

Abstract
It is well recognized that changes in the extracellular concentration of calcium ions influence the excitability of neurons, yet what mechanism(s) mediate these effects is still a matter of debate. Using patch‐clamp recordings from rat hippocampal CA1 pyramidal neurons, we examined the contribution of G‐proteins and intracellular calcium‐dependent signaling mechanisms to changes in intrinsic excitability evoked by altering the extracellular calcium concentration from physiological (1.2 mM) to a commonly used experimental (2 mM) level. We find that the inhibitory effect on intrinsic excitability of calcium ions is mainly expressed as an increased threshold for action potential firing (with no significant effect on resting membrane potential) that is not blocked by either the G‐protein inhibitor GDPβS or the calcium chelator BAPTA. Our results therefore argue that in the concentration range studied, G‐protein coupled calcium‐sensing receptors, non‐selective cation conductances, and intracellular calcium signaling pathways are not involved in mediating the effect of extracellular calcium ions on intrinsic excitability. Analysis of the derivative of the action potential, dV/dt versus membrane potential, indicates a current shift towards more depolarized membrane potentials at the higher calcium concentration. Our results are thus consistent with a mechanism in which extracellular calcium ions act directly on the voltage‐gated sodium channels by neutralizing negative charges on the extracellular surface of these channels to modulate the threshold for action potential activation.
Keywords: calcium, CaSR, hippocampus, NALCN, neuronal excitability
Although it is a well‐known fact that extracellular calcium has a suppressing effect on neuronal excitability, no consensus exists about the mechanism behind this phenomenon. Using patch‐clamp recordings from CA1 neurons from rat hippocampal acute slices, we investigate the literature‐proposed mechanisms and find that the effect of calcium on excitability does not depend on g‐protein signaling, excluding mechanisms involving the CaSR (calcium‐sensing receptor), neither does it require changes in the concentration of intracellular calcium. Our findings support the model of charge screen, where calcium ions neutralize negative charges on or adjacent to the voltage‐gated sodium channels.
Abbreviations
- BAPTA
1,2‐Bis(2‐aminophenoxy)ethane‐N,N,N′,N′‐tetraacetic acid
- CSF
cerebrospinal fluid
- CCh
carbachol
- CaSR
calcium‐sensing receptor
- GDPβS
Guanosine 5′‐[β‐thio]diphosphate
- NALCN
sodium leak channel non‐selective
- RMP
resting membrane potential
- RRID
Research Resource Identifier
1. INTRODUCTION
When Sydney Ringer in the late 1800s realized that he had used pipe water instead of distilled water when preparing saline solution to study the physiology of frog hearts, he realized that low concentration(s) of some ion(s), beside sodium and chloride, was necessary for normal contractility. He soon came to the conclusion that calcium plays an important role in the physiology of excitable tissues (Ringer, 1883). The concentration of intracellular calcium is very low at baseline (50–100 nM), but increases transiently when the neuron is activated (Zucker, 1996). While brief activity‐dependent rises in intracellular calcium have been thoroughly investigated, the concentration and neuromodulatory effect of extracellular calcium is, on the other hand, relatively understudied. Normal levels as well as fluctuations in the concentration of extracellular calcium remain poorly investigated even though some studies have indicated a role for altered concentrations in pathologies such as dementia with Lewi bodies (Boström et al., 2009) and epilepsy (Sood et al., 1993).
Ringer eventually used a concentration of 1.25 mM of calcium in his famous Ringer's solution, whereas in vitro experiments in the field of neurophysiology have traditionally used higher concentrations (2–4 mM) in the experimental medium in order to suppress excess activity in slice preparations and/or to facilitate synaptic transmission. Since the total concentration of calcium in human cerebrospinal fluid is about the same as in Ringer's original recipe (1.2 mM; Forsberg et al., 2019), this practice creates an unphysiological condition for the neurons and the results of such experiments may not be fully translatable to the in vivo situation. For example, recent experiments indicate that the common way of inducing long‐term potentiation in vitro, to study cellular mechanisms of learning and memory may not be plausible in the physiological concentration of calcium (Forsberg et al., 2019; Inglebert et al., 2020). To add another layer of complexity, the concentration of calcium may not be the same in the synaptic cleft as in the surrounding interstitial fluid or cerebrospinal fluid (Lopes & Cunha, 2019).
Studies on the mechanism(s) behind the effects of extracellular calcium on neuronal excitability have a long history, but are, however, still controversial (Martiszus et al., 2021). In the 1950s, Frankenhaeuser and Hodgkin described an inhibitory effect of extracellular calcium on the excitability of the squid giant axon (Frankenhaeuser & Hodgkin, 1957). They then proposed what later evolved to become the charge‐screening theory, which states that divalent cations are attracted to negative charges on the membrane (McLaughlin et al., 1971) or extracellular part of ion channels (Bennett et al., 1997; Recio‐Pinto et al., 1990), and by way of screening these negative charges affect the voltage sensing of voltage‐gated ion channels. The charge‐screening theory was later challenged by theories in which the calcium ions exert their effects on gating via more direct interactions with the channel pore (Armstrong, 1999; Armstrong & Cota, 1990, 1991, 1999). A heritage from the early experiments in sea living animals is that the concentrations of calcium and other divalent cations used in these experiments are quite extreme from a mammalian perspective, often 10‐fold changes of the concentrations are compared. Estimations of the effect in the span of 1–4 mM relevant for research conducted on mammalian cells suggest rather modest effects. Recent studies have, however, presented profound differences in spontaneous firing, excitability, and synaptic transmission in the relatively small span of 1–2 mM of extracellular calcium (Forsberg et al., 2019; Wang & Lu, 2023).
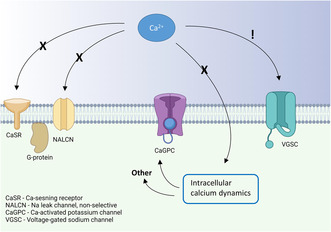
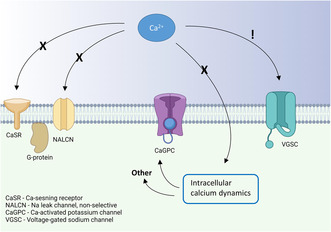
During the last two decades, studies have focused on mechanisms that can explain the inhibitory effect of extracellular calcium on neuronal excitability in this narrower concentration range. One such candidate mechanism is activation of the G‐protein coupled calcium‐sensing receptor (CaSR), first discovered in the parathyroid gland (Brown et al., 1993) and later found to be expressed in neurons (Chen et al., 2010; Gama et al., 2001; Vassilev et al., 1997). The CaSR has in multiple studies been implicated in the regulation of a non‐specific cation current (NSCC) (Chen et al., 2010; Ye et al., 1996), which may be carried by the sodium leak channel NALCN. Moreover, this channel has also been shown to be regulated by extracellular calcium without the CaSR present (Chua et al., 2020; Martiszus et al., 2021). Other studies have attributed the effect of extracellular calcium on neuronal excitability to altered activation of calcium‐dependent potassium channels (Vysotskaya et al., 2014), possibly via calcium‐induced calcium release (CICR; Segal, 2018), affecting the relative refractory period of the action potential (Roshchin et al., 2020).
Here we tested the involvement of above suggested mechanisms mediating the effects of extracellular calcium on the intrinsic excitability of hippocampal CA1 pyramidal neurons in the rat hippocampal slice, using concentrations spanning from the physiological level of 1.2–2 mM (the concentration often used in in vitro experiments). We find no evidence of G‐protein or intracellular calcium dependence (e.g. CaSR, NALCN, and CICR) in the effects of extracellular calcium ions on neuronal excitability in this concentration range. On the other hand, we found that the increase to 2 mM calcium was associated with a current shift during the action potential towards more depolarized membrane potentials, favoring surface charge neutralization on the extracellular part of voltage‐gated sodium channels as mechanistic explanation.
