaaacccttgtgtcgagg | This paper | N/A |
| 28S RT primer bc4 for H7-hESC monosome: CACGCACACACGCGCGgacaaacccttgtgtcgagg | This paper | N/A |
| 18S RT primer bc1 for H7-hESC heavy polysome: CACATATCAGAGTGCGtaatgatccttccgcaggttc | This paper | N/A |
| 18S RT primer bc2 for H7-hESC medium polysome: ACACACAGACTGTGAGtaatgatccttccgcaggttc | This paper | N/A |
| 18S RT primer bc3 for H7-hESC light polysome: ACACATCTCGTGAGAGtaatgatccttccgcaggttc | This paper | N/A |
| 18S RT primer bc4 for H7-hESC monosome: CACGCACACACGCGCGtaatgatccttccgcaggttc | This paper | N/A |
| 28S RT primer bc6 H7-hESC: CATATATATCAG CTGTgacaaacccttgtgtcgagg |
This paper | N/A |
| 28S RT primer bc20 H7-hESC: CACGACACGA CGATGTgacaaacccttgtgtcgagg |
This paper | N/A |
| 28S RT primer bc01 for DMS seq:/5Phos/CAC ATATCAGAGTGCGgacaaacccttgtgtcgagg |
This paper | N/A |
| Deposited data | ||
| Monosome and polysome sequencing of 18S and 28S from monosome and polysome fractions from H7-hESC cell line | This paper | BioProject ID PRJNA926787, SRA: SRR29419059 (monosomes), SRA: SRR29419058 (“light polysomes” with 2 or 3 ribosomes), SRA: SRR29419057 (“medium polysomes” with 4 or 5 ribosomes), SRA: SRR29419056 (“heavy polysomes” with 6 or more ribosomes) |
| Monosome and polysome sequencing of 18S and 28S from monosome and polysome fractions from K562 cell line | This paper | BioProject ID PRJNA926787, SRA: SRR29419055 (monosomes), SRA: SRR29419054 (“light polysomes” with 2 or 3 ribosomes), SRA: SRR29419053 (“medium polysomes” with 4 or 5 ribosomes), SRA: SRR29419052 (“heavy polysomes” with 6 or more ribosomes) |
| Whole-genome sequencing of H7-hESC | This paper | BioProject ID PRJNA926787, SRA: SRR23196516 |
| 28S rRNA sequencing after treatment with DMS in H7-hESC cell line | This paper | BioProject ID PRJNA926787, SRA: SRR29884466 |
| Software and algorithms | ||
| calc_word.py | Zielezinski et al.64 | Alfree tools (pip install alfpy) version 1.0.6 |
| Bowtie2 | Langmeade and Salzberg65 | Bowtie2 2.3.4.1 |
| Clustal Omega | Sievers et al.66 | Clustal Omega - 1.2.4 |
| Kallisto | Bray et al.67 | Kallisto 0.46.1 |
| Minimap2 | Li, H.68 | Minimap 2.17-r974-dirty |
| RGA method | This paper | Zenodo: https://zenodo.org/doi/10.5281/zenodo.11661415 |
| Samtools | Li et al.69 | Samtools 1.16.1 |
Resource availability
Lead contact
Maria Barna (mbarna@stanford.edu).
Materials availability
This study did not generate new unique reagents.
Data and code availability
The atlas is available as Extended Data to this publication (Data S1, S2, S3, S4, and S5). H7-hESC rDNA and rRNA, and K562 rRNA sequencing data is available under BioProject ID PRJNA926787. For the H7-hESC under accession numbers SRR23196516 (H7-hESC whole genome sequencing), SRR29419059 (H7-hESC 18S and 28S from monosomes), SRR29419058 (H7-hESC 18S and 28S from “light polysomes” with 2 or 3 ribosomes), SRR29419057 (H7-hESC 18S and 28S from “medium polysomes” with 4 or 5 ribosomes), SRR29419056 (H7-hESC 18S and 28S from “heavy polysomes” with 6 or more ribosomes), SRR29884466 (H7-hESC 28S treated with DMS). For the K562 under accession numbers SRR29419055 (K562 18S and 28S from monosomes), SRR29419054 (K562 18S and 28S from “light polysomes” with 2 or 3 ribosomes), SRR29419053 (K562 18S and 28S from “medium polysomes” with 4 or 5 ribosomes), SRR29419052 (K562 18S and 28S from “heavy polysomes” with 6 or more ribosomes).
Method details
Reference index for rDNA extraction
For calling variants we map against three rDNA references.
18S and 28S extraction from hESC, GIAB and T2T, 1KGP
rDNA calling - hESC and GIAB: Reads were extracted by mapping HiFi fastq long reads (Table S1 for HiFi sample list) to the “reference index for rDNA extraction” using minimap268 with “-N 20 -ax map-ont” parameters and processes with samtools.69
T2T rDNA calling: We used complete 219 rDNA copies with 18S and 28S annotation by T2T.
hESC rRNA calling: The RT with 3′ primers of 18S and 28S results in 18S and 28S rRNA reads.
For both rDNA and rRNA, we keep reads that we consider full or near full length as follows.
-
(1)
We split long reads to consecutive non-overlapping 50bp short reads and map the short reads to “reference index for rDNA extraction” using bowtie2 using default parameters.65.
-
(2)
To call a long read 18S, we demand a long read to have at least 18 short reads (which is the equivalent of ∼900bp) to map to the 18S gene. For 28S calling, we demand a read to have at least 50 short reads (which is the equivalent of ∼2500bp) to map to the 28S gene.
-
(3)
We keep 18S reads in the length range 1,500-2,100 bp and 28S reads in the length range 4,500-5,500 bp.
-
(4)
For calling deletion variants, in order to avoid calling variants where the RT stopped, we only considered reads that do not have deletions at the beginning at position 56 in the 18S and position 25 in the 28S and only report deletion atlas variants after these positions.
Reference Gap Alignment (RGA) method
Python implementation source code is available here:https://zenodo.org/doi/10.5281/zenodo.11661415
-
(1)
We classified sequences as either 18S or 28S followed by Needleman–Wunsch global sequence alignment71 of each sequence to one RNA45S5 reference (either 18S or 28S based on read classification).72.
-
(2)
We created a reference sequence that aligns with all other sequences that we call a gap-aligned reference. This gap-aligned reference has the same sequence as the reference, but at each nucleotide position, we extended a gap at the size of the maximal gap found by the global sequence alignment to all sequences. Importantly, this gap-aligned reference allows straightforward comparison among all sequences without requiring computationally expensive all-by-all pairwise sequence alignments.
-
(3)
We aligned all H7-hESC sequences to the gap-aligned reference using the previous global alignment with additional extended gaps at reference positions.
-
(4)
Lastly, we extracted all variants at a given position across all aligned sequences.
Notably, we have benchmarked the gap-penalty opening and extension which can affect the indel number (Table S4). Since benchmarked parameters yielded a similar total number of indels, we use the default Needleman–Wunsch parameters of high penalty of gap opening and low penalty of gap extension.
Nucleotide variant calling in long reads
We ran our four step RGA, algorithm on the reads that pass the criteria in “18S and, 28S extraction from 1KGP, GIAB, hESC, and T2T”
The output of this alignment are exact alignment of all reads against the 18S/28S reference. With this we extract all sequence variants of the 18S and 28S both in rRNA and rDNA.
