

Abstract

Technological advances and breakthrough developments in the pharmaceutical field are knocking at the door of the “undruggable” fortress with increasing insistence. Notably, the 21st century has seen the emergence of macrocyclic compounds, among which cyclic peptides are of particular interest. This new class of potential drug candidates occupies the vast chemical space between classic small-molecule drugs and larger protein-based therapeutics, such as antibodies. As research advances toward clinical targets that have long been considered inaccessible, macrocyclic peptides are well-suited to tackle these challenges in a post-rule of 5 pharmaceutical landscape. Facilitating their discovery is an arsenal of high-throughput screening methods that exploit massive randomized libraries of genetically encoded compounds. These techniques benefit from the incorporation of non-natural moieties, such as non- proteinogenic amino acids or stabilizing hydrocarbon staples. Exploiting these features for the strategic architectural design of macrocyclic peptides has the potential to tackle challenging targets such as protein–protein interactions, which have long resisted research efforts. This Review summarizes the basic principles and recent developments of the main high-throughput techniques for the discovery of macrocyclic peptides and focuses on their specific deployment for targeting undruggable space. A particular focus is placed on the development of new design guidelines and principles for the cyclization and structural stabilization of cyclic peptides and the resulting success stories achieved against well-known inaccessible drug targets.
1. Introduction: Undruggable Targets
The last two decades have seen a dramatic expansion of what is considered the “druggable” space.1−6 The emergence or explosion of new pharmaceutical modalities, such as antibodies, nucleic acids, or engineered proteins, has triggered a number of paradigm shifts and ushered in what could be called the “post-rule of 5” era.5,6 The well-known Lipinski rules, or rather guidelines, have been instrumental in the drug design for small molecules. However, as pharmaceutical research has expanded much beyond the small-molecule chemical space and into both larger proteins as well as intermediate-sized peptides, these design principles are no longer sine qua non conditions for drug development,7 and new “beyond-rule of 5” (bRo5) guidelines emerge.8 Consequently, the “undruggable” (or “less druggable”) frontier has been pushed ever further, to the point where the concept has shifted from pure impossibility to a more optimistic “yet-to-be-drugged” notion.1,9 Numerous diseases and proteins that were once considered fully out of reach for pharmaceutical chemists have been targeted with varying degrees of success. HIV, long considered an insurmountable challenge due to its high propensity to mutate and evolve resistance, has been largely tamed, although, notably, a vaccine still escapes scientists.10 Arguably the most iconic undruggable cancer-associated protein, KRas, saw a major breakthrough when its G12C mutant was successfully targeted by the Shokat group.11,12
These technological advances and target-specific breakthroughs have spurred hope that many other longstanding challenges can be addressed and given new momentum to innovative pharmaceutical research, both academic and industrial. Yet the undruggable space still represents formidable obstacles for scientists and requires tremendous amounts of effort and funding. While what constitutes an “undruggable” target is subject to debate and somewhat subjective appreciations, there is a consensus on certain groups (Figure 1).
Figure 1.

Traditional pocket-binding models have been successful for small-molecule drugs, but they do not apply to undruggable targets, including proteins without well-defined binding pockets, protein–protein interactions (PPIs), and intrinsically disordered proteins (IDPs). Macrocyclic peptides have long been considered promising, and they can be accessed through multiple high-throughput screening (HTS) methods and optimized following a variety of design strategies.
Proteins that lack well-defined hydrophobic-core-like binding pockets, such as small GTPases, are largely recognized as a major challenge.2 These proteins often play pivotal roles in critical biological processes but exhibit flat or featureless surfaces that are unsuitable for conventional ligand binding. The absence of a defined binding site makes it impossible to develop a binder through traditional rational and/or computer-assisted design approaches. Moreover, although high-throughput screening (HTS) methods may allow the identification of ligands, the emergence of functional binders remains challenging. In that sense, targeting protein–protein-interactions (PPIs) is a notably delicate endeavor.13−15 PPIs are central to numerous cellular processes governing signal transduction, gene expression, and protein function. However, targeting PPIs with small molecules is notoriously challenging due to the large, shallow interaction surfaces involved and the transient nature of many interactions. For this reason, peptides or small protein binders are ideal candidates, as their size is simply better suited for interaction with larger surfaces than small-molecule pharmacophores.16−19 However, transient interactions or conformational changes can make the development of binders difficult. The omnipresence of PPIs also typically raises concerns of off-target effects, as they are rarely specific to disease pathways but rather up- or down-regulated.
Another subset of proteins lacking well-defined binding pockets is intrinsically disordered proteins (IDPs).13,20−23 IDPs lack a stable three-dimensional structure under physiological conditions, existing instead as dynamic ensembles of interconverting conformations. IDPs play crucial roles in signaling, regulation, and molecular recognition but pose significant challenges for traditional drug discovery approaches. Their complex and often poorly understood dynamic nature renders the rational design of binders extremely difficult. Likewise, as screening techniques generally take place in simplified systems, identified binders may not translate once applied in physiological conditions where the overall environment is much more complex and the target’s dynamics are in full display.
This Review focuses on the use of nucleotide-encoded mass library screening (NELS) techniques to discover macrocyclic peptides targeting inaccessible targets. In this context, we will also consider intracellular targets as inaccessible. While such targets are not undruggable per se, they do remain a major challenge for peptide-based therapeutics, which often struggle considerably with membrane permeability. Additionally, due to the lack of publicly available data from industrial research, we will focus mainly on academic research outputs.
Peptide-based therapeutics effectively occupy the chemical space between small molecules and larger proteins.24,25 Their natural building-block composition provides them with a high interaction potential, essentially mimicking natural biological processes more closely than synthetic small molecules. Furthermore, their relatively modest size keeps them synthetically accessible. It has been well established that cyclizing peptides drastically enhances these desirable features, making macrocyclic peptides generally more pharmaceutically relevant than their linear counterparts.24
The constrained structure of macrocyclic peptides translates into better control of their active conformation, ensuring that the compounds discovered during the screening generally translate to further studies. Cyclization also enhances their stability, as it offers a degree of resistance toward proteolytic cleavage. Additionally, certain cyclization designs may even enhance membrane penetration.26,27
The first part of this Review summarizes the variety of high-throughput techniques that can be deployed for the discovery of cyclic peptides. For in depth discussion of these methods and their history, the reader is advised to selected dedicated reviews covering phage display,28 split intein-mediated circular ligation of peptides and proteins (SICLOPPS),29,30 mRNA display,31−33 ribosome display,34,35 yeast displays,36,37 and DNA-encoded libraries (DELs).38 Apart from the NELS method employed and the associated library design, the main tools to weaponize cyclic peptides against inaccessible targets are the choice of cyclic architecture and non-proteinogenic amino acids.24 Both enable control over the structural constraint and the interaction capabilities of the peptide sequence. Section 3 focuses on the different designs of cyclic peptides and how these architectural choices can be employed to tackle inaccessible targets. Recent non-proteinogenic amino acid developments will also be discussed. Section 4 will focus on the application of these techniques specifically for the discovery of macrocyclic peptides toward the previously defined inaccessible target groups and highlight recent breakthrough examples. Finally, we discuss potential avenues for further development. It should be noted that given the size of the field and the amount of related publications reported on a yearly basis, this review is not comprehensive; rather, it is a wide and up-to-date overview of the topic with a focus on the most relevant studies. Wherever a certain aspect has been the topic of a recent, in-depth review, the reader is advised to refer to the cited reference for a deeper dive into the particular topic.
2. Nucleotide-Encoded Mass Library Screening Techniques
2.1. Phage Display
Phage display is one of the most common high-throughput platforms for peptide discovery (Figure 2A).28 In this technique, a peptide-encoding cDNA is inserted into the coat protein gene of a bacteriophage (e.g., protein pIII). Subsequent expression in E. coli. then displays the peptide of interest in the phage surface. Using a library of randomized DNA thus leads to the expression of a randomized peptide library, which can be panned against an immobilized target for binding. As the expressed phenotype (i.e., the expressed peptide) is directly connected to its genetic signature, the top selected sequences can be identified and undergo iterative rounds of selection, ultimately leading to the enrichment of the strongest binding sequences.
Figure 2.

Summary of selected nucleotide-encoded library screening (NELS) techniques. (A) In phage display,28 the designed plasmid library is first transformed in E. coli. Helper phages are then used to infect the bacteria, creating a peptide library displayed on the phage coat protein. Cyclization occurs at this point according to the experimental design; here, a disulfide bridge serves as an example. The phage library is then screened against an immobilized target, and the hit sequences can be amplified and submitted to new rounds of selection before ultimate sequencing. (B) SICLOPPS is performed directly in cells,29,30 which allows coexpression of the plasmid library and the protein target of interest. Selection is performed through a variety of cell-based assays, and hits are identified by fluorescence, cell death, etc. The cyclization mechanism relies on intein splicing. (C) In mRNA display,31,32 a randomized mRNA library is ligated with a puromycin linker. This library is then translated and cyclized, typically via a thioether moiety. Affinity selection is then performed, sequences are amplified, and selection rounds are repeated until the suitable enrichment of top sequences. The combination of this technique with the FiT system to incorporate npAAs is called the RaPID method. (D) DNA-encoded libraries utilize a combinatorial chemistry approach.58 In each step, a building block is introduced and a DNA barcoded fragment is ligated. Repeating split-and-pool steps allow the construction of a diverse library, which can be cyclized through various methods before being panned against an immobilized target protein and identified through sequencing of the DNA barcodes.
Overall, the relatively straightforward setup and cost-effectiveness of this method have made it very popular. Cyclization is commonly achieved by a covalent linkage to cysteines in the linear peptide sequences, a strategy that allows the display of multicyclic peptides. A recent review by Chen et al. covers this topic in depth.39
Originally, as phage display relies on in vivo expression machinery, it was inherently limited to the use of the 20 proteinogenic amino acids,40 although selenocysteine and pyrrolysine have subsequently been incorporated.41 However, incorporation of non-proteinogenic residues has since been expanded. One strategy for such incorporation is to include the non-proteinogenic amino acid residue in the cyclizing scaffold. The residue of interest is fitted with a reactive organic handle such as a chloroacetyl or an alkene motif for nucleophilic substitution or Michael addition, respectively. Genetic code expansion has also been leveraged to incorporate non-proteinogenic amino acids (npAAs) into phage-displayed libraries. This relies on amber-codon suppression through the exploitation of aminoacyl-tRNA-synthetases (aaRS)/tRNA pairs that are orthogonal to the Escherichia coli system, such as the pyrrolysil-tRNA synthetase pair (PylRS)/tRNA pair. Using this technique, a variety of npAAs has been used in phage display.42,43
Recently, Oller-Salvia and Chin used two mutually orthogonal aminoacyl-tRNA synthetase (aaRS)/tRNA pairs to efficiently display peptides containing two different npAAs.42 The different npAAs thus introduced allowed them to dual-label the peptides with different probes in a one-pot manner.
The use of antisense codons can result in a negative bias during expression in E. coli, which naturally favors sense-containing sequences. To tackle this issue, Tharp et al. developed a technique called “superinfection-immunity-based selection”, which exploits the superinfection immunity property of filamentous phages (Ffs) to effectively remove from the phage library sequences that are only sense-containing (as well as those bearing deleterious mutations).44 Ultimately, this allows the efficient selection of peptide binding candidates assured to contain npAAs, thus alleviating unwanted bias and selection pressure. They went on to exploit this strategy to develop active site-directed ligand evolution, wherein the npAAs incorporated in the phage-displayed peptides serve as direct probes to the binding site of the target of interest.
Historically, the main drawback of phage display, compared to alternative methods developed afterward, is its relatively small library size of about 10.9 This is largely caused by the required transformation step, whose efficiency limits the workable size of the library and introduces certain biases, such as the inability to fold or toxicity of certain sequences. The Heinis group reported a significant expansion of the phage display library size, which they achieved by combining whole-plasmid PCR with smaller phagemid vectors, as opposed to the conventional phage vectors.45 The PCR technique enabled the more robust generation of high-quality vectors, while the smaller size of said vectors increased the infection efficiency compared with traditional techniques. They successfully panned this library against calprotectin to obtain a low-nanomolar-affinity peptide. There are alternative cell-surface display techniques that are conceptually similar to phage display. Notably, yeast display presents cyclic peptides on the surface of yeast and can also be leveraged for the acquisition of binders with high affinity and specificity for target proteins.36,37 Both phage and yeast display have been used by the van der Donk group to screen vast libraries of lanthipeptides.46 Other cells can be used in similar strategies,47,48 including mammalian cells.49,50 Generally, cells displaying hit peptides were identified by fluorescence-assisted cell sorting (FACS), enabling high-throughput identification of the relevant cultures to be submitted to further selection rounds.37
2.2. Split Intein-Mediated Circular Ligation of Peptides and Proteins (SICLOPPS)
SICLOPPS is another commonly used cell-based display system.29,30 The method relies on the use of inteins, i.e., self-excising proteins, that are encoded on both sides of randomized peptide-coding DNA sequences in the plasmid library (Figure 2B). Upon transcription and translation in a bacterial system, the two inteins interact to form an active species that splices and generates a cyclic peptide as a byproduct. Although the process is not limited by the size of the produced peptide, only up to about 6 randomized residues can be introduced for full coverage of the library (106 to 108 diversity). Furthermore, a cysteine or serine residue is required as the first amino acid of the peptide for the intein slicing to occur, not unlike other display methods where cysteines are often required for cyclization. Although it is largely limited to the use of proteinogenic amino acids, SICLOPPS has been combined with an orthogonal aaRS/tRNA pair for the incorporation of a single npAA.
A major advantage of SICLOPPS is the fact that the selection occurs intracellularly. Therefore, cyclic peptides can be screened for function rather than binding, thus ensuring the obtention of bioactive species over nonspecific binders, a drawback of in vitro methods such as mRNA display. Selection of the peptide through a complex intracellular environment system also favors lead compounds that can withstand such conditions as opposed to in vitro selected candidates whose behavior may differ significantly once applied to the target in its native environment. Another feature of SICLOPPS is its flexibility for combination with a wide variety of cell-based assays, which allows users to screen for functional binding with great ease and efficiency.
2.3. mRNA Display
In contrast to the above-mentioned cell-based techniques such as phage display and SICLOPPS, mRNA display (as well as the closely related but less commonly used ribosome display)34,35 occurs in an in vitro reconstituted-cell-free environment (Figure 2C).31−33 While this results in a more costly and complex technique, is also has a significant advantage: by eliminating the transformation step and its inherent biases and limitations, a much larger library size can be handled, typically around more than a trillion (>1012) different sequences. A larger library significantly increases the chances of finding superior binders. In mRNA display, each peptide contained in the library is covalently linked to its cognate mRNA by a puromycin–pC–pC (pC = phosphate–cytidine) poly(ethylene glycol) (PEG) linker, which allows PCR amplification of the selected peptides as well as sequencing of the genetic signature for identification. The design of the library is straightforward and flexible, and it can be prepared quickly by PCR using combinations of oligonucleotides with the desired degenerate codons. The peptide-encoding region is also flanked by fixed sequences, such as T7 transcriptase promoters as well as flexible spacer and binding sites for the ligation of a puromycin linker, which occurs after transcription of the library. The ribosome contained in the in vitro translation system reads through the mRNA to synthesize the peptide, ultimately stalling at the stop codon thanks to the omission of release factors from the system. Instead, the puromycin molecule gets covalently attached to the nascent peptidic chain, thus linking the peptide to its genetic signature before the whole complex is released from the ribosome. Cyclization is then performed either spontaneously or through chemical intervention, depending on the library design and experimental conditions. This is generally followed by a reverse transcription step, which gives rise to a hybrid mRNA/cDNA duplex. The resulting mRNA-displayed cyclic peptide library can then be panned against an immobilized target of interest, and top sequences are selected for affinity and amplified by PCR. Iterative rounds of selection are typically carried out to further enrich the strongest binders. The use of PCR is another advantage of mRNA display, as alternative techniques such as error-prone PCR can be used to introduce mutations, thus allowing the directed evolution of the amplified sequences.
Another major advantage of mRNA displays is its potent combination with genetic code reprogramming. A popular variant of mRNA display is the random nonstandard peptide integrated discovery (RaPID) system developed by the Suga group, which integrates genetic code reprogramming51 via the flexible in vitro translation (FIT) system.52−55
As both RaPID and FIT systems have been extensively reviewed,31,52,54 we will focus this discussion on the latest advances of these methods for the incorporation of npAA residues relevant to targeting inaccessible proteins. Briefly, in RaPID, npAA residues can be charged onto tRNAs using tRNA-aminoacylating artificial ribozymes known as flexizymes. As the mRNA display selection is performed in vitro, it is possible to fully control the mixture of amino acids and aaRS present in the system. Therefore, selected codons can be vacated and “reprogrammed” with npAAs that are precharged on the corresponding tRNAs and added to the translation system. RaPID then proceeds as a regular mRNA display selection, panning large libraries of peptides against an immobilized protein of interest, followed by amplification of the binding sequences by PCR and next-generation sequencing (NGS). In RaPID, the initiator codon is reprogrammed with an amino acid (usually tyrosine or tryptophan, in either l- or d-configuration) equipped with a chloroacetyl group. The last residue is fixed as a cysteine, which spontaneously reacts with the chloroacetyl group to form libraries of macrocyclic peptides. A huge variety of npAAs can be introduced into the RaPID system, which is discussed further in section 2.4. While extensive npAA compatibility and massive library size are two significant advantages of mRNA display, the technique also has some drawbacks, notably its propensity to yield nonspecific binders and the fact that it screens for affinity rather than functionality.31 Consequently, binders identified through mRNA display do not necessarily prove to be competent inhibitors, requiring thorough investigation of the hit compounds after selection.
2.4. Split-and-Pool Techniques
Alternative methods to the above-mentioned display technologies are derived from the realm of synthetic combinatorial chemistry. In split-and-pool techniques, an original reactive compound (traditionally immobilized on solid support) is first split into several portions, which are then reacted with a variety of compounds. The resulting mixtures are then pooled, mixed, split again, and submitted to new rounds of chemical reactions. As this approach relies on synthetic chemistry rather than translational machinery, an advantage of such techniques is the enormous chemical space that can be explored in a single split-and-pool campaign, incorporating proteinogenic and non-proteinogenic building blocks alike and directly benefiting from advances in synthetic research. Likewise, a very large number of diverse compounds can be produced through just a few steps of split-and-pool synthesis. This strategy is conceptually related to fragment-based drug design (FBDD), which has been successful in targeting inaccessible modalities.56,57
One popular approach to this concept is to use DNA-encoded-libraries (DELs, Figure 2D).58 In DELs, each synthetic step is concurrently performed with DNA-barcode ligation. This way, every fragment addition also adds a barcoded DNA signature, enabling the eventual identification of the hit compounds by NGS.38 Building blocks can be added through chemical or enzymatic synthesis, and the library can be constructed either with or without the use of a solid support. Much like display strategies, the library is then panned against an immobilized target for selection based on binding affinity, which has its limitations, as mentioned previously. In a recent report, Silvestri et. al performed DEL screening to identify an inhibitor of the MDM2/p53 PPI with an IC50 of 8.9 nM.59
Another approach is the one-bead-one-compound (OBOC) method.60 This technique relies on the use of beads that are manufactured to contain a single reactive handle, generally distanced from the bead surface through a PEG linker. Individual beads are then chemically linked to the desired starting materials, usually through a photocleavable moiety, and then submitted through several rounds of split-and-pool synthesis. As for DELs, the library is rapidly diversified and screened against an immobilized target. As they do not employ DNA barcodes, OBOC screenings are relatively straightforward and inexpensive. However, hit identification is more complex in the absence of a barcode-like traceability system and requires structure deconvolution through MS/MS analysis.61,62 Consequently, OBOC techniques generally limit their library size to a few thousand units (i.e., a few synthetic steps).
Traditionally, both DEL and OBOC screenings are performed on immobilized targets, which confers operational ease but suffers from the associated limitations (i.e., affinity-based selection rather than functional binding, relatively high rate of false -positive hits, and oversimplified target environment in vitro vs its natural in vivo context). For this reason, recent developments have focused on performing these strategies in vivo.63,64
All of the NELS methods discussed in this section come with their own set of advantages and drawbacks. Likewise, alternative techniques such as yeast display,36,37,46 ribosome display,34,35 and one-bead-two-compounds65 are less common but may well find situational use in particular scenarios. Often, a given laboratory will favor one method over the others based on their existing equipment and experience. For researchers who are new to the field and/or only need to deploy such an NELS technique occasionally for a given project, the available choices can be overwhelming, and we hope this Review helps them consider which techniques are best suited for their needs.
3. Cyclization and Design Strategies
3.1. Macrocyclization
Folding not only is crucial for binding but in many cases also increases the proteolytic stability and membrane permeability of the peptides. In this regard, macrocyclization is a common approach to rigidify the three-dimensional structure of the molecule. There are many approaches to peptide cyclization.26,27,39,66 Apart from the structural rigidity they confer, cyclization methods also must meet some practical considerations. In particular, they must be chemically feasible. This implies compatibility with coupling, protection, and deprotection steps and tolerance to polar solvents, as well as high yields and clean reaction profiles. Compatibility with the chosen NELS technique is another practical requirement. As the peptide libraries start off linearly, the cyclization occurs post-translationally, either enzymatically or chemically. Ideally, this post-translation cyclization should be complete and specific in order to avoid false-positive hits. Figure 3 showcases the most common cyclization strategies as well as new, recently published propositions. A very recent excellent review by He et al. covers the topic of biocompatible macrocyclization strategies in depth.66 While complete coverage of macrocyclization strategies is beyond the scope of this Review, we will introduce the main traditional techniques used and recent developments.
