

Abstract
INTRODUCTION
Reduced brain energy metabolism, mammalian target of rapamycin (mTOR) dysregulation, and extracellular amyloid beta (Aβ) oligomer (xcAβO) buildup are some well‐known Alzheimer's disease (AD) features; how they promote neurodegeneration is poorly understood. We previously reported that xcAβOs inhibit nutrient‐induced mitochondrial activity (NiMA) in cultured neurons. We now report NiMA disruption in vivo.
METHODS
Brain energy metabolism and oxygen consumption were recorded in heterozygous amyloid precursor protein knock‐in (APPSAA) mice using two‐photon fluorescence lifetime imaging and multiparametric photoacoustic microscopy.
RESULTS
NiMA is inhibited in APPSAA mice before other defects are detected in these Aβ‐producing animals that do not overexpress APP or contain foreign DNA inserts into genomic DNA. Glycogen synthase kinase 3 (GSK3β) signals through mTORC1 to regulate NiMA independently of mitochondrial biogenesis. Inhibition of GSK3β with TWS119 stimulates NiMA in cultured human neurons, and mitochondrial activity and oxygen consumption in APPSAA mice.
DISCUSSION
NiMA disruption in vivo occurs before plaques, neuroinflammation, and cognitive decline in APPSAA mice, and may represent an early stage in human AD.
Highlights
Amyloid beta blocks communication between lysosomes and mitochondria in vivo.
Nutrient‐induced mitochondrial activity (NiMA) is disrupted long before the appearance of Alzheimer's disease (AD) histopathology in heterozygous amyloid precursor protein knock‐in (APPSAA/+) mice.
NiMA is disrupted long before learning and memory deficits in APPSAA/+ mice.
Pharmacological interventions can rescue AD‐related NiMA disruption in vivo.
Keywords: amino acids, brain metabolism, insulin, mammalian target of rapamycin, tau
1. BACKGROUND
It is well accepted that reduced brain energy metabolism is a key feature of Alzheimer's disease (AD). Clinical studies tracking 2‐deoxy‐2‐[18F] fluoro‐d‐glucose (FDG) positron emission tomography (PET) uptake, a surrogate for glucose use in the brain, support the hypothesis that brain hypometabolism is one of the earliest AD biomarkers and is detectable years before symptom onset. 1 Although FDG PET screening has provided valuable information allowing the identification of patients at risk of AD and other brain disorders, the molecular mechanism(s) at fault remain poorly understood.
The amyloid cascade hypothesis 2 has guided much of our understanding of mechanisms of AD pathogenesis and most drug discovery efforts for AD. 3 , 4 , 5 Key players in the hypothesis are amyloid beta (Aβ) peptides, ≈ 40 amino acid long proteolytic fragments of the amyloid precursor protein (APP) and the principal amyloid plaque component. Although plaques, along with neurofibrillary tangles composed mainly of tau, are the defining histopathological lesions in AD, a preponderance of evidence indicates that the Aβ and tau species most responsible for the synapse loss and neuron death that together cause AD symptoms are soluble oligomers that accumulate in brain presymptomatically. 6
A few years ago, we reported a novel form of communication between lysosomes and mitochondria, a fundamental cell biological process that we named NiMA as an acronym for nutrient‐induced mitochondrial activity. 7 More specifically, NiMA represents the activation of the master regulatory protein kinase, mTORC1, 8 by insulin, or arginine and leucine (R+L), to trigger mitochondrial respiration within minutes in the absence of mitochondrial biogenesis. Importantly, we also found that extracellular Aβ oligomers (xcAβOs) block NiMA in cultured neurons. 7 The mechanism involves xcAβO‐mediated ectopic activation of mTORC1 at the neuronal plasma membrane 9 and upregulation of superoxide dismutase 1 (SOD1), a major regulator of cellular redox and a pathogenic factor in amyotrophic lateral sclerosis. 10 Whether NiMA is disrupted in the live AD brain, however, was not explored.
Glycogen synthase kinase 3 (GSK3β) is an evolutionarily conserved intracellular serine/threonine kinase. Originally found to control glucose metabolism 11 and later as a key downstream target of insulin signaling, 12 , 13 it has emerged as a key regulator of diverse cellular functions. GSK3β has been studied for several years in the context of AD. 14 It is a major tau kinase, 15 and studies in mouse models of AD strongly implicate hyperactive GSK3β in AD pathogenesis. 16 Additionally, GSK3β has been linked to the control of mitochondrial anterograde trafficking, 17 and feeding mice lithium, which inhibits GSK3β, 18 , 19 was shown to modulate brain energy metabolism. 20
Using two‐photon fluorescence lifetime imaging (2P‐FLIM) and multi‐parametric photoacoustic microscopy (MP‐PAM), we tracked mitochondrial activity and brain energy metabolism in the cortex of wild‐type (WT) and heterozygous APP knock‐in mouse littermates. The heterozygous knock‐ins, henceforth called APPSAA/+, express one copy of the endogenous WT mouse APP gene, with the second endogenous APP gene modified by homologous recombination to include a humanized Aβ1‐42 region containing the Swedish (KM670/671NL), Arctic (E693G), and Austrian (T714I) mutations. In contrast to nearly all other APP transgenic mouse strains, APPSAA mice do not overexpress APP, nor do they include any foreign transgene inserts that might toxically interrupt one or more endogenous mouse genes. The pathological progression of APPSAA mice has been extensively characterized, 21 allowing correlation of brain energy metabolic changes to disease progression.
We now show that NiMA inhibition occurs long before the accumulation of plaques, increased cerebrospinal fluid (CSF) tau, microgliosis, and cognitive decline recently reported for APPSAA/+mice. 21 Mechanistically, we found that GSK3β controls mitochondrial activity in neurons in culture and live mouse brains by a mechanism involving a GSK3β–mTORC1 signaling pathway. Importantly, pharmacological inhibition of GSK3β also stimulated mitochondrial activity and oxygen consumption in the cortex of the APPSAA/+ mice. Thus, disruption of NiMA in the brain could be a very early event in AD pathogenesis in humans.
2. METHODS
2.1. Cell culture and materials
Human embryonic kidney cells (Lenti‐X 293T Cell line from Takara, Cat no. 632180) were grown in DMEM/F12 media (GIBCO) supplemented with HyClone cosmic calf serum (GE Healthcare) and 50 µg/mL gentamycin (GIBCO).
Human neurons were differentiated from ReNcell VM neuronal precursor cells (EMD Millipore) as described earlier. 7 For 2D differentiation, the cells were plated into 35 mm glass‐bottom dishes with DMEM/F12 differentiation media (GIBCO) supplemented with 2 µg/mL heparin (ScienceCell), 2% (v/v) B27 neural supplement (GIBCO), and 50 µg/mL gentamycin (GIBCO) without growth factors. One‐half volume of the differentiation media was changed every 3 days until the cultures were used for experiments. 22
2.2. Mouse strain
The hAbetaSAA (APPSAA KI) knock‐in mouse model of AD was purchased from The Jackson Laboratory (Strain #:034711). Animals were housed in a pathogen‐free barrier facility with a 12‐hour light/12‐hour dark cycle, and ad libitum access to food and water under a protocol approved by the Institutional Animal Care and Use Committee (IACUC) of the University of Virginia. All pups were genotyped by quantitative polymerase chain reaction following The Jackson Laboratory's recommendations. Unless otherwise indicated, all experiments were performed in males heterozygous for APPSAA KI or WT littermates.
2.3. Lentivirus production and infection
Lentiviral particles for shGSK3β knockdowns were prepared as follows. The expression plasmids, pLKO.1 (Mission shRNA library from Sigma‐Aldrich), and the packaging vectors, pSPAX2 and pMD2.G (Addgene plasmids 12260 and 12259, respectively) were transfected using Lipofectamine 3000 (ThermoFisher) into HEK293T cells grown in 15 cm Petri dishes to ≈ 80% confluence in DMEM (GIBCO) supplemented with 10% HyClone cosmic calf serum. Each transfection was with 15 µg total DNA at a 50%/37.5%/12.5% ratio of expression vector/pSPAX2/pMD2G. The lentivirus‐conditioned medium was collected 24 and 48 hours after the start of transfection. Lentiviral particles were concentrated in a Beckman Coulter Optima XE‐90 ultracentrifuge for 2 hours at 90,353 x g, at 4°C in an SW32Ti rotor, resuspended in 400 µL Neurobasal medium, and stored at –80°C in 20 µL aliquots. Cultured neurons were transduced and incubated for 72 hours before assays were performed.
2.4. Human shRNA sequences
The following plasmids were purchased from The RNAi Consortium of the Broad Institute: (1) TRCN0000039564 (#1) targeting sequence CATGAAAGTTAGCAGAGACAA and TRCN0000039565, (#2) targeting sequence AGCAAATCAGAGAAATGAAC. These constructs are already cloned into pLKO.1, and lentivirus particles were prepared as described in a previous section.
RESEARCH IN CONTEXT
Systematic review: The search for presymptomatic diagnosis and disease‐modifying therapies for Alzheimer's disease (AD) depends on understanding the molecular mechanisms of AD pathogenesis. Impaired brain energy metabolism is a hallmark of aging and early AD, but its mechanisms, especially how it promotes synaptic dysfunction and neuron death, are not fully understood. This study was aimed at clarifying such pathogenic mechanisms.
