

Abstract
The reproducibility of chemical reactions, when obtaining protocols from literature or databases, is highly challenging for academicians, industry professionals and even now for the machine learning process. To synthesize the organic molecule under the photochemical condition, several years for the reaction optimization, highly skilled manpower, long reaction time etc. are needed, resulting in non-affordability and slow down the research and development. Herein, we have introduced the DigiChemTree backed with the artificial intelligence to auto-optimize the photochemical reaction parameter and synthesizing the on demand library of the molecules in fast manner. Newly, auto-generated digital code was further tested for the late stage functionalization of the various active pharmaceutical ingredient.
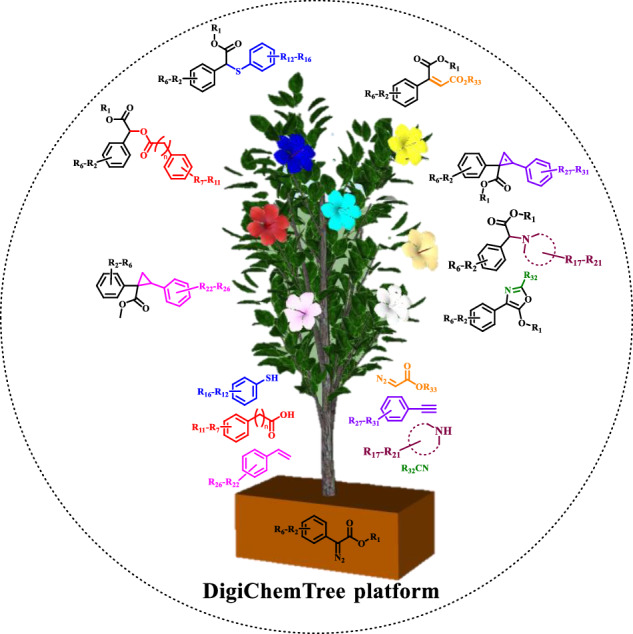
Subject terms: Light harvesting, Automation
Light-induced reactions of diazo compounds have become crucial in organic synthesis and drug discovery, however, optimization of reaction conditions is still very time-consuming. Here, the authors develop a DigiChemTree platform using artificial intelligence to auto-optimize the photochemical reaction parameters and rapidly synthesize an on-demand library of molecules.
Introduction
Light-induced reactions of diazo compound become crucial in organic synthesis and drug discovery1–4, facilitating fresh transformations and opening up unexplored chemical space for the biologically active molecule5. Highly reactive diazo compounds can undergo various chemical transformations, including C–H6, O–H7, Si–H8,9, N–H10,11, S–H bonds insertion12,13, Wolff rearranged product formation14, cyclopropanation15,16, cyclopropenation17, cycloaddition reaction17,18, etc. In the early phases of research, thermal-assisted conditions were commonly used to generate carbenes14. However, these conditions often resulted in low reactivity and selectivity, which hindered process development. Furthermore, the introduction of transition metal carbene has transformed the scenario due to their tunable electronic properties19 and stability (Scheme 1b)20–22 Moreover, strong coordination with expensive metal carbenoid23 and additional impurity generation limits the process for the molecule generation.
Scheme 1.

DigiChemTree: automated reaction optimization for accelerating the late-stage functionalization of biologically active molecules. 1a Biologically active compound, 1b Previously reported protocols for carbene generation, 1c Proposal of DigiChemTree.
Photochemical studies with diazo compounds have a rich legacy in organic synthesis24, primarily flourishing within academic research sectors1,25–28, with relatively limited application in industrial settings until recent times29,30. Light-induced carbene chemistry faces several challenges, including the need for high-energy UV irradiation31, costly monochromator filters, susceptibility to numerous side reactions, harsh reaction conditions, low quantum efficiency, and the requirement for specialized batch or flow reactors32 with significant spacing, thereby restricting the synthetic utility of these methods. On the other hand, many academic researchers, engineers, and professors tend to focus on intuition-driven time-consuming batch process experimentation, such as reaction discovery and exploring substrate scopes (12,523 molecules in last 60 years, specially in diazo acetate, see the supplementary data table S3).
However, numerous numerical parameters, such as light intensity, temperature, pressure, reaction time, quantum efficiency, molar concentration of substrate, catalyst loading, and reagent equivalents, as well as categorical parameters like additives, solvents, light sources, and reactor types, are essential for developing methodologies favorable for reproducible industrial applications33. But in a conventional laboratory an organic chemist can access only a limited subset of these parameters due to constrain of time and availability of material. In response to the challenge of rapidly optimizing reaction conditions34, we sought to develop a machine-based platform that facilitates self-optimization, intensification, and on-demand of chemical transformation (Scheme 1c)35. Recently, the reaction auto-optimization platform opens the new era for academia and industry to optimize the multivariable reaction condition at a time for a reaction36,37.
