

Abstract
Adipose tissue, which can provide adipokines and nutrients to tumors, plays a key role in promoting ovarian cancer (OvCa) metastatic lesions in peritoneal cavity. The adipokine Apelin promotes OvCa metastasis and progression through its receptor APJ, which regulates cell proliferation, energy metabolism and angiogenesis. The objective of this study was to investigate the functional role and mechanisms of the apelin-APJ pathway in OvCa metastasis, especially in context of tumor cell-adipocyte interactions. When co-cultured in the conditioned media (AdipoCM) derived from 3T3-L1 adipocytes, which express and secrete high apelin, human OvCa cells with high APJ expression showed significant increases in migration and invasion in vitro. We also found that cells expressing high levels of APJ had increased cell adhesion to omentum ex vivo, and preferentially ‘home-in’ on the omentum in vivo. These apelin-induced pro-metastatic effects were reversed by APJ antagonist F13A in a dose-dependent manner. Apelin-APJ activation increased lipid droplet accumulation in OvCa cells, which was further intensified in the presence of AdipoCM and reversed by F13A or APJ knockdown. Mechanistically, this increased lipid uptake was mediated by CD36 upregulation via APJ-STAT3 activation, and the lipids were utilized in promoting fatty acid oxidation via activation of AMPK-CPT1a axis. Together, our studies demonstrate that adipocyte-derived apelin activates APJ-expressing tumor cells in a paracrine manner, promoting lipid uptake and utilization and providing energy for OvCa cell survival at the metastatic sites. Hence, the apelin-APJ pathway presents a novel therapeutic target to curb OvCa metastasis.
Keywords: apelin/APJ, lipid droplets, fatty acid oxidation, ovarian cancer, metastasis
INTRODUCTION
Ovarian cancer (OvCa) is one of the most lethal gynecological malignancies associated with late-stage diagnosis and poor prognosis (1). Unlike other malignancies, OvCa exhibits a unique biological behavior wherein exfoliated tumor cells disseminate via peritoneal fluid, and implant on the mesothelial lining of the peritoneum to establish metastasis (2). Despite extensive research to identify vulnerabilities, high-grade serous ovarian carcinoma (HGSOC) remains the most lethal subtype with 5-year survival rate of 43% (3). This emphasizes the need to identify and better understand the key players and pathways that orchestrate malignant spread and growth, to aid in the development of novel therapeutic strategies.
Recently, we demonstrated that high apelin receptor (APJ) expression on tumor cells contributes to OvCa progression and is associated with worsened survival in patients with OvCa (4). APJ, a G-protein coupled receptor, is activated by endogenous ligand apelin, which is an adipokine. This pathway is important for numerous physiological processes, including cardiovascular regulation, energy metabolism, fluid homeostasis, angiogenesis, and neuroendocrine stress response. Further, its role in several pathologies including diabetes, obesity, and cancer are being recently recognized (5). Several studies have established that OvCa cells possess a specific metastatic predilection to the omentum (6–8), a visceral fat depot rich in adipocytes, blood and lymph vessels, stromal, and immune cells (9,10). The adipocytes are a major source of secreted adipokines, such as IL-6, IL-8, monocyte chemotactic protein 1 (MCP-1), leptin, and adiponectin. These adipokines contribute to tumor neovascularization, reprogrammed cell metabolism, enhanced inflammation, and cell growth in the ovarian tumor microenvironment (TME) (11–13). However, the role of apelin-mediated APJ activation in regulating OvCa cell metabolism at the tumor-adipocyte interface is unknown.
In the present study, we utilized a set of complementary in vitro, ex vivo, and in vivo model systems to investigate the functions and mechanisms of the apelin-APJ pathway, with respect to cancer cell-adipocyte interaction, in promoting metastasis of OvCa cells. Our studies revealed that adipocytes-derived apelin in the TME acts in a paracrine manner to increase various pro-metastatic phenotypes via activation of APJ, which is expressed on the tumor cells. Our findings highlight the novel role of activated APJ pathway in modulating lipid uptake and utilization, specifically fatty acid oxidation (FAO) in OvCa cells. This could potentially contribute to increased survival of tumor cells at the metastatic sites. Our study warrants for further investigation of this pathway as a potential druggable target in management of OvCa metastasis.
MATERIALS AND METHODS
Reagents and Cell Culture
Human OvCa cell lines OVCAR-5 and OVCAR-8 were purchased from the Division of Cancer Treatment and Diagnosis (DCTD) Tumor Repository, National Cancer Institute (NCI) at Frederick, Maryland. SKOV-3 cells were purchased from ATCC. Mouse fibroblast cell line (3T3-L1) were a generous gift from Dr. Ann Olson (OUHSC). The cell lines were profiled via short tandem repeat profiling to confirm their identity prior to receipt. The cell lines were cultured in RPMI (OVCAR-5 and OVCAR-8), McCoy’s (SKOV-3), and DMEM (3T3-L1) media supplemented with 10% fetal bovine serum (FBS) or fetal calf serum (FCS). Cells and media were periodically tested for mycoplasma using the MycoAlert™ Mycoplasma Detection Kit (Lonza), and if found positive, older freezes of mycoplasma-free cells were used. Experiments were performed on cells within 15 passages post thaw. APJ overexpressing stable cell lines (OVCAR-5-APJ) were established as described previously (4). SKOV-3 cells were transfected with APJ-pCMV3-SP-N-Myc (Sinobiologics, HG11477-NM) or empty vector (EV) using lipofectamine 2000 (Life Technologies), per manufacturer’s protocol. Two different shRNA constructs (shAPJ-1: 5’ GCGCTCAGCTGATATCTTCAT-3’ and shAPJ-2: 5’-GGCTTCTAGAAGGGAAGAAAT-3’) were inserted into RNAi-Ready pSIREN-Lenti vector at the BamHI and EcoRI restriction sites. OVCAR-8 cells were transduced and selected with 2 μg/ml puromycin. The inhibitors, F13A (Bachem, 4095877), STATTIC (MedChem Express, HY-13818), Etomoxir (Sigma-Aldrich, E1905), and Sulfo-N-succinimidyl Oleate sodium (Sigma-Aldrich, SML2148) were reconstituted according to manufacturer’s protocol.
Adipocyte Differentiation and Conditioned Media
3T3-L1 fibroblasts (pre-adipocytes) were induced to differentiate into adipocytes as described previously (14,15). Briefly, the preadipocytes were grown to confluence in DMEM medium containing 10% FCS. Subsequently, differentiation of these preadipocytes was induced by changing medium to DMEM medium containing 10% FBS and a mixture of 175 nM insulin, 1 μM dexamethasone, and 0.5 mM isobutyl-1-methylxanthine. After 4 days in this differentiation medium, cells were then changed to medium containing 10% FBS without the differentiation mixture and replenished again 3 days later (15). The adipocyte-derived conditioned media (AdipoCM) was collected after 9–10 days post differentiation, filtered through a 0.22 μm filter and stored at 4°C.
Migration Assay
Migration assays were performed using Transwell 8-μm cell culture inserts (BD Falcon, 353097). Briefly, 30,000–40,000 cells/well were plated in serum-free medium (SFM) on Transwell filter, and allowed to migrate to medium containing 10% FBS (control media) or AdipoCM. After 8 hours, cells from above the membrane were wiped with cotton swabs, and cells at the bottom were fixed in 10% formalin and stained with 0.05% crystal violet (CV). Cell migration was analyzed by counting cells using a bright field microscope (Leica) and ImageJ. F13A (0.5–4 ng/mL) and etomoxir (40 μM) was added to the cell culture insert at the time of plating.
In vivo Homing Assay
All animal studies were performed according to protocols reviewed and approved by the Institutional Animal Care and Use Committee at OUHSC. SKOV-3-APJ cells were labeled with CellTracker™ Red CMTPX Dye (Invitrogen, C34552), per manufacturers protocol. The labeled SKOV-3-APJ cells (4 × 106) were injected intraperitoneally in 8-to-10-week-old female athymic nude mice (Charles River). The omentum was removed 6 hours later, weighed and digested in 1% (v/v) NP-40 (Thermo Scientific, 85124), and fluorescence was measured using a plate reader (7).
Ex vivo Omental Adhesion Assay
Omenta were excised from 8-to-10-week-old female athymic nude mice and immediately attached to the membrane of Millicel culture inserts (EMD Millipore Sigma, PICM01250) using Cell-Tak cell and tissue adhesive (Corning™, 354240), as previously described (16). Briefly, 6 μL of the tissue adhesive was applied evenly on the membrane of the transwell insert. It was then air-dried and washed twice with 500 μL of sterile water. Each omentum was divided into two, weighed, and then placed on the transwell insert for a minute without media. The transwell inserts with attached omenta were then placed in a 12-well plate. One million OVCAR-5-APJ cells, labeled with CellTracker™ Green CMFDA Dye (Invitrogen, C7027) per manufacturer’s protocol, were suspended in 500 μL media and added to each insert. DMEM/F-12 media (2.5 mL) was placed around the transwell insert. After co-culturing with the cell suspension for 24 h at 37 °C in a 5% CO2 incubator, the omenta was removed and washed thrice with PBS. Images were then taken using a fluorescent microscope (Leica, CFD365-FX). Subsequently, omenta was digested in 1% (v/v) NP-40 (Thermo Scientific, 85124), and fluorescence was measured using a plate reader.
Invasion Assay
Invasion assays were performed using 8-μm Transwell cell culture inserts (BD Falcon, 353097), after coating the filters with 1:20 diluted Matrigel (Thermo Fisher Scientific, CB40230) in SFM. Cells (100,000/well) were plated on the Matrigel and allowed to invade the 10% FBS-medium (control media) or AdipoCM for 16 hours. F13A was added to the cells at the time of cell plating. Cell invasion was analyzed similar to Transwell migration assays.
Lipid Droplet Staining
Cells (20,000–30,000/ well) were seeded on sterile cover glasses in a 24-well plate and allowed to adhere overnight. Growth media was then replaced with either SFM or AdipoCM and cells were incubated for 24 hours. Cells were then fixed with 10% formalin and washed thrice with PBS. HCS LipidTOX™ Deep Red Neutral Lipid Stain (Invitrogen, H34477) was used to stain the lipid droplets per manufacturer’s protocol. Briefly, 500 μL LipidTOX, diluted in PBS (1:500) containing 0.01% saponin, was added to each well and incubated at room temperature for 45 minutes protected from light. The cells were washed with PBS followed by fixing with 10% formalin. Sytox green (Invitrogen, S7020) diluted in PBS (1:1000) was added and incubated for 2–3 minutes at room temperature. The cells were washed with PBS and the cover glass were transferred to slides by placing on top of mounting media (VECTASHIELD®, catalog no. H-1500). Images were taken using a fluorescence confocal microscope within 48 hours to avoid loss of signal.