Interpretation: Low levels of soluble amyloid beta oligomers interfere with the ability of insulin and amino acids to trigger mitochondrial activity (nutrient‐induced mitochondrial activity [NiMA]) in vivo presymptomatically in heterozygous amyloid precursor protein knock‐in APPSAA/+) mice, a model of human AD.
Future directions: Revealing further details of the mechanism(s) involved is a major goal. Examples include a refined understanding of (a) cell type–specific contributions to NiMA disruption in the brain, (b) how cerebrovascular dysfunction affects NiMA in health and disease, and (c) how to leverage new findings to improve AD diagnostics and therapeutics.
2.5. Mouse brain cell extracts
Brain cortex samples were placed in a Wheaton 1 mL Dounce homogenizer containing 1 mL of lysis buffer to cover the sample. Lysis buffer consisted of RIPA buffer (Bioworld, #42020024‐2), 1% HALT protease inhibitor cocktail, 1% phosphatase inhibitor cocktail 2 (Sigma Aldrich, #P5726), and 1% phosphatase inhibitor cocktail 3 (Sigma Aldrich, #P0044). Brain homogenates were placed in an Eppendorf 5415C centrifuge at 8160 x g for 10 minutes at 4°C. Finally, supernatants were transferred to new tubes and used as protein lysates for western blots.
2.6. Antibodies
The following antibodies were from Cell Signaling Technologies: rabbit anti‐GSK3β, catalog number 9832 (dilution 1:3000); mouse anti‐GSK3β phosphorylated on serine 9, catalog number 9323 (dilution 1:3000); mouse anti‐S6 ribosomal protein, catalog number 2317 (dilution 1:3000); and rabbit anti‐S6 ribosomal protein phosphorylated on serine 240/244, catalog number 5364. Antibodies from Proteintech included: mouse anti‐PGC1α, catalog number 66369‐1‐1 g (dilution 1:3000); and mouse anti‐TOM40, catalog number 66658‐1‐1 g (dilution 1:5000). Mouse anti‐human APP (clone 6E10) was from BioLegend (cat no. 803014; dilution 1:5000). The mouse anti‐total OXPHOS rodent antibody cocktail was from Abcam (catalog number ab110413; dilution 1:10,000).
2.7. Immunoblotting
Samples were resolved by sodium dodecyl sulfate polyacrylamide gel electrophoresis using either 10% or 12% acrylamide/bis‐acrylamide gels and transferred to 0.22 µm nitrocellulose (Bio‐Rad). Membranes were blocked with Odyssey blocking buffer (LI‐COR Biosciences) and were incubated with primary antibodies and secondary IRDye‐labeled antibodies diluted into antibody buffer (Odyssey blocking buffer diluted 1:1 with phosphate‐buffered saline [PBS]/0.1% Tween 20). All antibody incubations were overnight at 4°C, and three washes of 5 minutes each with PBS/0.1% Tween 20 were performed after each antibody step. Membranes were quantitatively scanned by an Odyssey imaging station (LI‐COR Biosciences).
2.8. Multiparametric photoacoustic microscopy
All animal procedures were approved by the IACUC at Washington University in St. Louis. After hair removal, a surgical incision was made on the scalp. The exposed skull was cleaned. Then, the mouse skull region over a 3 × 3 mm2 region of interest (ROI) was carefully removed to expose the cortex for topical application of leucine (0.8 mM) plus arginine (0.4 mM) or TWS119 (100 µM; Cayman Chemicals, catalog no. 10011251). After craniotomy, the anesthetized mouse was transferred to the MP‐PAM system. The animal body temperature was maintained at 37°C via a heating pad and the local brain temperature was also maintained at 37°C via a temperature‐controlled water tank. Ultrasound gel was applied between the open‐skull window and water tank for acoustic coupling. After the baseline imaging, the ultrasound gel was gently removed and a solution of either leucine (0.8 mM) plus arginine (0.4 mM) or TWS119 (100 µM) was applied topically. The exposed mouse cortex was treated with the corresponding solution for up to 80 minutes. Then, the ROI was covered again with ultrasound gel and subjected to post‐treatment MP‐PAM imaging. 23 MP‐PAM uses dual‐wavelength (i.e., 532 nm, 558 nm) and high‐repetition‐rate nanosecond‐pulsed laser excitation to achieve simultaneous pixel‐wise measurement of the oxygen saturation of hemoglobin (sO2) and blood flow speed. In the acquired MP‐PAM images, vessel segmentation was performed and the average values of sO2 and flow speed in main arteries and veins within the field of view were calculated in the vessel segments. Oxygen extraction fraction (OEF) is measured by calculating the relative difference in arterial and venous sO2.
2.9. Reduced Nicotinamide adenine dinucleotide (NADH) and Reduced Nicotinamide adenine dinucleotide phosphate (NADPH) measurements and In vivo 2P‐FLIM imaging
2.9.1. Time‐correlated single photon counting fluorescence lifetime imaging microscopy
Surgical preparation of animals and application of amino acids or TWS 119 were done as described in the previous paragraph. Live 2P‐FLIM was recorded on a Zeiss LSM‐780 NLO confocal/multiphoton microscopy system comprising an inverted Axio Observer Z1 microscope, an X‐Cite 120 PC Q mercury arc light source (Excelitas Technologies) for cell selection, a motorized stage for automated scanning, an IR Chameleon Vision‐II ultrafast Ti: sapphire laser for multiphoton excitation (Coherent), a Zeiss 40 × 1.3 NA oil immersion Planapo objective, an environmental chamber (PeCon GmbH) that envelops the microscope stage to control temperature and CO2 level, and a 3‐channel FLIM system based on three HPM‐100‐40 GaAsP‐based hybrid detectors and 3 SPC‐150 TCSPC boards (Becker & Hickl). The SPC‐150 boards are synchronized with the 2‐photon excitation laser and the Zeiss LSM‐780 NLO scan head signal. Ex 740 nm; Em450/50 nm.
2.9.2. Imaging
Human neuronal cultures were grown in 35‐mm glass‐bottom dishes, maintained at 37°C in 5% CO2/95% air on the stage of the Zeiss LSM‐980 NLO microscope. Cultures were developed in 20 to 25 days. Then, cells were treated with the indicated inhibitor for ≈ 80 minutes at 37°C. The laser was tuned to 740 nm with an average power of 7 mW at the specimen plane, and NAD(P)H fluorescence was collected using a 450 to 500 nm emission filter (Objective lens, 40 ×). For each experiment, 10 fields of view were recorded in the descanned mode, and then each field of view was subjected to a 40‐second acquisition in the non‐descanned mode. The laser power and acquisition time were selected to ensure enough photons per pixel while avoiding photodamage to cells.
2.9.3. Processing
FLIM images were processed with SPCImage software (v5; Becker & Hickl) at maximum‐likelihood fitting. All FLIM image parameter data were exported for further processing in Fiji software (https://hpc.nih.gov/apps/Fiji.html). Here, thresholded, normalized photon images selected pixel‐ROIs from the more prominent intensity of the mitochondrial morphology by a standard Fiji plugin. Mitochondrial ROIs were applied to all FLIM parameter data creating a mitochondria‐specific data pool by a custom Fiji plugin. Those results were analyzed by a custom code in Python software, in which all parameters were first examined and filtered for obvious outliers (insignificant numbers). The FLIM metric of interest—a2%, the fraction of enzyme‐bound NAD(P)H—an indicator for changes in the mitochondrial metabolic OXPHOS state was extracted. NAD(P)H‐a2% data were plotted for publication‐ready charts and statistics in Microsoft Excel.
2.10. Statistics
The paired t test was used for analyzing all 2P‐FLIM assays. Because thousands of ROIs were obtained per image, the average ROI for each field of view was calculated to reduce the sample size and thus the number of false positives. All data were assumed to be normally distributed. For MP‐PAM assays, vessel segmentations and quantitative analyses were done following our established protocol. 23 The paired t test was used to compare the hemodynamics and oxygen metabolism before and after the application of experimental compounds. A P value of < 0.05 was considered statistically significant.
3. RESULTS
3.1. Transient decrease in brain metabolism in APPSAA mice
We first measured baseline mitochondrial activity in the cortex of 2‐, 4‐, and 6‐month‐old APPSAA mice, and by comparison, in WT littermates. To do this, we used 2P‐FLIM to monitor NADH and NADPH fluorescence lifetimes and the degree to which these co‐enzymes associate with partner enzymes in mitochondria. 7 While the enzyme‐bound fraction of NADH, expressed as “a2%,” 24 is a surrogate for mitochondrial respiration, 7 the corresponding a2% measurement for NADPH reports activation of biosynthetic pathways. 25 Although the excitation and emission spectra of NADH and NADPH are identical, 2P‐FLIM allows measurement of their distinct fluorescence lifetimes. 25 Accordingly, we determined the relative contributions of each coenzyme to the total measured fluorescence lifetimes under baseline conditions. This approach revealed that on average, NADH accounted for 55% of the observed fluorescence lifetime signals, while the remaining 45% was due to NADPH. An ≈ 19% reduction in NADH and a corresponding increase in NADPH were observed in 2‐month‐old APPSAA/+ mice compared to WT littermates, but these differences did not persist as animals aged up to 6 months old (Figure 1A–C).
FIGURE 1.