For the 1KGP dataset, we report rDNA variants that were found in at least 5 reads and detected in 3 samples in Table S5 (out of 30 1KGP samples with HiFi reads). For GIAB 2 trio families, we report variants found in at least 5 reads and detected in 2 samples Tables S6 and S7 (For HiFi and ONT datasets). Since the Chinese Han father sample was not sequenced in ONT, we did not include this sample in the HiFi dataset and the total number of samples were 5: Ashkenazi mother, father, son, and Chinese Han mother and son.
Atlas variant calling in long-reads
We ran “Nucleotide, helix and ES variant calling” on both the hESC rDNA and the rRNA data and call a found variant an atlas variant if the variant is present in abundance greater than the HiFi read accuracy and are found in both rDNA and rRNA.
HiFi accuracy is expected to be greater than 99.9%.
For the hESC we obtained the following from “18S and, 28S extraction from hESC, GIAB and T2T” step:
From the hESC rDNA we have obtained 762 complete 18S sequences and 386 complete 28S sequences.
From rRNAs, we have obtained 7,454 and 51,040 complete 18S sequences from monosome and polysomes, and 5,834 and 8,596 complete 28S sequences from monosome and polysomes.
Then, assuming HiFi accuracy of 99.9%, we call atlas variants that satisfy.
-
(1)
Nucleotide variant was found in the rDNA at least twice.
-
(2)
Nucleotide variant abundance in rRNA is at least 59 for the 18S and 10 for 28S.
The raw read count for nucleotide variants are reported in the atlas. After finding nucleotide atlas variants, to call for atlas helix and ES variants, we use the helix/ES annotation (Tables S13 and S14) to aggregate nucleotide atlas variants at a given region and consider variants if they are found in both rDNA and rRNA. The raw read count for helix and ES resolution atlas is found in the names of the variants. There, the variant name ID indicates in the name the raw rDNA and rRNA read count.
The naming convention is “atlas_resolution:regional_variant:ID_Raw-rDNA-count_Raw-rRNA-count”. So for example, at the atlas resolution of expansion segments, the first regional variant of region es2s is named: ES:es2s:0_d730_r43137. In this example, this nucleotide sequence containing sequence variations was observed 730 in rDNA and 43,137 in rRNA.
Validation of atlas nucleotide variants using 1KGP short-read data
We use the common Bowtie2 mapper tool and map the short-reads data from the 30 individuals from the 1KGP for which we have long-read data and map short-reads to our atlas of expanded resolution which allows mapping short-reads against it. This atlas contains complete expansion segments and non-expansion segments (Data S3 and S5, ES/Non-ES marked in yellow and purple in Figure S11) which we also extend by 100 bases of the reference sequence to allow mapping to region ends. After mapping to this atlas, we only consider perfect matched reads. Afterward, for finding which indels and SNVs are detected, we convert variants found at ES resolution back to nucleotide variants.
In-cell DMS probing for long-read sequencing
Approximately 2 x 107 of H7-hESC were used for in-cell DMS probing. Cells were washed with pre-warmed DPBS (Gibco, 14040133) prior to dissociation with Accutase (Gibco, A1110501) for ∼5 min at 37°C. Then, cells were neutralized with mTeSR1 (StemCell Technologies, 85850) supplemented with 1μM thiazovivin (Tocris, 3845) and pelleted down by centrifuging at 200 x g at room temperature for 3 min. Cells were then resuspended in 2,800 μL of pre-warmed mTeSR1+Tv. Then, 800 μL of pre-warmed 1 M bicine (Fisher Scientific, ICN10100580) buffer (pH 8.3 at 37°C) was added, followed by 400 μL of 16% DMS (Sigma Aldrich, D186309) diluted in 100% ethanol (Gold Shield Distributors, 0412804-PINT). DMS labeling was done by incubating the mixture at 37°C for 5 min, prior to be quenched by adding 2,000 μL of ice-cold BME (Sigma Aldrich, M3148). Cells were pelleted down by centrifuging at 200 x g at 4°C for 3 min, and then lysed by resuspending them in 8 mL of cold TRIzolTM reagent (Invitrogen, 15596026). Solution was left at room temperature for 5 min prior to adding 600 μL chloroform (Fisher Scientific, C298-500). The tube was then shaken vigorously for 15 s or so and left at room temperature for 3 min. The sample was then centrifuged at 21,000 x g for 15 min at 4°C. A total of 4,440 μL aqueous phase was extracted and 4,440 μL of 100% ethanol was added before subjecting them into further cleanup and DNase digest using Zymo RNA Clean and Concentrator Kit-5 as elaborated in “polysome RNA extraction”.
50 μg of total RNA was used across ten 100 μL reactions. RT was done as described in “rRNA reverse transcription” section, with a few modifications. After RT, 0.4x beads by volume were used to size select cDNA. Ten reactions were then pooled together, and its cDNA concentration measured. For the second-strand synthesis, each reaction was done with a maximum of 500 ng of cDNA. Afterward, PacBio IsoSeq library was constructed as per described in “pacBio SMRT sequencing library preparation” section.
rRNA subtype DMS reactivity and structure calling
28S sequenced reads from “in-cell DMS probing for long-read sequencing” were binned intro rRNA subtypes as follows.
-
(1)
We ran RGA method on the DMS reads
-
(2)
We bin DMS reads to rRNA subtype groups based on the hESC subtypes nucleotide positions at 60, 2188, 3513, 4913
-
(3)
Per DMS read, at each nucleotide we mark sequence variants that are not in the atlas as modified.
-
(4)
For every rRNA subtype group, we calculate the rRNA subtype group reactivity as the average modification per nucleotide position across binned DMS reads.
-
(5)
Next we use 90% winsorization to set the DMS reactivity values from 0 to 1.
-
(6)
Lastly, we use the Biers MATLAB package with RNAstructure and Varna for plotting.73,74
28S full length sequence comparisons and visualization in PCoA
In the T2T genome we discovered that although there are only 62 reported rDNA variants, the high frequency rDNA variants in H7-hESC also appeared in high frequency in the T2T rDNA (Figure S9A, R = 0.8 Pearson correlation). In the GIAB samples, like in the H7-hESC, we found hundreds of variants with high agreement between their frequencies and H7-hESC frequencies (Figures S9B–S9D). We analyzed the linkage of the same positions in the T2T and GIAB, and found as found in the H7-hESC that es7l positions have low linkage, and es15l, es27l and es39l have relatively higher linkage within each region (Figure S10).
We compare the pairwise-distances between all hESC/GIAB 28S separately using 6-mer word base comparison with Alfree tools.64 Pairwise distances are then visualized by plotting the first two PCos of the and the Bray-Curtis PCoA (Figure 3D). Each 28S is colored by the haplotype of that 28S as defined by the 60, 3513 and 4913 positions.
Atlas relative abundance calling for RNA short-read datasets
Short read RNA-seq are mRNA targeted however we found that about 2% of reads mapped to rRNA in the GTEx and TCGA. For comparing relative abundance across samples, we rarefaction samples of GTEx dataset to 500,000 rRNA mapped reads and TCGA to 250,000 rRNA mapped reads and throw samples with less than 100,000 rRNA mapped reads.