To resolve the above-mentioned industrial favorable methodology issues, we thought to employ the integration of continuous flow reactions38–40 backed with artificial intelligence (AI) technology41–49. We report DigiChemTree for the automated optimization of light-induced carbene generation and its coupling with various nucleophiles (O-H, S-H, N-H), cycloaddition with alkenes, alkynes, C≡N, and cross-coupling with ethyl diazoester. The platform is also extended for late-stage functionalization and accelerated synthesis of innovative biologically active molecules50.
Results
Assembly of DigiChemTree and reaction auto-optimization
Our DigiChemTree platform is assembled with 9 liquid delivery pumps, PFA micro-flow tubular reactor, circular blue LED photo-light, controlled light power supply, affordable com port connectivity, 3D printer converted solution collector, and an in-line IR system. It uses a closed-loop BO approach to systematically explore the impact of light intensity and residence time on enhancing the efficiency of high-throughput flow reaction analysis51. Before conducting experiments, we conducted an In-line IR background analysis of each compound and further fed the data regarding the signature peak of the product moiety (details in supplementary methods).
We identified a distinctive alignment within the range of 1756 to 1775 cm−1 associated with compound 4a (C=O peak), designated as the signature peak for our analysis (details in supplementary data). We prepared a 0.1 M stock solution of compound 2a and a 0.2 M stock solution of reagent 3a, loaded into separate syringes connected to the syringe pump. After ensuring communication ports with attached instruments were operational, we set boundaries for the reaction mixture flow rate, power supply voltage, and current, and ran our accessible platform Python code (ab1, code in supplementary software 1). The experimental setup involved mixing the two solutions at a T-junction with the flow rate controlled by a Bayesian optimizer (BO). The resulting solution passed through the PFA tubular reactor in a stoichiometric ratio of 1:2 (compound 2a: reagent 3a), exposed to blue LED light at varied wattages. The closed-loop BO systematically explored these varying reaction conditions to maximize the yield percentage. Each optimization cycle comprised 62 experiments in a single day, with results tabulated in the optimization table (supplementary data Table S4). The initial phase involved three experiments, serving as a preliminary exploration (random search) of the reaction space. Subsequently, the BO algorithm suggested one new experimental condition at a time, aiming to maximize the yield percentage. We observed that at a flow rate of 50 μL min−1 each, a residence time of 10 min, and 68 ± 2 W under 3 bar pressure, we achieved a maximum conversion of 99% with a space-time yield of 0.0909 g mL−1 h−1 (Fig. 1a). Typically, the conventional batch process utilizes either metal catalyst52, or blue light24, with longer reaction time with a lower space-time yield 0.0007 g mL−1 h−1, which makes process unsuitable for the industrial applications. Given the broader scope of the carbene insertion reaction of diazo compounds in API synthesis (e.g., anti-tumor and anti-inflammatory agents) and chiral-enriched molecules, we are keen to leverage this platform to explore various substituted aromatic benzoic acids (3a-3o) and late-stage functionalization of bio-active molecules (3g-3j), resulting (4a-4p) in excellent yields 82-99%. Furthermore, a comprehensive literature survey revealed that an impressive 15 out of the 16 synthesized photo carboxylation products (4b-4p) are entirely recent additions compounds.
Fig. 1. Auto-optimization of various parameters for the carbene insertion reaction (code ab1–ab7).

a–c Stock solution 0.1: 0.2, 0.2 & 0.2 M of compound 2a: 3a, 5a, and 7a in EtOAc, respectively; exposure time (10.02, 5.7, & 11 min.) 1 mL reactor volume; d, e stock solution 0.1: 0.2, & 0.2 M of compound 2a: 9a & 11a, in DCE, respectively; exposure time (32 & 40 min) 4 mL reactor volume; f, g 0.1: 0.2, & 0.2 M of compound 2a, 13, & 15 in DCE; exposure time (16.6 & 12.3 min.); h represent the yield, 1 mL reactor volume; yields are based on standard IR analysis and cross-verified with crude NMR with ±1% error is acceptable; yield standard bar unit is same for all.