2. METHODS
2.1. Animals and slice preparation
Acute slices were prepared from Wistar rats of both sexes, P15–25 (RRID:RGD_13792727). A total of 27 animals were used in this study. All handling of animals was approved by an ethical committee (Göteborgs djurförsöksetiska nämnd #5.8.18‐06011/2019). The rats were bred in‐house, kept with the dam and 1–12 littermates in open, wire‐top‐type cages, kept on a 12:12 light: dark cycle, and had ad libitum access to food and water. On the day of the experiment, the rats were anesthetized by inhalation of isoflurane (suspended in air) until all reactivity to a painful stimulation had disappeared and then rapidly decapitated. The brain was removed from the scull and submerged in a cold (2–4°C) solution containing (in mM); 219 Glycerol, 2.5 KCl, 1.2 NaH2PO4, 1.2 CaCl2, 7 MgCl2, 26 NaHCO3 and 11 D‐glucose. The brain was cut to parasagittal slices, 350 μm wide, and the hippocampus was dissected out. The slices recovered for 15–30 min in 32°C in a solution containing (in mM): 92 NMDG, 2.5 KCl, 1.25 NaH2PO4, 30 NaHCO3, 20 HEPES, 2 thiourea, 5 ascorbate, 3 pyruvate, 0.5 CaCl2, 8 MgCl2, 25 glucose before resting in room temperature for 1–6 h in a solution containing (in mM): 129 NaCl, 20 NaHCO3, 1.29 NaH2PO4, 3 KCl, 4 MgCl2, 2 CaCl2, 0.5 ascorbic acid, 3 myo‐inositol, 4 L,D‐lactic acid, and 10 D‐glucose.
2.2. Electrophysiology
Whole‐cell patch‐clamp recordings were performed as described previously (Riebe & Hanse, 2012). The slices were transferred from the resting solution to the experimental setup and submerged in an aCSF at room temperature containing (in mM): 147.6 Na+, 2.79 K+, 129.27 Cl−, 1.14 Mg2+, 23 HCO3 −, 0.4 H2PO4 −, 3.66 D‐glucose and 1.2 or 2.0 Ca2+ as indicated. Pyramidal neurons in the CA1 area of the hippocampus were visualized with a CCD camera (Sony XC‐73 CE) and a differential interference contrast microscopy (Nikon E600FN). A viable cell was chosen based on visual appearance and patched with a borosilicate glass micropipette (resistance 3–6 MOhm) filled with a solution containing (in mM): 127 K‐gluconate, 8 KCl, 10 HEPES, 15 phosphocreatine, 4 Mg‐ATP, 0.3 Na‐GTP (pH ca. 7.3 and osmolality 280–300 mOsm). For the experiments investigating G‐protein dependence 0.3 mM GTP was exchanged for 0.3 mM GDPβS (see below). A liquid junction potential of −9 mV was measured and not corrected for. Following seal formation, the cells were allowed to recover for 5 min after opening after which data was collected with a sampling frequency of 10 kHz and filtered at 3 kHz by an EPC‐9 amplifier (HEKA Elektronik). Series resistance was monitored using a 20 ms 10 mV hyperpolarizing pulse and was not allowed to vary more than 20% else the cell, and data were discarded. All washin and washout of calcium and drugs were 10 min long and the evaluations were always performed in a paired manner.
All recordings were carried out in current‐clamp mode. For spontaneous activity, no current was injected and the frequency of action potential firing was determined over a period of 10–60 s depending on regularity and frequency. To measure the input–output relationship, an 800 ms long stepwise increasing current, starting at −30 pA and increasing by 30 pA in 15 steps up to +390 pA (see Figure 1a, inset) was injected and the resulting firing frequency was noted. To measure the input resistance, a slightly different stepwise increasing current protocol was used, 300 ms long currents starting at −100 pA and increasing in 15 steps of 10 pA. To measure the medium afterhyperpolarization (mAHP), single spikes were elicited by a brief (1 ms) current injection. mAHP was defined as the difference in membrane potential before and 100 ms after the current injection (see Figure 3a,b). Resting membrane potential was estimated in the zero current injection recordings of spontaneous activity by fitting a horizontal line to the baseline membrane potential between the action potentials (see Figure 4a). The action potential threshold was analyzed by plotting the first derivative of voltage over time (dV/dt) against membrane potential (phase‐plane plot) and quantified as a depolarization rate of 20 mV/ms. The first recorded action potential evoked at equal current injection amplitude in 1.2 or 2 mM calcium was used for threshold analysis.
FIGURE 1.
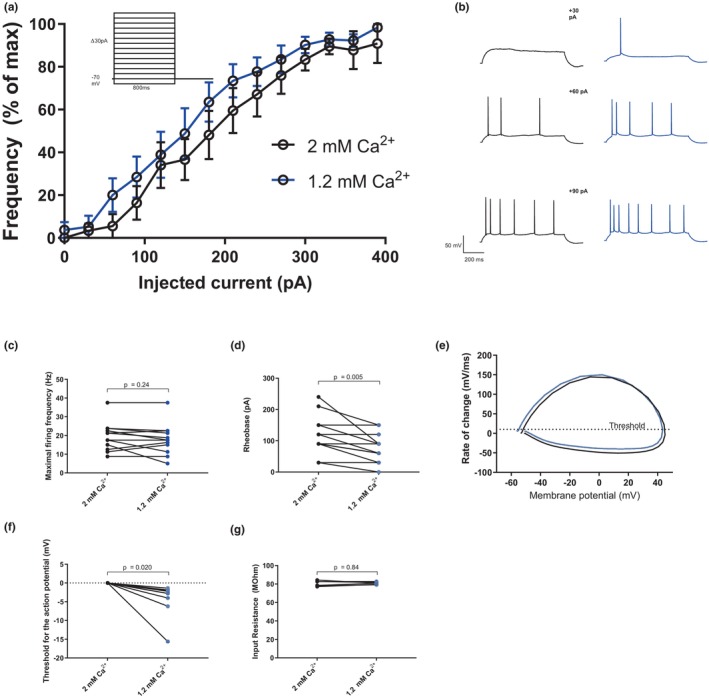
The excitability of CA1 pyramidal neurons is increased in 1.2 mM compared to 2 mM of extracellular calcium. (a) An input–output curve produced by a stepwise increasing current injection (inset) showing firing of action potentials with increasing frequency in 1.2 mM (blue) and 2 mM (black) extracellular calcium (n = 9 slices). The data from each cell are normalized to maximal firing frequency of that cell. (b) Example traces from (a), showing representative firing in 1.2 and 2 mM extracellular calcium in response to 30, 60, and 90 pA current injections. (c) and (d) show the maximal firing frequency and rheobase, respectively (two‐tailed paired t‐test, for (c); p = 0.24, t = 1.23, df = 12, n = 13 slices and for (d); p = 0.005, t = 3.42, df = 12, n = 13 slices). (e) An example phase‐plane plot. Dotted line indicates threshold for action potential, 10 mV/ms. (f) The threshold for the action potential, normalized to the threshold in 2 mM calcium (two‐tailed paired t‐test, p = 0.020, t = 2.91, df = 8, n = 9 slices). (g) The input resistance in response to a 300 ms hyperpolarizing current of −100 pA (two‐tailed paired t‐test, p = 0.84, t = 0.22, df = 5, n = 6)
FIGURE 3.