Figure 3.

Selected examples of macrocyclization strategies. (A) TBMB has been an extremely popular organic cross-linker, particularly for phage-displayed bicyclic libraries.68 In a recent variation, Bismuth was utilized for the cyclization of three cysteine residues in a similar way, enabling a faster and cleaner reaction.69−72 (B) mRNA display strategies generally perform spontaneous thioether cyclization between a cysteine and a reprogrammed initiator codon that contains a reactive chloroacetyl group.31 (C) Cysteine–proline–proline–cysteine units preferably cyclize with one another, enabling selective formation of disulfide bridges.73 (D) The Derda group used the orthogonal chemistry of cysteines and serines to control the design of their bicyclic library.77 (E) A carefully optimized nitrile-containing moiety reacts selectively with N-terminal cysteines, allowing the formation of polycyclic libraries in the mRNA display.80 (F) A tyrosinase was included in the RaPID system to catalyze the reaction of a cysteine and an unmodified tyrosine.82 (G) Lanthipeptides are a subclass of RiPPs that undergo a series of enzymatic reactions to yield cyclic peptides. A variety of enzymes and substrates exist that were simplified in this scheme for clarity.83
Cysteine residues are generally crucial for modern cyclization strategies, either through disulfide bond formation or cross-linking.39 Cyclization by disulfide bridging requires the least intervention, is widely found in natural macrocyclic compounds, and remains a common way of constraining a peptide’s structure. However, disulfide-bond-based cyclic peptides do suffer from a number of drawbacks. First, the number of possible cycles increases with the number of cysteines in the peptide sequences, which can dramatically complicate the lead sequence identification and peptide purification. Apart from their potential for complex polycyclization, disulfide bridges suffer from inherent vulnerability toward reducing conditions. Hence, cyclization by chemical cross-linking of cysteines has become an extremely common alternative.39 The nucleophilicity of the cysteine thiol group enables efficient and chemoselective cyclization by nucleophilic substitution reactions, which are controlled through the pH of the experimentally relevant buffers. This way, not only monocyclic but also multicyclic peptides can be formed with relative technical ease. While the resulting thioethers are more stable than disulfide bridges toward reducing conditions, they are prone to oxidation to sulfoxides and sulfones both in vitro and in vivo.
The traditional workhorse for the construction of bicyclic peptides is 1,3,5-tris(tribromomethyl)benzene (TBMB). While the chemistry itself was developed by Timmerman et al.,67 its use in phage display was first reported by the Winter group (Figure 3A).68 The thioether linkage formed is stable to reducing and oxidizing conditions and efficiently constrains the polycyclic peptide’s structure thanks to the compact benzene core. This approach has since been further expanded, and a multitude of alternative cross-linkers have been reported, allowing significant flexibility in the design and tuning of the cyclic peptide’s architecture and properties.39 A remarkable variation of this strategy was reported by Nitsche and co-workers, who employed bismuth(III) salts to form stable trivalent complexes with cysteine residues under biocompatible conditions (Figure 3A).69 Unlike traditional methods that rely on covalent bonds formed through reactions with electrophiles, bismuth complexation is reversible, minimizing unwanted side reactions with other cysteines that are not intended for cyclization, and tolerates phosphine-based reducing agents. Furthermore, Bi salts can be introduced without the need for an organic cosolvent and have been shown to be compatible with phage display technology.70,71 Peptides containing bismuth can cross mammalian cell membranes and possess low cytotoxicities, making them promising drug candidates; in particular, the α-emitter bismuth-213 could be utilized in radiotherapy applications.72
Another popular thioether-based cyclization is enabled by genetic code reprogramming, where the initiator codon normally encoding methionine is reprogrammed to include an N-terminal chloroacetyl group (typically born by a tyrosine or a tryptophan).31 Upon translation, this will spontaneously cyclize a cysteine to form a stable thioether macrocycle (Figure 3B). This is the most common way to generate macrocyclic peptide libraries in mRNA display, which is why most such libraries include a cysteine residue as the final amino acid in the peptide sequence. Furthermore, the Suga group showed that such cyclization preferentially occurs with the cysteine residue closest to the chloroacetyl group. Should there be more cysteines, they can form additional cycles by disulfide bond formation, or they can be fixed in strategic locations to be cross-linked with the TBMB (or similar) method.31
With regard to more classic disulfide bridge cyclization, other strategies can be employed to exert selectivity on the cyclic architecture. The Wu group used specific cysteine–proline–proline–cysteine (CPPC) sequences, which preferentially form disulfide bonds with one another rather than with isolated cysteines (Figure 3C).73 Exploiting this property allowed the authors to create tricyclic libraries where two cycles are formed from a pair of CPPC-based disulfide bonds and a third one results from two additional cysteine residues. Several peptides with this architecture were found to bind MDM2 in the low nanomolar range.73 Another way to exercise control over disulfide bond cyclization design is to use natural templates containing cysteine knots.39,74−76 Cysteine knot peptides (CKPs) are peptides in which one disulfide bridge is threaded through a cycle formed by two other such bridges, resulting in a constrained structure resembling a knot. Introducing randomized residues on this type of sequence enables the construction of CKP-derived cyclic peptides, which can then be screened by phage display.
Thioether-based cyclization does have drawbacks. First, it typically requires strategic positioning of the cysteine residues, which can complicate the library design. Preferably, cysteine residues should also be absent from the randomized positions to retain control over the cyclization architecture and avoid undesired mixtures of variously cyclized compounds. This largely means that cysteine cannot be used for target binding purposes. Additionally, in in vivo systems such as phage display, this means the expressed phage-coat protein must also be cysteine-free.30 Such engineered phages have been shown to suffer from a heavy negative infectivity bias compared to their wild type counterparts. Therefore, alternative non-cysteine-based cyclization methods have been developed, often resulting in the formation of heterocyclic linkages and relying on the installation of reactive organic side chains for click-chemistry, cross-coupling, or condensation reactions.39,66
Other amino acids can be used for cyclization with organic cross-linkers. Notably, N-terminal serines are readily oxidized to aldehydes and easily undergo a wide variety of chemical reactions. The Derda group recently used this strategy in combination with two fixed-cysteine residues and an organic cross-linker to produce bicyclic libraries in phage display that boasted enhanced serum stability (Figure 3D).77 Bicyclic peptides were obtained through this method which that showed low micromolar inhibitory activity toward the growth factor NODAL, a relevant target that is normally only present in embryonic tissue but can be found aberrantly expressed in tumors. Recently, the Mayer group also reported an asymmetric cross-linker that can react with a cysteine residue and an N-terminal alanine.78 Likewise, Lei and co-workers leveraged the reactivities of cysteine and lysine to generate asymmetrically cyclized phage display libraries.79 In a different approach, the Nitsche and Jongkees groups incorporated a meta-cyanopyridylalanine (mCNP) in mRNA display.80 This residue reacts selectively with N-terminal cysteines to form a thiazoline heterocycle (Figure 3E). Owing to the complete selectivity of the mCNP-N-terminal cysteine reaction, another cyclization can be performed using another cysteine residue and a classic chloroacetyl-substituted amino acid, thus generating mRNA-displayed bicyclic peptide libraries. They deployed this method to produce a peptide inhibitor of influenza hemeagglutinin with an IC50 of 10 nM. The reactivity of tailored non-proteinogenic amino acids was also utilized in a recent report by the Rovis group in a rhodium-catalyzed peptide macrocyclization strategy, although this has yet to be applied to NELS techniques.81
Apart from chemical strategies, peptide macrocyclization can also be achieved enzymatically with both in vivo and in vitro NELS systems.66 For example, the Bowers group added a post-translation enzymatic cyclization step in mRNA display (Figure 3F).82 The megTYR tyrosinase, isolated from Bacillus megaterium bacteria, catalyzed the oxidation of tyrosine residues to quinone moieties, which subsequently react with the nucleophilic cysteine side chain to cyclize the peptide, alleviating the need for reprogrammed chloroacetyl-containing residues. A NELS campaign using this method allowed them to identify multiple cyclic peptides with single-digit nM IC50 values against melanoma-associated antigen A4 (MAGE-A4). Enzymatic reaction cascades are also at play for lanthipeptide formation (Figure 3G). Lanthionine is formed upon the dehydration of a serine or threonine residue into dehydroalanine (Dha) or dehydrobutyrine (Dhb) respectively, followed by nucleophilic attack from a cysteine.83 By introducing the dehydrated moieties into the peptide sequence containing a cysteine, a cyclic “lanthipeptide” structure can be formed. Seebeck and van der Donk have used such lanthipeptides libraries in phage display and yeast display, respectively.46,84
3.2. Stapling and Other Fold Stabilizations
“Stapling” a peptide consists of stabilizing its structure by chemically linking its side chain—or more rarely, its backbone—to a hydrocarbon staple, thus forcing it to retain a more defined structural fold.85−87 This macrocyclization strategy differs from others in the sense that the staple can be designed to stabilize part of the peptide sequence rather than cyclize the whole sequence. As such, the resulting stapled peptide has a markedly different aspect compared to macrocylic or grafted peptides, i.e., it features a stabilizing cyclic motif flanked by the remainder of the sequence. Much like other macrocyclization strategies, stapling is performed post-translationally by chemical intervention. Staples can be designed not only to confer conformational rigidity but also to enhance membrane permeability or to block off proteolytically sensitive spots in the peptide sequence.88 Stapling is of particular interest to constrain peptides featuring well-defined helical secondary structures such as α-helices.89 Similar strategies have also been used to stabilize other secondary structures, such as β-hairpins. Both α-helices and β-hairpins are widely occurring motifs involved in PPIs.90−93
The strategies used to constrain the structure of α-helical peptides by stapling are chemically similar to those employed in classic macrocyclization approaches, i.e., they rely on side chains on the peptide sequence reacting with a cross-linking staple. Likewise, they should ideally be compatible with the conditions of the NELS method employed, as well as with solid-phase peptide systems (SPPS). A crucial point in this case is the location of the reactive stapling points, which must be carefully fixed in order to maintain the desired structure. Hence, most commonly the positioning follows an i,i + 4, i,i + 7, or i,i + 11 template for helical segments (where i denotes the position of the first stapling point).89
A plethora of staples have been reported, and a recent review by Hu and co-workers covers the topic in great depth.89 Generally, the four most common stapling strategies are lactamization, ring-closing metathesis (RCM), click-chemistry, and thioether formation. Of these four groups, thioether-based strategies have been reported to be the least competent in stabilizing helicity,94 but the thioether handle does provide interesting possibilities. For example, the Li group showed that oxidation of a thioether-stapled nonhelical peptide produced a helical sulfoxide-stapled compound.95 Interestingly, only one of the two sulfoxide enantiomers induced helicity, while the other remained unstructured, as did the overoxidized sulfone product. While the use of such compounds arguably becomes rather complicated, they could potentially be of use as redox-state-selective stapled compounds. Reversible and isomerizable staples have also been reported, e.g., by using photoswitchable azobenzene linkers, which provide a degree of spatial and temporal control over the helicity via light irradiation.96,97
Clearly the most popular and successful stapling strategies recently have been all-hydrocarbon-stapled peptides (Figure 4A) and hydrogen bond surrogate (HBS) based stapled peptides, both prepared through RCM, pioneered by the Verdine lab,98 which have yielded an impressive array of potent PPI inhibitors. Using this strategy, Grossmann and co-workers could develop a PPI inhibitor for the notoriously difficult to target β-catenin involved in the Wnt pathway.99,100 With their cell-permeable stapled peptide, they were able to show the inhibition of cell proliferation of Wnt-dependent cell lines, which even exceeded the activity of the known Wnt inhibitor XAV939.99
Figure 4.

Representative examples of common and emerging peptide stapling strategies. (A) All-hydrocarbon stapling via ring-closing metathesis resulted in a cell-permeable, potent β-catenin inhibitor.99 (B) Side chain bridging through lactamization circumventing the use of non-proteinogenic amino acids.101 (C) Strain-promoted catalyst-free click reaction leading to functionalized MDM2 inhibitors.102 (D) Dithioether formation via nucleophilic substitution yielded GABARAP binders able to inhibit autophagy.104 (E) Novel isonitrile–tetrazine–Ugi four-component reaction applied to an MDM2 inhibitory peptide.105 (F) Hydrogen bond surrogate stabilization increased the potency of β-catenin-binding β-hairpin mimics.110
MDM2 interacts with the tumor suppressor p53 via a PPI interface mediated by α-helical segments, which makes it an interesting target for stapled peptides. For example, it has been probed using lactamization and click chemistry approaches (Figure 4B and C).101,102 Compared with the all-hydrocarbon staple, the advantage of these methods is mainly their synthetic simplicity. The side-chain amide linkage can be installed using the proteinogenic amino acids lysine and aspartic/glutamic acid. The click approach, on the other hand, requires the use of azide moieties; however, with the strained dialkyne linker, no catalyst is needed to facilitate this reaction. Furthermore, it is possible to attach additional functional groups to the linker skeleton. The p53/MDM2 interface is also the target of the stapled peptide ALRN-6924, which has often been cited as the first and so far only stapled peptide to enter clinical trials, although many of these trials have now been terminated due to the lack of a consistent treatment response in the patient cohort despite a favorable safety profile.103
Another stapling strategy that can be implemented without the use of non-proteinogenic amino acids is cysteine-based stapling, predominantly via nucleophilic substitution, Michael addition, or disulfide bond formation. Kritzer et al. were able to utilize this approach in the design of helical peptides targeting GABARAP (Figure 4D).104 Interestingly, an unusual spacing of i,i + 5 yielded the highest binding affinities for the target, and the authors could show that the peptide adopted a 310 helical fold in the crystal structure. Further, introducing a β-branched cysteine analogue also had a beneficial effect, leading to low nanomolar binders against GABARAP that were able to reduce autophagic flux in cultured ovarian cancer cells and sensitize ovarian cancer cells to cisplatin. There is a vast amount of different stapling chemistries available. Next to the established approaches, new strategies are constantly being developed. An interesting recent addition to the arsenal of stapling chemistries, for example, is a multicomponent process introduced by the Bernardes group (Figure 4E).105 By combining the isonitrile–tetrazine (4 + 1) cycloaddition with an Ugi-type four-component reaction, they could establish a new way of introducing pyrazole-amide moieties while simultaneously cross-linking the side chains of the peptide. This approach allows the incorporation of four differently functionalized building blocks and could be successfully applied to create stapled peptides binding to MDM2, which showed cytotoxic effects on several cancer cell lines. Further, they employed machine learning to identify new targets of pyrazole amide derivatives, in particular, OATP1B3, which is also relevant to cancer formation, highlighting the significance of their novel scaffold. In addition to their inhibitor activity, such stapled peptides also boast enhanced proteolytic stability and membrane permeability, likely by blocking cleavable sites of the α-helix and masking polar residues.
In contrast to the thoroughly studied stapling of α-helices, the stabilization of β-hairpins remains more elusive.18 β-hairpin peptides commonly feature in PPIs and have also been reported as potent antibiotics.106 One difficulty lies in the location of the stabilizing unit, which may disrupt both the β-hairpin structure and the binding affinity more than in the case of α-helices. Several approaches have been reported. In an approach that also resembles grafting, the Meldal lab developed a rationally designed class of compounds termed “β-bodies”, wherein one face of the hairpin is stabilized by alternating threonine residues while the other face is free to engage in target binding.107 β-Bodies were further stabilized by a click-chemistry-generated thiazole, and they were used as antibody-mimetics. Notably, the binding regions were obtained through computational evolution rather than conventional phage-display screening.
The other main approach thus far is the HBS strategy.108,109 In this case, after obtention of the β-hairpin peptide candidate, the crucial residues must be identified in addition to the β-turn-inducing residues (typically proline analogues), since the ones involved in the binding must be conserved. Then, other residues can be replaced with suitable reactive handles to stabilize the structure through HBS via RCM. Arora and co-workers, for example, were able to use this approach in the design of β-hairpin mimics targeting the Tcf4/β-catenin interface. After introducing a de novo functionalizable strand to a known binding motif and stabilizing the whole sequence via thioether-mediated HBS, they could show a roughly 10-fold increase in potency compared to the linear analogues (Figure 4F).110
3.3. Conjugation to Functional Entities
The properties of potential peptide drug leads may also be optimized by covalently linking them to another functional entity like a small molecule, peptide, or biomolecule. In this regard, it is relatively straightforward to increase the membrane permeability of a compound by fusing it with a cell-penetrating peptide (CPP). This CPP conjugation was applied, for example, in the case of a stapled peptide targeting the β-catenin transcription factor PPI, which significantly improved its efficiency in a viability assay of Wnt-dependent cell lines compared to the non-CPP-conjugated control.99 However, this strategy is somewhat unpredictable as the mechanisms of cellular uptake of such conjugates are still poorly understood, although the Pei group very recently elucidated the uptake mechanism of three classic CPPs (R9, CPP12, and CPP17).111
Another issue of peptide hits derived from NELS methods is that their exact binding spot on the target protein is a priori unknown. Therefore, they might not show the originally intended inhibitory properties, or, for example, their disruption of a given PPI might not be strong enough to show satisfactory in vivo effects. This problem can be addressed by conjugating the peptides to a proteasome-directing entity, leading to proteolysis-targeting chimeras (PROTACs). Upon binding to the target protein, this chimera will direct the protein to the proteolytic machinery of the cell, ultimately leading to degradation of the target. Employing this strategy to the β-catenin-binding stapled peptides, the Chen group showed that the resulting compound is able to not only strongly inhibit Wnt signaling in cancer cells and APC–/– organoids but stall and even reduce tumor size in APCMin/+ mice, which is a common model with aberrant Wnt signaling used to simulate human familial adenomatous polyposis112 and colorectal tumors.113
A relatively new approach to macrocyclic peptides, grafting consists of tethering the peptide pharmacophore onto the loop of a protein scaffold, a topic we recently reviewed.114 This way, the resulting chimera inherits the properties of both the peptide and the scaffold, such as enhanced serum stability, water solubility, reduced renal clearance, etc. Furthermore, as the peptide cyclization now results from the scaffold protein structure rather than from chemical synthesis, the grafted compound can be genetically encoded and subsequently prepared through cellular expression protocols or even conceivably directly delivered to the target area for in situ drug expression.
As grafting is a relatively new concept, its definition can vary, and it has even been used interchangeably with the previously discussed stapling strategy.115 Herein, we consider grafting strictly as the fusion of a peptide motif into a protein scaffold, as opposed to stapling, which is rather the stabilization of a peptide’s structure by anchoring to a rigid organic skeleton.
The original approach developed by Craik is called “molecular grafting” and draws inspiration from natural compounds called cyclotides, a type of CKP.116 These compounds, such as trypsin inhibitor MCoTI-II or Möbius type Kalata B1, contain cysteine knots that strongly rigidify their structures and bestow them with a particularly stable and constrained conformation.74 Molecular grafting then consists of excising one of the cyclotide’s loop sequence and replacing it with the peptide sequence of interest, usually selected for its binding affinity, called an “epitope” (Figure 5A).116 Said epitope then benefits from the structural constraint brought about by the cyclotide scaffold and is able to better maintain its biologically relevant conformation, thus enhancing its therapeutic potential (as opposed to its linear counterpart). There is great flexibility as to the nature of the sequences that can be grafted this way, as selected target mutations have shown that only a few residues are crucial for proper folding.75,117,118 Mutated libraries can be expressed and screened in vivo,(117) and even randomized grafting sequences can be screened by NELS on certain CKP scaffolds.76 Similar approaches can be used to create antibody mimetics.119,120 By obtaining a lead peptide epitope (typically through phage display) to be grafted onto the variable regions of an antibody scaffold, the resulting chimera transfers the binding properties of the epitope onto the defined structure of the antibody scaffold derived from its constant regions. Using a variety of antibody scaffolds, this approach has given rise to a myriad class of compounds, such as monobodies,121 adnectins,122 designed-ankyrin-repeat proteins (DARPins),123 and affibodies.124
Figure 5.

Small proteins with peptide pharmacophore sequences. (A) Molecular grafting consists of excising a loop sequence out of a scaffold (here, two typical cyclotides are shown) and replacing it with the sequence of the peptide epitope, often derived from phage display.116 (B) Lasso-grafting spacer optimization of the U-body carried out by mRNA display.126 A RaPID-derived peptide epitope is encoded into an mRNA library, flanked by spacers that connect it to a scaffold protein, in this case, ubiquitin. Regular mRNA selection is then performed, and after suitable enrichment through multiple rounds of selections, lasso-grafted compounds are obtained with optimal spacers for the retention of the initial affinity.
Another approach termed “lasso-grafting” was recently developed through a collaborative effort from the Takagi, Matsumoto, and Suga groups.125 In this method, a macrocyclic peptide epitope is first obtained through RaPID selection. Then, the epitope is grafted on the exposed loop of a protein scaffold via a few residues called “spacers”. As the peptide sequence is anchored to the loop rather than substituting a native region of the scaffold, this results in the epitope protruding from the protein core in a lasso shape that strongly resembles the original cyclic peptide’s conformation. Many proteins can be used as scaffolds, including uteroglobin or adeno-associated virus capsids, but of particular interest are Fc and ubiquitin. Fc is the homodimeric crystallizable fragment of the immunoglobulin G antibody and features eight loops amenable to grafting. Using a RaPID-derived peptide for grafting on Fc yields constructs referred to as “Mirabodies”. These inherit both the binding affinity from the grafted peptide and Fc’s desirable properties such as excellent serum stability, bivalency, and compatibility with protein A-based immobilization techniques or immunostaining with anti-Fc antibodies. Furthermore, the presence of 8 loops allows for grafting of different epitopes to produce multispecific Mirabodies.