For short-reads, we use Kallisto67 tool for region relative abundance estimation in the following way:
Once made for all GTEx/TCGA samples. We create a Kallisto index67 with 18S and 28S variants in our ES/non-ES atlas with expanded 100bp reference (Data S5) using Kallisto default parameters.
Linux command line:
> kallisto index -i atlas_rRNA DataS5.atlas_expand100_region_edge.ES.fa.
This expanded reference version of the atlas is the same ES/non-ES atlas with additional flanking 100bp on both ends (3′ and 5′) of the relevant region with the reference sequence. With these expansions, short reads that map to the 5′ or 3′ end of a region are mapped to the variants (as opposed to unmapped without expansions).
Here we chose our ES/non-ES atlas reference, as ES/non-ES regions are longer than helices (Tables S16 and S17) but using the helix expanded reference atlas should give the same results (Data S4).
We quantify all-region abundances of a sample (in the example below named FASTQ-FILE) using Kallisto67 with default parameters.
Linux command line
> kallisto quant -i atlas_rRNA FASTQ-FILE -o OUTPUT_DIRECTORY
Then, to compare expression of a given ES/non-ES regional variant, we normalize read count by the region length and normalize to one every ES/non-ES region independently.
Python code
abundance = pd.read_csv(os.path.join(OUTPUT_DIRECTORY,'abundance.tsv'),sep = '\t',index_col = 0)[['eff_length','tpm']]
abundance = abundance['tpm']/abundance['eff_length']
ra = []
for group, group_df in abundance.groupby(lambda x: x.split(':')[1]):
ra.append(group_df/group_df.sum())
normalized_abundnces = pd.concat(ra)
normalized_abundnces in the above python code contains the relative abundances of variants after normalization by ES/non-ES region.
GTEx and TCGA sample handling
GTEx: Most individuals have multiple organs sequenced. To control for inter-individual variations when comparing tissues, we select one sample per individual in the GTEx dataset. For each compared tissue pair, individuals that have both tissues are randomly divided into two-halves, from the first group we keep one tissue and from the second group we keep the other tissue. This way we only have one tissue per individual when comparing tissues. In all analyses we compared tissues with at least 10 samples.
In the TCGA cancer/control comparison, we selected cancers with at least 50 samples.
Polysome RNA extraction
H7-hESCs were harvested with Accutase (Gibco), and the cell pellets were lysed in lysis buffer (20 mM Tris pH 7.5, 150 mM NaCl, 15 mM MgCl2, 100 μg/mL cycloheximide, 1 mM DTT, 0.5% Triton X-100, 0.1 mg/mL heparin, 8% glycerol, 20 U/ml TURBO DNase (Ambion, AM2238), 200 U/mL SUPERase In RNase Inhibitor (Ambion, AM2696), 1x Combined Protease and Phosphatase Inhibitor (Thermo Scientific, 78443)) at 4°C for 30 min with occasional vortexing. Lysates were sequentially centrifuged at 1800g for 5 min at 4°C and then at 10,000g for 5 min at 4°C, retaining the final supernatant as the cytoplasmic extract. Cytoplasmic extract was loaded on to a 10–45% sucrose gradient (20 mM Tris pH 7.5, 100 mM NaCl, 15 mM MgCl2, 100 μg/mL cycloheximide, made on a Biocomp Model 108 Gradient Master) and centrifuged in a Beckman SW41 rotor at 40,000 rpm for 2.5 h at 4°C. Gradients were then fractionated on a Density Gradient Fraction System (Brandel, BR-188) with continuous A260 measurements. To each fraction (which contained approximately 750 μL), 100 μL of 10% SDS was added and the tubes vortexed to mix, followed by the addition of 140 μL of 3 M sodium acetate (pH 5.5) and 200 μL of RNase-free water, vortexing to mix. For ribosomal populations that spanned multiple fractions, such as the polysomes, equal volumes of each corresponding fraction was pooled in a separate tube to a total volume of 900 μL. To 900 μL of fractionated sample, 900 μL of acid phenol chloroform was added and heated at 65°C for 5 min. The samples were then centrifuged at 21000g for 10 min at room temperature, and the aqueous phase transferred to a new tube. The aqueous phase was mixed with an equal volume of 100% ethanol and the RNA purified using the Zymo RNA Clean and Concentrator Kit-5 following manufacturer’s instructions. DNase treatment was performed using TURBO DNase (1 μL of 2 U/μL per 50 μL reaction) at 37°C for 30 min, and the RNA purified using the Zymo RNA Clean and Concentrator Kit-5 following manufacturer’s instructions.
400 μL of cold cytoplasmic lysis buffer was added to each cell pellet. Cells were mixed and lysed by repeated vortexing for 30 s, followed by cooling down on ice for 30 s, repeated for 3 times in total. Cells were then incubated on ice for 30 min, vortexing for approximately 10 s every 10 min for complete lysis. Afterward, cellular debris, organelles, and microsomes were removed with four serial centrifugations at 800 x g twice, 8,000 x g, and 21,300 x g for 5 min each at 4°C. RNA amount of the clarified cytoplasmic lysate was measured using nanodrop. Approximately 0.8–1 mg of RNA was set aside for sucrose gradient fractionation.
As the cells were being lysed, 10–45% sucrose gradient were prepared as follows: 50 mL of 10% and 45% sucrose buffers (20 mM Tris pH 7.5, 15 mM MgCl2, 150 mM NaCl, 1 mM DTT, 100 μg/mL cycloheximide, 5 gr (10% solution) and 22.5 gr of sucrose (45% solution) (Millipore 8510-OP), in nuclease free water) were prepared separately. Using SW 41 Ti rotor compatible ultracentrifuge tubes (Beckman Coulter 331372), the two sucrose gradient buffers were layered extremely gently, and then the gradient was established using a gradient maker (Biocomp Gradient Master 108).
The cytoplasmic lysate was then layered on top of the sucrose gradient. The tubes were then loaded into SW 41 Ti rotor and centrifuged at 40,000 rpm for 2.5 h at 4°C. Afterward, the gradient was then fractionated into 16 2 mL tubes every 30 s of ∼700 μL solution each using a fractionation system (Brandel BR-188). The A260 trace was used as a reference to determine where the free ribonucleoproteins, free subunits, monosomes, and polysomes were. 100 μL of 10% SDS (Invitrogen AM9820) and 200 μL of 1.5 M sodium acetate (Invitrogen AM9740) were added into each fraction.
RNA was extracted from each fraction by adding 500 μL of acid-phenol:chloroform, pH 4.5 with IAA (125:24:1) (Invitrogen AM9722). The fractions were then incubated at 65°C, 500 rpm thermomixer for 5 min. The RNA-containing aqueous phase, ∼700 μL, was separated from the organic phase by centrifugation at 21,300 x g for 15 min at 4°C. Further cleanup and trace DNA removal were done as described in the section “Whole-cell RNA extraction”.