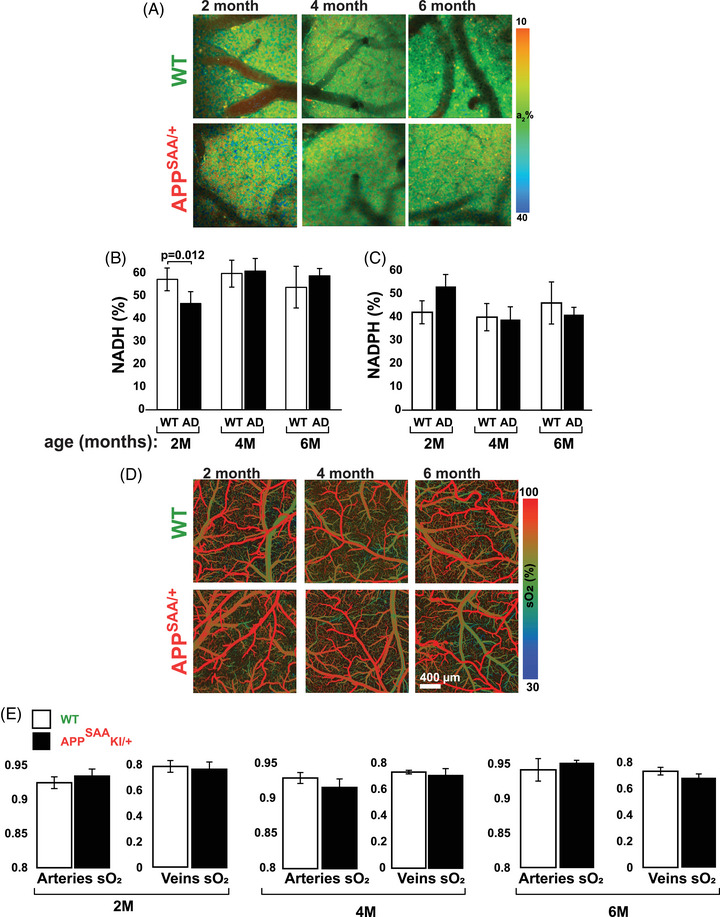
2P‐FLIM and MP‐PAM intravital imaging shows similar cortical energy metabolic activity in WT and APPSAA/+ mice. A–C, 2P‐FLIM imaging at baseline revealed a transient ≈ 19% decrease in enzyme‐bound NADH and the corresponding increase in enzyme‐bound NADPH in 2‐month‐old APPSAA/+ mice. D–E, MP‐PAM imaging at baseline revealed virtually identical levels of mitochondrial respiration irrespective of age or genotype. Four to six animals per group were used for NADH/NADPH measurements, and four animals per group were used for MP‐PAM. Student two‐tailed unpaired t tests were used to assess statistical significance, and error bars represent ± standard error of the mean. 2P‐FLIM, two‐photon fluorescence lifetime imaging; APPSAA/+, heterozygous amyloid precursor protein knock‐in; MP‐PAM, multi‐parametric photoacoustic microscopy; NADH, reduced nicotinamide adenine dinucleotide; NADPH, reduced nicotinamide adenine dinucleotide phosphate; WT, wild type.
In parallel experiments, we applied MP‐PAM to evaluate baseline levels of oxygen consumption in live mouse brains. 23 MP‐PAM imaging through a cranial window enables simultaneous, high‐resolution detection of total hemoglobin concentration (CHb), sO2, and blood flow. Notwithstanding the transient decrease in NADH detected by 2P‐FLIM, sO2 levels in the resting cortex were virtually constant irrespective of age or genotype (Figure 1D–E).
We also analyzed the expression levels of key proteins of the electron transport chain (ETC) in 2‐, 4‐, and 6‐month‐old mice. Western blots of cortical samples from WT and APPSAA/+ animals revealed no genetic‐dependent changes in the expression levels of NDUFB8, SDHB, UQCRC2, MTCO1, or ATP5A, all of which are subunits of corresponding complexes I to V of the ETC (Figure S1 in supporting information). Because APPSAA/+ mice do not develop amyloid plaques until ≈ 16 months of age but do have low levels of Aβ in the brain, CSF, and plasma by 4 months, 21 these results suggest that a low concentration of Aβ causes a transient decrease in energy metabolism that does not compromise steady‐state oxidative phosphorylation (OXPHOS) in the cortex. The data are consistent with observations made more than 30 years ago in the human brain that mitochondrial activity is normal at the early stages of AD. 26 Altogether, these observations suggest that APPSAA/+ mice can serve as a credible model for exploring mechanisms that regulate brain energy metabolism and might go awry in AD.
3.2. Early disruption of mitochondrial response to amino acids in APPSAA/+ mouse cortex
We recently reported NiMA inhibition in cultured mouse and human neurons exposed to xcAβOs, and in human neurons that were not exposed to xcAβOs but were co‐cultured with otherwise identical human neurons that express the K670N/M671L (Swedish) and V717I (London) APP mutations, and secrete Aβ. 7 , 10 To seek evidence for NiMA inhibition in vivo, we used 2P‐FLIM in the current study to compare mitochondrial activity in the cortex of WT versus APPSAA/+ mice. We topically applied R+L to the cortex through an open skull window, which caused the expected increase in OXPHOS (a2%) in 2‐month‐old WT mice. Increased mitochondrial activity was detected 30 minutes after the application of the amino acids, persisted for up to 80 minutes, and occurred regardless of the animals’ age (Figure 2A–C). In contrast, when the same approach was used for APPSAA/+ mice, the mitochondrial response to R+L was observed at 2 months (Figure 2D) but was completely absent in 4‐ (Figure 2E), 6‐ (Figure 2F), and 8‐month‐old (not shown) animals. In addition, the mitochondrial responses to amino acids in 4‐month‐old mice were not influenced by sex (Figure S2 in supporting information).
FIGURE 2.
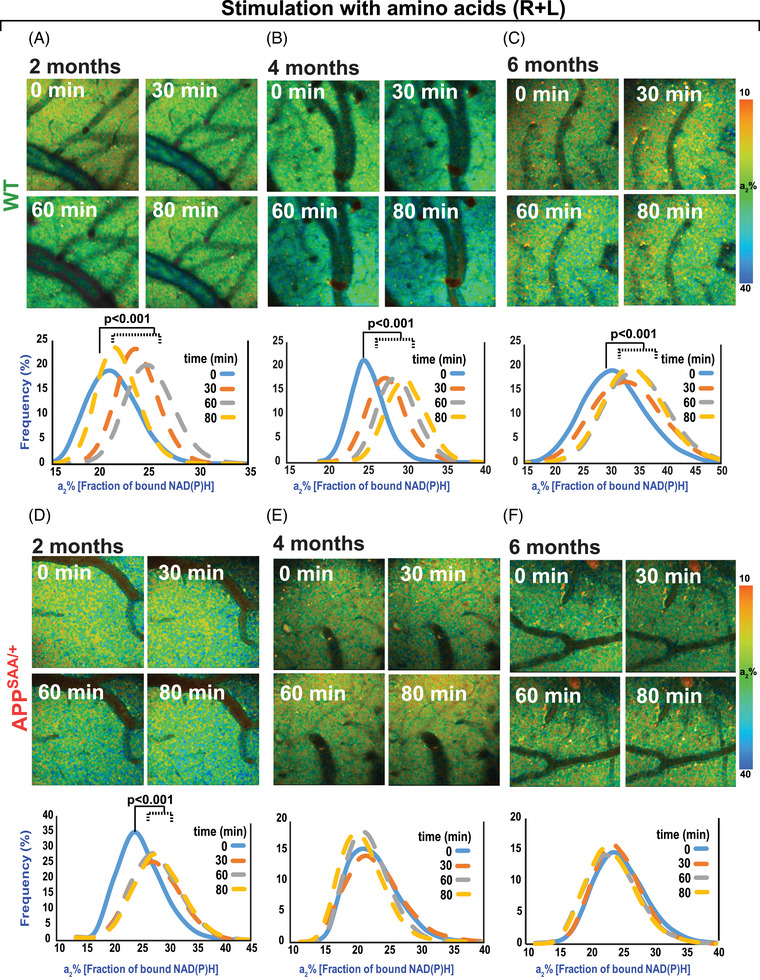
2P‐FLIM imaging reveals disruption of mitochondrial response to amino acids in the APPSAA/+ mouse brain. A–C, 2P‐FLIM imaging of WT mouse cortex through an open‐skull window after topical application of amino acids (R+L). An increase in a2%, indicating upregulation of mitochondrial activity, was observed 30 minutes after the initial application of amino acids and remained elevated for up to 80 minutes. A similar response was observed regardless of the age of the animals tested. D–F, 2P‐FLIM imaging of APPSAA/+ mice showed an increase in a2% in response to amino acids only in the 2‐month‐old group. Statistical analyses were performed using Student two‐tailed paired t test. Three to four animals/ages/genotypes were used in these experiments. 2P‐FLIM, two‐photon fluorescence lifetime imaging; APPSAA/+, heterozygous amyloid precursor protein knock‐in; R+L, arginine and leucine; WT, wild type.