After identifying and navigated the complexity of optimal conditions for photochemical reactions with carboxylate nucleophiles, we were interested check the feasibility with slightly weak thiophenol (5a) nucleophile as a coupling partner. Slight modification of the reaction condition system navigated the optimal condition (ab2 code condition: residence time 5.3 min., exposure light 68 ± 2, yield 98%, corresponding 0.148 g.mL−1 h−1 STY within 84 cycles of an experiment for C–S bond formation reaction. Further cross-verified the long-term suitability of the optimized condition, we have run the set-up for the 5 h of the reaction time for synthesis of compound 6a in gram scale. In contrast, a classical batch reaction required 24–48 h and the addition of a metal catalyst, resulting in a suboptimal to moderate yield of 35–89% for the same thiol derivative 6a (supplementary data Table S7)53. Next, the substrate scope of thiol and aryldiazoester were examined in the photo-flow coupling reaction. Surprisingly, all substrates yielded the corresponding products (6a-6h) in excellent yields, ranging from 75-98%. However, based on the literature survey, out of 8 synthesized photo α-thiocarbonyl products, 6 products (6b-6d, 6f-6h) were identified as recent additions.
After successful optimization of the acid and thiol, next we were interested to make the automated for the amines. Amines are key fragments in many natural products54,55, pharmaceuticals56,57, and agrochemicals58,59. For the long-term goal, we were working to synthesize the various amines in a fast manner but identified the long reaction time (80 min.) under the controlled white light photo-flow condition60. To further reduce the reaction time, we have introduced the AI system to identify the suitable reaction condition. Over a 24 h period, 44 experiments were systematically conducted under the selected boundary (watt 15–70, residence time 0.1–22.0 min). Remarkably, we observed that at a flow rate of 46 μL min−1 each, a residence time of 11 min, with 68 ± 2 W under 3 bar pressure, yielded a maximum 92% of 8a product with space-time yield of 0.058 g.mL−1 h−1. Having optimized photochemical C-N bond formation reaction python code (ab3), we explored various substituted aromatic amines (7a-7g) and aliphatic amines (7h-7j), resulting (8a-8j) in excellent yields ranging from 79–92%.
After validating the automated platform for single-objective optimization in carbene insertion reactions, the next goal is to explore its capacity for optimizing diverse photo-induced, highly stained ring formations. These strained cycloalkane or alkene rings, characterized by angle, torsional, and steric strains, release substantial energy upon combustion, making them valuable for high-energy materials61,62. The synthesis of highly stained molecules is challenging due to the high cost and significant waste associated with organometalloid complexes. Key optimization parameters include the concentration of compounds 2a and 9a, voltage, current, and residence time (see Table S10 in supplementary data). After conducting the series of experiments finally, we observed that at a flow rate of 62 μL min−1. each, residence time of 32 min. with 68 ± 2 W, under 3 bar pressure, yielded a maximum conversion of 95% with space-time yield of 0.025 g.mL−1h−1. These optimal conditions were then utilized for upscaling the transformation on a 0.01 mol scale within a 5 h timeframe. Notably, it’s worth to mention here that the conventional batch process for the identical reaction, even under excess use of metal catalyst and long reaction time (10–48 h)63,64, at higher temperatures (supplementary data Table S11). Various cyclopropanated products were obtained with excellent yields over 74–92% (Fig. 2, 10a–10i) in 32 min. of residence time by the fully automated photo-flow system with ab4 code and out of the 9 compounds, 3 (10b, 10c, & 10h) products were identified as recent additions.
Fig. 2. One code, one kind reaction.

Reaction condition: 69 ± 1 W light intensity; pressure 3 bar, AB1.py, AB2.py, & AB3.py; 0.1: 0.2, 0.2 & 0.2 M of compound 2, 3, 5, & 7 in EtOAc; exposure time (10.02, 5.7, & 11 min.); solution collection time & volume (5 h, 30 mL, 53 mL, 28 mL); 1 mL reactor volume; AB4.py & AB5.py; 0.1: 0.2, & 0.2 M of compound 2, 9, & 11 in DCE; exposure time (32 & 40 min); solution collection time & volume (5 h, 37 mL, 30 mL); 4 mL reactor volume; AB6.py & AB7.py; 0.1: 0.2, & 0.2 M of compound 2a, 13, & 15 in DCE; exposure time (16.6 & 12.3 min.); solution collection time & volume (5 h, 18 mL, 24 mL); 1 mL reactor volume; yields are based on isolated yield.
We next investigated phenyl acetylene nucleophile as a coupling partner but failed to get any desired product through the ab4 code. To access the cyclopropenated product, we have further developed the latest optimal condition through the same set-up by just slightly modifying the reaction time as shown in (Fig. 1e) and within 48 h period of the condition was optimized (supplementary data Table S12). We observed that a flow rate of 50 μL min−1 each, residence time of 40 min. with 68 ± 2 W, under 3 bar pressure, provided an excellent 96% yield with a space-time yield of 0.020 g.mL−1 h−1. The optimized condition with python code (ab5), various substituted aromatic phenyl acetylenes (11a-11h) were examined, which gave good to excellent yield (12a-12h) (75–96%) and out of 8 compounds 3 are latest (12g, 12f, & 12h). Further, the result was compared with previously reported literature, and it worth to mention here that convention batch process needs a longer reaction time with additional catalyst17,65,66.