Confocal Microscopy
Images were acquired as described previously (17), using the Nikon C1 confocal system on a Nikon TE2000U microscope with EZ-C1 (Version 3.6; Nikon) software. A minimum of five images per well from a minimum of three technical replicates were collected for each condition. Image collection parameters (neutral density filters, pinhole, and detector gains) were kept constant during image acquisition, to make reliable comparisons between different experimental conditions. Quantification of fluorescence intensity of lipid droplet stain was done as previously described (17). Briefly, 10 to 15 single-channel images were collected for each experimental condition. Total fluorescence intensity of each whole image (Field of view) was measured using the EZ-C1 software (Nikon) which was then normalized by the total number of cells per image to represent mean fluorescence intensity.
Quantitative Real-time Polymerase Chain Reaction
Total RNA was extracted using HP E.Z.N.A kit from Promega. cDNA was synthesized using Maxima cDNA synthesis kit (Thermofisher). qRT-PCR assays were performed using ssoFast Evagreen supermix (Bio-Rad). Analysis was performed using Bio-Rad CFX96. Primer sequences are listed in Supplementary Table S1.
ELISA Assay
The assay was performed following the manufacturer’s protocol for Extraction Free EIA Kit (Phoenix Pharmaceuticals, Burlingame, CA). In brief, 50 μL of conditioned media collected from pre-adipocytes and mature adipocytes was used, in duplicates. Concentration of apelin in the samples was determined from the standard curve of apelin ranging from 0.01–100 ng/ml.
Immunoblot Analysis
For the pre- and mature adipocytes, whole-cell lysates (WCL) were prepared in lysis buffer containing 20 mM HEPES, 1% Nonidet P-40, 2 mM EDTA, 10 mM sodium fluoride, 10 mM sodium pyrophosphate, 1 mM sodium orthovanadate, 1 mM molybdate, protease inhibitor mixture (Complete Mini EDTA-free protease inhibitor mixture, Roche), 1 mM PMSF, and 50 mM DTT [15].
For OvCa cell lines, RIPA (Sigma) lysis buffer was used to extract WCLs. Protein concentration was determined using DC Protein reagents (Bio-Rad). Equal amounts of lysates (15–30 μg) were electrophoresed and transferred to nitrocellulose membranes. Ponceau S Stain (Sigma) was used to stain total protein on membrane to obtain a nonspecific band. Membranes were blocked in 5% BSA in TBST for 1-hour post-transfer, and incubated overnight at 4°C with primary antibody. After secondary antibody incubation, membranes were analyzed using FluorChemFC2. The antibody sources and dilutions used are listed in Supplementary Table S2. Densitometry was performed using ImageJ.
Fatty Acid Oxidation (FAO) assay
Cells (15,000–20,000/well) were seeded in a 96-well microplate and incubated overnight to form a monolayer. Growth media was aspirated and the cells were cultured in nutrient limited media (DMEM, 0.5 mM glucose, 1mM GlutaMax, 0.5mM carnitine and 1%FBS) for 24 hours. Forty-five minutes prior to the assay, cells were washed with FAO assay media (KHB media supplemented with 2.5 mM glucose, 0.5 mM carnitine, and 5mM HEPES, pH 7.4). FAO assay media (135 μL) was added to each well. Oxygen consumption rate (OCR) was determined using Seahorse Extracellular Flux Analyzer XFe96 (Agilent, Santa Clara, CA). Injections of 2 μM oligomycin, 4 μM Carbonyl cyanide-4 (trifluoromethoxy) phenylhydrazone (FCCP), or 1 μM rotenone/ antimycin A were added to each well. Fifteen minutes prior to start of the assay, etomoxir (40 μM) was added to the designated wells to inhibit carnitine palmitoyl transferase 1 (CPT1) and incubated at 37°C in a non-CO2 incubator. Prior to start of assay, 30 μL of palmitate-BSA conjugate or BSA alone (Agilent Technologies, 102720–100) was added to the wells, according to the group assignment. The microplate was then inserted in the XFe96 Analyzer and protocol for XF Cell Mito Stress Test was run.
Mitochondrial Fuel Flexibility Assay
Mito Fuel Flex Test was performed using XFe96 Bioanalyzer (Agilent) wherein import of three major metabolic substrates (pyruvate, fatty acids, and/ or glutamine) is inhibited by using mitochondrial pyruvate carrier inhibitor UK5099 (2 μM; Sigma-Aldrich, PZ0160), CPT-1a inhibitor etomoxir (4 μM), or glutaminase inhibitor BPTES (3 μM; Sigma-Aldrich, SML0601). This test helps to determine cellular dependence and flexibility for each metabolite to fuel mitochondrial metabolism. Briefly, 15,000 cells/well were plated in the 96-well microplate and incubated overnight to form a monolayer. Following day, growth media was replaced with assay media (XF DMEM media containing 1 mM pyruvate, 2 mM glutamine, and 10 mM glucose). The cells were incubated in a non-CO2 incubator at 37°C for 1 h prior to the assay. The inhibitors were loaded on to the hydrated sensor cartridge according to the group assignments. The microplate was then inserted in the XFe96 Analyzer and protocol for XF Mito Fuel Flex Test was run.
Statistical Analysis
All data were expressed as mean ± SEM. Comparison between two groups were analyzed by t-test. Multiple groups and/or multiple time points were analyzed using ANOVA (GraphPad Prism version 8.0, San Diego, CA, USA), or repeated measures ANOVA (time × groups). Spearman correlation and Pearson correlation method were used to analyze The Cancer Genome Atlas (TCGA) dataset. A P value of <0.05 was denoted as statistical significance.
RESULTS
Adipocyte-derived apelin promotes OvCa cell homing-in and migration
To test the hypothesis that OvCa tumor cells with increased APJ expression/ activation show increased omental metastasis, we performed a homing study in-vivo. We observed a 4.5-fold increase in ‘homing-in’ of SKOV-3-APJ high expression tumor cells to the omentum compared to the empty vector control cells (Supplementary Fig. S1A). APJ is primarily activated by its endogenous ligand apelin, which is expressed and secreted by adipocytes (18). Therefore, we utilized conditioned media derived from differentiated 3T3-L1 mouse adipocytes to study the role of adipocyte-derived apelin-APJ activation in OvCa cell-adipocyte interaction. Adipocyte differentiation and maturation was confirmed by evaluating GLUT4 expression (Supplementary Fig. S1B). Using both western blot and ELISA, we first demonstrated that mature adipocytes express and secrete higher levels of apelin in their media compared to pre-adipocytes (Fig. 1A, B). This mature adipocyte-derived conditioned media (AdipoCM) containing apelin was used for subsequent in vitro experiments.
Fig 1. Apelin-derived from adipocytes promotes migration in APJ-dependent manner.
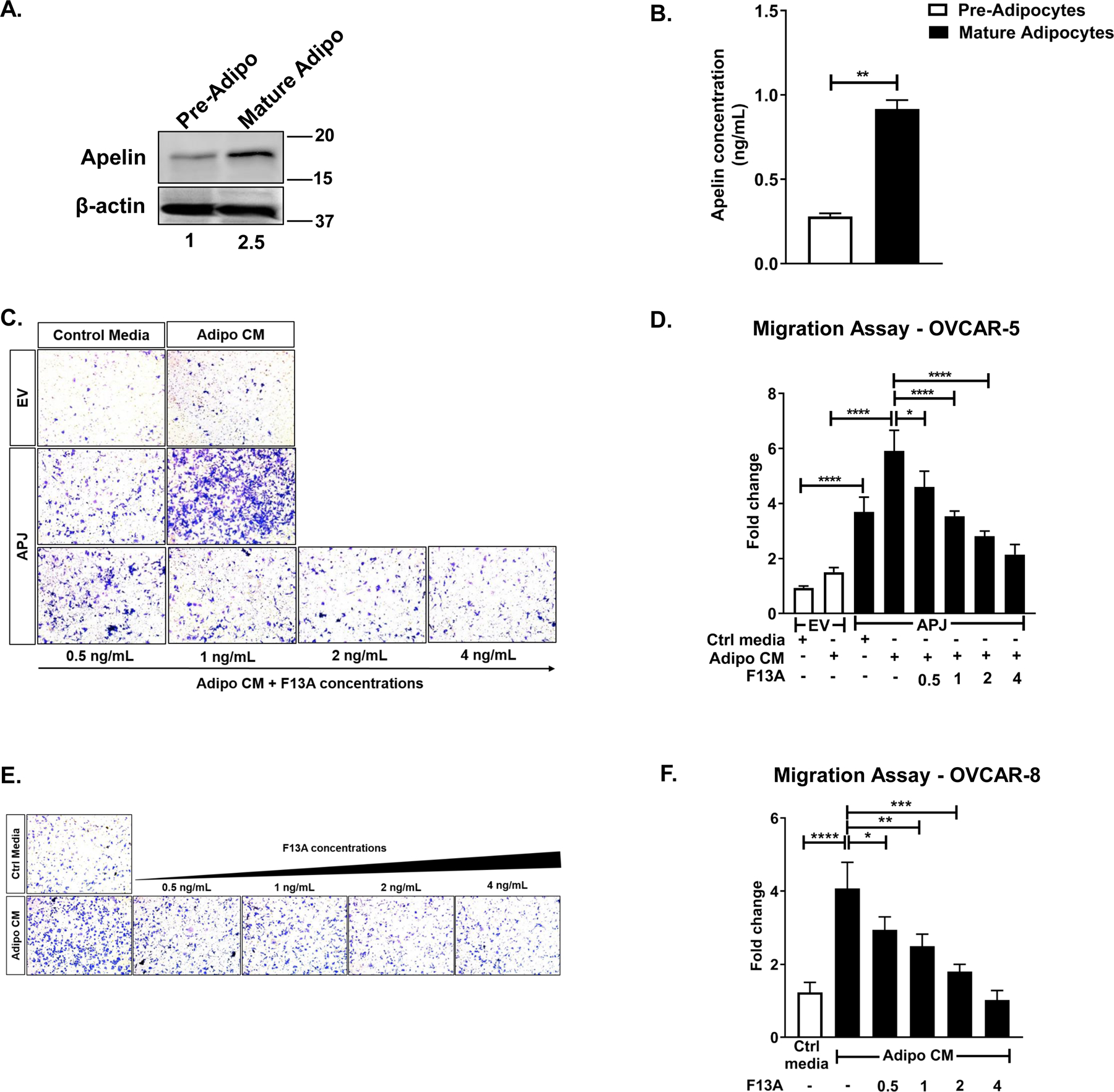
(A) Representative western blot for apelin performed on whole cell lysates obtained from pre-adipocytes and mature adipocytes. (B) Apelin secreted by pre-adipocytes and mature adipocytes into the medium quantified using ELISA. (C) Representative images of 8 hr-transwell migration assay performed in OVCAR-5-EV and APJ lines and (D) its quantification. (E) Representative images of 8 hr-transwell migration assay performed in OVCAR-8 cell and (F) its quantification. The cells were allowed to migrate to control media (10% FBS) or Adipocyte-conditioned media (Adipo CM), as indicated.