Lasso-grafting to loop 1 of ubiquitin has also been reported by the Suga group to produce protein–peptide hybrids called “U-bodies”. Ubiquitin is an interesting scaffold thanks to its favorable serum and thermal stability, small size, lack of cysteine residues, and ease of production in E. coli. As ubiquitin is also readily multimerized, multimeric U-bodies can be produced, which have been shown to feature enhanced properties compared to their monomeric U-body variants.126 In grafting technologies, the residues adjacent to the inserted sequence, called “spacers”, are of paramount importance.127 Grafting with naïve spacers can result in a significant loss of biological activity. This is likely due to the loss of conformation caused by unoptimized anchoring. In this regard, optimization of the spacer can be performed by mRNA display, as demonstrated with U-bodies (Figure 4).126 First, a peptide epitope is identified through RaPID selection. Then, a new mRNA library is constructed wherein the epitope is genetically encoded on loop 1 of ubiquitin and flanked by short randomized sequences. Iterative rounds of selection against the immobilized target of interest are then performed, leading to the enrichment of the most suitable spacer sequences. Interestingly, it was found that disulfide bridges in the spacers were not necessary for retention of the epitope’s affinity. Likewise, not only the length of the spacers but also the identity of the residues were found to converge, suggesting that the spacers are involved in stabilizing the binding event. Incremental optimization from the original cyclic peptide to the naïve-grafted U-body and then spacer-optimized and ultimately multimeric U-bodies provided increasingly serum-stable binders of c-MET. Although not tackling an inaccessible target, these U-bodies displayed superior affinity to cMET compared to its natural ligand, i.e., hepatocyte growth factor HGF, a known tight interaction. By fully separating the epitope discovery from the scaffold and spacer optimization, lasso-grafting provides the flexibility to transfer the binding properties of a peptide to any compatible scaffold. As these reports are still recent, it remains to be seen which protein scaffolds are most suitable for a given scenario and how challenges such as intracellular delivery can be overcome.
3.4. Incorporation of Non-Proteinogenic Amino Acids
Peptide drugs have the general problem of being susceptible to proteolytic degradation. If unmodified, they usually degrade quickly in vivo, seriously hampering their applicability.128 Therefore, improving their biological half-life is critical for the development of peptide drug leads. This can be accomplished through a variety of different strategies including sequence optimization, the use of npAAs, or stabilizing the peptide fold, for example, through macrocyclization or grafting to protein scaffolds. In NELS platforms that rely on chemical synthesis, such as DELs, the use of exotic building blocks is straightforward and only limited by the feasibility and compatibility of the available synthetic strategies. However, since they are not well recognized by the ribosomal machinery, the incorporation of npAAs in phage display and related technologies is difficult. One strategy to overcome this issue is the use of synthetic d-proteins in a mirror-image phage display (MIPD). The obtained l-peptide hits are equivalent to the respective d-peptides, which can bind to the l-protein target. Since the hydrolysis of the peptide bonds is mediated through enzymatic reactions that are optimized to recognize their natural l-amino acid substrates, the use of enantiomeric d-amino acids increases the biological stability of a peptide.129 Although this approach was already applied two decades ago, for example, to develop binders against the amyloid-Aβ plaques,130 only a handful of MIPD selections have been performed so far.131 The reason for this is the difficulty in obtaining d-proteins, as they cannot be recombinantly expressed and therefore have to be fully synthesized chemically. In this regard, the Pentelute lab recently published an extensive study of the production of several d-protein targets via single-shot fast-flow synthesis (Figure 6A). Using phage display, they were able to obtain different stapled d-peptides that were able to bind MDM2 in the low micromolar to nanomolar range.131
Figure 6.

Common strategies for the incorporation of non-proteinogenic amino acids into NELS of bioactive peptides. (A) Mirror image phage display method showing the selection workflow and an exemplary hit peptide against MDM2.131 In the X-ray structure, the peptide backbone is shown in ribbon representation, the staple is shown in purple, and side chains interacting with the protein are shown in blue. (B) Genetic code reprogramming to incorporate various cyclic β- and γ-amino acids.134 The left, right, and upper outside parts of the tables show the nucleobases of the mRNA codons, whereas the inside shows the respectively assigned amino acids in one-letter code. The hit peptide on the right was obtained from an mRNA display selection against the SARS-CoV2 main protease. (C) Strategy to implement cysteine reactive warhead moieties into mRNA-displayed peptides.146 Translation of the phenyl selenoether side chain containing peptides and oxidative treatment liberates the alkene warheads. After incubation with the target, a denaturation step ensures that only covalently linked peptides remain.
In vitro ribosomal systems are more amenable to the use of npAAs because they utilize isolated and recombinant elements for the translation step, allowing full control over the presence or absence of certain components, e.g., amino acids, tRNAs, or aaRSs. Through depriving the system of some of the proteinogenic amino acids and replacing them with non-proteinogenic ones, the Szostak group managed to create a library containing 12 non-proteinogenic amino acids and utilized it to obtain nanomolar binders against thrombin.132 However, this method is limited to the incorporation of npAAs that are recognized by natural aaRSs.
By using flexizymes to load npAAs on any tRNA, the FIT system enables the incorporation of a wide variety of residues.54,133 Of particular interest are npAAs that can strongly influence the secondary structures of cyclic peptides. For instance, cyclic β-amino acids are promising building blocks for peptide-based therapeutics due to their ability to induce strong helix/turn structures, enhance structural rigidity, and increase proteolytic resistance. Katoh et al. successfully translated peptides containing up to ten consecutive cyclic β-amino acids residues (Figure 6B).134 RaPID selection was performed using this new library to produce compounds with well-defined secondary structures and impressive serum stability. Likewise, α,α-disubstituted amino acids have been introduced into the FIT system and used in RaPID selection. Likewise, γ-amino acids are interesting building blocks that can act as unique helix turn inducers. A recent report highlights the efficient incorporation of cyclic γ-amino acids into the FiT system, which were screened in a RaPID campaign against the SARS-CoV-2 main protease (Mpro).133 The lead hit peptide GM4 showed very strong properties, with a 5.2 nM KD, 50 nM IC50, and 126 h half-life in serum. α,α-Disubstituted amino acids are also relevant as strong inducers of α-helicity and would benefit from further investigation, as they remain less common than other npAAs despite some examples of incorporation in RaPID.89,135,136N-Alkyl amino acids another area of interest, as they can confer structural constraints to the cyclic peptide as well as mask polar groups to improve membrane permeability. A large collection of N-alkylated amino acids have also been incorporated into the FIT systems through rational tRNA engineering.137−140 It should be noted that while some npAAs, such as N-methyl amino acids, are efficiently translated in the ribosome and are therefore wildly compatible with RaPID, their use in SPPS can be much more difficult. As this limits their potential to translate from NELS discovery to further development, efforts should be deployed to improve their synthetic chemistry.
Non-natural moieties can also be introduced or modified through bioorthogonal, post-translational chemical, or enzymatic modifications. In phage display, a reactive aldehyde handle can be installed via oxidative treatment of N-terminal serine or threonine residues, which has been used for conjugation to glycans, various pharmacophores, or biotin.141 Cysteine residues are commonly modified with electrophiles, Michael acceptors, or nitrile reagents in the case of N-terminal cysteines. This enables similar conjugation strategies but is mostly used for the creation of cyclic or stapled peptides or to install warheads. Next to chemical methods, enzymatic modification can serve as a complementary approach, albeit being limited by the sequence specificity of the enzymes. Biosynthetic pathways for known ribosomally synthesized and post-translationally modified peptides (RiPPs) could be utilized for the generation of partially randomized pseudo-natural products, for example, lanthipeptides,46,83 or thiopeptides.142 Another approach is the use of ligating enzymes, exemplified by the integration of a prenyltransferase into the FIT system, allowing the in vitro ribosomal synthesis of geranylated macrocyclic peptides.143
Lastly, the use of npAAs allows the incorporation of reactive functionalities that might be used as warheads. Selected covalent interactions with the target protein can drastically improve the specificity and efficiency of the cyclic peptide drug leads. For example, this strategy has been employed on the well-studied inhibition of the p53/MDM2 interface by stapling an α-helical MDM2-binding peptide with a bifunctional lysine reactive linker, resulting in a covalent inhibitor with nanomolar affinity.144 Generalization of warhead strategies is on the way, and they are increasingly included in display screening platforms.145 In a recent study, cysteine-reactive dehydroalanine residues were used in an mRNA display screening against two protein targets involved in PPIs: CIB1 and MAGE-A4 (Figure 6C).146 A denaturation step was included after incubation with the protein targets to ensure the selection of only covalently bound peptides, resulting in the discovery of nanomolar inhibitors for both targets.
4. NELS-Derived Cyclic Peptides for Inaccessible Targets
4.1. Targeting Intracellular Proteins
Arguably the largest barrier to clinical success for cyclic peptides is their notoriously poor cell membrane permeability. Despite the wide arsenal of NELS techniques, exotic building blocks, and structural modification strategies, many if not most of the cyclic peptide inhibitors reported in the literature fail to translate their promise in vivo simply because they cannot reach their target. Compounding this difficulty is the fact that many intracellular proteins also have one or more undruggable features (lacking well-defined binding pockets, PPIs, or IDPs). Consequently, the peptide community has dedicated vast amounts of efforts and resources to tackle the membrane permeability frontier, thus far with limited success. Furthermore, the penetration capabilities of cyclic peptides can be unpredictable. In this regard, efforts are being made to collect cell permeability data and construct computer models or build rational design principles to assist in the design of cell-penetrant peptides.147−151 In this section, we will present general considerations for targeting intracellular processes with cyclic peptides and examples thereof. These general principles can thus be considered to apply to the other specific categories of inaccessible targets discussed in further subsections.
The multitude of strategies to get cyclic peptides into the cell can be grouped into three general approaches: (1) conjugation with cell-penetrating motifs such as CPPs, (2) structure manipulation of the peptide architecture, e.g., by stapling or masking certain polar moieties (peptoids), and (3) vector-based delivery, e.g., with liposomes.
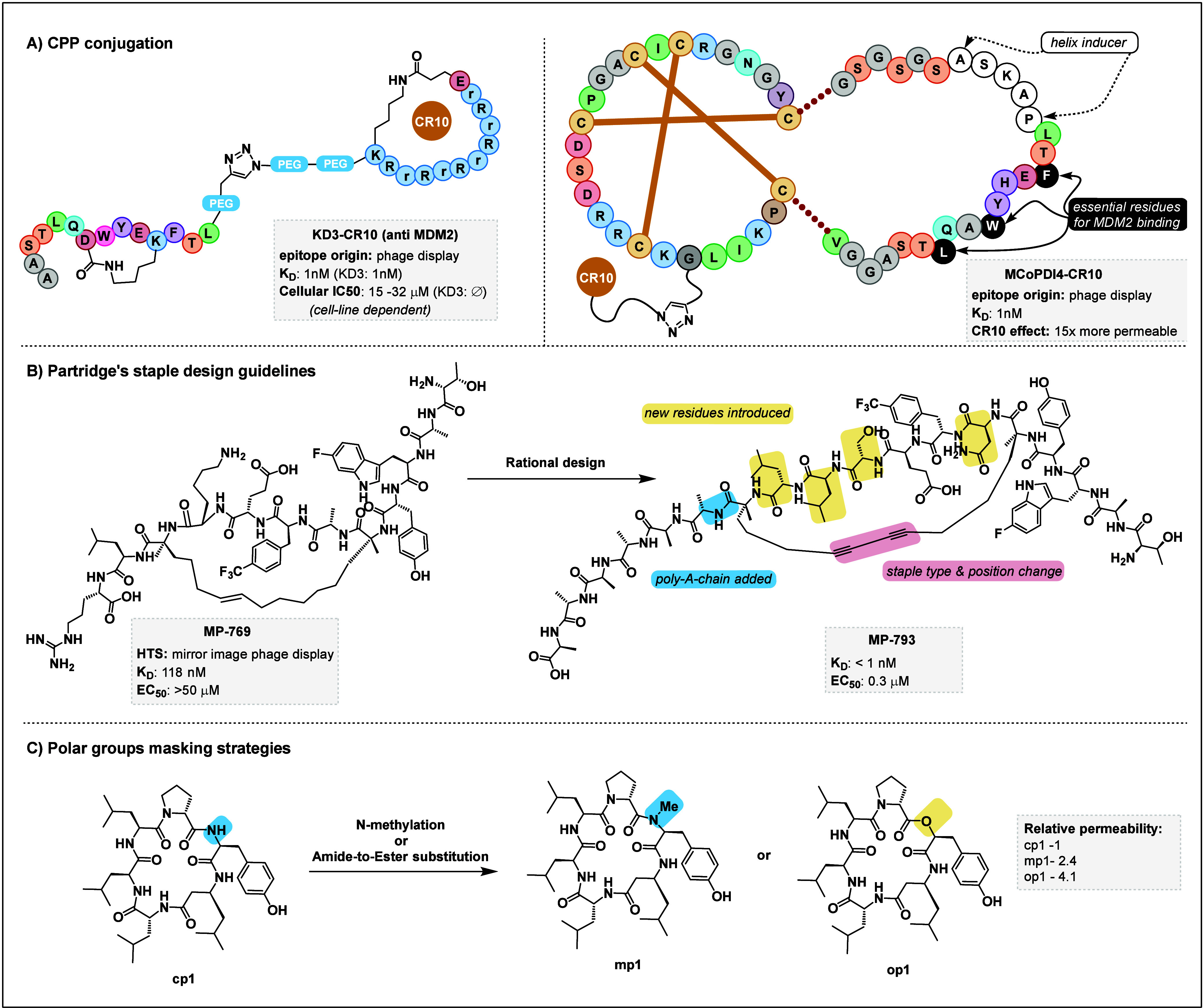
As mentioned in section 3.3, conjugation with CPPs has been popular as a relatively straightforward approach).152 However, as the resulting conjugated peptide may sometimes lose its affinity or its secondary structure, it requires careful characterization and optimization. Polyarginine chains have been used to grant cell permeability to promising but nonpenetrant drug candidates.153 Likewise, grafting of a CPP motif on exposed loops of engineered proteins has been reported, which could be combined with other grafting techniques to deliver grafted peptide binders.154,155 The Craik and Henriques groups recently reported the design of cyclic CPP-conjugated peptides called “angler peptides”.101 In this approach, a macrocyclic peptide inhibitor identified through phage display was conjugated to a cyclic CPP motif via a clicked triazole linker. The authors applied this approach to a p53/MDM2 nonpermeable stapled peptide inhibitor (Figure 7A, left panel). The resulting KD3-CR10 angler peptide retained a 1 nM KD and could restore p53 activity in cancer cells at the low micromolar range. This versatile and conceptually simple strategy could be applied to bestow cell permeability on other NELS-derived peptidic drug candidates, although it should be noted that the mechanism of action and the secondary structure of the peptide may be influenced strongly by conjugation to the cyclic CPP motif, thus requiring careful characterization.101 The same groups also extended the cyclic CPP conjugation strategy to a cyclotide grafted peptide (Figure 10 A, right panel).156 In this study, a peptide epitope originally identified from phage display was grafted on loop 6 of the MCoTI-II cyclotide scaffold. This peptide also included the apamin linker ASKAP sequence, which induces helicity, as an α-helix is required for proper binding to MDM2. With this design, the grafted and CR10-conjugated cyclotide presents the epitope in the relevant helical fold, with the three residues crucial for the interaction efficiently presented on the same face of the helix. The rather large grafted compound retains an excellent 1 nM binding affinity, and the CR10 conjugation enhances its cell-penetrating ability more than 10-fold. Other examples of CPP conjugation include a report from Schneider et al. on an mRNA-display-derived peptide, producing a cell-penetrating cyclic peptide conjugate inhibitor of MDM2 with an IC50 of ca. 1 nM, whereas the original peptide had no inhibitory action in cells despite a remarkable in vitro IC50 value of 0.18 nM.157 The Pei group also conjugated a CPP motif to otherwise impermeable peptide, also targeting the p53/MDM2 interaction as well as the β-catenin/TCF PPI.158
Figure 7.
Selected strategies for the improvement of cyclic peptide cell-permeability. (A) Left panel: Angler peptides combine a CPP motif and a stapled peptide for enhanced intracellular drug delivery.101 Right panel: CR10 conjugation was also used on a grafted cyclotide to deliver a potent MDM2 binder.156 (B) Stapling optimization guidelines enabled the rational optimization of an MDM2/p53 inhibitor.88 (C) Polar group masking can be done by including N-alkylated amino acids in the library design or through structural optimization as shown here.177 In this case, the amides of a small cyclic peptide were either methylated or substituted for ester functions. In all cases, improvements in the cell permeability as well as the proteolytic stability were observed.
Figure 10.

Alternative approaches for generating bioactive macrocyclic peptides. (A) A stereochemically diverse bicyclic peptide library was fitted with an alkyne for covalent “click” ligation of the MYC target protein upon binding by the peptide sequence.235,236 Complex downstream analysis is then performed to identify the correct binding sequences and their stereochemistry. Note that the overall design was simplified for clarity in this figure. (B) Proteomic-peptide phage display (ProP-PD) exploits the principle of classic phage display, but rather than displaying peptide binders, short-linear motifs (SLiMs), disordered regions from the proteome, are displayed.237−240 The library is then screened against a collection of high-quality bait proteins, enabling the identification of PPIs and mapping of the human interactome.
As described in section 3.2, in addition to stabilizing secondary structures, stapling has been proposed as a promising way to confer cell-permeability to cyclic peptides. In their review, the Hu group suggested that rather than developing new staples, research efforts should focus on elucidating the in vitro and in vivo biological activity, pointing to the dearth of stapled peptides advancing to clinical trials.89 A recent publication by the Partridge group followed up on this opinion and provided guidelines for the design and optimization of stapled peptides.88 They propose a five-step workflow: (1) acquisition of a peptide binder via NELS, (2) selection of the stapling points, (3) optimization of the peptide length, (4) optimization of staples, and (5) final sequence optimization. A key recommendation in the hit acquisition step is the removal of cationic residues that are not essential for binding to alleviate cell toxicity and concerns over false-positive hits.159 Regarding staple optimization, the guidelines suggest starting with an i,i + 7 design for optimal stabilization of the helicity and proteolytic resistance. As the amount of potentially available staples can be intimidating, they also recommend performing a “staple scan” restricted to a single chemistry group, e.g., RCM-based. Finally, final sequence optimization should focus on finding an appropriate balance between polar and apolar residues, preferably aiming for an amphipathic helical structure that is not excessively hydrophobic, as this can result in toxicity issues. Application of these guidelines to a peptide series targeting MDM2 resulted in a more than 100-fold improvement of in vivo potency (Figure 7B). The authors also note a strong correlation between peptide lipophilicity and membrane permeability and a weaker correlation with helicity, although improved aqueous helicity was also found to enhance permeability. While the authors do remark that these guidelines have not been tested toward other targets, and that they should not be construed as universal truths, they represent an important set of design principles that could be applied to stapling strategies in general, including those that are integrated with phage or mRNA display.160−162 The Verdine group also contributed an in depth analysis of the effect of stapling peptides on cell permeability.163 Their findings are in agreement with the above-mentioned guidelines, highlighting the importance of both staple type and formal charges for the membrane penetration potential. Although much focus has been placed on stapled α-helical peptides, stabilized β-hairpin structures are also relevant to cell penetration.164−167
Aside from stapling, other methods of architectural manipulation can be used to enhance cell permeability. Peptide backbone cyclization strategies have been reported to enhance cell permeability compared to side-chain cyclization, and their integration in NELS display systems is emerging.168,169 They may resemble stapling in that they can also integrate organic cross-linkers whose properties can enhance permeability further, as exemplified by a thioether-bipyridyl ring-closing unit reported by the Suga lab.170
Incorporation of non-proteinogenic residues in the peptide structure can also be leveraged to induce defined secondary structures. For example, foldamers, i.e., organic structures with a strong tendency to adopt and induce well-defined secondary structures, can be used.171 Huc and Suga used the RaPID system to reprogram the initiator codon in their mRNA library to encode an organic foldamer.172 The translated compounds then formed a dual-helical macrocyclic peptide library, which could be used to target PPIs known to favor α-helical conformations. Foldamers can also be obtained from peptoids, i.e., peptide-like compounds featuring N-functionalized amino acid residues.173 Peptoids may also adopt β-hairpin structures,174 or other defined shapes.150,175
N-Alkylation in particular has been shown to increase cell-permeability.176 In this regard, genetic code reprogramming by FIT has been widely expanded to incorporate N-alkylated amino acids in the RaPID systems. Subsequently, many RaPID selections have been performed for the identification of macrocyclic-peptide-containing N-functionalized amino acids.
Although N-alkylation is efficiently performed in RaPID, it is notoriously challenging in SPPS systems. Therefore, alternative ways of masking polar or sensitive moieties on the peptide chains are also desirable, as the increased lipophilicity/decreased overall formal charge correlates with enhanced membrane permeability.88 Inspired by natural peptides, the Sando group investigated the effect of substituting backbone amides with ester groups, a class of compound known as depsipeptides whose permeability properties had hitherto not been studied (Figure 7C).177 Evaluation of cell permeability by a PAMPA assay revealed that, overall, amide-to-ester substitution had a comparable or superior enhancement effect on cell penetration ability compared to N-methylation. Likewise, the Chatterjee group substituted amides for thioamides.178 Remarkably, this O to S single-atom replacement resulted in significant improvements in cell permeability, measured by both PAMPA and cellular assays. The thioamidated peptides also featured enhanced metabolic stability. Although in these studies the permeability enhancements and the structural impact of amide substitution could be unpredictable, patterns may emerge upon wider adoption of these alternative strategies to N-methylation.