Next, we attempted the sixth reaction using acetonitrile to form oxazole under previously reported photo-flow conditions (ab1-ab5), but the yield was disappointingly poor. To achieve maximum yield, we have established a fresh reaction condition with acetonitrile as the coupling partner. Notably, the 24 h period is sufficient to optimize the reaction for acetonitrile as the nucleophile within 44 experiments (supplementary data Table S14). We observed that at a flow rate of 31 μL min−1, residence time of 16 min. with 68 ± 2 W, under 3 bar pressure, yielded a maximum conversion of 91% with a space-time yield of 0.038 g.mL−1 h−1.
Similar pattern was applied to track the cross-coupling of aryldiazoacetates with ethyldiazoacetate reaction, using molar solutions of 2a and 15. A total of 44 experiments were conducted within a 24 h timeframe, with outcomes recorded in the optimization table (supplementary data Table S16). We observed that at a flow rate of 40 μL min−1, a residence time of 12 min, with 68 ± 2 W under 3 bar pressure, yielded a maximum conversion of 89% with a space-time yield of 0.058 g.mL−1 h−1 of compound 16a. Furthermore, the results for both reactions were compared with previously reported literature, and it is noteworthy that the conventional batch process requires a significantly longer reaction time67,68.
After successfully developing several optimized individual Python codes (AB1, AB2, AB3, AB4, AB5, AB6, AB7) for the synthesis of various cross-coupling products, we have now developed an integrated platform named “Innovative DigiChemTree.” This platform facilitates on-demand customized chemical synthesis, enabling both late-stage functionalization (LSF) and early-stage diversification of molecules. This capability is particularly beneficial for the de novo design and synthesis of drug molecules for structure-activity relationship (SAR) studies, eliminating the need for highly trained personnel69,70.
The pre-optimized codes (AB1 through AB7) were consolidated into a single Python code (see supplementary software 2), and the system components, such as the flow reactor, pump, in-line analysis, and solution collector, were systematically arranged as depicted in Fig. 3 (further details and video are available in the supplementary data and supplementary video). It is important to highlight that DigiChemTree can synthesize cross-coupling molecules (4a-16a) in excellent yields without human intervention.
Fig. 3. DigiChemTree machine process system for the man-free on-demand carbene inserted product.

Reaction condition: AB1.py, AB2.py, & AB3.py; 0.1: 0.2, 0.2 & 0.2 M of compound 2, 3, 5, & 7 in EtOAc; exposure time (10.02, 5.7, & 11 min.); AB4.py & AB5.py; 0.1: 0.2, & 0.2 M of compound 2, 9, & 11 in DCE; exposure time (32 & 40 min); AB6.py & AB7.py; 0.1: 0.2, & 0.2 M of compound 2a, 13, & 15 in DCE; exposure time (16.6 & 12.3 min.); yields are based on isolated yield.
Conclusion
In summary, we have developed a “one code one kind of reaction DigiChemTree” platform for the autonomous optimization and intensification of photochemical processes in a continuous flow manner. This innovative DigiChemTree platform demonstrated its efficacy through automated optimization and a high degree of adaptability for various types of photocatalytic reactions. Significant enhancements in yield were observed across all reactions, surpassing the performance achieved under-reported model reaction conditions. The rapid reaction auto-optimization and further utility in late-stage functionalization open fresh opportunities for medicinal chemistry to synthesize the on-demand library of the molecule. It’s also shorting out the highly trained organic chemist problem to make the innovative complex molecule. Human interface software work is going on in our laboratory, and soon we will report someplace.
Methods
Materials
Most of the reagents and chemicals are bought from Sigma-Aldrich and used as such without any further purification. Common organic chemicals and salts were purchased from AVRA Chemicals, India. The water used for the experiments was deionized water (18.2 mS conductivity). All work-up and purification procedures were carried out with reagent-grade solvents in the air. Analytical thin-layer chromatography (TLC) was performed using analytical chromatography silica gel 60 F254 precoated plates (0.25 mm). The developed chromatogram was analyzed by UV lamp (254 nm). PTFE (id = 500 μm) tubing, T-junction, high-purity PFA tubing was purchased from Upchurch IDEX HEALTH &SCIENCE. The photo-batch reactor bought from Lelesil Mumbai, India, was slightly modified for the continuous flow reaction. The visible light (Blue, Red, Green LED) reactor was bought from Smartchemsynth Machine Pvt. Ltd, Hyderabad.
