

Abstract
The development of potent, specific, and pharmacologically viable chemical probes and therapeutics is a central focus of chemical biology and therapeutic development. However, a significant portion of predicted disease-causal proteins have proven resistant to targeting by traditional small molecule and biologic modalities. Many of these so-called “undruggable” targets feature extended, dynamic protein–protein and protein–nucleic acid interfaces that are central to their roles in normal and diseased signaling pathways. Here, we discuss the development of synthetically stabilized peptide and protein mimetics as an everexpanding and powerful region of chemical space to tackle undruggable targets. These molecules aim to combine the synthetic tunability and pharmacologic properties typically associated with small molecules with the binding footprints, affinities and specificities of biologics. In this review, we discuss the historical and emerging platforms and approaches to design, screen, select and optimize synthetic “designer” peptidomimetics and synthetic biologics. We examine the inspiration and design of different classes of designer peptidomimetics: (i) macrocyclic peptides, (ii) side chain stabilized peptides, (iii) non-natural peptidomimetics, and (iv) synthetic proteomimetics, and notable examples of their application to challenging biomolecules. Finally, we summarize key learnings and remaining challenges for these molecules to become useful chemical probes and therapeutics for historically undruggable targets.
Graphical Abstract
1. INTRODUCTION: DESIGNING AND DEPLOYING DESIGNER PEPTIDES, PEPTIDOMIMETICS, AND PROTEOMIMETICS FOR “UNDRUGGABLE” TARGETS
Over the past few decades, rapid development in the fields of molecular biology and chemistry have ushered in an era of tremendous progress for drug discovery. Modern techniques for protein and nucleic acid purification along with data-drivenomics approaches have identified an ever-expanding list of disease-causal targets—typically proteins—that may serve as starting points for the development of chemical probes and therapeutics. Specific protein classes have proven to be intrinsically druggable by current modalities, including specific receptor families like ion channels and GPCRs, as well as intracellular enzymes and nuclear hormone receptor transcription factors (NHR-TFs).1,2 These protein targets are typically characterized by the presence of defined ligand binding sites and other hydrophobic pockets amenable to small molecule discovery and medicinal chemistry optimization.3 Other disease-associated protein targets are present on the surface of cells or are secreted into the extracellular space, thus enabling targeting by large, hydrophilic biologic modalities, including monoclonal antibodies (mAbs), recombinant proteins, and engineered peptides (Figure 1A). A significant number of disease-associated targets fall between these two druggable landscapes, with neither obvious ligand binding sites nor extracellular exposure lending themselves toward cell permeable small molecules or traditional biologics, respectively. This landscape of targets therefore has been referred to by many as being “undruggable” (Figure 1A).4,5 Additional characteristic features shared by many undruggable targets generally include the lack of enzymatic activity and reliance upon large, dynamic surfaces and disordered structures that mediate protein–protein interactions (PPIs) and/or protein–nucleic acid interactions integral to their function (Figure 1A). While all protein families contain disease-associated targets that could be considered undruggable, several families have emerged as particularly challenging, including small GTPases, phosphatases, transcription factors (TFs), epigenetic enzymes, and chromatin regulatory factors.3
Figure 1.

Designer peptides and peptidomimetics for undruggable targets. A) The vast majority of putative disease drivers lack structural features conventionally targeted by small molecules or antibodies. B) Molecular dynamics simulations (50 ns) of unmodified and Diels–Alder cyclized loop designer peptide, demonstrating significant stabilization of structural topology. C) Classes and respective characteristics of designer peptides and proteomimetics, discussed in this review.
Molecules that blend some of the physicochemical properties of synthetic small molecules with the higher energy and specificity of protein therapeutics could serve as attractive starting points for undruggable targets. Peptides naturally embody this molecular middle ground, owing to their larger binding footprint and propensity for specific interactions at PPI and protein–nucleic acid interfaces in myriad natural biological processes. Indeed, dozens of natural and engineered peptides are currently in the clinic for diverse disease indications.6,7 Relative to biologics, natural peptides are significantly smaller in size, which can confer increased stability, synthetic tractability, reduced manufacturing costs, and higher likelihood of developing peptide analogs with reasonable cell and tissue penetration for intracellular target engagement.8 However, as a class natural peptide sequences composed of linear α-amino acids generally exhibit poor cell membrane permeability, high in vivo clearance rates from blood, and susceptibility to proteolytic and other metabolic degradation outside and inside cells (Figure 1B).9-14 Therefore, considerable research effort has been focused upon chemical strategies to improve the physicochemical properties of peptides to overcome these limitations. A hallmark approach is to use synthetic modifications to stabilize local and global structural motifs, which can theoretically provide enhanced affinity and specificity for intended molecular targets, as well as vastly improved pharmacological properties (Figure 1B). These chemically stabilized molecules, generally termed peptidomimetics,15 have a rich history in drug discovery as ligands for various receptors or enzymes as described in several reviews.16-21 In this review, we consider these synthetically modified compounds “designer” peptides or peptidomimetics, as well as larger mimetics of protein domains, which encompass proteomimetics or “synthetic biologics.” In this review, we will specifically focus on the development for and application of these molecules to tackle undruggable targets (Figure 1C).
Designer peptides and peptidomimetics have previously been categorized into four classes.22 Class A represent minimally modified peptides wherein small changes to the backbone or side chains result in structurally stable or more active analogues. Class B are significantly modified with unnatural amino acids forming substantial backbone alterations such as foldamers. Class C are scaffolds that replace the peptide backbone entirely with an unnatural nonpeptidic scaffold but retain side chain functionality. Class D are mainly small molecule-like scaffolds that may mimic the mechanism of peptides but do not have a direct link to structure or design and therefore will not be discussed in this review and have been reviewed elsewhere.22-24
In this review, we will focus on designer peptides and peptidomimetics in four general classes, including: (i) natural product-like macrocyclic peptides (MCPs); (ii) side chain stabilized peptides; (iii) peptidomimetics containing nonnatural backbones; and (iv) synthetic proteomimetics and biologics (Figure 1C). Sections (i) and (ii) mainly cover class A mimetics, whereas section (iii) will discuss class B and class C mimetics. Section (iv) focuses on larger mimetics of protein tertiary and quaternary structures for expanded recognition properties suitable for challenging protein and nucleic acid targets. We will focus on the defining structural features that characterize each class, the breadth of synthetic strategies previously employed to construct each class as well as notable examples of their applications in modulating undruggable disease targets. While there are numerous small molecule and biologic-inspired modalities developed for undruggable targets that may share properties with the classes above, such as class D mimetics and engineered recombinant proteins, we find these to be outside the scope of the current review and direct readers to other excellent recent reviews.3,4,22-27 We will conclude our review with an outlook on the enormous potential of designer peptides, peptidomimetics, and synthetic biologics in targeting the “undruggable” as well as unmet requirements for advancing these promising modalities to the clinic.
2. MACROCYCLIC PEPTIDES
Peptide therapeutics are an attractive modality due to their synthetic accessibility and broad range of targetable biomolecules and interactions. However, naturally derived linear peptides suffer from pharmacological liabilities such as lack of cell entry and rapid degradation by cellular proteases, as well as reduced target affinity and specificity.12-14 Structural preorganization through cyclization—either “head-to-tail” or via interchain cross-links—has been shown to significantly enhance several or all of these pharmacological pitfalls. Indeed, nature has devised numerous routes to biologically active macrocyclic peptides, including head-to-tail cyclized molecules like cyclosporin A, amantins, and somatostatin or depsipeptides such as romidepsin, FK228, and leualacin, and hybrids of similar structure.28-30 Typically, these molecules are synthesized by nonribosomal peptide synthetase (NRPS) clusters and further modified with noncanonical peptide bonds, amino acid side chains, and other modifications.31 Researchers can take inspiration from the structure–function properties of these natural products to initiate campaigns to produce synthetic macrocyclic peptides (MCPs) with desirable properties.32 Due to their larger surface areas, arrayed amide backbone and chemically diverse side chains, synthetic MCPs could be developed to target traditional ligand binding pockets, as well as shallower allosteric pockets and grooves on the surface of or at the interface between proteins; these target features may be more prevalent in undruggable targets. Therefore, MCPs occupy a Goldilocks zone of physicochemical space to yield FDA-approved therapeutic molecules.33-36 In this review, we focus on synthetic MCPs containing a natural peptide backbone with the formation of at least one covalent cyclic linkage to induce conformational constraints. While natural product-derived and synthetic MCPs such as cyclotide scaffolds have been developed to target classically druggable targets such as enzymes and receptors,29,37-39 here we will focus primarily on their development for undruggable targets and surfaces.
2.1. Design Principles for MCPs
MCPs are generally constructed with ~5–20 amino acids to span a molecular weight range of ~0.5–3 kDa, making these one of the smallest classes of designer peptides (Figure 1C). This size confers small molecule character that is beneficial for biological applications. Conformational constraint and preorganization is often accomplished via head-to-tail cyclization with or without additional side chain linkages, which collectively introduce significant rigidity into MCP backbone conformations. In addition to protecting structures from metabolic degradation, the preorganization of active binding poses can reduce the entropic cost of folding during target binding. Common cyclization chemistries typically must proceed with high yield, chemoselectivity and operate under aqueous reaction environments. These include disulfide bridges, thioether linkages, amide bonds, unsaturated hydrocarbons, and cycloaddition adducts, for example through [3+2] azide–alkyne Huisgen “click” chemistry or [4+2] Diels–Alder cycloadditions (Figure 2).40-42 These common approaches each serve to provide unique properties that have advantages and disadvantages dependent upon application and design.
Figure 2.

Common macrocyclization adducts (right) generated from common synthons (left).
In general, the design, selection, and optimization of MCPs for undruggable targets should aim for several attributes including: (i) strong binding affinity and specificity for the protein of interest (POI); (ii) the ability to penetrate cellular membranes and access intracellular targets; (iii) high metabolic stability, and (iv) high bioavailability.6 Owing to their larger size and chemical properties, many FDA-approved MCPs33-36 do not adhere to Lipinski’s rule-of-5 or similar paradigms.43-46 Instead, many bioactive MCPs are thought to exhibit cell permeability and high bioavailability through their potential for “chameleon-like” structural character. In this theory, MCPs can adopt a conformation with a high polar surface area (PSA) and more exposed backbone NH bonds to retain solubility in aqueous conditions but adopt an alternative conformation minimizing PSA and exposed NH bonds when diffusing into hydrophobic membranes.47-50 Baker and colleagues explored this principle in examining MCPs from 6 to 12 amino acids in length, and found that internal hydrogen bonds and minimal exposed polar groups were critical to achieve cell entry.51 They designed cis/trans MCPs that isomerized around one peptide bond to achieve the chameleonic property, where the cis state is solvent exposed and the trans state leads to the formation of internal backbone hydrogen bonds. These findings agree with previous studies showing that backbone hydrogen bond matching as well as amide N-methylation and masking polarity contribute significantly to MCP cell uptake and oral bioavailability.52 Monovich and co-workers performed an extensive study assessing the properties of 6-, 7-, and 8-mer MCPs to determine that two factors are highly predictive of oral bioavailability and membrane permeability: (i) the lowest free energy of transition, ΔG*transfer, between aqueous and membrane states and (ii) the lowest number of solvent exposed backbone NH groups in an MCP.53 This study supported many others suggesting that the backbone plays a more critical role in permeability as opposed to the side chain, hence cell permeable MCP backbones with varied side chains could be used as a framework for large array library formats for discovering ligands to therapeutically relevant targets. For further discussion on MCP chameleonic design principles, refer to the excellent review by Monovich and co-workers.54
Although the smaller size and constrained structure of MCPs generally improves cell uptake, this does not directly translate for every design. Therefore, a simple and accessible method that many designer peptides have adopted is appending a cellpenetrating peptide (CPP). CPPs are short peptides that can cross the cell membrane, which facilitates the transport of various attached cargo.55-58 Typical CPP examples such as Tat,59 penetratin,60 or octa-arginine61 are popular choices for conjugating to MCPs, though amphipathic and anionic CPPs have also been reported.55,62 Physicochemical properties such as formal charge and hydrophobicity correlate with cell penetration, and CPPs are internalized through clathrin- and caveolin-independent, energy-dependent endocytosis pathways involving sulfated cell surface proteoglycans.56 Given that linear peptides suffer from protease digestion, cyclic CPPs have been developed that are more metabolically stable and membrane permeable than their linear counterparts and are nicely described in a recent review by Parangi, Tiwari, and co-workers.63 While appending CPPs to MCPs is useful for improving bioactivity, there have been no therapeutics containing a CPP approved by the FDA to date, likely due to potential for cellular toxicity, adverse immune response, and poor efficacy arising from the lack of specificity due to the CPP moiety.64
To create MCPs with desirable pharmacological properties without the need for external modifications such as CPPs, a team from Chugai Pharmaceuticals analyzed several properties inherent to MCPs that would yield the most pharmacologically viable molecules.65 Using naturally cell permeable cyclosporine A as a model, they designed and tested 553 analogs to understand the critical properties that drive potential therapeutic efficacy. They determined desirable properties for metabolic stability and cell uptake include high hydrophobicity, a high permeability coefficient, a medium-sized ring between 8 to 11 amino acids, extensive backbone amide N-alkylation of 6 or more residues, and reduced side chain polarity (Figure 3). They also found that, for medium-sized rings, oxidative metabolism is dominant over enzymatic hydrolysis, and MCPs of 8 residues, while having higher average membrane permeability than MCPs of 11 residues, have higher oxidative metabolism. Smaller rings more susceptible to oxidative metabolism are unlikely to overcome this limitation without decreasing lipophilicity necessary for membrane permeability. However, ring size and residue identity also play important roles in target recognition that must be balanced with desirable metabolic stability.13 While these studies greatly improved our knowledge of MCP design, general strategies for designing therapeutically viable MCPs remain elusive owing to the greater complexity afforded by larger and more diverse molecules and interactions. However, interest in MCPs as a drug modality is increasing exponentially with many biotechnology firms exploring MCP therapeutic discovery platforms,13,44 and an increased interest from large biopharma groups in exploring this modality for a range of targets.
Figure 3.

Summary of common structural features and modifications of drug-like MCPs. Representative features are highlighted on the structure of cyclosporin A, alongside a representative summary of structure–function correlations from a study65 that systematically explored cyclosporin A-derived macrocycle libraries (right).
2.2. Discovery, Design, and Synthesis of MCPs for Challenging Targets
MCPs are versatile and can be expressed by natural machinery or produced synthetically utilizing the diverse chemistries. Synthetic optimization typically follows derivatization from existing scaffolds or de novo selection from large combinatorial libraries in high-throughput screening. In the following section, we will discuss how MCPs are developed through rational design or selection from large libraries in multiwell screening, one-bead-one-compound, DNA-programmable libraries, phage display, mRNA display, and in-cell formats. We will also discuss the common synthetic methods to form the MCP libraries and provide key examples that have focused on generating chemical probes and therapeutics for undruggable targets.
2.2.1. Rational Design.
The majority of MCP rational design campaigns begin with the identification of specific binding epitopes within naturally occurring proteins and peptides that are potentially amenable to ligand development and optimization (Figure 4A). Early structure-blind approaches involved synthesis of cyclic analogs of complementarity determining regions (CDRs) of antibodies or endogenous peptides such as enkephalin to target various POIs.66-68 As more cocrystal structures of apo- or bound target structures have become publicly available, rational or structure-based drug design (SBDD) approaches have become more common. This methodology has been used to find MCPs that interfere with interactions such as TNKS-AXIN in WNT signaling,69 RGS4,70 and TGF-β-SMAD7.71 A notable recent example for a rationally designed MCP targeting an undruggable interaction is between the transcription factor TEAD1 and the coactivator YAP1, which represent a TF-co-activator complex that is central in the Hippo signaling pathway.72 The TEAD1/YAP1 interaction is highly implicated in many cancer types, and abolishing this interaction leads to the suppression of cell proliferation.73 Hu and co-workers examined the structure of the TEAD1/YAP1 interaction and identified a looped-coil segment of YAP1 that interacts with TEAD1.74 Using this structure as a guide, they generated a series of MCPs via disulfide bridging, and discovered optimized molecules that could bind to TEAD1 with greater potency than wild-type YAP1. Subsequent work has further focused on this interaction to identify small molecules that can disrupt this binding interaction,75 highlighting an important byproduct of identifying peptide leads to ultimately develop more drug-like small molecules. In a separate study, an MCP derived from the BB loop in MyD88 was developed to interrupt protein dimerization.76 MyD88 is an adaptor protein critical in the TLR and IL-1R signaling pathways that functions via dimerization and overexpression in cancers leads to overactivation of NF-κB and cancer metastasis.77 Nussbaum and co-workers identified a sequence from the BB loop that controls dimerization and synthesized an MCP via an amide linkage to form c(MyD 4-4).76 c(MyD 4-4) blocked the TLR/IL-1 signaling pathway both in vitro and in mice models. In a subsequent study, they sought to increase the bioactivity of c(MyD 4-4) and enable oral administration by myristoylating the MCP at the N-terminus.78 Myristolyation led to a significantly improved cellular bioactivity and in vivo oral bioavailability while still retaining activity in mice models. In addition to these PPIs, there are many MCPs that have been rationally designed against amyloid peptides, which have recently been covered in an excellent review by Khairnar et al.79
Figure 4.

Rational design of MCPs. A) MCP starting points can be directly extracted from key interacting loops discovered in natural protein–protein interactions. B) Structure of stonin2 (orange/green) bound to Eps15 (cyan), identified “hot loop” by LoopFinder highlighted in green. PDB: 2JXC. C) Chemical structure of Eps15 inhibitor ST1-oxy identified by LoopFinder. O-xylene macrocyclization linker is highlighted in green.81
One way that potentially druggable PPI interacting loops, also called “hot loops”, can be identified is through the program LoopFinder, designed by Kritzer and co-workers.80 Methodologically, LoopFinder identifies “hot loops” if the interacting loop fit one of three parameters: (i) three or more hot-spot residues (defined as ΔΔGresidue ≥ 1 kcal mol−1), (ii) two consecutive hot-spot residues, or (iii) the average ΔΔGresidue over the entire loop is greater than 1 kcal mol−1. Most loops identified in this approach were secondary structures such as β-turns and β-hairpins, though many had noncanonical structures. In the updated version, they improved on the energy calculations and benchmarked it against known PPIs to refine the selection criteria for an identified “hot loop” to (i) an average ΔΔGresidue ≥ 0.6 Rosetta Energy Units (REU, roughly equal to 1 kcal mol−1), (ii) three hot spot residues (ΔΔGresidue ≥ 0.6 REU), and (iii) a greater than 50% of binding energy contributed by the loop, represented as the sum of ΔΔGresidue.81 Loops that fulfilled all three criteria were seen as the best starting points for MCP development for a given target of interest. A close examination of the 210 “hot loops” that fulfilled all three criteria suggests that MCPs can only mimic some hot loops due to conformational constraints.82 It also suggests that, while larger MCPs can accommodate the native conformation of the hot loop, this must be balanced against the entropy loss of the larger MCP upon binding. One of the loops that fulfilled all three criteria is found in the interaction between Eps15 and stonin2, for which there are no known high affinity inhibitors (Figure 4B).81 Eps15 is a clathrin-associated protein and part of the endocytosis machinery83 and its interaction with stonin2 leads to the recruitment of AP-2 and endocytosis, which is implicated in Ebola infectivity and potentially other diseases.84 After identifying the loop on stonin2, cysteine or penicillamine residues were appended to both termini of the peptide sequence, and a panel of dithiol, bis-alkylation linkers were tested to cyclize the peptide by forming stable thioether linkages. The lead compound, ST1-oxy, was flanked by two cysteine residues and cyclized using dibromo-o-xylene to generate a selective and high affinity (Kd = 0.33 μM) ligand for Eps15 (Figure 4C). A 2D-NMR structure in water confirmed ST1-oxy recapitulated the structure of the stonin2 hot loop, displaying the powerful tools for computationally defining native protein structures for the discovery of designer peptide ligands.81
Expanding the discovery of cyclization-amenable loops at protein interfaces can identify starting points for chemical probe development. Building upon the LoopFinder work by the Kritzer group, Burgess and co-workers recently developed a virtual library approach termed backbone matching (BM) that is able to sample the conformations of cyclo-organopeptides composed of 4–10 residues cyclized with various organic elements through amide bonds.85 Compounds capable of adopting the proper low energy conformation in solution corresponding to the target backbone structure are prioritized for further evaluation and development. Using this approach, they identified and validated MCP inhibitors for interactions between iNOS/SPSB2 and uPA/uPAR. They also discovered candidates for 1245 of the 1398 hot loops screened, suggesting a plethora of other target interfaces that may be interesting to pursue. The continued alignment of computational methods with experimentally determined protein structures for discovery and validation of interacting loop structures has been and will continue to be a powerful approach to rationally design MCP probes and therapeutics.
MCPs derived from natural protein surfaces can also be designed based on more active linear epitopes, which can be cyclized then reversed to develop pro-drugs. For example, on disulfide linked MCPs, a reversible cyclization strategy could leverage intracellular glutathione to reduce the disulfide bridge and linearize the peptide upon cell uptake (Figure 5A). The cyclic peptide gains stability and cell permeability afforded by rigidification but retains the linear binding epitope once inside of cells. Using this strategy, Dougherty et al. designed an MCP derived from a section in the C-terminus of CFTR to inhibit the intracellular CAL-CFTR interaction to prevent lysosomal degradation of the CFTR ion channel, a potential treatment strategy for cystic fibrosis.86 Although symptomatic treatments using small molecules for dysregulated ion channels exist, direct modulation of intracellular CFTR mutant interactions remains challenging. In this design, a disulfide linked MCP, PGD97, containing a cell penetrating handle was generated that exhibited high affinity and specificity for the PDZ domain of CAL and an in vitro serum half-life over 24 h (Figure 5B). PGD97 acted as a stabilizer of F508del-CFTR and improved the CFTR function in cells. This strategy is also amenable to bicyclic peptides (BCPs). Qian et al. designed a disulfide BCP targeting the undruggable interaction between NEMO and IKK-β, which are part of the IKK kinase complex that mediates NF-κB signaling and is implicated in cancer and autoimmune diseases.87-91 Similar to the CAL-CFTR inhibitor, a cyclic recognition domain was connected to a cyclic CPP to ensure entry into the cell to generate a BCP. The recognition domain binds to NEMO in linear form but not in cyclic form, thus acting as a prodrug to only function in reducing environments.92 Although only achieving modest inhibition (IC50 ~20 μM), this strategy increases metabolic stability and cell permeability. Other approaches to create prodrug MCPs to function in reducing or oxidative environments take advantage of the redox lability of disulfides.93
Figure 5.
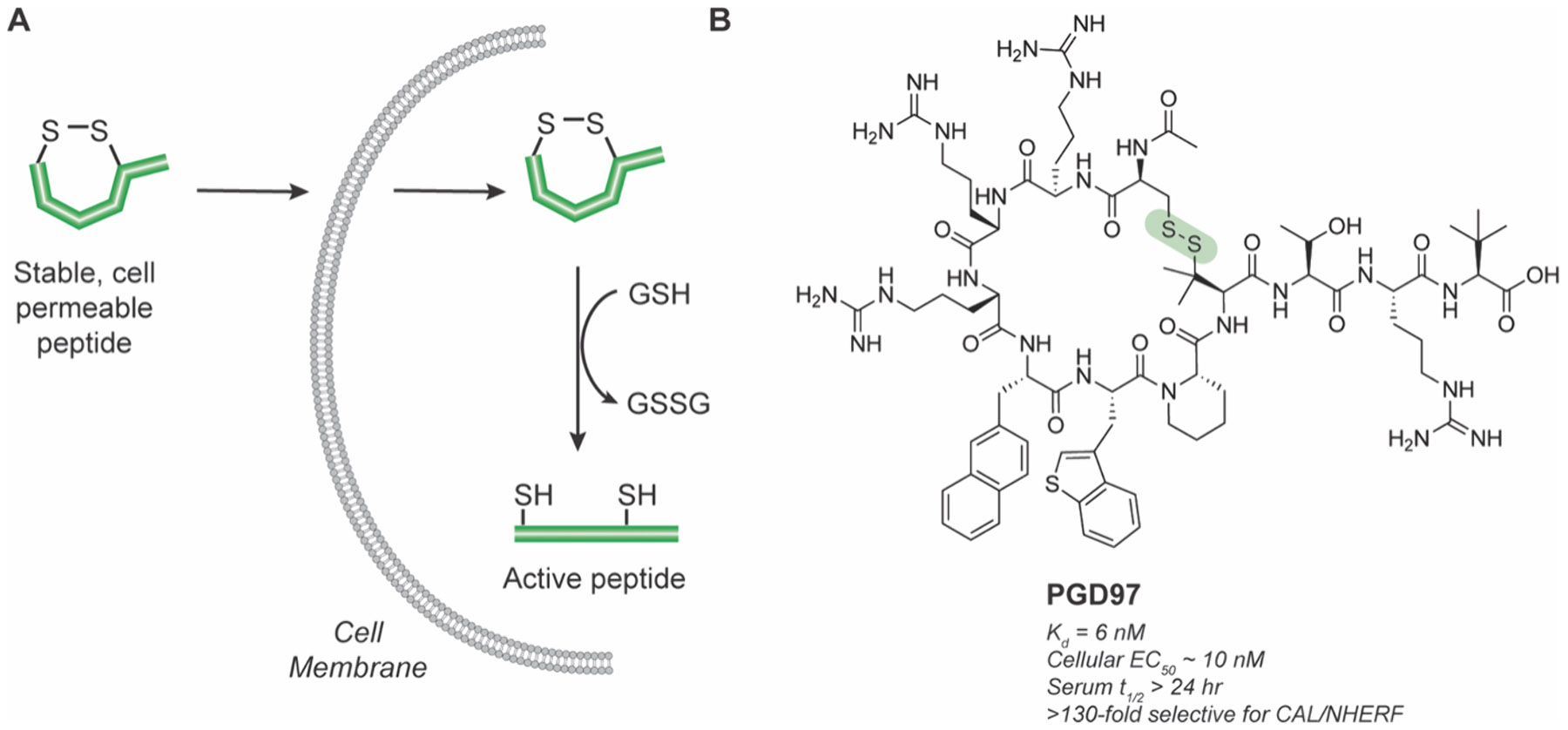
Pro-drug disulfide linked MCPs. A) Dithiol cyclization improves cell permeability and is reduced by glutathione to produce active linear peptide in cells. B) Structure of stable, cell permeable pro-drug CAL-CFTR inhibitor PGD97. The reversible disulfide bond is highlighted in green.86
Recent advances in computational approaches such as docking analysis and molecular dynamics simulations have contributed to the design of MCPs for challenging POIs. Baker and co-workers used computational analysis to design an MCP that could specifically bind HDAC6 over HDAC2, which is a notoriously difficult challenge to distinguish HDAC family members with small molecules or peptides.94 They started with an “anchor” moiety ((2S)-2-amino-7-sulfanylheptanoic acid; SHA), based on a pan-HDAC inhibitor that coordinated with the zinc ion in the HDAC active site to situate the model peptides to the HDAC surface (Figure 6A). Their first attempt used two peptides of known structure and a library of previously generated MCPs of 7–10 residues that were docked to the anchor.95 Using this method, they found an MCP that was decently potent in vitro (sub-μM IC50) but was not specific for either HDAC. They then added an additional tryptophan anchor because indole moieties are present in some known small molecule HDAC inhibitors before extending the peptide to form new MCPs. This MCP also displayed good affinity but little selectivity. From there, they sampled and optimized the torsional angle of the anchors. This led to an MCP designated des4.3.1 that was 88-fold selective for HDAC6 compared to HDAC2 (Figure 6B).94 Satisfying both binding and structural criteria is essential for generating de novo highly selective and efficacious MCP inhibitors. This and related studies are notable because they interrogate not only on-target95-99 but also propensity for off-target specificity, which is essential for the concurrent development of specific probes and therapeutic leads.
Figure 6.

Docking analysis and molecular simulations select for HDAC6-specific inhibitors. A) Crystal structure of des4.3.1 bound to HDAC6 anchored with SHA. PDB: 6WSJ. B) Chemical structure of HDAC6-specific inhibitor des4.3.1.94
The examples above demonstrate the key advantage of rational design: the lower barrier of entry to discover an MCP derived from a native protein structure. Identifying a potential interactor does not require lengthy and sometimes complicated screening approaches. However, there are a few limitations to the rational design approach: (i) requires prior knowledge of the target structure or interaction surface to identify potential ligands, (ii) identified ligands may not be amenable to structural stabilization after isolation, as proper folding may be dictated by interactions within the entire protein structure or interface,100 and (iii) identified ligands may not represent the optimal binders as not all combinatorial space is explored compared to screening approaches. Many of these shortcomings are being addressed with the integration of computational design and virtual screening, which has and will continue to explode in coming years with improvements to computational methods and technologies. For example, Des3PI101,102 and cPEPmatch,103 have been used to predict MCP binding to classically undruggable PPIs but have yet to be substantiated with experimental results. Improved structural prediction methods such as AlphaFold104 and RoseTTAFold105,106 provide further tools for predicting ligand interactions. For further reading on the quickly evolving computational approaches to design macrocyclic peptide interactions, we point readers to other reviews.107-110 While rational design approaches serve as solid starting points for MCP discovery particularly for identifying ligand-protein interactions, many undruggable targets are characterized by large, flat surfaces and disordered structures, and therefore unlikely to possess obvious domains from which to derive a potential ligand. For these challenging targets, large compound library screening is a more powerful approach for the de novo discovery of MCPs as described in the following sections.
2.2.2. Multiwell MCP Synthesis and Screening.
Multiwell screening involves plating a library of known compounds into a microtiter plate for subsequent biochemical or cellular testing, or concurrent synthesis and testing in multiwell formats. These methods are now anchor approaches to library screening in academia and pharmaceutical companies.111 Tranzyme Pharma demonstrated an early approach to multiwell screening of an MCP compound library, with the main scaffold containing a tripeptide with a tether moiety bridging the two termini via amide linkages (Figure 7).112 By diversifying the tethers used and incorporating ncAAs, they generated a 104 member library that was used to design MCPs to targets such as the motilin receptor and the ghrelin receptor and others that made it into clinical trials.113-117
Figure 7.

Scheme for the combinatorial synthesis of a diversified tripeptide MCP library in multiwell format, representing an early example reported by Tranzyme Pharma.112 Bts: benzothiazole-2-sulfonyl, Ddz: α,α-dimethyl-3,5-dimethoxybenzyloxycarbonyl; DIAD: diisopropyl azodicarboxylate, MP: microporous, PS: polystyrene.
Many alternative approaches for the synthesis and screening of diverse MCP libraries have been developed in recent years. For example, Heinis and co-workers have reported the generation of low molecular weight MCPs through the cyclization of tri- and tetrapeptides via thioether linkages.118-122 In one report they synthesized a core of Na-bromoacetyl-activated linear peptides, which were reacted with diverse primary amines.119 Reduction of a masked thiol and introduction of a reactive linker moiety thus enabled cyclization with the newly formed secondary amine for MCP generation. This approach used acoustic liquid transfer to generate macrocyclic peptides at a picomolar scale, which minimizes the use of reagents and decreases the cost of producing the macrocyclic library (Figure 8A,B). This library of 2700 MCPs was tested against the classically undruggable p53/MDM2 interaction, which is a critical interaction in numerous cancers and a bellwether target used by many studies for designer peptide synthesis.123,124 Screening the library in a competition format against a displacement peptide bound to MDM2 in a 384-well plate format resulted in the identification of several hit scaffolds with low-to-sub-μM inhibition, including the sulfone containing macrocycle 9 (Figure 8C).119
Figure 8.
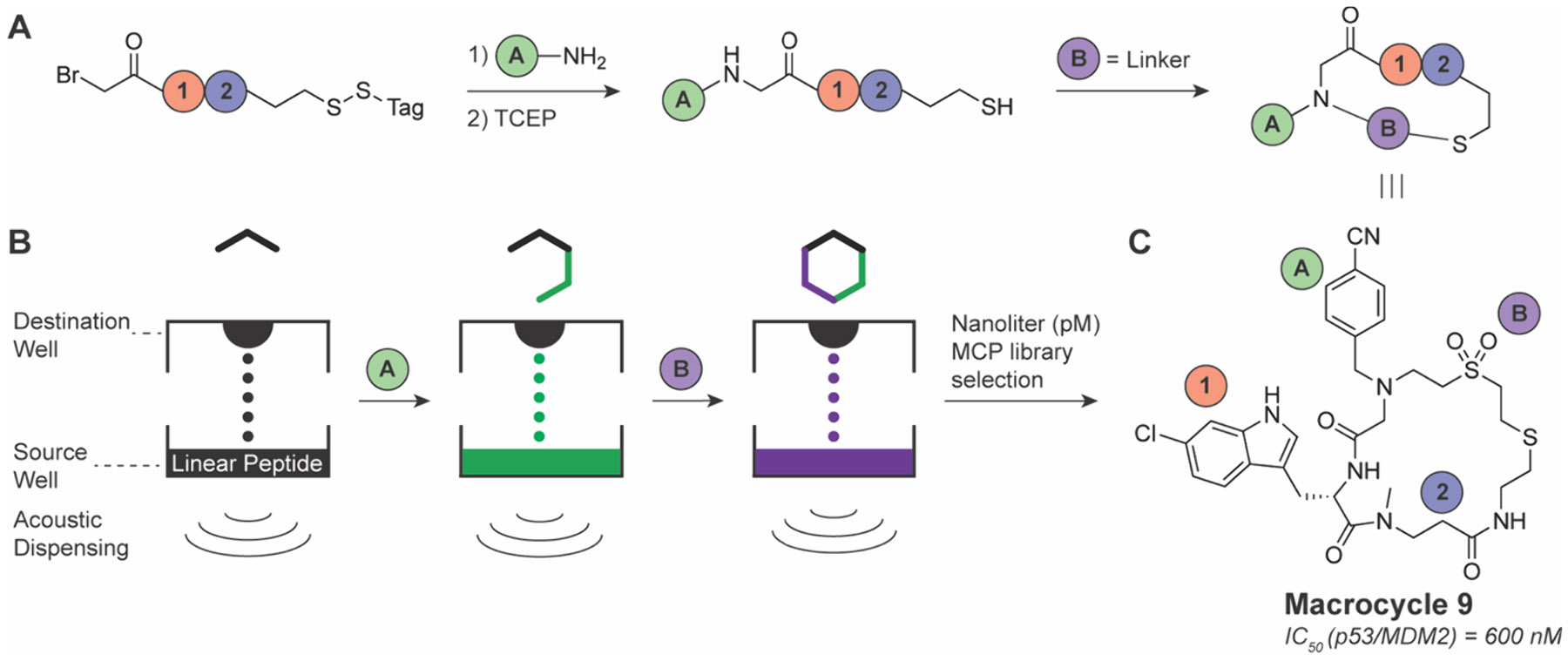
Nanoliter (pM scale) MCP library preparation using acoustic liquid transfer. A) General reaction scheme to prepare dipeptide library. B) Schematic of acoustic liquid transfer for small volume mixing. C) Chemical structure of macrocycle 9.119
Recently, a new “blind” screening strategy termed Peptide Exploration Platform with Tag-Free Intramolecular Chemistry (PEPTIC), was introduced for label-free MCP library synthesis.125 This platform uses “CyClick” chemistry to cyclize peptides, where an N-terminal amine reacts with an embedded C-terminal aldehyde to generate a 4-imidazolidinone cyclization group (Figure 9A).126 After screening for hit compounds binding to a POI, MCPs are then linearized using a DeClick approach, wherein hydrolysis of the 4-imidazolidinone linker occurs at high temperatures and low pH (pH 2–3). Remaining hits are then deconvoluted using liquid chromatography tandem mass spectrometry (LC-MS/MS) to determine the MCP sequence. This selection strategy is beneficial as it eliminates the need to append an additional biomolecule tag for identification, which may interfere with the intended interaction and lead to false-positive or false-negative results. As a proof-of-concept, this strategy was used to select an MCP that binds to an HIV-1 capsid protein that is critical in viral replication to prevent its interaction with other proteins.127 A generated hit molecule, cyFG-3, displayed submicromolar affinity for the capsid protein (Figure 9B).125
Figure 9.