Finally, a roundabout strategy is to bring the cyclic peptide to its intracellular target as a cargo via a suitable delivery vector, for example, via liposomes. For instance, Sauter and co-workers delivered the well-known cyclic peptide vancomycin intracellularly by encapsulation in liposomes that were modified with a polyarginine CPP motif to enhance their cellular uptake.179 A recent review Liu et al. provides an in-depth look into liposomes as drug delivery vehicles.180 They point out that while the field is still somewhat in its infancy and suffers from drawbacks, such as complex and costly manufacturing processes, reproducibility issues, and rapid blood clearance, there is considerable research effort being deployed on the topic, which is expected to mainstream this technology within the next decade.
4.2. Small GTPases
A significant challenge in drug discovery is posed by proteins that lack well-defined binding pockets, rendering traditional small-molecule inhibitors as ineffective. Among such proteins is a large collection of small GTPases, biological switches that upon converting GTP to GDP trigger a variety of signaling cascades. These proteins, often characterized by flat or featureless surfaces, present formidable obstacles for therapeutic intervention, in particular, as small molecules are generally inadequate for such targets. In the absence of hints for rational design, the NEL screening of large macrocyclic peptide libraries for these targets is particularly attractive.
Arguably the most famous protein target in this category is KRas, which has long been one of the “holy grails” of undruggable targets.3 The Ras family (which also includes HRas and NRAS) is involved in multiple downstream pathways essential for functional cell health. Ras mutations, however, are virulent drivers of cancers and typically associated with poor prognosis. Over the years, KRas has become the poster child of clinically attractive targets whose lack of deep-binding cavity impedes drugging efforts and has even been described as the “death star” in mainstream publications. There are two main reasons that KRas is a significant challenge: (1) as a GTPase, it has picomolar affinity for GTP, against which an inhibitor would be hard-pressed to compete, and (2) its surface does not feature notable binding sites that could be targeted. To get around these problems, researchers have instead targeted downstream effector of KRas such as P13K for which cyclic peptides have been identified via SYCLOPPS.181
The undruggable aura of KRas was famously shattered when the Shokat group reported a potent inhibitor of the KRas(G12C) mutant.11,12 Sotorasib and adagrasib have since been approved by the American Food and Drug Administration (FDA) for the treatment of KRas(G12C)-mutated non-small cell lung carcinoma.182 Crucially, the mutant cysteine residue was covalently targeted by small-molecule compounds, conferring an excellent mutant vs wild type selectivity and therefore limited off-target toxicity (an otherwise typical downside of cancer treatments). Follow-up studies now aim at enhancing this further to overcome rapidly developing drug resistance.183
Other mutations of KRas, including the most common G12D mutation, do not feature a cysteine residue vulnerable to covalent inhibition, and aspartate-targeting chemistry is much less common. However, earlier this year the Shokat group for the first time identified a malolactone compound capable of covalently modifying the acquired aspartate of KRas(G12D).184 This compound showed high potency and mutant selectivity, representing a remarkable breakthrough in targeting challenging KRas mutations.
With regards to cyclic peptides, in 2020 Shokat and Suga collaborated to identify macrocyclic peptide inhibitors of KRas(G12D).185 A key feature of this study was the modified RaPID selection deployed for the selection of binders (Figure 8A). A GTPase switch, KRas exists primarily in a GTP-bound “on” state and a GDP-bound “off” state. The G12D mutation results in a loss of GTPase activity, effectively locking KRas in its “on” state and leading to aberrant activity such as promoting tumor growth. With this in mind, the screening strategy focused on identifying a binder selective for the GTP-bound (“on”) state over the GDP-bound (“off”) state. This would result in primarily inhibiting the most abundant form of KRas(G12D) and also limiting interference with healthy cells, where a significant amount of KRas is found in the off-state. To this aim, the RaPID screening was modified to include a counter-selection. The cyclic peptide library is first screened against the immobilized GDP-bound KRas(G12D), thus removing off-state binders. The remaining library is then normally screened against the GTP-bound KRas(G12D), resulting in amplification in preferentially on-state binders. After several rounds of selection, three peptide sequences were identified that showed selective binding to the on-state of KRas(G12D). Of these, peptide KD2 (Figure 5B) was found to be the most potent, and cocrystallization revealed its binding to the switch II groove region of KRas(G12D)·GTP. One threonine residue in the peptide sequence, which showed direct interaction with the mutant aspartate, could be replaced with non-proteinogenic amino acids to generate more potent inhibitors with up to 10-fold increased binding affinity. However, these peptides failed to show cellular activity, likely due to having little-to-no membrane penetration capacity. This modified RaPID screening strategy was also deployed by the same groups to target another inaccessible GTPase,3 Gαs.153 Potent binders of both GTP- and GDP-bound states were identified (Figure 8B). Notably, in both studies, the peptides identified showed a strong in vitro affinity in the nanomolar range that did not translate to in vivo studies. This was again ascribed to the poor cell-membrane permeability of the peptides. Therefore, the Gαs binders were conjugated with a polyarginine tail, a well-known cell-penetrating-peptide motif. Although the improved compounds were found to penetrate the cell, their potency was still lacking, likely due to the more complex environment and competing PPIs of the cellular system as compared with the in vitro selection. While this may be improved by further optimization of the peptide’s structure, this also highlights a common drawback of mRNA display, i.e., the affinity of the selected peptide does not always translate into inhibitory activity once in the cell.
Figure 8.

Multiple KRAS inhibitors have been reported. (A) A modified RaPID screening can be employed, in which the library is first panned against the undesired state of the target (here, KRas(G12D)·GDP).185 The unwanted binders are thus removed, and the remainder of the library is screened against the desired state (KRas(G12D)·GTP). The rest of the selection proceeds as in a normal RaPID selection. With this strategy, lead compound KD2 was obtained, which was strongly selective for the GTP state of KRas(G12D) over its GDP state. (B) The same strategy was employed to identify the state selectivity of another GPCR, Gαs.153 (C) Bicyclic peptide KS-58 was derived from a hit obtained from phage display,190 then optimized for selectivity toward the G12D mutant over the wildtype.189 (D) Chugai Pharmaceuticals conducted a rational optimization of a hit KRas(G12D) inhibitor,186 whose initial hit precursor was identified through mRNA display.187
Two other peptide inhibitors of KRas(G12D) were reported recently.186−189 While both are derived from initial hits identified through phage190 or mRNA display,187 respectively, extensive SAR studies and classic medicinal chemistry campaigns were then conducted to obtain the final drug candidates. Sakamoto et al. reported a cyclic peptide derived from phage display that featured a notable bicyclic structure, including a double thioether and a backbone amide linkage (Figure 8C). Remarkably, this compound showed a 4× higher binding affinity to the G12 mutant over the wild type and even showed some membrane permeability, which the authors recently probed further through molecular dynamics.191 Chugai Pharmaceuticals also reported the discovery of LUNA18, a noncovalent KRas inhibitor.186 While the initial hit was obtained by mRNA display and boasted a high binding affinity, it had no cellular activity, likely due to an incapacity to cross the cell-membrane. They approached the hit-to-lead optimization in a more traditional fashion, focusing on side-chain optimization rather than scaffold-hopping (Figure 8D).187 The resulting LUNA18 improved its affinity over 20× to about 2 nM, with a remarkable 1.4 nM IC50. The authors argue that such strategies should be more widely considered for turning NELS-derived peptide into viable drugs, and they expect that as further patterns and guideline emerge from peptide discovery, semirational design will facilitate this approach and streamline the straightforward optimization of hit peptides for solubility, permeability, and inhibitory activity.
4.3. Protein–protein Interactions (PPIs)
Protein–protein interactions (PPIs) play fundamental roles in diverse biological processes governing signal transduction, gene regulation, and protein function. Dysregulated PPIs are implicated in numerous diseases, including cancer, neurodegenerative disorders, and infectious diseases, highlighting the therapeutic potential of targeting challenging protein interactions. Much like proteins lacking well-defined binding pockets, as PPIs typically involve large, flat interaction surfaces, they are particularly difficult to inhibit with small molecule drugs; in fact, many targets from the first category are also involved in PPIs. The proposed model of “hot spots” stipulates that, while the absence of a conventional binding pocket precludes standard druggability, compounds can be designed that target multiple hot-spots through binding fragments connected to some sort of scaffold (Figure 9A). This enabled the development of fragment-based drug discovery (FBDD), a technique analogous to HTS except that rather than screening libraries of complex compounds, smaller “fragments” libraries are screened instead.56,192 The binding fragments thus selected are then stitched together through rational design and optimized in a classic drug-discovery approach. This method has gained significant popularity57 over the last decades and, in fact, it was instrumental in the development of the KRas(G12C) inhibitors discussed in section 3.1.11,12,193 Famously, FBDD also led to the discovery of the leukemia drug venetoclax, a potent inhibitor of another classic inaccessible target, BCl-2.57 Alternatively, macrocyclic peptides are well-suited for PPI inhibition and have regularly been touted as a promising pharmaceutical development avenue.30,194,195 Their larger potential binding area is a better match for that of PPI surfaces, and amino acid residues (including non-proteinogenic ones) can interact with the hot spots in a manner that inherently mimics natural interactions. While peptides featuring defined secondary structures such as α-helices or β-hairpins are privileged scaffolds for PPI interactions, macrocyclic peptides devoid of these features are also viable PPI inhibitors. This is exemplified by the SICLOPPS-obtained hypoxia inducible factor 1 (HIF-1) compounds reported by the Tavassoli group, which showed micromolar-range inhibition in hypoxic U2OS and MCF7 cell lines. (Figure 9B).196,197 Another family of cyclic peptides has been reported to target resistant mutants of Bcl2 (Figure 8C).198 While these peptides lacked a membrane-penetration capacity, conjugation to the known cell-penetrating-peptide TAT enabled the intracellular delivery of lead compound cp1, resulting in a 27 μM IC50. New Bcl2 inhibitors are of particular interest, as since the approval of venetoclax resistance has started to emerge through mutations that threaten to thwart current therapeutic strategies. An alternative to structurally constrained PPI inhibitors is to screen for covalent binding.147 This approach, which is closer to traditional drug development strategies, has gained renewed interest following the “drugging” of KRAS(G12C) by a covalently bound small molecule. This emerging strategy relies on the installation of suitable electrophilic warheads on peptides that can react with a nucleophilic residue at the PPI interface. Chen et al. designed a phage-display library cyclized from two cysteine residues with a dichloroacetone linker.199 They then functionalized that linker further into either a vinyl sulfone or a diphenylphosphonate moiety, which reacts with cysteines or serines, respectively. By performing phage-display selections with these functionalized libraries, they were able to obtain cyclic peptides that covalently bound to the protein of interest selectively. This suggests that the sequence of the peptide is essential for the initial binding, while the covalent linkage occurs later on and stabilizes the bound complex. In another covalent approach, Gao and co-workers introduced an ortho-acetylphenylboronic acid (APBA) moiety on a cross-linker used to cyclize a phage-display library (Figure 9D).200 The APBA warhead selectively binds lysine residues by forming an iminoboronate, which is notably is reversible. The lead peptide from this study, S4-APBA3, showed low-micromolar-range inhibition of the sortase A (SrtA) of Staphilococcus aureus.
Figure 9.

(A) The hot-spot model presumes that there are punctual binding sites on the otherwise flat PPI interface. This makes it amenable to fragment-based drug design (FBDD) and macrocyclic peptide NELS screenings.192 In this example, the peptide’s structure is stabilized by stapling. (B) A small cyclic peptide with sub-micromolar activity against HIF-1 was identified by SICLOPPS.196 (C) A family of macrocyclic peptides exhibit potent activity against BCl2.198 (D) Covalent binding can be used in conjunction with other methods to enhance PPI inhibition. In this example, an acetylphenylboronic acid warhead installed on an organic staple specifically binds to lysine residues when brought in close proximity by the standard interaction of the peptide with the protein.200 (E) CKP grafting leveraging phage display and directed evolution yielded a potent CKP binder of LRP6 to inhibit the extracellular Wnt1/LRP6 PPI.76
There are multiple interesting PPIs in the Wnt signaling pathway, notably β-catenin/TCF and β-catenin/Bcl9, for which there are several reported small-molecule201,202 and macrocyclic peptide binders (particularly stapled peptides),203 although these were generally obtained from rational structure-based design rather than NELS campaigns.99,112,204−208 The onset of the Wnt signaling pathway also features a variety of interesting extracellular PPIs.100 Hansen et al. targeted one of these through molecular grafting onto a CKP, i.e., the squash trypsin inhibitor EETII-I (Figure 9E).76 They grafted a randomized sequence onto various loops of the scaffold and then submitted the randomized grafted library to phage display and directed evolution, which led to remarkable convergence of sequence motifs binding to the LRP6 partner of Wnt1 and identified both agonistic and antagonistic peptides. Lead grafted peptide Lr-EET-3.5 displayed an excellent binding affinity of 1 nM and an IC50 of 50 nM.
The p53/MDM2 interaction is another typical PPI example. p53 is a major tumor suppressor involved in DNA repair whose inactivation is extremely common in all sorts of cancers, leading to aberrant activity of downstream regulatory pathways. p53 can be inactivated in two ways either s a by a result of a mutation or through aberrant negative regulatory activity. MDM2 is a negative regulator of p53 that is commonly overexpressed in cancer cells, where p53 itself is not mutated. In these cases, restoration of the tumor-suppression activity could be achieved by inhibiting the p53/MDM2 interaction, which has made this PPI one of the most targeted inaccessible interactions, a topic that has been covered in several dedicated reviews.16,209−211
As with other PPIs, wherever structural information is available, a traditional approach has been to extract the natural binding epitopes of one of the PPI binding partners and turn them into smaller peptide inhibitors. In a study by Baek et al., the p53-derived peptide was stabilized with a i,i + 7 staple and successfully cocrystallized with MDM2.212 Interestingly, this study found that the staple itself was noninnocent in the binding to MDM2, further supporting the case that staple design is a crucial element to develop potent PPI inhibitors. Notably, successful reports of p53/MDM2 inhibitor peptides all featured compounds with an α-helical structure, which appears to be crucial for peptide binding affinity.213 The p53/MDM2 interaction has been studied extensively and is discussed further in section 4.4 from the standpoint of intracellular targets.
Aside from p53, helical requirements or favors are commonplace in PPIs.89,214,215 Inhibitors of the p53/MDM2 interaction have also been identified from a DEL screening59 or one-bead-one-sequence libraries.61 Other transcription factors have likewise been targeted with α-helical stapled peptides, including nuclear factor-γ (NF-γ)216,217 and X-chromosome encoded transcription factor FOXP3.218 β-Bairpins are also usual suspects in PPIs, although as discussed in section 2.3 their stabilization via stapling is not as well established; consequently, there are fewer examples of cyclic β-hairpin PPI inhibitors, and they generally emerge from epitope-derived rational design rather than from NEL screenings.219,220
4.4. Intrinsically Disordered Proteins
Intrinsically disordered proteins (IDPs) play essential roles in diverse biological processes, including signaling, regulation, and protein–protein interactions.20 As they not only lack standard binding pockets but also feature ill-defined structures and/or dynamic conformations, they constitute an undruggable challenge. Compounding this difficulty is the lack of available structural information due to little to no understanding of their dynamics in the cellular environment. This makes these targets particularly challenging not only for rational or computer-based design but also for in vitro display techniques such as mRNA display where the immobilized protein is not representative of its natural behavior. There are two prevalent strategies to target IDPs: natural binder-mimicry/hot-spot targeting221 and allosteric site hunting.222−224 Similarly to PPIs, known-epitope derivatization or fragment-based drug design has been used to target hot spots in disordered regions.13,21−23,221 Alternatively, circumventing the IDP in favor of targeting its binding partner can be done. Histones, located in the cell nucleus, are one such group of IDPs that are particularly difficult to target directly. To tackle histone deregulation225,226 or aberrant activity of histone mutants,227 researchers instead targeted histone deacetylases228 or demethylases, the latter having been inhibited by RaPID derived macrocyclic peptides.229,230
The most well-known example of inaccessible disordered protein is the transcription factor Myc, which is aberrantly active in over 75% of cancers and whose carcinogenic mechanism is yet unclear.231,232 Despite its status as one of the “most-wanted” cancer targets, Myc has remained undruggable thus far, although this year a miniprotein inhibitor has entered clinical trials; remarkably, this miniprotein is cell-permeable.233,234 Myc forms a heterodimer with its binding partner Max, with the resulting heterodimer binding DNA to regulate gene expression. Interestingly, the binding regions of both Myc and Max involved in their PPI are disordered on their own but organize themselves upon binding.22 Further complicating the targeting of Myc is the fast turnover of the Myc/Max heterodimer. Earlier this year, Li et al. identified bicyclic peptide inhibitors of Myc (Figure 10A).235,236 The design of their bicyclic library is remarkable for its unconventional cross-linker, which differs from more common heterocyclic or hydrophobic backbones. First, a norbornene cross-linker and two alkene side chains are reacted through a tandem ROM/RCM reaction, resulting in bicycles with diverse stereochemistries. This library is then immobilized and screened against the biotinylated peptide epitope of Myc, also labeled with an azide handle. As the library is fitted with an alkyne side chain, hit Myc binders can be “clicked” onto the epitope and stained using a biotin handle for identification, followed by sequencing by MS/MS analysis. The lead candidate NT-B2R was shown to directly effect Myc through a series of in vitro assays. While it was not cell-permeable, after intracellular delivery using liposomes, NT-BR2 also led to a decrease in proliferation and metabolic activity of the model Myc-dependent glioblastoma U87 with an EC50 of 1 μM. Although this breakthrough report does have limitations, including a relatively modest library size, a complex sequencing process, and challenges associated with stereochemical determination, it has already proven its potential for the development of cyclic peptides as inhibitors of Myc and other IDPs and could inspire further developments in other NELS methods.
In a different approach, the Ivarsson lab has leveraged a modified NELS technique termed proteomic-peptide phage display (ProP-PD, Figure 10B).237−240 They recognized that many IDPs act via short linear motifs (SLiMs) and that there is a severe dearth of information on the sequence identity, distribution, and interactivity of these motifs, leading to a significant gap in the mapping of the human interactome. In ProP-PD, a library is first computationally designed that comprises the entirety of a given proteome of interest. That library is then assembled through suitably designed oligonucleotides for phage display. The displayed SLiMs are then screened against an assembly of bait proteins to interrogate potential PPI interactions, and several rounds of selection are performed as usual to alleviate nonspecific binding issues. Sequence identification is then performed by NGS, and thorough data analysis enables mapping of PPI interactions to identified SLiMs. Although this method enables the high throughput analysis of virtually any proteome of interest, provided that the initial library be suitably designed, it has been limited by a few challenges. In particular, the requirement for high-quality bait proteins is not trivial. Furthermore, library design is complex, and analysis of the data obtained following selection and sequencing is not easy and requires both computational assistance and careful cross-checking, particularly to avoid wrongly mapping promiscuous SLiMs. With this in mind, the Ivarsson lab published workflow guidelines for the use of ProP-PD and made available a resource called PepTools that comprises a library of SLiMs derived from disordered proteomes, as well as a toolkit for data analysis.241 This streamlined workflow is expected to enhance the accessibility of ProP-PD and contribute to further filling the gap in the mapping of the human interactome and provide invaluable insights into IDPs. In this regard, the Lindorff-Larsen group recently developed a breakthrough computational method for the modeling of IDPs and made their results available as a large public database.242
5. Conclusion and Future Perspectives
This Review looks at recent additions to the arsenal of NELS methods that can be employed to target the undruggable space with cyclic peptides. Notably, an ever-expanding toolbox of npAAs, organic staples, cell-penetrating tags, and grafting scaffolds has enabled the identification of numerous cyclic peptides inhibiting inaccessible targets such as PPIs, intrinsically disordered regions, and proteins lacking well-defined binding pockets. Through the multitude of examples discussed, we believe a clear case is made that differently designed cyclic peptides have proven competent in targeting objectives previously deemed inaccessible. A focused look at some of the most famous undruggable targets, such as KRas and the p53/MDM2 interaction, reveals that a fairly large number of peptidic inhibitors have been identified through various methods. This begs the question: is the concept of “undruggable” outdated? It may still be too early to take that step. Although the frontiers of the undruggable are definitely being pushed further, formidable obstacles still remain. In particular, cyclic peptides still suffer from their poor membrane permeability, although major progress is being made, e.g., via stapling or backbone modifications. Intrinsically disordered targets also present a major hurdle, not only from their nature but also from the lack of available information. Additionally, beyond the undruggable characteristics discussed herein, traditional pharmaceutical concerns will remain, particularly with regard to the selective targeting of diseased vs healthy tissues, which is especially relevant for PPIs. Nonetheless, careful optimizm suggests that the “undruggable” space may be better seen as “yet to be drugged”.