In parallel experiments, we used MP‐PAM to test if fluctuations in a2% reflect changes in mitochondrial respiration in mouse brains. Topical application of R+L through an open skull window reduced sO2 in WT mice by ≈ 10% and 15% in arteries and veins, respectively (Figure 3), suggesting an increase in oxygen consumption due to mitochondrial activity in the tissue surrounding blood vessels. These changes were similar in magnitude irrespective of the age of the animals (Figure 3). When MP‐PAM microscopy was applied to APPSAA/+ mice, we observed that amino acids triggered a comparable reduction in sO2 in 2‐month‐old animals, but not in older mice (Figure 4). Altogether, this in vivo, label‐free imaging of mitochondrial energy metabolism in APPSAA/+ mice suggests that low levels of soluble Aβ peptides inhibit NiMA in the cortex.
FIGURE 3.
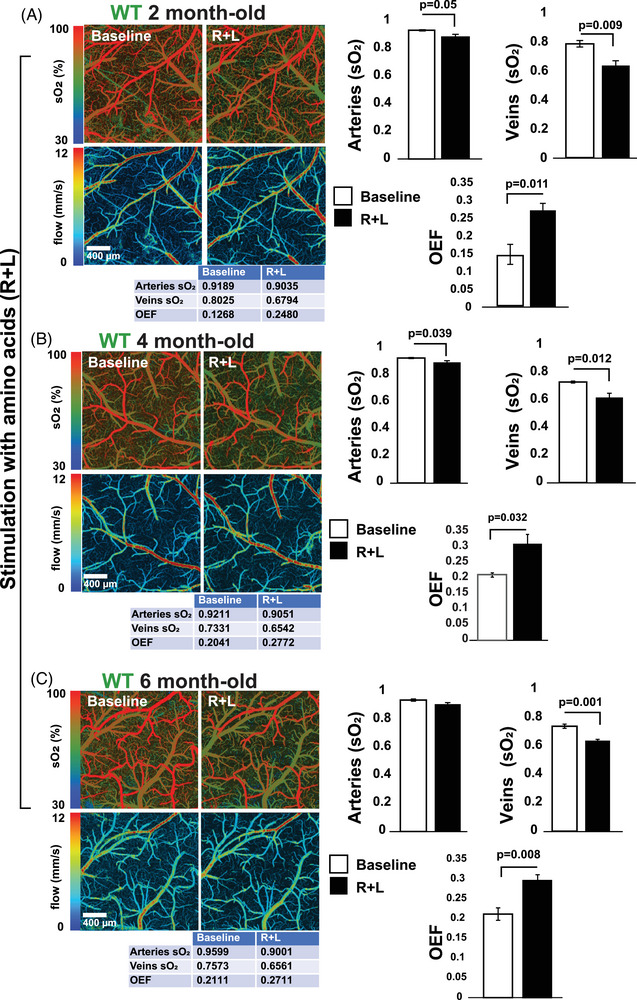
Amino acids (R+L) trigger oxygen consumption in the WT mouse brain. A–C, MP‐PAM imaging of WT mouse cortex through an open‐skull window before and 80 minutes after topical application of R+L. A decrease in sO2 in the cortical vasculature was observed at all ages, indicating elevated oxygen extraction and consumption due to the upregulation of mitochondrial activity. sO2 in arteries, veins, and OEF values for the experiments shown in the figure are depicted in the table below each image. The bar graphs show the quantification of four independent experiments. Statistical analyses were performed using Student two‐tailed unpaired t test. Error bars represent ± standard error of the mean. Four animals/age groups were used. MP‐PAM, multi‐parametric photoacoustic microscopy; OEF, oxygen extraction fraction; R+L, arginine and leucine; sO2, saturation of hemoglobin; WT, wild type.
FIGURE 4.
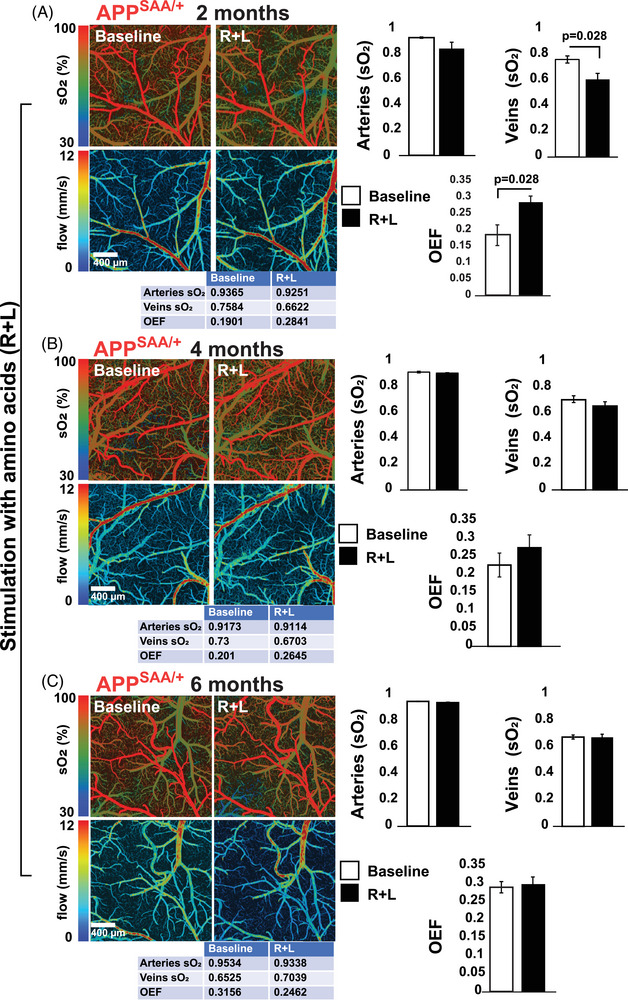
Amino acid‐induced oxygen consumption is blocked in the APPSAA/+ mouse brain. A–C, MP‐PAM imaging of APPSAA/+ mouse cortex through an open‐skull window before and 80 minutes after a topical application of amino acids (R+L). Decreases in cortical vasculature sO2 and OEF were observed only in the 2‐month‐old group. sO2 in arteries, veins, and OEF values for the experiments shown in the figure are depicted in the table below each image. The bar graphs show the quantification of four independent experiments. Statistical analyses were performed using Student two‐tailed unpaired t test. Error bars represent ± standard error of the mean. Four animals/age groups were used in these experiments. APPSAA/+, heterozygous amyloid precursor protein knock‐in; MP‐PAM, multi‐parametric photoacoustic microscopy; OEF, oxygen extraction fraction; R+L, arginine and leucine; sO2, saturation of hemoglobin.
3.3. GSK3β regulates NiMA in neurons in culture and live mouse brain
To test if GSK3β controls mitochondrial functioning, human neurons differentiated from the ReNcell VM line of human neuronal progenitors 7 were first treated for 80 minutes with increasing concentrations of TWS119, a GSK3β inhibitor. 27 Mitochondrial activity was monitored by 2P‐FLIM of neurons exposed to the inhibitor or vehicle control. The contributions of enzyme‐bound NADH and NADPH to the total fluorescence lifetime were extracted and plotted as described earlier. 7 , 25 TWS119 treatment at 500 nM had no effect until 18 hours (not shown), but 1 and 10 µM TWS119 increased the proportion of NADH over NADPH in a concentration‐dependent manner within 80 minutes (Figure 5A), indicating an increase in mitochondrial respiration. Under these experimental conditions, we observed the expected increase in phosphorylation of serine‐9 on GSK3β (Figure 5B), which inhibits its kinase activity. 28 Inhibition of GSK3β did not change the level of PGC1α, a major regulator of mitochondrial biogenesis (Figure 5B) and was paralleled by increased phosphorylation of the ribosomal S6 protein in residues Ser 240 and 244, a read‐out of mTORC1 activity (Figure 5B), without changes in the phosphorylation of S6 protein at residue Ser235, a downstream target of the Ras/ERK pathway (not shown). Thus, our results suggest that GSK3β controls NiMA through activation of mTORC1. We also tested if Li+, another well‐known GSK3β inhibitor, 18 , 19 triggered similar effects. As shown in Figure S3A in supporting information, treating human neuron cultures with LiCl increased both the fraction of NADH and the phosphorylation of GSK3β on Ser‐9. Then, we verified whether TWS119 increases mitochondrial activity in the live mouse brain. 2P‐FLIM assays revealed that topical application of TWS119 for 80 minutes through a cranial window also caused a ≥ 30% increase in a2% in WT mouse brain (Figure 5C).
FIGURE 5.