Tag-free PEPTIC selection strategy for MCPs. A) Schematic workflow of PEPTIC. B) Chemical structure of cyFG-3.125
2.2.3. One-Bead-One-Compound MCP Libraries.
A classic approach to MCP library generation and screening is the use of “one-bead-one-compound” (OBOC) libraries.128 Here, MCPs are synthesized through a split-and-pool model, where individual beads react with a single amino acid at a time before being split to react with another amino acid. This continues until a library of up to 109-members is created that can then be screened against various targets, after which hit compounds are sequenced via Edman degradation or mass spectrometry.129-131 An early example of OBOC used to select MCPs against intractable targets was the discovery of inhibitors specific to HDAC6, which is implicated in the development of several cancers and neurodegenerative disorders.132 Ghadiri and Olsen designed an OBOC MCP library based on a α3β tetrapeptide scaffold cyclized via head-to-tail cyclization.133 Initial attempts to use a fully α-amino acid tetrapeptide failed due to difficulty in lactam formation, presumably due to high ring strain.134 Including an additional backbone methylene in the single β-amino acid residue led to a much higher yield of lactamization.134,135 After two generations of focused OBOC selections determined by structure–activity relationship (SAR) analysis, a highly potent and HDAC6-selective MCP emerged, which was confirmed in cells by observing increased α-tubulin acetylation, a hallmark of HDAC6 inhibition. Finally, they explored inhibition of cell proliferation in HeLa cells and showed only compounds that also inhibited HDAC1 reduced cell growth, suggesting HDAC1 is much more important for cell viability than HDAC6.133 Since then, OBOC has been used to discover MCP inhibitors for other difficult-to-drug targets including the RGS4/Gα0 interaction,136 PTP1B,137 c-MYC,138,139 hLysRS/CA,140 and the calcineurin/NFAT interaction.141 For additional reading on OBOC and similar methods, we point readers to other reviews.142,143
The early version of OBOC had two significant limitations that hampered its widespread use: (i) high false positive rates and (ii) difficulty in sequencing MCPs.144 The first problem has been improved by lowering the density of peptide ligand on beads, testing diverse bead substrates and also requires ad hoc optimization based on the libraries and targets of interest. Solutions to the second identification problem have been approached through several strategies that incorporate a second molecular barcode onto each bead, including DNA, small molecules, and peptides themselves in a so-called one bead two compound (OBTC) approach. OBTC screens a linear peptide barcode that identifies the MCP sequence that is synthesized in the interior of the bead and does not interfere with selection (Figure 10).145-147 Additional innovations include DNA-encoded solid phase synthesis to encode peptide information into DNA barcodes read by sequencing.148 A seminal example using OBTC involved screening the Ras–Raf interaction, and more generally, the interaction of Ras with its effector proteins. The Ras family of proteins are a group of G-proteins (K-Ras, H-Ras, N-Ras) and are a hallmark of cancer, with 30% of human cancers having at least one Ras gene mutated.149 Ras has over 50 different effector proteins that interact with and modulate Ras signaling, with the most famous of these being Raf kinase. Pei and co-workers focused on a specific K-Ras mutation, G12V and discovered a peptide termed peptide 12, that has submicromolar in vitro IC50 values.150 It bound specifically to K-Ras(G12V) but did not have activity in cells due to poor cell uptake. To improve cell uptake and increase potency, a second-generation MCP library was generated with OBTC.151 Optimization of the cyclic peptide led to cyclorasin 9A5, which exhibited an in vitro IC50 value of 120 nM and an EC50 value of 3 μM in cells and prevented Ras signaling events such as phosphorylation of Akt. To further increase cell uptake, they rescreened using OBTC with a bicyclic peptide framework where one of the cycles was a cell-penetrating peptide (CPP).152 With this BCP framework, they generated peptide 49 that selectively bound to K-Ras(G12V) and exhibited good cell penetration (Figure 11). Peptide 49 inhibited Akt phosphorylation in cellulo and reduced proliferation of H1299 lung cancer cells with an EC50 value of 8 μM. However, peptide 49 displayed a higher affinity for inactive state Ras·GDP over active state Ras·GTP, leading to high concentrations required to inhibit cell proliferation. Following this, they sought to make a pan-K-Ras-GTP MCP inhibitor.153 Using OBOC based on a previous design, cyclorasin B3-4, they screened to find a BCP that preferentially bound to Ras-GTP. After a comprehensive optimization campaign, cyclorasin B4-27 was developed as a 10-fold selective inhibitor of K-Ras-GTP over K-Ras-GDP (Figure 11). B4-27 was successful in acting as a pan-Ras inhibitor against various Ras mutant cancer lines and reduced proliferation with modest potency (single-digit μM EC50 values). Importantly, B4-27 exhibited a > 24 h half-life in vitro in human serum and significantly reduced tumor growth in A549 xenograft models following subcutaneous administration.154
Figure 10.
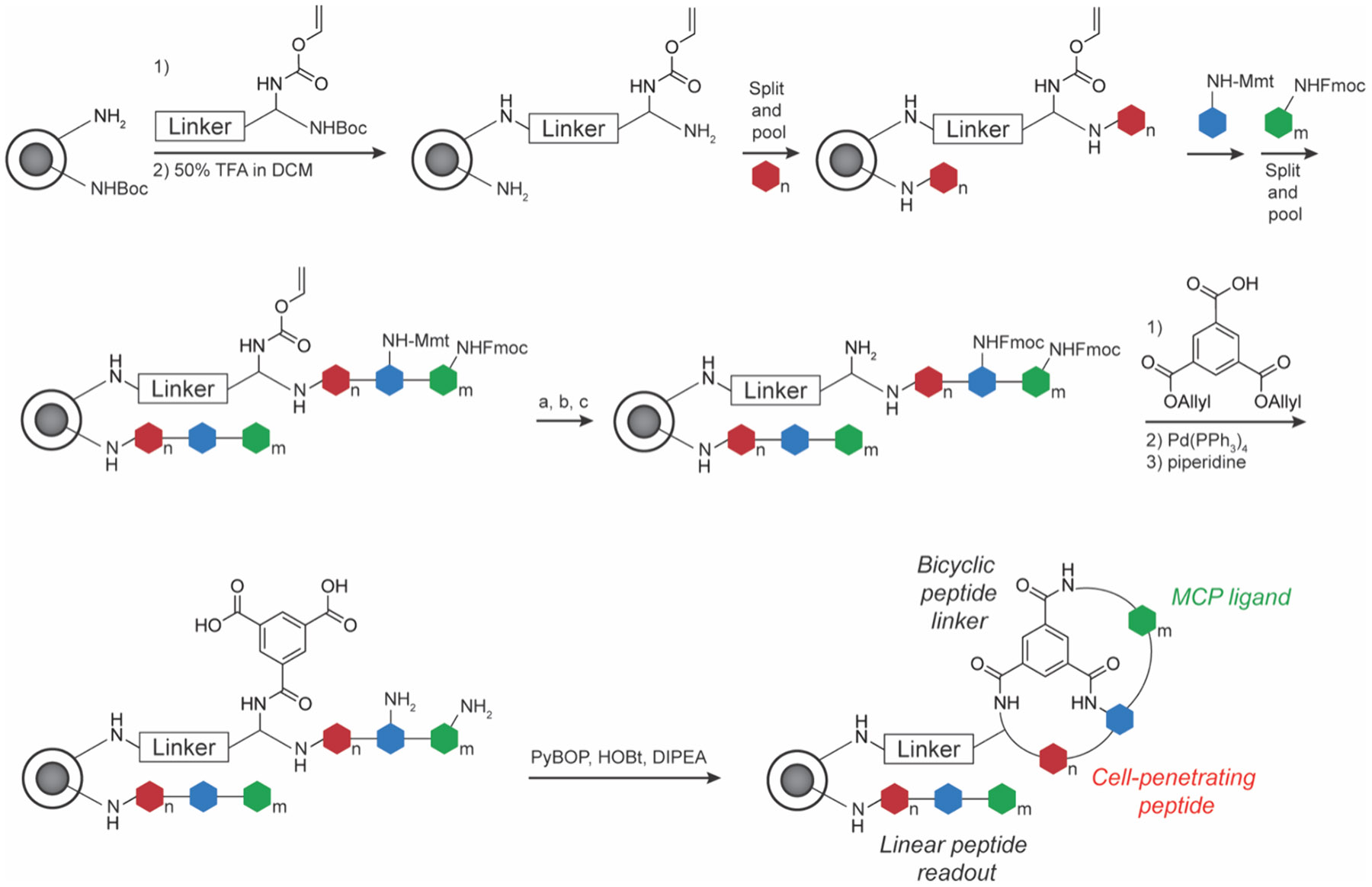
One-bead-two-compound BCP library preparation using standard SPPS chemistry and split-and-pool diversification. Peptide chain is extended on both inner and outer bead sites. Inner chain is used as the identified readout by LC-MS/MS. Outer chain is cyclized and interacts with POI. (a) 2% TFA in DCM; (b) Fmoc-OSu/DIPEA in DCM; (c) Pd(PPh3)4.147
Figure 11.
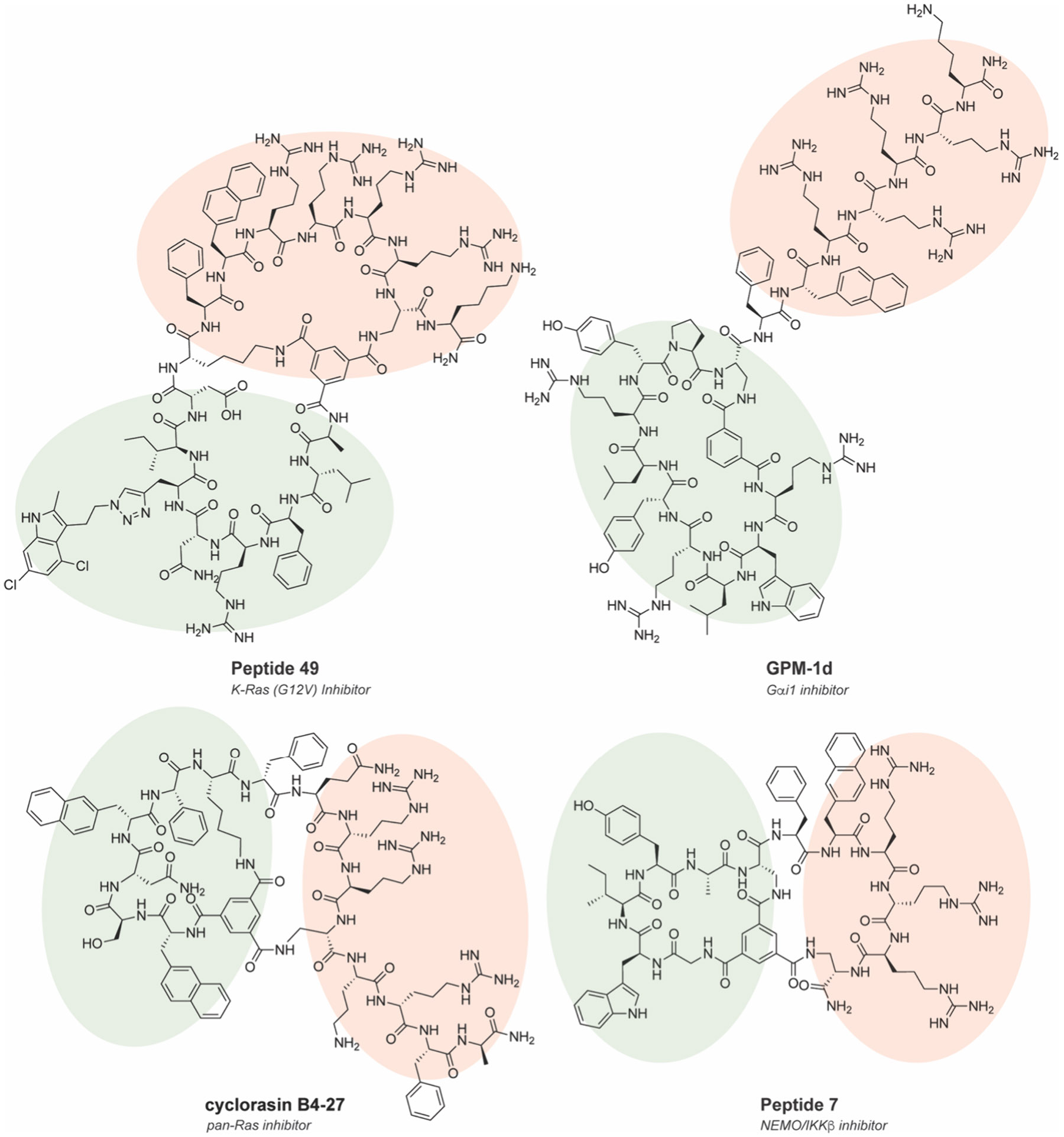
Chemical structures of CPP-MCPs selected from OBOC or OBTC libraries. Peptide 49152 and cyclorasin B4-27153 target Ras interactions, GPM-1d155 targets Gαi1, and Peptide 7156 targets NEMO/IKK interactions. CPP portion is highlighted in red. POI recognition portion is highlighted in green.
Another important example of using bead-based compound libraries was to target Gαi1 and Gαs, which are heterotrimeric G-protein subunits that function downstream of G-protein coupled receptors (GPCRs) to route signals from GPCRs to downstream effector pathways.157 This pathway has been tied to many different cancers, such as thyroid and adrenal tumors, and directly targeting G-proteins represents attractive alternatives to GPCRs, yet as a class G-proteins remain challenging for conventional small molecules.158 Following a previously discovered MCP binding motif for Gαi1 and Gαs that follows a guanine-nucleotide exchange modulator (GEM) motif of ΦζΦX[±]ΩL, (where Φ is a hydrophobic residue, ζ is a uncharged hydrophilic residue, X is any residue, [±] is any positively or negative charged amino acid, Ω is an aromatic residue, and L is a leucine),159-163 Nubbemeyer et al. generated and screened a OBOC library to discover a peptide, GPM-1, that potently binds to Gαi/s.155 Optimization of GPM-1 by cyclization and conjugation with a CPP to form GPM-1d increased the affinity, structural stability, and cell permeability to modulate the activity of Gαi/s in cell culture in a bifunctional GEM-like manner (Figure 11). Structural analysis using biomolecular simulations analyzed underlying MCP-protein interactions by identifying a network of important H-bonds that occur between the CPP and the protein at Arg14-Asn250 and Arg16-Asn251. In addition, the MCP has a stacking interaction between Tyr8 and the N-terminal Ipa, along with a hydrophobic interaction between Trp2 and binding site.155 A second-generation design identified GPM-3, which binds to active-state Gαi1 with a submicromolar binding constant and was shown to act as a GTPase-activating protein (GAP) modulator.164 In a separate example, Rhodes et al. developed a 2.4 × 109-member OBTC BCP compound library and incorporated cell penetration and cyclization into the activity screen against the NEMO-IKKβ interaction.156 Optimization of the CPP and binding interface produced a BCP, peptide 7, with low μM inhibition activity on viability in multiple ovarian cancer cell lines and reduced canonical NF-κB signaling (Figure 11). Interestingly, as in the previous example, the positively charged CPP portion contributed to the binding of negatively charged NEMO surface, suggesting a potential cooperative approach for improving the affinity and cell permeability for MCP-CPP constructs.156
2.2.4. DNA-Encoded and DNA-Programmed MCP Libraries.
DNA-programmable MCP libraries were first reported using DNA-templated synthesis (DTS) by the Liu lab in 2001.165 DTS uses synthetic oligonucleotide-small molecule conjugates to template ordered monomer coupling as a result of proximity induced DNA hybridization and either regio- or chemoselective reactivity. Sequential addition of DNA-fragment conjugates, which react in a sequence-specific manner based on DNA templates in solution, can generate diverse linear and cyclized peptide, peptide-like and small molecule libraries. This technology was first adapted to generate MCPs by amino acid coupling and cyclization by Wittig olefination to generate libraries ranging from 104 to 105 members (Figure 12A,C).166-168 These libraries were used to screen for bioactive compounds against phosphatases (DEP1, MEG2, PRL2), GTPases (Cdc42, H-Ras-V12, RhoA), antiapoptotic proteins (BCL-XL), nuclear receptors, PDZ and SH2 domains, along with assorted kinases and proteases.168,169 The most potent molecules derived from these screens were focused within kinase and protease families, including Src, VEGFR2, and IDE. Other DNA-programmed MCP libraries, such as those generated by teams at Ensemble Therapeutics and Bristol-Myers Squibb using azide–alkyne cycloaddition (~160,000 members; Figure 12C), have also been successfully screened against the caspase inhibitor proteins cIAPs and XIAP, and could be applied to other challenging protein targets.170 These screens produced highly potent compounds that exhibited modest pharmacokinetic properties (half-lives ~2 h and mean residence times ~2.5 h in mice), and bioactivity against MDA-MB-231 xenografts in mice, showcasing potential for these libraries to produce MCP therapies.
Figure 12.
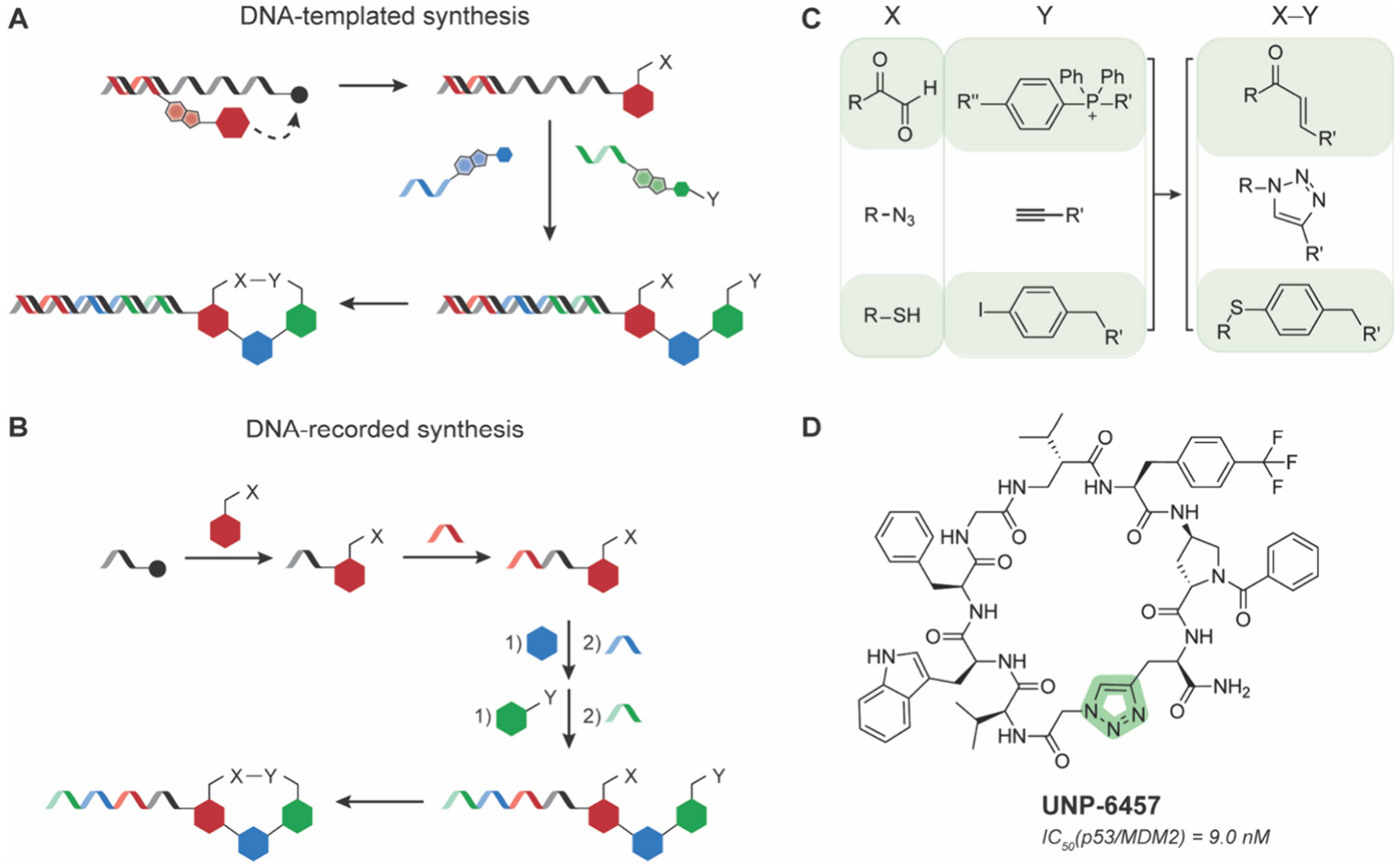
DNA-programmable MCP libraries. Schematics of MCP library preparation by A) DNA-templated synthesis or B) DNA-recorded synthesis. C) Common DNA-compatible chemistries used to prepare MCP DNA-libraries. D) Chemical structure of UNP-6457.176
A conceptually similar and widely used approach for library synthesis and screening is DNA-encoded library (DEL) technology. First proposed in 1992171 as “DNA-recorded synthesis,”172,173 DEL libraries have been used extensively to record the synthesis sequence of complex and large MCP and small molecule libraries, now commonly reaching ~106–109 members. DEL synthesis uses small molecule or amino acid fragments ligated onto DNA oligonucleotides, which can be subjected to linear or parallel split-pool synthesis rounds for further fragment coupling, which is accompanied by DNA-ligation to encode the identity of each fragment in each round (Figure 12B). This process repeats multiple times until the library is synthesized and each compound has a unique DNA barcode for identification. Chen and co-workers generated a 106-member MCP DEL by performing a DNA-compatible palladium-catalyzed S-arylation reaction to react the thiol groups on cysteine residues with noncanonical aryl iodide side chains.174 As a proof-of-concept, they screened the library against p300, a transcriptional coactivator integral to oncoproteins function,175 and discovered two MCPs with single-digit μM inhibition potency. As expected, the linear precursors to these MCPs showed very poor (>100 μM) inhibition of p300, highlighting the necessary conformational constraints to achieve high affinity binding. DELs have also been used to target the transcription factor p53 and its interaction with MDM2. Su and co-workers synthesized an MCP DEL library of 109 tetrapeptides cyclized via azide–alkyne triazole formation to pan for molecules that disrupted the interaction.176 They discovered a hit compound, UNP-6457, that binds potently (low nanomolar Kd) to MDM2 and projected similar hydrophobic groups into the surface groove that interacts with the key p53 residues Phe19, Trp23, and Leu26, further confirming these necessary interactions for potent binding (Figure 12D). This compound, while not passively permeable in cells, could be a scaffold for improving biophysical and pharmacokinetic properties down the line.
DNA-programmable libraries have many potential advantages for MCP synthesis and screening. First, the miniaturization of synthesis and screening is enabled by the robust, enzyme mediated amplification of reaction products, but also places specific requirements and limitations on the compatible chemistries that can be deployed. Furthermore, because no purification of library members is needed, entire libraries can be generated, screened, retrieved and reintroduced to subsequent “evolution” rounds all in solution. Collectively, these attributes make DEL- and DTS-like approaches very attractive for MCP generation and application to undruggable targets. The requirement for DNA tags does hinder the use of DEL libraries against some undruggable targets, for example large families of TFs and chromatin regulatory proteins that have intrinsic affinity for DNA. Also, advances in the chemistries that can be used in DEL construction could continue to advance the likelihood that MCP hits are likely to contain other necessary attributes like cell penetration and metabolic stability.177-179 For additional reading on DTS and DELs, we refer readers to other in-depth reviews.180-182
2.2.5. Phage Display MCP Libraries.
First conceived by Smith in 1985, phage display is a powerful in vitro selection method that uses a bacteriophage, commonly M13 or fd, to express and display a library of peptides by fusing them to a surface protein, commonly pIII.183,184 This library is used to screen for binding to the POI typically through affinity purification of the phage particles. Iterative rounds of selection can be performed to isolate high affinity interactors that are identified by sequencing the phage DNA. This technology has been adapted to select for mono- and bicyclic peptides (Figure 13). The earliest examples of using phage display to discover MCP ligands date back to the early 1990s and was used to identify integrin-binding MCPs containing the RGD motif.185-188 In 1995, phage display was used to identify an MCP ligand for DNA matrix associated regions (MARs), the first example of a nonclassical drug target.189 Since then, phage display has been adapted to discover MCPs against various difficult-to-drug protein and nucleic acid targets including TEAD4,190 the Grb7-HER3 interaction,191 interactions between βB2-Crystallin fibrils,192 and the LEDGF/p75-IN interaction.193 For further reading on phage display or other related technologies capable of selecting peptide ligands, such as yeast display, bacterial display, or mammalian display, we point readers to other reviews.194,195
Figure 13.

Representative schematic of bicyclic phage display for selection of BCPs. Chemical structure of β-catenin/ICAT inhibitor BC1 is shown.196
An important set of intractable targets is selectively inhibiting the highly conserved B-cell lymphoma-2 (BCL-2) family members. These proteins that include the antiapoptotic members BCL-2, BCL-XL, and MCL-1 regulate the intrinsic apoptosis pathway and are highly upregulated in cancer, thus represent important therapeutic targets.197-200 These proteins are very structurally similar and often perform the same role in the regulation of apoptotic cell death. Wu and co-workers used phage display to discover an MCP to target BCL-2.201 In their MCP design, they used 2-((alkylthio)(aryl)methylene) malononitrile (TAMM) conjugated with a chloroacetamide moiety as the scaffold molecule to cyclize via two included cysteine residues (Figure 14A).202 TAMM specifically reacts with the N-terminal cysteine residue to generate 2-aryl-4,5-dihydrothiazole (ADT) (Figure 14A). From this selection, they identified cp1, which selectively bound to BCL-2 19-fold more than BCL-XL (Figure 14 B,C).201 Interestingly, when the scaffold molecule TAMM is not present and the MCP is instead cyclized via a simple internal disulfide linkage, these MCPs had no binding affinity to BCL-2, suggesting the size and conformation of the ring plays a role in binding despite the exact same amino acid sequence or the scaffold molecule directly interacts with the protein. To look closer at the binding interactions of cp1 with BCL-2 over BCL-XL they solved a crystal structure and found that the hydrogen bond formed with BCL-2 at D111 and the backbone of cp1 at G6 is the critical differentiating factor (Figure 14C). In BCL-XL, A104, the closest residue to cp1 G6, is 7.2 Å away, precluding the formation of a hydrogen bond. Generating a BCL-2 D111A mutant led to significantly decreased cp1 binding. Another interesting finding was that cp1 binds to the BCL-2 surface without inducing conformational changes of the α3- or α4-helices, unlike small molecule or BH3-derived ligands. This unique binding mode allowed cp1 to interact with drugresistant BCL-2 mutants G101V and F104L. From here, a phage display library constructed from the cp1 sequence was prepared and used to discover MCPs specific for BCL-XL, which selected for cp3 that showed ~3-fold more selectivity for BCL-XL compared to BCL-2 (Figure 14D,E). Interestingly, they found that the residue at position 6 is key for affinity and selectivity, suggesting that highly similar surfaces require very few distinct interactions for selectivity. Unfortunately, poor membrane permeability even when tagged by the CPP Tat severely limited the MCPs efficacy in live cells, and significant efforts to produce clinically relevant compounds are needed.201 However, the ability to bind wild-type and drug-resistant mutant BCL-2 family proteins may provide starting points for additional ligand development for these challenging targets.
Figure 14.

Phage display selection of BCL-2 or BCL-XL-specific MCPs. A) Schematic of cyclization approach to generate MCPs with 2-((alkylthio)(aryl)methylene) malononitrile (TAMM) linker. B) Chemical structures of BCL-2 specific cp1 (left) and BCL-XL targeting cp3 (right). C) Crystal structure of cp1 bound to BCL-2. Key interacting H-bond between cp1(G6) and D114 of BCL-2 shown in yellow. PDB: 7Y90. D) Crystal structure of cp3 bound to BCL-XL. Key interacting residues between cp3(P6) and BCL-XL are shown. PDB: 7YAA.201
Another key family of undruggable targets to overcome are gain-of-function mutants of K-Ras. Sakamoto et al. used phage display and panned an MCP library to target K-Ras(G12D), the most common K-Ras mutation in human cancers.203 This screen identified a molecule, KRpep-2d, which contains a disulfide cyclization linkage and exhibited very high affinity (Kd = 51 nM) and more than 10-fold selectivity for K-Ras(G12D) over WT K-Ras, while the linear version of the peptide displayed no binding (Figure 15A). In cancer cell lines with the G12D mutant K-Ras, KRpep-2d suppressed cell proliferation and the Ras signaling cascade, suggesting the MCP may have some cell permeability and stability. This is unsurprising given that KRpep-2d contains 8 arginine residues in close proximity, which has previously been identified as a CPP motif.204,205 A 1.25 Å resolution crystal structure demonstrated that KR-pep2d recognizes a unique binding region not previously targeted by other small molecule inhibitors (Figure 15D).206 In a follow-up study by the same group, to increase the efficacy of KRpep-2d, they used SAR analysis to design a BCP that would have reduced molecular weight, not be susceptible to reducing conditions and remain protease-resistant.207 First, they started with the crystal structure of KRpep-2d binding to K-Ras(G12D) and introduced ncAA mutations at positions 6–14. They found that substituting linear aliphatic hydrophobic amino acids at positions 7 and 9, along with hydrophobic aromatic amino acids at positions 11 and 14 led to increased binding. In addition, (S)-2,3-diaminopropanoic acid (Dap) at position 10 forms a salt bridge with D69 of K-Ras(G12D), which increased binding approximately 5-fold. To eliminate bond cleavage under reductive conditions, they substituted the disulfide with an amide linkage between positions 5 and 15, which yielded a 0.6-fold weaker binding affinity. Combining these modifications led to KS-36, which bound to K-Ras(G12D) 30-fold stronger than KRpep-2d but had weaker than expected cell growth inhibition (Figure 15B). They identified the arginine-rich CPP region as the limiting factor due to its endocytosis-dependent uptake, so they eliminated the arginine residues and created BCPs by joining together positions 4 and 11, along with a head-to-tail amide linkage. By substituting in either Cys or d-Cys at positions 4 and 11, they determined that Cys4, d-Cys11, and a 1,4-diiodobutane linker led to the greatest cell uptake. Final optimization at positions 5 and 10 to introduce (S)-2-aminononanoic acid and a tryptophan residue, respectively, generated KS-58 (Figure 15C). Computational examination of KS-58’s mechanism for cell uptake revealed that it behaves like a chameleonic molecule, altering its polar surface area and internal hydrogen bond network depending on the solvent.208 KS-58 displayed improved K-Ras(G12D)-dependent cancer cell growth inhibition compared to KRpep-2d in both liver and lung cancer cells. This is likely because KS-58 inhibits the interactions of both SOS1 and B-Raf with K-Ras(G12D), whereas KRpep-2d only inhibits SOS1. In a PANC-1 mice xenograft model, administration of KS-58 led to a sizable decrease (~35%) of tumor growth. Co-administration with gemcitabine, a common chemotherapy drug, led to a larger decrease in tumor growth, showing similar efficacy in different xenograft models of colorectal and pancreatic cancer.209-211 However, KS-58 has low water solubility and requires frequent administration. To address both issues, KS-58 was made into a nanoformulation with a biocompatible surfactant, Cremophor EL, to generate a cell permeable nanoparticle.212 This formulation reduced the cancer cell growth rate in mice xenografts of both colorectal and pancreatic models. A separate team from Merck also looked to optimize KRpep-2d to improve its pharmacologic properties.213 Through SAR, they designed MCPs that are membrane-permeable, protease-resistant, and inhibit cell proliferation in K-Ras(G12D) cancer cells. However, these polyarginine-rich MCPs, which were included to improve cell uptake, induced mast cell degranulation (MCD) that in extreme cases can lead to patient death. They found that there was a strong correlation between the number of arginine residues and MCD, with MCPs containing greater than two arginine residues inducing MCD. Though these designs were not taken further, these studies demonstrated the potential to use MCP display approaches to identify high affinity and cell permeable ligands for a challenging and as-yet undrugged target like K-Ras (G12D).
Figure 15.

SAR optimization of KRpep-2d. Chemical structures and properties of KRpep-2d (A), KS-36 (B), and KS-58 (C). Cyclization linker is highlighted in green. D) Crystal structure of KRpep-2d bound to K-Ras (G12D). Key interacting residues are labeled. PDB: 5XCO.203,206,207
One of the biggest limitations of the first generation of phage display is the inability to incorporate ncAAs. Although ambercodon suppression can only incorporate the one ncAA, it can lead to a genetically encoded, intramolecular thioether linkage. A phage display library of MCPs that incorporated this design was used to discover MCPs with single-digit micromolar dissociation constant and in vitro IC50 values for HDAC8, a class I HDAC member.214 Macrocyclic organo-peptide hybrid phage display (MOrPH-PhD) developed by Owens et al. works in a similar vein, but incorporates O-(2-bromoethyl)-tyrosine as the ncAA.215 This new phage display technique was used to disrupt the KEAP1/NRF2 interaction, which is part of the stress response pathway.216,217 Owens et al. discovered an MCP that interacted strongly with KEAP1 (Kd = 40 nM) and the amino acid sequence closely matches the tip of the NRF2 β-hairpin that interacts with a natural cleft in KEAP1.215 Interestingly, though the most strongly binding sequences closely matched the NRF2 β-hairpin, there was one comparatively weak MCP (Kd = 2.8 μM) that contained a different sequence, suggesting that it interferes with the KEAP1/NRF2 interaction at a different location. Using a different variation of phage selection, Zheng et al. also generated an MCP against the KEAP1/NRF2 interaction.218 In this approach, two cysteine residues were introduced and cyclized with a special linker, 2-cyanobenzothiazole conjugated with an α-cyanoacrylamide. The N-terminal cysteine reacts specifically with the 2-cyanobenzothiazole while the other cysteine reacts with the α-cyanoacrylamide to generate the MCP, similar to the reaction with the TAMM scaffold.202 The highest affinity MCP (sub-μM Kd) also contained close sequence homology with the NRF2 β-hairpin as those selected by Owens et al. Interestingly, although using different linkers and ring sizes for generating MCPs, the top sequences between the two studies differed by only one residue within the β-hairpin sequence (DSETGE vs DIETGE), and obtained similar affinity. This suggests this sequence is likely highly specific for KEAP1 and can tolerate different substitutions, potentially to develop KEAP1-targeting MCPs with improved pharmacological properties.
To increase the diversity and versatility of phage display, a BCP selection strategy was developed by introducing three cysteines into the peptide sequence and adding 1,3,5-tris(bromomethyl)benzene (TBMB) to effectuate the production of BCPs via thioether bond formation.219 Using this strategy, BCPs have been selected against two different historically undruggable targets such as the β-catenin/ICAT interaction and Dcp2.196,220 β-catenin is a cell signaling molecule that is a central component of the WNT/β-catenin pathway, among other roles.221-223 Mutations in this pathway can lead to cancer, and β-catenin is seen as a key therapeutic target. Bertoldo et al. discovered a β-catenin-targeting BCP using phage display by testing three different scaffold molecules: TBMB, TATA (1,3,5-triacryloyl-1,3,5-triazinane), and N,N′,N”-(benzene-1,3,5-triyl)-tris(2-bromoacetamide) (TBAB). All three scaffold molecules yielded MCPs with single-digit micromolar dissociation constants; however, MCPs cyclized by TBMB and TBAB interfered with ICAT binding, which inactivates the WNT pathway by binding to β-catenin.196 In a separate study, Luo et al. selected a high affinity BCP (Kd = 116 nM) for Dcp2,220 which is an enzyme that removes the 5′ cap from a subset of mRNA molecules that is difficult to target due to an intrinsically disordered region (IDR) and intracellular delivery.224-226 This BCP engages the IDR of Dcp2 and inhibits its activity in cells. The BCP was also used to find novel mRNA sequences that are recognized by Dcp2 in the decapping process, demonstrating how MCPs can be used as chemical probes to elucidate novel biology surrounding undruggable targets.
Multiple different strategies have been reported to form BCPs and tricyclic peptides using phage display and are summarized in this recent excellent review.227 This strategy has yielded several bicyclic peptides now in clinical trials with Bicycle Therapeutics, such as BT8009 targeting nectin-4,228 BT5528 targeting the EphA2 receptor,229 BT1718 targeting the zinc metalloprotease MT1-MMP,230 and BCY12491 targeting the CD137 receptor.231 Even though none of these targets are classically undruggable, this selection strategy should be amenable to additional targets in the future. Further developments in incorporation of ncAAs may also yield MCPs with better pharmacological properties. Additional phage display technologies have been used to synthesize lanthipeptides, and these may be used to target classically undruggable proteins in the future.232 Finally, yeast selection, though used much less frequently than phage display, can also be used to find MCP inhibitors. First reported in 2012, Bowen et al. used yeast display to generate MCPs cyclized via lactam formation to target the WW-domain and the N-terminal region of YAP1, an intractable transcriptional coactivator in the Hippo signaling pathway.72,233,234
2.2.6. mRNA Display of MCPs.
Simultaneously designed by the Yanagawa lab and the Szostak lab in 1997, mRNA display involves transcribing a diverse DNA library into mRNAs before puromycin, an antibiotic that terminates translation in the ribosome, is conjugated at the 3′ end (Figure 16).235,236 Peptides translated from the mRNA are covalently attached to puromycin to terminate translation, leaving an mRNA-peptide conjugate that can be biopanned against various POIs. After identification of interacting peptides, RT-PCR is performed on the RNA to identify the peptide sequence. In a move away from endogenous expression systems, modern mRNA display methods largely rely on in vitro translation, an early example of which is the protein synthesis using recombinant elements (PURE) system developed in 2001 from purified aminoacyl-tRNA synthetases, initiation factors, elongation factors, release factors, RNA polymerase and ribosomes to reconstitute translation.237 This approach can produce relatively large libraries of 1012-members. mRNA display has been used to select MCPs against numerous intractable targets and interactions such as p53/MDM2,238 SIRT2,239 SIRT7,240 TET1,241,242 and KDM7,243 among others.244-247
Figure 16.

General schematic of mRNA display for MCP selection.
An early example of using mRNA display to select MCPs against undruggable targets was performed by Millward et al. to target Gαi1·GDP.159 The initial MCP, cycGiBP, exhibited high target affinity but was prone to protease cleavage. To overcome this limitation, they developed scanning unnatural protease resistant (SUPR) mRNA display (Figure 17A). In SUPR, the library incorporates ncAAs such as N-methylalanine and is exposed to a protease cocktail prior to affinity purification, thus only selecting for protease-resistant interactors.161 This method was used to identify Gα SUPR (Figure 17B), which maintained similar binding affinity to cycGiBP but exhibited a > 35-fold increase in serum stability a half-life of 115 h, thus highlighting the versatility of in vitro display selection approaches.161
Figure 17.

Scanning unnatural protease resistant (SUPR) mRNA display. A) Schematic of SUPR mRNA display selection for protease-resistant MCP ligands. B) Chemical structure of Gαi1 inhibitor Gα SUPR.161
Building off of the PURE in vitro translation system, the Suga lab designed the random nonstandard peptide integrated discovery (RaPID) system, where mRNA display is combined with a flexible in vitro translation (FIT) system to screen large (>1012-member) MCP libraries.248,249 Along with the other recombinant elements, FIT involves flexizymes, which are tRNA-acylation ribozymes that can install ncAAs on to tRNAs that have been programmed to recognize different codons during translation (Figure 18A). The benefit of this system is that ncAAs can easily be incorporated into MCPs for testing via mRNA display, which expands the chemical diversity used to discover interactors. The most popular strategy for cyclization using RaPID incorporates a chloroacetamide moiety in the form of an N-chloroacetyl amino acid located at the N-terminus that reacts with an embedded cysteine to form a thioether bond.250 This strategy has been used to find MCPs against difficult-to-drug targets such as K-Ras-(G12D),251 α-Synuclein amyloid fibrils,252 and the retromer complex involved in endosomal membrane trafficking.253 For further reading on mRNA display or other similar display technologies, such as ribosome display, we point readers to other relevant reviews.254-256
Figure 18.
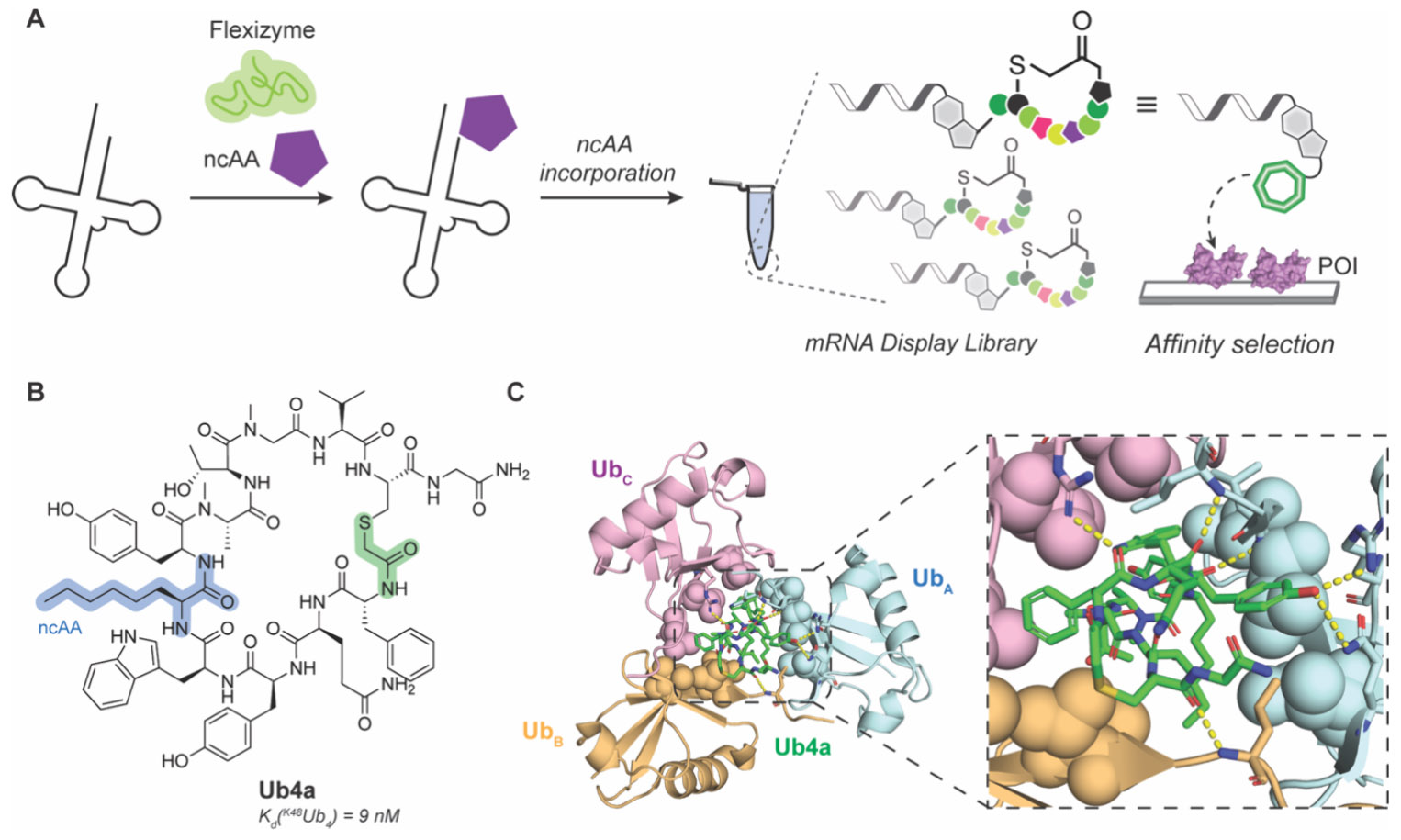
Random nonstandard peptide integrated discovery (RaPID). A) General schematic of RaPID incorporating flexizyme technology to include ncAAs during in vitro translation. B) Chemical structure of K48Ub4-selective MCP Ub4a generated by RaPID. Thioether linker used for head-to-tail cyclization is highlighted in green. C) Crystal structure of Ub4a bound to K48Ub3 displaying ring-like arrangement (left) and H-bonding network (right, insert, yellow dashes) for recognition. PDB: 8F1F.258,260
An interesting example of using RaPID to discover an MCP is for the recognition of poly ubiquitin (polyUb).257,258 Ubiquitination is a post-translational modification where a small protein (ubiquitin, Ub) is attached to mainly lysine side chains on cellular proteins to perform different functions including degradation by the proteasome.259 Interfering with the Ub-proteosome interaction is an attractive approach for cancer therapy. PolyUbs have diverse linkages that yield different conformations and diverse biological effects that are difficult to distinguish with small molecules like ubistatins. Using RaPID, Nawatha et al. specifically targeted K48-linked tetra-ubiquitin (K48Ub4), which is important for 26S proteosome recognition.257 They discovered a single MCP, Ub4ix, that was capable of distinguishing Ub linkage and chain size by targeting K48Ub4 with low nanomolar affinity. Ub4ix binding prevented USP2, a nonspecific deubiquitinase, from cleaving more than one Ub from the target, resulting in blocked deubiquitination of proteins like p53 and p27 that then accumulated in HeLa cells. To increase cell permeability and protease resistance, the same group performed another RaPID selection and incorporated ncAAs ClAc-d-Phe, N-methyl-Gly, N-methyl-Ala, N-methyl-Phe, d-Ala, d-Phe, and Aoc to replace Met, Glu, Asp, and Arg.258 This selection produced a cell permeable MCP, Ub4a, that bound to K48Ub4 with high affinity (Kd = 9 nM; Figure 18B). Interestingly, structural NMR analysis of Ub4a bound to K48Ub4 suggested that there is minimal interaction with the distal ubiquitin and that Ub4a is actually specific for K48Ub3, which may explain the mechanism of only allowing a single Ub cleavage similar to Ub4ix. This was later confirmed by a solved crystal structure showing tri-Ub wraps around Ub4a in a ring-like arrangement driven mainly by H-bonding and hydrophobic interactions (Figure 18C).260 Significantly, Ub4a demonstrated anticancer activity in a mouse model of CAG myeloma by reducing tumor burden in a manner similar to the approved 26S proteasome inhibitor medication bortezomib.258
RaPID can also be used to explore physicochemical properties of MCPs and the effects on target binding, as ring size and residue identity play important roles in target recognition. For example, Lin et al. used two separate RaPID libraries and compared 7–9 residue MCPs to 10–14 residue MCPs targeting the stimulator of interferon genes (STING) adaptor protein.261 These libraries were very diverse with 25 potential amino acids that could be incorporated. Surprisingly, they found that only 9-mer MCPs with 8-residue ring size exhibited measurable affinity using surface plasmon resonance (SPR). This is potentially due to the more thorough sampling of sequence space with fewer randomized positions or more likely the smaller loss of conformational entropy for shorter peptides to bind the target. Another key question is whether RaPID explores enough sequence space to select for the most efficient interactors. Suga and colleagues used RaPID to efficiently perform deep mutational scanning and explore the effects of ncAA incorporation for CP2, an MCP previously selected via RaPID to target the histone demethylase KDM4A-C.262-264 Including ncAAs changed properties such as charge, steric bulk, N-methylation, or number of hydrogen bonding groups. Interestingly, they found few mutations resulted in increased binding affinity, while many substitutions had little effect on binding energy. Although likely to be target-specific, many MCPs can potentially be screened and optimized through numerous modifications to gain desirable properties.
While small molecules have difficulty distinguishing between different conformational states, MCPs are more likely to behave as state-specific inhibitors. Dai, Hu, and Gao et al. showed this by using RaPID to select for MCPs that distinguish between GTP- or GDP-bound Gαs.265 To promote selectivity, MCP libraries were exposed to negative selection for the undesired alternate states. Using this approach, they developed MCPs that could target only the Gαs·GTP state (MCP termed GN13) or Gαs·GDP state (MCP termed GD20), which were determined to be at least 100-fold selective for their respective target state (Figure 19A and C). The MCPs bind to the Gαs switch II-α3 pocket, which adopts a different conformation dependent on nucleotide to discriminate between the different states. Analysis of crystal structures showed that GN13 interacts with active state Gαs through necessary and complex hydrogen-bonding network and hydrophobic interactions, whereas an expected steric clash of Gαs R232 in inactive-state switch II domain likely explains the conformationally selectivity (Figure 19B). Conversely, GD20 recognizes the inactive state Gαs through important electrostatic interactions, hydrogen bonding, and hydrophobic interactions, and likely sterically clashes with R231, R232, and W234 in the active state switch II domain (Figure 19D). In addition, both GN13 and GD20 demonstrated excellent specificity for Gαs over other structurally similar G-protein members regardless of state. Additional modifications to both MCPs improved cell permeability and bioactivity, resulting in compounds capable of in cellulo disruption of Gαs interactions and activity, demonstrating the power and versatility of RaPID and generated MCPs.265
Figure 19.
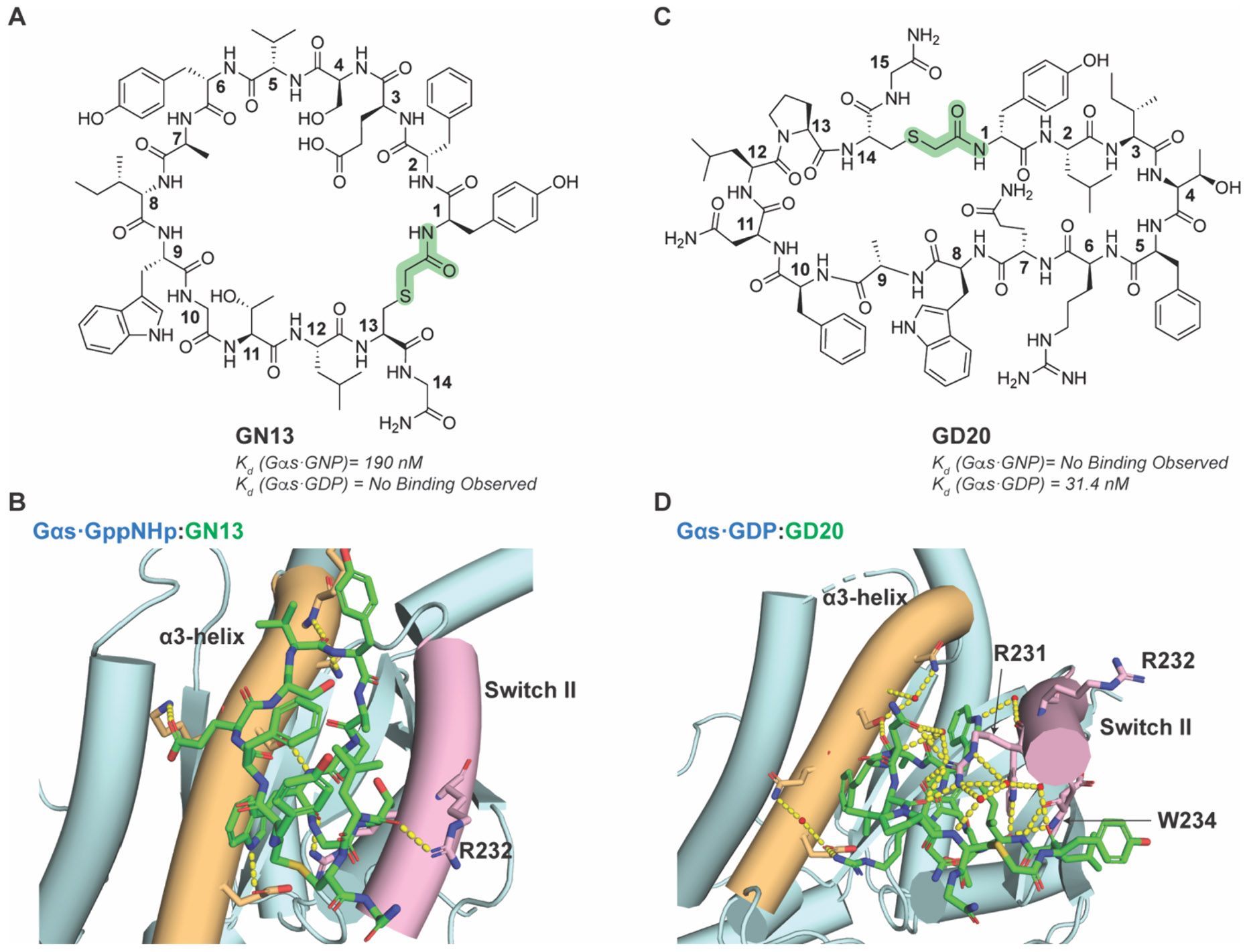
Discrimination of active and inactive Gαs GTPase by RaPID-selected MCP. A) Chemical structure of active Gαs-selective GN13. B) Crystal structure of GN13 bound to active-state Gαs. H-bond between GN13 and R232 is highlighted as yellow dashed line. PDB: 7BPH. C) Chemical structure of inactive Gαs-selective GD20. D) Crystal structure of GD20 bound to inactive-state Gαs. Key interactions between GD20 and R231 and W234 of Gαs are highlighted. PDB: 7E5E. Thioether cyclization linkage highlighted in green in chemical structures.265
The team at Chugai Pharmaceuticals who identified the previously described desirable MCP properties for drug-like molecules used their guidelines to generate an mRNA display library to select for inhibitors to K-Ras.65,266 Incorporating a high number ncAAs, particularly N-alkylated amino acids, and native chemical ligation for cyclization resulted in a library that could satisfy the criteria for drug-like MCPs. They discovered an 11-residue MCP, AP8784, that bound at the interface of KRas and SOS1 to potently inhibit the interaction with an IC50 of 54 nM but showed no activity in live cells (Figure 20). Following their guidelines and a crystal structure to improve the physicochemical properties of AP8784, they embarked on a comprehensive SAR campaign.266 The crystal structure revealed AP8784 interacted with the Switch II pocket on GDP-K-Ras(G12D), which identified positions 8 and 11 as likely to be amenable to modification without disrupting binding, while positions 2, 5, 7, and 10 made direct surface contacts. Positions 7 and 10 specifically occupied the SII-hole, and likely could be modified to enhance bioactivity. To summarize the outcome of the SAR analysis, positions 7 and 10 were optimized from 3-chloro-phenylalanine at position 7 and N-methyl-valine at position 10 to homophenylalanine(4-CF3,3,5-F2) and N-methyl-cyclopentyl for positions 7 and 10, respectively to improve bioactivity. Further improvement to cell permeability was achieved by altering positions 8 and 9 from tryptophan and N-methyl-phenylalanine to a proline and cycloleucine, respectively. Rigidity was increased by modifying positions 3 and 4 from N-methyl-glycine to N-methyl-alanine at position 3 and N-ethyl-azetidine-2-carboxylic acid at position 4. Finally, replacing position 1 to N-methyl-leucine, position 5 with an N-ethyl-4-methyl-phenylalanine, and N-alkylation of position 11 resulted in lead compound LUNA18 (Figure 20). This lead MCP contained desirable drug-like properties such as a high Clog P, and membrane permeability via Caco-2 assay without the need to significantly alter the scaffold. LUNA18 displayed a > 25-fold improvement to potency compared to AP8784 and exhibited low picomolar affinity for GDP-K-Ras-WT and G12C, G12D, and G12V mutants. LUNA18 selectively reduced proliferation in K-Ras-G12X-dependent cancer cell lines and showed dose-dependent antitumor activity after oral administration in mice.65 LUNA18 is currently undergoing a phase I clinical trial.
Figure 20.