We foresee that beyond the continuous development of new design scaffolds for macrocyclic peptides and technical improvements for NELS methods, the emergence of design guidelines will enhance the optimization of peptide hits to drug candidates. With the breadth of collected data, a deeper understanding of the influence of cyclic peptide architecture on solubility, the ability to target PPIs, and membrane permeability will undoubtedly facilitate the transfer of hits from in vitro conditions to cellular performance and beyond. Some research groups also advocate for alternative approaches, such as the retro-engineering of macrocyclic peptide hits into small-molecule-like compounds more amenable to PK/PD optimization.29,194,243 Still, today the main challenge to the adoption of cyclic peptides in pharmaceuticals is the membrane permeability issue. In this regard, stapled peptides, in particular, appear ripe for an imminent breakthrough. The more recent grafted peptides also hold promise, particularly if a genetic delivery vehicle can be devised for their in vivo on-target expression. Likewise, the development of improved ways to robustly estimate cell membrane permeability would considerably facilitate further development of both hits and the design principle. Continuous acquisition of information on the nature and interactions of PPIs or disordered regions is also highly desirable. Finally, the integration of all these advances into the various NELS techniques available would streamline access to cyclic peptides to break down the undruggable barrier.
Acknowledgments
We thank Dr. Gregoire Philippe, Dr. Alexis Richaud and Dr. Choi Yi Li for helpful and educational discussions.
Glossary
Abbreviations
- AA/aa
amino acid
- npAA
non-proteinogenic amino acid
- pAA
proteinogenic amino acid
- bRo5
beyond the rule of 5
- aaRS
aminoacyl-tRNA synthetase
- APBA
acetylphenylboronic acid
- Bcl-2
B-cell lymphoma 2
- Bcl-9
B-cell lymphoma 9
- ClAc
chloroacetyl
- CKP
cysteine-knot peptide
- Dha
dehydroalanine
- Dhb
dehydrobutyrine
- mCNP
meta-cyanopyridine
- CPP
cell-penetrating peptide
- DARPin
designed ankyrin repeat protein
- DEL
DNA-encoded chemical library
- FACS
fluorescence-assisted cell sorting
- FBDD
fragment-based drug discovery
- Fc
fragment crystallizable
- FDA
American Food and Drug Administration
- FIT
flexible in vitro translation
- GABARAP
γ-aminobutyric acid A receptor
- GTP/GDP
guanosine triphosphate/guanosine diphosphate
- HBS
hydrogen-bond surrogate
- HGF
hepatocyte growth factor
- HIF1
hypoxia-inducible factor
- HIV
human immunodeficiency virus
- HTS
high-throughput screening
- IDP
intrinsically disordered protein
- LRP6
low-density lipoprotein receptor-related protein 6
- MAGE-A4
melanoma associated antigen 4
- MIPD
mirror-image phage display
- NEL
nucleotide-encoded library
- NELS
nucleotide-encoded library screening
- NGS
next-generation sequencing
- pIII M13
phage protein III
- OBOC
one-bead-one-compound
- PCR
polymerase chain reaction
- PAMPA
parallel artificial membrane permeability assay
- PK/PD
pharmacokinetics/pharmacodynamics
- PEG
polyethlyene glycol
- PPI
protein–protein interaction
- ProP-PD
proteomic-peptide phage display
- PROTAC
proteolysis targeted chimera
- PylRS
pyrrolysil-tRNA synthetase
- RaPID
random nonstandard peptide integrated discovery
- RCM/ROM
ring-closing metathesis/ring-opening metathesis
- RiPP
ribosomally synthesized and post-translationally modified peptide
- SICLOPPS
split intein-mediated circular ligation of peptides and proteins
- SLiMS
short linear motifs
- SPPS
solid-phase peptide synthesis
- TBMB
tribromomethylbenzene
Biographies
Kilian Colas obtained his M.Sc. and Engineering degrees from the INSA of Rouen, his hometown in France, and then a Ph.D. in Organic Chemistry from the University of Stockholm, Sweden, where he studied main-group organometallic chemistry. After a postdoctoral stay in the Dyrager group in Uppsala, Sweden, on the study of fluorescent cancer probes for cancer diagnosis, he joined the Suga Laboratory, where he is currently a postdoctoral fellow.
Daniel Bindl received his B.Sc. and M.Sc. in Chemistry from the University of Regensburg and LMU in Munich. He conducted his Ph.D. in Supramolecular Chemistry in the Department of Pharmacy at LMU, where he studied the folding behavior of aromatic oligoamides. Afterwards, he joined the Laboratory of Prof. Suga, where he is currently working as a postdoctoral fellow on chemical post-translational modification strategies.
Hiroaki Suga received his Ph.D. degree in Chemistry in 1994 from the Massachusetts Institute of Technology. Subsequently, he was an Assistant Professor at the Department of Chemistry in the State University of New York at Buffalo from 1997 and was promoted to Associate Professor in 2002. He became Professor of the Research Center for Advanced Science and Technology at the University of Tokyo in 2003 and, in 2010, of the Department of Chemistry, Graduate School of Science. He is the recipient of the 2014 Akabori Memorial Award, the 2016 Japanese Peptide Society and Max-Bergmann gold medal, the 2017 Nagoya silver medal, a Wolf Prize Laureate in Chemistry in 2023, and various other awards.
Author Contributions
CRediT: Kilian Colas conceptualization, data curation, writing - original draft, writing - review & editing; Daniel Bindl data curation, writing - review & editing.
K.C. and D.B. received postdoctoral fellowship funding from the Japanese Society for the Promotion of Science (JSPS). H.S. currently received funding of the Japan Society for the Promotion of Science (JSPS) Grant-in-Aid for Specially Promoted Research (JP20H05618).
The authors declare no competing financial interest.
Special Issue
Published as part of Chemical Reviewsspecial issue “Drugging the Undruggable”.
References
- Dang C. V.; Reddy E. P.; Shokat K. M.; Soucek L. Drugging the ’undruggable’ cancer targets. Nat. Rev. Canc. 2017, 17, 502–508. 10.1038/nrc.2017.36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang G.; Zhang J.; Gao Y.; Li Y.; Li Y. Strategies for targeting undruggable targets. Expert Opin. Drug Discovery 2022, 17, 55–69. 10.1080/17460441.2021.1969359. [DOI] [PubMed] [Google Scholar]
- Xie X.; Yu T.; Li X.; Zhang N.; Foster L. J.; Peng C.; Huang W.; He G. Recent advances in targeting the “undruggable” proteins: from drug discovery to clinical trials. Signal Transduct. Target. Ther. 2023, 8, 335. 10.1038/s41392-023-01589-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gressel J. Perspective: It is time to consider new ways to attack unpesticidable (undruggable) target sites by designing peptide pesticides. Pest Manag. Sci. 2022, 78, 2108–2112. 10.1002/ps.6817. [DOI] [PubMed] [Google Scholar]
- Benet L. Z.; Hosey C. M.; Ursu O.; Oprea T. I. BDDCS, the Rule of 5 and drugability. Adv. Drug Delivery Rev. 2016, 101, 89–98. 10.1016/j.addr.2016.05.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shultz M. D. Two Decades under the Influence of the Rule of Five and the Changing Properties of Approved Oral Drugs. J. Med. Chem. 2019, 62, 1701–1714. 10.1021/acs.jmedchem.8b00686. [DOI] [PubMed] [Google Scholar]
- Tsomaia N. Peptide therapeutics: targeting the undruggable space. Eur. J. Med. Chem. 2015, 94, 459–70. 10.1016/j.ejmech.2015.01.014. [DOI] [PubMed] [Google Scholar]
- Tyagi M.; Begnini F.; Poongavanam V.; Doak B. C.; Kihlberg J. Drug Syntheses Beyond the Rule of 5. Chem.-Eur. J. 2020, 26, 49–88. 10.1002/chem.201902716. [DOI] [PubMed] [Google Scholar]
- Cagan R.; Shokat K. Drugging the undruggable: Ross Cagan interviews Kevan Shokat. Dis. Model. Mech 2022, 15, dmm049468. 10.1242/dmm.049468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pietrucci F.; Vargiu A. V.; Kranjc A. HIV-1 Protease Dimerization Dynamics Reveals a Transient Druggable Binding Pocket at the Interface. Sci. Rep. 2016, 5, 18555. 10.1038/srep18555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ostrem J. M.; Peters U.; Sos M. L.; Wells J. A.; Shokat K. M. K-Ras(G12C) inhibitors allosterically control GTP affinity and effector interactions. Nature 2013, 503, 548–551. 10.1038/nature12796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ostrem J. M. L.; Shokat K. M. Direct small-molecule inhibitors of KRAS: from structural insights to mechanism-based design. Nat. Rev. Drug Discovery 2016, 15, 771–785. 10.1038/nrd.2016.139. [DOI] [PubMed] [Google Scholar]
- Olah J.; Szenasi T.; Lehotzky A.; Norris V.; Ovadi J. Challenges in Discovering Drugs That Target the Protein-Protein Interactions of Disordered Proteins. Int. J. Mol. Sci. 2022, 23, 1550. 10.3390/ijms23031550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu H.; Zhou Q.; He J.; Jiang Z.; Peng C.; Tong R.; Shi J. Recent advances in the development of protein–protein interactions modulators: mechanisms and clinical trials. Signal Transduct. Target. Ther. 2020, 5, 213. 10.1038/s41392-020-00315-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sedov I. A.; Zuev Y. F. Recent Advances in Protein–Protein Interactions. Int. J. Mol. Sci. 2023, 24, 1282. 10.3390/ijms24021282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X.; Ni D.; Liu Y.; Lu S. Rational Design of Peptide-Based Inhibitors Disrupting Protein-Protein Interactions. Front. Chem. 2021, 9, 682675. 10.3389/fchem.2021.682675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gonzalez-Muniz R.; Bonache M. A.; Perez de Vega M. J. Modulating Protein-Protein Interactions by Cyclic and Macrocyclic Peptides. Prominent Strategies and Examples. Molecules 2021, 26, 445. 10.3390/molecules26020445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng J.; Zhou J.; Kong L.; Wang H.; Zhang Y.; Wang X.; Liu G.; Chu Q. Stabilized cyclic peptides as modulators of protein-protein interactions: promising strategies and biological evaluation. RSC Med. Chem. 2023, 14, 2496–2508. 10.1039/D3MD00487B. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buyanova M.; Pei D. Targeting intracellular protein-protein interactions with macrocyclic peptides. Trends Pharmacol. Sci. 2022, 43, 234–248. 10.1016/j.tips.2021.11.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saurabh S.; Nadendla K.; Purohit S. S.; Sivakumar P. M.; Cetinel S. Fuzzy Drug Targets: Disordered Proteins in the Drug-Discovery Realm. ACS Omega 2023, 8, 9729–9747. 10.1021/acsomega.2c07708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choudhary S.; Lopus M.; Hosur R. V. Targeting disorders in unstructured and structured proteins in various diseases. Biophys. Chem. 2022, 281, 106742. 10.1016/j.bpc.2021.106742. [DOI] [PubMed] [Google Scholar]
- Santofimia-Castano P.; Rizzuti B.; Xia Y.; Abian O.; Peng L.; Velazquez-Campoy A.; Neira J. L.; Iovanna J. Targeting intrinsically disordered proteins involved in cancer. Cell. Mol. Life Sci. 2020, 77, 1695–1707. 10.1007/s00018-019-03347-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ambadipudi S.; Zweckstetter M. Targeting intrinsically disordered proteins in rational drug discovery. Expert Opin. Drug Discovery 2016, 11, 65–77. 10.1517/17460441.2016.1107041. [DOI] [PubMed] [Google Scholar]
- Vinogradov A. A.; Yin Y.; Suga H. Macrocyclic Peptides as Drug Candidates: Recent Progress and Remaining Challenges. J. Am. Chem. Soc. 2019, 141, 4167–4181. 10.1021/jacs.8b13178. [DOI] [PubMed] [Google Scholar]
- Jing X.; Jin K. A gold mine for drug discovery: Strategies to develop cyclic peptides into therapies. Med. Res. Rev. 2020, 40, 753–810. 10.1002/med.21639. [DOI] [PubMed] [Google Scholar]
- Hayes H. C.; Luk L. Y. P.; Tsai Y. H. Approaches for peptide and protein cyclisation. Org. Biomol. Chem. 2021, 19, 3983–4001. 10.1039/D1OB00411E. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vu Q. N.; Young R.; Sudhakar H. K.; Gao T.; Huang T.; Tan Y. S.; Lau Y. H. Cyclisation strategies for stabilising peptides with irregular conformations. RSC Med. Chem. 2021, 12, 887–901. 10.1039/D1MD00098E. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song B. P. C.; Ch’ng A. C. W.; Lim T. S. Review of phage display: A jack-of-all-trades and master of most biomolecule display. Int. J. Biol. Macromol. 2024, 256, 128455. 10.1016/j.ijbiomac.2023.128455. [DOI] [PubMed] [Google Scholar]
- Tavassoli A. SICLOPPS cyclic peptide libraries in drug discovery. Curr. Opin. Chem. Biol. 2017, 38, 30–35. 10.1016/j.cbpa.2017.02.016. [DOI] [PubMed] [Google Scholar]
- Sohrabi C.; Foster A.; Tavassoli A. Methods for generating and screening libraries of genetically encoded cyclic peptides in drug discovery. Nat. Rev. Chem. 2020, 4, 90–101. 10.1038/s41570-019-0159-2. [DOI] [PubMed] [Google Scholar]
- Huang Y.; Wiedmann M. M.; Suga H. RNA Display Methods for the Discovery of Bioactive Macrocycles. Chem. Rev. 2019, 119, 10360–10391. 10.1021/acs.chemrev.8b00430. [DOI] [PubMed] [Google Scholar]
- Newton M. S.; Cabezas-Perusse Y.; Tong C. L.; Seelig B. In Vitro Selection of Peptides and Proteins-Advantages of mRNA Display. ACS Synth. Biol. 2020, 9, 181–190. 10.1021/acssynbio.9b00419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Helmling C.; Cunningham C. N., mRNA Display and Its Growing Potential in the Discovery of De Novo Therapeutic Peptide Candidates. In Approaching the Next Inflection in Peptide Therapeutics: Attaining Cell Permeability and Oral Bioavailability; ACS Symposium Series, Vol. 1417; American Chemical Society: Washington, DC, 2022; pp 27–53. [Google Scholar]
- Plückthun A. Ribosome Display: A Perspective. Methods Mol. Biol. 2012, 805, 3–28. 10.1007/978-1-61779-379-0_1. [DOI] [PubMed] [Google Scholar]
- Li R.; Kang G.; Hu M.; Huang H. Ribosome Display: A Potent Display Technology used for Selecting and Evolving Specific Binders with Desired Properties. Mol. Biotechnol. 2019, 61, 60–71. 10.1007/s12033-018-0133-0. [DOI] [PubMed] [Google Scholar]
- Teymennet-Ramírez K. V.; Martínez-Morales F.; Trejo-Hernández M. R. Yeast Surface Display System: Strategies for Improvement and Biotechnological Applications. Front. Mol. Biol. 2022, 9, 794742. 10.3389/fbioe.2021.794742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lopez-Morales J.; Vanella R.; Appelt E. A.; Whillock S.; Paulk A. M.; Shusta E. V.; Hackel B. J.; Liu C. C.; Nash M. A. Protein Engineering and High-Throughput Screening by Yeast Surface Display: Survey of Current Methods. Small Sci. 2023, 3, 2300095. 10.1002/smsc.202300095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gironda-Martínez A.; Donckele E. J.; Samain F.; Neri D. DNA-Encoded Chemical Libraries: A Comprehensive Review with Succesful Stories and Future Challenges. ACS Pharmacol. & Transl. Sci. 2021, 4, 1265–1279. 10.1021/acsptsci.1c00118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen F.-J.; Pinnette N.; Gao J. Strategies for the Construction of Multicyclic Phage Display Libraries. ChemBioChem. 2024, 25, e202400072 10.1002/cbic.202400072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ho J. M. L.; Miller C. A.; Smith K. A.; Mattia J. R.; Bennett M. R. Improved pyrrolysine biosynthesis through phage assisted non-continuous directed evolution of the complete pathway. Nat. Commun. 2021, 12, 3914. 10.1038/s41467-021-24183-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sandman K. E.; Benner J. S.; Noren C. J. Phage Display of Selenopeptides. J. Am. Chem. Soc. 2000, 122, 960–961. 10.1021/ja992462m. [DOI] [Google Scholar]
- Oller-Salvia B.; Chin J. W. Efficient Phage Display with Multiple Distinct Non-Canonical Amino Acids Using Orthogonal Ribosome-Mediated Genetic Code Expansion. Angew. Chem., Int. Ed. 2019, 58, 10844–10848. 10.1002/anie.201902658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tian F.; Tsao M.-L.; Schultz P. G. A Phage Display System with Unnatural Amino Acids. J. Am. Chem. Soc. 2004, 126, 15962–15963. 10.1021/ja045673m. [DOI] [PubMed] [Google Scholar]
- Tharp J. M.; Hampton J. T.; Reed C. A.; Ehnbom A.; Chen P. C.; Morse J. S.; Kurra Y.; Perez L. M.; Xu S.; Liu W. R. An amber obligate active site-directed ligand evolution technique for phage display. Nat. Commun. 2020, 11, 1392. 10.1038/s41467-020-15057-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carle V.; Kong X. D.; Comberlato A.; Edwards C.; Diaz-Perlas C.; Heinis C. Generation of a 100-billion cyclic peptide phage display library having a high skeletal diversity. Protein Eng. Des. Sel. 2021, 34, gzab018. 10.1093/protein/gzab018. [DOI] [PubMed] [Google Scholar]
- Hetrick K. J.; Walker M. C.; van der Donk W. A. Development and Application of Yeast and Phage Display of Diverse Lanthipeptides. ACS Cent. Sci. 2018, 4, 458–467. 10.1021/acscentsci.7b00581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Case M.; Navaratna T.; Vinh J.; Thurber G. Rapid Evaluation of Staple Placement in Stabilized α Helices Using Bacterial Surface Display. ACS Chem. Biol. 2023, 18, 905–914. 10.1021/acschembio.3c00048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Navaratna T.; Atangcho L.; Mahajan M.; Subramanian V.; Case M.; Min A.; Tresnak D.; Thurber G. M. Directed Evolution Using Stabilized Bacterial Peptide Display. J. Am. Chem. Soc. 2020, 142, 1882–1894. 10.1021/jacs.9b10716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- See K.; Kadonosono T.; Miyamoto K.; Tsubaki T.; Ota Y.; Katsumi M.; Ryo S.; Aida K.; Minegishi M.; Isozaki T.; Kuchimaru T.; Kizaka-Kondoh S. Antibody-guided design and identification of CD25-binding small antibody mimetics using mammalian cell surface display. Sci. Rep. 2021, 11, 22098. 10.1038/s41598-021-01603-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crook Z. R.; Sevilla G. P.; Friend D.; Brusniak M.-Y.; Bandaranayake A. D.; Clarke M.; Gewe M.; Mhyre A. J.; Baker D.; Strong R. K.; Bradley P.; Olson J. M. Mammalian display screening of diverse cystine-dense peptides for difficult to drug targets. Nat. Commun. 2017, 8, 2244. 10.1038/s41467-017-02098-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schultz P. Expanding the genetic code. Protein Sci. 2023, 32, e4488 10.1002/pro.4488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Passioura T.; Suga H. A RaPID way to discover nonstandard macrocyclic peptide modulators of drug targets. Chem. Commun. 2017, 53, 1931–1940. 10.1039/C6CC06951G. [DOI] [PubMed] [Google Scholar]
- Suga H. Max-Bergmann award lecture:A RaPID way to discover bioactive nonstandard peptides assisted by the flexizyme and FIT systems. J. Pept. Sci. 2018, 24, e3055. 10.1002/psc.3055. [DOI] [PubMed] [Google Scholar]