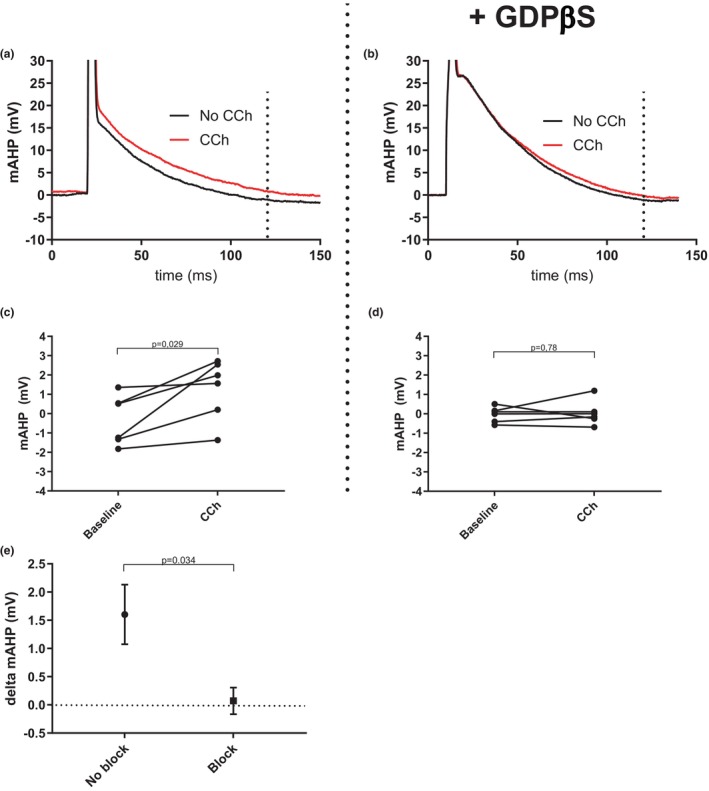
The G‐protein blocker GDPβS blocks the effect of carbachol (CCh) on the mAHP. (a) Example trace showing the mAHP at baseline (black) and after washin of CCh (40 μM, red) in a CA1 pyramidal neuron. Dotted line indicates 100 ms after action potential initiation, where the mAHP was measured. (b) Same as (a) but in the presence of the intracellular G‐protein blocker GDPβS. (c) Graph summarizing the effect of 40 μM CCh on the mAHP (two‐tailed paired t‐test, p = 0.029, t = 3.03, df = 5, n = 6 slices). (d) Same as (c) but in the presence of the intracellular G‐protein blocker GDPβS (two‐tailed paired t‐test, p = 0.78, t = 0.30, df = 5, n = 6 slices). (e) Graph summarizing the change in mAHP (in mV) after the addition of CCh in the presence and absence of G‐protein block (two‐tailed unpaired t‐tests with Welch correction, p = 0.034, t = 2.65, df = 6.91, n = 6 slices in both groups).
FIGURE 4.
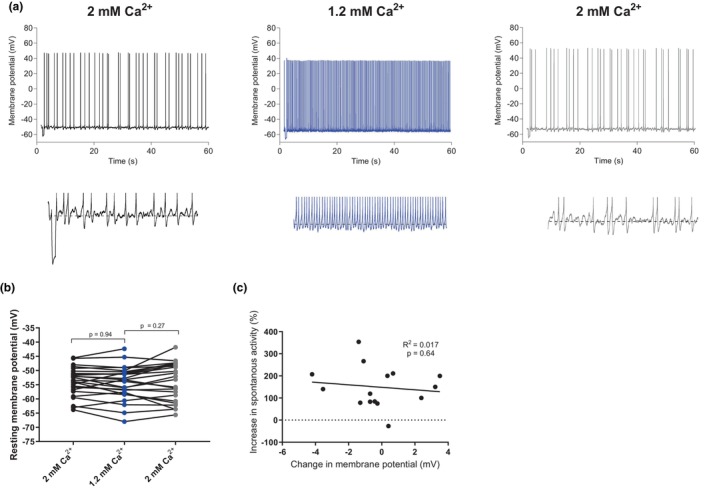
The resting membrane potential is not affected by changes in extracellular calcium. (a) Example traces of spontaneous firing in 2, 1.2, and 2 mM extracellular calcium. Under each graph are the same traces zoomed in to show the measured resting membrane potential, indicated by a dashed line. (b) Graph summarizing the resting membrane potential in 1.2 mM calcium compared to 2 mM calcium (Repeated measures one‐way ANOVA with Tukey's multiple comparisons test, F = 1.35, df (total) = 86, p = 0.27, n = 29 slices). (c) Graph showing the correlation between the relative change in spontaneous firing frequency and the change in resting membrane potential when changing extracellular calcium concentration from 2 to 1.2 mM (linear regression, R 2 = 0.017, F‐value = 0.22, p = 0.64, n = 16 slices).
2.3. Chemicals
All chemicals were purchased from Merck. To block G‐protein dependent mechanisms (Figures 2, 3) we used GDPβS (Guanosine 5′‐[β‐thio]diphosphate), a non‐hydrolysable competitive blocker G‐proteins. Previous studies have used this blocker in concentrations from 0.1–3 mM (Anazco et al., 2013; Simmons & Mather, 1991). A concentration of 0.3 mM GDPβS was determined as effective in control experiments where addition of 40 μM carbachol (D'Angelo et al., 2013) was applied, which is known to reduce the mAHP amplitude via a G‐protein dependent mechanism (Storm, 1989) (see Figure 3). To buffer intracellular calcium (Figure 5) we added 10 mM 1,2‐bis(o‐aminophenoxy)ethane‐N,N,N′,N′‐tetraacetic acid (BAPTA) to the pipette solution.
FIGURE 2.
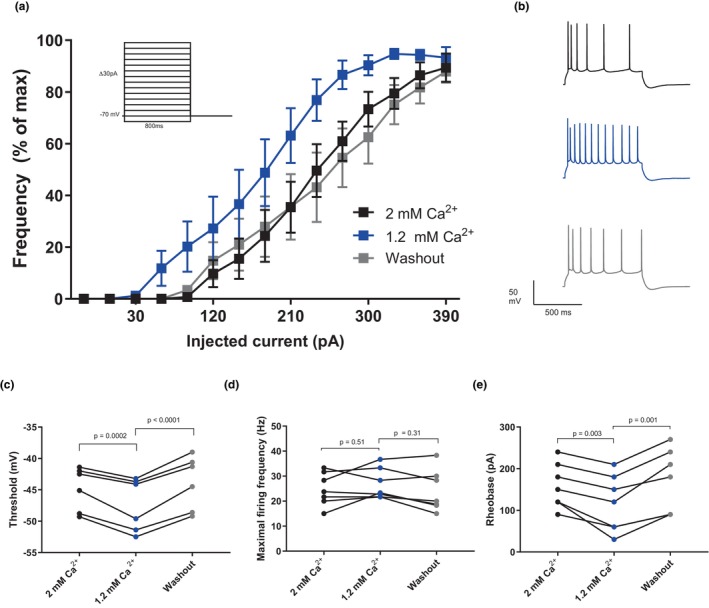
Intracellular G‐protein block has no effect on extracellular calcium‐dependent excitability changes in CA1 pyramidal neurons. (a) An input–output curve showing a left shift in physiological calcium (1.2 mM, blue) compared to high calcium (2 mM, black) and washout (2 mM calcium, gray) (n = 7 slices). Inset shows the stimulation (stepwise current injections increasing by 30 pA from −30 to 390 pA). The data from each cell are normalized to its maximal firing frequency. (b) Example traces from A, injected current is 300 pA. (c) Graph summarizing the effect on the action potential threshold as a result of decreasing the extracellular calcium concentration from 2 to 1.2 mM (Repeated measures one‐way ANOVA with Tukey's multiple comparisons test, F = 42.0, df (total) = 17, p < 0.0001, n = 7 slices). (d) Graph summarizing the effect on the maximal firing frequency as a result of decreasing the extracellular calcium concentration from 2 to 1.2 mM (Repeated measures one‐way ANOVA with Tukey's multiple comparisons test, F = 1.28, df (total) = 20, p = 0.32, n = 7 slices). (e) Graph summarizing the effect on the rheobase as a result of decreasing the extracellular calcium concentration from 2 to 1.2 mM (Repeated measures one‐way ANOVA with Tukey's multiple comparisons test, F = 14.6, df (total) = 20, p = 0.0006, n = 7 slices).