General method
For synthetic details and analytical data of all reaction compounds (4, 6, 8, 10, 12, 14, 16) see supplementary data and for NMR spectra see supplementary data 1.
General procedure for DigiChemTree
Before starting the experiments, 0.1 M stock solutions of compound 2a were prepared in both EtOAc and DCE, alongside 0.2 M solutions of compounds 3a, 5a, 7a, 9a, 11a, 13a, and 15a, which were stored in conical flasks and syringes connected to pumps. The integrated continuous flow setup, enhanced by AI, was activated to execute various experiment codes (AB1.py to AB7.py). Each experiment involved different pump configurations to dispense specific solutions at optimized flow rates, with the reaction mixtures traversing through a 1 mL PFA tubular reactor under blue LED illumination and controlled pressure. After each run, the resulting products were collected in individual test tubes for analysis, demonstrating successful outcomes in synthesizing diverse biologically active molecules through various photochemical carbene and coupling reactions (details in supplementary data).
Supplementary information
Description of additional supplementary files
Acknowledgements
Two of the authors (Abhilash Rana and Ruchi Chauhan) are thankful to the Department of Science and Technology (DST), New Delhi, for the INSPIRE Fellowship Award. The authors thank DIICT for providing the facilities. The project was partly funded by CSIR as part of the FTT project (MLP-0091). CSIR-IICT manuscript communication no. IICT/Pubs./2024/224. Part of this manuscript has been filed for the patent.
Author contributions
A.K.S. conceived the project. A.R., R.C. conducted the process experiments. A.K.S., A.R., and R.C. wrote the paper. A.M. and D.P.K. wrote the Python code for running the automated system.
Peer review
Peer review information
Communications Chemistry thanks the anonymous reviewers for their contribution to the peer review of this work. Peer reviewer reports are available.
Data availability
The authors declare that all other data supporting the findings of this study are available within the manuscript file and supplementary information files and also are available from the corresponding author upon request (supplementary data, supplementary data 1, supplementary movie, supplementary software 1 and 2). Source data are provided with the publication.
Code availability
The code used for the simulations is available from the authors upon request.
Competing interests
The authors declare no competing interests.
Footnotes
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
These authors contributed equally: Abhilash Rana, Ruchi Chauhan.
Supplementary information
The online version contains supplementary material available at 10.1038/s42004-024-01330-z.
References
- 1.Zhang, Z. & Gevorgyan, V. Visible light-induced reactions of diazo compounds and their precursors. Chem. Rev.124, 7214–7261 (2024). [DOI] [PubMed] [Google Scholar]
- 2.Durka, J., Turkowska, J. & Gryko, D. Lightening diazo compounds?. ACS Sustain. Chem. Eng.9, 8895–8918 (2021). [Google Scholar]
- 3.Hu, X., Cheng-Sánchez, I., Kong, W., Molander, G. A. & Nevado, C. Nickel-catalysed enantioselective alkene dicarbofunctionalization enabled by photochemical aliphatic C–H bond activation. Nat. Catal.7, 655–665 (2024). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Blakemore, D. C. et al. Organic synthesis provides opportunities to transform drug discovery. Nat. Chem.10, 383–394 (2018). [DOI] [PubMed] [Google Scholar]
- 5.Rohrbach, S. et al. Digitization and validation of a chemical synthesis literature database in the ChemPU. Science377, 172–180 (2022). [DOI] [PubMed] [Google Scholar]
- 6.Klimovica, K., Heidlas, J. X., Romero, I., Le, T. V. & Daugulis, O. Sandwich diimine‐copper catalysts for C−H functionalization by carbene insertion. Angew. Chem. Int. Ed.61, e202200334 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Li, Y. et al. Highly enantioselective O–H bond insertion reaction of α-alkyl-and α-alkenyl-α-diazoacetates with water. J. Am. Chem. Soc.142, 10557–10566 (2020). [DOI] [PubMed] [Google Scholar]