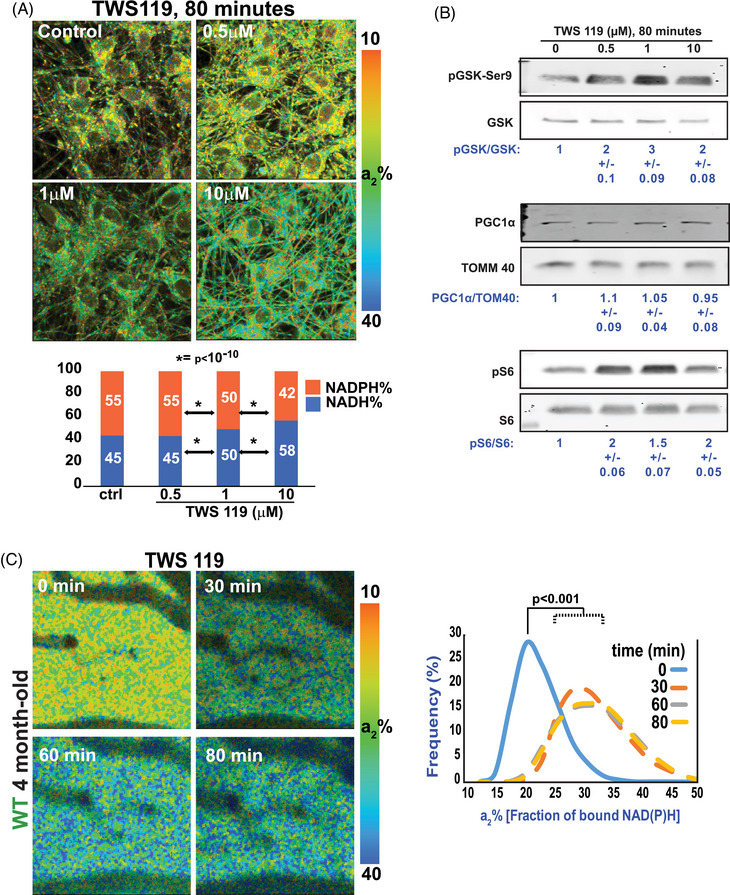
GSK3β inhibition stimulates mitochondrial activity in cultured human neurons and WT mouse brains. A, Neurons derived from the RenCell VM line of human neuronal progenitors were differentiated for 20 days. Then, one culture was left untreated (control) while others were treated with TWS119 at the indicated concentration for ≈ 80 minutes. 2P‐FLIM revealed that inhibition of GSK3β increased the fraction of bound NADH, indicating a rise in mitochondrial respiration. Micrographs are representative of three independent experiments. Statistical analyses were performed using Student two‐tailed unpaired t test. B, TWS119 efficiency was tested by western blots of protein samples from the same experiments used for 2P‐FLIM. TWS119 caused large increases in phosphorylation of the inhibitory site, serine‐9 on GSK3β, and in phosphorylation of the ribosomal protein S6, a readout of mTORC1 activity. Changes in mitochondrial activity occurred without changes in the expression of the mitochondrial transcription factor PGC1α. The experiment is representative of three independent assays. C, 2P‐FLIM imaging of WT mouse cortex through an open‐skull window after topical application of TWS119. An increase in a2% was observed for several minutes indicating upregulation of mitochondrial activity. Statistical analyses were performed using Student two‐tailed paired t test. Four animals were used in these experiments. 2P‐FLIM, two‐photon fluorescence lifetime imaging; GSK3β, glycogen synthase kinase 3; NADH, reduced nicotinamide adenine dinucleotide; WT, wild type.
FIGURE 6.
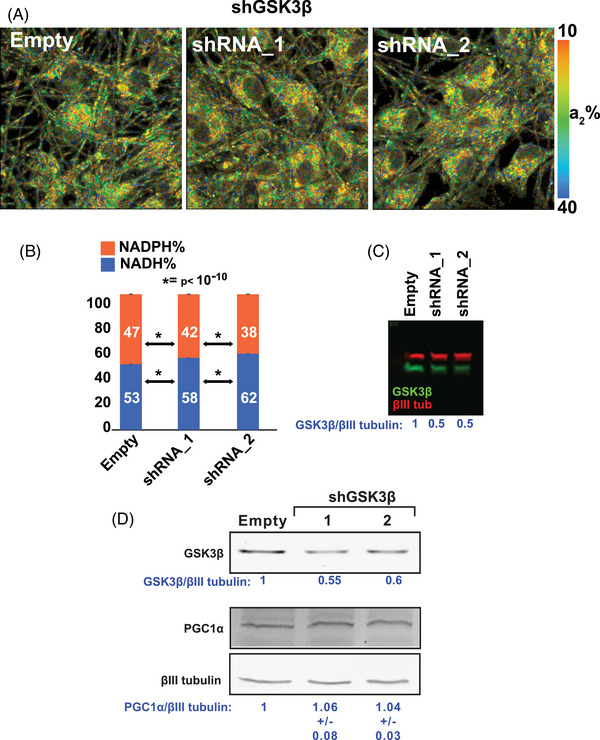
GSK3β reduction stimulates mitochondrial activity in cultured human neurons. A–B, GSK3β expression was reduced by lentiviral‐mediated delivery of antisense shRNA. 2P‐FLIM analysis in live cells revealed an increase of NADH, indicating a rise in mitochondrial respiration in GSK3β knock‐down cells. The experiment is representative of three independent assays. Statistical analyses were performed using Student two‐tailed unpaired t test. C–D, GSK3β knockdown efficiency in the 2P‐FLIM assays was ≈ 50%. Under similar experimental conditions, there were no changes in the expression of PGC1α. The experiment is representative of three independent assays. 2P‐FLIM, two‐photon fluorescence lifetime imaging; GSK3β, glycogen synthase kinase 3; NADH, nicotinamide adenine dinucleotide.
To corroborate that GSK3β was mediating the effect of TWS119 and lithium on mitochondrial respiration, we used lentiviral‐driven delivery of antisense shRNA to knock down GSK3β by ≈ 50% in human neurons. As we found for TWS119 (Figure 5) and LiCl (Figure S3), GSK3β knockdown increased the NADH/NADPH ratio in cultured human neurons (Figure 6A–C). Similar to TWS119 (Figure 5B) and LiCl (Figure S3B), GSK3β knockdown did not change the level of PGC1α (Figure 6D), fortifying our previously reported evidence that NiMA activation does not require an increase in mitochondrial number. 7 Collectively, the results for TWS119, LiCl, and GSK3β knockdown establish that GSK3β regulates mTORC1 activity in neurons and extend our prior finding that stimulation of lysosomal mTORC1 increases NADH and OXPHOS in neuronal mitochondria. 7 GSK3β is thus a newly identified upstream regulator of NiMA in neurons.
3.4. TWS119 rescues mitochondrial activity in the APPSAA/+ mice
We and others have shown that AβOs impair insulin/IGF1 signaling in neurons and insulin resistance is a major risk factor for developing AD. 29 Mechanistically these effects are partially explained by a mechanism including (1) AβOs‐induced internalization of insulin receptors; 30 (2) reduction in AKT activity, a downstream insulin receptor target; 10 and (3) increased association with lysosomes of the tuberous sclerosis complex, an mTORC1 inhibitor. 9 As AβOs also affect the anterograde trafficking of mitochondria through activation of GSK3β in a tau‐dependent manner, 17 it is possible to speculate that AβOs‐induced and tau‐dependent insulin resistance led to GSK3β activation in AD. Thus, inhibiting GSK3β in vivo at presymptomatic stages of AD could represent a potential treatment.
As TWS119 increased mitochondrial activity in WT live mouse brain (Figure 5C), we then tested TWS 119's ability to stimulate mitochondrial activity in the 4‐month‐old APPSAA/+ live brain (when NiMA is unresponsive). Like our observations in WT animals and cultured neurons (Figure 5), TWS119 application for 80 minutes through a cranial window also increased a2% in the APPSAA/+ mice (Figure 7A). This change in a2% was also paralleled by a partial increase in oxygen consumption as measured by MP‐PAM (Figure 7B).
FIGURE 7.
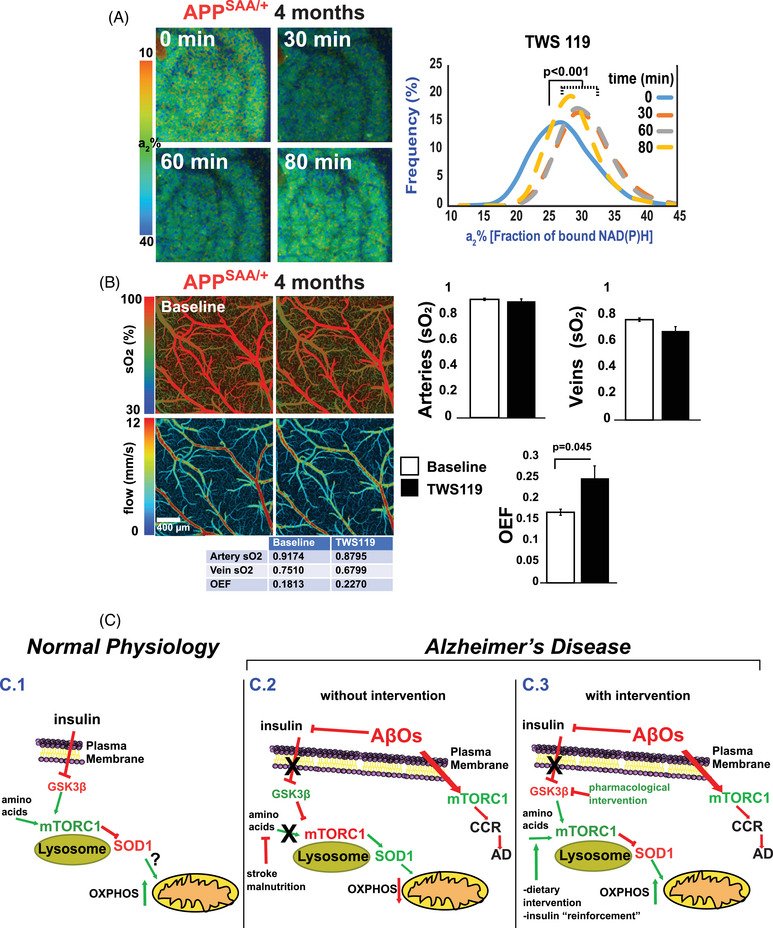
TWS119 triggers mitochondrial respiration in the AD mouse cortex. A, 2P‐FLIM imaging of the APPSAA/+ mouse cortex through an open‐skull window before and after topical application of TWS119. An increase in a2% was observed for several minutes indicating upregulation of mitochondrial activity. Statistical analyses were performed using Student two‐tailed paired t test. Three animals were used in these experiments. B, MP‐PAM imaging of APPSAA/+ mouse cortex through an open‐skull window before and 80 minutes after a topical application of TWS119. A small decrease in blood oxygenation of the cortical arteries and veins was observed but did not reach statistical significance. Still, there was a significant increase in the overall OEF. The bar graphs show the quantification of three independent experiments. Statistical analyses were performed using Student two‐tailed unpaired t test. Error bars represent ± standard error of the mean. C, Model for NiMA regulation by GSK3β in vivo in the brain:(C.1) In normal physiological conditions insulin and amino acids stimulate lysosomal mTORC1 7 by a mechanism involving inhibition of GSK3β. Then, mTORC1‐mediated phosphorylation of SOD1 at T40, 10 triggers mitochondrial respiration. (C.2) Insulin‐mediated inhibition of GSK3β is blocked by AβOs, decreasing mTORC1 activity on lysosomes and stimulating it instead at the plasma membrane, where mTORC1 kinase activity triggers CCR, a frequent prelude to cortical neuron death in AD. (C.3) Pharmacological treatments that block AβO‐induced activation of GSK3β might restore normal lysosomal mTORC1 activity in AD. 2P‐FLIM, two‐photon fluorescence lifetime imaging; AD, Alzheimer's disease; APPSAA/+, heterozygous amyloid precursor protein knock‐in; CCR, cell cycle re‐entry; GSK3β, glycogen synthase kinase 3; MP‐PAM, multi‐parametric photoacoustic microscopy; NiMA, nutrient‐induced mitochondrial activity; OEF, oxygen extraction fraction; SOD1, superoxide dismutase 1.