FIGURE 5.
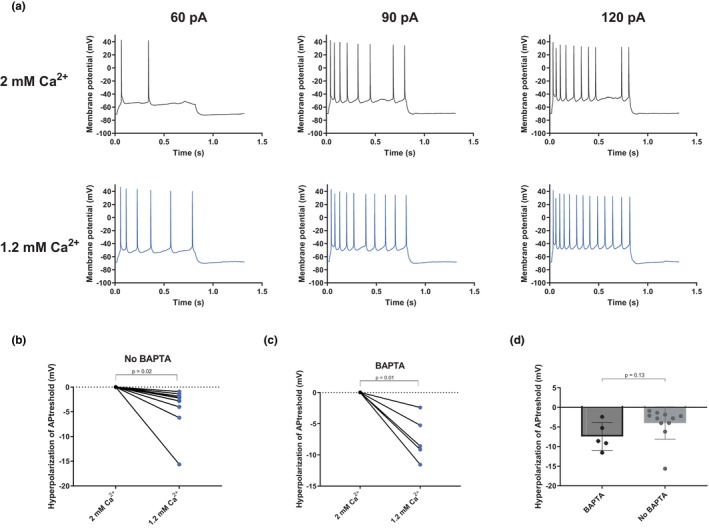
Intracellular application of the calcium chelator BAPTA (10 mM) does not block the effect of extracellular calcium on the action potential threshold. (a) Example traces of action potential firing evoked by an injected current in 2 and 1.2 mM of extracellular calcium with 10 mM BAPTA in the pipette solution. (b) Graph summarizing the average change in action potential threshold observed when changing the extracellular calcium concentration from 2 to 1.2 mM (two‐tailed paired t‐test, p = 0.018, t = 2.9, df = 9, n = 10 slices). (c) Same as (b) but in the presence of intracellular 10 mM BAPTA (two‐tailed paired t‐test, p = 0.020, t = 4.6, df = 4, n = 5 slices). (d) Graph summarizing the hyperpolarization of the threshold in 1.2 mM compared to in 2 mM calcium with and without BAPTA (two‐tailed unpaired t‐tests with Welch correction, p = 0.13, t = 1.68, df = 9.0, n = 10 slices in the non‐BAPTA group and n = 5 slices in the BAPTA group).
2.4. Data analysis
The collected whole‐cell data were analyzed in the software IGOR Pro (WaveMetrics, Inc. Portland, USA; RRID:SCR_000325). All statistical testing and graphical design were performed in GraphPad Prism 7.03 (GraphPad Software Inc, La Jolla, USA, RRID:SCR_002798). A p‐value <0.05 was considered significant. The sample sizes were based on the effect sizes seen in our previous study (Forsberg et al., 2019) but no formal power calculation was performed. No test for outliers was conducted and no assessment of normality because of the limited number of data points. The paired and highly controlled experimental design did nonetheless allow us to show clear effects, open to the readers’ own assessment by the display of every individual data point. For paired data sets, a two‐tailed paired test was used. For assessment of non‐paired data sets, two‐tailed unpaired t‐tests with Welch correction for unequal variances were used. For the assessment of data sets with more than two repeated measurements (Figures 2 and 4), a repeated measures one‐way ANOVA with Tukey's multiple comparisons test was used, and the p‐values for the ANOVA are stated in the text and figure text, whereas the p‐values for the Turkey's multiple comparison tests are shown in the graphs.
Sample sizes, p‐values, and statistical tests used are stated for each experiment in the figure or figure legends. Data are expressed as means ± SEM.
3. RESULTS
We have, in a previous study, thoroughly described the changes in excitability and synaptic transmission caused by a decrease in extracellular calcium from 2 mM to the physiological level of 1.2 mM (Forsberg et al., 2019). The aim of this study was to evaluate the relative contributions of various mechanisms described in the literature in mediating calcium‐dependent effects on the intrinsic excitability of CA1 pyramidal neurons. To first recapitulate the most striking aspects of the calcium effect we performed whole‐cell current clamp recordings from CA1 pyramidal neurons in acute hippocampal slices during perfusion with either 2 or 1.2 mM extracellular calcium. Half of the recordings started in 2 mM and half in 1.2 mM calcium to control for effects over time. Consistent with our previous findings, lower extracellular calcium increased action potential firing in these neurons when stimulated with a stepwise increasing current injection (Figure 1a,b). The increased firing frequency was associated with a lowered rheobase, i.e. the lowest current injection required to elicit at least one action potential which shifted from, on average 115 ± 17 pA in 2 mM extracellular calcium to 76 ± 13 pA in 1.2 mM (Figure 1d, p = 0.005). The maximal firing frequency was not affected by the change in extracellular calcium (19.5 ± 2.0 Hz vs. 18.2 ± 2.2 Hz in 2 mM vs. 1.2 mM, p = 0.24, Figure 1c). As a decreased rheobase may result from a hyperpolarizing shift in the action potential threshold we studied spontaneous action potentials (in current clamp mode with no applied current) and defined the threshold as a depolarization rate of 20 mV/ms. We created phase‐plane plots in which a single action potential is plotted with the membrane potential on the x‐axis and the change in membrane potential on the y‐axis (see Figure 1e). The threshold for the action potential was changed on average 4.0 ± 1.4 mV towards hyperpolarization in 1.2 mM compared with 2 mM extracellular calcium (−57.6 ± 2.0 mV vs. −53.8 ± 2.2 mV, p = 0.020, Figure 1f).
A decreased rheobase could also be a result of an increased input resistance, possibly because of effects on channels other than those specifically investigated in this article. To test for this possibility, we injected a hyperpolarizing current of 100 pA (300 ms) and calculated the input resistance by Ohms law. There was no significant difference in input resistance in cells in 2 and 1.2 mM of extracellular calcium (80.6 ± 1.2 MOhm in 2 mM vs. 80.8 ± 0.5 MOhm in 1.2 mM calcium, p = 0.84. Figure 1g). There was however a non‐significant trend towards a rundown in input resistance over time (data not shown), stressing the importance of controlling for wash‐out effects.
We next moved on to test whether the CaSR could mediate all or part of the effect via a G‐protein‐dependent pathway and repeated the same experiment with the G‐protein blocker GDPβS (0.3 mM) added to the patch pipette solution. Interestingly, at 1.2 mM extracellular calcium, the threshold of the action potential was increased from −57.6 ± 2.0 mV (n = 9) to −47.4 ± 1.7 mV (n = 7) when GDPβS was present in the pipette solution (p = 0.039). This result suggests that tonically active G‐protein‐mediated processes contribute to increase excitability in CA1 pyramidal neurons. To test the effect of extracellular calcium under conditions of blocked G‐protein dependent signaling, we always started these experiments in 2 mM extracellular calcium and effects over time were instead controlled for by a washout of 1.2 mM calcium back to 2 mM. The input–output curve showed a left shift similar to the one seen without the blocker (Figure 2a). This left shift was, as shown in Figure 1, mainly because of a decrease of the rheobase which decreased from 159 ± 20 pA in 2 mM calcium to 116 ± 26 pA in 1.2 mM calcium (Figure 2e, p = 0.0006). The threshold for action potential firing was hyperpolarized in 1.2 mM calcium (−47.4 ± 1.7 mV) compared to 2 mM calcium (−44.9 ± 1.4 mV; Figure 2c, p < 0.0001). The maximal firing frequency was not affected (26.8 ± 2.3 Hz in 1.2 mM vs. 24.8 ± 2.5 Hz in 2 mM calcium, p = 0.32, Figure 2d). All these observations correspond well to the effects of changing extracellular calcium in absence of intracellular G‐protein blocker.