SAR optimization of pan-K-Ras targeting MCPs. A) Chemical structure of AP8784. B) Chemical structure of LUNA18 after SAR optimization. Amide cyclization linkage is highlighted in green.266
mRNA display technologies are very powerful tools for selecting MCPs against various targets that have the potential to enter and succeed in clinical trials, key examples being FDA-approved zilucoplan267,268 and more recently orally available PCSK9 inhibitor MK-0616 currently in phase III.269-273 These examples and LUNA18 demonstrate how encodable designer peptides selected from large libraries can undergo synthetic optimization to produce viable clinical candidates. Including the ability of RaPID to incorporate multiple ncAAs during selection and SUPR to select for metabolically stable ligands, variations of mRNA display can be combined to generate MCPs with ideal pharmacological properties. Improvements to these technologies harness the power of computation to categorize the top hits into different binding modes on the POI, providing different starting points for further optimization.274,275 Given the relatively large library size, these technologies may generate more pharmacologically viable lead MCPs that function in cells or animals with less required modification and optimization.
2.2.7. Split Intein Circular Ligation of Peptides and Proteins.
Split Intein Circular Ligation of Peptides and Proteins (SICLOPPS) is a selection technique for discovering potent MCP inhibitors in live cells (Figure 21).276,277 In this approach, split inteins at the C- (IC) and N-terminus (IN) are used to catalyze head-to-tail peptide ligation (Figure 21B). After expression, trans-splicing of the peptide precursor occurs by the interaction of the termini to form a lariat structure, wherein the first residue at the N-terminus, usually a cysteine or serine, forms a thioester or ester linkage to cyclize the peptide, which is then spontaneously converted to the native lactam (Figure 21C). This naturally restricts the first residue of the MCP to either a serine or a cysteine but is amenable to various functional groups at the other positions in the MCP library. Since the cyclization process occurs in cellulo, the advantage of this selection is that it can be paired with bacterial or yeast reverse two-hybrid systems to select for MCPs that disrupt PPIs.276,277 For further reading on the properties and applications of SICLOPPS we point readers to other reviews.278,279
Figure 21.

SICLOPPS selection of macrocyclic peptides. A, B) Representative schematic of in cellulo selection strategy for cyclic peptides targeting HIF1α interactions. C) Representative example chemical structures of a cyclic peptide proceeding through SICLOPPS selection to produce HIF1α inhibitor cyclo-CLLFVY.285
The earliest use of SICLOPPS to identify an MCP against a target was in 2004 where it was used as a proof of principle to target HIV-1 protease and ribonuclease reductase.280 Since then, it has been adapted for several undruggable target interactions such as BCL6 dimerization,281 IDOL E3 ligase homodimerization,282 the interaction between HIV Gag protein and TSG101,283 between the GTPase Ras and p110α,284 and HIF1α/HIF1β heterodimerization.285,286 In the latter example, an MCP SICLOPPS synthesis and screening strategy was developed where HIF1α was fused with the P22 bacteriophage repressor, and HIF1β was fused with bacteriophage 434 repressor (Figure 21A). In this system, HIF1α and HIF1β heterodimerization, which is an interaction necessary for transactivation and activation of pro-oncogenic gene programs,287,288 controls the 434/P22 repressor binding to 434/P22 operator sites preventing transcription of selection genes necessary for survival. Disrupting this interaction with an MCP results in cell survival and propagation to be later sequenced to determine the MCP identity. After positively screening for activity and negatively screening against false positives and nonspecific inhibitors, they identified a peptide, Tat-cyclo-CLLFVY, that showed activity against HIF1α/β dimerization in two different cancer cell lines, MCF-7 and U2OS.285,289
In another example, Tavassoli and colleagues used SICLOPPS to target dimerization of the C-terminal binding protein (CtBP) transcriptional repressor.290 There are two CtBP proteins, CtBP1 and CtBP2, that can either homodimerize or heterodimerize under high NADH conditions.291,292 In their dimeric form, CtBPs associate with around 30 different transcription factors to recruit histone deacetylases and histone methyltransferase to silence gene expression at specific loci.293,294 CtBP dimerization has been implicated as regulators of highly glycolytic tumor cells.295 Using a bacterial reverse two-hybrid system similar to the previous example, they first probed MCPs that disrupted CtBP1 homodimerization before taking those hits to probe their activity in disrupting CtBP2 homodimerization. They found a cyclo-nonapeptide, CP61, that inhibited CtBP1 and CtBP2 homodimerization both in vitro and in cellulo.290
Although it is uniquely capable of screening for interactions in live prokaryotic or eukaryotic cells, a current limitation with the SICLOPPS approach is that it is limited to the 20 naturally occurring amino acids with a few ncAAs able to be incorporated. Additional future studies should also focus more on targeting diverse undruggable sites with MCPs of varied ring sizes to explore the relationship between size and selectivity.278 Increasing the chemical diversity and library sizes within SICLOPPS technology platforms remain open areas to increase the utility of this approach for undruggable targeting.
2.3. Summary and Outlook of Macrocyclic Peptides for Undruggable Targets
MCPs are well-established therapeutic modalities with desirable pharmacological properties capable of modulating interactions in a variety of undruggable targets. Approaches to design and generate MCPs in silico, in vitro, and in cellulo highlight the versatility for this molecular class. Rational design and selection methods from diverse libraries have produced metabolically stable and cell permeable MCPs. Genetically encoded libraries (GELs) such as mRNA display, phage display, and SICLOPPs are particularly powerful for efficiently screening large compound libraries with reduced time and cost.296 Recent advances in genetic code expansion and late stage functionalization297 greatly diversify library members for selection, such as including reactive electrophiles298,299 and multicyclization.227 Expanding these strategies for MCP generation provides novel avenues for targeting undruggable classes.
For MCPs to translate into therapeutically viable molecules they should bind tightly and specifically to the POI, permeate cell membranes, and retain activity and structure in cells and animals. The different strategies for the design and production of MCPs described have all been used to generate very potent MCPs for undruggable targets, but many are unable to enter cells without significant modification greatly limiting their therapeutic potential. Next steps would be to take these selection strategies and bias sampling toward MCPs that have chameleonic properties. This would increase the odds that MCPs selected would also have good pharmacological properties. An additional shortcoming is that the activity of many compounds found through in vitro methods does not translate directly to in cellulo or in vivo. Either improvements on in cellulo selection methods to incorporate ncAAs or changes in in vitro methods to select for only MCPs that perturb POI activity in a high throughput manner would help to address this issue.
MCPs can also serve a slightly different role: as a warhead for a PROTAC. Most PROTACs developed today use small molecules as warheads; however, a lack of selectivity for intractable targets may lead to off-target effects with potentially devastating responses. As MCPs and other designer peptides exhibit very specific interactions with target molecules, including these as PROTAC warheads could limit off-target effects. Though few MCP or designer peptide PROTACs have been reported,300-302 this is a potentially powerful space to explore, as eliminating an undruggable protein rather than simply blocking function may provide a greater therapeutic effect. Additionally, as MCPs discussed in this review retain peptide-like structure and may be susceptible to proteolytic degradation, efforts to rationally derive nonpeptide based compounds (e.g., class D mimetics) from peptide-based structures may result in improved PROTAC warheads.
Though the primary reason to discover and generate MCPs against targets of interest was to develop them as therapeutics, they can also serve to explore basic biology. As a couple of examples above demonstrate, MCPs are great tools to acutely and specifically disrupt biological pathways to study the biological role of the target without requiring significant external manipulation of the cells. As more MCPs are discovered, they can be used to further elucidate key pathways in disease states or to study how disrupting the formation of a complex affects protein expression and function.
3. SIDE CHAIN STABILIZED SECONDARY STRUCTURES: DESIGNER HELIX AND HAIRPIN PEPTIDES
The hotspot regions involved in protein–protein and protein–nucleic acid interactions are often found within well-defined secondary structure motifs such as α-helices, β-sheets and β-hairpins. In particular, a majority of interactions found in the PDB involve α-helices, which adopt an i, i+4 hydrogen bonding pattern in the amide backbone with an average of 3.6 residues per turn (Figure 22A).303 310 and π helices, which have i, i+3 and i, i+5 hydrogen bonding patterns, are also found in some protein interfaces. β-Sheets also have an extensive internal hydrogen bonding network between the amide backbone of adjacent protein strands that may run parallel or antiparallel. Smaller peptides generally form β-hairpins, consisting of two antiparallel strands where the i, i+2 side chains are oriented in the same direction (Figure 22B).304 Because these secondary structure motifs are common at interfaces, approaches to chemically mimic and stabilize these structures could serve as general methods for chemical probe development.
Figure 22.

Depiction of a typical α-helix, 310-helix, and β-hairpin. A) Top (left) and side (right) representation of an α-helix and 310-helix depicting hydrogen bonding patterns (dotted yellow lines), residue spacing, and helical turn distance. B) Representative β-hairpin showing hydrogen bonding pattern between antiparallel strands (dotted yellow lines).
Early attempts to stabilize peptide secondary structure folds relied on recreating the interactions used in native protein folding, namely hydrogen bonding, ionic and hydrophobic interactions, steric constraints, and covalent disulfide bridges.305-314 These approaches were soon supplanted with the emergence of more stable cross-linking reactions such as lactam formation,315-317 ring-closing metathesis,318,319 and azide–alkyne cycloadditions,320 among many other novel strategies to stabilize secondary structures. Collectively, this class of side chain stabilized secondary structure mimetics has become broadly referred to as “stapled peptides.”
3.1. Design Principles for Secondary Structure Stabilization
Since it is known that certain amino acids have a stronger propensity toward helix formation or β-turn induction, strategic additions and mutations to the peptide sequence can also help to stabilize the desired secondary structure. Namely, Ala, Glu, Lys, Met, and Leu are traditionally considered helix-promoting residues (Figure 22A).321 Conversely, residues like Gly, Pro, Ser and Thr generally disfavor α-helical structures, as Gly, Pro, and other non-natural turn inducing residues such as l-ornithine promote and stabilize β-hairpin structures (Figure 22B).322,323 However, these residues are still useful in the design of stabilized α-helices as they can form kinked helical structures and initiate/terminate helical segments,324 which have been shown to be beneficial for potency in certain contexts. Fuchs et al. demonstrated this in a ribosome display screen of LXXLL peptides selected for binding to ERα and ERβ.325 After several rounds of enrichment, the consensus sequences for both isoforms were capped on either side with Pro, and subsequent investigation revealed that these residues limited the helix length and oriented adjacent charge clamp residues appropriately. Additionally, α,α-disubstituted amino acids reduce rotational freedom due to the potential steric clashes, which can contribute to overall rigidity. α-Aminoisobutyric acid (Aib) is a disubstituted amino acid first discovered in some fungal peptides, and its geminal methyl groups contribute to a steric effect that strongly promotes both 310 and α-helix formation (Figure 22A).326,327 Its achirality also makes it suitable for inclusion in either l- or d-peptides. The α,α-disubstituted, olefin-containing amino acids used in hydrocarbon stapling (vide inf ra), S5 and R8, have a similar helix-inducing effect.319,328
The formation of secondary structure generally helps to overcome the pharmacological pitfalls of poor cell permeability and proteolytic stability. A combination of hydrophobicity and positive charge, particularly in amphipathic helices, broadly correlate with better cell uptake, but beyond a certain threshold, these properties also lead to membrane disruption and cell lysis.56,329 The presence of intramolecular hydrogen bonding, which typically shields the polar amide backbone from aqueous solvent interactions, makes entry into the lipid membrane more energetically favorable, so long as pendant side chains are appropriate for interaction with and diffusion through lipid bilayers.330 Balancing these properties is an area of great importance for designing and developing membrane permeable secondary structure mimetics for undruggable targets, as discussed above with MCPs. Consideration of these properties will be important in the discussion of exemplar stapled peptides and other stabilized mimetics in this section. Helix and hairpin motifs by definition have these internal hydrogen bond networks, and successful introduction of stabilizing mutations through rational structure optimization takes these patterns into account by incorporating constraints in positions that promote the desired hydrogen bonding pattern and “locking” the peptide into its proper conformation. This also reduces the exposure of the backbone to degradation by proteases and other metabolic enzymes. The amino acid positions that are covalently linked are generally ones that will be proximal to each other in 3D space when the peptide is folded in the desired conformation. For helical peptides, the most common staple positions are i, i+4 and i, i+7, as these positions span one and two turns of an α-helix, respectively (Figure 22A). For a hairpin structure, the staple is usually positioned across the two antiparallel strands or in some cases, across the termini to clamp the two strands together.331 However, there are important caveats to consider when making amino acid substitutions and selecting staple positions. For example, while the introduction of d-amino acids generally confers proteolytic stability, as enzymatic cleavage is stereospecific for l-amino acids, d-amino acids may also destabilize the helices of predominantly l-peptides.332 There is also a distinction between helix stabilization and introduction of structural rigidity. For example, a hydrogen exchange study of a small peptide library with i, i+4 hydrocarbon staples in various positions concluded that solvent exchange rates, which are directly correlated to the conformational freedom and solvent exposure of the amide protons on the backbone, were a better predictor of protease resistance than more classic measures of helicity.333 This highlights the fact that structural stabilization alone may not be enough to achieve ideal pharmacological properties, and side chain stabilization chemistry and placement must be carefully considered through iterative design and testing. We will consider these and other important elements of design and pharmacologic use for stabilized peptides throughout this section.
3.2. Early Side Chain Stabilization Strategies for Secondary Structure Mimetics
Early studies with model peptides and proteins demonstrated that properly positioned salt bridges could stabilize helical peptide stretches in solution. For example, studies with a short helical peptide derived from RNase A only exhibited helical character at the pH in which a protonated His residue at the i position interacts with the Glu carboxylate at the i+4 position, effectively stabilizing one helical turn.305 Similarly, attempts to form β-hairpin structures often used model peptides that normally adopted this conformation in the protein. Many early structural NMR studies and computational models reached the conclusion that the hairpin is nucleated by the presence of a β-turn that often utilizes specific residues such as Asn, Pro, and Gly.314,334,335 Other early studies recognized the potential for metal chelation on specific helical faces or between helix-turn-sheet motifs commonly found in zinc-finger domains, to be highly effective at nucleating and stabilizing peptide secondary structure.307,308,336-338 Another early stabilization chemistry explored was natural intra- and interchain covalent stabilization via disulfide bond formation. The short cysteine side chains are more practical for hairpin and loop structures, but some examples with i, i+3 spacing between d-Cys and l-Cys have been reported.339-341 In 1991, Jackson et al., reported a series of peptides with two orthogonally protected 2-amino-6-mercaptohexanoic acid residues at i, i+7 positions that were oxidized to form the disulfide in an aqueous solution (Figure 23A).309 CD spectroscopy confirmed an increase in the α-helical character for all peptides regardless of length. While the disulfide bridge spanned two helix turns in this system, it was shown to nucleate helix formation for longer sequences.
Figure 23.
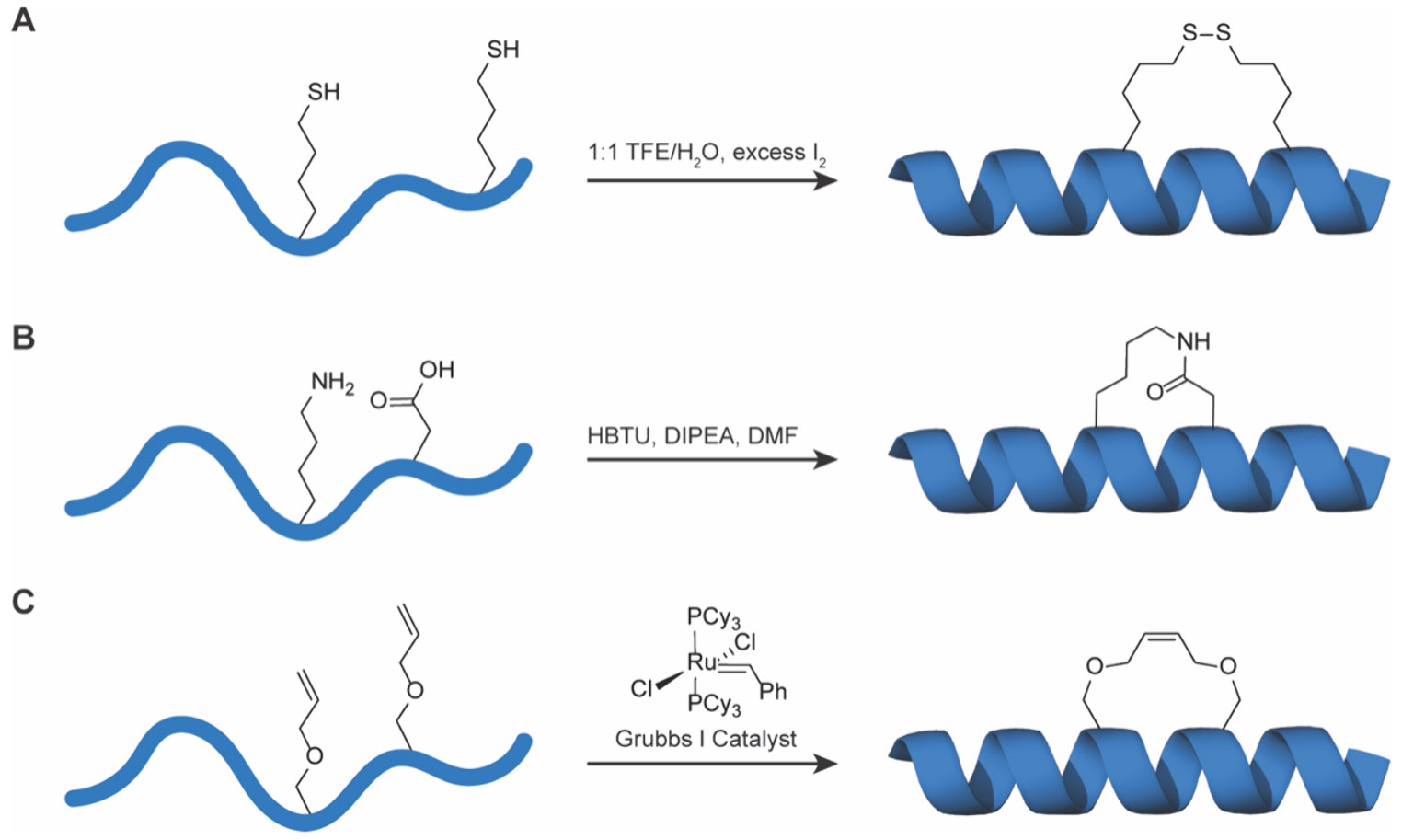
Early examples of helical stapling. A) Oxidation to i, i+7 disulfide bridge reported by Jackson et al.309 B) General scheme for side chain lactam formation. Any standard coupling reagent and base typically used in SPPS workflows can be used. C) Early example of olefin metathesis to stabilize 310 helix formation by Blackwell, Grubbs, and colleagues.318
Lactam formation between amino acid side chains had been extensively applied to macrocycle synthesis315 specifically used in early helix stabilization studies, usually forming amide bonds between Asp and Lys, or Glu and Lys, with the capability to form multiple staples using orthogonal protecting groups (Figure 23B).316 Phelan et al. took an alternate approach to this chemistry by using two acidic residues bridged with diamine linkers of variable length,317 which permitted exploration of the effect of different staple lengths on helical structure. Shorter methylene bridges resulted in some helical bending, while longer bridges allowed more conformational freedom leading to transient unwinding of the helix. While lactam staples are more stable against redox conditions in different cell compartments, neither it nor disulfide staples are completely biorthogonal and can be reversed in biological environments.
Lactam staples are still widely used for the development of peptide ligands for undruggable targets. For example, Wang et al. designed a stapled peptide derived from the histone H3K56 acetylated peptide site, which is found within an extended α-helix.342 They recapitulated a 12-residue stretch of this helix to contain a chelating hydroxamic acid warhead to target the HDAC active site Zn2+, and introduced a lactam staple between l-isoaspartic acid and 2,3-diaminopropionic acid as substitutes for the common Asp-Lys pair, a strategy they previously reported and validated in the context of ERα targeting.343 Molecules derived from these stable histone variants demonstrated antitumor activity in PA-1 xenograft models with intraperitoneal injection. Intriguingly, this lactam-stapled peptide outperformed vorinostat, an FDA-approved small molecule HDAC inhibitor, and did not exhibit toxicity.342 Other similar examples include lactam-stabilized helical fusion inhibitors for RSV.344
3.3. β-Hairpin Stabilization
A common motif that enables PPI formation is a β-hairpin or a β-turn, and like the MCPs designed via rational design discussed in Section 2.3.1, these motifs can be identified and cyclized to create β-hairpin peptides with programs such as LoopFinder.80,81 One of the common approaches toward designing β-hairpin peptides begins with identifying the key binding residues or a binding epitope and then grafting it onto a hairpin scaffold, where the signature turn is usually formed by a d-Pro-l-Pro segment or other turn-inducing structure, and the antiparallel strand may have a variable sequence that can be optimized as needed. This step is not entirely modular, as the introduction of the rigid turn may lead to backbone conformations that are not favorable to binding.345,346 The loop is often closed using one of the aforementioned crosslinking chemistries (Figure 2). The original binding epitope does not necessarily have to be a hairpin; for example, the key residues on the binding face of a helix can be strung together as one of the strands of the β-hairpin, leading to the same presentation of side chain interactions in 3D space as the original helix.
Hairpins generally rely on specific turn inducing templates that may include covalent cross-links to stabilize the structure.347-350 While it is known that the classic hairpin can be formed by introduction of a d-Pro-l-Pro segment, the introduction of the rigid turn may lead to backbone conformations that are not favorable to binding, so it is often important to sample multiple alternatives for the turn inducing residues in order to result in the ideal conformation, followed by staple incorporation. In one example reported by Pace et al., 4-mercaptoproline was cross-linked to another cysteine via meta-dibromomethylxylene to form an antiparallel β-hairpin structure that proved to be more resistant to proteolytic degradation compared to its unmodified counter-part.351 This peptide, 4MP-m was computationally designed, using a novel “staple-first” approach. After identifying a set of cross-links that were most compatible with the desired β-hairpin structure, they introduced the turn and grafted on the rest of the sequence. The resulting hairpin peptides exhibited a higher degree of ordered structure than the parent sequence. The crucial hydrogen bonding interactions should also be considered during the design process and may alleviate the need for a natural or synthetic β-turn altogether, as recently reported by Nazzaro et al. using a hydrogen bond surrogate (HBS) approach.352 However, natural hydrogen bonds within the turn may provide stability to the structure, as was the case in a report of a ULM peptide, a spliceosome assembly inhibitor, which naturally has a hydrogen bond between Glu340 and Arg337 residue supported by the Ser336 residue forming a hydrogen bond with Trp338 to form the β-turn. Therefore, these residues were left intact, while an adjacent Lys side chain was used to lactamize the ring with Glu to form the complete hairpin.353 Another hairpin example by Wendt et al. targets β-catenin/E-cadherin using dithiol and increasingly longer thioether linkers to stabilize a hairpin derived from E-cadherin.354 The structure–activity investigation through a series of FP assays revealed that the longest linkers showed higher inhibition, but only in certain positions along the hairpin, suggesting that not all staple positions are equally conducive to hairpin stabilization and binding affinity. In another example for this target, Blosser et al. report a β-hairpin targeting β-catenin/TCF4, using a Gly-Pro turn template and closing the loop with a thioether linkage.350 Computational modeling was used to select a linear strand that mimics TCF4 while the opposite strand is present for stabilization of β-hairpin structure. Because β-catenin has Cys residues near the TCF4 binding interface, it was possible to make a covalent peptide inhibitor using electrophilic groups on a Dap residue. Strømgaard and colleagues used a similar d-Pro-Gly turn closed with native chemical ligation to generate an nNOS/PSD-95 inhibitor.355 A positional heat map scan revealed which residues were most conducive to binding, followed by additional studies to determine binding mechanism. The resulting constrained hairpin exhibited nanomolar binding affinity and may have the potential to outperform linear peptides that are already in clinical development for targeting this interaction.356
A key example of a β-turn motif mediating recognition is in the interaction between KEAP1 and NRF2.217 Since the discovery of this DEETGE loop, many research groups have developed cyclized β-hairpins capable of interacting with KEAP1 with high affinity.357-360 However, most designs suffer from a lack of activity in cells due to low uptake. A general approach to improve permeability and activity is to reduce the size of the peptide required to bind to the target site. Ortet et al. examined the minimum structural motif required for a cyclic β-hairpin peptide to achieve the same affinity as wild-type NRF2, which has low nanomolar dissociation constants.361 Further studies suggested that the terminal residues only serve to stabilize a favorable conformation, but head-to-tail cyclization of a 7-mer, based on residues 76–82, resulted in a 10-fold decrease in binding. The addition of a β-amino acid, d-β-homoalanine, at residue 76 to expand the β-hairpin cycle, along with a E78P mutation that leads to better preorganization resulted in a lead compound 23, which closely mimics the conformation of the NRF2-derived peptide (Figure 24A,B). Similarly, Lopez et al. performed all-atom, explicit-solvent molecular dynamics simulations on the CDEETGEC sequence cyclized using various thioether-forming linker molecules for binding to KEAP1.362 To improve cell uptake of the NRF2 cyclic β-hairpin, Legre et al. studied various factors, including appending a CPP, appending a fatty acid chain, shortening the peptide, and masking the net negative charge.363 They found that by shortening the peptide from an 11-mer to a 9-mer and attaching a fatty acid tag induced transcription of the antioxidant response element (ARE) with an EC50 of 740 nM in BEAS-2B cells.
Figure 24.

β-turn stabilized peptides for targeting KEAP1. A) Chemical structure of compound 23. B) Crystal structure overlay of compound 23 (blue; PDB: 7K2S) and the natural loop of NRF276-84 (yellow; PDB: 2FLU) bound to KEAP1 (cyan).361
Beyond proteins, stabilized β-hairpins can target numerous biomolecular surfaces including nucleic acids. Initial studies targeted the transactivation binding response (TAR) RNA, which is found in the 5′ untranslated region (UTR) of mRNA of viruses such as HIV and bovine immunodeficiency virus (BIV).364 This loop interacts with a β-hairpin located in the Tat protein, and this β-hairpin was first stabilized to bind BIV TAR RNA in 2001 by Tok et al.365,366 Additional studies examined HIV TAR RNA and designed stabilized β-hairpin peptides that achieved subnanomolar binding affinity.346,367-370 Another way to design stabilized β-hairpin peptides are to employ evolved RNA-recognition motifs (RRMs) in a yeast display library using U1A RRM, a protein that contain multiple β-sheets, as a starting point.371,372 This approach has yielded many stabilized β-hairpin peptides against HIV TAR RNA, though the best β-hairpin peptides designed using this method has a dissociation constant of 800 nM, 3 orders of magnitude worse than that of Shortridge et al.373-375 Stapled β-hairpins have also been developed for miRNAs.376 Shortridge et al. first reported a β-hairpin mimic for targeting pre-miR-21.377 After screening a panel of existing stabilized β-hairpins, they identified a compound, L50, that bound to pre-miR-21 with a dissociation constant of 200 nM, which is still the strongest binding observed by a stabilized β-hairpin against an miRNA (Figure 25A). The crystal structure of L50 binding to pre-miR-21 was solved, which shows that the arginine residues (Arg3, Arg5, Arg8, Arg12) bind to the anionic miRNA backbone, whereas Dpr13/Pro14, along with Gly6/Lys7, form the β-turns (Figure 25B,C). L50 was shown to inhibit miR-21 formation both in vitro and in cells. Other miRNAs, such as pre-miRNA-20b, have also been targeted with stabilized β-hairpins, though with relatively weak affinity.378 A deeper examination of stabilized β-hairpins binding RNAs, along with other stabilized structures binding RNAs, have been covered in this excellent review by Pal and ‘t Hart.379
Figure 25.

β-Hairpin stabilized peptides targeting RNA. A) Chemical structure and binding properties of L50. B) Crystal structure of L50 (blue) bound to pre-miR21 (gray). PDB: 5UZZ. C) Insert highlighting the key residues for interacting with RNA (Arg3, Arg5, Arg8, and Arg21) or forming the β-turn (Dpr13/Pro14, Gly6/Lys7).377
3.4. Stabilized Helical Secondary Structures Using Hydrocarbon Stapling
A significant breakthrough in helical stabilization came with the application of ring closing metathesis for peptide stabilization as it introduced a stable, nonpolar stabilizing cross-link that could improve cellular uptake and activity of stapled peptides. Blackwell and Grubbs first reported the use of the ring closing metathesis (RCM) reaction to stabilize peptide helices in 1998, where they incorporated serine or homoserine O-allyl ether residues (Figure 23C).318 The sequence, even prior to substitution with the olefinic residues, showed helical character, likely due to the prevalence of amino acids with high propensity for helix formation. An X-ray crystal structure suggested a predominantly 310-helix conformation, due to the i, i+3 hydrogen bonding pattern and the torsional angles. While the helical character showed little change with substitution and metathesis, the bioorthogonality and hydrophobic nature of the cross-link was an attractive strategy to stabilize α-helical conformations.
Schafmeister et al. further investigated the potential of RCM for peptide stabilization by using disubstituted amino acids with both R and S configuration and various side chain length to determine which combinations would give the best yield as well as the most stabilizing effect on helix formation (Figure 26).319 Within a specific model system, they discovered that for i, i+7 staple placement between R- and S-amino acids, at least a 9-carbon cross-link was required to observe appreciable conversion of an RNase A derived peptide to the stapled product. For i, i+4, using Ri, Ri+4 and Si, Si+4 α-methyl amino acids, 8 carbons was the minimum length required for near complete conversion. From this library they concluded that an i, i+7 staple with R,S paired configuration and an 11-carbon cross-link resulted in the greatest increase in helicity, followed by the R,S paired i, i+7 staple with 12-carbon cross-link and S,S paired i, i+4 staple with 8-carbon cross-link.380 The proteolytic stability of these peptides was assessed using trypsin, and it was observed that the unstapled, disubstituted amino acids alone conferred some resistance, reducing cleavage rates 5-fold, which increased to 41-fold after metathesis.319 The metathesis reaction can result in a mixture of cis and trans isomers that will have differential effects on peptide structure and biological activity,381 though the general rules that govern this are not well understood.
Figure 26.
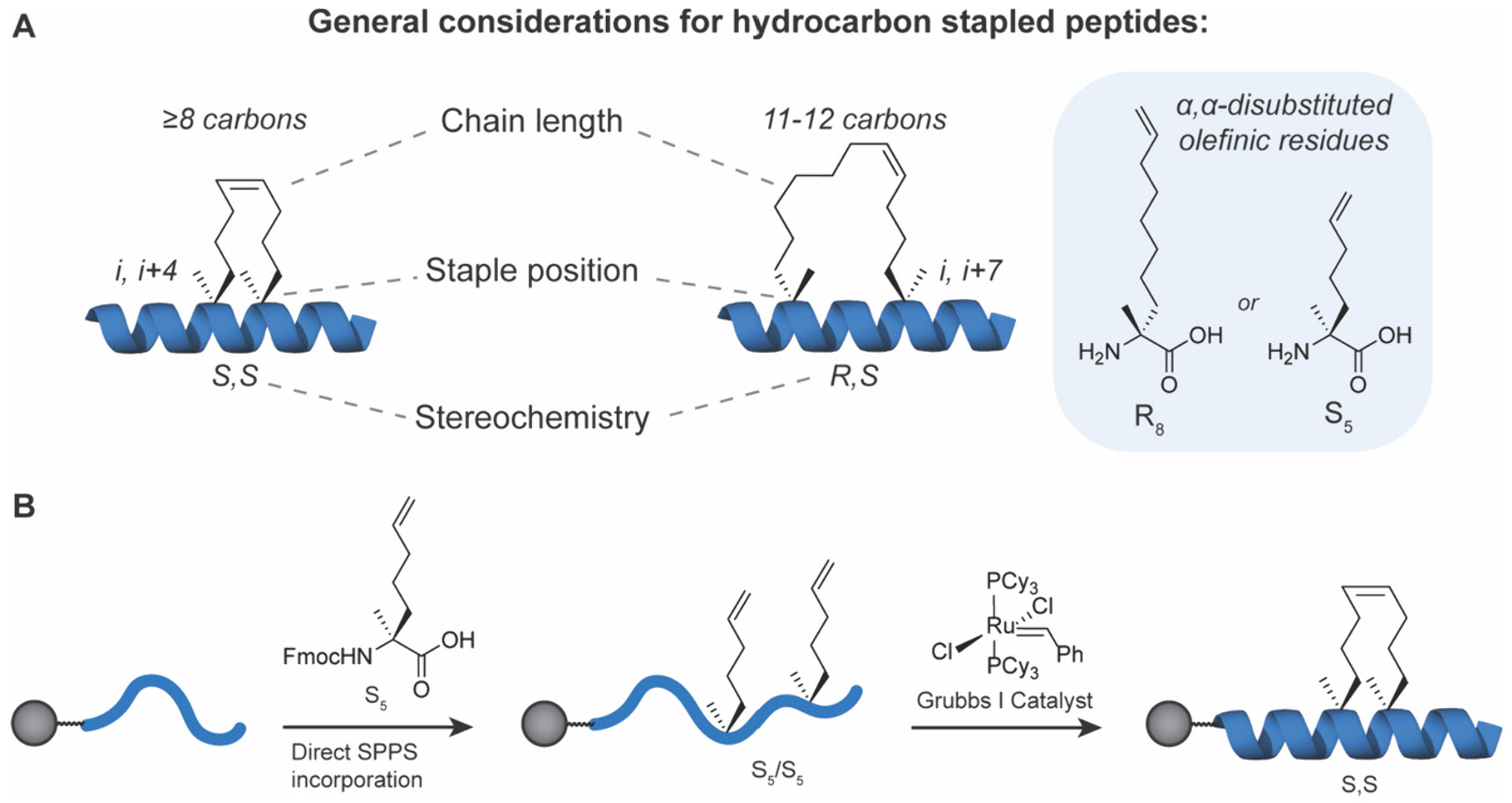
Design of bis-alkylated, hydrocarbon stapled peptides. A) Chain length, staple position, and stereochemistry are key considerations for generating helical hydrocarbon stapled peptides. B) Schematic of peptide functionalization using ring closing metathesis.
The initial protein family that was targeted using hydrocarbon-stapled peptides was the BCL-2 family, known for its central role in regulating apoptosis and ensuing importance in cancer.197-200 Within this family, the pro-apoptotic members contain a conserved BH3 α-helical domain required for binding to other BCL-2 members and subsequent initiation of the apoptotic cascade. This BH3 domain served as the template for the development of stapled peptides targeting various proteins in this family.382-388 In 2004, Yang et al. reported a lactam stapled peptide binder derived from BAK, a BCL-2 family member, and Walensky et al. described the development of a hydrocarbon-stapled peptide derived from the BH3 helix of BID, another member of the BCL-2 family, that bound BCL-XL and has potential to broadly interact with other BCL-2 family members (Figure 27A).389-391 In the case of the BAK-derived peptide, the i, i+4 lactam staple provided moderate helix stabilization, up to 56% in PBS.390 For the peptide derived from BID, a panel of analogs with varying positions of the i, i+4 olefin staple, including one with an i, i+7 staple, were synthesized and characterized for their helical characteristics, proteolytic stability, and binding affinity for BCL-2. All showed an improvement in helicity compared to the linear wild-type sequence. A lead compound, SAHBA with 87% helical content, exhibited significant improvements in serum half-life and showed evidence of triggering cytochrome c release for apoptosis in vitro. Additionally, it was shown to be cell-permeable via endocytosis to a much greater degree than its linear counterpart. Importantly, SAHBA suppressed growth of leukemia xenografts in vivo over the course of 7 days of treatment following IP administration, demonstrating for the first time that a hydrocarbon stapled peptide could be a viable therapeutic modality.384 Although the family wide BH3 binding motif is generally conserved among family members, selectivity can be attained by careful selection of the nonconserved residues. For example, a selective MCL-1 inhibitor based on the MCL-1 BH3 domain was designed by considering the unique residues present in MCL-1 and determining which mutations would abolish binding to other BCL-2 members. The MCL-1 BH3 domain contains a hydrophobic triad consisting of Leu213, Val216, and Val220, which are all crucial for binding to MCL-1 itself (Figure 27A-C). Treatment of cells with an MCL-1 designed SAHB in conjunction with other death receptor agonists showed that there was sensitization to the apoptotic cascade. The significance of this lies in the fact that MCL-1 overexpression is correlated to resistance against current drugs like Venetoclax that target the BCL-2 proteins, thus making selective and potent anti-MCL-1 inhibitors highly desirable. Following these initial studies, several groups reported BIM-derived sequences that also had selective binding for MCL-1, and Araghi et al. further modified these with i, i+4 and i, i+7 RCM staples to determine optimal positioning. Two of their compounds, SAH-MS1-14 and SAH-MS1-18 had binding affinities of 80 and 25 nM respectively. They verified that the lead compounds were not membrane lytic before proceeding to cell studies where they showed that treatment with SAH-MS1 peptides was only cytotoxic for MCL-1-dependent lines, despite the observation that SAH-MS1-14 also activated BAK to a certain extent.388 These stapled peptides have been applied for structural studies of the BCL-2 proteins as well. A BID SAH peptide binding to BAK was used as a tool for studying its activation mechanism, including NMR domain mapping and photo-cross-linking mass spectrometry.386,392
Figure 27.

Stapled peptides derived from BH3 domains. A) Primary sequences of peptides derived from BAK, BID, and MCL-1 showing staple positions in designer peptides. B) Representative helix from MCL-1 SAHBD. Hydrocarbon staple is highlighted in yellow, and key interacting residues are shown. C) Crystal structure of SAHBD bound to MCL-1 with interacting residues labeled. PDB 3MK8.384
It must be noted that simply introducing the RCM staple does not always lead to the desired properties. Across the panel of stapled peptides synthesized by Walensky and colleagues, some only had a slight increase in helical character, and not all showed good cell permeability.385 Monte Carlo folding simulations suggest the existence of “decoy” states, where the peptide is partially folded or bent in a manner inconsistent with its natural structure in the binding domain.393 This also explains why i, i+7 staples are only sometimes more beneficial than the i, i+4 for helix formation: the effects of the hydrocarbon staple are highly context dependent. The tradeoff between the residues being mutated out and the new olefin side chains must be considered with regards to its effect on overall charge, polarity, and the potential loss of existing helixstabilizing interactions. Because of the need for extensive structure optimization with RCM stapled peptides, there have been many studies with hydrocarbon peptides that attempt to compile a general list of guidelines that would shorten lead time toward successful compounds.329 Bird et al. report their findings from a library of BH3 hydrocarbon stapled peptides, which included several observations that the peptides that exhibit high cell uptake generally contain properties in common such as (i) the hydrophobic staple is located along the border of the hydrophilic and hydrophobic face of the helix, (ii) overall high degree of hydrophobic character between 61 and 86% and (iii) pI between 8.8 and 9.34. Earlier, Chu and Moellering et al. used a quantitative, high-throughput imaging approach to assess the productive permeability and distribution of approximately 200 stapled peptides from diverse target proteins.56 This study showed that peptides with net positive charge, though not hypercharged like many CPPs (e.g., Tat, Penetratin, and octa-arginine), were the most productively cell permeable and displayed distribution throughout cells. This was staple and context dependent and showed that much more penetrant and potent stapled CPPs could be developed, but that introducing staples alone was not always sufficient to improve cell uptake. Of course, the conclusions from these studies provided some general guidelines for active cellular uptake, but there are other design principles that must be balanced or even orthogonal to attain passive permeability.
Another example of a hydrocarbon stabilized peptide targeting a PPI in an undruggable protein involves targeting the NOTCH/CSL transcriptional complex. The Notch pathway involves activation of NOTCH transmembrane receptors (NOTCH1–4) that are cleaved in response to ligand binding, releasing its intracellular domain ICN1 that binds the transcription factor CSL and subsequently coactivator MAML, promoting downstream gene expression.394-396 Domain mapping studies identified an extended stretch of the MAML1 N-terminus that acted in a dominant negative fashion to block MAML1 binding and transactivation by ICN/CSL complexes.397 Structural studies by Nam et al. showed that this dominant negative stretch formed an extended α-helix that bound to the interfacial groove formed by ICN1/CSL398 (Figure 28). Moellering et al. designed a series of stapled peptides derived from various segments of dnMAML1 and utilized hydrocarbon staples with i, i+4 spacing to stabilize the α-helical conformation.396 While all stapled peptides showed an increase in helicity, not all were cell permeable or strong binders of the ICN1/CSL dimer. A lead compound, SAHM1, showed significant inhibition of NOTCH1-dependent gene expression in T-cell acute lymphoblastic leukemia (T-ALL) cells, which was further established by an early example of using Gene Set Enrichment Analysis (GSEA) for specificity analysis. Furthermore, SAHM1 inhibited NOTCH1-dependent T-ALL cell lines, but not other lines with known resistance to NOTCH inhibition. Finally, SAHM1 demonstrated inhibition of systemic T-ALL xenografts in mice, further suggesting that stapled peptides could be viable in vivo regulators of TF activity and that they could be developed to target protein interfaces.396
Figure 28.
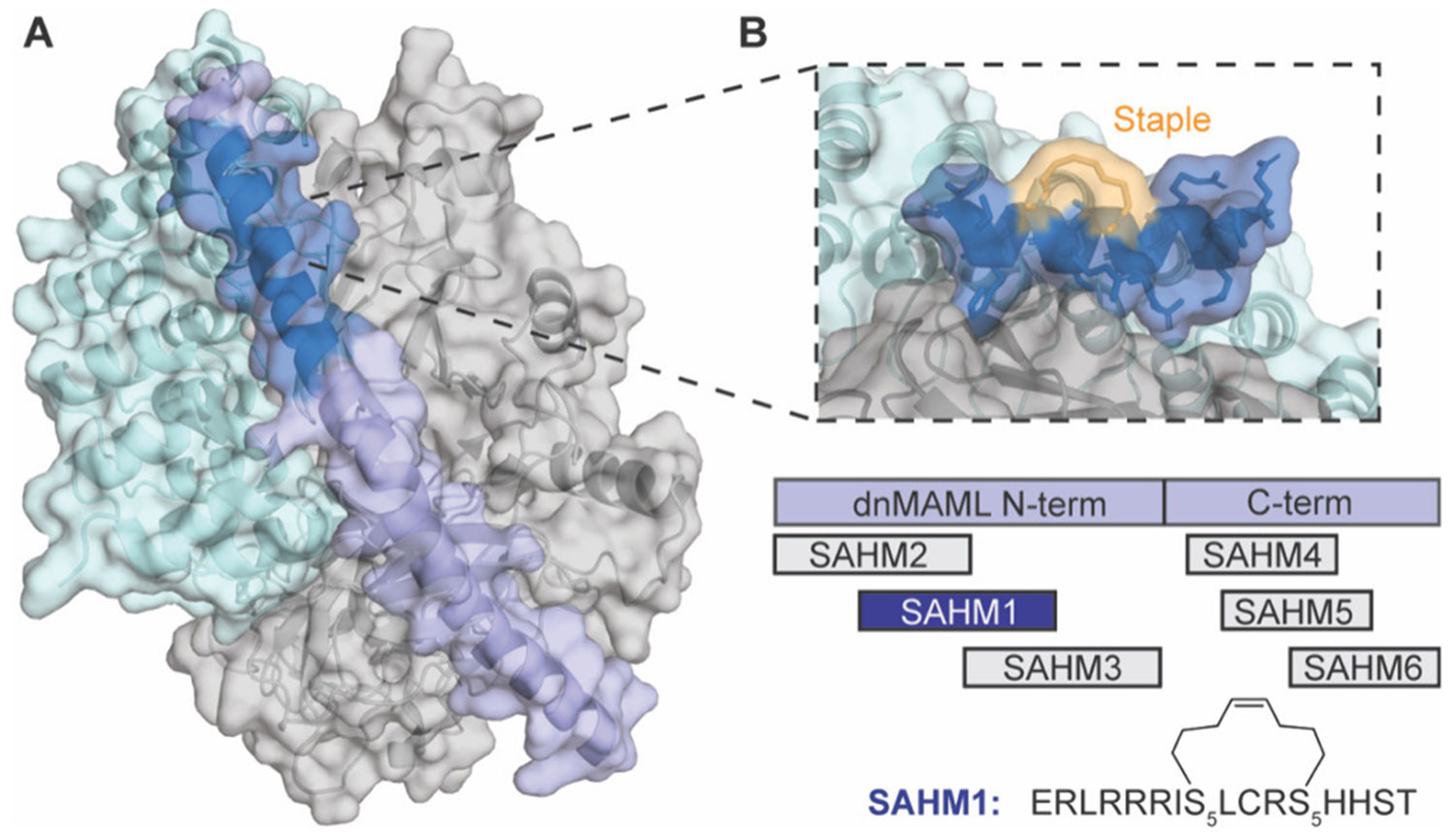
Design of stapled peptide NOTCH inhibitors. A) Crystal structure of the NOTCH1 ternary complex: CSL (gray), dnMAML1 (cyan), ICN1 (purple), MAML1 interacting domain (blue). PDB: 2F8X. B) Model of stapled peptide SAHM1 (blue) overlaid on CSL (gray) and dnMAML1 (cyan). Staple is shown in yellow. Underneath is a representation of the primary sequences of designed SAHM peptides against dnMAML1 and the primary sequence of SAHM1 showing the staple position.396
Another notable undruggable transcriptional regulatory pathway targeted by hydrocarbon stapled peptides is the WNT/β-catenin pathway. β-catenin is a central mediator of transcriptional regulation downstream of WNT signaling and does so via interactions with several proteins like BCL9, Axin and TCF transcription factors. Grossmann et al. directly targeted β-catenin using a series of Axin-derived peptides stabilized by hydrocarbon staples in both i, i+4 and i, i+7 positions and evaluated their helicity and binding affinity with CD spectroscopy and FP assays, respectively.399 A unique aspect of this effort was the integration of phage display screening to identify privileged consensus sequences to serve as starting points or optimizations of the WNT-Axin-derived sequences (Figure 29A). Improvements were made to initial stapled peptide leads by increasing the overall positive charge of the peptide by mutating nonessential positions of Asp or Glu and introducing Arg. Taking the staple design that worked best from the first rationally designed set of peptides, they stabilized the phage display-derived peptides (StAx peptides; Figure 29A) and showed cellular uptake by confocal fluorescence imaging. Several StAx peptides, including a lead StAx-35, inhibited TCF4-driven and endogenous target genes in colorectal cancer cell lines; however, these effects were not mediated by changes in β-catenin expression. This study also solved a cocrystal structure of StAx-35 peptide bound to β-catenin, highlighting conserved register within the Axin-targeted groove as well as new contacts introduced by phage-derived mutants (Figure 29B). Beyond Axin, several groups have reported stapled peptides derived from the BCL9 protein, which binds a unique site on the β-catenin armadillo repeat domain.400
Figure 29.