Results were obtained from 3 independent experiments (mean±SEM). Statistical analysis was performed using student’s t-test for B, and one-way ANOVA followed by Tukey post hoc test for D,F. *P< 0.05; **P< 0.01;***P<0.001; ****P<0.0001.
In order to mimic different steps involved in OvCa metastasis in vitro (19), we first performed a transwell migration assay using OVCAR-5 cells stably over-expressing APJ (OVCAR-5-APJ vs. its empty vector control OVCAR-5-EV) established in our laboratory and OVCAR-8 cells, which have high endogenous APJ expression (4). In the presence of control media (containing 10% FBS), OVCAR-5-APJ cells migrated more than the EV cells (Fig 1C, D), which was consistent with our previous study (4). When co-cultured in AdipoCM (collected from mature adipocytes), OVCAR-5-APJ cells showed further increased migration (Fig 1C, D) compared to EV cells. The magnitude of increase in cell migration was much greater in AdipoCM than in control medium. In case of OVCAR-8 cells, which endogenously express high levels of APJ, we observed a 4-fold increase in migration in presence of AdipoCM than in control medium (Fig 1E, F). As adipocytes secrete multiple inflammatory cytokines and proteins, e.g., IL-6, IL-8, leptin, adiponectin, which are also known to be pro-migratory (20), we used an APJ antagonist F13A to establish the fact that the observed increase was specific to APJ pathway. We found that the APJ inhibition reversed the increased migration in a dose-dependent manner in both OVCAR-5-APJ and OVCAR-8 cells, up to an extent similar to their corresponding controls (Fig 1D, F, Supplementary Fig. S1C). These data suggest that the observed phenotype is specific to the apelin-APJ pathway and adipocyte-derived apelin can act in a paracrine manner in OvCa cells to promote cell migration.
Apelin-APJ pathway promotes OvCa cell adhesion to omentum and invasion
OvCa cells, once detached, usually float in ascites as single cells or multicellular aggregates which interact with different adhesion molecules such as collagen types I and IV, fibronectin, and laminin in the basement membrane of peritoneal organs (21). To further investigate the role of apelin-APJ pathway in promoting OvCa cell adhesion to the omentum, we established a 3-dimensional ex vivo model of omental metastasis (16). This ex vivo metastasis model involves co-culture of the OvCa cells with omentum excised from mice under conditions which maintain physiological integrity of omental tissue. After 24 h incubation, we observed 4.5–5-fold increase in omental adhesion for OVCAR-5-APJ cells compared to EV cells (Fig 2A, B). This increase in adhesion was reversed by F13A treatment (Fig 2A, B), indicating the specificity to the apelin-APJ pathway.
Fig 2. Apelin-mediated APJ activation promotes omental adhesion & invasion.
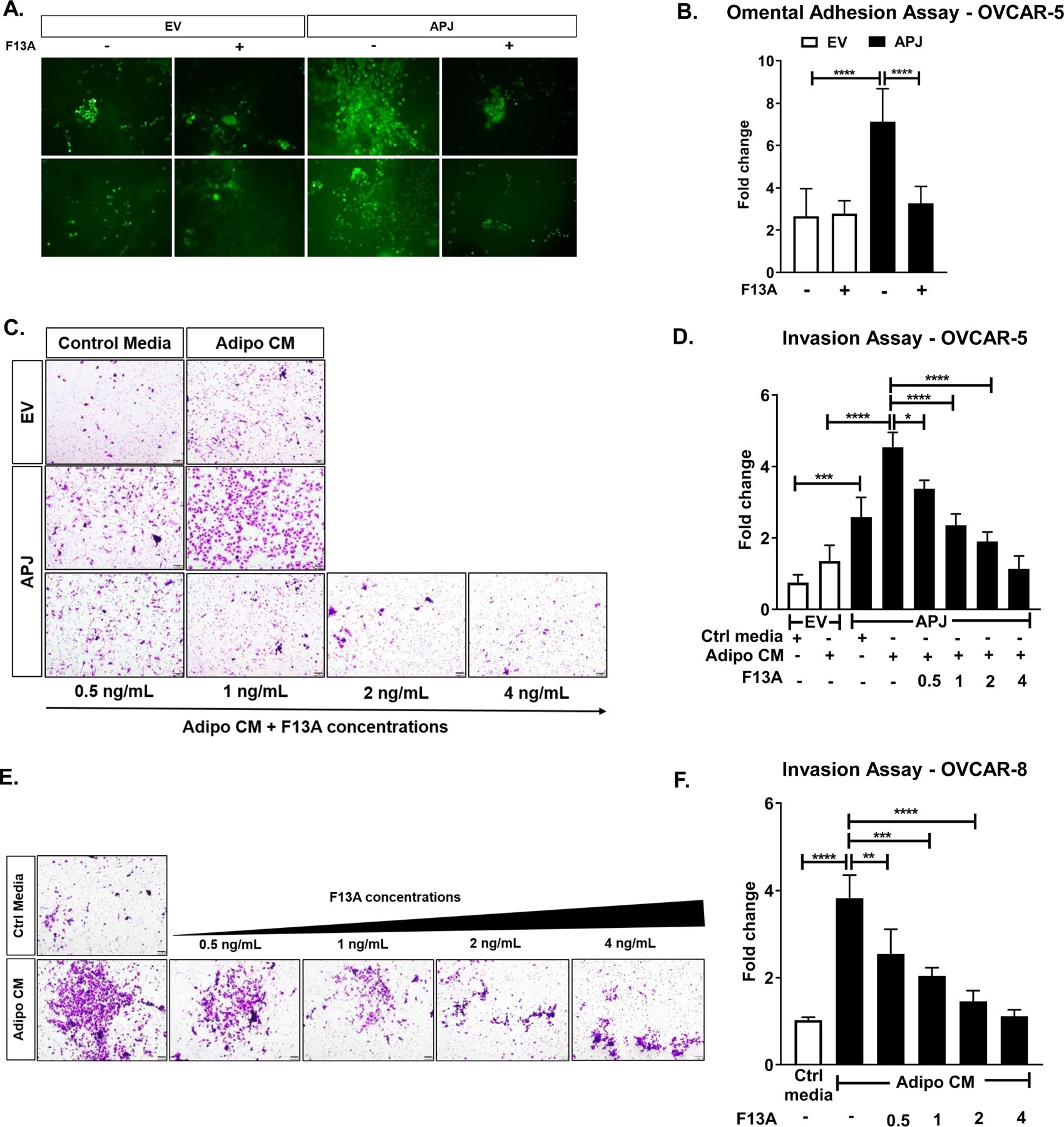
(A) Representative images of omental adhesion assay performed ex vivo in OVCAR-5-EV and APJ cell lines treated with F13A (4 ng/mL) and (B) its quantification. (C) Representative images of 16 hr-transwell invasion assay performed in OVCAR-5-EV and APJ cell and (D) its quantification. (E) Representative images of 16 hr-invasion assay performed in OVCAR-8 cell and (F) its quantification. The cells were allowed to invade towards control media (10% FBS) or Adipocyte-conditioned media (Adipo CM), as indicated.
Results were obtained from 2 independent experiments in A and 3 independent experiments in C,E (mean±SEM). Statistical analysis was performed using one-way ANOVA followed by Tukey post hoc test for B,D,F. *P< 0.05; **P< 0.01;***P<0.001; ****P<0.0001.
Next, using transwell chambers coated with matrigel, which mimics the in vivo basement membrane, we investigated the role of adipocyte-derived apelin in OvCa cell invasion. The assay was performed for 16 h to exclude the effects of APJ activation on cell proliferation. Compared to EV cells, OVCAR-5-APJ cells showed increased invasion in control medium (Fig 2C, D). In the presence of AdipoCM, the OVCAR-5-APJ cells also invaded significantly more compared to the EV cells (Fig 2C, D). Its magnitude was much higher under AdipoCM than control media. F13A treatment was able to reduce the invasion of OVCAR-5-APJ cells in a dose-dependent manner in presence of AdipoCM (Fig 2C, D). F13A at a dose of 4 ng/mL reduced cell invasion in both OVCAR-5-EV and APJ cells but to a much greater extent in the APJ high expression cells (Supplementary Fig. S1D). OVCAR-8 cells invaded more under AdipoCM than control media, and F13A was effective in blocking the apelin/APJ-induced invasion (Fig 2E, F). Together, these data indicate that APJ activation by adipocyte-derived apelin promotes omental adhesion and invasion of OvCa cells.
Adipocyte-derived apelin promotes lipid droplet accumulation in OvCa cells
Adipocytes can act as a source of energy-dense lipids to OvCa cells to support cell growth and metastasis (7). Further, abundant lipids have been observed at the adipocyte-cancer cell interface in tissues from ovarian cancer patients with omental metastasis (7), indicative of the important role of adipokines in cross-talk between tumor and fat cells. We hypothesized that APJ activation by adipocyte-derived apelin regulates lipid metabolism in tumor cells at the adipocyte-tumor cell interface. We investigated lipid droplet accumulation in OvCa cells with high APJ expression when co-cultured in AdipoCM. We found that OVCAR-5-APJ cells cultured in serum-free media (SFM) showed an increase in lipid droplets relative to the EV cells (Fig 3A, B). Furthermore, in the presence of AdipoCM the lipid droplet accumulation was further increased in APJ high expression cells relative to the control (Fig 3A, B). F13A treatment was effective in inhibiting lipid droplet accumulation in APJ high expression cells (Fig 3A, B). Similarly, under AdipoCM condition OVCAR-8 cells showed enhanced cytoplasmic lipid droplet accumulation which was reversed by F13A treatment (Fig 3C, D). Further, we used genetic perturbation of APJ using shRNA in OVCAR-8 cells (4). With APJ knockdown, we observed a significant decline in lipid droplet accumulation relative to the shNT controls when the cells were treated with AdipoCM (Supplementary Fig. S2A, B). APJ knockdown by itself did reduce the lipid droplets in SFM albeit not statistically significant (Supplementary Fig. S2B). Collectively, our data revealed that APJ pathway activation specifically enhances lipid droplet accumulation in OvCa cells.