The decreased excitability observed when GDPβS was present in the pipette solution indicates that GDPβS was actively inhibiting G‐protein‐mediated processes. Nevertheless, to confirm that G‐protein signaling was blocked in these experiments we used the general acetylcholine receptor agonist carbachol (CCh) as positive control. CCh is well known to decrease the mAHP amplitude in cortical pyramidal cells via M1 muscarinic acetylcholine receptor (and G‐protein)‐dependent inhibition of M‐current (Storm, 1989). In Figure 3, we compare the change in mAHP amplitude caused by addition of 40 μM CCh with and without GDPβS in the pipette solution. In the presence of CCh, but no GDPβS, the mAHP amplitude decreased by on average 1.60 ± 0.53 mV (from −0.3 ± 0.5 mV to 1.3 ± 0.6 mV, p = 0.029 Figure 3c) in agreement with results from earlier studies. In the presence of GDPβS, there was no significant decrease of the mAHP (−0.04 ± 0.2 mV at baseline compared with 0.03 ± 0.3 mV after the addition of CCh, p = 0.78, Figure 3d). When compared there was a significant difference in CCh effect between the blocked and unblocked group (p = 0.025, Figure 3e). We therefore conclude that inhibition of G‐protein signaling does not alter the neuromodulatory effect of extracellular calcium in these cells, which strongly argues against previously proposed mechanisms involving the CaSR.
Next, we examined whether modulation of NALCN, a non‐selective cation leak channel, was involved in the excitability changes induced by altering extracellular calcium. This channel has been reported to react to changes in extracellular calcium via both CaSR‐dependent (Chen et al., 2010; Lu et al., 2010) and independent (Martiszus et al., 2021) mechanisms, but its impact on the resting membrane potential (RMP) as a result of extracellular calcium changes in the 1–2 mM range is unknown. We recorded from cells in current clamp without any injected current from the pipette in order to examine the RMP. Our data showed no significant change in resting membrane potential in 1.2 mM calcium compared to 2 mM (−54.1 ± 1.1 mV vs. 53.7 ± 0.9 mV, p = 0.27 Figure 4b). It is possible that a small depolarization of the membrane potential in lower calcium could have affected excitability in some cells even if we could not find any significance in the difference of the groups, possibly because of variance in the membrane potential. When studying the individual data points in Figure 4b, some cells seem to depolarize in lower calcium, maybe these cells owe their increase in excitability to the depolarization of membrane potential (possibly because of an influence on the NALCN). To test this idea, we examined the correlation between the change in membrane potential between the recordings and the effect on excitability (defined as increase in spontaneous firing frequency in 1.2 mM compared to 2 mM extracellular calcium). There was no significant correlation between the two parameters (Figure 4c, R 2 = 0.017, p = 0.64). Not all cells were spontaneously active, which is why the number of included cells is smaller in 4C than in 4B. We thus conclude that modulation of the NALCN is not a feasible explanation for the modulatory effect of extracellular calcium on these cells.
Next, we used the calcium chelator BAPTA to investigate if the effect of extracellular calcium on excitability was dependent on intracellular calcium dynamics. Since BAPTA is a fast chelator of calcium, it will prevent increases in intracellular concentration from occurring upon calcium entrance via activated channels. We observed a trend (although not statistically significant) that adding BAPTA (10 mM) in the patch pipette solution increased the threshold of the action potential. In 1.2 mM extracellular calcium the threshold was −57.6 ± 2.0 mV (n = 10) in the control pipette solution and −55.6 ± 3.2 mV (n = 5; p = 0.61) in the BAPTA‐containing pipette solution and the corresponding values in 2 mM extracellular calcium were −53.8 ± 2.2 mV (n = 10) and −48.2 ± 3.3 mV (n = 5; p = 0.20). When increasing extracellular calcium concentration from 1.2 to 2 mM and having BAPTA in the pipette solution the threshold for the action potential was hyperpolarized by 7.4 ± 1.6 mV (n = 5, p = 0.021) (Figure 5c), an effect of similar magnitude as that seen in the absence of BAPTA (hyperpolarization of 4.0 ± 1.4 mV, p = 0.02, Figure 5b). The difference between the groups was not statistically significant (p = 0.13, Figure 5d) We therefore conclude that the mechanism behind the lowered action potential threshold following a switch from 2 to 1.2 mM extracellular calcium is unlikely to be mediated by changes in the intracellular calcium concentration.
The results so far do not support G‐protein coupled signaling, changes in intracellular calcium concentration, or a change of the resting membrane potential as an explanation for the decreased excitability when switching from 1.2 to 2 mM extracellular calcium. Other proposed mechanisms for extracellular calcium's excitability regulating effects include a pore block of voltage‐gated sodium channels and a hyperpolarizing shift of the activation of voltage‐gated sodium channels because of less neutralization of negative charges at the extracellular mouth of the channels (Armstrong & Cota, 1991, 1999; Hille, 1968; McLaughlin et al., 1971). To elucidate the plausibility of these two different mechanisms, we examined the phase‐plane plots of the action potentials in 1.2 and 2 mM calcium more closely. The derivative of action potential, dV/dt, can be used as a proxy for the ionic current during the action potential since the ionic current is opposite to the capacitive current (C × dV/dt) (Bean, 2007). In our matched situation when we record from the same neuron in 1.2 and 2 mM calcium, we can assume that cell capacitance is constant and therefore use dV/dt to compare the ionic currents in the two conditions. Subtracting dV/dt in 2 mM calcium from that in 1.2 mM calcium will thus reveal how the ionic current during the action potential differs at different membrane voltages. This procedure is illustrated in Figure 6 in which Figure 6 A shows examples of action potentials in 1.2 mM calcium (blue) and 2 mM calcium (black) and Figure 6b shows the corresponding phase‐plane plots. The subtracted phase‐plane plots, illustrated in Figure 6c, show that there is a shift with smaller currents in 2 mM calcium at membrane voltages more hyperpolarized than about −35 mV and larger currents at membrane voltages more depolarized than −35 mV. The average subtraction from 6 experiments, shown in Figure 6d, illustrates that this shift was a clear and general feature among the experiments. Such a shift with larger currents during the initial rising phase of the action potential and smaller currents during the remaining of the action potential does not seem compatible with a generally lower conductance of the voltage‐gated sodium channels, or a reduced voltage‐sensitive block of the channels. On the other hand, it seems compatible with a depolarizing shift of the activation and inactivation of these channels, which is expected from less screening of negative charges at the extracellular mouth of the channels.
FIGURE 6.