- Goto Y.; Suga H. The RaPID Platform for the Discovery of Pseudo-Natural Macrocyclic Peptides. Acc. Chem. Res. 2021, 54, 3604–3617. 10.1021/acs.accounts.1c00391. [DOI] [PubMed] [Google Scholar]
- Goto Y.; Katoh T.; Suga H. Flexizymes for genetic code reprogramming. Nat. Protoc. 2011, 6, 779–90. 10.1038/nprot.2011.331. [DOI] [PubMed] [Google Scholar]
- Bon M.; Bilsland A.; Bower J.; McAulay K. Fragment-based drug discovery—the importance of high-quality molecule libraries. Mol. Oncol. 2022, 16, 3761–3777. 10.1002/1878-0261.13277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mureddu L. G.; Vuister G. W. Fragment-Based Drug Discovery by NMR. Where Are the Successes and Where can It Be Improved?. Frontiers in molecular biosciences 2022, 9, 834453. 10.3389/fmolb.2022.834453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Plais L.; Scheuermann J. Macrocyclic DNA-encoded chemical libraries: a historical perspective. RSC Chem. Biol. 2022, 3, 7–17. 10.1039/D1CB00161B. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silvestri A. P.; Zhang Q.; Ping Y.; Muir E. W.; Zhao J.; Chakka S. K.; Wang G.; Bray W. M.; Chen W.; Fribourgh J. L.; Tripathi S.; He Y.; Rubin S. M.; Satz A. L.; Pye C. R.; Kuai L.; Su W.; Schwochert J. A. DNA-Encoded Macrocyclic Peptide Libraries Enable the Discovery of a Neutral MDM2–p53 Inhibitor. ACS Med. Chem. Lett. 2023, 14, 820–826. 10.1021/acsmedchemlett.3c00117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lam K. S.; Lehman A. L.; Song A.; Doan N.; Enstrom A. M.; Maxwell J.; Liu R. Synthesis and Screening of “One-Bead One-Compound” Combinatorial Peptide Libraries. Methods Enzymol. 2003, 369, 298–322. 10.1016/S0076-6879(03)69017-8. [DOI] [PubMed] [Google Scholar]
- Morimoto J.; Hosono Y.; Sando S. Isolation of a peptide containing d-amino acid residues that inhibits the α-helix-mediated p53–MDM2 interaction from a one-bead one-compound library. Bioorg. Med. Chem. Lett. 2018, 28, 231–234. 10.1016/j.bmcl.2018.01.001. [DOI] [PubMed] [Google Scholar]
- Blanc A.; Todorovic M.; Dude I.; Merkens H.; Bénard F.; Perrin D. M. Toward tryptathionine-stapled one-bead-one-compound (OBOC) libraries: solid phase synthesis of a bioactive octretoate analog. Org. Biomol. Chem. 2023, 21, 8112–8116. 10.1039/D3OB01378B. [DOI] [PubMed] [Google Scholar]
- Yan Y.-Q.; Wang J.-Q.; Zhang L.; Yang P.-P.; Ye X.-W.; Liu C.; Hou D.-Y.; Lai W.-J.; Wang J.; Zeng X.-Z.; Xu W.; Wang L. Localized Instillation Enables In Vivo Screening of Targeting Peptides Using One-Bead One-Compound Technology. ACS Nano 2023, 17, 1381–1392. 10.1021/acsnano.2c09894. [DOI] [PubMed] [Google Scholar]
- Petersen L. K.; Christensen A. B.; Andersen J.; Folkesson C. G.; Kristensen O.; Andersen C.; Alzu A.; Sløk F. A.; Blakskjær P.; Madsen D.; Azevedo C.; Micco I.; Hansen N. J. V. Screening of DNA-Encoded Small Molecule Libraries inside a Living Cell. J. Am. Chem. Soc. 2021, 143, 2751–2756. 10.1021/jacs.0c09213. [DOI] [PubMed] [Google Scholar]
- Meldal M. ‘One bead two compound libraries’ for detecting chemical and biochemical conversions. Curr. Opin. Chem. Biol. 2004, 8, 238–244. 10.1016/j.cbpa.2004.04.007. [DOI] [PubMed] [Google Scholar]
- He J.; Ghosh P.; Nitsche C. Biocompatible strategies for peptide macrocyclisation. Chem. Sci. 2024, 15, 2300–2322. 10.1039/D3SC05738K. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Timmerman P.; Beld J.; Puijk W. C.; Meloen R. H. Rapid and Quantitative Cyclization of Multiple Peptide Loops onto Synthetic Scaffolds for Structural Mimicry of Protein Surfaces. ChemBioChem. 2005, 6, 821–824. 10.1002/cbic.200400374. [DOI] [PubMed] [Google Scholar]
- Heinis C.; Rutherford T.; Freund S.; Winter G. Phage-encoded combinatorial chemical libraries based on bicyclic peptides. Nat. Chem. Biol. 2009, 5, 502–507. 10.1038/nchembio.184. [DOI] [PubMed] [Google Scholar]
- Voss S.; Rademann J.; Nitsche C. Peptide–Bismuth Bicycles: In Situ Access to Stable Constrained Peptides with Superior Bioactivity. Angew. Chem., Int. Ed. 2022, 61, e202113857 10.1002/anie.202113857. [DOI] [PubMed] [Google Scholar]
- Ullrich S.; Somathilake U.; Shang M.; Nitsche C. Phage-encoded bismuth bicycles enable instant access to targeted bioactive peptides. Commun. Chem. 2024, 7, 143. 10.1038/s42004-024-01232-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He R.-N.; Zhang M.-J.; Dai B.; Kong X.-D. Selection of Peptide–Bismuth Bicycles Using Phage Display. ACS Chem. Biol. 2024, 19, 1040–1044. 10.1021/acschembio.4c00099. [DOI] [PubMed] [Google Scholar]
- Voss S.; Adair L. D.; Achazi K.; Kim H.; Bergemann S.; Bartenschlager R.; New E. J.; Rademann J.; Nitsche C. Cell-Penetrating Peptide-Bismuth Bicycles. Angew. Chem., Int. Ed. 2024, 63, e202318615 10.1002/anie.202318615. [DOI] [PubMed] [Google Scholar]
- Lu S.; Wu Y.; Li J.; Meng X.; Hu C.; Zhao Y.; Wu C. Directed Disulfide Pairing and Folding of Peptides for the De Novo Development of Multicyclic Peptide Libraries. J. Am. Chem. Soc. 2020, 142, 16285–16291. 10.1021/jacs.0c06044. [DOI] [PubMed] [Google Scholar]
- Craik D. J.; Du J. Cyclotides as drug design scaffolds. Curr. Opin. Chem. Biol. 2017, 38, 8–16. 10.1016/j.cbpa.2017.01.018. [DOI] [PubMed] [Google Scholar]
- Jagadish K.; Camarero J. A. Cyclotides, a promising molecular scaffold for peptide-based therapeutics. Pept. Sci. 2010, 94, 611–616. 10.1002/bip.21433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hansen S.; Zhang Y.; Hwang S.; Nabhan A.; Li W.; Fuhrmann J.; Kschonsak Y.; Zhou L.; Nile A. H.; Gao X.; Piskol R.; de Sousa e Melo F.; de Sauvage F. J.; Hannoush R. N. Directed evolution identifies high-affinity cystine-knot peptide agonists and antagonists of Wnt/β-catenin signaling. Proc. Natl. Adac. Sci. 2022, 119, e2207327119 10.1073/pnas.2207327119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wong J. Y. K.; Mukherjee R.; Miao J.; Bilyk O.; Triana V.; Miskolzie M.; Henninot A.; Dwyer J. J.; Kharchenko S.; Iampolska A.; Volochnyuk D. M.; Lin Y.-S.; Postovit L.-M.; Derda R. Genetically-encoded discovery of proteolytically stable bicyclic inhibitors for morphogen NODAL. Chem. Sci. 2021, 12, 9694–9703. 10.1039/D1SC01916C. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oppewal T. R.; Jansen I. D.; Hekelaar J.; Mayer C. A Strategy to Select Macrocyclic Peptides Featuring Asymmetric Molecular Scaffolds as Cyclization Units by Phage Display. J. Am. Chem. Soc. 2022, 144, 3644–3652. 10.1021/jacs.1c12822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiang H.; Bai L.; Zhang X.; Dan T.; Cheng P.; Yang X.; Ai H.; Li K.; Lei X. A facile strategy for the construction of a phage display cyclic peptide library for the selection of functional macrocycles. Chem. Sci. 2024, 15, 11847–11855. 10.1039/D4SC03207A. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu M.; Morewood R.; Yoshisada R.; Pascha M. N.; Hopstaken A. J. P.; Tarcoveanu E.; Poole D. A.; de Haan C. A. M.; Nitsche C.; Jongkees S. A. K. Selective thiazoline peptide cyclisation compatible with mRNA display and efficient synthesis. Chem. Sci. 2023, 14, 10561–10569. 10.1039/D3SC03117A. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lamartina C. W.; Chartier C. A.; Hirano J. M.; Shah N. H.; Rovis T. Crafting Unnatural Peptide Macrocycles via Rh(III)-Catalyzed Carboamidation. J. Am. Chem. Soc. 2024, 146, 20868–20877. 10.1021/jacs.4c05248. [DOI] [PubMed] [Google Scholar]
- Fleming M. C.; Bowler M. M.; Park R.; Popov K. I.; Bowers A. A. Tyrosinase-Catalyzed Peptide Macrocyclization for mRNA Display. J. Am. Chem. Soc. 2023, 145, 10445–10450. 10.1021/jacs.2c12629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Repka L. M.; Chekan J. R.; Nair S. K.; van der Donk W. A. Mechanistic Understanding of Lanthipeptide Biosynthetic Enzymes. Chem. Rev. 2017, 117, 5457–5520. 10.1021/acs.chemrev.6b00591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hofmann F. T.; Szostak J. W.; Seebeck F. P. In Vitro Selection of Functional Lantipeptides. J. Am. Chem. Soc. 2012, 134, 8038–8041. 10.1021/ja302082d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verdine G. L.; Hilinski G. J. Stapled peptides for intracellular drug targets. Methods Enzymol. 2012, 503, 3–33. 10.1016/B978-0-12-396962-0.00001-X. [DOI] [PubMed] [Google Scholar]
- Ali A. M.; Atmaj J.; Van Oosterwijk N.; Groves M. R.; Domling A. Stapled Peptides Inhibitors: A New Window for Target Drug Discovery. Comput. Struc. Biotechnol. J. 2019, 17, 263–281. 10.1016/j.csbj.2019.01.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moiola M.; Memeo M. G.; Quadrelli P. Stapled Peptides-A Useful Improvement for Peptide-Based Drugs. Molecules 2019, 24, 3654. 10.3390/molecules24203654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chandramohan A.; Josien H.; Yuen T. Y.; Duggal R.; Spiegelberg D.; Yan L.; Juang Y.-C. A.; Ge L.; Aronica P. G.; Kaan H. Y. K.; Lim Y. H.; Peier A.; Sherborne B.; Hochman J.; Lin S.; Biswas K.; Nestor M.; Verma C. S.; Lane D. P.; Sawyer T. K.; Garbaccio R.; Henry B.; Kannan S.; Brown C. J.; Johannes C. W.; Partridge A. W. Design-rules for stapled peptides with in vivo activity and their application to Mdm2/X antagonists. Nat. Commun. 2024, 15, 489. 10.1038/s41467-023-43346-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li X.; Chen S.; Zhang W.-D.; Hu H.-G. Stapled Helical Peptides Bearing Different Anchoring Residues. Chem. Rev. 2020, 120, 10079–10144. 10.1021/acs.chemrev.0c00532. [DOI] [PubMed] [Google Scholar]
- Pelay-Gimeno M.; Glas A.; Koch O.; Grossmann T. N. Structure-Based Design of Inhibitors of Protein–Protein Interactions: Mimicking Peptide Binding Epitopes. Angew. Chem., Int. Ed. 2015, 54, 8896–8927. 10.1002/anie.201412070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watkins A. M.; Wuo M. G.; Arora P. S. Protein–Protein Interactions Mediated by Helical Tertiary Structure Motifs. J. Am. Chem. Soc. 2015, 137, 11622–11630. 10.1021/jacs.5b05527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watkins A. M.; Arora P. S. Anatomy of β-Strands at Protein–Protein Interfaces. ACS Chem. Biol. 2014, 9, 1747–1754. 10.1021/cb500241y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gavenonis J.; Sheneman B. A.; Siegert T. R.; Eshelman M. R.; Kritzer J. A. Comprehensive analysis of loops at protein-protein interfaces for macrocycle design. Nat. Chem. Biol. 2014, 10, 716–722. 10.1038/nchembio.1580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Araujo A. D.; Hoang H. N.; Kok W. M.; Diness F.; Gupta P.; Hill T. A.; Driver R. W.; Price D. A.; Liras S.; Fairlie D. P. Comparative α-Helicity of Cyclic Pentapeptides in Water. Angew. Chem., Int. Ed. 2014, 53, 6965–6969. 10.1002/anie.201310245. [DOI] [PubMed] [Google Scholar]
- Zhang Q.; Jiang F.; Zhao B.; Lin H.; Tian Y.; Xie M.; Bai G.; Gilbert A. M.; Goetz G. H.; Liras S.; Mathiowetz A. A.; Price D. A.; Song K.; Tu M.; Wu Y.; Wang T.; Flanagan M. E.; Wu Y.-D.; Li Z. Chiral Sulfoxide-Induced Single Turn Peptide α-Helicity. Sci. Rep. 2016, 6, 38573. 10.1038/srep38573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flint D. G.; Kumita J. R.; Smart O. S.; Woolley G. A. Using an Azobenzene Cross-Linker to Either Increase or Decrease Peptide Helix Content upon Trans-to-Cis Photoisomerization. Chem. & Biol. 2002, 9, 391–397. 10.1016/S1074-5521(02)00109-6. [DOI] [PubMed] [Google Scholar]
- Woolley G. A. Photocontrolling Peptide α Helices. Acc. Chem. Res. 2005, 38, 486–493. 10.1021/ar040091v. [DOI] [PubMed] [Google Scholar]
- Kim Y.-W.; Grossmann T. N.; Verdine G. L. Synthesis of all-hydrocarbon stapled α-helical peptides by ring-closing olefin metathesis. Nat. Protoc. 2011, 6, 761–771. 10.1038/nprot.2011.324. [DOI] [PubMed] [Google Scholar]
- Dietrich L.; Rathmer B.; Ewan K.; Bange T.; Heinrichs S.; Dale T. C.; Schade D.; Grossmann T. N. Cell Permeable Stapled Peptide Inhibitor of Wnt Signaling that Targets β-Catenin Protein-Protein Interactions. Cell Chem. Biol. 2017, 24, 958–968.e5. 10.1016/j.chembiol.2017.06.013. [DOI] [PubMed] [Google Scholar]
- Liu J.; Xiao Q.; Xiao J.; Niu C.; Li Y.; Zhang X.; Zhou Z.; Shu G.; Yin G. Wnt/β-catenin signalling: function, biological mechanisms, and therapeutic opportunities. sig.nal Transduct. Target. Ther. 2022, 7, 3. 10.1038/s41392-021-00762-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Philippe G. J. B.; Mittermeier A.; Lawrence N.; Huang Y.-H.; Condon N. D.; Loewer A.; Craik D. J.; Henriques S. T. Angler Peptides: Macrocyclic Conjugates Inhibit p53:MDM2/X Interactions and Activate Apoptosis in Cancer Cells. ACS Chem. Biol. 2021, 16, 414–428. 10.1021/acschembio.0c00988. [DOI] [PubMed] [Google Scholar]
- Sharma K.; Strizhak A. V.; Fowler E.; Xu W.; Chappell B.; Sore H. F.; Galloway W. R. J. D.; Grayson M. N.; Lau Y. H.; Itzhaki L. S.; Spring D. R. Functionalized Double Strain-Promoted Stapled Peptides for Inhibiting the p53-MDM2 Interaction. ACS Omega 2020, 5, 1157–1169. 10.1021/acsomega.9b03459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guerlavais V.; Sawyer T. K.; Carvajal L.; Chang Y. S.; Graves B.; Ren J.-G.; Sutton D.; Olson K. A.; Packman K.; Darlak K.; Elkin C.; Feyfant E.; Kesavan K.; Gangurde P.; Vassilev L. T.; Nash H. M.; Vukovic V.; Aivado M.; Annis D. A. Discovery of Sulanemadlin (ALRN-6924), the First Cell-Permeating, Stabilized α-Helical Peptide in Clinical Development. J. Med. Chem. 2023, 66, 9401–9417. 10.1021/acs.jmedchem.3c00623. [DOI] [PubMed] [Google Scholar]
- Brown H.; Chung M.; Üffing A.; Batistatou N.; Tsang T.; Doskocil S.; Mao W.; Willbold D.; Bast R. C. Jr.; Lu Z.; Weiergräber O. H.; Kritzer J. A. Structure-Based Design of Stapled Peptides That Bind GABARAP and Inhibit Autophagy. J. Am. Chem. Soc. 2022, 144, 14687–14697. 10.1021/jacs.2c04699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Méndez Y.; Vasco A. V.; Ivey G.; Dias A. L.; Gierth P.; Sousa B. B.; Navo C. D.; Torres-Mozas A.; Rodrigues T.; Jiménez-Osés G.; Bernardes G. J. L. Merging the Isonitrile-Tetrazine (4 + 1) Cycloaddition and the Ugi Four-Component Reaction into a Single Multicomponent Process. Angew. Chem., Int. Ed. 2023, 62, e202311186 10.1002/anie.202311186. [DOI] [PubMed] [Google Scholar]
- Panteleev V. P.; Balandin V. S.; Ivanov T. V.; Ovchinnikova V. T. A Therapeutic Potential of Animal β-hairpin Antimicrobial Peptides. Curr. Med. Chem. 2017, 24, 1724–1746. 10.2174/0929867324666170424124416. [DOI] [PubMed] [Google Scholar]
- Hu H.; Kofoed C.; Li M.; Gonçalves J. P. L.; Hansen J.; Wolfram M.; Hansen A. K.; Friis Hansen C. H.; Diness F.; Schoffelen S.; Meldal M. Computational Evolution of Threonine-Rich β-Hairpin Peptides Mimicking Specificity and Affinity of Antibodies. ACS Cent. Sci. 2019, 5, 259–269. 10.1021/acscentsci.8b00614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sawyer N.; Arora P. S. Hydrogen Bond Surrogate Stabilization of β-Hairpins. ACS Chem. Biol. 2018, 13, 2027–2032. 10.1021/acschembio.8b00641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Obrecht D.; Chevalier E.; Moehle K.; Robinson J. A. β-Hairpin protein epitope mimetic technology in drug discovery. Drug Discovery Today Technol. 2012, 9, e63–e69. 10.1016/j.ddtec.2011.07.006. [DOI] [PubMed] [Google Scholar]
- Blosser S. L.; Sawyer N.; Maksimovic I.; Ghosh B.; Arora P. S. Covalent and Noncovalent Targeting of the Tcf4/β-Catenin Strand Interface with β-Hairpin Mimics. ACS Chem. Biol. 2021, 16, 1518–1525. 10.1021/acschembio.1c00389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sahni A.; Ritchey J. L.; Qian Z.; Pei D. Cell-Penetrating Peptides Translocate across the Plasma Membrane by Inducing Vesicle Budding and Collapse. J. Am. Chem. Soc. 2024, 146, 25371–25382. 10.1021/jacs.4c10533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liao H.; Li X.; Zhao L.; Wang Y.; Wang X.; Wu Y.; Zhou X.; Fu W.; Liu L.; Hu H.-G.; Chen Y.-G. A PROTAC peptide induces durable β-catenin degradation and suppresses Wnt-dependent intestinal cancer. Cell Discovery 2020, 6, 35. 10.1038/s41421-020-0171-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ren J.; Sui H.; Fang F.; Li Q.; Li B. The application of ApcMin/+ mouse model in colorectal tumor researches. J. Cancer Res. Clin. Oncol. 2019, 145, 1111–1122. 10.1007/s00432-019-02883-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Imai M.; Colas K.; Suga H. Protein Grafting Techniques: From Peptide Epitopes to Lasso-Grafted Neobiologics. ChemPlusChem. 2024, 89, e202400152 10.1002/cplu.202400152. [DOI] [PubMed] [Google Scholar]
- Sun S.; Compañón I.; Martínez-Sáez N.; Seixas J. D.; Boutureira O.; Corzana F.; Bernardes G. J. L. Enhanced Permeability and Binding Activity of Isobutylene-Grafted Peptides. ChemBioChem. 2018, 19, 48–52. 10.1002/cbic.201700586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muratspahić E.; Koehbach J.; Gruber C. W.; Craik D. J. Harnessing cyclotides to design and develop novel peptide GPCR ligands. RSC Chem. Biol. 2020, 1, 177–191. 10.1039/D0CB00062K. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Austin J.; Wang W.; Puttamadappa S.; Shekhtman A.; Camarero J. A. Biosynthesis and Biological Screening of a Genetically Encoded Library Based on the Cyclotide MCoTI-I. ChemBioChem. 2009, 10, 2663–2670. 10.1002/cbic.200900534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simonsen S. M.; Sando L.; Rosengren K. J.; Wang C. K.; Colgrave M. L.; Daly N. L.; Craik D. J. Alanine Scanning Mutagenesis of the Prototypic Cyclotide Reveals a Cluster of Residues Essential for Bioactivity. J. Biol. Chem. 2008, 283, 9805–9813. 10.1074/jbc.M709303200. [DOI] [PubMed] [Google Scholar]
- Binz H. K.; Amstutz P.; Plückthun A. Engineering novel binding proteins from nonimmunoglobulin domains. Nat. Biotechnol. 2005, 23, 1257–1268. 10.1038/nbt1127. [DOI] [PubMed] [Google Scholar]
- Simeon R.; Chen Z. In vitro-engineered non-antibody protein therapeutics. Protein Cell 2018, 9, 3–14. 10.1007/s13238-017-0386-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sha F.; Salzman G.; Gupta A.; Koide S. Monobodies and other synthetic binding proteins for expanding protein science. Protein Sci. 2017, 26, 910–924. 10.1002/pro.3148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lipovšek D. Adnectins: engineered target-binding protein therapeutics. Prot. Eng. Des. Sel. 2011, 24, 3–9. 10.1093/protein/gzq097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Plückthun A. Designed Ankyrin Repeat Proteins (DARPins): Binding Proteins for Research, Diagnostics, and Therapy. Ann. Rev. Pharmacol. Toxicol. 2015, 55, 489–511. 10.1146/annurev-pharmtox-010611-134654. [DOI] [PubMed] [Google Scholar]