- 8.Jagannathan, J. R., Fettinger, J. C., Shaw, J. T. & Franz, A. K. Enantioselective Si–H insertion reactions of diarylcarbenes for the synthesis of silicon-stereogenic silanes. J. Am. Chem. Soc.142, 11674–11679 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Yang, L.-L. et al. Enantioselective diarylcarbene insertion into Si–H bonds induced by electronic properties of the carbenes. J. Am. Chem. Soc.142, 12394–12399 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Zhang, X.-G., Yang, Z.-C., Pan, J.-B., Liu, X.-H. & Zhou, Q.-L. Enantioselective synthesis of chiral amides by carbene insertion into amide N–H bond. Nat. Commun.15, 4793 (2024). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Li, M.-L., Yu, J.-H., Li, Y.-H., Zhu, S.-F. & Zhou, Q.-L. Highly enantioselective carbene insertion into N–H bonds of aliphatic amines. Science366, 990–994 (2019). [DOI] [PubMed] [Google Scholar]
- 12.Wang, P. et al. Solvent-free, B(C6F5)3-catalyzed S−H insertion of thiophenols and thiols with α-diazoesters. Chem. Asian J.17, e202200465 (2022). [DOI] [PubMed] [Google Scholar]
- 13.Qin, L.-Z. et al. Visible-light-mediated S−H bond insertion reactions of diazoalkanes with cysteine residues in batch and flow. Adv. Syn. Catal.362, 5093–5104 (2020). [Google Scholar]
- 14.Pan, J. B., Yang, Z. C., Zhang, X. G., Li, M. L. & Zhou, Q. L. Enantioselective synthesis of chiral amides by a phosphoric acid catalyzed asymmetric wolff rearrangement. Angew. Chem. Int. Ed.62, e202308122 (2023). [DOI] [PubMed] [Google Scholar]
- 15.Bao, M. et al. Photo-cycloaddition reactions of vinyldiazo compounds. Nat. Commun.15, 4574 (2024). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Damiano, C., Sonzini, P. & Gallo, E. Iron catalysts with N-ligands for carbene transfer of diazo reagents. Chem. Soc. Rev.49, 4867–4905 (2020). [DOI] [PubMed] [Google Scholar]
- 17.Stefkova, K., Heard, M. J., Dasgupta, A. & Melen, R. L. Borane catalysed cyclopropenation of arylacetylenes. Chem. Commun.57, 6736–6739 (2021). [DOI] [PubMed] [Google Scholar]
- 18.Su, Y. L., Dong, K., Zheng, H. & Doyle, M. P. Generation of diazomethyl radicals by hydrogen atom abstraction and their cycloaddition with alkenes. Angew. Chem. Int. Ed.133, 18484–18488 (2021). [DOI] [PubMed] [Google Scholar]
- 19.Yang, D. et al. Electrochemical oxidative difunctionalization of diazo compounds with two different nucleophiles. Nat. Commun.14, 1476 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Epping, R. F. J., Vesseur, D., Zhou, M. & de Bruin, B. Carbene radicals in transition-metal-catalyzed reactions. ACS Catal.13, 5428–5448 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Zhao, J. et al. A heterogeneous iridium single-atom-site catalyst for highly regioselective carbenoid O–H bond insertion. Nat. Catal.4, 523–531 (2021). [Google Scholar]
- 22.Lee, W.-C. C., Wang, D.-S., Zhu, Y. & Zhang, X. P. Iron(III)-based metalloradical catalysis for asymmetric cyclopropanation via a stepwise radical mechanism. Nat. Chem.15, 1569–1580 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Liu, L. & Zhang, J. Gold-catalyzed transformations of α-diazocarbonyl compounds: selectivity and diversity. Chem. Soc. Rev.45, 506–516 (2016). [DOI] [PubMed] [Google Scholar]
- 24.Jurberg, I. D. & Davies, H. M. L. Blue light-promoted photolysis of aryldiazoacetates. Chem. Sci.9, 5112–5118 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Rana, A., Malviya, B. K., Jaiswal, D. K., Srihari, P. & Singh, A. K. A solar tracker integrated microreactor for real-time sunlight induced ketene formation and API synthesis. Green. Chem.24, 4794–4799 (2022). [Google Scholar]
- 26.Timmann, S. & Alcarazo, M. α-Diazo-λ3-iodanes and α-diazo sulfonium salts: the umpolung of diazo compounds. Chem. Commun.59, 8032–8042 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hu, J. et al. Photocatalyzed dual strain release of [1.1.1]propellane with diazo compounds. ACS Catal.14, 5481–5490 (2024). [Google Scholar]
- 28.Su, Y.-L. et al. Radical cascade multicomponent minisci reactions with diazo compounds. ACS Catal.12, 1357–1363 (2022). [Google Scholar]
- 29.Buglioni, L., Raymenants, F., Slattery, A., Zondag, S. D. A. & Noël, T. Technological innovations in photochemistry for organic synthesis: flow chemistry, high-throughput experimentation, scale-up, and photoelectrochemistry. Chem. Rev.122, 2752–2906 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Zondag, S. D., Mazzarella, D. & Noël, T. Scale-up of photochemical reactions: transitioning from lab scale to industrial production. Annu. Rev. Chem. Biom. Eng.14, 283–300 (2023). [DOI] [PubMed] [Google Scholar]