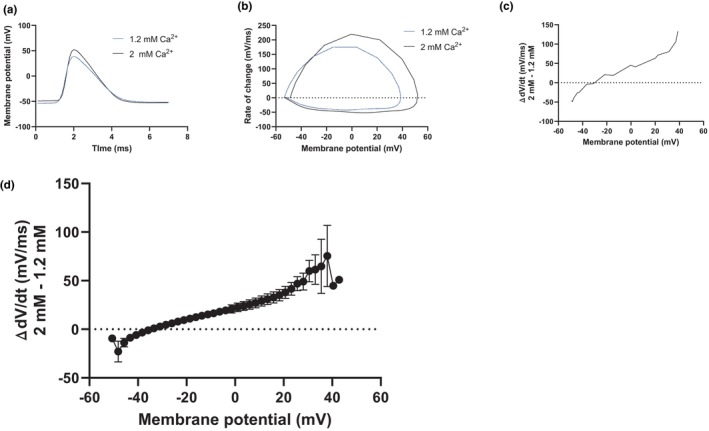
Changes in the extracellular calcium concentration shift the current during the action potential. (a) Example traces of spontaneous action potentials in 2 mM (black) and 1.2 mM (blue) extracellular calcium. (b) Phase‐plane plots of the action potentials shown in (a). (c) Subtraction of the phase‐plane plots shown in (b) showing the difference in dV/dt between 1.2 and 2 mM calcium (rate in 2 mM subtracted from rate in 1.2 mM). (d) Average difference in dV/dt between 1.2 and 2 mM calcium (n = 6 slices, rate in 2 mM subtracted from rate in 1.2 mM).
4. DISCUSSION
In this study, we evaluated the contribution of G‐protein signaling and changes in intracellular calcium in extracellular calcium‐dependent regulation of intrinsic excitability in CA1 pyramidal neurons. We focused on a deviation from physiological levels of extracellular calcium to 2 mM calcium simply because this calcium concentration (or higher) is often used in in vitro experiments. We show that the effect of extracellular calcium in the 1–2 mM range does not require functioning G‐protein signaling, strongly arguing against mechanisms involving the CaSR. The effect is also not associated with a change in the resting membrane potential, as would be expected from involvement of NALCN or other leak cation channels. The effect of extracellular calcium in the concentration range here studied also persists when the concentration of intracellular calcium was held steady by a high concentration of the calcium chelator BAPTA, which strongly argues against involvement of intracellular calcium signaling, e.g. via calcium‐dependent potassium channels. Analysis of the current flow during the action potential shows a shift towards larger currents at more depolarized, and smaller currents at more hyperpolarized, membrane potentials at 2 mM calcium compared to 1.2 mM calcium. Together, these findings lead us to conclude that, in the range of 1–2 mM, neutralization of surface charges on voltage‐gated sodium channels is likely to account for most of the inhibitory effect of extracellular calcium ions on intrinsic excitability.
A problem when studying changes of extracellular divalent cations with the patch‐clamp method is the unspecific impact on the patch leak current (Boone et al., 2014). Boone and colleagues showed that calcium, among other divalent cations, can act as sealants between the membrane patch and the patch pipette, thereby reducing the leak current. Therefore, an increased leak current caused by reducing calcium in the extracellular medium could possibly be mistaken for an activation of a non‐selective cation conductance, as seen in many publications (Hablitz et al., 1986; Smith et al., 2004; Xiong et al., 1997). Attempts to counter this effect by elevating magnesium to compensate for the reduced calcium concentration are unlikely to alleviate the problem since magnesium and calcium ions have similar capacity to counter both the leak current and the increased neuronal excitability. Is it thus possible that the effect of calcium on excitability (in this concentration range) is altogether an artifact associated with the patch‐clamp method? There are several arguments against this. A clinical argument is that it is well known that hypocalcemia in patients leads to hyperexcitability and in severe cases induces seizures (Juan, 1979). Moreover, the effect of changes in extracellular calcium concentration on excitability is also observed in intact axons, for example using extracellular recordings of the compound action potential (Owen et al., 2021; Turner et al., 1982). Another example is our previous observation (Forsberg et al., 2019) that the frequency of spontaneous inhibitory post‐synaptic currents (IPSCs) and excitatory post‐synaptic currents (EPSCs) is rather constant when changing the extracellular calcium concentration from 1.2 to 2 mM. This increase in extracellular calcium concentration augments the synaptic release probability (as indicated by a reduction of the paired‐pulse ratio), which would increase the frequency of spontaneous IPSCs/EPSCs. The most likely explanation for the constant frequency of spontaneous IPSCs and EPSCs is that the increased release probability is counteracted by fewer presynaptic axons reaching the threshold for action potential caused by a calcium‐dependent decrease of excitability in intact axons. Finally, our present observation of an increase in spontaneous firing (Figure 4a,b) when lowering the extracellular calcium concentration from 2 to 1.2 mM was not associated with any significant change of the membrane potential, despite no compensatory injection of current. These results argue that the observed increased excitability is not caused by an increased cation conductance. This conclusion is also supported by our finding that we did not detect any change in input resistance when changing the calcium concentration from 1.2 to 2 mM. Nevertheless, based on the Boone et al study (Boone et al., 2014), we pose the question of whether the many patch‐clamp studies that have implicated an increase of non‐selective cation conductance as an explanation for the increased excitability caused by hypocalcemia (Hablitz et al., 1986; Smith et al., 2004; Xiong et al., 1997) might be based on artifactual data as a result of an inadvertently increased leak current.
Lu et al. (2010) argue against the charge‐screening effect of extracellular calcium by making a strong case for a mechanism that involves the G‐protein coupled CaSR inhibiting the UNC79‐UNC80‐NALCN cation complex. They show that neurons from NALCN and UNC79 knock‐out mice are insensitive to changes in extracellular calcium. It is important to note, however, that the effect they observed was in the 0.01–1 mM range of extracellular calcium. We have restricted our study to the 1.2–2 mM range of extracellular calcium, and in this range, we did not observe any effect on the membrane potential or by the G‐protein blocker GDPβS in the patch pipette. Thus, our tentative conclusion is that CaSR‐mediated inhibition of the NALCN is close to saturated above 1 mM levels of extracellular calcium, leaving no room for contribution of this mechanism to excitability changes in the concentration range we have examined. It is worth noting that our conclusion of negligible contribution of G‐protein mediated effects of the CaSR is solely based on the examination of the action potential threshold in the 1.2–2 mM range and not on other potential effects of the CaSR such as modulation of synaptic release (Chen et al., 2010).
Another explanation for the inhibitory calcium effect was proposed by Segal (Segal, 2018). He instead argued for a mechanism involving calcium‐induced calcium release from internal stores and activation of calcium‐activated potassium channels. This study was carried out in cultured hippocampal neurons in an extracellular medium completely devoid of calcium. He showed a decrease in number of spikes and a depolarization of the threshold for action potentials when a puff of calcium‐containing medium was applied with pressure near the cell soma. This is in contrast to our finding that the calcium effect in the 1–2 mM range on the action potential threshold is independent of changes in the concentration of intracellular calcium. The difference could again be because of a variance of the concentration span involved, which is difficult to estimate with the method used by Segal. Indeed, when examined in presynaptic terminals of mouse neocortical neurons, increasing extracellular calcium concentration from 1.1 to 6 mM in the perfusion fluid resulted in little, or no, change of the intracellular calcium concentration (Vyleta & Smith, 2011). Another related confounder may be effects of prolonged exposure to calcium‐free medium and the accompanying hyperexcitability. As indicated above there are reasons to believe that the CaSR‐coupled mechanism is most active in the 0–1 mM concentration span, and could thus constitute a signaling mechanism involved in the cellular response to hypocalcemia.