- Ståhl S.; Gräslund T.; Eriksson Karlström A.; Frejd F. Y.; Nygren P.-Å.; Löfblom J. Affibody Molecules in Biotechnological and Medical Applications. Trends Biotechnol. 2017, 35, 691–712. 10.1016/j.tibtech.2017.04.007. [DOI] [PubMed] [Google Scholar]
- Mihara E.; Watanabe S.; Bashiruddin N. K.; Nakamura N.; Matoba K.; Sano Y.; Maini R.; Yin Y.; Sakai K.; Arimori T.; Matsumoto K.; Suga H.; Takagi J. Lasso-grafting of macrocyclic peptide pharmacophores yields multi-functional proteins. Nat. Commun. 2021, 12, 1543. 10.1038/s41467-021-21875-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawakami N.; Sato H.; Terasaka N.; Matsumoto K.; Suga H. MET-Activating Ubiquitin Multimers. Angew. Chem., Int. Ed. 2023, 62, e202307157 10.1002/anie.202307157. [DOI] [PubMed] [Google Scholar]
- Yin H.; Craik D. J.; Wang C. K. Anchor Residues Guide Form and Function in Grafted Peptides. Angew. Chem., Int. Ed. 2019, 58, 7652–7656. 10.1002/anie.201901572. [DOI] [PubMed] [Google Scholar]
- Muttenthaler M.; King G. F.; Adams D. J.; Alewood P. F. Trends in peptide drug discovery. Nat. Rev. Drug Discovery 2021, 20, 309–325. 10.1038/s41573-020-00135-8. [DOI] [PubMed] [Google Scholar]
- Feng Z.; Xu B. Inspiration from the mirror: D-amino acid containing peptides in biomedical approaches. Biomol. Concepts 2016, 7, 179–187. 10.1515/bmc-2015-0035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wiesehan K.; Buder K.; Linke R. P.; Patt S.; Stoldt M.; Unger E.; Schmitt B.; Bucci E.; Willbold D. Selection of D-Amino-Acid Peptides That Bind to Alzheimer’s Disease Amyloid Peptide Aβ1–42 by Mirror Image Phage Display. ChemBioChem. 2003, 4, 748–753. 10.1002/cbic.200300631. [DOI] [PubMed] [Google Scholar]
- Callahan A. J.; Gandhesiri S.; Travaline T. L.; Reja R. M.; Lozano Salazar L.; Hanna S.; Lee Y.-C.; Li K.; Tokareva O. S.; Swiecicki J.-M.; Loas A.; Verdine G. L.; McGee J. H.; Pentelute B. L. Mirror-image ligand discovery enabled by single-shot fast-flow synthesis of D-proteins. Nat. Commun. 2024, 15, 1813. 10.1038/s41467-024-45634-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guillen Schlippe Y. V.; Hartman M. C. T.; Josephson K.; Szostak J. W. In Vitro Selection of Highly Modified Cyclic Peptides That Act as Tight Binding Inhibitors. J. Am. Chem. Soc. 2012, 134, 10469–10477. 10.1021/ja301017y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miura T.; Malla T. R.; Owen C. D.; Tumber A.; Brewitz L.; McDonough M. A.; Salah E.; Terasaka N.; Katoh T.; Lukacik P.; Strain-Damerell C.; Mikolajek H.; Walsh M. A.; Kawamura A.; Schofield C. J.; Suga H. In vitro selection of macrocyclic peptide inhibitors containing cyclic γ2,4-amino acids targeting the SARS-CoV-2 main protease. Nat. Chem. 2023, 15, 998–1005. 10.1038/s41557-023-01205-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katoh T.; Sengoku T.; Hirata K.; Ogata K.; Suga H. Ribosomal synthesis and de novo discovery of bioactive foldamer peptides containing cyclic β-amino acids. Nat. Chem. 2020, 12, 1081–1088. 10.1038/s41557-020-0525-1. [DOI] [PubMed] [Google Scholar]
- Kato T.; Kita Y.; Iwanari K.; Asano A.; Oba M.; Tanaka M.; Doi M. Synthesis of six-membered carbocyclic ring α,α-disubstituted amino acids and arginine-rich peptides to investigate the effect of ring size on the properties of the peptide. Bioorg. Med. Chem. 2021, 38, 116111. 10.1016/j.bmc.2021.116111. [DOI] [PubMed] [Google Scholar]
- Hirano M.; Saito C.; Goto C.; Yokoo H.; Kawano R.; Misawa T.; Demizu Y. Rational Design of Helix-Stabilized Antimicrobial Peptide Foldamers Containing α,α-Disubstituted Amino Acids or Side-Chain Stapling. ChemPlusChem. 2020, 85, 2731–2736. 10.1002/cplu.202000749. [DOI] [PubMed] [Google Scholar]
- Iwane Y.; Kimura H.; Katoh T.; Suga H. Uniform affinity-tuning of N-methyl-aminoacyl-tRNAs to EF-Tu enhances their multiple incorporation. Nucleic Acids Res. 2021, 49, 10807–10817. 10.1093/nar/gkab288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawakami T.; Ishizawa T.; Murakami H. Extensive Reprogramming of the Genetic Code for Genetically Encoded Synthesis of Highly N-Alkylated Polycyclic Peptidomimetics. J. Am. Chem. Soc. 2013, 135, 12297–12304. 10.1021/ja405044k. [DOI] [PubMed] [Google Scholar]
- Katoh T.; Suga H. Ribosomal incorporation of negatively charged d-α- and N-methyl-l-α-amino acids enhanced by EF-Sep. Philos. Trans Royal Soc. B. Biol. Sci. 2023, 378, 20220038. 10.1098/rstb.2022.0038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sigal M.; Matsumoto S.; Beattie A.; Katoh T.; Suga H. Engineering tRNAs for the Ribosomal Translation of Non-proteinogenic Monomers. Chem. Rev. 2024, 124, 6444–6500. 10.1021/acs.chemrev.3c00894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hampton J. T.; Liu W. R. Diversification of Phage-Displayed Peptide Libraries with Noncanonical Amino Acid Mutagenesis and Chemical Modification. Chem. Rev. 2024, 124, 6051–6077. 10.1021/acs.chemrev.4c00004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vinogradov A. A.; Zhang Y.; Hamada K.; Chang J. S.; Okada C.; Nishimura H.; Terasaka N.; Goto Y.; Ogata K.; Sengoku T.; Onaka H.; Suga H. De Novo Discovery of Thiopeptide Pseudo-natural Products Acting as Potent and Selective TNIK Kinase Inhibitors. J. Am. Chem. Soc. 2022, 144, 20332–20341. 10.1021/jacs.2c07937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y.; Hamada K.; Nguyen D. T.; Inoue S.; Satake M.; Kobayashi S.; Okada C.; Ogata K.; Okada M.; Sengoku T.; Goto Y.; Suga H. LimF is a versatile prenyltransferase for histidine-C-geranylation on diverse non-natural substrates. Nat. Catal. 2022, 5, 682–693. 10.1038/s41929-022-00822-2. [DOI] [Google Scholar]
- Charoenpattarapreeda J.; Tan Y. S.; Iegre J.; Walsh S. J.; Fowler E.; Eapen R. S.; Wu Y.; Sore H. F.; Verma C. S.; Itzhaki L.; Spring D. R. Targeted covalent inhibitors of MDM2 using electrophile-bearing stapled peptides. Chem. Commun. 2019, 55, 7914–7917. 10.1039/C9CC04022F. [DOI] [PubMed] [Google Scholar]
- Lan T.; Peng C.; Yao X.; Chan R. S. T.; Wei T.; Rupanya A.; Radakovic A.; Wang S.; Chen S.; Lovell S.; Snyder S. A.; Bogyo M.; Dickinson B. C. Discovery of Thioether-Cyclized Macrocyclic Covalent Inhibitors by mRNA Display. J. Am. Chem. Soc. 2024, 146, 24053–24060. 10.1021/jacs.4c07851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iskandar S. E.; Chiou L. F.; Leisner T. M.; Shell D. J.; Norris-Drouin J. L.; Vaziri C.; Pearce K. H.; Bowers A. A. Identification of Covalent Cyclic Peptide Inhibitors in mRNA Display. J. Am. Chem. Soc. 2023, 145, 15065–15070. 10.1021/jacs.3c04833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paulussen F. M.; Grossmann T. N. Peptide-based covalent inhibitors of protein–protein interactions. J. Pept. Sci. 2023, 29, e3457 10.1002/psc.3457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhardwaj G.; O’Connor J.; Rettie S.; Huang Y.-H.; Ramelot T. A.; Mulligan V. K.; Alpkilic G. G.; Palmer J.; Bera A. K.; Bick M. J.; Di Piazza M.; Li X.; Hosseinzadeh P.; Craven T. W.; Tejero R.; Lauko A.; Choi R.; Glynn C.; Dong L.; Griffin R.; van Voorhis W. C.; Rodriguez J.; Stewart L.; Montelione G. T.; Craik D.; Baker D. Accurate de novo design of membrane-traversing macrocycles. Cell 2022, 185, 3520–3532.e26. 10.1016/j.cell.2022.07.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mizuno-Kaneko M.; Hashimoto I.; Miyahara K.; Kochi M.; Ohashi N.; Tsumura K.; Suzuki K.; Tamura T. Molecular Design of Cyclic Peptides with Cell Membrane Permeability and Development of MDMX-p53 Inhibitor. ACS Med. Chem. Lett. 2023, 14, 1174–1178. 10.1021/acsmedchemlett.3c00102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Faris J. H.; Adaligil E.; Popovych N.; Ono S.; Takahashi M.; Nguyen H.; Plise E.; Taechalertpaisarn J.; Lee H.-W.; Koehler M. F. T.; Cunningham C. N.; Lokey R. S. Membrane Permeability in a Large Macrocyclic Peptide Driven by a Saddle-Shaped Conformation. J. Am. Chem. Soc. 2024, 146, 4582–4591. 10.1021/jacs.3c10949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Batistatou N.; Kritzer J. A. Recent advances in methods for quantifying the cell penetration of macromolecules. Curr. Opin. Chem. Biol. 2024, 81, 102501. 10.1016/j.cbpa.2024.102501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gayraud F.; Klußmann M.; Neundorf I. Recent Advances and Trends in Chemical CPP–Drug Conjugation Techniques. Molecules 2021, 26, 1591. 10.3390/molecules26061591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dai S. A.; Hu Q.; Gao R.; Blythe E. E.; Touhara K. K.; Peacock H.; Zhang Z.; von Zastrow M.; Suga H.; Shokat K. M. State-selective modulation of heterotrimeric Gαs signaling with macrocyclic peptides. Cell 2022, 185, 3950–3965.e25. 10.1016/j.cell.2022.09.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen K.; Pei D. Engineering Cell-Permeable Proteins through Insertion of Cell-Penetrating Motifs into Surface Loops. ACS Chem. Biol. 2020, 15, 2568–2576. 10.1021/acschembio.0c00593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee Y.; Kim K.-M.; Nguyen D. L.; Jannah F.; Seong H.-J.; Kim J.-M.; Kim Y.-P. Cyclized proteins with tags as permeable and stable cargos for delivery into cells and liposomes. Int. J. Biol. Macromol. 2023, 252, 126520. 10.1016/j.ijbiomac.2023.126520. [DOI] [PubMed] [Google Scholar]
- Philippe G. J.-B.; Huang Y.-H.; Mittermeier A.; Brown C. J.; Kaas Q.; Ramlan S. R.; Wang C. K.; Lane D.; Loewer A.; Troeira Henriques S.; Craik D. J. Delivery to, and Reactivation of, the p53 Pathway in Cancer Cells Using a Grafted Cyclotide Conjugated with a Cell-Penetrating Peptide. J. Med. Chem. 2024, 67, 1197–1208. 10.1021/acs.jmedchem.3c01682. [DOI] [PubMed] [Google Scholar]
- Schneider A. F. L.; Kallen J.; Ottl J.; Reid P. C.; Ripoche S.; Ruetz S.; Stachyra T.-M.; Hintermann S.; Dumelin C. E.; Hackenberger C. P. R.; Marzinzik A. L. Discovery, X-ray structure and CPP-conjugation enabled uptake of p53/MDM2 macrocyclic peptide inhibitors. RSC Chem. Biol. 2021, 2, 1661–1668. 10.1039/D1CB00056J. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dougherty P. G.; Wen J.; Pan X.; Koley A.; Ren J.-G.; Sahni A.; Basu R.; Salim H.; Appiah Kubi G.; Qian Z.; Pei D. Enhancing the Cell Permeability of Stapled Peptides with a Cyclic Cell-Penetrating Peptide. J. Med. Chem. 2019, 62, 10098–10107. 10.1021/acs.jmedchem.9b00456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ng S.; Juang Y.-C.; Chandramohan A.; Kaan H. Y. K.; Sadruddin A.; Yuen T. Y.; Ferrer-Gago F. J.; Lee X. E. C.; Liew X.; Johannes C. W.; Brown C. J.; Kannan S.; Aronica P. G.; Berglund N. A.; Verma C. S.; Liu L.; Stoeck A.; Sawyer T. K.; Partridge A. W.; Lane D. P. De-risking Drug Discovery of Intracellular Targeting Peptides: Screening Strategies to Eliminate False-Positive Hits. ACS Med. Chem. Lett. 2020, 11, 1993–2001. 10.1021/acsmedchemlett.0c00022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diderich P.; Bertoldo D.; Dessen P.; Khan M. M.; Pizzitola I.; Held W.; Huelsken J.; Heinis C. Phage Selection of Chemically Stabilized α-Helical Peptide Ligands. ACS Chem. Biol. 2016, 11, 1422–1427. 10.1021/acschembio.5b00963. [DOI] [PubMed] [Google Scholar]
- Anananuchatkul T.; Tsutsumi H.; Miki T.; Mihara H. hDM2 protein-binding peptides screened from stapled α-helical peptide phage display libraries with different types of staple linkers. Bioorg. Med. Chem. Lett. 2020, 30, 127605. 10.1016/j.bmcl.2020.127605. [DOI] [PubMed] [Google Scholar]
- Li K.; Tokareva O. S.; Thomson T. M.; Wahl S. C. T.; Travaline T. L.; Ramirez J. D.; Choudary S. K.; Agarwal S.; Walkup W. G.; Olsen T. J.; Brennan M. J.; Verdine G. L.; McGee J. H. De novo mapping of α-helix recognition sites on protein surfaces using unbiased libraries. Proc. Natl. Adac. Sci. 2022, 119, e2210435119 10.1073/pnas.2210435119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chu Q.; Moellering R. E.; Hilinski G. J.; Kim Y.-W.; Grossmann T. N.; Yeh J. T. H.; Verdine G. L. Towards understanding cell penetration by stapled peptides. MedChemComm 2015, 6, 111–119. 10.1039/C4MD00131A. [DOI] [Google Scholar]
- Miller S. E.; Schneider J. P. The effect of turn residues on the folding and cell-penetrating activity of β-hairpin peptides and applications toward protein delivery. Pept. Sci. 2020, 112, e24125 10.1002/pep2.24125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hymel H. C.; Anderson J. C.; Liu D.; Gauthier T. J.; Melvin A. T. Incorporating a β-hairpin sequence motif to increase intracellular stability of a peptide-based PROTAC. Biochem. Eng. J. 2023, 199, 109063. 10.1016/j.bej.2023.109063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Safa N.; Anderson J. C.; Vaithiyanathan M.; Pettigrew J. H.; Pappas G. A.; Liu D.; Gauthier T. J.; Melvin A. T. CPProtectides: Rapid uptake of well-folded β-hairpin peptides with enhanced resistance to intracellular degradation. Pept. Sci. 2019, 111, e24092 10.1002/pep2.24092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moxam J.; Naylon S.; Richaud A. D.; Zhao G.; Padilla A.; Roche S. P. Passive Membrane Permeability of Sizable Acyclic β-Hairpin Peptides. ACS Med. Chem. Lett. 2023, 14, 278–284. 10.1021/acsmedchemlett.2c00486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shinbara K.; Liu W.; van Neer R. H. P.; Katoh T.; Suga H. Methodologies for Backbone Macrocyclic Peptide Synthesis Compatible With Screening Technologies. Front. Chem. 2020, 8, 447. 10.3389/fchem.2020.00447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takatsuji R.; Shinbara K.; Katoh T.; Goto Y.; Passioura T.; Yajima R.; Komatsu Y.; Suga H. Ribosomal Synthesis of Backbone-Cyclic Peptides Compatible with In Vitro Display. J. Am. Chem. Soc. 2019, 141, 2279–2287. 10.1021/jacs.8b05327. [DOI] [PubMed] [Google Scholar]
- Chen H.; Katoh T.; Suga H. Macrocyclic Peptides Closed by a Thioether–Bipyridyl Unit That Grants Cell Membrane Permeability. ACS Biol. & Med. Chem. Au 2023, 3, 429–437. 10.1021/acsbiomedchemau.3c00027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kato T. Foldamer. In Cell-Penetrating. Peptides 2023, 79–107. 10.1002/9783527835997.ch7. [DOI] [Google Scholar]
- Dengler S.; Howard R. T.; Morozov V.; Tsiamantas C.; Huang W.-E.; Liu Z.; Dobrzanski C.; Pophristic V.; Brameyer S.; Douat C.; Suga H.; Huc I. Display Selection of a Hybrid Foldamer–Peptide Macrocycle. Angew. Chem., Int. Ed. 2023, 62, e202308408 10.1002/anie.202308408. [DOI] [PubMed] [Google Scholar]
- Shin S. B. Y.; Kirshenbaum K. Conformational Rearrangements by Water-Soluble Peptoid Foldamers. Org. Lett. 2007, 9, 5003–5006. 10.1021/ol702207n. [DOI] [PubMed] [Google Scholar]
- Zhang S.; Prabpai S.; Kongsaeree P.; Arvidsson P. I. Poly-N-methylated α-peptides: synthesis and X-ray structure determination of β-strand forming foldamers. Chem. Commun. 2006, 497–499. 10.1039/B513277K. [DOI] [PubMed] [Google Scholar]
- Morimoto J.; Fukuda Y.; Kuroda D.; Watanabe T.; Yoshida F.; Asada M.; Nakamura T.; Senoo A.; Nagatoishi S.; Tsumoto K.; Sando S. A Peptoid with Extended Shape in Water. J. Am. Chem. Soc. 2019, 141, 14612–14623. 10.1021/jacs.9b04371. [DOI] [PubMed] [Google Scholar]
- Li Y.; Li W.; Xu Z. Improvement on Permeability of Cyclic Peptide/Peptidomimetic: Backbone N-Methylation as A Useful Tool. Marine Drugs 2021, 19, 311. 10.3390/md19060311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hosono Y.; Uchida S.; Shinkai M.; Townsend C. E.; Kelly C. N.; Naylor M. R.; Lee H.-W.; Kanamitsu K.; Ishii M.; Ueki R.; Ueda T.; Takeuchi K.; Sugita M.; Akiyama Y.; Lokey S. R.; Morimoto J.; Sando S. Amide-to-ester substitution as a stable alternative to N-methylation for increasing membrane permeability in cyclic peptides. Nat. Commun. 2023, 14, 1416. 10.1038/s41467-023-36978-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghosh P.; Raj N.; Verma H.; Patel M.; Chakraborti S.; Khatri B.; Doreswamy C. M.; Anandakumar S. R.; Seekallu S.; Dinesh M. B.; Jadhav G.; Yadav P. N.; Chatterjee J. An amide to thioamide substitution improves the permeability and bioavailability of macrocyclic peptides. Nat. Commun. 2023, 14, 6050. 10.1038/s41467-023-41748-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uhl P.; Bajraktari-Sylejmani G.; Witzigmann D.; Bay C.; Zimmermann S.; Burhenne J.; Weiss J.; Haefeli W. E.; Sauter M. A Nanocarrier Approach for Oral Peptide Delivery: Evaluation of Cell-Penetrating-Peptide-Modified Liposomal Formulations in Dogs. Adv. Ther. 2023, 6, 2300021. 10.1002/adtp.202300021. [DOI] [Google Scholar]
- Liu P.; Chen G.; Zhang J. A Review of Liposomes as a Drug Delivery System: Current Status of Approved Products, Regulatory Environments, and Future Perspectives. Molecules 2022, 27 (m), 1372. 10.3390/molecules27041372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ismail M.; Martin S. R.; George R.; Houghton F.; Kelly G.; Chaleil R. A. G.; Anastasiou P.; Wang X.; O’Reilly N.; Federico S.; Joshi D.; Nagaraj H.; Cooley R.; Hui N. S.; Molina-Arcas M.; Hancock D. C.; Tavassoli A.; Downward J. Characterisation of a cyclic peptide that binds to the RAS binding domain of phosphoinositide 3-kinase p110α. Sci. Rep. 2023, 13, 1889. 10.1038/s41598-023-28756-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tria S. M.; Burge M. E.; Whitehall V. L. J. The Therapeutic Landscape for KRAS-Mutated Colorectal Cancers. Cancers 2023, 15, 2375. 10.3390/cancers15082375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Z.; Rohweder P. J.; Ongpipattanakul C.; Basu K.; Bohn M. F.; Dugan E. J.; Steri V.; Hann B.; Shokat K. M.; Craik C. S. A covalent inhibitor of K-Ras(G12C) induces MHC class I presentation of haptenated peptide neoepitopes targetable by immunotherapy. Cancer Cell 2022, 40, 1060–1069.e7. 10.1016/j.ccell.2022.07.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng Q.; Zhang Z.; Guiley K. Z.; Shokat K. M. Strain-release alkylation of Asp12 enables mutant selective targeting of K-Ras-G12D. Nat. Chem. Biol. 2024, 20, 1114–1122. 10.1038/s41589-024-01565-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Z.; Gao R.; Hu Q.; Peacock H.; Peacock D. M.; Dai S.; Shokat K. M.; Suga H. GTP-State-Selective Cyclic Peptide Ligands of K-Ras(G12D) Block Its Interaction with Raf. ACS Cent. Sci. 2020, 6, 1753–1761. 10.1021/acscentsci.0c00514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanada M.; Tamiya M.; Matsuo A.; Chiyoda A.; Takano K.; Ito T.; Irie M.; Kotake T.; Takeyama R.; Kawada H.; Hayashi R.; Ishikawa S.; Nomura K.; Furuichi N.; Morita Y.; Kage M.; Hashimoto S.; Nii K.; Sase H.; Ohara K.; Ohta A.; Kuramoto S.; Nishimura Y.; Iikura H.; Shiraishi T. Development of Orally Bioavailable Peptides Targeting an Intracellular Protein: From a Hit to a Clinical KRAS Inhibitor. J. Am. Chem. Soc. 2023, 145, 16610–16620. 10.1021/jacs.3c03886. [DOI] [PubMed] [Google Scholar]