Hydrocarbon stapled peptide inhibitors of β-catenin selected from phage display. A) Schematic of phage display to select for consensus sequences of peptide ligands that are later stabilized by stapling. B) Crystal structure of StAx-35 peptide (blue) bound to β-catenin (cyan). Inset shows key interacting residues and staple (yellow).399
Perhaps the most studied “undruggable” target for which stapled peptides have been developed is tumor suppressor protein p53 and its negative regulators.23 As one of the most mutated proteins in cancer and therefore desirable targets in oncology, many approaches have been taken to prevent suppression of p53 protein levels and transactivation by oncoproteins like the ubiquitin ligases MDM2 and MDMX. In 2007, Bernal et al. reported the development of SAH-p53-8, an α-helical peptide stabilized using RCM, derived from the MDM2 binding region of p53.401 Since the initial designs were not cell permeable, they mutated negatively charged residues and specific residues known to contribute to ubiquitinylation or nuclear export, namely K24 and L14. The lead compounds were shown to bind MDM2 with high affinity and reactivate p53 expression. Three years later, it was shown that SAH-p53-8 was a dual inhibitor of MDM2 and MDMX, overcoming the limitation of Nutlin-3, a small molecule p53 activator that only targeted MDM2, and were therefore ineffective in MDMX overexpressing cancer models.402 The emergence of crystal structures and phage display libraries yielded other potential peptide sequences that could disrupt binding of p53 and MDM2, which could then be structurally stabilized using a variety of chemistries.403-406 Brown et al. used the crystal structure of p53 bound to MDM2 as well as the phage-derived sequence, PMI, reported by Pazgier et al. to design stapled peptides with the help of computational simulations to guide further mutations.405,407 Their lead compound, sMTide-02, later called PM2, showed p53-activation in T22 reporter assays, outperforming SAH-p53-8 in the presence of serum. It also showed good potency in viability assays benchmarked against both Nutlin and SAH-p53-8.407 In mouse models, PM2 was tested in combination with radiotherapy and it was discovered that this regimen prolonged survival of HCT116 tumor bearing mice in a synergistic manner without adverse effects observed in treatment with small molecule MDM2 antagonists.408
Another phage display-derived peptide, pDI, became the core of perhaps one of the most successful stapled peptides and the first one to reach clinical trials, sulanemadlin.404,409 While initial attempts were built around the SAH-p53-8 sequence, separate introduction of i, i+7 staples into the pDI sequence obtained from phage display by Hu et al. afforded significant increases in binding affinity. Notably, pDI preserved the Trp/Leu/Phe core binding residues but nearly every other residue was different from the native p53 sequence, which shows the advantage of peptide libraries in offering unexpected solutions that are often overlooked during rational design. Additionally, solubility and cell permeability were improved with the introduction of noncanonical amino acids and C-terminal extensions. The lead compound, termed ATSP-7041, was able to suppress tumor growth with a daily 15 mg/kg dose in SJSA-1 xenograft models, which has high expression of MDM2. Additional pharmacokinetic studies in mice, rats, and monkeys all showed a relatively low rate of clearance.410 Before moving to clinical trials, some non-natural amino acids were reverted to the natural residue as a practical consideration for larger scale synthesis and a polyalanine tail was appended to the C-terminus for extension of the α-helix. This modification improved solubility as well, which was one of the disadvantages of the precursor ATSP-7041. The new and improved ALRN-6924, also called sulanemadlin, was found to be metabolized in vivo, via proteolytic cleavage of the polyalanine tail (Figure 30).409,411 However, this metabolite was a 10-fold more potent binder of MDM2 and MDMX but was less cell permeable when used directly in cell proliferation assays leading to lower efficacy. Thus, ALRN-6924 functions as a cell permeable precursor drug that is metabolized to a more potent form in situ. ALRN-6924 was shown to have chemoprotective effects at low doses by inhibiting DNA synthesis and reduced cell viability at higher doses. In numerous clinical trials it also had favorable clearance rates and safety profile.409 Ultimately, the chemoprotective effects and prevention of cancer progression was not statistically significant and consistent efficacy was not observed, leading to the end of the sulanemadlin development. The potential benefits of stapled peptides, namely, the excellent safety profile and stability compared to larger biologics, were clearly demonstrated and serve as a promising first attempt for stapled peptide translation to patients. Other interesting attempts to protect p53 have emerged, such as all-D stapled peptides, where the original sequence was derived from the PMI sequence,412 and single, double, and stitched RCM staples were introduced in various positions and assessed for helicity, binding affinity, and p53 activation, with some of the analogs achieving comparable results to ATSP-7041, validating the “left-handed” approach to creating selective and highly stable peptide therapeutics.413
Figure 30.

Hydrocarbon stapled peptide inhibitors of p53/MDM2. ALRN-6924 behaves as a cell permeable prodrug that is metabolized to more potent ALRN-8714 in cells. Crystal structure of ALRN-6924 in complex with MDM2. Inset shows key residues F19, W23, and L26, as well as staple (yellow) forming hydrophobic contacts with MDM2 surface. PDB: 8GJS.409
Initially developed by Arora, ring closing metathesis has also been used as a hydrogen bond surrogate (HBS) to stabilize α-helical peptides in many notable examples.414-417 While technically distinct from side-chain stabilization, the HBS approach uses orthogonal chemistry to mimic the N-terminal hydrogen bonding pattern and nucleate α-helical structure. As such, it is powerful alone or in combination with other layered stabilization chemistries (discussed further below). For example, Arora and colleagues designed a stabilized helical peptide that mimics the p300/CBP coactivator of HIF1α with the purpose of inhibiting HIF1α driven transcription.415,416 The construct was designed to disrupt the HIF1α-p300-CH1 interaction and subsequently downregulate hypoxia driven gene expression. It was stabilized using the hydrogen bond surrogate strategy, cross-linking a terminal 4-penetenoic acid to an N-allylalanine residue two amino acids away via RCM.416 The p300/CBP coactivator regulates other transcription factors as well, so future work will likely involve building in specificity for a narrower mechanism of action. In one example against HIF1α, it was shown that 1 μM of the constrained peptide derived from CH1 domain of p300 could decrease VEGFA levels comparably to 200 nM chetomin, a molecule previously shown to antagonize HIF1α activity. Interestingly, the peptide was not cell permeable until the addition of an Arg residue on the C-terminus, which both improved helicity and permeability, presumably because of a potential salt bridge between it and a Glu i+4 residues away that further aided helix folding.415
In a separate study targeting Ras, Patgiri et al. designed an α helical mimic of the interacting αH motif using an olefin metathesis linked HBS between i, i+4 residues on the N-terminus.418 Nonessential residues were mutated to optimize solubility and improve helix propensity. The resulting compound had only moderate helicity but was shown to slow the Ras nucleotide exchange of GDP to GTP in vitro. NMR titration studies showed shifts for residues known to be part of the binding interaction and fluorescence microscopy confirmed cellular uptake. They also found that addition of the constrained peptide to HeLa cells attenuated ERK phosphorylation, which is triggered by Ras activation.418 The activation of Ras is mediated by the SOS protein, which interacts with Ras via an α-helical hairpin motif. Another stapled peptide targeting the Ras-SOS interaction using the same helix template was reported by Leshchiner et al., also using olefin metathesis but through side chain cross-linking rather than the HBS approach.419 They showed that their lead compound SAH-SOS1a, which had a central i, i+4 hydrocarbon staple, was able to bind both wild-type and mutant K-Ras, in both the GDP-loaded and GTP-loaded conformations. When incubated with K-Ras concomitantly with a fluorescent GTP analog, SAH-SOS1a inhibited GTP binding. They also showed that treatment with SAH-SOS1a reduced phosphorylation of downstream proteins both in cells and in vivo. These examples show the potential of targeting this PPI for broad applicability to the various mutant K-Ras variants that make it so difficult to drug by traditional means.
Beyond Ras family proteins, small GTPases represent a large, challenging family of proteins that are commonly considered undruggable. The first application of stabilized peptide approaches to a RAB-family small GTPase, RAB8a, was derived from the R6IP protein. An early inhibitor, StRIP3, was developed and bound RAB8a with a Kd of approximately 0.03 mM.420,421 It was not proteolytically stable or cell permeable in its linear form, so an alanine scan was performed to assess which positions would be suitable for mutation to stapling residues. Additionally, an arginine scan was completed to identify compatible positions to introduce positively charged residues. While nearly all positions were sensitive to the mutations, the least perturbing positions were chosen, and a disubstituted Aib residue was strategically introduced in place of one of the key Leu residues, which improved affinity. After incorporation of single and double i, i+4 and i, i+7 staples, the peptides were again evaluated for affinity and proteolytic stability. StRIP14 containing two i, i+4 staples on the same helix face and an overall net negative charge of −4 was the most stable, but in order to reduce the negative charge, some Asp and Glu residues were mutated to Asn and Gln, respectively. The resultant StRIP16 still had an overall −2 charge but was able to colocalize with Rab8a in cells and demonstrated cell uptake comparable to the cell penetrating Tat peptide. In a separate study, Mitra et al., used hydrocarbon stapling to develop inhibitors derived from FIP-family adaptor proteins to target the constitutively active endosomal sorting proteins RAB11 and RAB25.422 The latter protein, RAB25, is implicated as both an oncogene and tumor suppressor in specific tumor contexts like ovarian and breast cancers.423,424 The FIP interacting domain regulates GTPase activity and binds as an extended α-helix-turn-310 helix pair, requiring a unique approach to design stabilized antagonists with desirable properties. A panel of FIPα-helix-derived peptides were synthesized, scanned for ideal staple positions, then further optimized with residue mutations to remove anionic residues for better cell uptake, thermal stability and oxidative stability.422 In addition to stable helical character, the peptides showed pronounced phenotypic effects on RAB25-driven processes such as cell migration that were context specific to breast and ovarian tumor models, mimicking the effect of RAB25 overexpression or knockdown in these cellular systems. For example, RAB25 overexpression increases migration in MCF7 but decreases it in MDA-MD-231. The treatment of RFP14 peptide, which was the most RAB25 specific, led to the reversal of the respective RAB25 phenotypic effects in both cell lines.422
Beyond many of the classically undruggable protein classes discussed above, notable studies have developed stabilized peptides for many other challenging targets and interfaces. These include cell surface receptors,425 cytoskeletal proteins,426,427 chromatin modifying enzymes,428 viral proteins429-431 and even nonprotein targets like RNA and DNA.432 A notable example of stapled peptide development for viral targets include the development of NYAD-1, which was based on a previously identified peptide sequence that is known to interfere with HIV capsid particle assembly but is not a viable treatment due to lack of cell permeability.429 Using an i, i+4 hydrocarbon staple with S5 residues, Zhang et al. showed that constrained peptides were more α-helical and cell permeable and able to bind to the capsid protein but not able to prevent HIV-1 entry altogether.429 In follow-up studies, they tested i, i+7 variants of NYAD-1, which demonstrated some impaired HIV-1 infectivity in treated cells.430,431 Another notable study by Walensky and colleagues used stapled peptides to help stabilize target epitopes from HIV for the development of adaptive immunity. This represents a novel example of using structural stabilization as a viral vaccine.433
Epigenetic enzymes, such as EZH2, have also served as targets for stapled peptide development. Dysregulation and mutation of EZH2, a histone methyltransferase, has been linked to the proliferation and metastasis of various cancers.434 While methyltransferases can be targeted with small molecules, selectivity is a concern in targeting, for example, the SAM-binding active site. Kim et al. reported a selective peptide inhibitor of the EZH2/EED complex that utilized hydrocarbon staples in an i, i+4 pattern.428 This sequence was derived from the EED-binding domain of EZH2, which adopts an α-helical conformation. The staples were strategically placed away from the interacting helix face, and different sequence lengths were assessed in vitro for EED binding. It was determined that an IQPV motif at the C-terminal end of the peptide was crucial for binding. The lead 27-residue peptide was further optimized for cellular uptake by mutating out negatively charged residues that were not expected to be crucial for binding to EED.428
Protein kinases are a large family of proteins that have been extensively studied for small molecule drug development, especially in oncology. While active-site directed small molecules have been enormously successful drugs for a subset of kinases, the ability to create selective inhibitors remains a challenge. Targeting allosteric sites could offer another path for selective modulation, and several stabilized peptides have been developed to regulate protein kinase effector binding as well as the conformational state. For example, Schepartz and colleagues developed hydrocarbon stapled peptides that allosterically inhibit EGFR kinase function by preventing the coiled coil interaction that occurs upon ligand binding.425,435,436 The sequence was derived from the juxtamembrane (JM) segment that forms the coiled coil dimer and stapled in various positions opposite the binding interface of the helix. They identified multiple sequences that decreased cell viability in EGFR expressing cell lines, the most potent being peptide E1S with a central i, i+7 hydrocarbon staple.425 Interestingly, this peptide exhibited potent bioactivity in its unstapled form close to that of the stapled E1S. The unstapled peptide had greater helicity than the wild-type sequence but less than its stapled counterpart, which is not entirely unprecedented due to the previous studies of α,α-disubstituted amino acids promoting helix formation.435 Further structural studies revealed that the peptide functioned by interacting with both the JM segment and C-terminal tail of the kinase domain.436
In another example, hydrocarbon-stapled peptides were developed to target effector interactions with protein kinase A (PKA) and protein kinase C (PKC). A-kinase anchoring proteins (AKAPs) are a family of proteins that act as a scaffold to spatiotemporally control PKA activity.437 Disrupting the AKAP-PKA interaction in an isoform-selective manner has long been seen as a viable therapeutic strategy, with early peptides derived from the α-helical domain of AKAP that either specifically prevent AKAP interaction with PKA-R1438 or PKA-R2; however, these native peptides had poor pharmacologic properties.439,440 Kennedy and co-workers designed hydrocarbon-stapled peptides by incorporating S5 residues at i, i+4 positions in sequences from the α-helical AKB domain in different AKAP proteins.441 The best hit, STAD-2, targeted PKA-RIIα with 40-fold selectivity over PKA-RIα and inhibited phosphorylation of PKA targets in cells. STAD-2 decreased parasitemia in malaria-infected red blood cells via cell lysis, with single-digit micromolar IC50 efficacy.442 Subsequent work demonstrated improved cellular uptake443 as well as design of compounds targeting AKAP-PKC interaction.444
The examples described showcase the immense potential of hydrocarbon stapling for stabilizing helical secondary structures and developing chemical probes and therapeutics for undruggable targets. Several aspects have to be considered to successfully incorporate hydrocarbon staples into a peptide as many designs are context and target specific. These include peptide sequence as well as staple linker length, stereochemistry, and position along the structure to impart desired properties. It is often necessary to sample the sequence space of the progenitor protein or peptide structure to discover the most active ligands as observed for the reported NOTCH inhibitors.396 It is also important to scan the staple placement across the sequence to identify the optimal stapling positions while ensuring the orientation of key interacting residues remains intact. This can greatly affect binding affinity and specificity to the target of interest and help reduce off-target effects, as well as generate highly specific mimetics capable of distinguishing closely related targets, for example the BCL-2 family. Future strategies may include library generation of hydrocarbon stapled peptides with the emergence of compatible chemistries445 that will become powerful strategies for screening against numerous challenging targets.
3.5. Thiol-Mediated Staples
While RCM chemistry has proven incredibly versatile and useful for the stabilization of α-helices, the requirement of custom, potentially toxic catalysts and relatively expensive amino acids could be a practical hindrance for scaling some probes and therapeutics. The thioether linkage is also widely used, especially in the macrocycle space, but its utility for secondary structure stabilization has been proven as well (Figure 31). It avoids the labile nature of the disulfide linkage, and can utilize natural cysteines, or cysteine analogs. Two of the common reactions are Michael addition to an α,β-unsaturated carbonyl446 or an SN2 reaction with an α-halo acetamide.447 These electrophilic groups can be incorporated as non-natural amino acid side chains or as substrates that are added to dithiol peptides for a two-component staple. Jo et al. reported the use of thiol bis-alkylation to stabilize an α-helical calpain inhibitor.448 They screened several potential electrophilic cross-linking reagents to determine which would lead to the lowest energy conformations of the peptide. Benzyl halides performed the best, with dibromo-m-xylene yielding the highest conversion with the fewest side products and the greatest improvement in helicity, suggesting that yield and helical propensity are correlated, as in many other examples in the literature. They proceeded to apply this chemistry toward a calpastatin fragment sequence and showed dose-dependent inhibitory activity of the peptide.448 Dibromo-substituted xylenes remain a common choice for cysteine cross-linking, especially for use with phage display449,450 and mRNA display libraries as demonstrated by Hartman and co-workers.451 In this approach, they hypothesized that one reason library-generated peptides stapled with thiol-based linkers exhibit lower affinity than their hydrocarbon stapled counterparts was due to the loss of the stabilizing effect provided by the α,α-disubstituted amino acid. Thus, they synthesized a series of peptides using either hydrocarbon staples or thiol-alkylated staples via an α-methyl cysteine to examine the effects. The results showed that incorporating the disubstituted α-methyl cysteine at both ends of the staple with a dibromoxylene linker yielded a peptide with affinity close to that of the hydrocarbon stapled version. They then confirmed that this modified cysteine was able to be incorporated into an mRNA display library, which opens the possibility of more diverse libraries with potential to discover higher affinity hits. In addition, the diversity of bromo-substituted linkers allows for easy sampling of various peptide conformations. Sometimes this results in lead compounds with unusual staple patterns, such as the i, i+3 staple in the DD5-o autophagic activator peptide reported by Peraro et al., among other examples.452,453 The less common spacing for helical stabilization may be uniquely compatible with d-amino acids.339,452,454
Figure 31.

Representative example of the synthesis of bis-alkylated thioether stapled peptides using common building blocks.
A relatively new class of stabilized α-helices that utilize thioether stapling chemistry are “helicons,” which were originally reported by Li et al.455 They are generated and selected through phage display, with cysteines incorporated at i, i+7 positions in the helix (Figure 32A). The α-helix is stabilized with the addition of N,N’-(1,4-phenylene)bis(2-bromoacetamide). In this first report, a stabilized phage library was panned against β-catenin to both validate prior known α-helix binding sites as well as discover de novo sites. Through phage display selection, they discovered that helicons were able to recapitulate native α-helix interactions such as those known with Axin. An example from the library is Helicon FP01567, which binds in the same location as the α-helix Axin466-482 and interfered with the β-catenin-Axin interaction with low micromolar affinity (Figure 32B). In addition, novel α-helical binding sites have been discovered that interferes with the ICAT binding site. In a follow-up study, Tokareva et al. developed a method to study helicon trimerizers, where the helicon facilitates interactions between two different proteins.456 They were able to form a novel PPI between E3 ligase CHIP and enzyme PPIA, CHIP and transcription factor TEAD4, and E3 ligase MDM2 and transcriptional coactivator β-catenin. To do so, they first screen for helicons that bind the E3 ligase, before performing a second phage display screen that is biased for residues that bind the relevant E3 ligase but retains flexibility in other residues. Only helicons that require both the E3 ligase and the relevant protein to bind were synthesized. An example is Helicon H330 (Figure 32B), which captured critical interactions H330-D18 with β-catenin H578 and R612, H330-D19 with β-catenin R582 and MDM2 H96, and the interaction between MDM2 V109 and β-catenin K433, collectively stabilizing the ternary complex.456 As the helicon technology has shown, phage display can select for stabilized α-helical peptides that can recognize novel sites on undruggable targets.
Figure 32.

Helicons selected by phage display for targeting diverse POIs. A) Schematic of phage display selection for Helicons targeting β-catenin. After translation and prior to affinity selection, displayed peptides are stapled between separate cysteine residues using a bifunctional alkyl bromide linker. B) Middle: Crystal structures of Helicon H330 (PDB: 8EIC) and Helicon FP01567 (PDB: 7UWI) bound to β-catenin (cyan) and MDM2 (gray). Left insert: Cooperative interactions promoted by Helicon H330 (blue) to the interface of MDM2 (gray) and β-catenin (cyan). Right insert: Overlay of FP01567 (blue) and Axin466-482 (pink; PDB: 1QZ7) bound to β-catenin (cyan). Staples are shown in yellow.456
Another demonstration of dithiol stapling includes a study by Brown et al. with inhibitors of GABARAP. Here, they developed novel peptides that have unnatural modes of inhibition arising from a sampling of diverse structures, most commonly compatible with thiol mediated cross-links, rather than starting with a template derived from natural ligands and subsequently making modifications. The K1 peptide was found to be an inhibitor of GABARAP through phage display screening.457 It binds GABARAP and LC3 as a single 310-helical turn with high affinity, although the mode of binding is entirely different than natural interactors of GABARAP. In order to improve the pharmacokinetics of K1, various thiolreactive cross-linkers were tested with different spacing.458 The i, i+5 staples proved the most effective in improving binding affinity for GABARAP, and introducing penicillamine further enhanced affinity and specificity, yielding two peptides with completely different binding modes for GABARAP as revealed by crystal structures (Figure 33A-C). Pen3 mimicked K1, while Pen8, having the penicillamine on the opposite end of the peptide, more closely mirrored the natural ligands. In both cases, the staple itself was also involved in hydrophobic interactions between the peptide and GABARAP. Swapping the penicillamine for 4-mercaptoproline yielded a higher affinity peptide with better biological stability and generally improved membrane permeability.
Figure 33.

Stapled peptides targeting GABARAP. A) Primary sequence and staple locations of Pen3-ortho (left) and Pen8-ortho (right). Peptides were stapled between cysteine and penicillamine (denoted “X”) residues using ortho-dimethylbenzene linker. B) Crystal structure of Pen3-ortho (blue) bound to GABARAP (cyan). PDB: 7ZKR. C) Crystal structure of Pen8-ortho (purple) bound to GABARAP (cyan). PDB: 7ZL7. Interacting residues are labeled.458
Other variants of the thiol-based staples have been reported, such as the thiol–ene reactions and ketone-mediated modifications.459-461 One interesting inversion of this chemistry involves incorporating a fluoroacetamide onto the peptide and using a benzyl thiol as the linker for proximity driven fluoride-thiol displacement, which can be done in an aqueous solution on unprotected peptides.462 Another method that is compatible with unprotected peptides is perfluoroarylation.463 Jimenez-Macias et al. used this strategy to stabilize a CPP for cisplatin delivery to brain tumors.464 Additionally, it has been shown that introducing chiral centers into the staple linker leads to different effects on the helicity and thus, the cell permeability and stability of the peptides.465-468 In a proof-of-concept study using thiol–ene chemistry to target ERα and MDM2, the authors introduced a chiral center on the alkene residue side chain and unexpectedly observed different structural and activity properties from unique diastereomers.465 A similar example was demonstrated by Speltz et al., where a γ-methyl substituent was incorporated into the hydrocarbon staple to mimic the branched side chain of an Ile residue involved in the native ligand interaction between SRC2 and ERα.469 This study demonstrated that the chiral staples slightly destabilized helix propensity, but bound ERα with higher affinity than the achiral hydrocarbon staple. These examples demonstrate ways in which additional functionality can be built into staple chemistries, furthering their potential for improving the pharmacological properties of peptide therapeutics.
A structurally interesting example of a RbAp48 inhibitor was reported by Hart et al., where a central trisalkylated linker templates a short one and a half turn helix, followed by a proline turn and an extended loop (Figure 34).470 The sequence was derived from MTA1, a scaffold protein that recruits RbAp48 into a histone modifying complex. Despite truncating two residues that were previously reported to have hydrophobic interactions with RbAp48, the weakened binding affinity was overcome by rigidifying the peptide using various thioether linkers, notably, 1,3,5-tris(bromomethyl)benzene, to form a bicycle peptide that formed a stabilized α-helix with two of the thioether links with i, i+4 spacing and a large constrained loop, resulting in compound 33, with a Kd value of 8.56 nM.470 Another recent example reported by Tennet et al. showcased the utility of thioether-mediated staples by stabilizing a less common polyproline II helix conformation.453 The highest affinity compound targeting the EphB2 receptor was stabilized via a 2,7-dimethylnaphthyl linker bridging two cysteines with i, i+3 spacing. However, trypsin digestion assays suggested the hydrophobicity of the linker increased susceptibility to degradation compared to lactam stapled analogs. This highlights the fact that potency may not always correlate with favorable pharmacologic properties. The use of thiol-based staples and others to stabilize structurally unique conformations such as constrained helix-loops or polyproline II helices opens new avenues for designer peptides beyond highly abundant α-helix or β-structures.471
Figure 34.

Peptide stapling using trifunctional linker to generate bicyclic Peptide 33 to form a stabilized α-helix and constrained loop to increase affinity to RbAp48. Crystal structure of Peptide 33 bound to RbAp48 is shown. PDB: 6ZRD.470
3.6. Azide–Alkyne Click Reaction Staples
Cycloaddition click reactions such as CuAAC or RuAAC have the advantage of being bioorthogonal with fast reaction kinetics. RuAAC is not limited to terminal alkynes as in CuAAC and can include a variety of substituted alkynes (Figure 35A).472 Ingale et al. reported a method for the incorporation of intramolecular CuAAC on-resin to stabilize α-helical peptides and used an HIV-1 gp41-derived sequence for proof-of-concept studies.320 In an ELISA against HIV-1 neutralizing antibodies, the peptides showed moderate binding. Strain-promoted cycloadditions (SPAAC) have been utilized for azide–alkyne cross-links as well, and the lack of a required metal catalyst enables these reactions to be biocompatible. Lau et al., demonstrated this advantage by reporting the in situ stapling and selection of peptides in cell culture.473 Using a p53-derived peptide functionalized with two azide containing residues, they treated p53 reporter cells and cotreated with the strained diyne that enabled SPAAC stapling. Reporter assays showed a dose-responsive increase in p53 activation for the stapled peptides (cotreated with diyne) while the same peptides without addition of the diyne showed no activation even at the highest dose. A crystal structure of the lead compound E1 in complex with MDM2 showed that the cycloadduct was part of the binding interface, possibly contributing to the interaction (Figure 35B).473 In a separate study, Kawamoto and co-workers identified two solvent-exposed residues on the β-catenin binding helix of BCL9 from the crystal structure that seemed suitable for modification with a triazole staple.474 The effect of the length of the azide side chain and location of the triazole itself was examined, and they found that minute differences caused significant changes in binding affinity, solubility and helix stabilization. Additionally, they designed and tested “double-stapled” peptides and showed that this dramatically improved the helical character. They also discussed the formation of “mixed staples” and how positioning of the azide and alkyne modified residues favors or disfavors these types of staples.474 This is a unique attribute of asymmetric staple chemistries.
Figure 35.

Synthesis of stapled peptides using azide–alkyne cycloaddition reactions. A) Schematic of peptide functionalization using Cu- or Ru-catalyzed or strain-promoted cycloaddition (SPAAC). B) Crystal structure of peptide E1 stapled through SPAAC using a strained dicyclooctyne linker (blue; staple shown in yellow) bound to MDM2 (cyan). PDB: 5AFG.473
While staples primarily serve to lock the peptide into a particular conformation, two-component staples are well-suited to the modular creation of diversified libraries that attempt to build the desired properties directly into the stapling chemistry and minimize necessary structural optimization to the primary sequence. The alkyne linkers for azide–alkyne cycloadditions can be functionalized in a number of ways to fine-tune properties such as polarity, steric, and electronic effects. A recent report from Sharma et al. created a library of functionalized diynes to investigate such effects on the p53 peptide bioactivity.475 Lau et al. report the use of dialkynyl linkers that are conjugated to a diazido peptide via CuAAC chemistry while having a third substituent, which can be a peptidic sequence, a fluorophore, or a small molecule.476 The practical application of this approach was demonstrated by multiple groups using the p53/MDM2/MDMX PPI for proof-of-concept. Wu et al. used azide–alkyne click chemistry to staple a p53-derived sequence in an i, i+7 fashion, and used diverse dialkyne linkers conjugated to CPPs as well as small molecule carriers.477 Despite similar binding affinities for MDM2 in fluorescence polarization assays, there was greater variability for p53 activation in cell based reporter assays, presumably due to differential uptake. Similarly, Charoenpattarapreeda et al. used a diazidopeptide clicked with a dialkyne linker containing an electrophilic, lysine reactive moiety.478 After confirming the helical character of the peptide via CD spectroscopy, they showed that a p53-derived peptide stapled in this way exhibited selective and covalent binding to MDM2 in vitro.
3.7. Diels–Alder Staples
Another cycloaddition in the toolbox of stabilizing chemistries is the well-known Diels–Alder reaction, favored for the spontaneous reaction occurring at room temperature or with mild heating with no catalyst and compatibility with a wide range of solvents and substitutions. In the interest of using diverse stapling chemistries to sample different peptide conformations, as discussed previously by Kritzer and others,452 Diels–Alder chemistry is well-suited for this application, as it can utilize a variety of diene and dienophile combinations and has already proved its value as a bioorthogonal, biostable conjugation chemistry.479,480 Montgomery et al. reported the use of Diels–Alder cycloaddition for stabilizing peptides.481 The dienes or dienophiles were incorporated onto cysteine or lysine side chains postsynthesis or were built into the SPPS workflow using amino acid building blocks that already had the desired moiety. As the first proof-of-concept experiment, stabilization of an RGD loop was carried out using the Diels–Alder cycloaddition of hexadiene and maleimide, which was shown to increase trypsin resistance and enhance bioactivity. This was followed by the stabilization of the p53-derived α-helix in an i, i+7 fashion using furan and maleimide, which readily underwent near-complete cyclization in 15 min and showed increased helicity in an aqueous solution. The same strategy was applied to the development of an ERα inhibitor. ERα is a transcription factor with a ligand binding domain that contains a druggable pocket for estrogen binding that is typically targeted by selective estrogen receptor modulators (SERMs).482 However, common point mutations such as Y537S are known to confer drug resistance by stabilizing the agonist conformation independent of estrogen binding, resulting in constitutively active ERα.483 An alternative strategy is to disrupt the binding of coactivator SRC2 to ERα, which binds via a hydrophobic helix with the LXXLL motif. Many groups have reported stapled peptides to target ERα, particularly by developing an SRC2-derived peptide sequence spanning the LXXLL motif that is responsible for the SRC2 coactivator binding and using various staple chemistries to stabilize the helix.340,469,481,484-487 Montgomery et al. used this well studied PPI as a proof-of concept for Diels–Alder peptide stabilization (Figure 36A).481 During the SPPS workflow, furylalanine and lysyl-maleimide residues in i, i+4 positions were reacted on resin in DMSO. A small panel of Diels–Alder cyclized (DAC) peptides identified improved helical character and binding for several members. The mode of binding was confirmed by obtaining a crystal structure of ERα in complex with one lead compound, DAC6. Interestingly, although one of the Leu residues responsible for the binding interaction was substituted with the furylalanine, this was compensated by the interaction of the cycloadduct with the hydrophobic shelf of ERα, showing that Diels–Alder staples could not only stabilize secondary structure, but contribute to novel binding interactions (Figure 36B). Taking into consideration the modularity of the Diels–Alder reaction, this stapling approach could prove to be highly versatile for library synthesis and in combination with other stapling chemistries.481 In addition, reverse Diels–Alder reactions have proved very useful for bioorthogonal crosslinking. The diene of choice is usually tetrazine, which can rapidly undergo Inverse Electron Demand Diels–Alder (IEDDA) followed by nitrogen loss, making the process irreversible and entropically favored.488,489
Figure 36.

Diels–Alder stabilized peptides targeting ERα. A) Schematic of DAC peptide functionalization on resin. B) Crystal structure of DAC6 (blue; staple shown in yellow) in complex with ERα (cyan) bound to estradiol (red). PDB: 6PIT.481
3.8. Staple Properties and Layered Stabilization Approaches
The emergence of complementary and orthogonal chemistry approaches for secondary structure stabilization allows the sampling of various strategies to achieve the optimal scaffold architectures and improvements in affinity, metabolic stability, and membrane permeability.41,490 In a notable comparative study of several common stapling chemistries (lactamization, hydrocarbon, CuAAC, thiol alkylation, and perfluorarylation) using CD, NMR, and molecular dynamics of pentapeptides in water revealed that lactamization resulted in the most α-helical character, while the hydrocarbon staples and CuAAC staples showed characteristics of both α- and 310-helices.491 Recently, Strømgaard and colleagues performed a comprehensive analysis of several stapling approaches by varying the position, chemistry, and linker length using amyloid precursor protein (APP) dodecamer peptide targeting Mint2 as a template.492 They systematically tested lactam cyclization, hydrocarbon stapling, triazole stapling through CuAAC, and cysteine crosslinking, using various side chain substituents to alter the linker length for each approach. In general, increasing the linker length also increased affinity, and all stapled variants tested displayed increased resistance to tryptic digestion. Hydrocarbon stapling provided the most structured peptide in solution by NMR, followed by triazole and cysteine crosslinked peptides. While the hydrocarbon stapled peptides exhibited the highest affinity, cysteine cross-linked peptides displayed the greatest cell permeability, whereas the lactam and triazole linked peptides suffered from a lack of uptake and stability. Finally, metabolic stability and affinity was closely linked to the degree of structural order, and hydrocarbon stapled peptides most closely mimicked the native bound structure of APP to Mint2. A similar study by de Araujo et al. explored the effects of staple identity on the hMD2 binding peptide derived from p53.493 In this study, cysteine crosslinked peptides displayed the lowest protection from degradation, whereas lactam and hydrocarbon staples provided exceptional stabilization. However, the use of more hydrophilic linkers in lactams or triazoles greatly reduced cell uptake, as only modifications that increased hydrophobic surface area such as aromatic thioether and hydrocarbon staples led to increased permeability. These studies and others494 demonstrate the importance of systematically testing different scaffolds to balance affinity, stability, and permeability in a case-by-case basis, although in general it appears that hydrocarbon stapling enhances desirable properties.
Orthogonal stapling chemistries can also be used to layer multiple staples in a single scaffold to better improve structural stability. Showcasing the utility of layering multiple hydrocarbon staples, Bird et al. developed a double stapled peptide based off of the gp41 HR2 fusion domain, resulting in SAHgp41, which exhibited greater antiviral activity and better proteolytic stability compared to FDA-approved enfuvirtide, as well as some indication of oral bioavailability.328 Likewise, the concept of “stitched” peptides first reported by Hilinksi et al.495 These scaffolds utilized α,α-disubstituted residues with two olefin side chains to generate dual hydrocarbon staples to greatly improve the thermal and metabolic stability, as well as cell penetrating ability compared to single stapled counterparts (Figure 37C). Due to the requirement for two regioselective metathesis reactions, specific combinations of amino acids and spacing must be used; even with these designs a mixture of side products can be obtained, highlighting the value of complementary layered chemistries.
Figure 37.

Functionalization of peptides using stitched or layered stapling. Synthesis of peptide stapling using hydrocarbon and Diels–Alder (A), hydrocarbon and lactam (B), or dual hydrocarbon stapling (C).
We demonstrated an example of orthogonal and layered staple chemistries using hydrocarbon and Diels–Alder stapling in a double stapled peptide derived from SRC2 (Figure 37A).481 The double stapled peptide displayed a strong α-helical signal by CD with classic minima at 208 and 222 nm, and suggests that this combination of chemistries could be developed further for hyperstabilized or larger stabilized peptides. Speltz et al. used a similar template to combine hydrocarbon and lactam stapling (Figure 37B).486 The bicyclic peptide exhibited increased α-helicity and proteolytic stability but comparable binding affinity to unmodified and single stapled counterparts. Fairlie and co-workers also explored dual hydrocarbon and lactam staples in a BH3 domain peptide.496 Including a lactam staple to induce stronger α-helicity and a hydrocarbon staple to improve cell permeability led to a cell permeable, metabolically stable dual inhibitor selective for BCL2A1 and MCL-1. We expect that as additional chemistries are added to the stapled peptide toolbox, more options will become available for layered synthesis of more stable and efficacious peptide probes and therapeutics.
3.9. Summary and Outlook for Side Chain Stabilized Secondary Structure Mimetics
Side chain stabilized secondary structure mimetics have shown immense promise in the development of chemical probes and therapeutics. Indeed, several “stapled peptides” have advanced to the clinic and have shown good safety profiles as a class in advancing the field of therapeutic biologics. However, despite the incredible breadth of studies performed to examine the types of staples, layering chemistries, and biophysical properties of various secondary structure mimetics, side chain stabilized peptides have failed to advance through late stage clinical trials. For example, hydrocarbon staples have been extensively applied to increase the cell permeability and affinity for peptide therapeutics and have shown good promise but have yet to be incorporated into a market drug. The consensus of the literature in this area seems to indicate that while some generalizations can be made regarding staple effects on pharmacologic properties (e.g., linker identity, length, and stereochemistry), there are no hard and fast rules applied to all cases. Alternatively, the field is moving further into the generation of diverse stapled peptide libraries, such as hydrocarbon DELs445 and phage display libraries.497,498 Developing more compatible ways to incorporate stapling earlier into library generation and screening will be highly valuable going forward in identifying potential therapeutic candidates.
The number of useful reactions that can be used to stabilize secondary structures also enable the construction of larger combinations of motifs, or even different modalities. Peptides are easily conjugated to small molecules, other peptides, or larger biologics. Chemical stabilization enables them to maintain their structure and functionality in a range of environments, an advantage over larger natural biologics. As a testament to their versatility, many stapled peptides have been used in the development of PROTACs against difficult protein targets.301,343,499,500 Peptide library technologies are expanding to accommodate the selection of specific structural elements as well.449 While there are still many hurdles to be overcome in terms of pharmacokinetic properties, stabilized peptides generally have a good safety profile, and it has been repeatedly shown that with the right amount of tuning, better permeability, bioavailability, and efficacy in vivo is possible.
4. NON-NATURAL PEPTIDOMIMETICS
4.1. Non-natural Peptidomimetics Integrate Changes to the Peptide Backbone to Improve Activity
A common goal in the application of designer peptidomimetics is to recapitulate the folding or secondary structure of native peptides and protein domains. This is particularly important for binding to the large and disordered surfaces characteristic of undruggable targets. However, even with a well characterized ligand or surface, naturally derived peptides can still suffer from poor biostability and cellular uptake, which may stem partly from the peptide backbone core itself.501,502 To alter the biophysical properties of peptides, many non-natural backbone analogs have been developed and implemented to create myriad structures that retain the ability to form helical or sheet-like structures. Most importantly, these non-natural backbones are not recognized by native hydrolytic enzymes, which can significantly improve biological stability (Figure 38).501,502 Additionally, inclusion of substituents along different portions of the backbone can greatly improve solubility and cellular delivery, resulting in potentially improved pharmacological properties.502
Figure 38.
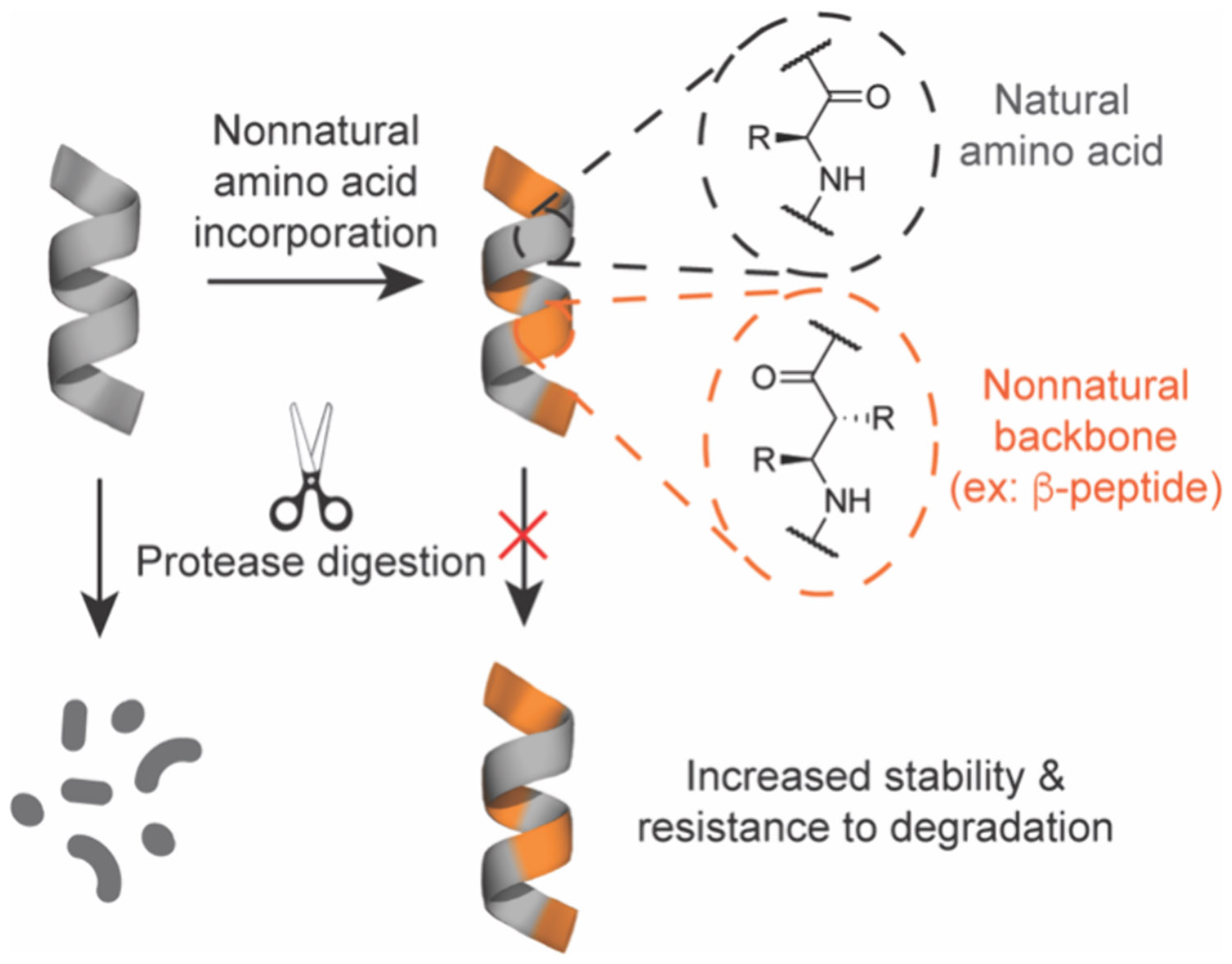
Non-natural backbones mimic native peptide secondary structure and provide resistance to proteolytic degradation.
This section covers non-natural peptidomimetics that may include features of both cyclic peptides and side chain stabilized peptides, but also incorporate changes to the peptide backbone that yield non-native species and structures. This includes foldamers, where a significant portion of the backbone is replaced by non-natural residues (class B mimetics), and structural mimetics that replace the entire backbone with nonpeptidic scaffolds that still retain side chain functionality and conformation (class C mimetics).22,503-505 These modalities benefit from a repeating backbone unit or pattern for enhancing stability while maintaining side chain identity and position to promote interactions, as compared to side chain stabilized secondary structures that require key positions for modification. We will discuss the design and characteristics of these different backbone modifications and highlight examples for targeting undruggable classes.
4.2. Design and Characteristics of Non-natural, Backbone Modified Peptidomimetics
4.2.1. β-Peptides.
The addition of a methylene group to create a repeating aminoethyl carboxyamide backbone affords a β-peptide and is the most thoroughly studied backbone modification for non-natural peptidomimetics (Figure 39A,B).22,506 Easily incorporated into SPPS protocols, β-amino acid monomers contain multiple locations for side chain substitution at the β2- and β3-positions, which can provide increased molecular recognition vectors and contribute to overall structure stabilization. Additionally, β-peptides have shown a propensity to fold into several unique helical conformations in both aqueous and organic environments, such as the 10/12-helix, 12-helix (2.5 residues per turn and 12 atoms between H-bonds), and 14-helix (3 residues per turn and 14 atoms between H-bonds) (Figure 39C). The position and stereochemistry of the side chains in a β-peptide play a strong role in the handedness and helix structure.506 In a 10/12-helix, alternating monosubstituted β2- and β3-positions result in the characteristic intertwined 10- and 12-membered hydrogen-bonded rings.507 12- and 14-helix conformations are favorable with substituents at the β3-position and including cyclic β-amino acids such as ACPC and ACHC can induce conformational preference to 12- or 14-helix geometries, respectively (Figure 39B). Cyclic β-amino acids also increase the tendency of β-peptides to fold into helices in aqueous environments, which is important to maintain activity in biological settings. For additional in-depth discussion of β-peptide folding and synthesis refer to several excellent reviews.22,506,508-512
Figure 39.