4. DISCUSSION
Brain hypometabolism is one of the earliest signs of AD, 31 , 32 , 33 , 34 and here we present new mechanistic evidence for how this pernicious decline in brain glucose consumption occurs. Using mitochondrial metabolic imaging of a live mouse brain, we demonstrate the disruption of the NiMA pathway in APPSAA/+ mice. The relevance of this process is two‐fold. First, this Aβ‐driven phenomenon coincides with the earliest detection of soluble Aβ1‐40/42 in the brain and CSF and occurs months before plaques and other key AD features emerge in this transgenic mouse model. 21 Second, NiMA disruption does not impact overall mitochondrial metabolism, which is regulated by a plethora of inputs including a diverse source of nutrients, 35 but specifically interferes with mitochondrial respiration triggered by amino acids and likely insulin as well.
The physiological significance of NiMA inhibition for human AD is underscored by the genotype of the APPSAA/+ mice used for this study. APP is expressed in these animals exclusively by the two endogenous murine APP genes, one of which is unmodified and the other of which was altered by homologous recombination to contain a humanized Aβ1‐42 region encoding the Swedish (KM670/671NL), Arctic (E693G), and Austrian (T714I) mutations in place of the corresponding region of WT mouse APP. 21 Consequently, and in sharp contrast to nearly all other transgenic APP mouse models of AD, APPSAA/+ mice do not overexpress APP, nor do they contain numerous mutant human APP cDNAs that incorporate into mouse genomic DNA at random sites and thereby potentially interfere with normal expression of multiple mouse genes. Just as importantly, the NiMA response to amino acid stimulation is normal in 2‐month‐old APPSAA/+ mice but is completely inhibited by the time animals reach 4 months of age (Figure 2). The simplest explanation for this observation is that the single mutated APP gene drove Aβ production to levels sufficient to block NiMA a year before the first plaques appeared in these mice at 16 months of age. 21 It follows naturally that NiMA inhibition, which includes reduced adenosine triphosphate (ATP) production by mitochondria in response to nutrient stimulation, might occur in the brains of human AD patients far in advance of the histopathological features and cognitive decline that define the disease. This soluble Aβ‐induced deficiency in the fundamental cellular fuel, ATP, could underlie the myriad aspects of neuronal function that deteriorate as AD progresses from its silent, prodromal stage to full‐blown symptoms.
How does NiMA disruption set the stage for AD pathogenesis? Central to this story are three well‐known AD players: AβOs, mTORC1, and GSK3β. Moreover, we previously found that AβO‐mediated inhibition of NiMA required the expression of tau, 7 , 10 the building block of neurofibrillary tangles. The physiological mechanisms controlling mTORC1 functions have been widely studied and include central roles for insulin and the amino acids (R+L) in activating mTORC1 activity on lysosomes. There, mTORC1 regulates key processes, such as mRNA translation, autophagy, gene expression, and mitochondrial activity. 7 , 8 At the cellular level, mTORC1‐mediated regulation of protein synthesis is required for synaptic function 36 and removal of misfolded proteins and organelles via autophagy. 37 Mitophagy, the removal of defective mitochondria by autophagic mechanisms, is partially regulated by mTORC1 38 and has been shown to reduce Aβ and tau pathology, and cognitive decline in AD. 39 However, much less is known about the mechanisms leading to mTORC1 dysregulation in AD. Work by our group has shown that AβOs may work as a two‐edged sword. At one level, AβOs trigger ectopic activation of mTORC1 at the plasma membrane by a mechanism that is dependent on tau expression and thereby causes the re‐entry of neurons into the cell cycle, a prelude to most neuron death in AD. 9 At a second, parallel level, AβOs reduce mTORC1 activity on lysosomes by inhibiting insulin signaling. 10 It is reasonable to speculate that during AD progression the combination of Aβ production and insulin resistance leads to an imbalance in mTORC1 activity favoring its ectopic and toxic activity at the plasma membrane over its normal physiological functions on lysosomes. By extension, these events also lead to NiMA disruption, as we previously shown for cultured neurons, 7 , 10 and as reported in the current study, in vivo as well. This hypothesis is consistent with the facts that mTORC1 activity is elevated in the AD brain, 40 and that decreasing mTOR expression in an AD mouse model ameliorates AD‐like pathology and behavior. 41 Thus, NiMA is a fundamental biological process that integrates several key players in AD pathogenesis, including tau, mTOR, and autophagy.
The collective findings reported here have therapeutic implications for AD. For example, we show that low doses of lithium, an FDA‐approved treatment for mood disorders, increase mitochondrial activity in neurons in culture (Figure S3). We also found that TWS119, a GSK3β inhibitor, increased mitochondrial respiration in both cultured neurons and mouse brains (Figure 5). Just as importantly, we found that TWS119 also increased mitochondrial activity in the APPSAA/+ mouse cortex (Figure 7A and B). Tideglusib, another GSK3β inhibitor, 42 has been tested in clinical trials for AD, and although found to be safe it showed no beneficial effects. 43 , 44 Interestingly, in our hands, using corresponding isomolar concentrations of tideglusib was also ineffective at stimulating mitochondrial respiration in cultured neurons and mouse brains (not shown). Thus, although GSK3β is a promising target for AD therapy, a deeper understanding of its diverse mechanisms of action and cellular effects of GSK3β inhibitors will improve the outcome of future clinical trials directed at this protein kinase.
Several studies have investigated the association between Aβ deposition and local brain energy metabolism, but multimodal FDG PET and amyloid PET aimed at comparing plaque deposition to changes in local glucose metabolism studies have provided conflicting results. 45 , 46 , 47 , 48 , 49 , 50 , 51 Although informative, amyloid PET is limited by resolution and its inability to detect AβOs. Still, amyloid PET studies have suggested that brain areas loaded with plaques are “protected” from Aβ insult 52 , 53 , 54 and represent in vivo support for the idea that soluble forms of Aβ are disruptive of cellular energy metabolism. A recent study provided evidence that AβOs are likely initiators of brain hypometabolism 55 and are in line with our findings shown here that AβOs disrupt NiMA in vivo in the brain.
Although strongly supported by data from rare autosomal dominant mutations in APP, or presenilins 1 and 2 (known as familial AD, or fAD), the amyloid cascade hypothesis is limited because most AD cases are sporadic (sAD). From a cell biological point of view, sAD likely involves alterations in lysosomal and autophagosome functioning, disruption of Ca++ homeostasis, failure of neuronal cell cycle control, and neuroinflammation, 3 which can be exacerbated by metabolic conditions such as type 2 diabetes and reduced insulin signaling. These perturbations, in turn, can provoke the accumulation of toxic Aβ species. 56 Interestingly, growing evidence shows that mutations in mitochondrial DNA, oxidative damage, and disrupted brain glucose and energy metabolism occur in the brain decades before disease onset, 1 strongly suggesting that mitochondrial dysfunction is an early and key factor in the etiology of AD. 57 The results presented here argue that a communication glitch between lysosomes and mitochondria occurs when AβO reaches a certain threshold, becoming evident long before disease onset. Importantly, disruption of NiMA may also occur in other disease contexts. Previously we reported NiMA disruption in dermal fibroblast from patients afflicted by tuberous sclerosis, a benign form of cancer caused by inactivating mutations in TSC1 or TSC2. 7 , 10 Thus, disruption of neuronal NiMA could be a converging point between genetic and metabolic alterations leading to mitochondrial and neuronal dysfunction in AD.