- 31.Ciszewski, Ł. W., Rybicka-Jasińska, K. & Gryko, D. Recent developments in photochemical reactions of diazo compounds. Org. Biomol. Chem.17, 432–448 (2019). [DOI] [PubMed] [Google Scholar]
- 32.Rehm, T. H. Reactor technology concepts for flow photochemistry. ChemPhotoChem4, 235–254 (2020). [Google Scholar]
- 33.Porta, R., Benaglia, M. & Puglisi, A. Flow chemistry: recent developments in the synthesis of pharmaceutical products. Org. Process Res. Dev.20, 2–25 (2016). [Google Scholar]
- 34.González-Esguevillas, M. et al. Rapid optimization of photoredox reactions for continuous-flow systems using microscale batch technology. ACS Cent. Sci.7, 1126–1134 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Slattery, A. et al. Automated self-optimization, intensification, and scale-up of photocatalysis in flow. Science383, eadj1817 (2024). [DOI] [PubMed] [Google Scholar]
- 36.Taylor, C. J. et al. A brief introduction to chemical reaction optimization. Chem. Rev.123, 3089–3126 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Torres, J. A. G. et al. A multi-objective active learning platform and web app for reaction optimization. J., Am. Chem. Soc.144, 19999–20007 (2022). [DOI] [PubMed] [Google Scholar]
- 38.Chauhan, R., Rana, A., Ghosh, S., Srihari, P. & Singh, A. K. Micro-total process system machine (μ-TPSM) for rapid synthesis of antiretroviral darunavir. React. Chem. Eng.8, 908–916 (2023). [Google Scholar]
- 39.Purwa, M., Rana, A. & Singh, A. K. The assembly of integrated continuous flow platform for on-demand rosiglitazone and pioglitazone synthesis. React. Chem. Eng.7, 2084–2092 (2022). [Google Scholar]
- 40.Rana, A., Mahajan, B., Ghosh, S., Srihari, P. & Singh, A. K. Integrated multi-step continuous flow synthesis of daclatasvir without intermediate purification and solvent exchange. React. Chem. Eng.5, 2109–2114 (2020). [Google Scholar]
- 41.Shields, B. J. et al. Bayesian reaction optimization as a tool for chemical synthesis. Nature590, 89–96 (2021). [DOI] [PubMed] [Google Scholar]
- 42.Ha, T. et al. AI-driven robotic chemist for autonomous synthesis of organic molecules. Sci. Adv.9, eadj0461 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Ahn, G.-N. et al. Exploring ultrafast flow chemistry by autonomous self-optimizing platform. Chem. Eng. J.453, 139707 (2023). [Google Scholar]
- 44.Sheng, H. et al. Autonomous closed-loop mechanistic investigation of molecular electrochemistry via automation. Nat. Commun.15, 2781 (2024). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Aand, D., Rana, A., Mottafegh, A., Kim, D.-P. & Singh, A. K. Autonomous closed-loop photochemical reaction optimization for the synthesis of various angiotensin II receptor blocker molecules. React. Chem. Eng.9, 2427–2435 (2024). [Google Scholar]
- 46.Coley, C. W. et al. A robotic platform for flow synthesis of organic compounds informed by AI planning. Science365, eaax1566 (2019). [DOI] [PubMed] [Google Scholar]
- 47.Nambiar, A. M. K. et al. Bayesian optimization of computer-proposed multistep synthetic routes on an automated robotic flow platform. ACS Cent. Sci.8, 825–836 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Burger, B. et al. A mobile robotic chemist. Nature583, 237–241 (2020). [DOI] [PubMed] [Google Scholar]
- 49.Dunlap, J. H. et al. Continuous flow synthesis of pyridinium salts accelerated by multi-objective Bayesian optimization with active learning. Chem. Sci.14, 8061–8069 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Guillemard, L., Kaplaneris, N., Ackermann, L. & Johansson, M. J. Late-stage C–H functionalization offers new opportunities in drug discovery. Nat. Rev. Chem.5, 522–545 (2021). [DOI] [PubMed] [Google Scholar]
- 51.Mennen, S. M. et al. The evolution of high-throughput experimentation in pharmaceutical development and perspectives on the future. Org. Process Res. Dev.23, 1213–1242 (2019). [Google Scholar]
- 52.Tan, F. et al. Asymmetric catalytic insertion of α-diazo carbonyl compounds into O–H bonds of carboxylic acids. ACS Catal.6, 6930–6934 (2016). [Google Scholar]