Fig 3. Apelin/APJ Pathway Promotes Lipid Droplet Accumulation in OvCa cells.
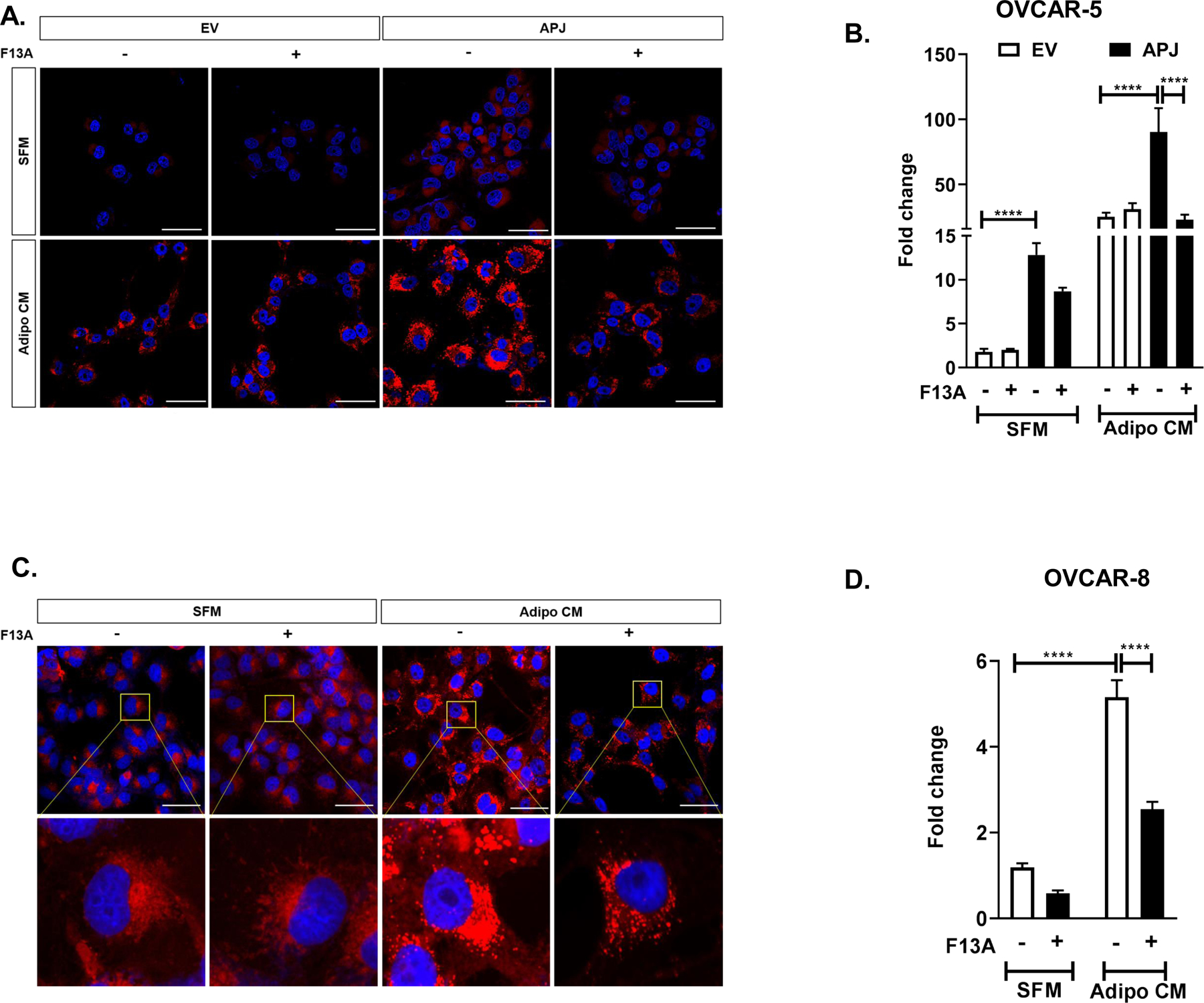
(A) Representative images of 24 hr lipid droplet accumulation in OVCAR-5-EV and APJ cells treated with F13A by confocal microscopy and (B) its quantification. (C) Representative images of 24 hr lipid droplet accumulation in OVCAR-8 cells treated with F13A by confocal microscopy and (D) its quantification. The cells were cultured either in serum free medium (SFM) or in adipocyte conditioned medium (Adipo CM) as indicated. F13A dose used in these experiments ranged from 10–25 ng/mL.
Results were obtained from 3 independent experiments (mean±SEM). Statistical analysis was performed using one-way ANOVA followed by Tukey post hoc test for B,D. ****P<0.0001. Scale bar: 50μm.
Apelin-APJ pathway promotes lipid uptake by upregulating CD36 via STAT3 activation
Fatty acids are absorbed by the cells either through passive diffusion or saturable, protein-facilitated transporters such as fatty acid transport proteins (FATP), plasma membrane fatty acid-binding protein (FABPpm), and CD36 (fatty acid translocase [FAT]) (22,23). To identify the mechanism by which APJ activation promotes lipid uptake, we investigated the expression levels of different fatty acid transporters, namely FABP-4 and CD36, in our APJ overexpression and knockdown systems. We found no correlation between APJ and FABP-4 expression, indicating that other players might be involved in regulation of FABP-4 in OvCa cells (Supplementary Fig. S3A, B).
OVCAR-5-APJ cells showed a significant increase in CD36 expression not only in AdipoCM but also under SFM conditions (Fig 4A, C) which could contribute to increased lipid droplets seen in these cells when cultured in SFM. This increase was inhibited by F13A treatment (Fig 4A, C). In OVCAR-8 cells we also observed a significant increase in CD36 expression which was reversed by F13A treatment (Fig 4B, D), indicating that the increase was specific to apelin-mediated activation of APJ. Additionally, we performed a time course study which showed that CD36 expression in OVCAR-5-APJ and OVCAR-8 cells increased over time with maximum levels reaching in 24 to 48 h (Supplementary Fig. S3C, D). With APJ knockdown (shAPJ-1 and shAPJ-2) in OVCAR-8 cells there was a decrease in CD36 expression compared to shNT cells (control) in presence of AdipoCM (Supplementary Fig. S3E).
Fig 4. CD36 is upregulated in OvCa cells via STAT3 activation in apelin/APJ-dependent manner.
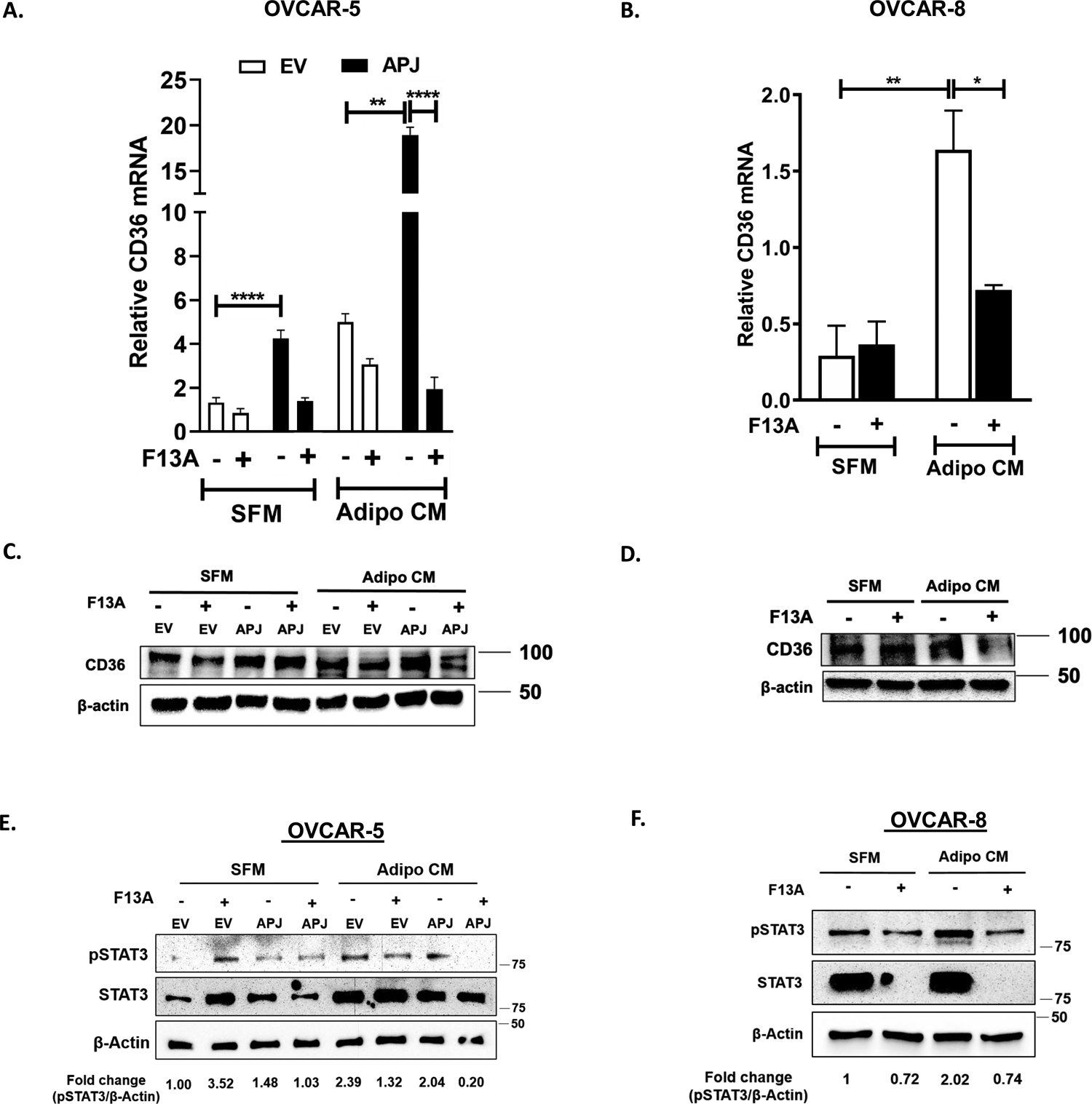
(A,B) qRT-PCR performed using cDNA obtained from (A) OVCAR-5-EV and APJ cells and (B) OVCAR-8 cells in the absence and presence of F13A treatment. (C, D) Western blots for CD36 performed on whole cell lysates (WCLs) obtained from (C) OVCAR-5-EV and APJ cells, and (D) OVCAR-8 cells. (E,F) Representative western blots and quantification for p-STAT3 and total STAT3 levels performed on WCLs obtained from (E) OVCAR-5-EV and APJ cells, and (F) OVCAR-8 cells. The cells were cultured either in serum free medium (SFM) or in adipocyte conditioned medium (Adipo CM) as indicated. F13A dose used in these experiments ranged from 10–25 ng/mL.