A) Chemical structures of α-, β-, γ-peptide, oligourea, and peptoid backbones. B) Structures of helix-inducing cyclic β-amino acids. C) Hydrogen bonding patterns of β-peptide helices.
While the geometry and properties of homogeneous β-peptides are well characterized and provide excellent resistance to proteolytic degradation, it is more straightforward to design initial leads from natural peptide or protein (i.e., α-amino acid mediated) epitopes.511,513 Thus, incorporating β-amino acids strategically within α-peptides, known as α/β-peptides, emerged as an alternative to mimic the native geometry and still provide enhanced resistance to proteolysis.514 In these designs, α-amino acids are used for surface recognition, whereas β-amino acids typically induce and stabilize secondary structure conformations.515 Many other conformations and properties depend upon the ratio and identity of included α- and β-amino acids.22,511,514,516 Most importantly, replacing even a small portion of a peptide mimetic with β-amino acids has been shown to increase metabolic stability in serum or in vivo half-life by orders of magnitude, both desirable traits necessary for development of useful therapeutics or chemical probes.501,509,517,518
Early work by the Schepartz lab using β-peptides as antagonists of undruggable targets aimed to inhibit the interaction between the transcription factor p53 and the E3 ubiquitin ligase hDM2.519 This interaction, which is broadly implicated in cancer, has become one of the hallmark models for designing and testing non-natural peptidomimetics. In addition to being an attractive therapeutic target, this is also due to the short α-helical motif from p53 that binds to a hydrophobic cleft on hDM2 involving three key hydrophobic residues: Phe19, Trp23, and Leu26, which can be incorporated into foldamers attempting to mimic this structure.520 Schepartz and co-workers utilized this motif to generate β3-peptides that adopt a left-handed 14-helix structure capable of binding hDM2 with submicromolar affinity.519 Later use of computational modeling,521 inclusion of non-natural or cationic side chains,522,523 and stabilization by side chain stapling,524 led to β3-peptides with improved affinity for hDM2/X and cell permeability that may provide access to more active compounds in physiological disease settings.
Another common target for designing non-natural peptidomimetic antagonists is the highly conserved BCL-2 family of proteins.200 Gellman and co-workers first discovered a hybrid α/β-peptide that could mimic an α-helical BH3 domain necessary for many interactions involving BCL-2 family proteins.525,526 A solved crystal structure of α/β-1 bound to BCL-2 indicated this architecture effectively mimics the display of side chains with good overlap of conserved hydrophobic residues h1–h4 compared to native BH3 domains (α-Bim), showcasing the potential for this design to specifically target protein–protein interaction interfaces using sequence-based design (Figure 40A,B).527-529 This architecture was further improved by including chemical stabilization strategies such as side chain stapling to improve cell uptake and activity, while retaining the affinity and improved proteolytic resistance even over analogous stapled α-peptides.529 Other β-peptide and α/β-peptide mimetics have been developed to target viral infection and replication, showcasing the extraordinary potential for modulating biological processes involving PPIs and protein–nucleic acid interactions.530-533 However, shortcomings including poor intrinsic cell permeability and bioavailability have limited the translation into clinically relevant compounds for targeting undruggable classes. Lessons learned from the immense research on structural stability as well as more recent studies to achieve prolonged half-life and efficacy in vivo against viral infection and modulating receptor activity may help to provide guidelines for adapting these structures toward undruggable intracellular targets in more complex disease settings.533,534
Figure 40.

α/β-Peptides derived from BH3 domains for targeting BCL-2. A) Crystal structure of stapled α/β-1 (orange; staple shown in green) bound to BCL-2 (cyan). PDB: 5AGW. B) Overlay of reported α-Bim analogue (PDB: 3FDL), a stapled analogue α-1 (PDB 2YQ6), designed α/β-1 (PDB: 5AGW), and α/β-1-LIN (PDB: 5AGX) analogues showing close overlap of conserved hydrophobic residues h1–h4. Adapted from ref 529. Copyright 2015 American Chemical Society.
4.2.2. γ-Peptides.
The retention of the peptide-like backbone structure analogous to α-peptides but increasing the backbone by an additional two methylene groups yields a γ-peptide (Figure 39A). Similar to β-peptides, γ-peptides are easily incorporated into standard SPPS protocols and offer additional functionality along the backbone to fold into various conformations.509,516,535-537 Promising results in folding of homogeneous γ-peptides as short as tetramers into helical structures535 led to the creation of heterogeneous α/γ-, β/γ-, and α/β/γ-peptides to mimic protein secondary structure.516,538-543 Although they are less widely used and studied than β-peptides, the additional functionality and complexity afforded by γ-peptides may offer significant opportunities for structural optimization and applications for targeting diverse biomolecular surfaces.539,544 Additional information on mixed homologous α-peptides, β-peptides, and γ-peptides can be found in several interesting studies and reviews.509,516,536,541
Although there are few applications in literature of using γ-peptides and hybrids, these analogues have been reported to target interactions of p53/hDM2 and amyloid formation.540,545 Aitken and co-workers reported an α/β/γ-peptide foldamer as a selective inhibitor of the p53/hDM2 interaction. This α-helix mimetic contains alternating trans-2-aminocyclobutanecarboxylic acid (tACBC) β-amino acids and γ4-amino acids that adopt a 12,13-helix conformation capable of projecting the key hotspot residues involved in the natural p53 interacting helix (Figure 41A-B).540 Although this structure only achieved modest binding affinity and inhibitory activity, there was a significant increase in proteolytic stability. More recently, the Maillard group developed conformationally constrained heterocyclic γ-amino acids composed of 4-amino(methyl)-1,3-thiazole-5-carboxylic acids termed ATC monomers (Figure 41C). This design folds to adopt a 9-helix structure analogous to the native 310-helix (Figure 41D).546 They surmised that this architecture could be used to target transient 310-helices involved in amyloidogenesis to prevent aggregation. Using this design, they generated γ-ATC-peptides capable of modulating aggregation of both Aβ1–42 and human islet amyloid polypeptide (hIAPP), which are desirable drug targets for Alzheimer’s disease and type 2 diabetes, respectively.545 They explored different side chain substitutions for effects on affinity and solubility, and found that ATC oligomers with at least 6 units containing cationic side chains to increase solubility was necessary for activity. This study demonstrated the importance for designs capable of binding large, disordered surfaces and the necessary requirements for balancing biophysical properties such as size and solubility to attain active compounds.545 For additional information on recent γ-peptide monomers and structures, refer to the excellent review by Legrand and Maillard.536
Figure 41.

Helical conformations of γ-amino acid containing foldamers. A) Hydrogen bonding patterns of α/β/γ-peptide mimicking p53 helix. α-Residues are highlighted in gray. β-tACBC-residues are highlighted in blue. γ-residues are highlighted in orange. Key residues Phe, Trp, and Leu are labeled. B) Molecular modeling simulation overlay of p5316–29 (red) with α/β/γ-peptide mimic (yellow); RMSD = 0.89 Å for α-carbons. Adapted from ref 540. Copyright 2016 The Authors under CC BY https://creativecommons.org/licenses/by/4.0/.540 C) Hydrogen bonding pattern of a homogeneous γ-ATC 9-helix. D) Overlay of a native 310-helix (yellow/orange) and γ-ATC 9-helix (green/red) showing structural overlap of key residue positions. Adapted from ref 545. Copyright 2020 Wiley-VCH GmbH.
4.2.3. Oligourea Peptidomimetics.
Aliphatic N,N’-linked oligoureas are composed of a similar length backbone unit as γ-peptides but contain an additional nitrogen replacement of a carbon within the backbone to form the urea bond (Figure 39A).547 As in backbone modifications described above, oligourea foldamers are attractive mimetic scaffolds due to their synthetic accessibility, broad functionality, and folding propensity. Monomers are synthesized from available chiral substituted ethylene diamine or α-amino acid building blocks, and span a diverse chemical space beyond that of α- or β-amino acids.548 Oligoureas have been produced by both solution and solid phase methods; however, significant solubility issues resulted in solid phase becoming the routine approach.549,550 Guichard and co-workers extensively studied the synthesis of oligoureas and eventually discovered a microwave-assisted solid phase protocol using azido-terminated succinimidyl-activated carbamate monomers that efficiently produced oligoureas and oligourea–peptide hybrids (Figure 42A).551,552
Figure 42.

Synthesis and properties of oligoureas. A) Solid-phase synthesis of oligoureas using azide-terminated succinimidyl-activated carbamate monomers with microwave assistance. B) Hydrogen bonding pattern of homogeneous oligourea 12- ,14-helix. C) Crystal structure of p53-derived oligourea–peptide hybrid bound to MDM2 (cyan). Key interacting residues are labeled. PDB: 6HFA.556 D) Crystal structure of oligourea–peptide hybrid bound to ASF1 (cyan) with key interacting residues labeled. PDB: 6ZUF.557 Oligourea monomers are orange and α-amino acids are blue.
Oligourea peptidomimetics containing homogeneous or heterogeneous backbones with incorporated α-amino acids can adopt helical confirmations closely akin to an α-helix. This is independent of a primary sequence553,554 and is caused by additional backbone conformational restriction and intramolecular hydrogen bonding stemming from the ethylene diamine moiety resulting in the presence of linked 12- and 14-membered hydrogen bonded rings (Figure 42B).555 This propensity to fold into helical conformations regardless of side chain identity make oligourea foldamers highly desirable for their potential to become active, biostable, and conformationally controlled therapeutics. For more detailed information regarding oligourea structure and synthesis, refer to the excellent perspective by Yoo et al.548
Although first synthesized and studied as foldamers in 2002, only a few examples of oligoureas targeting intractable PPIs have been reported, albeit nearly two decades later. Guichard and co-workers generated peptide–oligourea hybrids to target the transcription factors p53 and vitamin D receptor (VDR).556 Using the hallmark p53/MDM2 system and a peptide previously identified from a phage display library, PMI,405 as a blueprint, oligourea residues were gradually incorporated into the structure. Five or six residues on the C-terminus of PMI, of which contain the key interacting residues Leu and Trp, were replaced with a short oligourea triad, resulting in a minimal ~2-fold decrease in activity compared to PMI by a time-resolved fluorescence energy transfer assay. A solved crystal structure deduced that key hydrophobic contacts are maintained and the oligourea backbone closely mimics the contacts formed by the cognate peptide (Figure 42C). However, the position and spacing of side chains in the oligourea portion was critical to properly mimic the α-helix geometry, stemming in part from the larger diameter of the oligourea helix.556 The increased diversity for side chain positions available to oligourea peptidomimetics may provide more opportunities to improve structural mimicry.
The same group employed a similar approach to target an epigenetic modifying protein, antisilencing function 1 (ASF1).557 ASF1 is a histone chaperone protein involved in regulating gene expression that is overexpressed in some forms of cancer.558 Depletion of ASF1 paralogs A and B reduced cancer cell proliferation and increased sensitivity to some chemotherapeutic agents.559 ASF1 binds to the H3/H4 heterodimer over a large surface area containing several shallow pockets, which are key features involved in undruggable targets.560 A previously identified 26-residue peptide, ip4, binds to ASF1 with nanomolar affinity through four hotspot residues Lys2, Leu6, Arg9, and Ile10 derived from a helix on H3.561 Mbianda et al. replaced the six central residues of this helical portion with four ureido residues (Figure 42D).557 This design was only able to recapitulate the positioning of three out of four key anchoring residues (Leu6, Arg9, and Ile10), resulting in a greater than 10-fold decrease in affinity (Kd = 2.7 μM vs 0.13 μM). However, the oligoureapeptide hybrid displayed much greater resistance to degradation in serum compared to the parent peptide, which agrees with other studies showing prolonged half-lives of oligourea peptidomimetics.557,562,563
4.2.4. N-Alkylated Peptoids.
Peptoids are α-amino acids developed in the late 1980s that contain a side chain on the amide nitrogen of the backbone to produce oligomers of N-substituted glycines (Figure 39A).564 This substitution confers improved cell permeability and resistance to proteolysis.565-567 Short oligomer peptoids closely mimic small molecules while providing peptide-like functional contacts and folds. Ramachandran-type plot calculations suggested peptoids contain more available conformational states compared to that of natural peptides.564 Structure-guided designs have shown that peptoids are capable of folding into helical and sheet-like conformations, closely mimicking the secondary structures seen in natural proteins.564,568 Interestingly, these structures were unaffected by heat or urea denaturation, suggesting that steric forces drive structural stabilization.569 This is much different from the hydrogen bonding forces that apply for native and non-natural helices previously described. Additional strategies to conformationally constrain peptoids, such as the inclusion of chiral side chains or cyclization may improve activity.570-572 This relationship between chemical structure and conformational preference is nicely outlined in a recent review by Eastwood et al.573
While peptoid monomers are capable of being incorporated into conventional SPPS methods for synthesis, a significant drawback to these standard protocols is the need for suitable quantities of protected building blocks. Moos and co-workers developed an alternative approach that uses readily available submonomers that sidesteps the need for backbone protecting groups (Figure 43A).574 Instead, synthesis proceeds by backbone elongation using haloacetic acid derivatives followed by introduction of side chains with a primary amine, thus greatly expanding the chemical diversity. Using this simple synthetic setup and a broad selection of substituents, large and chemically diverse peptoid libraries can be produced and screened against therapeutically relevant targets.575-577
Figure 43.
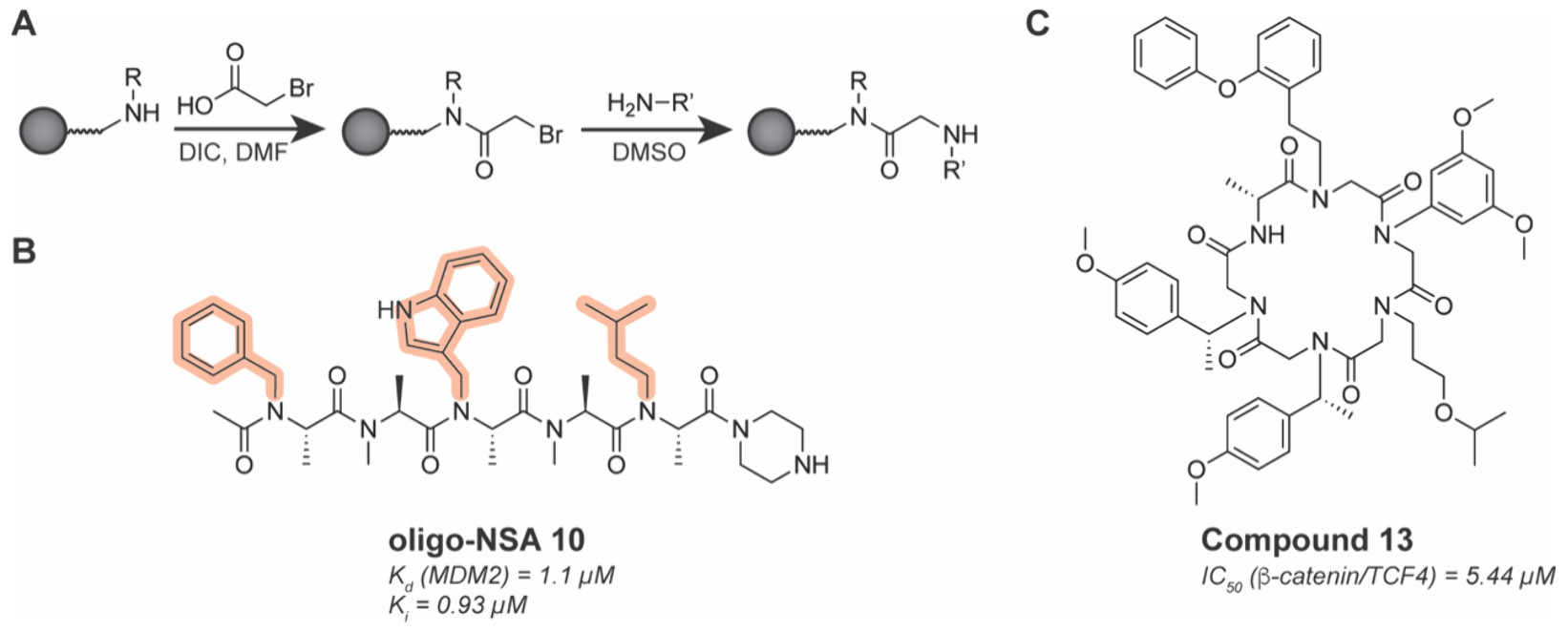
Synthesis and structures of peptoids. A) Solid-phase synthesis of peptoids using a submonomer strategy. B) Chemical structure of constrained peptoid targeting MDM2. Key residues Phe, Trp, and Leu are highlighted in orange. C) Chemical structure of β-catenin/TCF interaction inhibitor macrocyclic peptide-peptoid hybrid compound 13.578
Peptoids have a rich history in drug discovery as described by Zuckerman in the commentary “Peptoid Origins”.568 Original designs centered around combinatorial libraries screened against various receptors.579,580 However, the discovery of peptoids that are able to fold into higher order structures inspired interest in rational targeting approaches.581 For example, the Sando group designed a conformationally constrained peptoid ligand for MDM2 by using an α-carbon substituted backbone to constrain the inherent flexibility through steric hindrance (Figure 43B).582 This design provides a scaffold to display multiple functional groups in well-defined positions. Strategic placement of the hotspot residues Phe, Trp, and Leu, resulted in oligo-NSA 10, a peptoid capable of binding to MDM2 and competing with the native p53 helix peptide TAD. Importantly, they show that conformationally constraining the peptoid was essential for achieving efficient protein binding.582 In a subsequent study, the group introduced side chain modifications to increase affinity and cell permeability, highlighting the programmability of peptoids for improvement via SAR analysis.583
In another study, Logan and co-workers applied computational design protocols to generate in silico cyclic peptoid–peptide hybrids to inhibit the β-catenin/TCF interaction, which plays a critical role in the cancer associated WNT signaling pathway.578,584 Their identified macrocyclic hybrid, compound 13, was capable of inhibiting WNT-dependent luciferase activity in live cells better than commercially available WNT inhibitors, and decreased viability of WNT-dependent prostate cancer cell lines (Figure 43C). Compound 13 was confirmed to disrupt the β-catenin/TCF interaction in live cells by coimmunoprecipitation, suggesting inhibition of this PPI to the be main mechanism of action. Compound 13 was also shown to be efficacious in vivo, where it down-regulated WNT-dependent pathways to restore phenotypes in a zebrafish model. This example portrays the immense potential for peptoids and related compounds to address the myriad undruggable interactions implicated in human disease with the goal to advance therapies into the clinic. In fact, studies into the in vivo efficacy and oral bioavailability of cyclic peptoid hybrids are already showing promising results.585
4.2.5. γ-AApeptides.
Developed by the Cai group in 2011, γ-AApeptides are oligomers of γ-substituted N-acylated-N-aminoethyl amino acids (Figure 44A).586 Originally synthesized based on the γ-chiral peptide nucleic acid (PNA) backbone containing proteinaceous side chains,587-589 this backbone modification is amenable to SPPS protocols with many different scaffolds and substituents.590 Importantly, the repeating unit projects an equivalent number of side chains comparable to repeating di-α-peptides, thus containing potential for mimicking natural structures. However, because of the side chain locations on the aminoethyl unit and proximal tertiary amide, significant differences in hydrogen bonding patterns, conformational flexibility, and side chain orientation creates a challenge in predicting the folding patterns of these mimetics. Therefore, homogeneous sulfono-γ-AApeptides and hybrid α/sulfono-γ-AApeptides were developed as helical mimetics capable of adopting left- and right-handed geometries.591-593 These constructs adopt a few different helical conformations dependent upon monomer ratio, including a homogeneous left-handed 14-helix (Figure 44B) and a right-handed 1:1 l-α/l-sulfono-γ-AA 13-helix that most closely resembles a native α-helix (Figure 44C).
Figure 44.
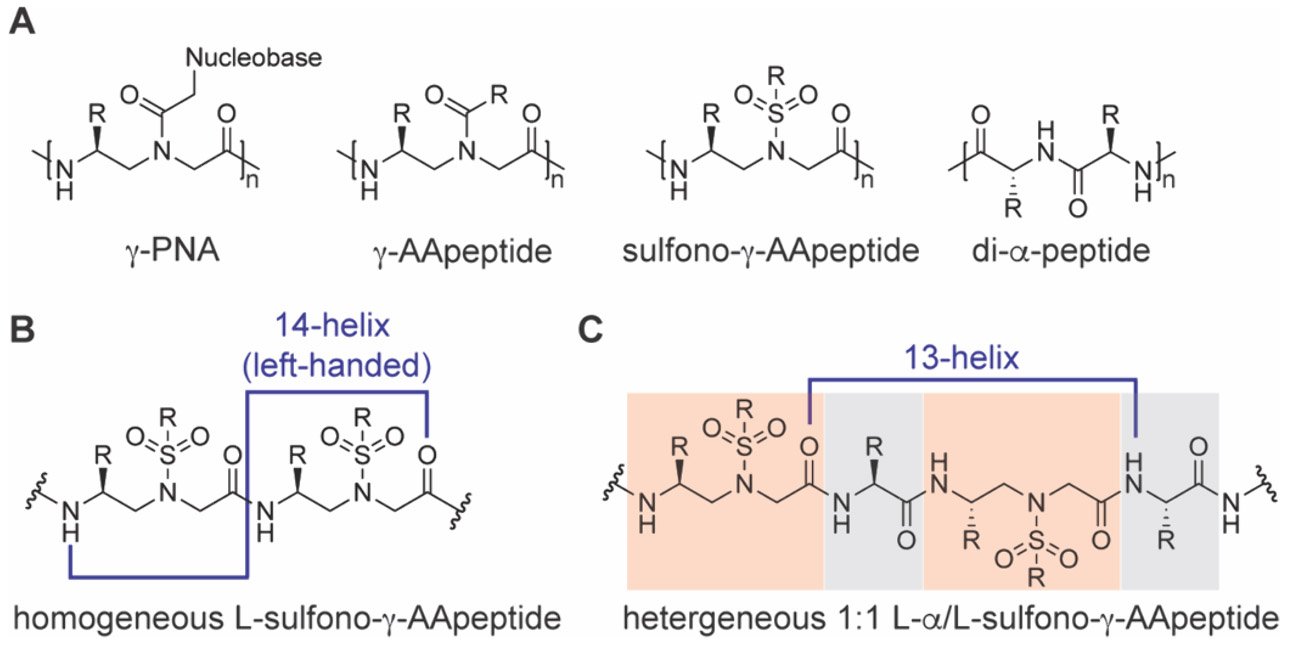
A) Chemical structures of PNA, γ-AApeptide, sulfono-γ-AApeptide, and di-α-peptide backbones. B) Hydrogen bonding pattern of a homogeneous l-sulfono-γ-AApeptide left-handed 14-helix. C) Hydrogen bonding pattern of a heterogeneous 1:1 l-α/l-sulfono-γ-AA 13-helix. Sulfono-γ-AA monomers are highlighted in orange. l-α-amino acids are highlighted in gray.
Cai and co-workers generated γ-AApeptides to target the canonical p53/MDM2 interaction as a case study.586 Although only slight inhibition was achieved (IC50 > 50 μM), the chemical diversity afforded to this design may provide better inhibitors in the future. Indeed, the group later developed a series of sulfono-γ-AApeptides, which have a greater helical propensity, and identified more potent inhibitors of p53/MDM2 interaction with submicromolar potency by fluorescence polarization (IC50 = 0.891 μM).594 Computational simulations suggest this architecture retains good overlap with the key residues of the p53 peptide when bound to MDM2 (Figure 45A,B). In a separate study, the same group developed sulfono-γ-AApeptide inhibitors of the difficult-to-drug target β-catenin based on a helical interacting motif in BCL9.595 In this work, cell uptake of FITC-labeled sulfono-γ-AApeptides and associated activity in live cells was achieved, suggesting primary sequence may be important for cell permeability of this class of peptidomimetics, as also observed in a recent study targeting amyloid β (Aβ42).596 Modeling studies again showed that a homogeneous sulfono-γ-AApeptide can project functional side chains and interact with a surface in a similar manner to native helical peptides (Figure 45C,D). Most notably, in all studies γ-AApeptides or sulfono-γ-AApeptides were shown to be stable against proteolytic degradation.594,595,597
Figure 45.

Sulfono-γ-AApeptides recapitulate native helical peptides to bind surfaces. A) Overlay of side chains on a sulfono-γ-AApeptide mimic (red) with native Phe19, Trp23, and Lue26 of p53 (green). B) Crystal structure of sulfono-γ-AApeptide bound to MDM2 (CCDC: 1841094). C) Overlay of sulfono-γ-AApeptide mimic (purple) with critical residues of native BCL9 helix (red). D) Overlay of sulfono-γ-AApeptide mimic (purple) with BCL9 helix (red) on the binding surface of β-catenin (PDB: 2GL7). A,B Adapted from ref 594. Copyright 2020 American Chemical Society. C,D Adapted from ref 595.
Due to difficulties in predicting the γ-AApeptide structure as well as the large chemical space available, de novo selection screens using OBOC libraries could be necessary for compound identification, and were piloted for undruggable targets such as amyloid β and the transcription factor STAT3.598,599 In the latter example, the selected γ-AApeptide inhibitors did not disrupt STAT3 dimerization but prevented STAT3-DNA binding, suggesting potential for this class to disrupt protein–nucleic acid interactions. Additional characterization of folding patterns and biological potential of γ-AApeptides for in vivo applications is necessary to further develop this class toward undruggable targets.600
4.2.6. Nonpeptidic Helix-Mimicking Scaffolds.
Many foldamers induce a secondary structure using backbone modifications that retain peptidic character; however, arguably the most important aspect of specifically targeting surfaces with high affinity is the position and orientation of the side chains to interact with key “hotspot” residues.601 In 2001, Hamilton and co-workers envisioned replacing the peptide-like backbone with an entirely nonpeptidic scaffold that would mimic the side chain projection of an α-helix. Using a functionalized terphenyl scaffold, they showed that relative orientation of side chains from the i, i+3/4, and i+7 positions in an α-helix are closely mimicked (Figure 46A,B).602 By functionalizing the aromatic rings at the ortho position, steric interactions induced a nonplanar, staggered conformation that mimics two turns of an α-helix.603 This low molecular weight scaffold with small molecule character could provide improved pharmacological properties such as proteolytic stability and oral availability. However, tedious synthetic routes and low solubility have thus far limited their use. To improve upon the design and production, many alternative nonpeptidic backbones were developed to include heterocyclic substituents and more accessible oligomerization strategies (Figure 46B). For a complete overview of nonpeptidic structural mimetics, refer to the corresponding reviews.504,604-607
Figure 46.

Nonpeptidic backbone mimetics. A) Nonpeptidic backbones mimic the side chain orientation of natural α-helices. B) Chemical structures of common helix-inducing nonpeptidic backbones.
Initial designs by Hamilton and co-workers using terphenyl and terephthalamide derivatives focused on recapitulating the binding residues of p53/MDM2 and BCL-XL/Bak.603,608,609 They showed that this class had the potential to target these interfaces with submicromolar efficacy and activity in cells.610 Since then, many different designs and improvements to nonpeptidic scaffolds have been developed for numerous undruggable targets including transcription factors,611-614 GTPases,615 and interactions governing amyloid aggregation,616-620 antiapoptotic proteins,621,622 and nucleic acids.623-625 A scaffold that has generated much interest is based on aromatic oligoamides (Figure 46B). This design exhibits predictable folding patterns controlled by intermonomer hydrogen bonding and ortho substituent interactions and can be adapted to both solution and solid phase synthesis for library preparation.613,626,627 The Wilson group generated a library of N-alkylated aromatic oligoamide helix mimetics and discovered potent and selective inhibitors of p53/hDM2 and MCL-1/NOXA-B interactions.628 Rather than screening for binding, they used a series of cell-based assays to look for phenotypic effects caused by library members, thus selecting for active compounds. Cell uptake, binding to hDM2 and disruption of the p53/hDM2 interaction were later confirmed for lead members. Interestingly, some lead compounds also bound to MCL-1, but not to structurally similar BCL-XL, suggesting potential for specific dual inhibitors, though this may also give rise to off target effects and the source of specificity remains incompletely understood. The main purpose of the study was to determine relationship between biophysical and cellular potency and shows that it is important to evaluate the interplay between potency for binding and actual mechanisms in cellulo, taking into consideration both on- and off-target effects.628
Another scaffold that has shown potential for targeting undruggable space is based on the oligooxopiperazine backbone developed by Arora and co-workers (Figure 46B).629 This backbone is derived from chiral α-amino acids, which are more effective for recognizing chiral protein pockets and easily diversified with myriad functional groups. The nonpeptidic chiral scaffold was initially designed based on a helix derived from the transcription factor HIF1α that interacts with the CH1 domain of p300/CBP (Figure 47A,B).614 By mimicking three key residues—Leu818, Leu822, and Gln824—oxopiperazine compounds (OHMs) were produced with submicromolar affinity for p300 by fluorescence polarization. Treatment with lead OHMs decreased gene expression associated with HIF1α targets in MDA-MB-231 cells. Importantly, this compound was shown to be well tolerated in vivo and modestly reduced tumor growth in a mouse xenograft model by intraperitoneal delivery, exhibiting the remarkable potential for nonpeptidic scaffolds as relevant therapeutics.614 Incorporating computational design strategies show potential to improve oligooxopiperazines for targeting HIF1α/p300 and other therapeutically relevant PPIs.630
Figure 47.

Oligooxopiperazines designed to inhibit HIF1α/p300 interactions. A) Model of HIF1α bound to the CH1 domain of p300/CBP (PDB 1L8C). Key residues of HIF1α CTAD Leu818, Leu822, and Gln824 in the binding pocket of the CH1 domain are highlighted and presented with an overlay of OHM 1. Adapted from ref 614. B) Chemical structure of lead OHM 1 compound.
The modular structural and synthetic strategies within this class of mimetics provide many avenues to improve activity including generating multivalent ligands and inducing native protein interactions among other functional attributes.631-634 The small molecule character and large chemical diversity afforded to nonpeptidic scaffolds situate this class for future development into inhibitors for a wide range of undruggable interactions. Further investigation into structural stability, specificity, bioavailability, and mechanisms in relevant in vivo disease settings is necessary. Many nonpeptidic scaffolds beyond those highlighted here exist and are in development as PPI or protein–nucleic acid interaction inhibitors, which are broadly covered in a recent review by Algar et al.607
4.3. Summary and Outlook for Non-natural Peptidomimetics
Non-natural peptidomimetics incorporate significant changes to the peptidic backbone yet retain the ability to fold into defined secondary structures. These peptidic and nonpeptidic mimetics are capable of folding into discrete architectures that portray key interacting residues in defined three-dimensional space, allowing the recognition of large, diverse surfaces characteristic of undruggable interactions. Incorporating nonnatural backbones, even in a small portion of the overall oligomer, leads to substantial improvement in metabolic stability, typically by preventing proteolytic degradation. Difficulties in solubility and production can be overcome by developing new building blocks and synthetic strategies as seen in microwave-assisted oligomerization approaches.552,635,636 Additional studies exploring the pharmacological properties and efficacy of these mimetics in vivo would greatly improve the knowledge and design principles necessary to generate therapeutic compounds.637 This information, along with further improvements through chemical stabilization strategies or the development of new scaffolds to improve cell permeability could lead to improved mimetics capable of targeting various intracellular interactions associated with disease.
5. SYNTHETIC PROTEOMIMETICS AND BIOLOGICS
The vast majority of synthetic peptidomimetics developed to date have been focused on secondary structure stabilization to target relatively compact hotspot surfaces on protein targets. Indeed, the work reviewed in Sections 2-4 is almost entirely made up of secondary structure mimetics. However, many protein–protein and protein–nucleic acid interaction surfaces utilize extended, noncontiguous protein sequences for high affinity and specific binding. Therefore, the ability to move beyond peptide secondary structure to tertiary and quaternary structure stabilization is an important and still emerging challenge for undruggable targets. Proteomimetics that function through tertiary or quaternary folding analogous to protein domains would be able to achieve specific interactions of high affinity with myriad biomolecular surfaces. In this section, we discuss proteomimetics that are synthetically tractable and incorporate chemical modification to stabilize higher-ordered structures (Figure 48). We will specifically focus on the application of biorthogonal chemistries for the total or semisynthesis of protein mimetics that target large “undruggable” protein and nucleic acid surfaces. Engineered proteins and protein mimetics that are genetically encoded, including monobodies, affibodies, nanobodies, DARPins, and related scaffolds are outside of the synthetic scope of this review and have been extensively reviewed elsewhere.27,638-644 Instead, our goal here is to focus on designs that include side chain functionality or backbone alterations to stabilize natural or non-natural tertiary or quaternary protein structure. In particular, we will focus on the role of these design elements in both biochemical and physicochemical properties required for increased stability and physiologic activity in targeting undruggable biomolecules.
Figure 48.

Designer synthetic proteomimetics utilize layered chemistries to stabilize higher-order tertiary and quaternary structure. Beyond encoding three-dimensional structure, synthetic modifications can significantly improve metabolic stability for cellular and in vivo applications. Structure derived from PDB:7RCU.
5.1. Design and Characteristics of Synthetic Proteomimetics and Biologics
Proteomimetics, or synthetic biologics, contain synthetically enhanced tertiary and/or quaternary structures that recreate features of a protein to drive function. In many applications, target synthetic biologics occupy a region of molecular space between short peptides and full length proteins; accessing these molecules therefore presents synthetic and pharmacologic challenges (Figure 1C). Research on stabilized secondary structures has shown how structural constraints can enhance pharmacologic stability and cell uptake.56,329,502 Likewise, recent efforts have been made to produce structurally stable tertiary or quaternary mimetics that contain desirable biophysical properties. This can be accomplished through partial or complete artificial backbone replacements, altered side chain interactions that promote defined interfacial contacts, as well as the incorporation of non-natural interdomain covalent cross-linking and bridging to define tertiary structure (Figure 48).645 Early work focused on structure stabilization through the use of biselectrophilic agents for cross-linking amines on lysine side chains to explore tertiary folding and stability.646,647 However, the abundance of these residues in natural protein domains reduces inherent selectivity, severely limiting amenable designs. Alternatively, total chemical synthesis of proteins emerged to site-selectively incorporate extensive natural and non-natural building blocks for defined intermolecular ligation,648 utilizing secondary structures such as sheets or helices to create higher order structures (Figure 49). These designs, which we refer to as synthetic biologics or proteomimetics, are more amenable to extensive chemical modifications to enhance biochemical and pharmacologic activity.
Figure 49.
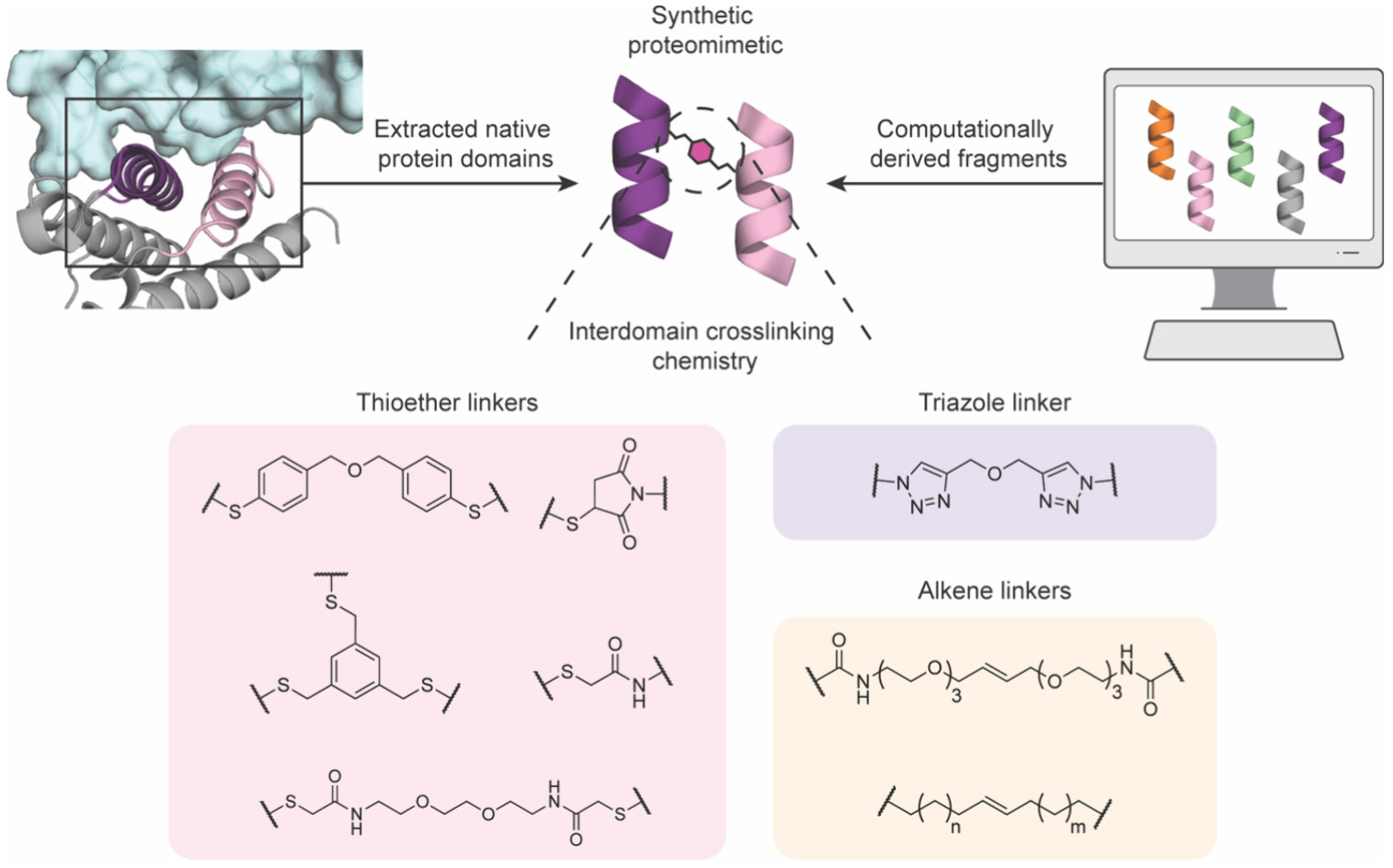
General design and production of synthetic proteomimetics and biologics. Chemical structures of selected common linker chemistries for induced tertiary or quaternary formation are shown. Structure derived from PDB 1IAR.
Synthetic proteomimetics or biologics are assembled through convergent synthesis from multiple native or modified peptide domains to create higher order structures with defined local and global three-dimensional structures. These extracted domains or smaller fragments can include stabilized β-sheet649 and coiled-coil motifs650,651 present across many protein–protein and protein–nucleic acid interfaces (Figure 49).652 For example, Arora and colleagues identified the Nervy homology two (NHR2) domain of the AML1-ETO-containing transcription factor complex as a necessary coiled-coil motif that interacts with NHR2-binding (N2B) domains of E-proteins. They extracted the native primary sequence and synthetically produced the individual helices then covalently ligated the helix dimer to produce a synthetic proteomimetic capable of adopting a defined tertiary structure. Maintaining the key interacting residues resulted in a coiled-coil mimetic capable of binding to N2B peptides with much greater affinity than the native NHR2 domain.650 Alternatively, computational methods can produce entirely artificial domains and folds as seen with the CovCore approach (Figure 49).653 In many of these designs, intra- or intermolecular covalent linkages as well as optimized interfacial contacts between domains are essential for obtaining minimal structures that retain high affinity and stability.654 Common interdomain linking chemistries include those previously described in peptide macrocyclization and secondary structure stabilization such as thioether formation between bis- or triselectrophiles or Michael acceptors and the side chain of cysteine,651,653-655 bis-triazole formation via azide–alkyne cycloaddition,650 and alkene formation from ring closing metathesis649,656 (Figure 49). In these examples, synthetic tractability is paramount for including site-selective conjugation through layered bioorthogonal chemistries to produce defined higher order structures, which is a hallmark property of synthetic proteomimetics less feasible with larger engineered proteins. Additional linkages such as amide or lactam formation657 and more recently Diels–Alder cycloadditions658 may also prove useful as selective conjugation strategies.
In addition to interdomain conjugation approaches, partial or complete replacement of the backbone can result in structurally and metabolically stable tertiary and quaternary mimetics.645,659 The challenging endeavor of producing protein-like folding patterns from non-natural architectures hints at the prospect of creating structures and functions currently unknown to nature. However, the difficulties associated with the requisite near de novo design have thus far limited the applications of non-natural synthetic proteomimetics to just a few structural classes, including zinc finger,660-662 amyloid,663 and Z-domain mimetics.664 Moreover, most examples of tertiary domain mimetics have been focused on extracellular targets.645,657,665,666
5.2. Designer Synthetic Biologics for Undruggable Targets
Many intracellular undruggable targets require high affinity, high specificity, cell permeable and metabolically stable chemical probes, and therapeutics for efficacy. Identifying chemical scaffolds with these properties has remained a challenge, and only a few examples exist within the class of synthetic biologics and proteomimetics. For example, a recent study by Sadek and Wuo et al. aimed to disrupt the interaction between NEMO, which is a scaffolding protein necessary for downstream NF-κB signaling, and the viral protein vFLIP (Figure 50A).667 Screening of a 40,000 member small molecule library against the extended coiled-coil NEMO/vFLIP binding interface identified only compounds with modest binding inhibition (IC50 < 65 μM). These compounds also showed highly active submicromolar inhibition in a cell proliferation assay; however, there was no specificity between vFLIP-dependent and control cell types, suggesting nonspecific off-target effects. To develop more specific NEMO/vFLIP inhibitors, they rationally designed linear and HBS-constrained peptides to mimic the key hotspot residues on helix two of NEMO. Unfortunately, neither design inhibited this interaction at 100 μM, likely due to the requirement of the coiled coil partner to properly situate the hotspot residues for binding. They then turned to the cross-linked helix dimers (CHD) approach to generate a synthetic coiled coil mimetic.654 Design parameters obtained by computation and experimental characterization informed the creation of CHD3NEMO, which incorporates both helix one and two of NEMO ligated by a bis-triazole linker with natural and noncanonical amino acids to potently and specifically disrupt the NEMO/vFLIP interaction (Ki = 6.9 μM) (Figure 50B). This synthetic proteomimetic also displayed increased resistance to thermal denaturation and proteolysis. Importantly, this design was cell permeable through an undetermined active uptake mechanism and capable of dose-dependent inhibition of vFLIP-mediated NF-κB activation and cell proliferation. Additional experiments in vivo resulted in delayed tumor growth and improved survival in a primary effusion lymphoma xenograft mouse model, showcasing the therapeutic potential for synthetic proteomimetics.667
Figure 50.