Many cellular functions disrupted by AβOs are known to be dependent on the expression of tau. 58 Among them, disruption of synaptic functioning and neurodegeneration has been recognized for a long time, leading to the concept that AD is a “synaptic disease.” 59 The fact that disruption of NiMA was detected in the cortex of APPSAA/+ mice months before cognitive decline 21 leads us to propose that chronic metabolic disruption caused by an AβO‐induced, tau‐dependent mechanism represents a very early event in human AD.
AUTHOR CONTRIBUTIONS
Andrés Norambuena conceived, designed, and initiated the study; performed all the studies of neurons in culture; and quantified the expression levels of complex I‐V in mouse brain samples. He also led the entire study, wrote the first draft of the paper, and along with George S. Bloom served as the main editor of the original text. Shagufta Rehman Alam and Prakash Raut did an optimization of the 2P‐FLIM assay in the live mouse brain. Vijay Kumar Sagar and Prakash Raut performed 2P‐FLIM experiments in the live mouse brain. Horst Wallrabe and Vijay Kumar Sagar analyzed 2P‐FLIM experiments in live cells and mouse brains. Ammasi Periasamy provided extensive technical support for 2P‐FLIM. Evelyn Pardo and Taylor Kim took care of the animal colony, performed q‐PCR, and along with Andrés Norambuena and Vijay Kumar Sagar coordinated live mouse experiments. Zhuoying Wang, Ziang Feng, and Song Hu performed and analyzed experiments involving MP‐PAM. George S. Bloom analyzed data and provided intellectual feedback throughout the study. All other authors read, edited, and approved the submission of the manuscript.
CONFLICT OF INTEREST STATEMENT
The authors have no conflicts of interest to report. Author disclosures are available in the supporting information.
CONSENT STATEMENT
Consent was not necessary.
Supporting information
Supporting Information
Supporting Information
Supporting Information
Supporting Information
ACKNOWLEDGMENTS
We would like to thank Dr. Anthony Spano for providing the βIII‐tubulin antibody (Tuj1). Funding for this work was provided by NIH/NIA Grant R01AG067048 (to A.N.); the Owens Family Foundation (G.S.B.); Cure Alzheimer's Fund (G.S.B.); Rick Sharp Alzheimer's Foundation (G.S.B.); and the NIH/Office of the Director for funds to purchase a Zeiss 780 and 980 microscopes used in this study (OD016446 and NIH‐OD 025156 to A.P.).
Norambuena A, Sagar VK, Wang Z, et al. Disrupted mitochondrial response to nutrients is a presymptomatic event in the cortex of the APPSAA knock‐in mouse model of Alzheimer's disease. Alzheimer's Dement. 2024;20:6844–6859. 10.1002/alz.14144
Andrés Norambuena dedicates this paper to the memory of Edward P. Owens. Ed, during his years of association with the Bloom Lab, has been most supportive of my work in Alzheimer's research. I fondly remember his friendly and hopeful outlook that one day we will find a complete answer to this dreadful disease.
REFERENCES
- 1. Wang W, Zhao F, Ma X, Perry G, Zhu X. Mitochondria dysfunction in the pathogenesis of Alzheimer's disease: recent advances. Mol Neurodegener. 2020;15(1):30. doi: 10.1186/s13024-020-00376-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Hardy JA, Higgins GA. Alzheimer's disease: the amyloid cascade hypothesis. Science. 1992;256:184‐185. [DOI] [PubMed] [Google Scholar]
- 3. Herrup K. The case for rejecting the amyloid cascade hypothesis. Nat Neurosci. 2015;18:794‐799. doi: 10.1038/nn.4017 [DOI] [PubMed] [Google Scholar]
- 4. Musiek ES, Holtzman DM. Three dimensions of the amyloid hypothesis: time, space and “wingmen”. Nat Neurosci. 2015;18:800‐806. doi: 10.1038/nn.4018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Reardon S. FDA approves Alzheimer's drug lecanemab amid safety concerns. Nature. 2023;613:227‐278. doi: 10.1038/d41586-023-00030-3 [DOI] [PubMed] [Google Scholar]
- 6. Hardy J, Selkoe DJ. The amyloid hypothesis of Alzheimer's disease: progress and problems on the road to therapeutics. Science. 2002;297:353‐356. doi: 10.1126/science.1072994 [DOI] [PubMed] [Google Scholar]
- 7. Norambuena A, Wallrabe H, Cao R, et al. A novel lysosome‐to‐mitochondria signaling pathway disrupted by amyloid‐β oligomers. EMBO J. 2018;37:e100241. doi: 10.15252/embj.2018100241 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Saxton RA, Sabatini DM. mT OR Signaling in growth, metabolism, and disease. Cell. 2017;168:960‐976. doi: 10.1016/j.cell.2017.02.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Norambuena A, Wallrabe H, McMahon L, et al. mTOR and neuronal cell cycle reentry: how impaired brain insulin signaling promotes Alzheimer's disease. Alzheimers Dement. 2017;13:152‐167. doi: 10.1016/j.jalz.2016.08.015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Norambuena A, Sun X, Wallrabe H, et al. SOD1 mediates lysosome‐to‐mitochondria communication and its dysregulation by amyloid‐β oligomers. Neurobiol Dis. 2022;169:105737. doi: 10.1016/j.nbd.2022.105737 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Embi N, Rylatt DB, Cohen P. Glycogen synthase kinase‐3 from rabbit skeletal muscle. Separation from cyclic‐AMP‐dependent protein kinase and phosphorylase kinase. Eur J Biochem. 1980;107:519‐527. [PubMed] [Google Scholar]
- 12. Welsh GI, Proud CG. Glycogen synthase kinase‐3 is rapidly inactivated in response to insulin and phosphorylates eukaryotic initiation factor eIF‐2B. Biochem J. 1993;294:625‐629. doi: 10.1042/bj2940625 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Hughes K, Ramakrishna S, Benjamin WB, Woodgett JR. Identification of multifunctional ATP‐citrate lyase kinase as the α ‐isoform of glycogen synthase kinase‐3. Biochem J. 1992;288:309‐314. doi: 10.1042/bj2880309 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Hooper C, Killick R, Lovestone S. The GSK3 hypothesis of Alzheimer's disease. J Neurochem. 2008;104:1433‐1439. doi: 10.1111/j.1471-4159.2007.05194.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Hanger DP, Hughes K, Woodgett JR, Brion JP, Anderton BH. Glycogen synthase kinase‐3 induces Alzheimer's disease‐like phosphorylation of tau: generation of paired helical filament epitopes and neuronal localisation of the kinase. Neurosci Lett. 1992;147:58‐62. doi: 10.1016/0304-3940(92)90774-2 [DOI] [PubMed] [Google Scholar]
- 16. Muyllaert D, Kremer A, Jaworski T, et al. Glycogen synthase kinase‐3β, or a link between amyloid and tau pathology? Genes Brain Behav. 2008;7:57‐66. doi: 10.1111/j.1601-183X.2007.00376.x [DOI] [PubMed] [Google Scholar]
- 17. Vossel KA, Xu JC, Fomenko V, et al. Tau reduction prevents Aβ‐induced axonal transport deficits by blocking activation of GSK3β. J Cell Biol. 2015;209:419‐433. doi: 10.1083/jcb.201407065 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Hedgepeth CM, Conrad LJ, Zhang J, Huang HC, Lee VMY, Klein PS. Activation of the Wnt Signaling Pathway: a Molecular Mechanism for Lithium Action. Dev Biol. 1997;185:82‐91. doi: 10.1006/dbio.1997.8552 [DOI] [PubMed] [Google Scholar]
- 19. Stambolic V, Ruel L, Woodgett JR. Lithium inhibits glycogen synthase kinase‐3 activity and mimics Wingless signalling in intact cells. Curr Biol. 1996;6:1664‐1669. doi: 10.1016/S0960-9822(02)70790-2 [DOI] [PubMed] [Google Scholar]
- 20. Martin SA, Souder DC, Miller KN, et al. GSK3β regulates brain energy metabolism. Cell Rep. 2018;23:1922‐1931.e4. doi: 10.1016/j.celrep.2018.04.045 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Xia D, Lianoglou S, Sandmann T, et al. Novel App knock‐in mouse model shows key features of amyloid pathology and reveals profound metabolic dysregulation of microglia. Mol Neurodegener. 2022;17:41. doi: 10.1186/s13024-022-00547-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Choi SH, Kim YH, Hebisch M, et al. A three‐dimensional human neural cell culture model of Alzheimer's disease. Nature. 2014;515:274‐278. doi: 10.1038/nature13800 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Cao R, Li J, Ning B, et al. Functional and oxygen‐metabolic photoacoustic microscopy of the awake mouse brain. Neuroimage. 2017;150:77‐87. doi: 10.1016/j.neuroimage.2017.01.049 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Lakowicz JR. Principles of Fluorescence Spectroscopy Springer; 2006. doi: 10.1007/978-0-387-46312-4 [DOI] [Google Scholar]