Polysome fractions collection
We collected and sequenced RNA from ribosome containing fractions: ribosomes (monosomes) and polysomes (Figure S1 for H7-hESC A260 trace).
rRNA reverse transcription
Reverse transcription was done using TGIRT-III enzyme (InGex TGIRT50) with modified buffer and reaction conditions to increase enzyme processivity against highly structured and modified rRNA. To start, 4 μL of 100 μM pooled barcoded RT primers were added into 4.3 μL of 1 μg of RNA. RNA-primer mix was then denatured at 65°C for 5 min. 2.5 μL of 8x RT buffer (600 mM KCl (Invitrogen AM9640G), 160 mM Tris pH 7.5, 80 mM MgCl2) was then added at 65°C, and the reaction then cooled to 25°C. Subsequently, 8.7 μL of enzyme mix (12.2% of PEG 8000, 12.2 mM of DTT, 2.44 M of Betaine, 4.88 U/μL of TGIRT-III, 12.2 U/μL of SUPERaseIn RNAse Inhibitor) was added into each reaction, followed by 30 min incubation at 25°C. Afterward, 1 μL of 25 mM dNTP was added prior to incubating the samples at 60°C for 2 h. The final concentration of the reagents in the 20 μL RT reaction are: 20 mM Tris HCl, 75 mM KCl, 10 mM MgCl2, 5% PEG 8000, 5 mM DTT, 1 M Betaine, 2 U/μL TGIRT-III, 1 U/μL SUPERaseIN RNAse Inhibitor, and 10 μM pooled RT primers for 1 μg of RNA. After the RT, the RNA template is hydrolyzed by adding 1 μL of 2.5 M NaOH at 95°C for 3 min. After cooling down to 4°C, the reaction was neutralized by adding 1 μL of 2.5 M HCl and 1 μL of 500 mM Tris pH 7.5.
SPRISelect magnetic beads (Beckman Coulter B23319) were used for cDNA cleanup following the manufacturer’s protocol. Beads were washed to remove contaminants that elute simultaneously with the DNA and interfere with polymerase binding in PacBio Sequel IIe system. In brief, for every 500 μL of SPRISelect beads in a low binding tube (Eppendorf 0030108442), the beads were centrifuged down at 21,300 x g for 1 min. The tube was then placed in a magnetic rack, and the supernatant was aspirated and kept aside. The beads were then washed with 1 mL of nuclease-free water, vortexed, centrifuged at 21,300 x g for 1 min, and placed in a magnetic rack to remove the water wash. The wash was repeated a total of five times. Afterward, the beads were washed similarly with 1 mL of Qiagen buffer EB (Qiagen 19086) instead. The beads were then resuspended with the original supernatant reserved previously, and could be kept at 4°C for at most a week.
To clean up the single-stranded cDNA with the washed beads, a 2.2x beads-to-sample volume ratio was added. The beads then were washed with freshly prepared 85% ethanol for 1 min while the tubes were attached on the magnetic rack. cDNA was then eluted in 40 μL of nuclease-free water for second-strand synthesis.
For each reaction, 1 μL of 12.5 mM dNTP (Thermo Scientific R0181) and 1 μL of 2 μM random hexamer (Thermo Scientific SO181) were added. Making sure that all components were kept on ice, 26 μL of nuclease-free water, and 4 μL of second-strand synthesis enzyme with 8 μL of 10x buffer (NEB E6111S) were added for a total of 80 μL reaction volume. The sample was then incubated at 16°C in the thermocycler with the lid heating turned off. Double-stranded cDNA was then cleaned up using SPRIselect following the manufacturer’s protocol, with a 0.82x beads-to-reaction ratio instead. Washing was done twice with freshly prepared 85% ethanol, and the cDNA was then eluted in 50 μL of nuclease-free water. Cleanup was repeated once again with the same bead ratio to enrich for full-length reverse-transcribed rRNA molecules, with the final elution done in 30 μL of nuclease free water. cDNA concentration was then measured with qubit.
PacBio SMRT sequencing library preparation
Multiplexed library was made as described in “Iso-Seq Express Template Preparation for Sequel and Sequel II Systems” using SMRTbell prep kit 3.0. cDNA amplification was skipped to prevent possible amplification bias against highly structured or repetitive sequences. In brief: equal amounts of cDNA from each barcode was pooled for a total of ∼200 ng prior to DNA damage repair step. After pooling, cDNA was concentrated with 1x volume of SPRIselect beads and eluted in 48 μL of nuclease-free water. DNA damage repair, end-repair/A-tailing, overhang adapter ligation, and the final library cleanup were performed according to the protocol mentioned above, substituting the ProNex beads with the washed SPRIselect beads.
HeLa cell culture
The Human HeLa cell line was sourced from ATCC(CCL-2), and subsequent culturing was performed in DMEM supplemented with 10% FBS at 37°C and 5% CO2.
In situ rRNA sequencing experimental procedure
Glass-bottom 12-well plates were treated as follows: Oxygen plasma treatment was applied for 5 min (Anatech Barrel Plasma System, 100W, 40% O2), followed by sequential incubation with 1% methacryloxypropyltrimethoxysilane (Bind-Silane) 88% ethanol, 10% acetic acid, and 1% H2O at room temperature for 1 h and 0.1 mg/mL Poly-D-lysine solution at room temperature for an additional hour. Micro cover glasses underwent a pretreatment step with Gel Slick at room temperature for 15 min and were then air-dried.
HeLa cells were cultured in treated 12-well plates, and after rinsing with 1× PBS, they were fixed with 1 mL of 1.6% PFA (Electron Microscope Sciences, 15710-S) in PBS buffer at room temperature for 15 min. Following fixation, the cells underwent permeabilization by treatment with 1 mL of pre-chilled (−20°C) methanol and incubation at −20°C for an hour. Thereafter, HeLa cells were transferred from the −20°C fridge to room temperature for 5 min, and then washed twice with PBSTR (0.1% Tween 20, 0.1 U/μL RNaseOUT in PBS) for 5 min each.
For the reverse transcription (RT) process, primers were prepared by dissolving them at a concentration of 250 μM in ultrapure RNase-free water, followed by pooling. All probes were manufactured by Integrated DNA Technologies (IDT). The probe mixture was subjected to heating at 90°C for 5 min, followed by cooling to room temperature. The samples were then treated with 300 μL of template switching mixture, which included 1× template switching buffer, 250 μM dNTP mix, 40 μM 5-(3-aminoallyl)-dUTP, 2.5 μM RT primer, 0.4 U/μL RNaseOUT, 3.3 μM template switching oligo, and 1× template switching RT enzyme mix. This mixture was incubated at 4°C for 15 min, followed by an overnight placement in a 42°C humidified oven with gentle shaking.
The following day, the samples underwent three washes with 500 μL PBST (0.1% Tween 20 in PBS) for 5 min each. To cross-link cDNA molecules containing aminoallyl-dUTP, the specimens were incubated with 5 mM BS(PEG)9 in PBST for 1 h at room temperature, followed by a wash with PBST at room temperature for 5 min. The cross-linking reaction was quenched by treating the samples with 0.1 M Glycine in PBST at room temperature for 30 min. To degrade residual RNA and generate single-stranded cDNA, the specimens were incubated for 2 h at 37°C with an RNA digestion mixture, composed of 0.25 U/μL RNase H, 1 mg/mL RNase A, and 10 U/μL RNase T1 in 1× RNaseH buffer. The samples were then washed twice with PBST for 5 min each. After the final PBST wash, the samples were incubated with 300 μL of splint ligation mixture containing 0.2 mg/mL BSA, 2.5 μM splint ligation primer, and 0.1 U/μL T4 DNA ligase in 1× T4 DNA ligase buffer at room temperature for 4 h with gentle shaking. Subsequently, they were washed three times with 500 μL PBST for 5 min each.