Results were obtained from 3 independent experiments (mean±SEM). Statistical analysis was performed using one-way ANOVA followed by Tukey post hoc test for A,B. *P< 0.05; **P< 0.01; ****P<0.0001.
A study has shown that STAT3 transcriptionally regulates CD36 gene expression in chronic lymphocytic leukemia cells by binding to the promoter region of the CD36 gene (24) and we have previously demonstrated that STAT3 is a downstream effector of APJ activation in OvCa cells (4). Thus, we determined whether CD36 expression is regulated via APJ mediated STAT3 activation. In the presence of AdipoCM, STAT3 and pSTAT3 levels were higher in both EV and APJ cells, compared to SFM conditions (Fig. 4E). The pSTAT3 level was dramatically reduced in the APJ cells upon treatment with F13A (Fig. 4E, Supplementary Fig. S3F). Interestingly, F13A had the opposite effect under SFM condition on EV cells, which showed an increase in pSTAT3 with F13A treatment. For OVCAR-8 cells, there was an increase in STAT3 phosphorylation under AdipoCM which was ameliorated by F13A treatment (Fig. 4F, Supplementary Fig. S3G). Of note, we were unable to detect total STAT3 levels in OVCAR-8 cells treated with F13A (Fig. 4F). Further, treatment with SSO (CD36 inhibitor) reduced lipid droplet accumulation in OVCAR-5-APJ cells (Supplementary Fig. S4A, B), confirming that the lipid accumulation was indeed mediated via CD36 in these cells. Lastly, we observed that the inhibition of STAT3 activation (stattic) prevented the increase in CD36 mRNA levels in OVCAR-5-APJ cells, compared to the control cells (Supplementary Fig. S4C). Collectively, our data demonstrates that APJ activation upregulates CD36 via STAT3 activation to facilitate fatty acid uptake from the extracellular environment.
APJ pathway activation promotes lipid utilization in OvCa cells
Recent studies have shown that apelin is implicated in modulating glucose and lipid metabolism (25–27). To determine the impact of APJ activation on energy generation in OvCa cells, we first tested the dependency and flexibility of OvCa cells in oxidizing nutrients by mitochondrial respiration. In OVCAR-5 cells, with high APJ expression we observed an increased dependency on fatty acid oxidation (FAO) compared to the EV cells (Fig. 5A). In addition, the cells were also dependent on glucose oxidation; however, this dependency was independent of APJ expression. Interestingly, we did not observe any dependency on glutamine for OVCAR-5 cell lines. With APJ high expression, the cells showed a significant increase in mitochondrial flexibility for FAO, but not for glucose or glutamine oxidation (Fig. 5B).
Fig 5. APJ pathway promotes fatty acid utilization in OvCa cells.
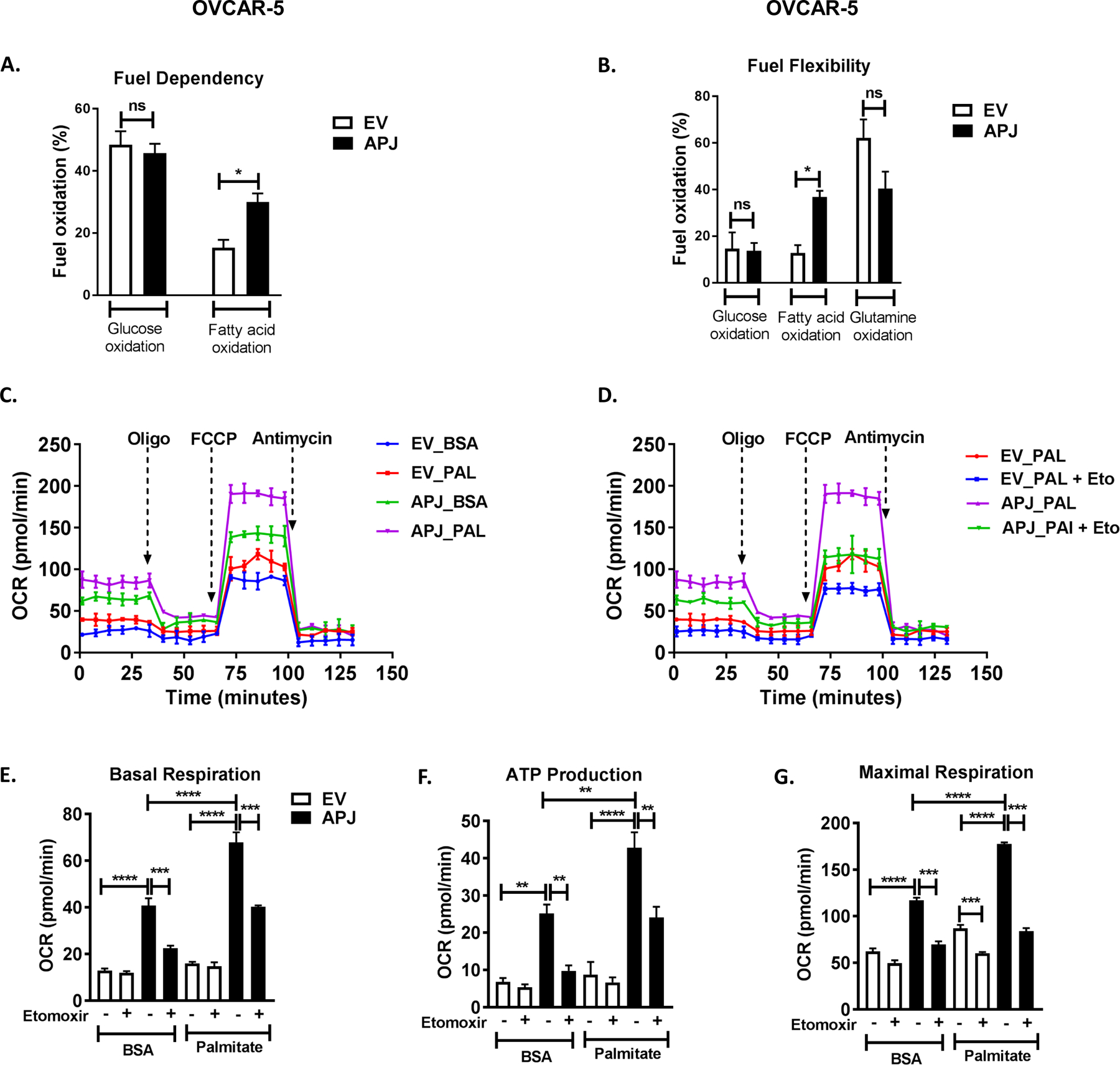
(A) Fuel dependency for oxidation of glucose, glutamine, and fatty acids in OVCAR-5-EV and APJ cells as quantified by Seahorse Metabolic Analyzer. (B) Fuel flexibility for oxidation of glucose, glutamine, and fatty acids in OVCAR-5-EV and APJ cells. (C,D) Oxygen consumption rate (OCR) in OVCAR-5-EV and APJ cells when treated with (C) BSA and palmitate, and (D) BSA and palmitate in presence of CPT1a inhibitor etomoxir (40 μM) as quantified by Seahorse Metabolic Analyzer. Quantification of (E) Basal respiration rates, (F) Mitochondrial ATP production rate (oligomycin [oligo]-sensitive respiration), and (G) Maximal respiration rate (induced by FCCP, uncoupler of mitochondrial oxidative phosphorylation [OXPHOS]) in OVCAR-5-APJ cells in presence of etomoxir (40 μM).
Results were obtained from 2 independent experiments (mean±SEM). Statistical analysis was performed using one-way ANOVA followed by Tukey post hoc test for A,B,E-G. *P< 0.05; **P< 0.01; ***P< 0.001; ****P<0.0001.
Tumor cells utilize fatty acids by FAO for ATP production and cell proliferation. The first and rate-limiting step in FAO is governed by a shuttle CPT1, which transports long chain fatty acids from the cytoplasm to the mitochondria (28). These fatty acids are utilized to generate acetyl CoA, which is eventually used via Krebs cycle to generate ATP. We hypothesized that APJ pathway regulates mitochondrial FAO in OvCa cells, contributing to the observed pro-metastatic phenotypes. To test this, we determined mitochondrial bioenergetics of OVCAR-5 cells, when treated with fatty acid substrate (palmitate conjugated to BSA) and CPT1a inhibitor, etomoxir. We observed that the palmitate addition increased basal respiration rate in EV cells, indicating that the cells were able to effectively utilize the exogenously added fatty acids (Fig. 5C, E). With abundant palmitates, the high APJ-expressing cells showed a much greater increase in their basal and maximal respiration rates and ATP production, indicating that the APJ pathway promotes fatty acid utilization in these cells (Fig. 5C, E, G). The inhibition of CPT1a using etomoxir, however, significantly decreased mitochondrial respiration and ATP production in OVCAR-5-APJ cells, indicating that these cells were able to utilize both endogenous and exogenous fatty acids (Fig. 5D–G). Further, APJ knockdown in OVCAR-8 cells showed a significant decrease in respiration (basal and maximal) and ATP production, indicating that APJ pathway is both, sufficient and necessary to modulate FAO in OvCa cells (Supplementary Fig. S5A–E).