- Ohta A.; Tanada M.; Shinohara S.; Morita Y.; Nakano K.; Yamagishi Y.; Takano R.; Kariyuki S.; Iida T.; Matsuo A.; Ozeki K.; Emura T.; Sakurai Y.; Takano K.; Higashida A.; Kojima M.; Muraoka T.; Takeyama R.; Kato T.; Kimura K.; Ogawa K.; Ohara K.; Tanaka S.; Kikuchi Y.; Hisada N.; Hayashi R.; Nishimura Y.; Nomura K.; Tachibana T.; Irie M.; Kawada H.; Torizawa T.; Murao N.; Kotake T.; Tanaka M.; Ishikawa S.; Miyake T.; Tamiya M.; Arai M.; Chiyoda A.; Akai S.; Sase H.; Kuramoto S.; Ito T.; Shiraishi T.; Kojima T.; Iikura H. Validation of a New Methodology to Create Oral Drugs beyond the Rule of 5 for Intracellular Tough Targets. J. Am. Chem. Soc. 2023, 145, 24035–24051. 10.1021/jacs.3c07145. [DOI] [PubMed] [Google Scholar]
- Sakamoto K.; Lin B.; Nunomura K.; Izawa T.; Nakagawa S. The K-Ras(G12D)-inhibitory peptide KS-58 suppresses growth of murine CT26 colorectal cancer cell-derived tumors. Sci. Rep. 2022, 12, 8121. 10.1038/s41598-022-12401-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakamoto K.; Masutani T.; Hirokawa T. Generation of KS-58 as the first K-Ras(G12D)-inhibitory peptide presenting anti-cancer activity in vivo. Sci. Rep. 2020, 10, 21671. 10.1038/s41598-020-78712-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakamoto K.; Kamada Y.; Sameshima T.; Yaguchi M.; Niida A.; Sasaki S.; Miwa M.; Ohkubo S.; Sakamoto J.-i.; Kamaura M.; Cho N.; Tani A. K-Ras(G12D)-selective inhibitory peptides generated by random peptide T7 phage display technology. Biochem. Biophys. Res. Commun. 2017, 484, 605–611. 10.1016/j.bbrc.2017.01.147. [DOI] [PubMed] [Google Scholar]
- Sakamoto K.; Hirokawa T. Lipid bilayer membrane permeability mechanism of the K-Ras(G12D)-inhibitory bicyclic peptide KS-58 elucidated by molecular dynamics simulations. Bioorg. Med. Chem. Lett. 2024, 100, 129649. 10.1016/j.bmcl.2024.129649. [DOI] [PubMed] [Google Scholar]
- Kirsch P.; Hartman A. M.; Hirsch A. K. H.; Empting M. Concepts and Core Principles of Fragment-Based Drug Design. Molecules 2019, 24, 4309. 10.3390/molecules24234309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen L.; Zhuang C.; Lu J.; Jiang Y.; Sheng C. Discovery of Novel KRAS-PDEδ Inhibitors by Fragment-Based Drug Design. J. Med. Chem. 2018, 61, 2604–2610. 10.1021/acs.jmedchem.8b00057. [DOI] [PubMed] [Google Scholar]
- Philippe G. J. B.; Craik D. J.; Henriques S. T. Converting peptides into drugs targeting intracellular protein–protein interactions. Drug Discovery Today 2021, 26, 1521–1531. 10.1016/j.drudis.2021.01.022. [DOI] [PubMed] [Google Scholar]
- Vázquez Torres S.; Leung P. J. Y.; Venkatesh P.; Lutz I. D.; Hink F.; Huynh H.-H.; Becker J.; Yeh A. H.-W.; Juergens D.; Bennett N. R.; Hoofnagle A. N.; Huang E.; MacCoss M. J.; Expòsit M.; Lee G. R.; Bera A. K.; Kang A.; De La Cruz J.; Levine P. M.; Li X.; Lamb M.; Gerben S. R.; Murray A.; Heine P.; Korkmaz E. N.; Nivala J.; Stewart L.; Watson J. L.; Rogers J. M.; Baker D. De novo design of high-affinity binders of bioactive helical peptides. Nature 2024, 626, 435–442. 10.1038/s41586-023-06953-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ball A. T.; Mohammed S.; Doigneaux C.; Gardner R. M.; Easton J. W.; Turner S.; Essex J. W.; Pairaudeau G.; Tavassoli A. Identification and Development of Cyclic Peptide Inhibitors of Hypoxia Inducible Factors 1 and 2 That Disrupt Hypoxia-Response Signaling in Cancer Cells. J. Am. Chem. Soc. 2024, 146, 8877–8886. 10.1021/jacs.3c10508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miranda E.; Nordgren I. K.; Male A. L.; Lawrence C. E.; Hoakwie F.; Cuda F.; Court W.; Fox K. R.; Townsend P. A.; Packham G. K.; Eccles S. A.; Tavassoli A. A Cyclic Peptide Inhibitor of HIF-1 Heterodimerization That Inhibits Hypoxia Signaling in Cancer Cells. J. Am. Chem. Soc. 2013, 135, 10418–10425. 10.1021/ja402993u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li F.; Liu J.; Liu C.; Liu Z.; Peng X.; Huang Y.; Chen X.; Sun X.; Wang S.; Chen W.; Xiong D.; Diao X.; Wang S.; Zhuang J.; Wu C.; Wu D. Cyclic peptides discriminate BCL-2 and its clinical mutants from BCL-XL by engaging a single-residue discrepancy. Nat. Commun. 2024, 15, 1476. 10.1038/s41467-024-45848-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen S.; Lovell S.; Lee S.; Fellner M.; Mace P. D.; Bogyo M. Identification of highly selective covalent inhibitors by phage display. Nat. Biotechnol. 2021, 39, 490–498. 10.1038/s41587-020-0733-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng M.; Chen F.-J.; Li K.; Reja R. M.; Haeffner F.; Gao J. Lysine-Targeted Reversible Covalent Ligand Discovery for Proteins via Phage Display. J. Am. Chem. Soc. 2022, 144, 15885–15893. 10.1021/jacs.2c07375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanton H.; Sewastianik T.; Seo H.-S.; Remillard D.; Pierre R. S.; Bala P.; Aitymbayev D.; Dennis P.; Adler K.; Geffken E.; Yeoh Z.; Vangos N.; Garbicz F.; Scott D.; Sethi N.; Bradner J.; Dhe-Paganon S.; Carrasco R. D. A novel β-catenin/BCL9 complex inhibitor blocks oncogenic Wnt signaling and disrupts cholesterol homeostasis in colorectal cancer. Sci. Adv. 2022, 8, eabm3108 10.1126/sciadv.abm3108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McHenry M. W.; Shi P.; Camara C. M.; Cohen D. T.; Rettenmaier T. J.; Adhikary U.; Gygi M. A.; Yang K.; Gygi S. P.; Wales T. E.; Engen J. R.; Wells J. A.; Walensky L. D. Covalent inhibition of pro-apoptotic BAX. Nat. Chem. Biol. 2024, 20, 1022–1032. 10.1038/s41589-023-01537-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang N.; Shen H.; Chen B.; Hu H.; Liu C.; Chen Y.; Cong W. The recent progress of peptide regulators for the Wnt/β-catenin signaling pathway. Front. Med. 2023, 10, 1164656. 10.3389/fmed.2023.1164656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takada K.; Zhu D.; Bird G. H.; Sukhdeo K.; Zhao J.-J.; Mani M.; Lemieux M.; Carrasco D. E.; Ryan J.; Horst D.; Fulciniti M.; Munshi N. C.; Xu W.; Kung A. L.; Shivdasani R. A.; Walensky L. D.; Carrasco D. R. Targeted Disruption of the BCL9/β-Catenin Complex Inhibits Oncogenic Wnt Signaling. Sci. Transl. Med. 2012, 4, 148ra117. 10.1126/scitranslmed.3003808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feng M.; Jin J. Q.; Xia L.; Xiao T.; Mei S.; Wang X.; Huang X.; Chen J.; Liu M.; Chen C.; Rafi S.; Zhu A. X.; Feng Y. X.; Zhu D. Pharmacological inhibition of β-catenin/BCL9 interaction overcomes resistance to immune checkpoint blockades by modulating Treg cells. Sci. Adv. 2019, 5, eaau5240 10.1126/sciadv.aau5240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schneider J. A.; Craven T. W.; Kasper A. C.; Yun C.; Haugbro M.; Briggs E. M.; Svetlov V.; Nudler E.; Knaut H.; Bonneau R.; Garabedian M. J.; Kirshenbaum K.; Logan S. K. Design of Peptoid-peptide Macrocycles to Inhibit the β-catenin TCF Interaction in Prostate Cancer. Nat. Commun. 2018, 9, 4396. 10.1038/s41467-018-06845-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grossmann T. N.; Yeh J. T. H.; Bowman B. R.; Chu Q.; Moellering R. E.; Verdine G. L. Inhibition of oncogenic Wnt signaling through direct targeting of β-catenin. Proc. Natl. Adac. Sci. 2012, 109, 17942–17947. 10.1073/pnas.1208396109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X.; Dong L.; Cheng J.; Verdine G. L.; Lin A.; Chu Q. Targeted β-catenin ubiquitination and degradation by multifunctional stapled peptides. J. Pept. Sci. 2022, 28, e3389 10.1002/psc.3389. [DOI] [PubMed] [Google Scholar]
- Henchey L. K.; Porter J. R.; Ghosh I.; Arora P. S. High Specificity in Protein Recognition by Hydrogen-Bond-Surrogate α-Helices: Selective Inhibition of the p53/MDM2 Complex. ChemBioChem. 2010, 11, 2104–2107. 10.1002/cbic.201000378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lao B. B.; Drew K.; Guarracino D. A.; Brewer T. F.; Heindel D. W.; Bonneau R.; Arora P. S. Rational Design of Topographical Helix Mimics as Potent Inhibitors of Protein–Protein Interactions. J. Am. Chem. Soc. 2014, 136, 7877–7888. 10.1021/ja502310r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teveroni E.; Lucà R.; Pellegrino M.; Ciolli G.; Pontecorvi A.; Moretti F. Peptides and peptidomimetics in the p53/MDM2/MDM4 circuitry - a patent review. Expert Opin. Ther. Pat. 2016, 26, 1417–1429. 10.1080/13543776.2017.1233179. [DOI] [PubMed] [Google Scholar]
- Baek S.; Kutchukian P. S.; Verdine G. L.; Huber R.; Holak T. A.; Lee K. W.; Popowicz G. M. Structure of the Stapled p53 Peptide Bound to Mdm2. J. Am. Chem. Soc. 2012, 134, 103–106. 10.1021/ja2090367. [DOI] [PubMed] [Google Scholar]
- Kussie P. H.; Gorina S.; Marechal V.; Elenbaas B.; Moreau J.; Levine A. J.; Pavletich N. P. Structure of the MDM2 Oncoprotein Bound to the p53 Tumor Suppressor Transactivation Domain. Science 1996, 274, 948–953. 10.1126/science.274.5289.948. [DOI] [PubMed] [Google Scholar]
- Wilson A. J., α-Helix Mimetics as Inhibitors of Protein–Protein Interactions. In Inhibitors of Protein–Protein Interactions; Tavassoli A., Ed.; The Royal Society of Chemistry, 2020; pp 305–335. [Google Scholar]
- Roy S.; Ghosh P.; Ahmed I.; Chakraborty M.; Naiya G.; Ghosh B. Constrained α-Helical Peptides as Inhibitors of Protein-Protein and Protein-DNA Interactions. Biomedicines 2018, 6, 118. 10.3390/biomedicines6040118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jeganathan S.; Wendt M.; Kiehstaller S.; Brancaccio D.; Kuepper A.; Pospiech N.; Carotenuto A.; Novellino E.; Hennig S.; Grossmann T. N. Constrained Peptides with Fine-Tuned Flexibility Inhibit NF-Y Transcription Factor Assembly. Angew. Chem., Int. Ed. 2019, 58, 17351–17358. 10.1002/anie.201907901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Durukan C.; Arbore F.; Klintrot R.; Bigiotti C.; Ilie I. M.; Vreede J.; Grossmann T. N.; Hennig S. Binding Dynamics of a Stapled Peptide Targeting the Transcription Factor NF–Y. ChemBioChem. 2024, 25, e202400020 10.1002/cbic.202400020. [DOI] [PubMed] [Google Scholar]
- Hawley K. M.; Eclov R. J.; Schnorenberg M. R.; Tian Y.; Shah R. N.; Thomas-Toth A. T.; Fefferman M.; Bird G. H.; Walensky L. D.; Tirrell M. V.; LaBelle J. L. Inhibition of FOXP3 by stapled alpha-helical peptides dampens regulatory T cell function. Proc. Natl. Acad. Sci. U. S. A. 2022, 119, e2209044119 10.1073/pnas.2209044119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pham T. L.; Thomas F. Design of Functional Globular β-Sheet Miniproteins. ChemBioChem. 2024, 25, e202300745 10.1002/cbic.202300745. [DOI] [PubMed] [Google Scholar]
- Wendt M.; Bellavita R.; Gerber A.; Efrém N.-L.; van Ramshorst T.; Pearce N. M.; Davey P. R. J.; Everard I.; Vazquez-Chantada M.; Chiarparin E.; Grieco P.; Hennig S.; Grossmann T. N. Bicyclic β-Sheet Mimetics that Target the Transcriptional Coactivator β-Catenin and Inhibit Wnt Signaling. Angew. Chem., Int. Ed. 2021, 60, 13937–13944. 10.1002/anie.202102082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh A.; Kumar P.; Sarvagalla S.; Bharadwaj T.; Nayak N.; Coumar M. S.; Giri R.; Garg N. Functional inhibition of c-Myc using novel inhibitors identified through “hot spot”; targeting. J. Biol. Chem. 2022, 298, 101898. 10.1016/j.jbc.2022.101898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berlow R. B.; Dyson H. J.; Wright P. E. Expanding the Paradigm: Intrinsically Disordered Proteins and Allosteric Regulation. J. Mol. Biol. 2018, 430, 2309–2320. 10.1016/j.jmb.2018.04.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pérez-Jover I.; Rochon K.; Hu D.; Mahajan M.; Madan Mohan P.; Santos-Pérez I.; Ormaetxea Gisasola J.; Martinez Galvez J. M.; Agirre J.; Qi X.; Mears J. A.; Shnyrova A. V.; Ramachandran R. Allosteric control of dynamin-related protein 1 through a disordered C-terminal Short Linear Motif. Nat. Commun. 2024, 15, 52. 10.1038/s41467-023-44413-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wen B.; Zhang W.; Zhang Y.; Lei H.; Cao Y.; Li W.; Wang W. Self-Effected Allosteric Coupling and Cooperativity in Hypoxic Response Regulation with Disordered Proteins. J. Phys. Chem. Lett. 2022, 13, 9201–9209. 10.1021/acs.jpclett.2c02065. [DOI] [PubMed] [Google Scholar]
- Watanabe H.; Soejima K.; Yasuda H.; Kawada I.; Nakachi I.; Yoda S.; Naoki K.; Ishizaka A. Deregulation of histone lysine methyltransferases contributes to oncogenic transformation of human bronchoepithelial cells. Cancer Cell Int. 2008, 8, 15. 10.1186/1475-2867-8-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maleszewska M.; Kaminska B. Deregulation of histone-modifying enzymes and chromatin structure modifiers contributes to glioma development. Future Oncol. 2015, 11, 2587–2601. 10.2217/fon.15.171. [DOI] [PubMed] [Google Scholar]
- Qiu L.; Hu X.; Jing Q.; Zeng X.; Chan K.-M.; Han J. Mechanism of cancer: Oncohistones in action. J. Genet. Genomics 2018, 45, 227–236. 10.1016/j.jgg.2018.04.004. [DOI] [PubMed] [Google Scholar]
- Hosseinzadeh P.; Watson P. R.; Craven T. W.; Li X.; Rettie S.; Pardo-Avila F.; Bera A. K.; Mulligan V. K.; Lu P.; Ford A. S.; Weitzner B. D.; Stewart L. J.; Moyer A. P.; Di Piazza M.; Whalen J. G.; Greisen P. Jr.; Christianson D. W.; Baker D. Anchor extension: a structure-guided approach to design cyclic peptides targeting enzyme active sites. Nat. Commun. 2021, 12, 3384. 10.1038/s41467-021-23609-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawamura A.; Münzel M.; Kojima T.; Yapp C.; Bhushan B.; Goto Y.; Tumber A.; Katoh T.; King O. N. F.; Passioura T.; Walport L. J.; Hatch S. B.; Madden S.; Müller S.; Brennan P. E.; Chowdhury R.; Hopkinson R. J.; Suga H.; Schofield C. J. Highly selective inhibition of histone demethylases by de novo macrocyclic peptides. Nat. Commun. 2017, 8, 14773. 10.1038/ncomms14773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coleman O. D.; Macdonald J.; Thomson B.; Ward J. A.; Stubbs C. J.; McAllister T. E.; Clark S.; Amin S.; Cao Y.; Abboud M. I.; Zhang Y.; Sanganee H.; Huber K. V. M.; Claridge T. D. W.; Kawamura A. Cyclic peptides target the aromatic cage of a PHD-finger reader domain to modulate epigenetic protein function. Chem. Sci. 2023, 14, 7136–7146. 10.1039/D2SC05944D. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Madden S. K.; de Araujo A. D.; Gerhardt M.; Fairlie D. P.; Mason J. M. Taking the Myc out of cancer: toward therapeutic strategies to directly inhibit c-Myc. Mol. Cancer 2021, 20, 3. 10.1186/s12943-020-01291-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whitfield J. R.; Beaulieu M.-E.; Soucek L. Strategies to Inhibit Myc and Their Clinical Applicability. Front. Cell Dev. Biol. 2017, 5, 10. 10.3389/fcell.2017.00010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garralda E.; Beaulieu M.-E.; Moreno V.; Casacuberta-Serra S.; Martínez-Martín S.; Foradada L.; Alonso G.; Massó-Vallés D.; López-Estévez S.; Jauset T.; Corral de la Fuente E.; Doger B.; Hernández T.; Perez-Lopez R.; Arqués O.; Castillo Cano V.; Morales J.; Whitfield J. R.; Niewel M.; Soucek L.; Calvo E. MYC targeting by OMO-103 in solid tumors: a phase 1 trial. Nat. Med. 2024, 30, 762–771. 10.1038/s41591-024-02805-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Massó-Vallés D.; Soucek L. Blocking Myc to Treat Cancer: Reflecting on Two Decades of Omomyc. Cells 2020, 9, 883. 10.3390/cells9040883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Z.; Huang Y.; Hung T. I.; Sun J.; Aispuro D.; Chen B.; Guevara N.; Ji F.; Cong X.; Zhu L.; Wang S.; Guo Z.; Chang C.-e.; Xue M. MYC-Targeting Inhibitors Generated from a Stereodiversified Bicyclic Peptide Library. J. Am. Chem. Soc. 2024, 146, 1356–1363. 10.1021/jacs.3c09615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Z.; Shao S.; Ren X.; Sun J.; Guo Z.; Wang S.; Song M. M.; Chang C.-e. A.; Xue M. Construction of a Sequenceable Protein Mimetic Peptide Library with a True 3D Diversifiable Chemical Space. J. Am. Chem. Soc. 2018, 140, 14552–14556. 10.1021/jacs.8b08338. [DOI] [PubMed] [Google Scholar]
- Sundell G.; Ivarsson Y. Interaction Analysis through Proteomic Phage Display. BioMed. Res. Int. 2014, 2014, 176172. 10.1155/2014/176172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davey N. E.; Simonetti L.; Ivarsson Y. ProP-PD for proteome-wide motif-mediated interaction discovery. Trends Biotechnol. Sci. 2022, 47, 547–548. 10.1016/j.tibs.2022.01.005. [DOI] [PubMed] [Google Scholar]
- Ivarsson Y.; Arnold R.; McLaughlin M.; Nim S.; Joshi R.; Ray D.; Liu B.; Teyra J.; Pawson T.; Moffat J.; Li S. S.-C.; Sidhu S. S.; Kim P. M. Large-scale interaction profiling of PDZ domains through proteomic peptide-phage display using human and viral phage peptidomes. Proc. Natl. Adac. Sci. 2014, 111, 2542–2547. 10.1073/pnas.1312296111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ali M.; Simonetti L.; Ivarsson Y. Screening Intrinsically Disordered Regions for Short Linear Binding Motifs. Methods Mol. Biol. 2020, 529–552. 10.1007/978-1-0716-0524-0_27. [DOI] [PubMed] [Google Scholar]
- Benz C.; Ali M.; Krystkowiak I.; Simonetti L.; Sayadi A.; Mihalic F.; Kliche J.; Andersson E.; Jemth P.; Davey N. E.; Ivarsson Y. Proteome-scale mapping of binding sites in the unstructured regions of the human proteome. Mol. Syst. Biol. 2022, 18, e10584 10.15252/msb.202110584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tesei G.; Trolle A. I.; Jonsson N.; Betz J.; Knudsen F. E.; Pesce F.; Johansson K. E.; Lindorff-Larsen K. Conformational ensembles of the human intrinsically disordered proteome. Nature 2024, 626, 897–904. 10.1038/s41586-023-07004-5. [DOI] [PubMed] [Google Scholar]
- Hayward D.; Beekman A. M. Strategies for converting turn-motif and cyclic peptides to small molecules for targeting protein-protein interactions. RSC Chem. Biol. 2024, 5, 198–208. 10.1039/D3CB00222E. [DOI] [PMC free article] [PubMed] [Google Scholar]