Synthetic proteomimetic inhibitor of NEMO/vFLIP interaction. A) Thus far, small molecules and single helix mimics have proven incapable of inhibiting the NEMO/vFLIP association. PDB: 3CL3. B) Structure and sequence of tertiary NEMO mimic CDH3NEMO, which was shown to be a potent inhibitor of vFLIP.667
As the study by Sadek and Wuo et al. shows, affinity and specificity are important to develop robust inhibitors, but metabolic stability and cell permeability are equally if not more necessary for bioactive molecules. The larger size of synthetic proteomimetics complicates the delivery to intracellular organelles, as it is unlikely to permeate cells through passive diffusion, but rather active uptake mechanisms such as endocytosis or macropinocytosis. Fortunately, these mechanisms are often upregulated in disease such as cancer, which may provide selective uptake to certain cell types.668 Hong and Yoo et al. developed a synthetic proteomimetic pan-Ras inhibitor derived from the interacting helix–loop–helix domain of SOS that engages the nucleotide-bound Ras.669 In addition to increased conformational and proteolytic stability as well as high affinity target engagement, they also observed increased uptake into cell lines harboring Ras mutations, presumably through amplified macropinocytosis as previously shown.668 This suggests certain cancer cell lines are more amenable to proteomimetic therapeutics and that specificity can be achieved through selective uptake mechanisms. In a separate study, Yang, Zhang, and You et al. developed a BCL9/β-catenin interaction inhibitor based on the helix–loop–helix domain of ECRV stabilized by a thioether linker.670 To overcome poor cellular uptake and promote endosomal escape, the cECRV proteomimetic was attached to a poly-l-lysine coated gold nanoparticle (pAuNP). The pAuNP-cECRV construct induced apoptosis of the WNT-hyperactive cancer cell line HCT116, whereas free cECRV mimetic and cold pAuNPs showed no effect, highlighting the use of nanocarriers to overcome membrane permeability limitations for some synthetic proteomimetic designs.
5.2.1. Synthetic Biologics as Direct Modulators of Transcription Factor Activity.
Transcription factors (TFs) remain one of the most challenging classes of undruggable targets due to the reliance of TFs on extended PPI and protein–nucleic acid interactions to carry out their function in regulating gene expression. Aside from nuclear receptors, which can often be regulated by small molecule binding, many classes of TFs lack defined druggable pockets and function mainly through complex regulatory networks involving large surface interactions with highly disordered domains.671,672 This makes most small molecules and small peptidomimetic designs based solely on secondary structure unlikely to retain necessary affinity and specificity to disrupt relevant TF interactions. On the other hand, the larger size and three-dimensional structural complexity of suitably designed synthetic proteomimetics could be promising for targeting undruggable TFs. Furthermore, tertiary domain mimetics derived from native TFs could be developed with two modes of action to antagonize TF function. First, they could be developed to mimic and/or block PPIs that are necessary for effector complex formation. Second, synthetic biologics could be developed to mimic the DNA binding domain of a TF, thereby enabling direct binding of target DNA sequences and inhibition of TF-dependent gene regulation at those sites. Displacement of TF-DNA interactions remains a sought-after approach for TF inhibition. However, TFs bind and recruit many protein cofactors to influence gene expression, and disrupting these interactions may provide a means to regulate transcription. The inherent complexity of these interactions involving flat and flexible protein surfaces reduces the efficacy of secondary structure mimics. Adihou et al. showcased this in the development of a two-helix synthetic proteomimetic to disrupt the TEAD/VGL4 interaction, wherein both helices derived from VGL4 were necessary for potent binding.673 Chemical cross-linking via lactam formation of the distinct helices considerably increased helicity in solution, affinity for hTEAD, and proteolytic stability. Unfortunately, this design showed poor cellular uptake, likely due to the overall negative charge. Subsequent terminal modification with a cell penetrating peptide, Tat, led to a bioactive mimetic that upregulated TEAD target genes and restored Hippo signaling pathway phenotypes.673 The versatility of synthetic proteomimetics extends beyond native helical domains, but also can incorporate stabilized sheet structures500 and non-natural domains. For example, Wilson and co-workers produced a synthetic proteomimetic derived from the C-terminal transactivation domain of an oncogenic bHLH member, HIF1α, that interacts with p300 to regulate transcription. They previously identified two helical domains that are required for the HIF1α/p300 interaction674 and developed a potent oligoamide non-natural peptidomimetic (helix mimetic 1).612 However, helix mimetic 1 also potently inhibited p53/hDM2 suggesting a nonspecific component to inhibition.675 To increase specificity, they synthetically fused the oligoamide helix mimetic and a necessary adjacent HIF1α-derived helix, resulting in a peptide–oligoamide mimetic hybrid. Although this construct resulted in reduced potency, it was accompanied by a significant increase in selectivity,675 and the natural/non-natural hybrid nature exhibits the extensive modularity afforded to synthetic proteomimetic designs.
A defining feature of TFs is the recognition and binding to distinct genomic DNA motifs to regulate gene expression. Early studies into the modularity of TF families demonstrated that TF DNA-binding domains (DBDs) could retain affinity and specificity for target DNA even when separated from the rest of the native TF protein.676,677 This work inspired the creation of truncated DBDs derived from different TF families such as zinc fingers,676 homeodomains,678,679 basic leucine zipper domains (bZIP),680 and basic helix–loop–helix domains (bHLH).681,682 However, as discussed extensively in this review, the larger native peptide sequences in many early designs suffer from poor cellular uptake and significant proteolytic susceptibility. Coupled with the need to compete with high affinity endogenous TF-DNA interactions (usually in the low nM range), natural peptide or protein sequences struggle as pharmacologically viable chemical probes and therapeutics targeting TFs.677
An exemplar case study in the development of a natural TF-mimetic involves the exploration of the dominant negative, MYC-derived molecule Omomyc. A key attribute of many TFs is the requirement for homo- or heterodimerization to assemble the competent DBD. This is true, for example, in the basic leucine zipper (bZIP) and basic-helix–loop–helix (bHLH) TF families, the latter which holds the oncogenic protein MYC. MYC is a bHLH-family protein that requires heterodimerization with a partner MAX in order to assemble the intact DBD, bind “E-box” containing DNA motifs and regulate the expression of proliferative gene programs. Therefore, inhibition of dimerization interactions to prevent complex assembly of TFs like MYC and MAX is an approach that has garnered a lot of interest over the years. In 1998, Soucek et al. explored why MYC was incapable of homodimerization but could bind to the obligate partner MAX. By mutating only four residues in the leucine zipper helix of MYC, a structural analog termed Omomyc was developed that could homodimerize or heterodimerize with MYC or MAX to prevent DNA binding.683 Although initially expressed using natural protein machinery in E. coli, a team at Merck demonstrated a feasible solid-phase and native chemical ligation synthesis of the 91-amino acid miniprotein, albeit in limited quantities (10–15 mg) and yields (8–10% yield).684 Several classic studies in peptide total synthesis681 as well as recent approaches using improved SPPS and cross-coupling chemistries have also described total synthesis of “native-like” MYC-derived protein mimetics685 with biological activity. Despite its large size, disordered structure and proteolytic susceptibility, Omomyc was unexpectedly shown to be internalized into cells by fluorescence microscopy, likely by clathrin-mediated endocytosis or macropinocytosis.686 In subsequent in vivo studies, it was rapidly cleared from mouse plasma where it was distributed to the liver and kidneys and quickly degraded, thus highlighting expected limitations of miniproteins derived from entirely native peptides.687 However, a separate study showed Omomyc was able to distribute to tumor tissue and antagonize MYC-dependent signaling.688 Nevertheless, nearly 30 years after its inception, Omomyc (OMO-103) has successfully completed a phase I clinical trial, which is a promising endeavor for synthetic miniproteins aimed at an important undruggable target.689
To overcome many of the existing challenges with natural polypeptide-derived miniproteins aimed at TFs, our lab recently developed a new class of modular synthetic biologics we termed synthetic transcriptional repressors (STRs).690 We aimed to created compact, fully synthetic and chemically stabilized TF DNA binding domain mimetics that could directly target TF-defined DNA binding sites, thereby blocking downstream gene expression (Figure 51A). As a first target, we developed a non-natural architecture to mimic the tertiary and quaternary bHLH domain structure of MAX. We wanted to avoid inefficient synthesis of long linear polypeptides that span the necessary regions of a functional bHLH domain (>80 AAs in MYC or MAX), which would further hinder the incorporation of non-natural amino acids and structural modifications. We identified minimal regions of the basic (B) and leucine zipper (Z) helices from MAX that, when ligated together at appropriate sites, could retain the potential for homodimerization to form a minimal, yet highly functional bHLH domain structure (Figure 51A). Intriguingly, we found that this dimerization could be preserved and promoted by covalently tethering B- and Z-helices from opposing dimerization partners, requiring a “sandwich-like” dimerization event to form the bHLH tetrahelix bundle, significantly departing from the scissor-like dimerization by natural bHLH polypeptides (Figure 51C,D). To stabilize these structures, we identified compatible, layered chemistries for local secondary structure stabilization within α-helical structures (e.g., side chain stapling mediated by RCM) and interdomain covalent construction of tertiary domain structure by orthogonally masked thiol-maleimide ligation, which can take place under facile aqueous conditions for relatively high yielding synthetic biologic construction (Figure 51B). These positions and chemistries were identified using structure-based design and iterative biochemical profiling, ultimately identifying representative lead compounds with low nM affinity and high specificity for canonical E-box DNA sequences (5′-NCACGTGN-3′). Importantly, these attributes enable direct competition with full-length MYC/MAX and MAX/MAX bHLH domains for DNA binding, the first such compounds with this activity and clear mechanism of action. In parallel, we demonstrated that synthetic stabilization confers significant protease resistance and enhanced cellular uptake, allowing lead STRs to bind genomic E-box DNA sites and inhibit expression of MYC-dependent proteins in MYC-driven Burkitt’s Lymphoma cell lines.690 This work demonstrated how synthetic optimization and layering of both secondary and higher-order stabilization strategies can simultaneously enhance biophysical and pharmacological properties of non-natural proteomimetics.
Figure 51.

Synthetic transcriptional repressors (STRs) are modular proteomimetics capable of sequence-specific DNA-binding. A) Convergent synthetic approach to prepare STRs derived from MAX. B) Convergent synthetic scheme to produce stabilized STRs purified and characterized by HPLC-MS. C) Crystal structure of bHLH STR on E-box DNA displaying the sandwich-like assembly. PDB: 7RCU. D) Overlay of crystal structures of STR (blue/pink) and MAX:MAX (orange/yellow) homodimers. PDB: 7RCU and 1HLO, respectively. E) Structure-blind design of STRs derived from distinct bHLH domains. Adapted from ref 690.
A second goal of this study was to determine whether the inherent modulatory properties found in natural bHLH domains (of which there are >100 in the human proteome)691 could be retained in the STR architecture. Crystal structures of MAX-derived STR homodimerized and bound to E-box DNA confirmed the intended, non-natural “sandwich cross-dimer” fashion, yet faithful retention of overall domain register and specific side chain interactions with target nucleotides (Figure 51C,D). This shows the synthetic STR architecture mimics the DNA-binding specificity and structure of native bHLH domains and can be adapted to target other sequences. We therefore used the modular STR structure to recapitulate the binding activities of two other intractable oncogenic TFs, TFAP4 and OLIG2. Using a structure-blind design, we synthesized and ligated Z- and B-helices based on the primary sequence of TFAP4 or OLIG2 and showed that model STRs can approach or even exceed the DNA-binding specificity of native TF domains (Figure 51E).690
Recently, we extended the STR platform approach to mimic bZIP TFs, starting with the stress-responsive TF X-box binding protein 1 (XBP1).692 While bZIP TFs bind DNA through homo- or heterodimerized helices that intercalate the major groove, the bHLH-derived binding approach deviates substantially enough from that of bZIPs to prevent bHLH-derived STRs from mimicking a bZIP TF. Thus, we developed a bZIP-mimicking STR architecture that involved (i) identification of the minimal, linear basic and leucine zipper domain residues required for DNA binding after (ii) covalent ligation of two DNA binding helix monomers (Figure 52). We further found that modification of helix–helix contact residues and incorporation of secondary structure-stabilizing macrocycles was necessary to create high affinity and high specificity XBP1-derived mimetics. Beyond development of a bZIP-derived architecture, there were several notable biochemical and biological discoveries in this work. First, we recognized that the target motif for XBP1—the UPRE—encompasses the canonical hypoxia-response element (HRE) motif bound by HIF1α and HIF2α in response to low oxygen levels (Figure 52A). Thus, we reasoned that an XBP1-derived STR should be able to bind and regulate hypoxia-regulated target genes and potentially oppose the action of oncogenic HIF proteins (Figure 52C). Indeed, we demonstrated that a lead XBP1-derived STR retains improved pharmacologic properties and is able to directly compete with hypoxia-dependent target gene binding (via ChIP-qPCR and ChIP-seq) and gene activation through global RNA-seq analyses. Importantly, these STRs could be delivered via intravenous injections where they modulated hypoxia-dependent gene expression in established 4T1, triple-negative breast cancer (TNBC) tumors. Repeated treatments did not exhibit overt toxicity or immunogenicity, suggesting in general that chemically stabilized proteomimetics may be designed without the worry of inherent immunogenic properties that may come from non-natural or non-human peptide sequences. Finally, hypoxia-targeting STRs showed significant inhibition of TNBC xenografts as well as spontaneous metastasis in syngeneic TNBC tumor models. Collectively, this work extends the synthetic biologic space to the bZIP family of TFs, demonstrates that TFs from distinct families can be targeted simultaneously and confirms that the pharmacologic stabilization of larger proteomimetics can engender in vivo activity for seemingly “undruggable” targets like XBP1 and HIF1α.692
Figure 52.