- 25. Blacker TS, Mann ZF, Gale JE, et al. Separating NADH and NADPH fluorescence in live cells and tissues using FLIM. Nat Commun. 2014;5:3936. doi: 10.1038/ncomms4936 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Hoyer S, Oesterreich K, Wagner O. Glucose metabolism as the site of the primary abnormality in early‐onset dementia of Alzheimer type? J Neurol. 1988;235:143‐148. doi: 10.1007/BF00314304 [DOI] [PubMed] [Google Scholar]
- 27. Ding S, Wu TYH, Brinker A, et al. Synthetic small molecules that control stem cell fate. Proc Natl Acad Sci USA. 2003;100:7632‐7637. doi: 10.1073/pnas.0732087100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Sutherland C, Leighton IA, Cohen P. Inactivation of glycogen synthase kinase‐3 β by phosphorylation: new kinase connections in insulin and growth‐factor signalling. Biochem J. 1993;296:15‐19. doi: 10.1042/bj2960015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. De Felice FG, Gonçalves RA, Ferreira ST. Impaired insulin signalling and allostatic load in Alzheimer disease. Nat Rev Neurosci. 2022;23:215‐230. doi: 10.1038/s41583-022-00558-9 [DOI] [PubMed] [Google Scholar]
- 30. Zhao W, De Felice FG, Fernandez S, et al. Amyloid beta oligomers induce impairment of neuronal insulin receptors. FASEB J. 2008;22:246‐260. doi: 10.1096/fj.06-7703com [DOI] [PubMed] [Google Scholar]
- 31. Gordon BA, Blazey TM, Su Y, et al. Spatial patterns of neuroimaging biomarker change in individuals from families with autosomal dominant Alzheimer's disease: a longitudinal study. Lancet Neurol. 2018;17:241‐250. doi: 10.1016/S1474-4422(18)30028-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Croteau E, Castellano CA, Fortier M, et al. A cross‐sectional comparison of brain glucose and ketone metabolism in cognitively healthy older adults, mild cognitive impairment and early Alzheimer's disease. Exp Gerontol. 2018;107:18‐26. doi: 10.1016/j.exger.2017.07.004 [DOI] [PubMed] [Google Scholar]
- 33. Crane PK, Walker R, Hubbard RA, et al. Glucose levels and risk of dementia. N Engl J Med. 2013;369:540‐548. doi: 10.1056/NEJMoa1215740 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Kapogiannis D, Mattson MP. Disrupted energy metabolism and neuronal circuit dysfunction in cognitive impairment and Alzheimer's disease. Lancet Neurol. 2011;10:187‐198. doi: 10.1016/S1474-4422(10)70277-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Yassine HN, Self W, Kerman BE, et al. Nutritional metabolism and cerebral bioenergetics in Alzheimer's disease and related dementias. Alzheimers Dement. 2023;19:1041‐1066. doi: 10.1002/alz.12845 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Stoica L, Zhu PJ, Huang W, Zhou H, Kozma SC, Costa‐Mattioli M. Selective pharmacogenetic inhibition of mammalian target of Rapamycin complex I (mTORC1) blocks long‐term synaptic plasticity and memory storage. Proc Natl Acad Sci. 2011;108:3791‐3796. doi: 10.1073/pnas.1014715108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Aman Y, Schmauck‐Medina T, Hansen M, et al. Autophagy in healthy aging and disease. Nat Aging. 2021;1:634‐650. doi: 10.1038/s43587-021-00098-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Bartolomé A, García‐Aguilar A, Asahara S‐I, et al. MTORC1 regulates both general autophagy and mitophagy induction after oxidative phosphorylation uncoupling. Mol Cell Biol. 2017;37:e00441‐e00447. doi: 10.1128/MCB.00441-17 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Fang EF, Hou Y, Palikaras K, et al. Mitophagy inhibits amyloid‐β and tau pathology and reverses cognitive deficits in models of Alzheimer's disease. Nat Neurosci. 2019;22:401‐412. doi: 10.1038/s41593-018-0332-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Oddo S. The role of mTOR signaling in Alzheimer disease. Front Biosci. 2012;4:941‐952. doi: 10.2741/s310 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Caccamo A, De Pinto V, Messina A, Branca C, Oddo S. Genetic reduction of mammalian target of rapamycin ameliorates Alzheimer's disease‐like cognitive and pathological deficits by restoring hippocampal gene expression signature. J Neurosci. 2014;34:7988‐7998. doi: 10.1523/JNEUROSCI.0777-14.2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Domínguez JM, Fuertes A, Orozco L, del Monte‐Millán M, Delgado E, Medina M. Evidence for irreversible inhibition of glycogen synthase kinase‐3β by tideglusib. J Biol Chem. 2012;287:893‐904. doi: 10.1074/jbc.M111.306472 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. del Ser T, Steinwachs KC, Gertz HJ, et al. Treatment of Alzheimer's disease with the GSK‐3 inhibitor tideglusib: a pilot study. J Alzheimers Dis. 2012;33:205‐215. doi: 10.3233/JAD-2012-120805 [DOI] [PubMed] [Google Scholar]
- 44. Lovestone S, Boada M, Dubois B, et al. A phase II trial of tideglusib in Alzheimer's disease. J Alzheimers Dis. 2015;45:75‐88. doi: 10.3233/JAD-141959 [DOI] [PubMed] [Google Scholar]
- 45. Li Y, Rinne JO, Mosconi L, et al. Regional analysis of FDG and PIB‐PET images in normal aging, mild cognitive impairment, and Alzheimer's disease. Eur J Nucl Med Mol Imaging. 2008;35:2169‐2181. doi: 10.1007/s00259-008-0833-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Rabinovici GD, Furst AJ, Alkalay A, et al. Increased metabolic vulnerability in early‐onset Alzheimer's disease is not related to amyloid burden. Brain. 2010;133:512‐528. doi: 10.1093/brain/awp326 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Furst AJ, Rabinovici GD, Rostomian AH, et al. Cognition, glucose metabolism and amyloid burden in Alzheimer's disease. Neurobiol Aging. 2012;33:215‐225. doi: 10.1016/j.neurobiolaging.2010.03.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Cohen AD, Price JC, Weissfeld LA, et al. Basal cerebral metabolism may modulate the cognitive effects of Aβ in mild cognitive impairment: an example of brain reserve. J Neurosci Nurs. 2009;29:14770‐14778. doi: 10.1523/JNEUROSCI.3669-09.2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Edison P, Archer HA, Hinz R, et al. Amyloid, hypometabolism, and cognition in Alzheimer disease: an [11C]PIB and [18F]FDG PET study. Neurology. 2007;68:501‐508. doi: 10.1212/01.wnl.0000244749.20056.d4 [DOI] [PubMed] [Google Scholar]
- 50. Lowe VJ, Weigand SD, Senjem ML, et al. Association of hypometabolism and amyloid levels in aging, normal subjects. Neurology. 2014;82:1959‐1967. doi: 10.1212/WNL.0000000000000467 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Engler H, Forsberg A, Almkvist O, et al. Two‐year follow‐up of amyloid deposition in patients with Alzheimer's disease. Brain. 2006;129:2856‐2866. doi: 10.1093/brain/awl178 [DOI] [PubMed] [Google Scholar]
- 52. Altmann A, Ng B, Landau SM, Jagust WJ, Greicius MD; Alzheimer's Disease Neuroimaging Initiative . Regional brain hypometabolism is unrelated to regional amyloid plaque burden. Brain. 2015;138:3734‐3746. doi: 10.1093/brain/awv278 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Lee H, Casadesus G, Zhu X, Takeda A, Perry G, SMITH MA. Challenging the amyloid cascade hypothesis: senile plaques and amyloid‐β as protective adaptations to Alzheimer disease. Ann N Y Acad Sci. 2004;1019:1‐4. doi: 10.1196/annals.1297.001 [DOI] [PubMed] [Google Scholar]
- 54. Cuajungco MP, Goldstein LE, Nunomura A, et al. Evidence that the β‐amyloid plaques of Alzheimer's disease represent the redox‐silencing and entombment of Aβ by zinc. J Biol Chem. 2000;275:19439‐19442. doi: 10.1074/jbc.C000165200 [DOI] [PubMed] [Google Scholar]
- 55. Malkov A, Popova I, Ivanov A, et al. Aβ initiates brain hypometabolism, network dysfunction and behavioral abnormalities via NOX2‐induced oxidative stress in mice. Commun Biol. 2021;4:1054. doi: 10.1038/s42003-021-02551-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. De Felice FG, Ferreira ST. Inflammation, defective insulin signaling, and mitochondrial dysfunction as common molecular denominators connecting type 2 diabetes to Alzheimer Disease. Diabetes. 2014;63:2262‐2272. doi: 10.2337/db13-1954 [DOI] [PubMed] [Google Scholar]
- 57. Swerdlow RH, Burns JM, Khan SM. The Alzheimer's disease mitochondrial cascade hypothesis: progress and perspectives. Biochim Biophys Acta. 2014;1842:1219‐1231. doi: 10.1016/j.bbadis.2013.09.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Bloom GS. Amyloid‐β and tau: the trigger and bullet in Alzheimer's disease pathogenesis. JAMA Neurol. 2014;71:505‐508. doi: 10.1001/jamaneurol.2013.5847 [DOI] [PubMed] [Google Scholar]
- 59. Selkoe DJ. Alzheimer's disease is a synaptic failure. Science. 2002;298:789‐791. doi: 10.1126/science.1074069 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting Information
Supporting Information
Supporting Information
Supporting Information