To create nanoballs of cDNA (amplicons) containing multiple copies of the original cDNA sequence, each cDNA circle undergoes linear amplification through rolling-circle amplification (RCA). This is achieved by immersing the cDNA in a 300 μL RCA mixture consisting of 0.2 U/μL Phi29 DNA polymerase, 250 μM dNTP, 40 μM 5-(3-aminoallyl)-dUTP, and 0.2 mg/mL BSA in 1× Phi29 buffer at 30°C for 4 h with gentle shaking. Following RCA, the samples were subjected to two washes with PBST. Subsequently, they were incubated with 20 mM methacrylic acid N-hydroxysuccinimide ester in 100 mM sodium bicarbonate buffer at room temperature for 1 h, followed by two additional washes with PBST for 5 min each. The samples then experience a 10-min incubation in 500 μL monomer buffer containing 4% acrylamide and 0.2% bis-acrylamide in 2× SSC at 4°C. Following the aspiration of the buffer, a 35 μL polymerization mixture, made of 0.2% ammonium persulfate and 0.2% tetramethylethylenediamine dissolved in monomer buffer, is placed at the core of the sample and is promptly covered with a Gel Slick-coated coverslip. The polymerization is then carried out inside an N2 enclosure for 90 min at room temperature. Afterward, the sample is washed three times with PBST, each time for 5 min.
Several iterative sequencing experiments were conducted to decode the rRNA identity. For each iteration, the sample initially underwent treatment with a stripping buffer containing 60% formamide and 0.1% Triton X-100 at room temperature twice for 10 min each, followed by a triple wash in PBST, each lasting 5 min. Then the samples were incubated with a 300 μL sequencing mixture containing 0.2 U/μL T4 DNA ligase, 0.2 mg/mL BSA, 10 μM reading probe, and 5 μM fluorescent decoding oligos in 1× T4 DNA ligase buffer for at least 3 h at room temperature. Post-incubation, the samples were thrice washed with a washing and imaging buffer made of 10% formamide in 2× SSC buffer, each wash lasting for 10 min. Following the washing steps, the samples were immersed in the washing and imaging buffer for imaging. DAPI was dissolved in the wash and imaging buffer and performed following manufacturer’s instruction for nuclei staining for 20 min. Images were captured using a Leica TCS SP8 confocal microscope equipped with a 40× oil immersion objective (NA 1.3) and an acquisition voxel size of 142 nm × 142 nm × 500 nm.
Acknowledgments
We thank Rhiju Das for interpreting DMS results. We thank Xiangling Meng, Craig Kerr, Ali Wilkening, and Michael Montgomery for help with early experiments that were not later followed in this study. We thank the Barna and Pritchard group members for discussions. M.B. is supported by the New York Stem Cell Foundation and the National Institutes of Health grant R01HD086634. J.K.P. is supported by RO1 HG008140. D.R. is supported by ALTF 1042-2019 EMBO, LT000218/2020-L HFSP, and McCormick postdoctoral scholarships. T.T.S. is supported by a National Science Scholarship (PhD) from the Agency for Science, Technology and Research. M.B. is an NYSCF Robertson Investigator. X.W. is supported by an Edward Scolnick Professorship, an Ono Pharma Breakthrough Science Initiative Award, a Merkin Institute Fellowship, and an NIH DP2 New Innovator Award 1DP2GM146245-01.
Author contributions
D.R. conceived the project, designed experimental and computational analyses, conducted all computational analyses, interpreted the results, and wrote the manuscript. T.T.S. designed and conducted all polysome and DMS sequencing experiments, interpreted the results, and wrote the manuscript. X.S. developed, optimized, and performed SWITCH-seq experiments. X.W. supervised the in situ sequencing experiment. J.P.S. helped in computational analyses. N.R.G. helped in experimental data collection. N.S.-A. helped interpret GTEx data. R.R. helped in DMS analyses. M.B. and J.K.P. conceived and directed the project and analyses, designed the analyses, interpreted the results, and wrote the manuscript.
Declaration of interests
X.W. is a scientific co-founder of Stellaromics. X.W. and X.S. are inventors on pending patent applications related to SWITCH-seq. D.R., J.K.P., M.B., and T.T.S. are inventors on a pending patent related to rRNA variation in cancer.
Published: August 6, 2024
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.xgen.2024.100629.
Supplemental information
This includes all of the rRNA variants found both in rDNA and rRNA in translating ribosomes in the H7-hESC cell line.
This is the same as Data S1 with the exception that variants are grouped to the resolution of rRNA helices. Helix annotations can be found in Table S16 and Figure S11.
This is the same as Data S1 with the exception that variants are grouped to the resolution of rRNA expansion segments. Expansion segment annotations can be found in Table S17 and Figure S11.
This is the same as Data S2 with flanking reference sequence on both 3′ and 5′ ends. This atlas is useful for estimating variant frequencies in short-read data. For usage example, see “atlas relative abundance calling for RNA short-read datasets.”
This is the same as Data S3 with flanking reference sequence on both 3′ and 5′ ends. This atlas is useful for estimating variant frequencies in short-read data. For usage example, see “atlas relative abundance calling for RNA short-read datasets.”