STRs derived from bZIP TF domains. A) Structures of bHLH TF HIF1α/ARNT and bZIP TF XBP1u DNA binding domains (DBD). The HRE DNA motif recognized by the bHLH transcription factor HIF1α is overlapped by the UPRE motif recognized by the bZIP transcription factor XBP1. Structures derived from PDB 4ZPK and 2H7H, respectively. B) Design and structural considerations for stabilized bZIP STRs derived from bZIP DBDs. C) bZIP STR-mediated HIF1α inhibition in the hypoxic tumor microenvironment prevents cancer cell survival and metastasis. Adapted from ref 692.
5.3. Summary and Outlook for Synthetic Proteomimetics and Biologics
Interactions driving many undruggable targets and processes mainly rely on highly dynamic and disordered surfaces for which small molecules and secondary structure mimetics struggle to attain affinity and specificity. Alternatively, proteins have precise recognition capabilities spanning large areas owing to tertiary or quaternary folding, but significant drawbacks including stability, cell permeability, and immunogenicity limit what can be feasibly targeted.693 Synthetic proteomimetics take advantage of the recognition abilities of proteins, but also have increased affinity and targetability, enhanced biostability and cellular uptake, and potentially improved pharmacology. The synthetic nature allows the reconstruction of stabilized higher-order domains to tune the biophysical properties and improve function. Altering the structural topology by site-specifically introducing backbone modifications or side chain interactions can recapitulate natural folding patterns or generate new structures not observed in nature yet retain ability to modulate biomolecular interactions. Although drug-like MCPs and secondary structure mimetics discussed above possess some advantages over larger proteomimetics for entering the clinic, these are not amenable to a wide variety of undruggable target classes. For example, certain PPIs, protein–nucleic acid interfaces and direct targeting of DNA motifs (a long-hypothesized approach with many potential advantages) are likely to require larger and potentially more polar binding footprints for efficacy. Thus, efforts to develop peptidomimetics or proteomimetics for these targets will require a balance of potency, specificity and pharmacokinetic and physicochemical properties. Stabilized proteomimetics may be the ideal, or perhaps the only solution for some targets. Future work to elucidate the mechanisms governing cellular uptake and pharmacologic properties for more examples of this class of molecules is needed to improve their design and hopefully contribute to their translation as therapeutics.
Although not a focus in this review, designer proteins based on structural scaffolds produced using genetically encoded methods are potentially promising tools for targeting undruggable classes. We recognize the immense potential of these engineered proteins, particularly through the genetic encoding of noncanonical amino acids, yet this is still restricted by the orthogonality and number of modifications that can be installed, thus limiting natural and engineered proteins in biological applications. Advances in synthetic approaches such as microwave-assisted solid phase synthesis, native chemical ligation, and alternative bioconjugation strategies, will allow these engineered scaffolds such as affibodies to be synthetically improved after selection strategies.694 Allied advances in computational de novo protein design will provide novel synthetic scaffolds and elucidate proteomimetic structures capable of disrupting previously intractable interactions.695,696 The synthetic modularity associated with improved biophysical properties afforded to synthetic proteomimetics present exciting opportunities for this emerging class of molecules.
6. SUMMARY AND PERSPECTIVES
The ability to target truncal drivers of disease ultimately begins and ends with the ability to discover drug-like molecules that act on specific targets. While small molecules and biologics represent the two largest classes of therapeutics, a molecular middle ground presents immense promise for new chemical probe and therapeutic development. The peptidomimetics covered in this review have evolved from the historic success of bioactive peptides like insulin,697 the ~100 FDA approved peptide-based drugs, and the recent blockbuster status of “designer” peptides for extracellular targets like GLP-1 agonists.6 However, the hard-wired physical and chemical properties of natural peptides and biologics largely precludes their utility to target intracellular undruggable biomolecules.
Here, we have focused on four overlapping classes of synthetic, stabilized peptide- and protein-derived mimetics that draw upon structural and functional properties of small molecules and biologics. The combination of natural and non-natural amino acids with biorthogonal chemistry has enabled the development of molecules with augmented properties needed for therapeutics, namely: target affinity, specificity, metabolic stability, cell membrane permeability, and in vivo bioavailability. The unique ability to recognize extended, diverse surfaces with high affinity and specificity and ease of functionalization for enhancing desired properties situate this class of compounds suitably for targeting challenging biomolecules. This review covers a broad list of targets with some containing examples represented by most classes highlighted here, such as β-catenin and p53/MDM2, while others like small GTPases are overrepresented by MCPs and side chain stabilized peptides (Table 1). In the case of p53/MDM2, many approaches have been utilized to recapitulate the interacting domain of p53 to disrupt MDM2 binding. In general, mimetics stemming directly from native protein structures (e.g., MCPs and side chain stabilized peptides) displayed greater affinity and inhibition in the single-digit nanomolar range compared to non-natural mimetics, although these still achieved mid-to-high nanomolar potency. However, non-natural backbones generally confer increased resistance to proteolysis even compared to stapled counterparts,529 suggesting there may be a trade-off between potency, stability, and cell permeability that should be considered when designing and pursuing architectures for developing chemical probes and therapeutics.
Table 1.
Representative Undruggable Protein/Pathway Targets and Exemplary Application to Create Stabilized Peptidomimetics or Proteomimetic Modulators
| Target | Biological Significance or Role |
Target Mechanism of Action | Macrocyclic Peptides |
Side Chain Stabilized Peptides |
Non-natural Peptidomimetics |
Synthetic Proteomimetics |
|---|---|---|---|---|---|---|
| Amyloid proteins | Fibrillar protein | Prevent aggregation and fibril formation | 252 | 545, 596, 598, 616-620 | 663 | |
| BCL-2 family | Apoptosis regulator | Promote pro-apoptotic programs | 201 | 383-391 | 525-529, 608, 609, 621, 622, 628 | |
| β-catenin | Transcription factor | Inhibition of Wnt signaling pathway | 196 | 350, 354, 399, 400, 456, 474 | 578, 595 | 670 |
| c-MYC | Transcription factor | Prevent Max dimerization; E-box DNA binding | 138, 139 | 611 | 683, 685-690 | |
| ERα | Transcription factor | Block agonist functon; co-activator binding; DNA binding | 325, 343, 465, 469, 481, 485, 486 | |||
| Gα G-proteins | GTPase subunit protein | Prevent nucleotide exchange and enzymatic activity | 136, 155, 162-164, 265 | |||
| HDAC family | Histone deacetylase | Inhibit enzymatic lysine deacetylase activity | 94, 132, 133, 214 | 342 | ||
| HIF1α | Transcription factor | Prevent ARNT dimerization; HRE DNA binding | 285, 286 | 415, 416 | 612, 614 | 675, 692 |
| KDM family | Histone demethylase | Inhibit enzymatic lysine demethylase activity | 243, 262-264 | |||
| NRF2 | Transcription factor | Prevent KEAP1-mediated degradation of NRF2 | 215, 218 | 357-363 | ||
| NEMO | Inhibitor of IKK kinase | Inhibit activation of NF-κB pathway | 92, 156 | 667 | ||
| NOTCH complex | Transcription factor | Inhibit MAML co-activator binding; NOTCH-CSL binding; DNA binding | 396 | |||
| p53 | Transcription factor | Prevent E3 ligase-mediated degradation of p53; promote stability and DNA binding of mutant p53 | 119, 176, 238 | 401-413, 465, 473, 475-478, 481, 493, 498 | 519, 521-524, 540, 556, 582, 586, 594, 603, 613, 628 | |
| Ras family | Small GTPase | Inhibit enzymatic activity | 150-154, 203, 206-208, 211, 212, 251, 266, 284 | 418, 419 | 669 | |
| TEAD proteins | Transcription factor | Inhibit YAP/YAZ/TEAD interactions and Hippo pathway signaling | 74, 190 | 456 | 673 |
While tremendous progress has been made across these categories, and molecules in these classes have even entered human clinical trials, there are still several challenges and horizons in the field to be addressed. First, the overarching limitation for many of these molecules is the fact the larger polar surface area, and therefore hydration, of peptides and protein mimetics limits their passive diffusion through cellular membranes, which is required for intracellular targets. Additional experimental and computational data sets should continue to refine our ability to hone-in on regions of molecular space that are likely to result in more permeable molecules, yet these regions are likely to be distinct between MCPs and synthetic biologics, for example. For molecules that may need to rely on active cellular uptake pathways, such as macropinocytosis and other endocytosis related mechanisms, the ability to properly traffic and access the cytosol is of paramount importance (Figure 53). For one, developing assays that quantitatively measure productive cellular uptake and intact distribution to specific cellular compartments can help in this regard. Methods like the CAPA assay698 and NanoClick699 assays represent recent advances that complement imaging-based approaches. Many studies, including our own recent work with synthetic mimics of TF DNA binding domains690,692 have highlighted the importance of concurrent survival of cell permeable species, as many seemingly permeable peptides are rapidly degraded in cells and would thus be inactive.
Figure 53.
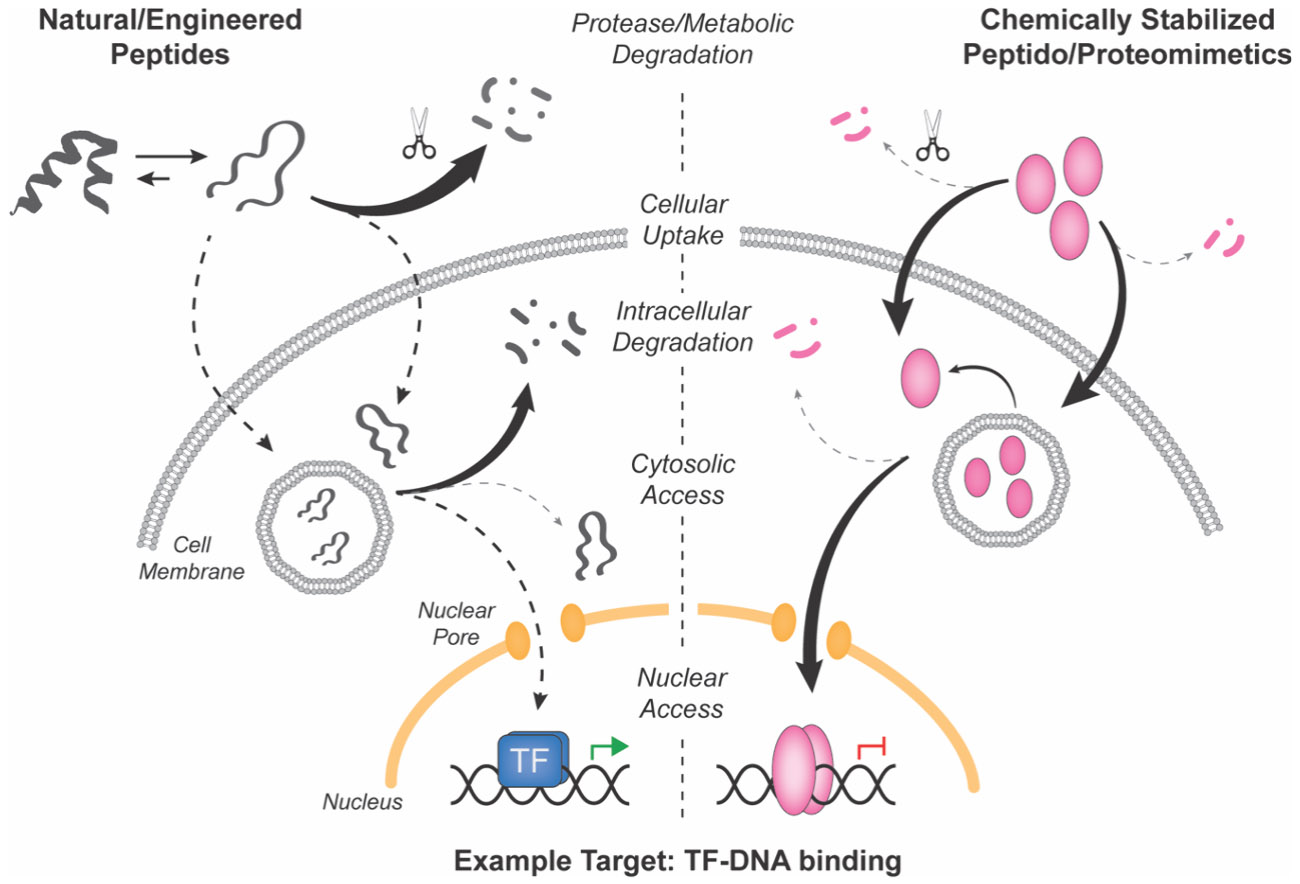
Designer peptides and peptidomimetics overcome limitations of unmodified peptides for modulating intracellular targets, such as transcription factor-mediated protein-protein and protein- DNA interactions.
Taking a page from the focus in macrocyclic peptide optimization, work in optimizing hydrocarbon stapled peptides is likely to advance to more active compounds through focused tracking of the mechanism(s) underlying cell permeability as well as the efficiency of this process. For example, several reported stapled peptides that are active in cells and in vivo utilize active uptake pathways, which then requires monitoring of compound stability within and through the endolysosomal pathway. Alternatively, trying to identify molecules that are permeable via passive membrane diffusion is more likely to bring cellular activity in line with biochemical potency. However, this may require the continued development and deployment of larger scale screening technologies, predictive computational models and identification of more compact binding epitopes. Balancing these properties and choosing which may be possible for a given target of interest will and should remain at the center of stabilized peptide and proteomimetic medicinal chemistry efforts and hopefully identification of development candidates and therapeutics.
Beyond cell permeability, recent research has begun to elucidate the necessary properties for bioavailability and efficacy in vivo;65 however, much more work is needed to establish general rules and guidelines for effective designer peptide and peptidomimetic therapeutics.303 This includes looking into pharmacokinetic profiles in various models to elucidate serum stability and biodistribution, as well as determining optimal delivery routes and vehicles.700 The advances of nanoparticle delivery for nucleic acids has rapidly expanded the effective reach of genetically encoded modalities recently, and it is likely that opportunities exist to augment the in vivo pharmacokinetic profiles of stabilized peptides and proteomimetics targeting undruggable targets.701
Another area of exploration could come in expanding the current target landscape for designer peptide and protein mimetics, as well as their mechanism of action. While many targets have been reviewed here, there is a common refrain of just a few prototypical protein targets across classes, such as the p53/MDM2 interface and the BCL2-family of apoptotic regulators (Table 1). This makes sense as these were early targets for the space; however, there is a large universe of undruggable targets and target biomolecules to be explored. For example, a less explored class of undruggable targets are epigenetic modifying proteins such as histone deacetylases and DNA methyltransferases. While a few peptidomimetic analogs targeting this class are reviewed here, such as the HDAC family and other chromatin regulators, these are mainly designs within MCPs or side chain stabilized peptide classes. This is not entirely unexpected due to the accessibility for these designs compared to wholly unnatural mimetics or larger synthetic biologics. However, expanding the target scope for these latter classes may yield molecules with better activity, which has been observed for targeting diverse classes of TFs.599,650,690,692 In this vein, further expansion to RNA and DNA as targets for upstream regulation of protein expression or activation of entire gene programs represents an exciting and perhaps fruitful landscape for these modalities. In parallel, the ability to target larger protein surfaces presents opportunities to create protein stabilizers, molecular glues and bifunctional modalities that could enhance the efficacy and scope of these molecules. Additionally, the capabilities of peptido- and proteomimetics extend beyond highly selective target interactors, and recent designs for generating artificial protein structures and biocatalysts has garnered much interest.702,703 Enhancing designer peptidomimetics with additional functionality may serve to create chemical probes and therapeutics with a wide range of functions to modulate currently undruggable targets and pathways.
Finally, dramatic advances in structural biology and computation,704 for example with AI and ML modeling, have tremendous potential for the discovery and selection of target interfaces as well as the guided optimization of synthetic designer peptide- and protein-mimetics. These approaches coupled with synthetic stabilization, optimization and experimental validation of peptido- and proteomimetic constructs greatly improve upon the therapeutic potential of these modalities. Allied advances in computation and modeling to predict folding patterns and ligandability of biomolecule surfaces to elucidate targetable areas will help jumpstart the development of peptidomimetic ligands.705,706 Alongside evergrowing advances in other therapeutic modalities, the molecules reviewed here hold tremendous promise to further reclassify and ultimately overcome a long list of “undruggable” targets in disease.
ACKNOWLEDGMENTS
We thank S. Ahmadiantehrani for assistance with figure and text editing. We are grateful for the financial support of this work from the National Institutes of Health: R01CA292876 and R01CA289378 (R.E.M.) and F32GM148062 (C.S.S), the V. Foundation for Cancer Research (R.E.M.), American Cancer Society-North Central Research Scholar Grant RSG-17-150-01-CDD (R.E.M.), the Alfred P. Sloan Foundation FG-2020-12839 (R.E.M.), and the Ullman Family Team Science Award (R.E.M.).
ABBREVIATIONS
- ACHC
aminocyclohexanecarboxylic acid
- ACN
acetonitrile
- ACPC
aminocyclopentanecarboxylic acid
- ADT
2-aryl-4,5-dihydrothiazole
- AI
artificial intelligence
- Aib
α-aminoisobutyric acid
- AKAP
A-kinase-anchoring protein
- AKB
protein kinase A binding domain
- Akt
protein kinase B
- Ala
Alanine
- AML1-ETO
acute myeloid leukemia 1 protein-ETO fusion protein
- Aoc
aminooctanoic acid
- APP
amyloid precursor protein
- Arg
arginine
- ARNT
aryl hydrocarbon receptor nuclear translocator
- ASF1
antisilencing function 1 protein
- Asp
aspartic acid
- ATC
4-amino(methyl)-1,3-thiazole-5-carboxylic acids
- BAD
BCL-2 associated agonist of cell death
- BAK
BCL-2 homologous antagonist killer protein
- BCL-2
B-cell lymphoma 2
- BCL9
B-cell CLL/lymphoma 9 protein
- BCL-XL
B-cell lymphoma extra large
- BCP
bicyclic peptide
- BEAS-2B
human bronchial epithelium cell line, non-tumorigenic
- BH3
BCL-2 homology domain 3
- bHLH
basic helix–loop–helix
- BID
BH3 interacting-domain death agonist
- BIM
BCL-2 interacting mediator of cell death
- Boc
tert-butyl carbamate
- B-Raf
serine/threonine protein kinase
- Bts
benzothiazole-2-sulfonyl
- bZIP
basic leucine zipper
- CAL
CFTR-associated ligand
- cAMP
adenosine 3,5-cyclic monophosphate
- CBP
CREB binding protein
- CD
circular dichroism
- CD137
tumor necrosis factor receptor
- CDR
complementarity determining regions
- CFTR
cystic fibrosis transmembrane conductance regulator
- CHD
cross-linked helix dimers
- ChIP
chromatin immunoprecipitation
- CHIP
C-terminus of Hsc70 interacting protein
- cIAP1
cellular inhibitor of apoptosis protein 1
- CPP
cell-penetrating peptide
- CSL
CBF1, suppressor of hairless, Lag-1 (transcription factor in the Notch pathway)
- CTAD
C-terminal transactivation domain
- CtBP
C-terminal binding protein transcriptional repressor
- CuAAC
copper catalyzed azide alkyne cycloaddition
- Cys
cysteine
- Dap
(S)-2,3-diaminopropanoic acid
- DARPins
designed ankyrin repeat proteins
- DBD
DNA binding domain
- DCM
dichloromethane
- Dcp2
mRNA decapping enzyme
- Ddz
α,α-dimethyl-3,5-dimethoxybenzylcarbonyl
- DEL
DNA-encoded library
- DEP1
receptor-like protein tyrosine phosphatase
- DIAD
diisopropyl azodicarboxylate
- DIC
N,N’-diisopropylcarbodiimide
- DIPEA
N,N’-diisopropylethylamine
- DMF
dimethylformamide
- DMSO
dimethyl sulfoxide
- DNA
DNA
- DTS
DNA templated synthesis
- E-Box
enhancer box
- ECRV
E-cadherin region V
- EED
embryonic ectoderm development protein
- EGFR
epidermal growth factor receptor
- ELISA
enzyme linked immunosorbent assay
- EphA2
Ephrin type-A receptor 2
- Eps15
epidermal growth factor receptor pathway substrate 15
- ERK
extracellular signal regulated kinase
- ERα
estrogen receptor alpha
- EZH2
enhancer of zeste homologue 2
- FIP
Rab11-family interacting proteins
- FIT
flexible in vitro translation
- FITC
fluorescein isothiocyanate
- Fmoc
fluorenylmethoxycarbonyl
- FP
fluorescence polarization
- GABARAP
GABA type A receptor associated protein
- Gag
group-specific antigen
- GAP
GTPase activating protein
- GEM
guanine-nucleotide exchange modulator
- Gln
glutamine
- GLP-1
glucagon like peptide 1
- Glu
glutamate
- Gly
glycine
- Gly-mal
glycine maleimide
- gp41
glycoprotein 41
- GPCR
G protein coupled receptor
- GTP
guanosine-5′-triphosphate
- HBS
hydrogen bond surrogate
- HBTU
hexafluorophosphate benzotriazole tetramethyl uronium
- HCT116
human epithelial cell line
- HCTU
O-(1H-6-Chlorobenzotriazole-1-yl)-1,1,3,3-tetramethyluronium hexafluorophosphate
- HDAC
histone deacetylase
- hDM2
human double minute 2 protein (E3 ubiquitin ligase)
- HER3
human epidermal growth factor receptor 3
- hIAPP
human islet amyloid polypeptide
- HIF1α
hypoxia induced factor 1 subunit alpha
- HIF1β
hypoxia induced factor 1 subunit beta, encoded by ARNT
- HIV-1
human immunodeficiency virus
- HOBt
hydroxybenzotriazole
- HPLC-MS
high performance liquid chromatography - mass spectrometry
- HR2
heptad repeat 2
- HRE
hypoxia-response element
- hTEAD
human transcriptional enhanced associate domain
- ICAT
inhibitor of β-catenin and TCF-4 protein
- ICN1
intracellular domain of Notch1
- IDE
insulin degrading enzyme
- IDOL
inducible degrader of low-density lipoprotein receptor (LDLR)
- IDR
intrinsically disordered region
- IEDDA
inverse electron demand Diels–Alder
- Ipa
isophthalic acid
- IKK-β
inhibitor of nuclear factor kappa B subunit beta
- IL-1
interleukin 1
- IN
integrase
- JM
juxtamembrane
- KDM7
lysine demethylase 7
- KEAP1
Kelch-like ECH-associated protein 1
- KOTMS
potassium trimethylsilanolate
- K-Ras
Kirsten rat sarcoma virus
- LC-MS
liquid chromatography-mass spectrometry
- LEDGF
lens epithelium-derived growth factor
- Leu
leucine
- Lys
lysine
- mAb
monoclonal antibody
- MAML1
mastermind like transcriptional coactivator 1
- MAR
matrix associated region
- MAX
MYC-associated factor X
- MCD
mast cell degranulation
- MCL-1
myeloid leukemia cell differentiation protein
- MCP
macrocyclic peptides
- MDA-MB-231
triple negative breast cancer cell line
- MDM2
mouse double minute 2
- MEG2
protein tyrosine phosphatase
- Met
methionine
- ML
machine learning
- Mmt
monomethoxytrityl
- MOrPH-PhD
macrocyclic organo-peptide hybrid phage display
- MP
microporous
- mRNA
messenger ribonucleic acid
- MT1-MMP
membrane type 1 - matrix metalloproteinase
- MTA1
metastasis-associated protein 1
- MYC
bHLH transcription factor, oncogene
- MyD88
Myeloid differentiation primary response 88
- N2B
NHR2-binding motif
- ncAA
noncanonical amino acids
- NCKAP1
NCK associated protein 1
- NEMO
NF-kappa-B essential modulator
- NFAT
nuclear factor of activated T cells
- NHR2
Nervy homology two domain
- NHR-TF
nuclear hormone receptor transcription factor
- NK-κB
nuclear factor kappa B
- NMR
nuclear magnetic resonance
- iNOS
inducible nitric oxide synthetase
- nNOS
neuronal nitric oxide synthetase
- NOTCH1
neurogenic locus notch homologue protein 1
- NOXA-2
phorbol-12-myristate-13-acetate-induced protein 1 (pro-apoptotic BH3 only protein)
- NRF2
nuclear factor erythroid 2-related factor
- NRPS
nonribosomal peptide synthetase
- OBOC
one bead one compound
- OBTC
one bead two compound
- OHM
oxopiperazine compounds
- OLIG2
oligodendrocyte transcription factor
- OSu
oxysuccinimide
- p300
histone acetyltransferase
- p53
tumor suppressor
- PA-1
ovarian tetracarcinoma cell line
- PANC-1
pancreatic carcinoma cell line
- pAuNP
poly-l-lysine coated gold nanoparticle
- PBS
phosphate buffered saline
- PCSK9
proprotein convertase subtilisin/kexin type 9
- PDB
protein data bank
- PDZ
structural domain
- PEPTIC
peptide exploration platform with Tag-free intramolecular chemistry
- Phe
phenylalanine
- PKA
protein kinase A
- PKC
protein kinase C
- PLSR
partial least-squares regression
- PNA
peptide nucleic acid
- POI
protein of interest
- PolyUb
polyubiquitin
- PPI
protein–protein interaction
- PPIA
peptidylprolyl cis–trans isomerase
- PRL2
phosphatase of regenerating liver 2
- Pro
proline
- PROTAC
proteolysis targeting chimera
- PS
polystyrene
- PSA
polar surface area
- PSD-95
postsynaptic density protein
- PTP1B
protein tyrosine phosphatase
- PURE
protein synthesis using recombinant elements
- Rab
Ras associated binding protein
- RaPID
random nonstandard peptide integrated discovery
- RCM
ring closing metathesis
- REU
Rosetta energy units
- RGS4
regulator of G protein signaling 4
- RMSD
root-mean-square deviation
- RNA
ribonucleic acid
- RRM
RNA-recognition motifs
- RSV
respiratory syncytial virus
- RT-PCR
real time polymerase chain reaction
- RuAAC
ruthenium catalyzed azide alkyne cycloaddition
- SAH
stabilized α helix
- SAR
structure activity relationship
- SBDD
structure-based drug design
- Seq
sequencing
- Ser
serine
- SH2
Src homology 2
- SHA
shavenoid
- SICLOPPS
split intein circular ligation of peptides and proteins
- SIRT2
sirtuin 2
- SJSA-1
osteosarcoma fibroblast
- SMAD7
Mothers against decapentaplegic homologue 7
- SOS1
son of sevenless homologue 1
- SPAAC
strain promoted azide alkyne cycloaddition
- SPPS
solid phase peptide synthesis
- SPR
surface plasmon resonance
- SPSB2
SPRY domain-containing SOCS box protein 2
- SRC2
steroid receptor coactivator 2
- STAT3
signal transducer and activator of transcription 3
- STING
stimulator of interferon genes adaptor protein
- STR
synthetic transcriptional repressors
- SUPR
scanning unnatural protease resistant
- tACBC
trans-2-aminocyclobutanecarboxylic acid
- TAMM
2-((alkylthio)(aryl)methylene)malononitrile
- TAR
transactivation binding response
- TATA
1,3,5-triacryloyl-1,3,5-triazinane
- TBAB
N,N′,N”-(benzene-1,3,5-triyl)-tris(2-bromoacetamide)
- TBMB
1,3,5-tris(bromomethyl)benzene
- TCEP
tris(2-carboxyethyl)phosphine
- TCF
T-cell factor
- TEAD1
transcriptional enhancer factor 1
- TET1
ten-11 translocation methylcytosine dioxygenase1
- TF
transcription factor
- TFA
trifluoroacetic acid
- TFAP4
transcription factor AP-4
- TGF-β
transforming growth factor beta
- THF
tetrahydrofuran
- Thr
threonine
- TIS
triisopropylsilane
- TLR
toll-like receptor
- TNBC
triple negative breast cancer
- TNKS
tankyrase
- Trp
tryptophan
- TSG101
tumor susceptibility gene
- ULM
U2AF ligand motifs
- uPA
urokinase-type plasminogen activator
- uPAR
uPA receptor
- UPRE
unfolded protein response element
- UTR
untranslated region
- Val
valine
- VDR
vitamin D receptor
- VEGF
vascular endothelial growth factor
- vFLIP
viral FLICE inhibitory proteins
- VGL4
vestigial-like protein 4
- WNT
wingless/Int1 signaling pathway
- XBP1
X-box binding protein 1
- XIAP
X-linked inhibitor of apoptosis protein
- YAP1
Yes1 associated transcriptional regulator
- γ-AA
γ-substituted N-acylated-N-aminoethyl amino acids
Biographies
Colin S. Swenson received his B.S. in Chemistry from the University of Utah in 2015. He then moved to Emory University where he completed his Ph.D. in Chemistry in 2020 with Prof. Jennifer Heemstra investigating amphiphilic peptide nucleic acids as stimuli-responsive biomaterials. In 2021, he joined the lab of Prof. Raymond Moellering at the University of Chicago as a NIH NRSA postdoctoral fellow, where he focuses on developing small molecule probes and synthetic biologics to study and target transcription factor activity.
Gunasheil Mandava received his combined B.S./M.S. degree in Molecular Biophysics and Biochemistry from Yale University as magna cum laude and earned the departmental award, the Sigler Prize, in 2021. He worked with Prof. Nikhil Malvankar to investigate the usage of electrically conductive bacterial nanowires in biomaterial applications. During his gap years, he continued his work in Prof. Nikhil Malvankar’s lab before moving to Prof. Sarah Slavoff’s lab in 2022 to design bicyclic peptides as viable PROTAC warheads to target undruggable proteins. He joined Prof. Raymond Moellering’s lab in 2023 as a graduate student in chemistry at the University of Chicago, where his current focus is to improve pharmacological properties of synthetic biologics that target transcription factor activity.
Deborah M. Thomas received her B.S. in Chemistry from Florida Atlantic University in 2019. She then moved to the University of Chicago in 2020 and is currently pursuing a Ph.D. in Chemistry with Prof. Raymond E. Moellering. Her work focuses on the development of novel chemistries for peptide stabilization, stabilized peptide therapeutics for transcription factors, and theranostic peptides for tumor imaging and treatment.
Ray Moellering is a Professor of Chemistry at the University of Chicago. He obtained Bachelor’s degrees in Chemistry and Biochemistry and Molecular Biophysics from the University of Arizona. He then earned a Ph.D. in Chemistry at Harvard University with Prof. Gregory L. Verdine as an American Association for Cancer Research Centennial Fellow, followed by postdoctoral training with Prof. Benjamin F. Cravatt, III as a Damon Runyon Postdoctoral Fellow at The Scripps Research Institute. Prof. Moellering started his independent program at UChicago in 2015, where his laboratory is focused on developing novel chemical probes and complementary technologies to expose and exploit novel signaling mechanisms in diseases like cancer, diabetes, and immunological disorders. Dr. Moellering has contributed to the development of stabilized peptide and protein mimetics as chemical probes and prototype therapeutics for diverse undruggable proteins in cancer.
Footnotes
The authors declare the following competing financial interest(s): R.E.M. is a founder, director, and consultant for ReAx Biotechnologies and Ama Therapeutics. All other authors declare no conflicts of interest.
REFERENCES
- (1).Radoux CJ; Vianello F; McGreig J; Desai N; Bradley AR The druggable genome: Twenty years later. Front Bioinform. 2022, 2, 958378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (2).Hopkins AL; Groom CR The druggable genome. Nat. Rev. Drug Discovery 2002, 1, 727–730. [DOI] [PubMed] [Google Scholar]
- (3).Xie X; Yu T; Li X; Zhang N; Foster LJ; Peng C; Huang W; He G Recent advances in targeting the “undruggable” proteins: from drug discovery to clinical trials. Signal Transduct. Target Ther 2023, 8, 335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (4).Coleman N; Rodon J Taking Aim at the Undruggable. Am. Soc. Clin. Oncol. Educ. Book 2021, 41, e145–e152. [DOI] [PubMed] [Google Scholar]
- (5).Dang CV; Reddy EP; Shokat KM; Soucek L Drugging the ’undruggable’ cancer targets. Nat. Rev. Cancer 2017, 17, 502–508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).Wang L; Wang N; Zhang W; Cheng X; Yan Z; Shao G; Wang X; Wang R; Fu C Therapeutic peptides: current applications and future directions. Signal Transduct. Target Ther 2022, 7, 48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).Muttenthaler M; King GF; Adams DJ; Alewood PF Trends in peptide drug discovery. Nat. Rev. Drug Discovery 2021, 20, 309–325. [DOI] [PubMed] [Google Scholar]
- (8).Tsomaia N. Peptide therapeutics: targeting the undruggable space. Eur. J. Med. Chem 2015, 94, 459–470. [DOI] [PubMed] [Google Scholar]
- (9).McGregor DP Discovering and improving novel peptide therapeutics. Curr. Opin. Pharmacol 2008, 8, 616–619. [DOI] [PubMed] [Google Scholar]
- (10).Pollaro L; Heinis C Strategies to prolong the plasma residence time of peptide drugs. MedChemComm 2010, 1, 319–324. [Google Scholar]
- (11).Mathur D; Prakash S; Anand P; Kaur H; Agrawal P; Mehta A; Kumar R; Singh S; Raghava GPS PEPlife: A Repository of the Half-life of Peptides. Sci. Rep 2016, 6, 36617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Katoh T; Goto Y; Reza MS; Suga H Ribosomal synthesis of backbone macrocyclic peptides. Chem. Commun 2011, 47, 9946–9958. [DOI] [PubMed] [Google Scholar]
- (13).Vinogradov AA; Yin Y; Suga H Macrocyclic Peptides as Drug Candidates: Recent Progress and Remaining Challenges. J. Am. Chem. Soc 2019, 141, 4167–4181. [DOI] [PubMed] [Google Scholar]
- (14).Wang W; Khojasteh SC; Su D Biosynthetic Strategies for Macrocyclic Peptides. Molecules 2021, 26, 3338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Farmer PS; Ariëns EJ Speculations on the design of nonpeptidic peptidomimetics. Trends Pharmacol. Sci 1982, 3, 362–365. [Google Scholar]
- (16).Gante J. Peptidomimetics—Tailored Enzyme Inhibitors. Angew. Chem., Int. Ed 1994, 33, 1699–1720. [Google Scholar]
- (17).Giannis A; Kolter T Peptidomimetics for Receptor Ligands—Discovery, Development, and Medical Perspectives. Angew. Chem., Int. Ed 1993, 32, 1244–1267. [Google Scholar]
- (18).Ripka AS; Rich DH Peptidomimetic design. Curr. Opin. Chem. Biol 1998, 2, 441–452. [DOI] [PubMed] [Google Scholar]
- (19).Vagner J; Qu H; Hruby VJ Peptidomimetics, a synthetic tool of drug discovery. Curr. Opin. Chem. Biol 2008, 12, 292–296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).Trabocchi A; Guarna A Peptidomimetics in organic and medicinal chemistry: the art of transforming peptides in drugs; John Wiley and Sons, Ltd, 2014. [Google Scholar]
- (21).Gokhale AS; Satyanarayanajois S Peptides and peptidomimetics as immunomodulators. Immunotherapy 2014, 6, 755–774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).Pelay-Gimeno M; Glas A; Koch O; Grossmann TN Structure-Based Design of Inhibitors of Protein-Protein Interactions: Mimicking Peptide Binding Epitopes. Angew. Chem., Int. Ed 2015, 54, 8896–8927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).Mabonga L; Kappo AP Peptidomimetics: A Synthetic Tool for Inhibiting Protein-Protein Interactions in Cancer. Int. J. Pept. Res. Ther 2020, 26, 225–241. [Google Scholar]
- (24).Li Petri G; Di Martino S; De Rosa M Peptidomimetics: An Overview of Recent Medicinal Chemistry Efforts toward the Discovery of Novel Small Molecule Inhibitors. J. Med. Chem 2022, 65, 7438–7475. [DOI] [PubMed] [Google Scholar]
- (25).Milroy L-G; Grossmann TN; Hennig S; Brunsveld L; Ottmann C Modulators of Protein–Protein Interactions. Chem. Rev 2014, 114, 4695–4748. [DOI] [PubMed] [Google Scholar]
- (26).Yan C; Higgins PJ Drugging the undruggable: transcription therapy for cancer. Biochim. Biophys. Acta 2013, 1835, 76–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (27).Gebauer M; Skerra A Engineered Protein Scaffolds as Next-Generation Therapeutics. Annu. Rev. Pharmacol. Toxicol 2020, 60, 391–415. [DOI] [PubMed] [Google Scholar]
- (28).Furumai R; Matsuyama A; Kobashi N; Lee K-H; Nishiyama M; Nakajima H; Tanaka A; Komatsu Y; Nishino N; Yoshida M; et al. FK228 (Depsipeptide) as a Natural Prodrug That Inhibits Class I Histone Deacetylases1. Cancer Res. 2002, 62, 4916–4921. [PubMed] [Google Scholar]
- (29).Abdalla MA; McGaw LJ Natural Cyclic Peptides as an Attractive Modality for Therapeutics: A Mini Review. Molecules 2018, 23, 2080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (30).Alonzo DA; Schmeing TM Biosynthesis of depsipeptides, or Depsi: The peptides with varied generations. Protein Sci. 2020, 29, 2316–2347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (31).Sieber SA; Marahiel MA Learning from nature’s drug factories: nonribosomal synthesis of macrocyclic peptides. J. Bacteriol 2003, 185, 7036–7043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (32).Hewitt WM; Leung SSF; Pye CR; Ponkey AR; Bednarek M; Jacobson MP; Lokey RS Cell-Permeable Cyclic Peptides from Synthetic Libraries Inspired by Natural Products. J. Am. Chem. Soc 2015, 137, 715–721. [DOI] [PubMed] [Google Scholar]
- (33).Faulds D; Goa KL; Benfield P Cyclosporin. Drugs 1993, 45, 953–1040. [DOI] [PubMed] [Google Scholar]
- (34).Lamberts SWJ; Hofland LJ ANNIVERSARY REVIEW: Octreotide, 40 years later. Eur. J. Endocrinol 2019, 181, R173–R183. [DOI] [PubMed] [Google Scholar]
- (35).Zhang H; Chen S Cyclic peptide drugs approved in the last two decades (2001–2021). RSC Chem. Biol 2022, 3, 18–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (36).Costa L; Sousa E; Fernandes C Cyclic Peptides in Pipeline: What Future for These Great Molecules? Pharmaceuticals 2023, 16, 996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (37).Aboye T; Kuang Y; Neamati N; Camarero JA Rapid Parallel Synthesis of Bioactive Folded Cyclotides by Using a Tea-Bag Approach. ChemBioChem. 2015, 16, 827–833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (38).Jagadish K; Camarero JA Cyclotides, a promising molecular scaffold for peptide-based therapeutics. Biopolymers 2010, 94, 611–616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (39).de Veer SJ; Kan M-W; Craik DJ Cyclotides: From Structure to Function. Chem. Rev 2019, 119, 12375–12421. [DOI] [PubMed] [Google Scholar]
- (40).Hayes HC; Luk LYP; Tsai Y-H Approaches for peptide and protein cyclisation. Org. Biomol. Chem 2021, 19, 3983–4001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (41).Bechtler C; Lamers C Macrocyclization strategies for cyclic peptides and peptidomimetics. RSC Med. Chem 2021, 12, 1325–1351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (42).Chow HY; Zhang Y; Matheson E; Li X Ligation Technologies for the Synthesis of Cyclic Peptides. Chem. Rev 2019, 119, 9971–10001. [DOI] [PubMed] [Google Scholar]
- (43).Lipinski CA; Lombardo F; Dominy BW; Feeney PJ Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings 1PII of original article: S0169–409X(96)00423–1. The article was originally published in Advanced Drug Delivery Reviews 23 (1997). Adv. Drug Delivery Rev 2001, 46, 3–26. [DOI] [PubMed] [Google Scholar]
- (44).Garcia Jimenez D; Poongavanam V; Kihlberg J Macrocycles in Drug Discovery—Learning from the Past for the Future. J. Med. Chem 2023, 66, 5377–5396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (45).Doak BC; Over B; Giordanetto F; Kihlberg J Oral Druggable Space beyond the Rule of 5: Insights from Drugs and Clinical Candidates. Chem. Biol 2014, 21, 1115–1142. [DOI] [PubMed] [Google Scholar]
- (46).Degoey DA; Chen H-J; Cox PB; Wendt MD Beyond the Rule of 5: Lessons Learned from AbbVie’s Drugs and Compound Collection. J. Med. Chem 2018, 61, 2636–2651. [DOI] [PubMed] [Google Scholar]
- (47).Wang CK; Swedberg JE; Harvey PJ; Kaas Q; Craik DJ Conformational Flexibility Is a Determinant of Permeability for Cyclosporin. J. Phys. Chem. B 2018, 122, 2261–2276. [DOI] [PubMed] [Google Scholar]
- (48).Danelius E; Poongavanam V; Peintner S; Wieske LHE; Erdélyi M; Kihlberg J Solution Conformations Explain the Chameleonic Behaviour of Macrocyclic Drugs. Chem. Eur. J 2020, 26, 5231–5244. [DOI] [PubMed] [Google Scholar]
- (49).Sethio D; Poongavanam V; Xiong R; Tyagi M; Duy Vo D; Lindh R; Kihlberg J Simulation Reveals the Chameleonic Behavior of Macrocycles. J. Chem. Inf. Model 2023, 63, 138–146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (50).Ramelot TA; Palmer J; Montelione GT; Bhardwaj G Cell-permeable chameleonic peptides: Exploiting conformational dynamics in de novo cyclic peptide design. Curr. Opin. Struct. Biol 2023, 80, 102603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (51).Bhardwaj G; O’Connor J; Rettie S; Huang Y-H; Ramelot TA; Mulligan VK; Alpkilic GG; Palmer J; Bera AK; Bick MJ; et al. Accurate de novo design of membrane-traversing macrocycles. Cell 2022, 185, 3520–3532.e3526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (52).Vorherr T; Lewis I; Berghausen J; Desrayaud S; Schaefer M Modulation of Oral Bioavailability and Metabolism for Closely Related Cyclic Hexapeptides. Int. J. Pept. Res. Ther 2018, 24, 35–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (53).Golosov AA; Flyer AN; Amin J; Babu C; Gampe C; Li J; Liu E; Nakajima K; Nettleton D; Patel TJ; et al. Design of Thioether Cyclic Peptide Scaffolds with Passive Permeability and Oral Exposure. J. Med. Chem 2021, 64, 2622–2633. [DOI] [PubMed] [Google Scholar]
- (54).Golosov AA; Flyer AN; Monovich LG Design and Discovery of Orally Bioavailable Macrocycles: Toward Orally Bioavailable Peptide Therapeutics. In Approaching the Next Inflection in Peptide Therapeutics: Attaining Cell Permeability and Oral Bioavailability; ACS Symposium Series, Vol. 1417; American Chemical Society, 2022; pp 199–222. [Google Scholar]
- (55).Bottens RA; Yamada T Cell-Penetrating Peptides (CPPs) as Therapeutic and Diagnostic Agents for Cancer. Cancers (Basel) 2022, 14, 5546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (56).Chu Q; Moellering RE; Hilinski GJ; Kim Y-W; Grossmann TN; Yeh JTH; Verdine GL Towards understanding cell penetration by stapled peptides. MedChemComm 2015, 6, 111–119. [Google Scholar]
- (57).Zorko M; Langel Ü Cell-penetrating peptides: mechanism and kinetics of cargo delivery. Adv. Drug Delivery Rev 2005, 57, 529–545. [DOI] [PubMed] [Google Scholar]
- (58).Heitz F; Morris MC; Divita G Twenty years of cellpenetrating peptides: from molecular mechanisms to therapeutics. Br. J. Pharmacol 2009, 157, 195–206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (59).Gump JM; Dowdy SF TAT transduction: the molecular mechanism and therapeutic prospects. Trends Mol. Med 2007, 13, 443–448. [DOI] [PubMed] [Google Scholar]
- (60).Derossi D; Joliot AH; Chassaing G; Prochiantz A The third helix of the Antennapedia homeodomain translocates through biological membranes. J. Biol. Chem 1994, 269, 10444–10450. [PubMed] [Google Scholar]
- (61).Futaki S. Membrane-permeable arginine-rich peptides and the translocation mechanisms. Adv. Drug Delivery Rev 2005, 57, 547–558. [DOI] [PubMed] [Google Scholar]
- (62).Kalafatovic D; Giralt E Cell-Penetrating Peptides: Design Strategies beyond Primary Structure and Amphipathicity. Molecules 2017, 22, 1929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (63).Park SE; Sajid MI; Parang K; Tiwari RK Cyclic Cell-Penetrating Peptides as Efficient Intracellular Drug Delivery Tools. Mol. Pharmaceutics 2019, 16, 3727–3743. [DOI] [PubMed] [Google Scholar]
- (64).Xie J; Bi Y; Zhang H; Dong S; Teng L; Lee RJ; Yang Z Cell-Penetrating Peptides in Diagnosis and Treatment of Human Diseases: From Preclinical Research to Clinical Application. Front. Pharmacol 2020, 11, 697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (65).Ohta A; Tanada M; Shinohara S; Morita Y; Nakano K; Yamagishi Y; Takano R; Kariyuki S; Iida T; Matsuo A; et al. Validation of a New Methodology to Create Oral Drugs beyond the Rule of 5 for Intracellular Tough Targets. J. Am. Chem. Soc 2023, 145, 24035–24051. [DOI] [PubMed] [Google Scholar]
- (66).DiMaio J; Schiller PW A cyclic enkephalin analog with high in vitro opiate activity. Proc. Natl. Acad. Sci. U.S.A 1980, 77, 7162–7166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (67).Saragovi HU; Fitzpatrick D; Raktabutr A; Nakanishi H; Kahn M; Greene MI Design and Synthesis of a Mimetic from an Antibody Complementarity-Determining Region. Science 1991, 253, 792–795. [DOI] [PubMed] [Google Scholar]
- (68).Chen S; Chrusciel RA; Nakanishi H; Raktabutr A; Johnson ME; Sato A; Weiner D; Hoxie J; Saragovi HU; Greene MI Design and synthesis of a CD4 beta-turn mimetic that inhibits human immunodeficiency virus envelope glycoprotein gp120 binding and infection of human lymphocytes. Proc. Natl. Acad. Sci. U.S.A 1992, 89, 5872–5876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (69).Xu W; Lau YH; Fischer G; Tan YS; Chattopadhyay A; De La Roche M; Hyvönen M; Verma C; Spring DR; Itzhaki LS Macrocyclized Extended Peptides: Inhibiting the Substrate-Recognition Domain of Tankyrase. J. Am. Chem. Soc 2017, 139, 2245–2256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (70).Jin Y; Zhong H; Omnaas JR; Neubig RR; Mosberg HI Structure-based design, synthesis, and pharmacologic evaluation tf peptide RGS4 inhibitors. J. Pept. Res 2004, 63, 141–146. [DOI] [PubMed] [Google Scholar]
- (71).Zhong B; Zhang C; Guo S; Zhang C Rational design of cyclic peptides to disrupt TGF-B/SMAD7 signaling in heterotopic ossification. J. Mol. Graph. Model 2017, 72, 25–31. [DOI] [PubMed] [Google Scholar]
- (72).Pan D The hippo signaling pathway in development and cancer. Dev. Cell 2010, 19, 491–505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (73).Sawada A; Kiyonari H; Ukita K; Nishioka N; Imuta Y; Sasaki H Redundant Roles of Tead1 and Tead2 in Notochord Development and the Regulation of Cell Proliferation and Survival. Mol. Cell. Biol 2008, 28, 3177–3189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (74).Zhang Z; Lin Z; Zhou Z; Shen HC; Yan SF; Mayweg AV; Xu Z; Qin N; Wong JC; Zhang Z; et al. Structure-Based Design and Synthesis of Potent Cyclic Peptides Inhibiting the YAP—TEAD Protein-Protein Interaction. ACS Med. Chem. Lett 2014, 5, 993–998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (75).Furet P; Bordas V; Le Douget M; Salem B; Mesrouze Y; Imbach-Weese P; Sellner H; Voegtle M; Soldermann N; Chapeau E; et al. The First Class of Small Molecules Potently Disrupting the YAP-TEAD Interaction by Direct Competition. ChemMedChem. 2022, 17, e202200303. [DOI] [PubMed] [Google Scholar]
- (76).Dishon S; Schumacher A; Fanous J; Talhami A; Kassis I; Karussis D; Gilon C; Hoffman A; Nussbaum G Development of a Novel Backbone Cyclic Peptide Inhibitor of the Innate Immune TLR/IL1R Signaling Protein MyD88. Sci. Rep 2018, 8, 9476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (77).Chen L; Zheng L; Chen P; Liang G Myeloid Differentiation Primary Response Protein 88 (MyD88): The Central Hub of TLR/IL-1R Signaling. J. Med. Chem 2020, 63, 13316–13329. [DOI] [PubMed] [Google Scholar]
- (78).Dishon S; Schumacher-Klinger A; Gilon C; Hoffman A; Nussbaum G Myristoylation Confers Oral Bioavailability and Improves the Bioactivity of c(MyD 4–4), a Cyclic Peptide Inhibitor of MyD88. Mol. Pharmaceutics 2019, 16, 1516–1522. [DOI] [PubMed] [Google Scholar]
- (79).Khairnar BD; Jha A; Rajwade JM Rationally designed cyclic peptides and nanomaterials as ‘next-generation’ anti-amyloid therapeutics. J. Mater. Sci 2023, 58, 9834–9860. [Google Scholar]
- (80).Gavenonis J; Sheneman BA; Siegert TR; Eshelman MR; Kritzer JA Comprehensive analysis of loops at protein-protein interfaces for macrocycle design. Nat. Chem. Biol 2014, 10, 716–722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (81).Siegert TR; Bird MJ; Makwana KM; Kritzer JA Analysis of Loops that Mediate Protein–Protein Interactions and Translation into Submicromolar Inhibitors. J. Am. Chem. Soc 2016, 138, 12876–12884. [DOI] [PubMed] [Google Scholar]
- (82).Huang H; Damjanovic J; Miao J; Lin Y-S Cyclic peptides: backbone rigidification and capability of mimicking motifs at protein–protein interfaces. Phys. Chem. Chem. Phys 2021, 23, 607–616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (83).Rumpf J; Simon B; Jung N; Maritzen T; Haucke V; Sattler M; Groemping Y Structure of the Eps15-stonin2 complex provides a molecular explanation for EH-domain ligand specificity. EMBO J. 2008, 27, 558–569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (84).Bhattacharyya S; Warfield KL; Ruthel G; Bavari S; Aman MJ; Hope TJ Ebola virus uses clathrin-mediated endocytosis as an entry pathway. Virology 2010, 401, 18–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (85).Mi T; Siriwibool S; Burgess K Streamlined Protein-Protein Interface Loop Mimicry. Angew. Chem., Int. Ed 2023, 62, e202307092. [DOI] [PubMed] [Google Scholar]
- (86).Dougherty PG; Wellmerling JH; Koley A; Lukowski JK; Hummon AB; Cormet-Boyaka E; Pei D Cyclic Peptidyl Inhibitors against CAL/CFTR Interaction for Treatment of Cystic Fibrosis. J. Med. Chem 2020, 63, 15773–15784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (87).Qian Z; Rhodes CA; McCroskey LC; Wen J; Appiah-Kubi G; Wang DJ; Guttridge DC; Pei D Enhancing the Cell Permeability and Metabolic Stability of Peptidyl Drugs by Reversible Bicyclization. Angew. Chem., Int. Ed 2017, 56, 1525–1529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (88).Rothwarf DM; Zandi E; Natoli G; Karin M IKK-γ is an essential regulatory subunit of the IκB kinase complex. Nature 1998, 395, 297–300. [DOI] [PubMed] [Google Scholar]
- (89).Yamaoka S; Courtois G; Bessia C; Whiteside ST; Weil R; Agou F; Kirk HE; Kay RJ; Israël A Complementation Cloning of NEMO, a Component of the IκB Kinase Complex Essential for NF-κB Activation. Cell 1998, 93, 1231–1240. [DOI] [PubMed] [Google Scholar]
- (90).Cildir G; Low KC; Tergaonkar V Noncanonical NF-κB Signaling in Health and Disease. Trends Mol. Med 2016, 22, 414429. [DOI] [PubMed] [Google Scholar]
- (91).Herrington FD; Carmody RJ; Goodyear CS Modulation of NF-κB Signaling as a Therapeutic Target in Autoimmunity. SLAS Discovery 2016, 21, 223–242. [DOI] [PubMed] [Google Scholar]
- (92).May MJ; D’Acquisto F; Madge LA; Glöckner J; Pober JS; Ghosh S Selective Inhibition of NF-κB Activation by a Peptide That Blocks the Interaction of NEMO with the IκB Kinase Complex. Science 2000, 289, 1550–1554. [DOI] [PubMed] [Google Scholar]
- (93).Ernst C; Heidrich J; Sessler C; Sindlinger J; Schwarzer D; Koch P; Boeckler FM Switching Between Bicyclic and Linear Peptides — The Sulfhydryl-Specific Linker TPSMB Enables Reversible Cyclization of Peptides. Front. Chem 2018, 6, 484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (94).Hosseinzadeh P; Watson PR; Craven TW; Li X; Rettie S; Pardo-Avila F; Bera AK; Mulligan VK; Lu P; Ford AS; et al. Anchor extension: a structure-guided approach to design cyclic peptides targeting enzyme active sites. Nat. Commun 2021, 12, 3384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (95).Hosseinzadeh P; Bhardwaj G; Mulligan VK; Shortridge MD; Craven TW; Pardo-Avila F; Rettie SA; Kim D-E; Silva DA; Ibrahim YM; et al. Comprehensive computational design of ordered peptide macrocycles. Science 2017, 358, 1461–1466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (96).Slough DP; McHugh SM; Cummings AE; Dai P; Pentelute BL; Kritzer JA; Lin Y-S Designing Well-Structured Cyclic Pentapeptides Based on Sequence–Structure Relationships. J. Phys. Chem. B 2018, 122, 3908–3919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (97).Mulligan VK; Kang CS; Sawaya MR; Rettie S; Li X; Antselovich I; Craven TW; Watkins AM; Labonte JW; DiMaio F; et al. Computational design of mixed chirality peptide macrocycles with internal symmetry. Protein Sci. 2020, 29, 2433–2445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (98).Miao J; Descoteaux ML; Lin Y-S Structure prediction of cyclic peptides by molecular dynamics + machine learning. Chem. Sci 2021, 12, 14927–14936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (99).Duffy FJ; Verniere M; Devocelle M; Bernard E; Shields DC; Chubb AJ CycloPs: Generating Virtual Libraries of Cyclized and Constrained Peptides Including Nonnatural Amino Acids. J. Chem. Inf. Model 2011, 51, 829–836. [DOI] [PubMed] [Google Scholar]
- (100).Houk KN; Leach AG; Kim SP; Zhang X Binding Affinities of Host–Guest, Protein–Ligand, and Protein–Transition-State Complexes. Angew. Chem., Int. Ed 2003, 42, 4872–4897. [DOI] [PubMed] [Google Scholar]
- (101).Delaunay M; Ha-Duong T Des3PI: a fragment-based approach to design cyclic peptides targeting protein–protein interactions. J. Comput. Aided Mol. Des 2022, 36, 605–621. [DOI] [PubMed] [Google Scholar]
- (102).Delaunay M; Ha-Duong T Computational design of cyclic peptides to inhibit protein-peptide interactions. Biophys. Chem 2023, 296, 106987. [DOI] [PubMed] [Google Scholar]
- (103).Santini BL; Zacharias M Rapid in silico Design of Potential Cyclic Peptide Binders Targeting Protein-Protein Interfaces. Front. Chem 2020, 8, 537259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (104).Jumper J; Evans R; Pritzel A; Green T; Figurnov M; Ronneberger O; Tunyasuvunakool K; Bates R; Žídek A; Potapenko A; et al. Highly accurate protein structure prediction with AlphaFold. Nature 2021, 596, 583–589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (105).Baek M; DiMaio F; Anishchenko I; Dauparas J; Ovchinnikov S; Lee GR; Wang J; Cong Q; Kinch LN; Schaeffer RD; et al. Accurate prediction of protein structures and interactions using a three-track neural network. Science 2021, 373, 871–876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (106).Yang J; Anishchenko I; Park H; Peng Z; Ovchinnikov S; Baker D Improved protein structure prediction using predicted interresidue orientations. Proc. Natl. Acad. Sci. U.S.A 2020, 117, 1496–1503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (107).Mulligan VK; Hosseinzadeh P Computational Design of Peptide-Based Binders to Therapeutic Targets. In Approaching the Next Inflection in Peptide Therapeutics: Attaining Cell Permeability and Oral Bioavailability; ACS Symposium Series, Vol. 1417; American Chemical Society, 2022; pp 55–102. [Google Scholar]
- (108).Kazmirchuk TDD; Bradbury-Jost C; Withey TA; Gessese T; Azad T; Samanfar B; Dehne F; Golshani A Peptides of a Feather: How Computation Is Taking Peptide Therapeutics under Its Wing. Genes 2023, 14, 1194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (109).Mulligan VK The emerging role of computational design in peptide macrocycle drug discovery. Expert Opin. Drug Discovery 2020, 15, 833–852. [DOI] [PubMed] [Google Scholar]
- (110).Wang T; Wang L; Zhang X; Shen C; Zhang O; Wang J; Wu J; Jin R; Zhou D; Chen S; et al. Comprehensive assessment of protein loop modeling programs on large-scale datasets: prediction accuracy and efficiency. Brief. Bioinform 2023, 25, bbad486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (111).Pereira DA; Williams JA Origin and evolution of high throughput screening. Br. J. Pharmacol 2007, 152, 53–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (112).Marsault E; Hoveyda HR; Gagnon R; Peterson ML; Vézina M; Saint-Louis C; Landry A; Pinault J-F; Ouellet L; Beauchemin S; et al. Efficient parallel synthesis of macrocyclic peptidomimetics. Bioorg. Med. Chem. Lett 2008, 18, 4731–4735. [DOI] [PubMed] [Google Scholar]
- (113).Hoveyda HR; Marsault E; Gagnon R; Mathieu AP; Vézina M; Landry A; Wang Z; Benakli K; Beaubien S; Saint-Louis C; et al. Optimization of the Potency and Pharmacokinetic Properties of a Macrocyclic Ghrelin Receptor Agonist (Part I): Development of Ulimorelin (TZP-101) from Hit to Clinic. J. Med. Chem 2011, 54, 8305–8320. [DOI] [PubMed] [Google Scholar]
- (114).Hoveyda HR; Fraser GL; Marsault E; Gagnon R; Peterson ML Optimization of a Macrocyclic Ghrelin Receptor Agonist (Part II): Development of TZP-102. In Practical Medicinal Chemistry with Macrocycles: Design, Synthesis, and Case Studies; John Wiley & Sons, Inc., 2017; pp 545–558. [Google Scholar]
- (115).Marsault E; Hoveyda HR; Peterson ML; Saint-Louis C; Landry A; Vézina M; Ouellet L; Wang Z; Ramaseshan M; Beaubien S; et al. Discovery of a New Class of Macrocyclic Antagonists to the Human Motilin Receptor. J. Med. Chem 2006, 49, 7190–7197. [DOI] [PubMed] [Google Scholar]