Lipid oxidation/catabolism is being recognized as an important component of cancer metabolism along with aerobic glycolysis and lipogenesis (29,30). However, the role of lipid oxidation in promoting the metastatic cascade is largely unknown. Thus, as a proof-of-principle, we tested the effect of etomoxir (FAO inhibitor) on cell migration phenotype in OVCAR-5-APJ cells when cultured in AdipoCM. We observed that etomoxir treatment reduced cell migration in OVACR-5-APJ cells, compared to EV cells (Supplementary Fig. S6A, B), when used at a dose sufficient to inhibit FAO as demonstrated by FAO assay. Collectively, our data indicates that high APJ expression augments FAO in OvCa cells. This enhanced lipid oxidation in OvCa cells potentially contributes to the increased cell migration phenotype.
Activation of AMPK-CPT1a axis downstream of APJ mediates fatty acid oxidation
AMPK is an energy sensor of the cell and known to modulate FAO (31). Using a phosphokinase array, we have previously shown that AMPKα is activated downstream of APJ (4). AMPK upon activation is known to favor energy producing processes by inhibiting lipogenesis and activating lipid oxidation (32). This switch in metabolism is regulated by phosphorylation of acetyl-CoA carboxylase (ACC). AMPK phosphorylates ACC, which results in decrease in cellular malonyl-CoA. The increased malonyl-CoA inhibits CPT1, which is responsible for FA transport into the mitochondria for β-oxidation (33). Thus, we investigated whether AMPK-CPT1a is involved in the increase in FAO by APJ activation. Using western blot analyses, we observed increased AMPK activation (i.e., higher pAMPK levels) in OVCAR-5-APJ cells compared to EV cells when cultured in AdipoCM (Fig. 6A, Supplementary Fig. S7A). OVCAR-8 cells also showed increased pAMPK levels when cultured in AdipoCM compared to in SFM (Fig. 6B, Supplementary Fig. S7B). In both cell lines these increases were suppressed by F13A treatment (Fig. 6A, B, and Supplementary Fig. S7A, B), confirming that apelin-mediated activation of APJ increases AMPK activity. Further, we observed a positive correlation between CPT1a and APJ expression in our in vitro systems. Increased CPT1a mRNA levels were consistently observed in APJ high expression cells (OVCAR-5-APJ and OVCAR-8), as compared to their corresponding control (Fig. 6C, D). Pharmacological inhibition by F13A was able to reverse these changes (Fig. 6C, D). The significant decrease in CPT1a expression by APJ knockdown in OVACR-8 cells, as compared to control, further supports CPT1a regulation by APJ pathway (Supplementary Fig. S7C). Together, our data indicates that APJ pathway possibly regulates fatty acid utilization in OvCa cells by the activation of AMPK-CPT1a axis.
Fig 6. APJ activation promotes fatty acid utilization via AMPK-CPT1a pathway.
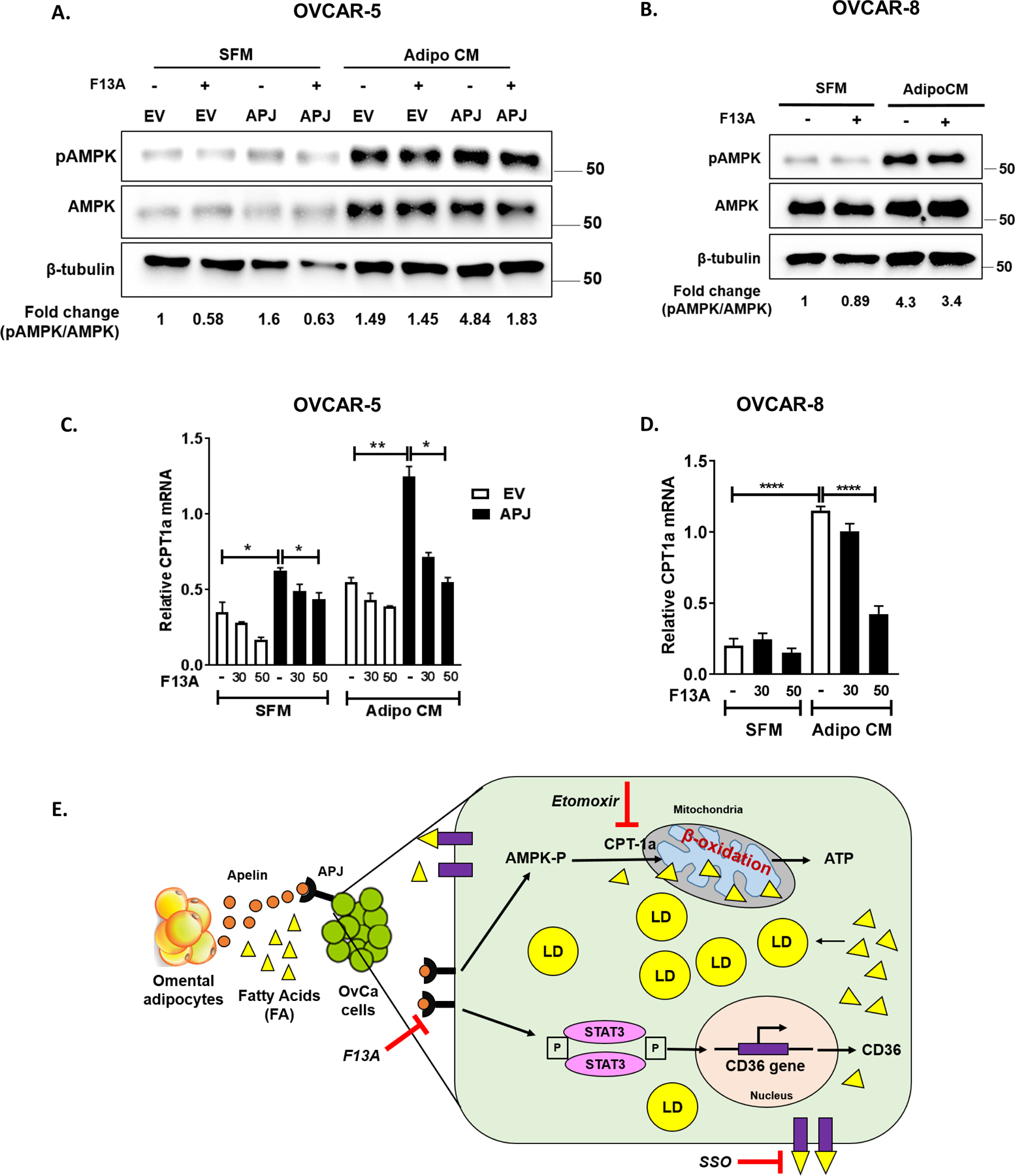
(A,B) Western blots performed for pAMPK and total AMPK levels on whole cell lysates (WCLs) obtained from (A) OVCAR-5-EV and APJ cells, and (B) OVCAR-8 cells in the absence and presence of F13A treatment. (C,D) qRT-PCR performed for CPT1a expression using cDNA obtained from (C) OVCAR-5-EV and APJ cells and (D) OVCAR-8 cells with F13A treatment. The cells were cultured either in serum free medium (SFM) or in adipocyte conditioned medium (Adipo CM) as indicated. F13A dose used in these experiments ranged from 10–50 ng/mL. (E) Graphical summary of the paper illustrating that adipocyte-derived apelin promotes metastasis by modulating fatty acid uptake and utilization in OvCa cells.
Results were obtained from 3 independent experiments (mean±SEM). Statistical analysis was performed using one-way ANOVA followed by Tukey post hoc test for C,D. *P< 0.05; **P< 0.01; ****P<0.0001.
In summary, our study demonstrated that adipocytes are a major source of apelin in the ovarian tumor microenvironment (TME) which activates APJ expressed predominantly on tumor cells. APJ activation in OvCa cells promotes fatty acid uptake via CD36 which are stored in lipid droplets. Apelin-mediated APJ activation also regulates β-oxidation of fatty acids in tumor cells which provides energy in for ATP for various cellular processes that promote tumor metastasis and progression (Fig. 6E).
DISCUSSION
Ovarian carcinoma is a serious health problem with the highest case-to-fatality ratio where majority (69%) of the patients succumb to their disease. This is mainly attributed to late-stage diagnosis (stage III and IV) wherein cancer cells have already metastasized to the peritoneum and/or distant organs with subsequent ascites formation (8). Tumorous transformation of omentum, an organ primarily composed of adipocytes, is one of the hallmark features of advanced HGSOC. Adipocytes of both subcutaneous and visceral origin have been shown to secrete chemokines and adipokines that not only supports OvCa progression but also enhances chemoresistance (34). Therefore, it is imperative to better understand the cellular and molecular mechanisms that govern adipocyte-OvCa cells interplay which can help develop novel treatment strategies, and compensate for the current, often ineffective standard-of-care treatments for OvCa.
Our previous work (4) showed that APJ is highly expressed on ovarian tumor cells and its increased activation promotes OvCa tumor growth and metastasis. We also identified that OvCa cells by themselves secrete apelin but at low concentrations (< 0.3 ng/ml) (4) as compared to the apelin level derived from mature adipocyte (Fig. 1B). In our current study, we demonstrated that adipocytes are an abundant source of apelin in the TME which can act in a paracrine manner to attract OvCa cells to the secondary metastasis sites. Using pharmacological and molecular inhibition of APJ and various model systems, we demonstrated that the increased pro-metastatic phenotypes of OvCa cells which include migration, omental adhesion, and invasion, were specific to activation of APJ by adipocyte-derived apelin.
In OvCa patients, the cancer cells at the adipocyte-cancer cell interface accumulate high amounts of lipids (7), and excessive lipid accumulation is a hallmark for cancer aggressiveness (35–37). Interestingly, we observed that activation of APJ modulates fatty acid uptake from extracellular environment, increasing lipid droplets in cancer cells. Several studies (7,38–40) have demonstrated that some cancer cells (e.g., ovarian, prostate, breast, etc.) scavenge lipids from the TME using different chaperones like FABP-4/5, CD36 etc. Our study shows that APJ expression consistently, positively correlated with CD36 expression but not with FABP-4. We observed that FABP-4 expression increased with increasing APJ expression and in presence of AdipoCM but with APJ knockdown FABP-4 had no significant changes relative to control (Supplementary Fig. S3A, B). This indicated that signaling pathways other than APJ might be involved in regulating FABP-4 expression in OvCa cells. In fact, miR-409–3p has been shown to be a negative regulator of FABP-4 in OvCa wherein miR-409–3p mimic significantly inhibited tumor metastasis in vivo (41).