References
- 1.Henderson A.S., Warburton D., Atwood K.C. Location of ribosomal DNA in the human chromosome complement. Proc. Natl. Acad. Sci. USA. 1972;69:3394–3398. doi: 10.1073/pnas.69.11.3394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Arnheim N., Southern E.M. Heterogeneity of the ribosomal genes in mice and men. Cell. 1977;11:363–370. doi: 10.1016/0092-8674(77)90053-8. [DOI] [PubMed] [Google Scholar]
- 3.Kurylo C.M., Parks M.M., Juette M.F., Zinshteyn B., Altman R.B., Thibado J.K., Vincent C.T., Blanchard S.C. Endogenous rRNA Sequence Variation Can Regulate Stress Response Gene Expression and Phenotype. Cell Rep. 2018;25:236–248.e6. doi: 10.1016/j.celrep.2018.08.093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Genomes P.C., Abecasis G.R., Altshuler D., Auton A., Brooks L.D., Durbin R.M., Gibbs R.A., Hurles M.E., McVean G.A. A map of human genome variation from population-scale sequencing. Nature. 2010;467:1061–1073. doi: 10.1038/nature09534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Parks M.M., Kurylo C.M., Dass R.A., Bojmar L., Lyden D., Vincent C.T., Blanchard S.C. Variant ribosomal RNA alleles are conserved and exhibit tissue-specific expression. Sci. Adv. 2018;4 doi: 10.1126/sciadv.aao0665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Fan W., Eklund E., Sherman R.M., Liu H., Pitts S., Ford B., Rajeshkumar N.V., Laiho M. Widespread genetic heterogeneity of human ribosomal RNA genes. RNA. 2022;28:478–492. doi: 10.1261/rna.078925.121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Clark C.G., Tague B.W., Ware V.C., Gerbi S.A. Xenopus laevis 28S ribosomal RNA: a secondary structure model and its evolutionary and functional implications. Nucleic Acids Res. 1984;12:6197–6220. doi: 10.1093/nar/12.15.6197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Wakeman J.A., Maden B.E. 28 S ribosomal RNA in vertebrates. Locations of large-scale features revealed by electron microscopy in relation to other features of the sequences. Biochem. J. 1989;258:49–56. doi: 10.1042/bj2580049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Barbitoff Y.A., Abasov R., Tvorogova V.E., Glotov A.S., Predeus A.V. Systematic benchmark of state-of-the-art variant calling pipelines identifies major factors affecting accuracy of coding sequence variant discovery. BMC Genom. 2022;23:155. doi: 10.1186/s12864-022-08365-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Gibbons J.G., Branco A.T., Godinho S.A., Yu S., Lemos B. Concerted copy number variation balances ribosomal DNA dosage in human and mouse genomes. Proc. Natl. Acad. Sci. USA. 2015;112:2485–2490. doi: 10.1073/pnas.1416878112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Paredes S., Branco A.T., Hartl D.L., Maggert K.A., Lemos B. Ribosomal DNA deletions modulate genome-wide gene expression: “rDNA-sensitive” genes and natural variation. PLoS Genet. 2011;7 doi: 10.1371/journal.pgen.1001376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Gibbons J.G., Branco A.T., Yu S., Lemos B. Ribosomal DNA copy number is coupled with gene expression variation and mitochondrial abundance in humans. Nat. Commun. 2014;5:4850. doi: 10.1038/ncomms5850. [DOI] [PubMed] [Google Scholar]
- 13.Nelson J.O., Watase G.J., Warsinger-Pepe N., Yamashita Y.M. Mechanisms of rDNA Copy Number Maintenance. Trends Genet. 2019;35:734–742. doi: 10.1016/j.tig.2019.07.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Xu B., Li H., Perry J.M., Singh V.P., Unruh J., Yu Z., Zakari M., McDowell W., Li L., Gerton J.L. Ribosomal DNA copy number loss and sequence variation in cancer. PLoS Genet. 2017;13 doi: 10.1371/journal.pgen.1006771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Malinovskaya E.M., Ershova E.S., Golimbet V.E., Porokhovnik L.N., Lyapunova N.A., Kutsev S.I., Veiko N.N., Kostyuk S.V. Copy number of human ribosomal genes with aging: unchanged mean, but narrowed range and decreased variance in elderly group. Front. Genet. 2018;9:306. doi: 10.3389/fgene.2018.00306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wang M., Lemos B. Ribosomal DNA copy number amplification and loss in human cancers is linked to tumor genetic context, nucleolus activity, and proliferation. PLoS Genet. 2017;13 doi: 10.1371/journal.pgen.1006994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Chen C., Feng L., Chen J., Shen J., Lin L. Ribosomal DNA copy number alteration in blood sample from gastric cancer patients. Mol. Biol. Rep. 2023;50:7155–7160. doi: 10.1007/s11033-023-08630-y. [DOI] [PubMed] [Google Scholar]
- 18.Stults D.M., Killen M.W., Williamson E.P., Hourigan J.S., Vargas H.D., Arnold S.M., Moscow J.A., Pierce A.J. Human rRNA gene clusters are recombinational hotspots in cancer. Cancer Res. 2009;69:9096–9104. doi: 10.1158/0008-5472.CAN-09-2680. [DOI] [PubMed] [Google Scholar]
- 19.Valori V., Tus K., Laukaitis C., Harris D.T., LeBeau L., Maggert K.A. Human rDNA copy number is unstable in metastatic breast cancers. Epigenetics. 2020;15:85–106. doi: 10.1080/15592294.2019.1649930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Udugama M., Sanij E., Voon H.P.J., Son J., Hii L., Henson J.D., Chan F.L., Chang F.T.M., Liu Y., Pearson R.B., et al. Ribosomal DNA copy loss and repeat instability in ATRX-mutated cancers. Proc. Natl. Acad. Sci. USA. 2018;115:4737–4742. doi: 10.1073/pnas.1720391115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Feng L., Du J., Yao C., Jiang Z., Li T., Zhang Q., Guo X., Yu M., Xia H., Shi L., et al. Ribosomal DNA copy number is associated with P53 status and levels of heavy metals in gastrectomy specimens from gastric cancer patients. Environ. Int. 2020;138 doi: 10.1016/j.envint.2020.105593. [DOI] [PubMed] [Google Scholar]
- 22.Nurk S., Koren S., Rhie A., Rautiainen M., Bzikadze A.V., Mikheenko A., Vollger M.R., Altemose N., Uralsky L., Gershman A., et al. The complete sequence of a human genome. Science. 2022;376:44–53. doi: 10.1126/science.abj6987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Rodriguez-Algarra F., Seaborne R.A.E., Danson A.F., Yildizoglu S., Yoshikawa H., Law P.P., Ahmad Z., Maudsley V.A., Brew A., Holmes N., et al. Genetic variation at mouse and human ribosomal DNA influences associated epigenetic states. Genome Biol. 2022;23:54. doi: 10.1186/s13059-022-02617-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Sims J., Sestini G., Elgert C., von Haeseler A., Schlögelhofer P. Sequencing of the Arabidopsis NOR2 reveals its distinct organization and tissue-specific rRNA ribosomal variants. Nat. Commun. 2021;12:387. doi: 10.1038/s41467-020-20728-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Poplin R., Chang P.-C., Alexander D., Schwartz S., Colthurst T., Ku A., Newburger D., Dijamco J., Nguyen N., Afshar P.T., et al. A universal SNP and small-indel variant caller using deep neural networks. Nat. Biotechnol. 2018;36:983–987. doi: 10.1038/nbt.4235. [DOI] [PubMed] [Google Scholar]
- 26.Luo R., Wong C.-L., Wong Y.-S., Tang C.-I., Liu C.-M., Leung C.-M., Lam T.-W. Exploring the limit of using a deep neural network on pileup data for germline variant calling. Nat. Mach. Intell. 2020;2:220–227. doi: 10.1038/s42256-020-0167-4. [DOI] [Google Scholar]