The clear depolarization of the action potential threshold caused by an increase of extracellular calcium indicates a mechanism directly involving the voltage‐gated sodium channels. To elucidate that possibility, we compared phase‐plane plots of the action potential in 1.2 and 2 mM calcium under the assumptions that the such a change in extracellular calcium does not change the cell capacitance. We found that the change in extracellular calcium induced a shift in the current flow during the action potential such that during the higher calcium concentration reduced the current at membrane potentials more hyperpolarized than about −35 mV and increased the currents at membrane potentials more depolarized than about −35 mV. Such a shift of the currents suggests a shift of the activation and inactivation of voltage‐gated sodium channels towards more depolarized membrane potentials, which argues for a charge‐screening effect of extracellular calcium (Hille, 2001). On the other hand, a shift with smaller and larger currents at different phases of the action potential would not seem compatible with a general decrease in channel conductance, or an increased voltage‐dependent pore block of the channels by extracellular calcium. The precise mechanism for a charge‐screening effect remains uncertain, but likely involves neutralization of negative charges on the extracellular side of the ion channel (Elinder & Århem, 2003). Different voltage‐gated ion channels have different density of such negative surface charges and voltage‐gated sodium channels have generally much higher density than voltage‐gated potassium channels (Elinder & Århem, 2003). Martiszus et al, examining cultured cortical neurons and a lowering of extracellular calcium and magnesium from 1.1 to 0.2 mM, showed a large shift of the activation of voltage‐gated sodium channels, but almost no effect on voltage‐gated potassium channels. Although focusing their study on CaSR and NALCN, these authors conclude, in conformity with our conclusion, that the dominant effect of extracellular calcium on excitability relates to a shift of the activation of voltage‐gated sodium channels (Martiszus et al., 2021).
It is also noteworthy that our experiments with GDPβS and BAPTA in the patch pipette suggest that tonically active G‐protein‐mediated processes, as well as possibly resting levels of intracellular calcium concentrations, contribute to keep up excitability in CA1 pyramidal neurons. The ion channel mechanisms involved in these tonically active reductions of the action potential threshold are unknown, but our results suggest that they did not interact with the effect of extracellular calcium on the action potential threshold. This finding seems consistent with a charge‐screening effect on voltage‐gated sodium channels of extracellular calcium, since it is unlikely that modulation of the threshold by G‐proteins, or intracellular calcium, acts by changing surface charges.
Our results underscore the importance of choosing a relevant concentration of extracellular calcium when conducting in vitro research. In this study, we chose to examine the effect on action potential threshold when changing extracellular calcium from 1.2 to 2 mM and our results favor an effect on negative charges on the extracellular part of voltage‐gated sodium channels. We, however, also propose that other mechanisms may contribute outside this concentration range. In particular, activation of NALCN via the CaSR may contribute to extracellular calcium concentrations below the physiological.
AUTHOR CONTRIBUTIONS
My Forsberg: Conceptualization; investigation; funding acquisition; writing – original draft; visualization; writing – review and editing; project administration; formal analysis; resources. Dinna Zhou: Investigation; writing – review and editing. Shadi Jalali: Investigation; writing – review and editing. Giorgia Faravelli: Investigation; writing – review and editing. Henrik Seth: Conceptualization; resources; writing – review and editing. Andreas Björefeldt: Conceptualization; writing – original draft; writing – review and editing. Eric Hanse: Conceptualization; resources; writing – review and editing; writing – original draft; supervision; funding acquisition.
CONFLICT OF INTEREST STATEMENT
The authors declare that they have no competing interests.
PEER REVIEW
The peer review history for this article is available at https://www.webofscience.com/api/gateway/wos/peer‐review/10.1111/jnc.16209.
ACKNOWLEDGEMENTS
MF, SJ, DZ, and GF conducted the experiments. MF designed and performed the analysis and designed the figures. MF, AB, and EH wrote the paper. All authors participated in designing the experiments, and all authors read and approved the manuscript. The authors would like to thank Wilhelm and Martina Lundgrens Vetenskapsfond (#2018‐2616), Göteborgs Läkaresällskap (#GLS‐878091, the Swedish Research Council (VR 00986)), Alzheimerfonden (AF‐640391 and AF‐993056), and the Swedish State Support for Clinical Research (ALFGBG 427611) for financial support.
Forsberg, M. , Zhou, D. , Jalali, S. , Faravelli, G. , Seth, H. , Björefeldt, A. , & Hanse, E. (2025). Evaluation of mechanisms involved in regulation of intrinsic excitability by extracellular calcium in CA1 pyramidal neurons of rat. Journal of Neurochemistry, 169, e16209. 10.1111/jnc.16209
DATA AVAILABILITY STATEMENT
The data that support the findings of this study are available from the corresponding author upon reasonable request.
REFERENCES
- Anazco, C. , Pena‐Munzenmayer, G. , Araya, C. , Cid, L. P. , Sepulveda, F. V. , & Niemeyer, M. I. (2013). G protein modulation of K2P potassium channel TASK‐2: A role of basic residues in the C terminus domain. Pflügers Archiv, 465, 1715–1726. [DOI] [PubMed] [Google Scholar]
- Armstrong, C. M. (1999). Distinguishing surface effects of calcium ion from pore‐occupancy effects in Na+ channels. Proceedings of the National Academy of Sciences of the United States of America, 96, 4158–4163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Armstrong, C. M. , & Cota, G. (1990). Modification of sodium channel gating by lanthanum. Some effects that cannot be explained by surface charge theory. The Journal of General Physiology, 96, 1129–1140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Armstrong, C. M. , & Cota, G. (1991). Calcium ion as a cofactor in Na channel gating. Proceedings of the National Academy of Sciences of the United States of America, 88, 6528–6531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Armstrong, C. M. , & Cota, G. (1999). Calcium block of Na+ channels and its effect on closing rate. Proceedings of the National Academy of Sciences of the United States of America, 96, 4154–4157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bean, B. P. (2007). The action potential in mammalian central neurons. Nature Reviews. Neuroscience, 8, 451–465. [DOI] [PubMed] [Google Scholar]
- Bennett, E. , Urcan, M. S. , Tinkle, S. S. , Koszowski, A. G. , & Levinson, S. R. (1997). Contribution of sialic acid to the voltage dependence of sodium channel gating. A possible electrostatic mechanism. The Journal of General Physiology, 109, 327–343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boone, A. N. , Senatore, A. , Chemin, J. , Monteil, A. , & Spafford, J. D. (2014). Gd3+ and calcium sensitive, sodium leak currents are features of weak membrane‐glass seals in patch clamp recordings. PLoS One, 9, e98808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boström, F. , Hansson, O. , Gerhardsson, L. , Lundh, T. , Minthon, L. , Stomrud, E. , Zetterberg, H. , & Londos, E. (2009). CSF Mg and Ca as diagnostic markers for dementia with Lewy bodies. Neurobiology of Aging, 30, 1265–1271. [DOI] [PubMed] [Google Scholar]
- Brown, E. M. , Gamba, G. , Riccardi, D. , Lombardi, M. , Butters, R. , Kifor, O. , Sun, A. , Hediger, M. A. , Lytton, J. , & Hebert, S. C. (1993). Cloning and characterization of an extracellular Ca(2+)‐sensing receptor from bovine parathyroid. Nature, 366, 575–580. [DOI] [PubMed] [Google Scholar]