Cancer cells are known to acquire lipids through two mechanisms i.e., uptake and de novo synthesis (lipogenesis) (42). Chen et al (43) demonstrated that OvCa cells upregulate fatty acid synthase which is a key enzyme in lipid biosynthesis. However, in our model systems we identified that increased CD36 expression contributed to the increased lipid droplets seen in APJ high expression cells both under SFM and AdipoCM condition. We further showed that APJ regulates CD36 expression levels via STAT3 phosphorylation. From gene expression profiles in TCGA, we found that APJ expression is correlated with CD36 and STAT3 expressions in serous ovarian cancers, particularly a strong positive relationship with CD36 (Supplementary Fig. S8), which is in line with our findings. Interestingly, we also observed increase in STAT3 protein in presence of AdipoCM in OVCAR-5 cells, which is consistent with the previous findings wherein STAT3 mRNA and protein levels increased in breast cancer cell lines under AdipoCM (44); however, the underlying mechanism is unclear. On the other hand, OVCAR-8 cells showed an inconsistent pattern wherein we were unable to detect STAT3 with F13A treatment in both SFM and AdipoCM conditions. This could be attributed to either post-translational modifications or alteration of protein stability in these cells following F13A treatment.
The role of lipid catabolism in the development and maintenance of malignant phenotypes in cancer is being slowly recognized (45,46). Studies show that cancer cells with this “lipolytic phenotype” rely on fatty acid oxidation for metastatic progression, survival, stemness and drug resistance (47–49). Our study reveals that APJ high expression cells were able to effectively utilize both exogenous and endogenous fatty acids via mitochondrial fatty acid oxidation to generate ATP, when bioenergetic stress is placed on the cells. This increase was abrogated by using etomoxir (fatty acid oxidation inhibitor) (50,51) and also in APJ knockdown cells. Our data shows that APJ pathway modulates fatty acid oxidation in OvCa cells; however, the importance of the use of oxidative substrates other than fatty acids, like glucose or glutamine is still under investigation.
Further, we found that apelin-derived from adipocytes was able to activate AMPK and increase CPT1a expression in OvCa cells, indicating that the increased fatty acid utilization by APJ activation was possibly mediated via the activation of AMPK-CPT1a axis. AMPK is a key energy sensor of the cell and its role in regulating fatty acid oxidation is well-recognized (52). Our findings are similar to the previous study where acute and chronic apelin treatment promoted AMPK-dependent fatty acid oxidation in obese and insulin-resistant mice (26). F13A was able to inhibit AMPK activation and CPT1a upregulation but higher doses of the drug were required, indicating a differential sensitivity of F13A. In our studies, we observed antagonistic effects with F13A treatment, though some studies have occasionally described partial agonistic activity of F13A in the absence of apelin (53–55). This can possibly explain the increased pSTAT3 and pAMPK levels we observed in EV cells when treated with F13A under the SFM condition. As APJ is a G protein coupled receptor, it can form homo- and/or heterodimers in different cell systems which could affect ligand binding and receptor activation and possibly explain the contradictory effects of F13A reported in different studies (56). More efficacious APJ antagonists such as MM54 are currently available and could be used instead in future studies. Our study have some limitations including the use of (1) differentiated mouse 3T3-L1 cells as a source AdipoCM instead of using human primary adipocytes, and (2) OVCAR-5 cells, which has recently been classified as metastatic gastrointestinal cancer (57), but our study indeed highlights the importance of APJ pathway as a viable target in OvCa metastasis and progression.
To our knowledge, our study is the first to demonstrate the role of adipokine apelin-APJ pathway in modulating lipid uptake and utilization, promoting metastasis of OvCa cells. Further studies are warranted to validate these findings in vivo to exploit these apelin-APJ mediated lipid metabolism-related dependencies in OvCa as a viable therapeutic strategy.
Supplementary Material
Implications:
Targeting the APJ pathway in HGSOC is a novel strategy to inhibit peritoneal metastasis.
ACKNOWLEDGEMENTS
The authors thank Dr. Muralidharan Jayaraman for his help with the metabolic experiments using Seahorse, and the OUHSC Functional Genomics Core for their technical assistance. This work was supported in part by research grants P20GM103639 (SW) from the National Institute of General Medical Sciences, NIH, DHHS, and Research Scholar Grant RSG-16-006-01-CCE (SW) from the American Cancer Society. The results shown here are in whole or part based upon data generated by the TCGA Research Network: https://www.cancer.gov/tcga.
Footnotes
COMPETING INTERESTS:
The authors have declared that no conflict of interest exists.
REFERENCES
- 1.Coburn SB, Bray F, Sherman ME, Trabert B. International patterns and trends in ovarian cancer incidence, overall and by histologic subtype. Int J Cancer 2017;140(11):2451–60 doi 10.1002/ijc.30676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Naora H, Montell DJ. Ovarian cancer metastasis: integrating insights from disparate model organisms. Nat Rev Cancer 2005;5(5):355–66 doi 10.1038/nrc1611. [DOI] [PubMed] [Google Scholar]
- 3.Torre LA, Trabert B, DeSantis CE, Miller KD, Samimi G, Runowicz CD, et al. Ovarian cancer statistics, 2018. CA Cancer J Clin 2018;68(4):284–96 doi 10.3322/caac.21456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Neelakantan D, Dogra S, Devapatla B, Jaiprasart P, Mukashyaka MC, Janknecht R, et al. Multifunctional APJ Pathway Promotes Ovarian Cancer Progression and Metastasis. Mol Cancer Res 2019;17(6):1378–90 doi 10.1158/1541-7786.MCR-18-0989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Yang Y, Lv SY, Ye W, Zhang L. Apelin/APJ system and cancer. Clin Chim Acta 2016;457:112–6 doi 10.1016/j.cca.2016.04.001. [DOI] [PubMed] [Google Scholar]
- 6.Motohara T, Masuda K, Morotti M, Zheng Y, El-Sahhar S, Chong KY, et al. An evolving story of the metastatic voyage of ovarian cancer cells: cellular and molecular orchestration of the adipose-rich metastatic microenvironment. Oncogene 2019;38(16):2885–98 doi 10.1038/s41388-018-0637-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Nieman KM, Kenny HA, Penicka CV, Ladanyi A, Buell-Gutbrod R, Zillhardt MR, et al. Adipocytes promote ovarian cancer metastasis and provide energy for rapid tumor growth. Nat Med 2011;17(11):1498–503 doi 10.1038/nm.2492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lengyel E Ovarian cancer development and metastasis. Am J Pathol 2010;177(3):1053–64 doi 10.2353/ajpath.2010.100105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Meza-Perez S, Randall TD. Immunological Functions of the Omentum. Trends Immunol 2017;38(7):526–36 doi 10.1016/j.it.2017.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Clark R, Krishnan V, Schoof M, Rodriguez I, Theriault B, Chekmareva M, et al. Milky spots promote ovarian cancer metastatic colonization of peritoneal adipose in experimental models. Am J Pathol 2013;183(2):576–91 doi 10.1016/j.ajpath.2013.04.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Maury E, Ehala-Aleksejev K, Guiot Y, Detry R, Vandenhooft A, Brichard SM. Adipokines oversecreted by omental adipose tissue in human obesity. Am J Physiol Endocrinol Metab 2007;293(3):E656–65 doi 10.1152/ajpendo.00127.2007. [DOI] [PubMed] [Google Scholar]
- 12.Vona-Davis L, Gibson LF. Adipocytes as a critical component of the tumor microenvironment. Leuk Res 2013;37(5):483–4 doi 10.1016/j.leukres.2013.01.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Himbert C, Delphan M, Scherer D, Bowers LW, Hursting S, Ulrich CM. Signals from the Adipose Microenvironment and the Obesity-Cancer Link-A Systematic Review. Cancer Prev Res (Phila) 2017;10(9):494–506 doi 10.1158/1940-6207.CAPR-16-0322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Eyster CA, Duggins QS, Olson AL. Expression of constitutively active Akt/protein kinase B signals GLUT4 translocation in the absence of an intact actin cytoskeleton. J Biol Chem 2005;280(18):17978–85 doi 10.1074/jbc.M409806200. [DOI] [PubMed] [Google Scholar]
- 15.Jackson RM, Griesel BA, Gurley JM, Szweda LI, Olson AL. Glucose availability controls adipogenesis in mouse 3T3-L1 adipocytes via up-regulation of nicotinamide metabolism. J Biol Chem 2017;292(45):18556–64 doi 10.1074/jbc.M117.791970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lungchukiet P, Sun Y, Kasiappan R, Quarni W, Nicosia SV, Zhang X, et al. Suppression of epithelial ovarian cancer invasion into the omentum by 1alpha,25-dihydroxyvitamin D3 and its receptor. J Steroid Biochem Mol Biol 2015;148:138–47 doi 10.1016/j.jsbmb.2014.11.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Patel MM, Behar AR, Silasi R, Regmi G, Sansam CL, Keshari RS, et al. Role of ADTRP (Androgen-Dependent Tissue Factor Pathway Inhibitor Regulating Protein) in Vascular Development and Function. J Am Heart Assoc 2018;7(22):e010690 doi 10.1161/JAHA.118.010690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Boucher J, Masri B, Daviaud D, Gesta S, Guigne C, Mazzucotelli A, et al. Apelin, a newly identified adipokine up-regulated by insulin and obesity. Endocrinology 2005;146(4):1764–71 doi 10.1210/en.2004-1427. [DOI] [PubMed] [Google Scholar]
- 19.Thibault B, Castells M, Delord JP, Couderc B. Ovarian cancer microenvironment: implications for cancer dissemination and chemoresistance acquisition. Cancer Metastasis Rev 2014;33(1):17–39 doi 10.1007/s10555-013-9456-2. [DOI] [PubMed] [Google Scholar]