- 27.Fairley S., Lowy-Gallego E., Perry E., Flicek P. The International Genome Sample Resource (IGSR) collection of open human genomic variation resources. Nucleic Acids Res. 2020;48:D941–D947. doi: 10.1093/nar/gkz836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wilm A., Aw P.P.K., Bertrand D., Yeo G.H.T., Ong S.H., Wong C.H., Khor C.C., Petric R., Hibberd M.L., Nagarajan N. LoFreq: a sequence-quality aware, ultra-sensitive variant caller for uncovering cell-population heterogeneity from high-throughput sequencing datasets. Nucleic Acids Res. 2012;40:11189–11201. doi: 10.1093/nar/gks918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Benjamin D.I., Sato T., Cibulskis K., Getz G., Stewart C., Lichtenstein L. Calling Somatic SNVs and Indels with Mutect2. BioRxiv. 2019 doi: 10.1101/861054. Preprint at. [DOI] [Google Scholar]
- 30.Bose P., Hermetz K.E., Conneely K.N., Rudd M.K. Tandem repeats and G-rich sequences are enriched at human CNV breakpoints. PLoS One. 2014;9 doi: 10.1371/journal.pone.0101607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Dohm J.C., Peters P., Stralis-Pavese N., Himmelbauer H. Benchmarking of long-read correction methods. NAR Genom. Bioinform. 2020;2:lqaa037. doi: 10.1093/nargab/lqaa037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Wenger A.M., Peluso P., Rowell W.J., Chang P.-C., Hall R.J., Concepcion G.T., Ebler J., Fungtammasan A., Kolesnikov A., Olson N.D., et al. Accurate circular consensus long-read sequencing improves variant detection and assembly of a human genome. Nat. Biotechnol. 2019;37:1155–1162. doi: 10.1038/s41587-019-0217-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lal A., Brown M., Mohan R., Daw J., Drake J., Israeli J. Improving Long-Read Consensus Sequencing Accuracy with Deep Learning. bioRxiv. 2021 doi: 10.1101/2021.06.28.450238. Preprint at. [DOI] [Google Scholar]
- 34.Benjamini Y., Speed T.P. Summarizing and correcting the GC content bias in high-throughput sequencing. Nucleic Acids Res. 2012;40 doi: 10.1093/nar/gks001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Taoka M., Nobe Y., Yamaki Y., Sato K., Ishikawa H., Izumikawa K., Yamauchi Y., Hirota K., Nakayama H., Takahashi N., Isobe T. Landscape of the complete RNA chemical modifications in the human 80S ribosome. Nucleic Acids Res. 2018;46:9289–9298. doi: 10.1093/nar/gky811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Gerbi S.A. Expansion segments: regions of variable size that interrupt the universal core secondary structure of ribosomal RNA. Ribosomal RNA—Structure, evolution, processing, and Function in Protein Synthesis. 1996;71:87. [Google Scholar]
- 37.Ramesh M., Woolford J.L. Eukaryote-specific rRNA expansion segments function in ribosome biogenesis. RNA. 2016;22:1153–1162. doi: 10.1261/rna.056705.116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Morgan D.G., Ménétret J.F., Radermacher M., Neuhof A., Akey I.V., Rapoport T.A., Akey C.W. A comparison of the yeast and rabbit 80 S ribosome reveals the topology of the nascent chain exit tunnel, inter-subunit bridges and mammalian rRNA expansion segments. J. Mol. Biol. 2000;301:301–321. doi: 10.1006/jmbi.2000.3947. [DOI] [PubMed] [Google Scholar]
- 39.van Nues R.W., Venema J., Planta R.J., Raué H.A. Variable region V1 of Saccharomyces cerevisiae 18S rRNA participates in biogenesis and function of the small ribosomal subunit. Chromosoma. 1997;105:523–531. doi: 10.1007/BF02510489. [DOI] [PubMed] [Google Scholar]
- 40.Houge G., Robaye B., Eikhom T.S., Golstein J., Mellgren G., Gjertsen B.T., Lanotte M., Døskeland S.O. Fine mapping of 28S rRNA sites specifically cleaved in cells undergoing apoptosis. Mol. Cell Biol. 1995;15:2051–2062. doi: 10.1128/MCB.15.4.2051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Burge S., Parkinson G.N., Hazel P., Todd A.K., Neidle S. Quadruplex DNA: sequence, topology and structure. Nucleic Acids Res. 2006;34:5402–5415. doi: 10.1093/nar/gkl655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Bing T., Zheng W., Zhang X., Shen L., Liu X., Wang F., Cui J., Cao Z., Shangguan D. Triplex-quadruplex structural scaffold: a new binding structure of aptamer. Sci. Rep. 2017;7 doi: 10.1038/s41598-017-15797-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Mestre-Fos S., Penev P.I., Suttapitugsakul S., Hu M., Ito C., Petrov A.S., Wartell R.M., Wu R., Williams L.D. G-Quadruplexes in Human Ribosomal RNA. J. Mol. Biol. 2019;431:1940–1955. doi: 10.1016/j.jmb.2019.03.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Fujii K., Susanto T.T., Saurabh S., Barna M. Decoding the function of expansion segments in ribosomes. Mol. Cell. 2018;72:1013–1020.e6. doi: 10.1016/j.molcel.2018.11.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Knorr A.G., Schmidt C., Tesina P., Berninghausen O., Becker T., Beatrix B., Beckmann R. Ribosome-NatA architecture reveals that rRNA expansion segments coordinate N-terminal acetylation. Nat. Struct. Mol. Biol. 2019;26:35–39. doi: 10.1038/s41594-018-0165-y. [DOI] [PubMed] [Google Scholar]
- 46.Halic M., Becker T., Pool M.R., Spahn C.M.T., Grassucci R.A., Frank J., Beckmann R. Structure of the signal recognition particle interacting with the elongation-arrested ribosome. Nature. 2004;427:808–814. doi: 10.1038/nature02342. [DOI] [PubMed] [Google Scholar]
- 47.Qu L.H., Nicoloso M., Bachellerie J.P. Phylogenetic calibration of the 5’ terminal domain of large rRNA achieved by determining twenty eucaryotic sequences. J. Mol. Evol. 1988;28:113–124. doi: 10.1007/BF02143502. [DOI] [PubMed] [Google Scholar]
- 48.Qu L.H., Nicoloso M., Bachellerie J.P. A sequence dimorphism in a conserved domain of human 28S rRNA. Uneven distribution of variant genes among individuals. Differential expression in HeLa cells. Nucleic Acids Res. 1991;19:1015–1019. doi: 10.1093/nar/19.5.1015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Guarracino A., Buonaiuto S., de Lima L.G., Potapova T., Rhie A., Koren S., Rubinstein B., Fischer C., Human Pangenome Reference Consortium. Gerton J.L., et al. Recombination between heterologous human acrocentric chromosomes. Nature. 2023;617:335–343. doi: 10.1038/s41586-023-05976-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Zook J.M., Catoe D., McDaniel J., Vang L., Spies N., Sidow A., Weng Z., Liu Y., Mason C.E., Alexander N., et al. Extensive sequencing of seven human genomes to characterize benchmark reference materials. Sci. Data. 2016;3 doi: 10.1038/sdata.2016.25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Hori Y., Shimamoto A., Kobayashi T. The human ribosomal DNA array is composed of highly homogenized tandem clusters. Genome Res. 2021;31:1971–1982. doi: 10.1101/gr.275838.121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Bray J.R., Curtis J.T. An Ordination of the Upland Forest Communities of Southern Wisconsin. Ecol. Monogr. 1957;27:325–349. doi: 10.2307/1942268. [DOI] [Google Scholar]