- Chen, W. , Bergsman, J. B. , Wang, X. , Gilkey, G. , Pierpoint, C. R. , Daniel, E. A. , Awumey, E. M. , Dauban, P. , Dodd, R. H. , Ruat, M. , & Smith, S. M. (2010). Presynaptic external calcium signaling involves the calcium‐sensing receptor in neocortical nerve terminals. PLoS One, 5, e8563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chua, H. C. , Wulf, M. , Weidling, C. , Rasmussen, L. P. , & Pless, S. A. (2020). The NALCN channel complex is voltage sensitive and directly modulated by extracellular calcium. Science Advances, 6, eaaz3154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- D'Angelo, E. , Solinas, S. , Garrido, J. , Casellato, C. , Pedrocchi, A. , Mapelli, J. , Gandolfi, D. , & Prestori, F. (2013). Realistic modeling of neurons and networks: Towards brain simulation. Functional Neurology, 28, 153–166. [PMC free article] [PubMed] [Google Scholar]
- Elinder, F. , & Århem, P. (2003). Metal ion effects on ion channel gating. Quarterly Reviews of Biophysics, 36, 373–427. [DOI] [PubMed] [Google Scholar]
- Forsberg, M. , Seth, H. , Bjorefeldt, A. , Lyckenvik, T. , Andersson, M. , Wasling, P. , Zetterberg, H. , & Hanse, E. (2019). Ionized calcium in human cerebrospinal fluid and its influence on intrinsic and synaptic excitability of hippocampal pyramidal neurons in the rat. Journal of Neurochemistry, 149, 452–470. [DOI] [PubMed] [Google Scholar]
- Frankenhaeuser, B. , & Hodgkin, A. L. (1957). The action of calcium on the electrical properties of squid axons. The Journal of Physiology, 137, 218–244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gama, L. , Wilt, S. G. , & Breitwieser, G. E. (2001). Heterodimerization of calcium sensing receptors with metabotropic glutamate receptors in neurons. The Journal of Biological Chemistry, 276, 39053–39059. [DOI] [PubMed] [Google Scholar]
- Hablitz, J. J. , Heinemann, U. , & Lux, H. D. (1986). Step reductions in extracellular Ca2+ activate a transient inward current in chick dorsal root ganglion cells. Ec Ca Blockar NSCC. Biophysical Journal, 50, 753–757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hille, B. (1968). Charges and potentials at the nerve surface. Divalent ions and pH. The Journal of General Physiology, 51, 221–236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hille, B. (2001). Ion channels of excitable membranes. Sinauer. [Google Scholar]
- Inglebert, Y. , Aljadeff, J. , Brunel, N. , & Debanne, D. (2020). Synaptic plasticity rules with physiological calcium levels. Proceedings of the National Academy of Sciences of the United States of America, 117, 33639–33648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Juan, D. (1979). Hypocalcemia: Differential diagnosis and mechanisms. Archives of Internal Medicine, 139, 1166–1171. [DOI] [PubMed] [Google Scholar]
- Lopes, J. P. , & Cunha, R. A. (2019). What is the extracellular calcium concentration within brain synapses?: An editorial for 'Ionized calcium in human cerebrospinal fluid and its influence on intrinsic and synaptic excitability of hippocampal pyramidal neurons in the rat' on page 452. Journal of Neurochemistry, 149, 435–437. [DOI] [PubMed] [Google Scholar]
- Lu, B. , Zhang, Q. , Wang, H. , Wang, Y. , Nakayama, M. , & Ren, D. (2010). Extracellular calcium controls background current and neuronal excitability via an UNC79‐UNC80‐NALCN cation channel complex. Neuron, 68, 488–499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martiszus, B. J. , Tsintsadze, T. , Chang, W. , & Smith, S. M. (2021). Enhanced excitability of cortical neurons in low‐divalent solutions is primarily mediated by altered voltage‐dependence of voltage‐gated sodium channels. eLife, 10, e67914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McLaughlin, S. G. , Szabo, G. , & Eisenman, G. (1971). Divalent ions and the surface potential of charged phospholipid membranes. The Journal of General Physiology, 58, 667–687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Owen, B. , Woode, F. , & Grover, L. M. (2021). Effects of divalent cations on Schaffer collateral axon function. Experimental Brain Research, 239, 3045–3057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Recio‐Pinto, E. , Thornhill, W. B. , Duch, D. S. , Levinson, S. R. , & Urban, B. W. (1990). Neuraminidase treatment modifies the function of electroplax sodium channels in planar lipid bilayers. Neuron, 5, 675–684. [DOI] [PubMed] [Google Scholar]
- Riebe, I. , & Hanse, E. (2012). Development of synaptic connectivity onto interneurons in stratum radiatum in the CA1 region of the rat hippocampus. BMC Neuroscience, 13, 14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ringer, S. (1883). A further contribution regarding the influence of the different constituents of the blood on the contraction of the heart. The Journal of Physiology, 4(29–42), 23–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roshchin, M. V. , Ierusalimsky, V. N. , Balaban, P. M. , & Nikitin, E. S. (2020). Ca(2+)‐activated KCa3.1 potassium channels contribute to the slow afterhyperpolarization in L5 neocortical pyramidal neurons. Scientific Reports, 10, 14484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Segal, M. (2018). Calcium stores regulate excitability in cultured rat hippocampal neurons. Journal of Neurophysiology, 120, 2694–2705. [DOI] [PubMed] [Google Scholar]
- Simmons, M. A. , & Mather, R. J. (1991). Selectivity of the effects of guanosine‐5'‐O‐(2‐thiodiphosphate) on agonist inhibition of the M‐current in amphibian sympathetic neurons. The Journal of Neuroscience, 11, 2130–2134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith, S. M. , Bergsman, J. B. , Harata, N. C. , Scheller, R. H. , & Tsien, R. W. (2004). Recordings from single neocortical nerve terminals reveal a nonselective cation channel activated by decreases in extracellular calcium. Neuron, 41, 243–256. [DOI] [PubMed] [Google Scholar]
- Sood, A. K. , Handa, R. , Malhotra, R. C. , & Gupta, B. S. (1993). Serum, CSF, RBC & urinary levels of magnesium & calcium in idiopathic generalised tonic clonic seizures. The Indian Journal of Medical Research, 98, 152–154. [PubMed] [Google Scholar]
- Storm, J. F. (1989). An after‐hyperpolarization of medium duration in rat hippocampal pyramidal cells. The Journal of Physiology, 409, 171–190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turner, R. W. , Baimbridge, K. G. , & Miller, J. J. (1982). Calcium‐induced long‐term potentiation in the hippocampus. Neuroscience, 7, 1411–1416. [DOI] [PubMed] [Google Scholar]
- Vassilev, P. M. , Ho‐Pao, C. L. , Kanazirska, M. P. , Ye, C. , Hong, K. , Seidman, C. E. , Seidman, J. G. , & Brown, E. M. (1997). Cao‐sensing receptor (CaR)‐mediated activation of K+ channels is blunted in CaR gene‐deficient mouse neurons. Neuroreport, 8, 1411–1416. [DOI] [PubMed] [Google Scholar]
- Vyleta, N. P. , & Smith, S. M. (2011). Spontaneous glutamate release is independent of calcium influx and tonically activated by the calcium‐sensing receptor. The Journal of Neuroscience, 31, 4593–4606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vysotskaya, Z. V. , Moss, C. R., 2nd , Gilbert, C. A. , Gabriel, S. A. , & Gu, Q. (2014). Modulation of BK channel activities by calcium‐sensing receptor in rat bronchopulmonary sensory neurons. Respiratory Physiology & Neurobiology, 203, 35–44. [DOI] [PubMed] [Google Scholar]
- Wang, H. , & Lu, Y. (2023). High calcium concentrations reduce cellular excitability of mouse MNTB neurons. Brain Research, 1820, 148568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiong, Z. , Lu, W. , & MacDonald, J. F. (1997). Extracellular calcium sensed by a novel cation channel in hippocampal neurons. Proceedings of the National Academy of Sciences of the United States of America, 94, 7012–7017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ye, C. , Kanazirska, M. , Quinn, S. , Brown, E. M. , & Vassilev, P. M. (1996). Modulation by polycationic Ca(2+)‐sensing receptor agonists of nonselective cation channels in rat hippocampal neurons. Biochemical and Biophysical Research Communications, 224, 271–280. [DOI] [PubMed] [Google Scholar]
- Zucker, R. S. (1996). Exocytosis: A molecular and physiological perspective. Neuron, 17, 1049–1055. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
The data that support the findings of this study are available from the corresponding author upon reasonable request.