- 20.Duong MN, Geneste A, Fallone F, Li X, Dumontet C, Muller C. The fat and the bad: Mature adipocytes, key actors in tumor progression and resistance. Oncotarget 2017;8(34):57622–41 doi 10.18632/oncotarget.18038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Yeung TL, Leung CS, Yip KP, Au Yeung CL, Wong ST, Mok SC. Cellular and molecular processes in ovarian cancer metastasis. A Review in the Theme: Cell and Molecular Processes in Cancer Metastasis. Am J Physiol Cell Physiol 2015;309(7):C444–56 doi 10.1152/ajpcell.00188.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Abumrad NA, Sfeir Z, Connelly MA, Coburn C. Lipid transporters: membrane transport systems for cholesterol and fatty acids. Curr Opin Clin Nutr Metab Care 2000;3(4):255–62. [DOI] [PubMed] [Google Scholar]
- 23.Abumrad N, Coburn C, Ibrahimi A. Membrane proteins implicated in long-chain fatty acid uptake by mammalian cells: CD36, FATP and FABPm. Biochim Biophys Acta 1999;1441(1):4–13 doi 10.1016/s1388-1981(99)00137-7. [DOI] [PubMed] [Google Scholar]
- 24.Rozovski U, Harris DM, Li P, Liu Z, Jain P, Ferrajoli A, et al. STAT3-activated CD36 facilitates fatty acid uptake in chronic lymphocytic leukemia cells. Oncotarget 2018;9(30):21268–80 doi 10.18632/oncotarget.25066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Bertrand C, Valet P, Castan-Laurell I. Apelin and energy metabolism. Front Physiol 2015;6:115 doi 10.3389/fphys.2015.00115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Attane C, Foussal C, Le Gonidec S, Benani A, Daviaud D, Wanecq E, et al. Apelin treatment increases complete Fatty Acid oxidation, mitochondrial oxidative capacity, and biogenesis in muscle of insulin-resistant mice. Diabetes 2012;61(2):310–20 doi 10.2337/db11-0100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Attane C, Daviaud D, Dray C, Dusaulcy R, Masseboeuf M, Prevot D, et al. Apelin stimulates glucose uptake but not lipolysis in human adipose tissue ex vivo. J Mol Endocrinol 2011;46(1):21–8 doi 10.1677/JME-10-0105. [DOI] [PubMed] [Google Scholar]
- 28.Duman C, Yaqubi K, Hoffmann A, Acikgoz AA, Korshunov A, Bendszus M, et al. Acyl-CoA-Binding Protein Drives Glioblastoma Tumorigenesis by Sustaining Fatty Acid Oxidation. Cell Metab 2019;30(2):274–89 e5 doi 10.1016/j.cmet.2019.04.004. [DOI] [PubMed] [Google Scholar]
- 29.Schlaepfer IR, Rider L, Rodrigues LU, Gijon MA, Pac CT, Romero L, et al. Lipid catabolism via CPT1 as a therapeutic target for prostate cancer. Mol Cancer Ther 2014;13(10):2361–71 doi 10.1158/1535-7163.MCT-14-0183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.van Weverwijk A, Koundouros N, Iravani M, Ashenden M, Gao Q, Poulogiannis G, et al. Metabolic adaptability in metastatic breast cancer by AKR1B10-dependent balancing of glycolysis and fatty acid oxidation. Nat Commun 2019;10(1):2698 doi 10.1038/s41467-019-10592-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hardie DG, Ross FA, Hawley SA. AMPK: a nutrient and energy sensor that maintains energy homeostasis. Nat Rev Mol Cell Biol 2012;13(4):251–62 doi 10.1038/nrm3311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Wang W, Guan KL. AMP-activated protein kinase and cancer. Acta Physiol (Oxf) 2009;196(1):55–63 doi 10.1111/j.1748-1716.2009.01980.x. [DOI] [PubMed] [Google Scholar]
- 33.Ke R, Xu Q, Li C, Luo L, Huang D. Mechanisms of AMPK in the maintenance of ATP balance during energy metabolism. Cell Biol Int 2018;42(4):384–92 doi 10.1002/cbin.10915. [DOI] [PubMed] [Google Scholar]
- 34.Yang J, Zaman MM, Vlasakov I, Roy R, Huang L, Martin CR, et al. Adipocytes promote ovarian cancer chemoresistance. Sci Rep 2019;9(1):13316 doi 10.1038/s41598-019-49649-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Yue S, Li J, Lee SY, Lee HJ, Shao T, Song B, et al. Cholesteryl ester accumulation induced by PTEN loss and PI3K/AKT activation underlies human prostate cancer aggressiveness. Cell Metab 2014;19(3):393–406 doi 10.1016/j.cmet.2014.01.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Bozza PT, Viola JP. Lipid droplets in inflammation and cancer. Prostaglandins Leukot Essent Fatty Acids 2010;82(4–6):243–50 doi 10.1016/j.plefa.2010.02.005. [DOI] [PubMed] [Google Scholar]
- 37.Abramczyk H, Surmacki J, Kopec M, Olejnik AK, Lubecka-Pietruszewska K, Fabianowska-Majewska K. The role of lipid droplets and adipocytes in cancer. Raman imaging of cell cultures: MCF10A, MCF7, and MDA-MB-231 compared to adipocytes in cancerous human breast tissue. Analyst 2015;140(7):2224–35 doi 10.1039/c4an01875c. [DOI] [PubMed] [Google Scholar]
- 38.Ladanyi A, Mukherjee A, Kenny HA, Johnson A, Mitra AK, Sundaresan S, et al. Adipocyte-induced CD36 expression drives ovarian cancer progression and metastasis. Oncogene 2018;37(17):2285–301 doi 10.1038/s41388-017-0093-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.DeFilippis RA, Chang H, Dumont N, Rabban JT, Chen YY, Fontenay GV, et al. CD36 repression activates a multicellular stromal program shared by high mammographic density and tumor tissues. Cancer Discov 2012;2(9):826–39 doi 10.1158/2159-8290.CD-12-0107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Watt MJ, Clark AK, Selth LA, Haynes VR, Lister N, Rebello R, et al. Suppressing fatty acid uptake has therapeutic effects in preclinical models of prostate cancer. Sci Transl Med 2019;11(478) doi 10.1126/scitranslmed.aau5758. [DOI] [PubMed] [Google Scholar]
- 41.Gharpure KM, Pradeep S, Sans M, Rupaimoole R, Ivan C, Wu SY, et al. FABP4 as a key determinant of metastatic potential of ovarian cancer. Nat Commun 2018;9(1):2923 doi 10.1038/s41467-018-04987-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Cheng C, Geng F, Cheng X, Guo D. Lipid metabolism reprogramming and its potential targets in cancer. Cancer Commun (Lond) 2018;38(1):27 doi 10.1186/s40880-018-0301-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Chen RR, Yung MMH, Xuan Y, Zhan S, Leung LL, Liang RR, et al. Targeting of lipid metabolism with a metabolic inhibitor cocktail eradicates peritoneal metastases in ovarian cancer cells. Commun Biol 2019;2:281 doi 10.1038/s42003-019-0508-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Gyamfi J, Lee YH, Min BS, Choi J. Niclosamide reverses adipocyte induced epithelial-mesenchymal transition in breast cancer cells via suppression of the interleukin-6/STAT3 signalling axis. Sci Rep 2019;9(1):11336 doi 10.1038/s41598-019-47707-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kuo CY, Ann DK. When fats commit crimes: fatty acid metabolism, cancer stemness and therapeutic resistance. Cancer Commun (Lond) 2018;38(1):47 doi 10.1186/s40880-018-0317-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Ma Y, Temkin SM, Hawkridge AM, Guo C, Wang W, Wang XY, et al. Fatty acid oxidation: An emerging facet of metabolic transformation in cancer. Cancer Lett 2018;435:92–100 doi 10.1016/j.canlet.2018.08.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Pike LS, Smift AL, Croteau NJ, Ferrick DA, Wu M. Inhibition of fatty acid oxidation by etomoxir impairs NADPH production and increases reactive oxygen species resulting in ATP depletion and cell death in human glioblastoma cells. Biochim Biophys Acta 2011;1807(6):726–34 doi 10.1016/j.bbabio.2010.10.022. [DOI] [PubMed] [Google Scholar]
- 48.Shao H, Mohamed EM, Xu GG, Waters M, Jing K, Ma Y, et al. Carnitine palmitoyltransferase 1A functions to repress FoxO transcription factors to allow cell cycle progression in ovarian cancer. Oncotarget 2016;7(4):3832–46 doi 10.18632/oncotarget.6757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Pascual G, Avgustinova A, Mejetta S, Martin M, Castellanos A, Attolini CS, et al. Targeting metastasis-initiating cells through the fatty acid receptor CD36. Nature 2017;541(7635):41–5 doi 10.1038/nature20791. [DOI] [PubMed] [Google Scholar]
- 50.Raud B, Roy DG, Divakaruni AS, Tarasenko TN, Franke R, Ma EH, et al. Etomoxir Actions on Regulatory and Memory T Cells Are Independent of Cpt1a-Mediated Fatty Acid Oxidation. Cell Metab 2018;28(3):504–15 e7 doi 10.1016/j.cmet.2018.06.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Yao CH, Liu GY, Wang R, Moon SH, Gross RW, Patti GJ. Identifying off-target effects of etomoxir reveals that carnitine palmitoyltransferase I is essential for cancer cell proliferation independent of beta-oxidation. PLoS Biol 2018;16(3):e2003782 doi 10.1371/journal.pbio.2003782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Thomson DM, Winder WW. AMP-activated protein kinase control of fat metabolism in skeletal muscle. Acta Physiol (Oxf) 2009;196(1):147–54 doi 10.1111/j.1748-1716.2009.01973.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Yang P, Kuc RE, Brame AL, Dyson A, Singer M, Glen RC, et al. [Pyr(1)]Apelin-13(1–12) Is a Biologically Active ACE2 Metabolite of the Endogenous Cardiovascular Peptide [Pyr(1)]Apelin-13. Front Neurosci 2017;11:92 doi 10.3389/fnins.2017.00092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Fan X, Zhou N, Zhang X, Mukhtar M, Lu Z, Fang J, et al. Structural and functional study of the apelin-13 peptide, an endogenous ligand of the HIV-1 coreceptor, APJ. Biochemistry 2003;42(34):10163–8 doi 10.1021/bi030049s. [DOI] [PubMed] [Google Scholar]
- 55.Medhurst AD, Jennings CA, Robbins MJ, Davis RP, Ellis C, Winborn KY, et al. Pharmacological and immunohistochemical characterization of the APJ receptor and its endogenous ligand apelin. J Neurochem 2003;84(5):1162–72 doi 10.1046/j.1471-4159.2003.01587.x. [DOI] [PubMed] [Google Scholar]
- 56.Chaves-Almagro C, Castan-Laurell I, Dray C, Knauf C, Valet P, Masri B. Apelin receptors: From signaling to antidiabetic strategy. Eur J Pharmacol 2015;763(Pt B):149–59 doi 10.1016/j.ejphar.2015.05.017. [DOI] [PubMed] [Google Scholar]
- 57.Blayney JK, Davison T, McCabe N, Walker S, Keating K, Delaney T, et al. Prior knowledge transfer across transcriptional data sets and technologies using compositional statistics yields new mislabelled ovarian cell line. Nucleic Acids Res 2016;44(17):e137 doi 10.1093/nar/gkw578. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.