- 53.Taskesen E., Reinders M.J.T. 2D Representation of Transcriptomes by t-SNE Exposes Relatedness between Human Tissues. PLoS One. 2016;11 doi: 10.1371/journal.pone.0149853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Donovan M.K.R., D’Antonio-Chronowska A., D’Antonio M., Frazer K.A. Cellular deconvolution of GTEx tissues powers discovery of disease and cell-type associated regulatory variants. Nat. Commun. 2020;11:955. doi: 10.1038/s41467-020-14561-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Liu J., Lichtenberg T., Hoadley K.A., Poisson L.M., Lazar A.J., Cherniack A.D., Kovatich A.J., Benz C.C., Levine D.A., Lee A.V., et al. An Integrated TCGA Pan-Cancer Clinical Data Resource to Drive High-Quality Survival Outcome Analytics. Cell. 2018;173:400–416.e11. doi: 10.1016/j.cell.2018.02.052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Schlesinger S., Selig S., Bergman Y., Cedar H. Allelic inactivation of rDNA loci. Genes Dev. 2009;23:2437–2447. doi: 10.1101/gad.544509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.van Sluis M., van Vuuren C., Mangan H., McStay B. NORs on human acrocentric chromosome p-arms are active by default and can associate with nucleoli independently of rDNA. Proc. Natl. Acad. Sci. USA. 2020;117:10368–10377. doi: 10.1073/pnas.2001812117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Grob A., Colleran C., McStay B. Construction of synthetic nucleoli in human cells reveals how a major functional nuclear domain is formed and propagated through cell division. Genes Dev. 2014;28:220–230. doi: 10.1101/gad.234591.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Wild K., Aleksić M., Lapouge K., Juaire K.D., Flemming D., Pfeffer S., Sinning I. MetAP-like Ebp1 occupies the human ribosomal tunnel exit and recruits flexible rRNA expansion segments. Nat. Commun. 2020;11:776. doi: 10.1038/s41467-020-14603-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Shankar V., Rauscher R., Reuther J., Gharib W.H., Koch M., Polacek N. rRNA expansion segment 27Lb modulates the factor recruitment capacity of the yeast ribosome and shapes the proteome. Nucleic Acids Res. 2020;48:3244–3256. doi: 10.1093/nar/gkaa003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Ohashi R., Umezu H., Sato A., Abé T., Kondo S., Daigo K., Sato S., Hara N., Miyashita A., Ikeuchi T., et al. Frequent germline and somatic single nucleotide variants in the promoter region of the ribosomal RNA gene in Japanese lung adenocarcinoma patients. Cells. 2020;9 doi: 10.3390/cells9112409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Therizols G., Bash-Imam Z., Panthu B., Machon C., Vincent A., Ripoll J., Nait-Slimane S., Chalabi-Dchar M., Gaucherot A., Garcia M., et al. Alteration of ribosome function upon 5-fluorouracil treatment favors cancer cell drug-tolerance. Nat. Commun. 2022;13:173. doi: 10.1038/s41467-021-27847-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Thomson J.A., Itskovitz-Eldor J., Shapiro S.S., Waknitz M.A., Swiergiel J.J., Marshall V.S., Jones J.M. Embryonic stem cell lines derived from human blastocysts. Science. 1998;282:1145–1147. doi: 10.1126/science.282.5391.1145. [DOI] [PubMed] [Google Scholar]
- 64.Zielezinski A., Vinga S., Almeida J., Karlowski W.M. Alignment-free sequence comparison: benefits, applications, and tools. Genome Biol. 2017;18:186. doi: 10.1186/s13059-017-1319-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Langmead B., Salzberg S.L. Fast gapped-read alignment with Bowtie 2. Nat. Methods. 2012;9:357–359. doi: 10.1038/nmeth.1923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Sievers F., Wilm A., Dineen D., Gibson T.J., Karplus K., Li W., Lopez R., McWilliam H., Remmert M., Söding J., et al. Fast, scalable generation of high-quality protein multiple sequence alignments using Clustal Omega. Mol. Syst. Biol. 2011;7:539. doi: 10.1038/msb.2011.75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Bray N.L., Pimentel H., Melsted P., Pachter L. Near-optimal probabilistic RNA-seq quantification. Nat. Biotechnol. 2016;34:525–527. doi: 10.1038/nbt.3519. [DOI] [PubMed] [Google Scholar]
- 68.Li H. Minimap2: pairwise alignment for nucleotide sequences. Bioinformatics. 2018;34:3094–3100. doi: 10.1093/bioinformatics/bty191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Li H., Handsaker B., Wysoker A., Fennell T., Ruan J., Homer N., Marth G., Abecasis G., Durbin R., 1000 Genome Project Data Processing Subgroup The Sequence Alignment/Map format and SAMtools. Bioinformatics. 2009;25:2078–2079. doi: 10.1093/bioinformatics/btp352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Goujon M., McWilliam H., Li W., Valentin F., Squizzato S., Paern J., Lopez R. A new bioinformatics analysis tools framework at EMBL-EBI. Nucleic Acids Res. 2010;38:W695–W699. doi: 10.1093/nar/gkq313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Needleman S.B., Wunsch C.D. A general method applicable to the search for similarities in the amino acid sequence of two proteins. J. Mol. Biol. 1970;48:443–453. doi: 10.1016/0022-2836(70)90057-4. [DOI] [PubMed] [Google Scholar]
- 72.Tweedie S., Braschi B., Gray K., Jones T.E.M., Seal R.L., Yates B., Bruford E.A. Genenames.org: the HGNC and VGNC resources in 2021. Nucleic Acids Res. 2021;49:D939–D946. doi: 10.1093/nar/gkaa980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Tian S., Cordero P., Kladwang W., Das R. High-throughput mutate-map-rescue evaluates SHAPE-directed RNA structure and uncovers excited states. RNA. 2014;20:1815–1826. doi: 10.1261/rna.044321.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Kladwang W., VanLang C.C., Cordero P., Das R. A two-dimensional mutate-and-map strategy for non-coding RNA structure. Nat. Chem. 2011;3:954–962. doi: 10.1038/nchem.1176. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
This includes all of the rRNA variants found both in rDNA and rRNA in translating ribosomes in the H7-hESC cell line.
This is the same as Data S1 with the exception that variants are grouped to the resolution of rRNA helices. Helix annotations can be found in Table S16 and Figure S11.
This is the same as Data S1 with the exception that variants are grouped to the resolution of rRNA expansion segments. Expansion segment annotations can be found in Table S17 and Figure S11.
This is the same as Data S2 with flanking reference sequence on both 3′ and 5′ ends. This atlas is useful for estimating variant frequencies in short-read data. For usage example, see “atlas relative abundance calling for RNA short-read datasets.”
This is the same as Data S3 with flanking reference sequence on both 3′ and 5′ ends. This atlas is useful for estimating variant frequencies in short-read data. For usage example, see “atlas relative abundance calling for RNA short-read datasets.”
Data Availability Statement
The atlas is available as Extended Data to this publication (Data S1, S2, S3, S4, and S5). H7-hESC rDNA and rRNA, and K562 rRNA sequencing data is available under BioProject ID PRJNA926787. For the H7-hESC under accession numbers SRR23196516 (H7-hESC whole genome sequencing), SRR29419059 (H7-hESC 18S and 28S from monosomes), SRR29419058 (H7-hESC 18S and 28S from “light polysomes” with 2 or 3 ribosomes), SRR29419057 (H7-hESC 18S and 28S from “medium polysomes” with 4 or 5 ribosomes), SRR29419056 (H7-hESC 18S and 28S from “heavy polysomes” with 6 or more ribosomes), SRR29884466 (H7-hESC 28S treated with DMS). For the K562 under accession numbers SRR29419055 (K562 18S and 28S from monosomes), SRR29419054 (K562 18S and 28S from “light polysomes” with 2 or 3 ribosomes), SRR29419053 (K562 18S and 28S from “medium polysomes” with 4 or 5 ribosomes), SRR29419052 (K562 18S and 28S from “heavy polysomes” with 6 or more ribosomes).