- 53.Keipour, H., Jalba, A., Tanbouza, N., Carreras, V. & Ollevier, T. α-Thiocarbonyl synthesis via the FeII-catalyzed insertion reaction of α-diazocarbonyls into S–H bonds. Org. Biomol. Chem.17, 3098–3102 (2019). [DOI] [PubMed] [Google Scholar]
- 54.Cabré, A., Verdaguer, X. & Riera, A. Recent advances in the enantioselective synthesis of chiral amines via transition metal-catalyzed asymmetric hydrogenation. Chem. Rev.122, 269–339 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Li, M., Jin, Y., Chen, Y., Wu, W. & Jiang, H. Palladium-catalyzed oxidative amination of unactivated olefins with primary aliphatic amines. J. Am. Chem. Soc.145, 9448–9453 (2023). [DOI] [PubMed] [Google Scholar]
- 56.Maity, S., Potter, T. J. & Ellman, J. A. α-Branched amines by catalytic 1,1-addition of C–H bonds and aminating agents to terminal alkenes. Nat. Catal.2, 756–762 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Zawodny, W. & Montgomery, S. L. Evolving new chemistry: biocatalysis for the synthesis of amine-containing pharmaceuticals. Catalysts12, 595 (2022). [Google Scholar]
- 58.Huang, Y., Law, J. C.-F. & Leung, K. S.-Y. The quest for metabolic biomarkers of agrochemicals exposure via in vitro studies and suspect screening. Sci. Total Environ.861, 160701 (2023). [DOI] [PubMed] [Google Scholar]
- 59.Ricci, A. & Bernardi, L. Methodologies in Amine Synthesis: Challenges and Applications. ISBN: 978-3-527-82619-3 (WILEY‐VCH GmbH, 2021).
- 60.Rana, A., Chauhan, R. & Singh, A. K. Thermal/photochemical micro-flow probe system for direct C–H bond functionalization of biologically active molecules. React. Chem. Eng.9, 1313–1319 (2024). [Google Scholar]
- 61.Liu, Y. et al. Zeolite catalytic simmons–smith cyclopropanation of alkenes for the synthesis of high-energy-density fuels. Ind. Eng. Chem. Res.63, 6985–6994 (2024). [Google Scholar]
- 62.Li, G. et al. Production of renewable hydrocarbon biofuels with lignocellulose and its derivatives over heterogeneous catalysts. Chem. Rev.124, 2889–2954 (2024). [DOI] [PubMed] [Google Scholar]
- 63.Sharland, J. C. et al. Asymmetric synthesis of pharmaceutically relevant 1-aryl-2-heteroaryl- and 1,2-diheteroarylcyclopropane-1-carboxylates. Chem. Sci.12, 11181–11190 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Mancinelli, J. P. & Wilkerson-Hill, S. M. Tris(pentafluorophenyl)borane-catalyzed cyclopropanation of styrenes with aryldiazoacetates. ACS Catal.10, 11171–11176 (2020). [Google Scholar]
- 65.Chen, L., Leslie, D., Coleman, M. G. & Mack, J. Recyclable heterogeneous metal foil-catalyzed cyclopropenation of alkynes and diazoacetates under solvent-free mechanochemical reaction conditions. Chem. Sci.9, 4650–4661 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Hommelsheim, R., Guo, Y., Yang, Z., Empel, C. & Koenigs, R. M. Blue-light-induced carbene-transfer reactions of diazoalkanes. Angew. Chem. Int. Ed.58, 1203–1207 (2019). [DOI] [PubMed] [Google Scholar]
- 67.Saha, A. et al. Photoinduced [3+2] cycloaddition of carbenes and nitriles: a versatile approach to oxazole synthesis. Angew. Chem. Int. Ed.62, e202308916 (2023). [DOI] [PubMed] [Google Scholar]
- 68.Xiao, T., Mei, M., He, Y. & Zhou, L. Blue light-promoted cross-coupling of aryldiazoacetates and diazocarbonyl compounds. Chem. Commun.54, 8865–8868 (2018). [DOI] [PubMed] [Google Scholar]
- 69.Liu, C. et al. Automated synthesis of prexasertib and derivatives enabled by continuous-flow solid-phase synthesis. Nat. Chem.13, 451–457 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Cernak, T., Dykstra, K. D., Tyagarajan, S., Vachal, P. & Krska, S. W. The medicinal chemist’s toolbox for late stage functionalization of drug-like molecules. Chem. Soc. Rev.45, 546–576 (2016). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Description of additional supplementary files
Data Availability Statement
The authors declare that all other data supporting the findings of this study are available within the manuscript file and supplementary information files and also are available from the corresponding author upon request (supplementary data, supplementary data 1, supplementary movie, supplementary software 1 and 2). Source data are provided with the publication.
The code used for the simulations is available from the authors upon request.