- (116).Zampaloni C; Mattei P; Bleicher K; Winther L; Thäte C; Bucher C; Adam J-M; Alanine A; Amrein KE; Baidin V; et al. A novel antibiotic class targeting the lipopolysaccharide transporter. Nature 2024, 625, 566–571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (117).Guenther A; Millar L; Messer A; Giraudon M; Patel K; Deurloo EJ; Lobritz M; Gloge A 2126. Safety, Tolerability, and Pharmacokinetics (PK) in Healthy Participants Following Single Dose Administration of Zosurabalpin, a Novel Pathogen-Specific Antibiotic for the Treatment of Serious Acinetobacter Infections. Open Forum Infectious Diseases 2023, 10, ofad500.1749. [Google Scholar]
- (118).Kale SS; Bergeron-Brlek M; Wu Y; Kumar MG; Pham MV; Bortoli J; Vesin J; Kong X-D; Machado JF; Deyle K; et al. Thiol-to-amine cyclization reaction enables screening of large libraries of macrocyclic compounds and the generation of subkilodalton ligands. Sci. Adv 2019, 5, eaaw2851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (119).Sangouard G; Zorzi A; Wu Y; Ehret E; Schuüttel M; Kale S; Díaz-Perlas C; Vesin J; Bortoli Chapalay J; Turcatti G; et al. Picomole-Scale Synthesis and Screening of Macrocyclic Compound Libraries by Acoustic Liquid Transfer. Angew. Chem., Int. Ed 2021, 60, 21702–21707. [DOI] [PubMed] [Google Scholar]
- (120).Habeshian S; Merz ML; Sangouard G; Mothukuri GK; Schuüttel M; Bognár Z; Díaz-Perlas C; Vesin J; Bortoli Chapalay J; Turcatti G; et al. Synthesis and direct assay of large macrocycle diversities by combinatorial late-stage modification at picomole scale. Nat. Commun 2022, 13, 3823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (121).Merz ML; Habeshian S; Li B; David J-AGL; Nielsen AL; Ji X; Il Khwildy K; Duany Benitez MM; Phothirath P; Heinis C De novo development of small cyclic peptides that are orally bioavailable. Nat. Chem. Biol 2024, 20, 624–633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (122).Nielsen AL; Bognar Z; Mothukuri GK; Zarda A; Schuttel M; Merz ML; Ji X; Will E; Chinellato M; Bartling CRO; et al. Large Libraries of Structurally Diverse Macrocycles Suitable for Membrane Permeation. Angew. Chem., Int. Ed 2024, 63, e202400350. [DOI] [PubMed] [Google Scholar]
- (123).Bhatia N; Khator R; Kulkarni S; Singh Y; Kumar P; Thareja S Recent Advancements in the Discovery of MDM2/ MDM2-p53 Interaction Inhibitors for the Treatment of Cancer. Curr. Med. Chem 2023, 30, 3668–3701. [DOI] [PubMed] [Google Scholar]
- (124).Brummer T; Zeiser R The role of the MDM2/p53 axis in anti-tumor immune responses. Blood 2024, 143, 2701–2709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (125).Bruce A; Adebomi V; Czabala P; Palmer J; Bhardwaj G; Raj M; McFadden WM; Lorson ZC; Slack RL; Sarafianos SG A Tag-Free Platform for Synthesis and Screening of Cyclic Peptide Libraries. Angew. Chem., Int. Ed 2024, 63, e202320045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (126).Adebomi V; Cohen RD; Wills R; Chavers HAH; Martin GE; Raj M CyClick Chemistry for the Synthesis of Cyclic Peptides. Angew. Chem., Int. Ed 2019, 58, 19073–19080. [DOI] [PubMed] [Google Scholar]
- (127).Carnes SK; Sheehan JH; Aiken C Inhibitors of the HIV-1 capsid, a target of opportunity. Curr. Opin. HIV AIDS 2018, 13, 359–365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (128).Komnatnyy VV; Nielsen TE; Qvortrup K Bead-based screening in chemical biology and drug discovery. Chem. Commun 2018, 54, 6759–6771. [DOI] [PubMed] [Google Scholar]
- (129).Furka Á; Sebestyén F; Asgedom M; Dibó G General method for rapid synthesis of multicomponent peptide mixtures. Int. J. Pept. Protein Res 1991, 37, 487–493. [DOI] [PubMed] [Google Scholar]
- (130).Lam KS; Salmon SE; Hersh EM; Hruby VJ; Kazmierski WM; Knapp RJ A new type of synthetic peptide library for identifying ligand-binding activity. Nature 1991, 354, 82–84. [DOI] [PubMed] [Google Scholar]
- (131).Lau DH; Guo L; Liu R; Song A; Shao C; Lam KS Identifying peptide ligands for cell surface receptors using cell-growth-on-bead assay and one-bead one-compound combinatorial library. Biotechnol. Lett 2002, 24, 497–500. [Google Scholar]
- (132).Geurs S; Clarisse D; Baele F; Franceus J; Desmet T; De Bosscher K; D’Hooghe M Identification of mercaptoacetamide-based HDAC6 inhibitors via a lean inhibitor strategy: screening, synthesis, and biological evaluation. Chem. Commun 2022, 58, 6239–6242. [DOI] [PubMed] [Google Scholar]
- (133).Olsen CA; Ghadiri MR Discovery of Potent and Selective Histone Deacetylase Inhibitors via Focused Combinatorial Libraries of Cyclic α3β-Tetrapeptides. J. Med. Chem 2009, 52, 7836–7846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (134).Glenn MP; Kelso MJ; Tyndall JDA; Fairlie DP Conformationally Homogeneous Cyclic Tetrapeptides: Useful New Three-Dimensional Scaffolds. J. Am. Chem. Soc 2003, 125, 640–641. [DOI] [PubMed] [Google Scholar]
- (135).Norgren AS; Buüttner F; Prabpai S; Kongsaeree P; Arvidsson PI β2-Amino Acids in the Design of Conformationally Homogeneous cyclo-Peptide Scaffolds. J. Org. Chem 2006, 71, 6814–6821. [DOI] [PubMed] [Google Scholar]
- (136).Roof RA; Sobczyk-Kojiro K; Turbiak AJ; Roman DL; Pogozheva ID; Blazer LL; Neubig RR; Mosberg HI Novel Peptide Ligands of RGS4 from a Focused One-Bead. One-Compound Library. Chem. Biol. Drug Des 2008, 72, 111–119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (137).Lian W; Jiang B; Qian Z; Pei D Cell-Permeable Bicyclic Peptide Inhibitors against Intracellular Proteins. J. Am. Chem. Soc 2014, 136, 9830–9833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (138).Li Z; Shao S; Ren X; Sun J; Guo Z; Wang S; Song MM; Chang C-EA; Xue M Construction of a Sequenceable Protein Mimetic Peptide Library with a True 3D Diversifiable Chemical Space. J. Am. Chem. Soc 2018, 140, 14552–14556. [DOI] [PubMed] [Google Scholar]
- (139).Li Z; Huang Y; Hung TI; Sun J; Aispuro D; Chen B; Guevara N; Ji F; Cong X; Zhu L; et al. MYC-Targeting Inhibitors Generated from a Stereodiversified Bicyclic Peptide Library. J. Am. Chem. Soc 2024, 146, 1356–1363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (140).Dewan V; Liu T; Chen K-M; Qian Z; Xiao Y; Kleiman L; Mahasenan KV; Li C; Matsuo H; Pei D; et al. Cyclic Peptide Inhibitors of HIV-1 Capsid-Human Lysyl-tRNA Synthetase Interaction. ACS Chem. Biol 2012, 7, 761–769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (141).Liu T; Qian Z; Xiao Q; Pei D High-Throughput Screening of One-Bead-One-Compound Libraries: Identification of Cyclic Peptidyl Inhibitors against Calcineurin/NFAT Interaction. ACS Comb. Sci 2011, 13, 537–546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (142).Martínez-Ceron CM; Giudicessi LS; Saavedra LS; Gurevich-Messina MJ; Erra-Balsells R; Albericio F; Cascone O; Camperi AS Latest Advances in OBOC Peptide Libraries. Improvements in Screening Strategies and Enlarging the Family From Linear to Cyclic Libraries. Curr. Pharm. Biotechnol 2016, 17, 449–457. [DOI] [PubMed] [Google Scholar]
- (143).Furka Á. Forty years of combinatorial technology. Drug Discovery Today 2022, 27, 103308. [DOI] [PubMed] [Google Scholar]
- (144).Pei D; Appiah Kubi G Developments with bead-based screening for novel drug discovery. Expert Opin. Drug Discovery 2019, 14, 1097–1102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (145).Liu R; Marik J; Lam KS A Novel Peptide-Based Encoding System for “One-Bead One-Compound” Peptidomimetic and Small Molecule Combinatorial Libraries. J. Am. Chem. Soc 2002, 124, 7678–7680. [DOI] [PubMed] [Google Scholar]
- (146).Joo SH; Xiao Q; Ling Y; Gopishetty B; Pei D High-Throughput Sequence Determination of Cyclic Peptide Library Members by Partial Edman Degradation/Mass Spectrometry. J. Am. Chem. Soc 2006, 128, 13000–13009. [DOI] [PubMed] [Google Scholar]
- (147).Lian W; Upadhyaya P; Rhodes CA; Liu Y; Pei D Screening Bicyclic Peptide Libraries for Protein–Protein Interaction Inhibitors: Discovery of a Tumor Necrosis Factor-α Antagonist. J. Am. Chem. Soc 2013, 135, 11990–11995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (148).MacConnell AB; McEnaney PJ; Cavett VJ; Paegel BM DNA-Encoded Solid-Phase Synthesis: Encoding Language Design and Complex Oligomer Library Synthesis. ACS Comb. Sci 2015, 17, 518–534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (149).Kolch W; Berta D; Rosta E Dynamic regulation of RAS and RAS signaling. Biochem. J 2023, 480, 1–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (150).Wu X; Upadhyaya P; Villalona-Calero MA; Briesewitz R; Pei D Inhibition of Ras–effector interactions by cyclic peptides. MedChemComm 2013, 4, 378–382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (151).Upadhyaya P; Qian Z; Selner NG; Clippinger SR; Wu Z; Briesewitz R; Pei D Inhibition of Ras Signaling by Blocking Ras–Effector Interactions with Cyclic Peptides. Angew. Chem., Int. Ed 2015, 54, 7602–7606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (152).Trinh TB; Upadhyaya P; Qian Z; Pei D Discovery of a Direct Ras Inhibitor by Screening a Combinatorial Library of Cell-Permeable Bicyclic Peptides. ACS Comb. Sci 2016, 18, 75–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (153).Upadhyaya P; Qian Z; Habir NAA; Pei D Direct Ras inhibitors identified from a structurally rigidified bicyclic peptide library. Tetrahedron 2014, 70, 7714–7720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (154).Buyanova M; Cai S; Cooper J; Rhodes C; Salim H; Sahni A; Upadhyaya P; Yang R; Sarkar A; Li N; et al. Discovery of a Bicyclic Peptidyl Pan-Ras Inhibitor. J. Med. Chem 2021, 64, 13038–13053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (155).Nubbemeyer B; Paul George AA; Kühl T; Pepanian A; Beck MS; Maghraby R; Shetab Boushehri M; Muehlhaupt M; Pfeil EM; Annala SK; et al. Targeting Gαi/s Proteins with Peptidyl Nucleotide Exchange Modulators. ACS Chem. Biol 2022, 17, 463–473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (156).Rhodes CA; Dougherty PG; Cooper JK; Qian Z; Lindert S; Wang Q-E; Pei D Cell-Permeable Bicyclic Peptidyl Inhibitors against NEMO-IκB Kinase Interaction Directly from a Combinatorial Library. J. Am. Chem. Soc 2018, 140, 12102–12110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (157).Gerwert K; Mann D; Kötting C Common mechanisms of catalysis in small and heterotrimeric GTPases and their respective GAPs. Biol. Chem 2017, 398, 523–533. [DOI] [PubMed] [Google Scholar]
- (158).Weinstein LS; Chen M; Xie T; Liu J Genetic diseases associated with heterotrimeric G proteins. Trends Pharmacol. Sci 2006, 27, 260–266. [DOI] [PubMed] [Google Scholar]
- (159).Millward SW; Fiacco S; Austin RJ; Roberts RW Design of Cyclic Peptides That Bind Protein Surfaces with Antibody-Like Affinity. ACS Chem. Biol 2007, 2, 625–634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (160).Howell SM; Fiacco SV; Takahashi TT; Jalali-Yazdi F; Millward SW; Hu B; Wang P; Roberts RW Serum Stable Natural Peptides Designed by mRNA Display. Sci. Rep 2014, 4, 6008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (161).Fiacco SV; Kelderhouse LE; Hardy A; Peleg Y; Hu B; Ornelas A; Yang P; Gammon ST; Howell SM; Wang P; et al. Directed Evolution of Scanning Unnatural-Protease-Resistant (SUPR) Peptides for in Vivo Applications. ChemBioChem. 2016, 17, 1643–1651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (162).de Opakua AI; Parag-Sharma K; DiGiacomo V; Merino N; Leyme A; Marivin A; Villate M; Nguyen LT; de la Cruz-Morcillo MA; Blanco-Canosa JB; et al. Molecular mechanism of Gαi activation by non-GPCR proteins with a Gα-Binding and Activating motif. Nat. Commun 2017, 8, 15163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (163).Ghosh P; Rangamani P; Kufareva I The GAPs, GEFs, GDIs and⋯now, GEMs: New kids on the heterotrimeric G protein signaling block. Cell Cycle 2017, 16, 607–612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (164).Pepanian A; Binbay FA; Roy S; Nubbemeyer B; Koley A; Rhodes CA; Ammer H; Pei D; Ghosh P; Imhof D Bicyclic Peptide Library Screening for the Identification of Gαi Protein Modulators. J. Med. Chem 2023, 66, 12396–12406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (165).Gartner ZJ; Liu DR The Generality of DNA-Templated Synthesis as a Basis for Evolving Non-Natural Small Molecules. J. Am. Chem. Soc 2001, 123, 6961–6963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (166).Gartner ZJ; Tse BN; Grubina R; Doyon JB; Snyder TM; Liu DR DNA-Templated Organic Synthesis and Selection of a Library of Macrocycles. Science 2004, 305, 1601–1605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (167).Tse BN; Snyder TM; Shen Y; Liu DR Translation of DNA into a Library of 13 000 Synthetic Small-Molecule Macrocycles Suitable for in Vitro Selection. J. Am. Chem. Soc 2008, 130, 15611–15626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (168).Usanov DL; Chan AI; Maianti JP; Liu DR Second-generation DNA-templated macrocycle libraries for the discovery of bioactive small molecules. Nat. Chem 2018, 10, 704–714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (169).Kleiner RE; Dumelin CE; Tiu GC; Sakurai K; Liu DR In Vitro Selection of a DNA-Templated Small-Molecule Library Reveals a Class of Macrocyclic Kinase Inhibitors. J. Am. Chem. Soc 2010, 132, 11779–11791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (170).Seigal BA; Connors WH; Fraley A; Borzilleri RM; Carter PH; Emanuel SL; Fargnoli J; Kim K; Lei M; Naglich JG; et al. The Discovery of Macrocyclic XIAP Antagonists from a DNA-Programmed Chemistry Library, and Their Optimization To Give Lead Compounds with in Vivo Antitumor Activity. J. Med. Chem 2015, 58, 2855–2861. [DOI] [PubMed] [Google Scholar]
- (171).Brenner S; Lerner RA Encoded combinatorial chemistry. Proc. Natl. Acad. Sci. U.S.A 1992, 89, 5381–5383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (172).Needels MC; Jones DG; Tate EH; Heinkel GL; Kochersperger LM; Dower WJ; Barrett RW; Gallop MA Generation and screening of an oligonucleotide-encoded synthetic peptide library. Proc. Natl. Acad. Sci. U.S.A 1993, 90, 10700–10704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (173).Nielsen J; Brenner S; Janda KD Synthetic methods for the implementation of encoded combinatorial chemistry. J. Am. Chem. Soc 1993, 115, 9812–9813. [Google Scholar]
- (174).Yang P; Wang X; Li B; Yang Y; Yue J; Suo Y; Tong H; He G; Lu X; Chen G Streamlined construction of peptide macrocycles via palladium-catalyzed intramolecular S-arylation in solution and on DNA. Chem. Sci 2021, 12, 5804–5810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (175).Iyer NG; OÖzdag H; Caldas C p300/CBP and cancer. Oncogene 2004, 23, 4225–4231. [DOI] [PubMed] [Google Scholar]
- (176).Silvestri AP; Zhang Q; Ping Y; Muir EW; Zhao J; Chakka SK; Wang G; Bray WM; Chen W; Fribourgh JL; et al. DNA-Encoded Macrocyclic Peptide Libraries Enable the Discovery of a Neutral MDM2–p53 Inhibitor. ACS Med. Chem. Lett 2023, 14, 820–826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (177).Li H; Li J; Chao J; Zhang Z; Qin C Head-to-tail cyclization for the synthesis of naturally occurring cyclic peptides on organophosphorus small-molecular supports. Org. Chem. Front 2022, 9, 946–952. [Google Scholar]
- (178).Pham MV; Bergeron-Brlek M; Heinis C Synthesis of DNA-Encoded Disulfide- and Thioether-Cyclized Peptides. Chem-BioChem. 2020, 21, 543–549. [DOI] [PubMed] [Google Scholar]
- (179).Onda Y; Bassi G; Elsayed A; Ulrich F; Oehler S; Plais L; Scheuermann J; Neri D A DNA-Encoded Chemical Library Based on Peptide Macrocycles. Chem. Eur. J 2021, 27, 7160–7167. [DOI] [PubMed] [Google Scholar]
- (180).Plais L; Scheuermann J Macrocyclic DNA-encoded chemical libraries: a historical perspective. RSC Chem. Biol 2022, 3, 7–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (181).Wang Y; Zhang K; Zhao Y; Li Y; Su W; Li S Construction and Applications of Mammalian Cell-Based DNA-Encoded Peptide/Protein Libraries. ACS Synth. Biol 2023, 12, 1874–1888. [DOI] [PubMed] [Google Scholar]
- (182).Kunig VBK; Potowski M; Klika Škopić M; Brunschweiger A Scanning Protein Surfaces with DNA-Encoded Libraries. ChemMedChem. 2021, 16, 1048–1062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (183).Smith GP Filamentous Fusion Phage: Novel Expression Vectors That Display Cloned Antigens on the Virion Surface. Science 1985, 228, 1315–1317. [DOI] [PubMed] [Google Scholar]
- (184).Sidhu SS Engineering M13 for phage display. Biomol. Eng 2001, 18, 57–63. [DOI] [PubMed] [Google Scholar]
- (185).Hart SL; Knight AM; Harbottle RP; Mistry A; Hunger HD; Cutler DF; Williamson R; Coutelle C Cell binding and internalization by filamentous phage displaying a cyclic Arg-Gly-Asp-containing peptide. J. Biol. Chem 1994, 269, 12468–12474. [PubMed] [Google Scholar]
- (186).O’Neil KT; Hoess RH; Jackson SA; Ramachandran NS; Mousa SA; Degrado WF Identification of novel peptide antagonists for GPIIb/IIIa from a conformationally constrained phage peptide library. Proteins 1992, 14, 509–515. [DOI] [PubMed] [Google Scholar]
- (187).Koivunen E; Gay DA; Ruoslahti E Selection of peptides binding to the alpha 5 beta 1 integrin from phage display library. J. Biol. Chem 1993, 268, 20205–20210. [PubMed] [Google Scholar]
- (188).Koivunen E; Wang B; Ruoslahti E Isolation of a highly specific ligand for the alpha 5 beta 1 integrin from a phage display library. J. Cell Biol 1994, 124, 373–380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (189).Wang B; Dickinson LA; Koivunen E; Ruoslahti E; Kohwi-Shigematsu T A Novel Matrix Attachment Region DNA Binding Motif Identified Using a Random Phage Peptide Library. J. Biol. Chem 1995, 270, 23239–23242. [DOI] [PubMed] [Google Scholar]
- (190).Wan X-C; Zhang Y-N; Zhang H; Chen Y; Cui Z-H; Zhu W-J; Fang G-M Asparaginyl Endopeptidase-Mediated Peptide Cyclization for Phage Display. Org. Lett 2024, 26, 2601–2605. [DOI] [PubMed] [Google Scholar]
- (191).Watson GM; Kulkarni K; Sang J; Ma X; Gunzburg MJ; Perlmutter P; Wilce MCJ; Wilce JA Discovery, Development, and Cellular Delivery of Potent and Selective Bicyclic Peptide Inhibitors of Grb7 Cancer Target. J. Med. Chem 2017, 60, 9349–9359. [DOI] [PubMed] [Google Scholar]
- (192).Ghaffari Sharaf M; Cetinel S; Semenchenko V; Damji KF; Unsworth LD; Montemagno C Peptides for targeting βB2-Crystallin fibrils. Exp. Eye Res 2017, 165, 109–117. [DOI] [PubMed] [Google Scholar]
- (193).Desimmie BA; Humbert M; Lescrinier E; Hendrix J; Vets S; Gijsbers R; Ruprecht RM; Dietrich U; Debyser Z; Christ F Phage display-directed discovery of LEDGF/p75 binding cyclic peptide inhibitors of HIV replication. Mol. Ther 2012, 20, 2064–2075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (194).Jaroszewicz W; Morcinek-Orłowska J; Pierzynowska K; Gaffke L; Węgrzyn G Phage display and other peptide display technologies. FEMS Microbiol. Rev 2022, 46, fuab052. [DOI] [PubMed] [Google Scholar]
- (195).Teymennet-Ramírez KV; Martínez-Morales F; Trejo-Hernández MR Yeast Surface Display System: Strategies for Improvement and Biotechnological Applications. Front. Bioeng. Biotechnol 2022, 9, 794742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (196).Bertoldo D; Khan MMG; Dessen P; Held W; Huelsken J; Heinis C Phage Selection of Peptide Macrocycles against β-Catenin To Interfere with Wnt Signaling. ChemMedChem. 2016, 11, 834–839. [DOI] [PubMed] [Google Scholar]
- (197).Youle RJ; Strasser A The BCL-2 protein family: opposing activities that mediate cell death. Nat. Rev. Mol. Cell. Biol 2008, 9, 47–59. [DOI] [PubMed] [Google Scholar]
- (198).Kale J; Osterlund EJ; Andrews DW BCL-2 family proteins: changing partners in the dance towards death. Cell Death Differ. 2018, 25, 65–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (199).Shamas-Din A; Kale J; Leber B; Andrews DW Mechanisms of action of Bcl-2 family proteins. Cold Spring Harb. Perspect. Biol 2013, 5, No. a008714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (200).Hardwick JM; Soane L Multiple functions of BCL-2 family proteins. Cold Spring Harb. Perspect. Biol 2013, 5, No. a008722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (201).Li F; Liu J; Liu C; Liu Z; Peng X; Huang Y; Chen X; Sun X; Wang S; Chen W; et al. Cyclic peptides discriminate BCL-2 and its clinical mutants from BCL-XL by engaging a single-residue discrepancy. Nat. Commun 2024, 15, 1476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (202).Zheng X; Li Z; Gao W; Meng X; Li X; Luk LYP; Zhao Y; Tsai Y-H; Wu C Condensation of 2-((Alkylthio)(aryl)-methylene)malononitrile with 1,2-Aminothiol as a Novel Bioorthogonal Reaction for Site-Specific Protein Modification and Peptide Cyclization. J. Am. Chem. Soc 2020, 142, 5097–5103. [DOI] [PubMed] [Google Scholar]
- (203).Sakamoto K; Kamada Y; Sameshima T; Yaguchi M; Niida A; Sasaki S; Miwa M; Ohkubo S; Sakamoto J-I; Kamaura M; et al. K-Ras(G12D)-selective inhibitory peptides generated by random peptide T7 phage display technology. Biochem. Biophys. Res. Commun 2017, 484, 605–611. [DOI] [PubMed] [Google Scholar]
- (204).Nakase I; Niwa M; Takeuchi T; Sonomura K; Kawabata N; Koike Y; Takehashi M; Tanaka S; Ueda K; Simpson JC; et al. Cellular Uptake of Arginine-Rich Peptides: Roles for Macropinocytosis and Actin Rearrangement. Mol. Ther 2004, 10, 1011–1022. [DOI] [PubMed] [Google Scholar]
- (205).Brock R. The Uptake of Arginine-Rich Cell-Penetrating Peptides: Putting the Puzzle Together. Bioconjugate Chem. 2014, 25, 863–868. [DOI] [PubMed] [Google Scholar]
- (206).Sogabe S; Kamada Y; Miwa M; Niida A; Sameshima T; Kamaura M; Yonemori K; Sasaki S; Sakamoto J.-i.; Sakamoto K Crystal Structure of a Human K-Ras G12D Mutant in Complex with GDP and the Cyclic Inhibitory Peptide KRpep-2d. ACS Med. Chem. Lett 2017, 8, 732–736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (207).Sakamoto K; Masutani T; Hirokawa T Generation of KS-58 as the first K-Ras(G12D)-inhibitory peptide presenting anti-cancer activity in vivo. Sci. Rep 2020, 10, 21671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (208).Sakamoto K; Hirokawa T Lipid bilayer membrane permeability mechanism of the K-Ras(G12D)-inhibitory bicyclic peptide KS-58 elucidated by molecular dynamics simulations. Bioorg. Med. Chem. Lett 2024, 100, 129649. [DOI] [PubMed] [Google Scholar]
- (209).O’Reilly EM; Ko AH; Friedberg JW Flashback Foreword: Gemcitabine for Advanced Pancreatic Cancer. J. Clin. Oncol 2023, 41, 5479–5480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (210).Heinemann V. Gemcitabine in metastatic breast cancer. Expert Rev. Anticancer Ther 2005, 5, 429–443. [DOI] [PubMed] [Google Scholar]
- (211).Sakamoto K; Lin B; Nunomura K; Izawa T; Nakagawa S The K-Ras(G12D)-inhibitory peptide KS-58 suppresses growth of murine CT26 colorectal cancer cell-derived tumors. Sci. Rep 2022, 12, 8121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (212).Sakamoto K; Qi Y; Miyako E Nanoformulation of the K-Ras(G12D)-inhibitory peptide KS-58 suppresses colorectal and pancreatic cancer-derived tumors. Sci. Rep 2023, 13, 518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (213).Lim S; Boyer N; Boo N; Huang C; Venkatachalam G; Angela Juang Y-C; Garrigou M; Kaan HYK; Duggal R; Peh KM; et al. Discovery of cell active macrocyclic peptides with on-target inhibition of KRAS signaling. Chem. Sci 2021, 12, 15975–15987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (214).Seto E; Yoshida M Erasers of histone acetylation: the histone deacetylase enzymes. Cold Spring Harb. Perspect. Biol 2014, 6, No. a018713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (215).Owens AE; Iannuzzelli JA; Gu Y; Fasan R MOrPH-PhD: An Integrated Phage Display Platform for the Discovery of Functional Genetically Encoded Peptide Macrocycles. ACS Cent. Sci 2020, 6, 368–381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (216).Baird L; Yamamoto M The Molecular Mechanisms Regulating the KEAP1-NRF2 Pathway. Mol. Cell. Biol 2020, 40, e00099–00020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (217).Canning P; Sorrell FJ; Bullock AN Structural basis of Keap1 interactions with Nrf2. Free Radic. Biol. Med 2015, 88, 101–107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (218).Zheng M; Haeffner F; Gao J N-Terminal cysteine mediated backbone-side chain cyclization for chemically enhanced phage display. Chem. Sci 2022, 13, 8349–8354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (219).Heinis C; Rutherford T; Freund S; Winter G Phage-encoded combinatorial chemical libraries based on bicyclic peptides. Nat. Chem. Biol 2009, 5, 502–507. [DOI] [PubMed] [Google Scholar]
- (220).Luo Y; Schofield JA; Na Z; Hann T; Simon MD; Slavoff SA Discovery of cellular substrates of human RNA-decapping enzyme DCP2 using a stapled bicyclic peptide inhibitor. Cell Chem. Biol 2021, 28, 463–474.e7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (221).Pai SG; Carneiro BA; Mota JM; Costa R; Leite CA; Barroso-Sousa R; Kaplan JB; Chae YK; Giles FJ Wnt/beta-catenin pathway: modulating anticancer immune response. J. Hematol. Oncol 2017, 10, 101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (222).Liu J; Xiao Q; Xiao J; Niu C; Li Y; Zhang X; Zhou Z; Shu G; Yin G Wnt/β-catenin signalling: function, biological mechanisms, and therapeutic opportunities. Signal Transduct. Target Ther 2022, 7, 3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (223).MacDonald BT; Tamai K; He X Wnt/β-Catenin Signaling: Components, Mechanisms, and Diseases. Dev. Cell 2009, 17, 9–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (224).Li Y; Dai J; Song M; Fitzgerald-Bocarsly P; Kiledjian M Dcp2 decapping protein modulates mRNA stability of the critical interferon regulatory factor (IRF) IRF-7. Mol. Cell. Biol 2012, 32, 1164–1172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (225).Maciej VD; Mateva N; Schwarz J; Dittmers T; Mallick M; Urlaub H; Chakrabarti S Intrinsically disordered regions of tristetraprolin and DCP2 directly interact to mediate decay of ARE-mRNA. Nucleic Acids Res. 2022, 50, 10665–10679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (226).Ziemniak M; Mugridge JS; Kowalska J; Rhoads RE; Gross JD; Jemielity J Two-headed tetraphosphate cap analogs are inhibitors of the Dcp1/2 RNA decapping complex. RNA 2016, 22, 518–529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (227).Chen F-J; Pinnette N; Gao J Strategies for the Construction of Multicyclic Phage Display Libraries. ChemBioChem. 2024, 25, e202400072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (228).Baldini C; Goldschmidt V; Brana I; Doger B; Italiano A; Cousin S; Falchook GS; Necchi A; Reig O; Carter L; et al. BT8009–100: A phase I/II study of novel bicyclic peptide and MMAE conjugate BT8009 in patients (pts) with advanced malignancies associated with nectin-4 expression, including urothelial cancer (UC). J. Clin. Oncol 2023, 41, 498–498. [Google Scholar]
- (229).Bennett G; Brown A; Mudd G; Huxley P; Van Rietschoten K; Pavan S; Chen L; Watcham S; Lahdenranta J; Keen N MMAE Delivery Using the Bicycle Toxin Conjugate BT5528. Mol. Cancer Ther 2020, 19, 1385–1394. [DOI] [PubMed] [Google Scholar]
- (230).Bennett G; Lutz R; Park P; Harrison H; Lee K Abstract 1167: Development of BT1718, a novel Bicycle Drug Conjugate for the treatment of lung cancer. Cancer Res. 2017, 77, 1167–1167. [Google Scholar]
- (231).Upadhyaya P; Lahdenranta J; Hurov K; Battula S; Dods R; Haines E; Kleyman M; Kristensson J; Kublin J; Lani R; et al. Anticancer immunity induced by a synthetic tumor-targeted CD137 agonist. J. Immunother. Cancer 2021, 9, No. e001762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (232).Urban JH; Moosmeier MA; Aumuüller T; Thein M; Bosma T; Rink R; Groth K; Zulley M; Siegers K; Tissot K; et al. Phage display and selection of lanthipeptides on the carboxyterminus of the gene-3 minor coat protein. Nat. Commun 2017, 8, 1500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (233).Ishii J; Yoshimoto N; Tatematsu K; Kuroda S. i.; Ogino C; Fukuda H; Kondo A Cell Wall Trapping of Autocrine Peptides for Human G-Protein-Coupled Receptors on the Yeast Cell Surface. PLoS One 2012, 7, e37136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (234).Bowen J; Schneible J; Bacon K; Labar C; Menegatti S; Rao BM Screening of Yeast Display Libraries of Enzymatically Treated Peptides to Discover Macrocyclic Peptide Ligands. Int. J. Mol. Sci 2021, 22, 1634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (235).Nemoto N; Miyamoto-Sato E; Husimi Y; Yanagawa H In vitro virus: Bonding of mRNA bearing puromycin at the 3′-terminal end to the C-terminal end of its encoded protein on the ribosome in vitro. FEBS Lett. 1997, 414, 405–408. [DOI] [PubMed] [Google Scholar]
- (236).Roberts RW; Szostak JW RNA-peptide fusions for the in vitro selection of peptides and proteins. Proc. Natl. Acad. Sci. U.S.A 1997, 94, 12297–12302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (237).Shimizu Y; Inoue A; Tomari Y; Suzuki T; Yokogawa T; Nishikawa K; Ueda T Cell-free translation reconstituted with purified components. Nat. Biotechnol 2001, 19, 751–755. [DOI] [PubMed] [Google Scholar]
- (238).Schneider AFL; Kallen J; Ottl J; Reid PC; Ripoche S; Ruetz S; Stachyra T-M; Hintermann S; Dumelin CE; Hackenberger CPR; et al. Discovery, X-ray structure and CPP-conjugation enabled uptake of p53/MDM2 macrocyclic peptide inhibitors. RSC Chem. Biol 2021, 2, 1661–1668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (239).Morimoto J; Hayashi Y; Suga H Discovery of Macrocyclic Peptides Armed with a Mechanism-Based Warhead: Isoform-Selective Inhibition of Human Deacetylase SIRT2. Angew. Chem., Int. Ed 2012, 51, 3423–3427. [DOI] [PubMed] [Google Scholar]
- (240).Bolding JE; Nielsen AL; Jensen I; Hansen TN; Ryberg LA; Jameson ST; Harris P; Peters GHJ; Denu JM; Rogers JM; et al. Substrates and Cyclic Peptide Inhibitors of the Oligonucleotide-Activated Sirtuin 7**. Angew. Chem., Int. Ed 2023, 62, e202314597. [DOI] [PubMed] [Google Scholar]
- (241).Nishio K; Belle R; Katoh T; Kawamura A; Sengoku T; Hanada K; Ohsawa N; Shirouzu M; Yokoyama S; Suga H Thioether Macrocyclic Peptides Selected against TET1 Compact Catalytic Domain Inhibit TET1 Catalytic Activity. ChemBioChem. 2018, 19, 979–985. [DOI] [PubMed] [Google Scholar]
- (242).Šimelis K; Saracç H; Salah E; Nishio K; McAllister TE; Corner TP; Tumber A; Belle R; Schofield CJ; Suga H; et al. Selective targeting of human TET1 by cyclic peptide inhibitors: Insights from biochemical profiling. Biorg. Med. Chem 2024, 99, 117597. [DOI] [PubMed] [Google Scholar]
- (243).Coleman OD; Macdonald J; Thomson B; Ward JA; Stubbs CJ; McAllister TE; Clark S; Amin S; Cao Y; Abboud MI; et al. Cyclic peptides target the aromatic cage of a PHD-finger reader domain to modulate epigenetic protein function. Chem. Sci 2023, 14, 7136–7146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (244).Cremosnik GS; Mesrouze Y; Zueger P; Furkert D; Grandjean F; Argoti D; Mermet-Meillon F; Bauer MR; Brittain S; Rogemoser P; et al. mRNA Display Identifies Potent, Paralog-Selective Peptidic Ligands for ARID1B. ACS Chem. Biol 2024, 19, 1142–1150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (245).Song X; Lu L-Y; Passioura T; Suga H Macrocyclic peptide inhibitors for the protein-protein interaction of Zaire Ebola virus protein 24 and karyopherin alpha 5. Org. Biomol. Chem 2017, 15, 5155–5160. [DOI] [PubMed] [Google Scholar]
- (246).Morgan M; Ikenoue T; Suga H; Wolberger C Potent macrocycle inhibitors of the human SAGA deubiquitinating module. Cell Chem. Biol 2022, 29, 544–554.e544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (247).Haberman VA; Fleming SR; Leisner TM; Puhl AC; Feng E; Xie L; Chen X; Goto Y; Suga H; Parise LV; et al. Discovery and Development of Cyclic Peptide Inhibitors of CIB1. ACS Med. Chem. Lett 2021, 12, 1832–1839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (248).Yamagishi Y; Shoji I; Miyagawa S; Kawakami T; Katoh T; Goto Y; Suga H Natural Product-Like Macrocyclic N-Methyl-Peptide Inhibitors against a Ubiquitin Ligase Uncovered from a Ribosome-Expressed De Novo Library. Chem. Biol 2011, 18, 1562–1570. [DOI] [PubMed] [Google Scholar]
- (249).Goto Y; Katoh T; Suga H Flexizymes for genetic code reprogramming. Nat. Protoc 2011, 6, 779–790. [DOI] [PubMed] [Google Scholar]
- (250).Goto Y; Ohta A; Sako Y; Yamagishi Y; Murakami H; Suga H Reprogramming the Translation Initiation for the Synthesis of Physiologically Stable Cyclic Peptides. ACS Chem. Biol 2008, 3, 120–129. [DOI] [PubMed] [Google Scholar]
- (251).Zhang Z; Gao R; Hu Q; Peacock H; Peacock DM; Dai S; Shokat KM; Suga H GTP-State-Selective Cyclic Peptide Ligands of K-Ras(G12D) Block Its Interaction with Raf. ACS Cent. Sci 2020, 6, 1753–1761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (252).Ikenoue T; Oono M; So M; Yamakado H; Arata T; Takahashi R; Kawata Y; Suga H A RaPID Macrocyclic Peptide That Inhibits the Formation of α-Synuclein Amyloid Fibrils. ChemBioChem. 2023, 24, e202300320. [DOI] [PubMed] [Google Scholar]
- (253).Chen K-E; Guo Q; Hill TA; Cui Y; Kendall AK; Yang Z; Hall RJ; Healy MD; Sacharz J; Norwood SJ; et al. De novo macrocyclic peptides for inhibiting, stabilizing, and probing the function of the retromer endosomal trafficking complex. Sci. Adv 2021, 7, eabg4007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (254).Kamalinia G; Grindel BJ; Takahashi TT; Millward SW; Roberts RW Directing evolution of novel ligands by mRNA display. Chem. Soc. Rev 2021, 50, 9055–9103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (255).Li R; Kang G; Hu M; Huang H Ribosome Display: A Potent Display Technology used for Selecting and Evolving Specific Binders with Desired Properties. Mol. Biotechnol 2019, 61, 60–71. [DOI] [PubMed] [Google Scholar]
- (256).Galán A; Comor L; Horvatić A; KuleŠ J; Guillemin N; Mrljak V; Bhide M Library-based display technologies: where do we stand? Mol. Biosyst 2016, 12, 2342–2358. [DOI] [PubMed] [Google Scholar]
- (257).Nawatha M; Rogers JM; Bonn SM; Livneh I; Lemma B; Mali SM; Vamisetti GB; Sun H; Bercovich B; Huang Y; et al. De novo macrocyclic peptides that specifically modulate Lys48-linked ubiquitin chains. Nat. Chem 2019, 11, 644–652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (258).Rogers JM; Nawatha M; Lemma B; Vamisetti GB; Livneh I; Barash U; Vlodavsky I; Ciechanover A; Fushman D; Suga H; et al. In vivo modulation of ubiquitin chains by N-methylated non-proteinogenic cyclic peptides. RSC Chem. Biol 2021, 2, 513–522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (259).Song L; Luo Z-Q Post-translational regulation of ubiquitin signaling. J. Cell Biol 2019, 218, 1776–1786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (260).Lemma B; Zhang D; Vamisetti GB; Wentz BG; Suga H; Brik A; Lubkowski J; Fushman D Mechanism of selective recognition of Lys48-linked polyubiquitin by macrocyclic peptide inhibitors of proteasomal degradation. Nat. Commun 2023, 14, 7212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (261).Lin C-W; Harner MJ; Douglas AE; Lafont V; Yu F; Lee VG; Poss MA; Swain JF; Wright M; Lipovšek D A Selection of Macrocyclic Peptides That Bind STING From an mRNA-Display Library With Split Degenerate Codons. Angew. Chem., Int. Ed 2021, 60, 22640–22645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (262).Rogers JM; Passioura T; Suga H Nonproteinogenic deep mutational scanning of linear and cyclic peptides. Proc. Natl. Acad. Sci. U.S.A 2018, 115, 10959–10964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (263).Kawamura A; Muünzel M; Kojima T; Yapp C; Bhushan B; Goto Y; Tumber A; Katoh T; King ONF; Passioura T; et al. Highly selective inhibition of histone demethylases by de novo macrocyclic peptides. Nat. Commun 2017, 8, 14773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (264).Passioura T; Bhushan B; Tumber A; Kawamura A; Suga H Structure-activity studies of a macrocyclic peptide inhibitor of histone lysine demethylase 4A. Biorg. Med. Chem 2018, 26, 1225–1231. [DOI] [PubMed] [Google Scholar]
- (265).Dai SA; Hu Q; Gao R; Blythe EE; Touhara KK; Peacock H; Zhang Z; von Zastrow M; Suga H; Shokat KM State-selective modulation of heterotrimeric Gαs signaling with macrocyclic peptides. Cell 2022, 185, 3950–3965.e3925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (266).Tanada M; Tamiya M; Matsuo A; Chiyoda A; Takano K; Ito T; Irie M; Kotake T; Takeyama R; Kawada H; et al. Development of Orally Bioavailable Peptides Targeting an Intracellular Protein: From a Hit to a Clinical KRAS Inhibitor. J. Am. Chem. Soc 2023, 145, 16610–16620. [DOI] [PubMed] [Google Scholar]
- (267).Howard JF Jr; Vissing J; Gilhus NE; Leite MI; Utsugisawa K; Duda PW; Farzaneh-Far R; Murai H; Wiendl H Zilucoplan: An Investigational Complement C5 Inhibitor for the Treatment of Acetylcholine Receptor Autoantibody–Positive Generalized Myasthenia Gravis. Expert Opin. Investig. Drugs 2021, 30, 483–493. [DOI] [PubMed] [Google Scholar]
- (268).Shirley M Zilucoplan: First Approval. Drugs 2024, 84, 99–104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (269).Alleyne C; Amin RP; Bhatt B; Bianchi E; Blain JC; Boyer N; Branca D; Embrey MW; Ha SN; Jette K; et al. Series of Novel and Highly Potent Cyclic Peptide PCSK9 Inhibitors Derived from an mRNA Display Screen and Optimized via Structure-Based Design. J. Med. Chem 2020, 63, 13796–13824. [DOI] [PubMed] [Google Scholar]
- (270).Tucker TJ; Embrey MW; Alleyne C; Amin RP; Bass A; Bhatt B; Bianchi E; Branca D; Bueters T; Buist N; et al. A Series of Novel, Highly Potent, and Orally Bioavailable Next-Generation Tricyclic Peptide PCSK9 Inhibitors. J. Med. Chem 2021, 64, 16770–16800. [DOI] [PubMed] [Google Scholar]
- (271).Brousseau ME; Clairmont KB; Spraggon G; Flyer AN; Golosov AA; Grosche P; Amin J; Andre J; Burdick D; Caplan S; et al. Identification of a PCSK9-LDLR disruptor peptide with in vivo function. Cell Chem. Biol 2022, 29, 249–258.e245. [DOI] [PubMed] [Google Scholar]
- (272).Ballantyne CM; Banka P; Mendez G; Garcia R; Rosenstock J; Rodgers A; Mendizabal G; Mitchel Y; Catapano AL Phase 2b Randomized Trial of the Oral PCSK9 Inhibitor MK-0616. J. Am. Coll. Cardiol 2023, 81, 1553–1564. [DOI] [PubMed] [Google Scholar]
- (273).Johns DG; Campeau L-C; Banka P; Bautmans A; Bueters T; Bianchi E; Branca D; Bulger PG; Crevecoeur I; Ding F-X; et al. Orally Bioavailable Macrocyclic Peptide That Inhibits Binding of PCSK9 to the Low Density Lipoprotein Receptor. Circulation 2023, 148, 144–158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (274).Bhushan B; Granata D; Kaas CS; Kasimova MA; Ren Q; Cramer CN; White MD; Hansen AMK; Fledelius C; Invernizzi G; et al. An integrated platform approach enables discovery of potent, selective and ligand-competitive cyclic peptides targeting the GIP receptor. Chem. Sci 2022, 13, 3256–3262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (275).Smith TP; Bhushan B; Granata D; Kaas CS; Andersen B; Decoene KW; Ren Q; Liu H; Qu X; Yang Y; et al. Identification and engineering of potent cyclic peptides with selective or promiscuous binding through biochemical profiling and bioinformatic data analysis. RSC Chem. Biol 2024, 5, 12–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (276).Scott CP; Abel-Santos E; Wall M; Wahnon DC; Benkovic SJ Production of cyclic peptides and proteins in vivo. Proc. Natl. Acad. Sci. U.S.A 1999, 96, 13638–13643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (277).Scott CP; Abel-Santos E; Jones AD; Benkovic SJ Structural requirements for the biosynthesis of backbone cyclic peptide libraries. Chem. Biol 2001, 8, 801–815. [DOI] [PubMed] [Google Scholar]
- (278).Tavassoli A SICLOPPS cyclic peptide libraries in drug discovery. Curr. Opin. Chem. Biol 2017, 38, 30–35. [DOI] [PubMed] [Google Scholar]
- (279).Lennard KR; Tavassoli A Peptides Come Round: Using SICLOPPS Libraries for Early Stage Drug Discovery. Chem. Eur. J 2014, 20, 10608–10614. [DOI] [PubMed] [Google Scholar]
- (280).Horswill AR; Savinov SN; Benkovic SJ A systematic method for identifying small-molecule modulators of protein–protein interactions. Proc. Natl. Acad. Sci. U.S.A 2004, 101, 15591–15596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (281).Osher EL; Castillo F; Elumalai N; Waring MJ; Pairaudeau G; Tavassoli A A genetically selected cyclic peptide inhibitor of BCL6 homodimerization. Biorg. Med. Chem 2018, 26, 3034–3038. [DOI] [PubMed] [Google Scholar]
- (282).Leitch EK; Elumalai N; Fridén-Saxin M; Dahl G; Wan P; Clarkson P; Valeur E; Pairaudeau G; Boyd H; Tavassoli A Inhibition of low-density lipoprotein receptor degradation with a cyclic peptide that disrupts the homodimerization of IDOL E3 ubiquitin ligase. Chem. Sci 2018, 9, 5957–5966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (283).Lennard KR; Gardner RM; Doigneaux C; Castillo F; Tavassoli A Development of a Cyclic Peptide Inhibitor of the p6/UEV Protein-Protein Interaction. ACS Chem. Biol 2019, 14, 1874–1878. [DOI] [PubMed] [Google Scholar]
- (284).Ismail M; Martin SR; George R; Houghton F; Kelly G; Chaleil RAG; Anastasiou P; Wang X; O’Reilly N; Federico S; et al. Characterisation of a cyclic peptide that binds to the RAS binding domain of phosphoinositide 3-kinase p110α. Sci. Rep 2023, 13, 1889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (285).Miranda E; Nordgren IK; Male AL; Lawrence CE; Hoakwie F; Cuda F; Court W; Fox KR; Townsend PA; Packham GK; et al. A Cyclic Peptide Inhibitor of HIF-1 Heterodimerization That Inhibits Hypoxia Signaling in Cancer Cells. J. Am. Chem. Soc 2013, 135, 10418–10425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (286).Ball AT; Mohammed S; Doigneaux C; Gardner RM; Easton JW; Turner S; Essex JW; Pairaudeau G; Tavassoli A Identification and Development of Cyclic Peptide Inhibitors of Hypoxia Inducible Factors 1 and 2 That Disrupt Hypoxia-Response Signaling in Cancer Cells. J. Am. Chem. Soc 2024, 146, 8877–8886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (287).Giaccia A; Siim BG; Johnson RS HIF-1 as a target for drug development. Nat. Rev. Drug Discovery 2003, 2, 803–811. [DOI] [PubMed] [Google Scholar]
- (288).Harris AL Hypoxia — a key regulatory factor in tumour growth. Nat. Rev. Cancer 2002, 2, 38–47. [DOI] [PubMed] [Google Scholar]
- (289).Forsythe JA; Jiang B-H; Iyer NV; Agani F; Leung SW; Koos RD; Semenza GL Activation of Vascular Endothelial Growth Factor Gene Transcription by Hypoxia-Inducible Factor 1. Mol. Cell. Biol 1996, 16, 4604–4613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (290).Birts CN; Nijjar SK; Mardle CA; Hoakwie F; Duriez PJ; Blaydes JP; Tavassoli A A cyclic peptide inhibitor of C-terminal binding protein dimerization links metabolism with mitotic fidelity in breast cancer cells. Chem. Sci 2013, 4, 3046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (291).Fjeld CC; Birdsong WT; Goodman RH Differential binding of NAD + and NADH allows the transcriptional corepressor carboxyl-terminal binding protein to serve as a metabolic sensor. Proc. Natl. Acad. Sci. U.S.A 2003, 100, 9202–9207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (292).Nardini M; Spanò S; Cericola C; Pesce A; Massaro A; Millo E; Luini A; Corda D; Bolognesi M CtBP/BARS: a dual-function protein involved in transcription co-repression and Golgi membrane fission. EMBO J. 2003, 22, 3122–3130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (293).Quinlan KGR; Nardini M; Verger A; Francescato P; Yaswen P; Corda D; Bolognesi M; Crossley M Specific Recognition of ZNF217 and Other Zinc Finger Proteins at a Surface Groove of C-Terminal Binding Proteins. Mol. Cell. Biol 2006, 26, 8159–8172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (294).Shi Y; Sawada J. -i.; Sui G; Affar EB; Whetstine JR; Lan F; Ogawa H; Po-Shan Luke M; Nakatani Y; Shi Y Coordinated histone modifications mediated by a CtBP co-repressor complex. Nature 2003, 422, 735–738. [DOI] [PubMed] [Google Scholar]
- (295).Bergman LM; Blaydes JP C-terminal binding proteins: Emerging roles in cell survival and tumorigenesis. Apoptosis 2006, 11, 879–888. [DOI] [PubMed] [Google Scholar]
- (296).Iskandar SE; Haberman VA; Bowers AA Expanding the Chemical Diversity of Genetically Encoded Libraries. ACS Comb. Sci 2020, 22, 712–733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (297).Ekanayake AI; Sobze L; Kelich P; Youk J; Bennett NJ; Mukherjee R; Bhardwaj A; Wuest F; Vukovic L; Derda R Genetically Encoded Fragment-Based Discovery from Phage-Displayed Macrocyclic Libraries with Genetically Encoded Unnatural Pharmacophores. J. Am. Chem. Soc 2021, 143, 5497–5507. [DOI] [PubMed] [Google Scholar]
- (298).Chen S; Lovell S; Lee S; Fellner M; Mace PD; Bogyo M Identification of highly selective covalent inhibitors by phage display. Nat. Biotechnol 2021, 39, 490–498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (299).Iskandar SE; Chiou LF; Leisner TM; Shell DJ; Norris-Drouin JL; Vaziri C; Pearce KH; Bowers AA Identification of Covalent Cyclic Peptide Inhibitors in mRNA Display. J. Am. Chem. Soc 2023, 145, 15065–15070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (300).Chen H; Katoh T; Suga H Macrocyclic Peptides Closed by a Thioether-Bipyridyl Unit That Grants Cell Membrane Permeability. ACS Bio & Med. Chem. Au 2023, 3, 429–437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (301).Liao H; Li X; Zhao L; Wang Y; Wang X; Wu Y; Zhou X; Fu W; Liu L; Hu H-G; et al. A PROTAC peptide induces durable β-catenin degradation and suppresses Wnt-dependent intestinal cancer. Cell Discovery 2020, 6, 35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (302).Chen S; Li X; Li Y; Yuan X; Geng C; Gao S; Li J; Ma B; Wang Z; Lu W; et al. Design of stapled peptide-based PROTACs for MDM2/MDMX atypical degradation and tumor suppression. Theranostics 2022, 12, 6665–6681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (303).Atangcho L; Navaratna T; Thurber GM Hitting Undruggable Targets: Viewing Stabilized Peptide Development through the Lens of Quantitative Systems Pharmacology. Trends Biochem. Sci 2019, 44, 241–257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (304).Hill TA; Shepherd NE; Diness F; Fairlie DP Constraining cyclic peptides to mimic protein structure motifs. Angew. Chem., Int. Ed 2014, 53, 13020–13041. [DOI] [PubMed] [Google Scholar]
- (305).Bierzynski A; Kim PS; Baldwin RL A salt bridge stabilizes the helix formed by isolated C-peptide of RNase A. Proc. Natl. Acad. Sci. U.S.A 1982, 79, 2470–2474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (306).Marqusee S; Robbins VH; Baldwin RL Unusually stable helix formation in short alanine-based peptides. Proc. Natl. Acad. Sci. U.S.A 1989, 86, 5286–5290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (307).Ghadiri MR; Fernholz AK Peptide Architecture. Design of Stable α-Helical Metallopeptides via a Novel Exchange-Inert Ru111 Complex. J. Am. Chem. Soc 1990, 112, 9633–9635. [Google Scholar]
- (308).Ruan F; Chen Y; Hopkins PB Metal ion-enhanced helicity in synthetic peptides containing unnatural, metal-ligating residues. J. Am. Chem. Soc 1990, 112, 9403–9404. [Google Scholar]
- (309).Jackson DY; King DS; Chmielewski J; Singh S; Schultz PG General Approach to the Synthesis of Short a-helical Peptides. J. Am. Chem. Soc 1991, 113, 9391–9392. [Google Scholar]
- (310).Albert JS; Hamilton AD Stabilization of helical domains in short peptides using hydrophobic interactions. Biochemistry 1995, 34, 984–990. [DOI] [PubMed] [Google Scholar]
- (311).Gilbertson SR; Wang X Synthesis of (dicyclohexylphosphino) serine, its incorporation into a dodecapeptide, and the coordination of rhodium. J. Org. Chem 1996, 61, 434–435. [DOI] [PubMed] [Google Scholar]
- (312).Blanco FJ; Jimenez MA; Herranz J; Rico M; Santoro J; Nieto JL NMR evidence of a short linear peptide that folds into a.beta.-hairpin in aqueous solution. J. Am. Chem. Soc 1993, 115, 5887–5888. [Google Scholar]
- (313).Takeuchi Y; Marshall GR Conformational Analysis of Reverse-Turn Constraints by N-Methylation and N-Hydroxylation of Amide Bonds in Peptides and Non-Peptide Mimetics. J. Am. Chem. Soc 1998, 120, 5363–5372. [Google Scholar]
- (314).Stanger HE; Gellman SH Rules for Antiparallel β-Sheet Design: d-Pro-Gly Is Superior to l-Asn-Gly for β-Hairpin Nucleation1. J. Am. Chem. Soc 1998, 120, 4236–4237. [Google Scholar]
- (315).FELIX AM; HEIMER EP; WANG C-T; LAMBROS TJ; FOURNIER A; MOWLES TF; MAINES S; CAMPBELL RM; WEGRZYNSKI BB; TOOME V; et al. Synthesis, biological activity and conformational analysis of cyclic GRF analogs. Int. J. Pept. Protein Res 1988, 32, 441–454. [DOI] [PubMed] [Google Scholar]
- (316).Bracken C; Gulyas J; Taylor JW; Baum J Synthesis and Nuclear Magnetic Resonance Structure Determination of an.alpha.-Helical, Bicyclic, Lactam-Bridged Hexapeptide. J. Am. Chem. Soc 1994, 116, 6431–6432. [Google Scholar]
- (317).Phelan JC; Skelton NJ; Braisted AC; McDowell RS A general method for constraining short peptides to an α-helical conformation. J. Am. Chem. Soc 1997, 119, 455–460. [Google Scholar]
- (318).Blackwell HE; Grubbs RH Highly Efficient Synthesis of Covalently Cross-Linked Peptide Helices by Ring-Closing Metathesis. Angew. Chem., Int. Ed 1998, 37, 3281–3284. [DOI] [PubMed] [Google Scholar]
- (319).Schafmeister CE; Po J; Verdine GL An all-hydrocarbon cross-linking system for enhancing the helicity and metabolic stability of peptide. J. Am. Chem. Soc 2000, 122, 5891–5892. [Google Scholar]
- (320).Ingale S; Dawson PE On resin side-chain cyclization of complex peptides using CuAAC. Org. Lett 2011, 13, 2822–2825. [DOI] [PubMed] [Google Scholar]
- (321).Pace CN; Scholtz JM A helix propensity scale based on experimental studies of peptides and proteins. Biophys. J 1998, 75, 422–427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (322).Nowick JS; Brower JO A New Turn Structure for the Formation of β-Hairpins in Peptides. J. Am. Chem. Soc 2003, 125, 876–877. [DOI] [PubMed] [Google Scholar]
- (323).Nowick JS; Chung DM; Maitra K; Maitra S; Stigers KD; Sun Y An Unnatural Amino Acid that Mimics a Tripeptide β-Strand and Forms β-Sheetlike Hydrogen-Bonded Dimers. J. Am. Chem. Soc 2000, 122, 7654–7661. [Google Scholar]
- (324).Kim MK; Kang YK Positional preference of proline in α-helices. Protein Sci. 1999, 8, 1492–1499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (325).Fuchs S; Nguyen HD; Phan TT; Burton MF; Nieto L; de Vries-van Leeuwen IJ; Schmidt A; Goodarzifard M; Agten SM; Rose R; et al. Proline primed helix length as a modulator of the nuclear receptor-coactivator interaction. J. Am. Chem. Soc 2013, 135, 4364–4371. [DOI] [PubMed] [Google Scholar]
- (326).Balaram P Non-standard amino acids in peptide design and protein engineering. Curr. Opin. Struct. Biol 1992, 2, 845–851. [Google Scholar]
- (327).Karle IL; Flippen-Anderson JL; Uma K; Balaram P Apolar peptide models for conformational heterogeneity, hydration, and packing of polypeptide helices: Crystal structure of hepta- and octapeptides containing α-aminoisobutyric acid. Proteins 1990, 7, 62–73. [DOI] [PubMed] [Google Scholar]
- (328).Bird GH; Madani N; Perry AF; Princiotto AM; Supko JG; He X; Gavathiotis E; Sodroski JG; Walensky LD Hydrocarbon double-stapling remedies the proteolytic instability of a lengthy peptide therapeutic. Proc. Natl. Acad. Sci. U.S.A 2010, 107, 14093–14098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (329).Bird GH; Mazzola E; Opoku-Nsiah K; Lammert MA; Godes M; Neuberg DS; Walensky LD Biophysical determinants for cellular uptake of hydrocarbon-stapled peptide helices. Nat. Chem. Biol 2016, 12, 845–852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (330).Rezai T; Yu B; Millhauser GL; Jacobson MP; Lokey RS Testing the conformational hypothesis of passive membrane permeability using synthetic cyclic peptide diastereomers. J. Am. Chem. Soc 2006, 128, 2510–2511. [DOI] [PubMed] [Google Scholar]
- (331).Sawyer N; Arora PS Hydrogen Bond Surrogate Stabilization of β-Hairpins. ACS Chem. Biol 2018, 13, 2027–2032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (332).Krause E; Bienert M; Schmieder P; Wenschuh H The Helix-Destabilizing Propensity Scale of d-Amino Acids: The Influence of Side Chain Steric Effects. J. Am. Chem. Soc 2000, 122, 4865–4870. [Google Scholar]
- (333).Shi XE; Wales TE; Elkin C; Kawahata N; Engen JR; Annis DA Hydrogen exchange-mass spectrometry measures stapled peptide conformational dynamics and predicts pharmacokinetic properties. Anal. Chem 2013, 85, 11185–11188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (334).Dyson HJ; Rance M; Houghten RA; Lerner RA; Wright PE Folding of immunogenic peptide fragments of proteins in water solution: I. Sequence requirements for the formation of a reverse turn. J. Mol. Biol 1988, 201, 161–200. [DOI] [PubMed] [Google Scholar]
- (335).Blanco FJ; Rivas G; Serrano L A short linear peptide that folds into a native stable β-hairpin in aqueous solution. Nat. Struct. Biol 1994, 1, 584–590. [DOI] [PubMed] [Google Scholar]
- (336).Siedlecka M; Goch G; Ejchart A; Sticht H; Bierzyǹski A α-Helix nucleation by a calcium-binding peptide loop. Proc. Natl. Acad. Sci. U.S.A 1999, 96, 903–908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (337).Benson DR; Hart BR; Zhu X; Doughty MB Design, Synthesis, and Circular Dichroism Investigation of a Peptide-Sandwiched Mesoheme. J. Am. Chem. Soc 1995, 117, 8502–8510. [Google Scholar]
- (338).Beyer RL; Hoang HN; Appleton TG; Fairlie DP Metal Clips Induce Folding of a Short Unstructured Peptide into an α-Helix via Turn Conformations in Water. Kinetic versus Thermodynamic Products. J. Am. Chem. Soc 2004, 126, 15096–15105. [DOI] [PubMed] [Google Scholar]
- (339).Galande AK; Bramlett KS; Trent JO; Burris TP; Wittliff JL; Spatola AF Potent inhibitors of LXXLL-based protein-protein interactions. ChemBioChem. 2005, 6, 1991–1998. [DOI] [PubMed] [Google Scholar]
- (340).Leduc A-M; Trent JO; Wittliff JL; Bramlett KS; Briggs SL; Chirgadze NY; Wang Y; Burris TP; Spatola AF Helix-stabilized cyclic peptides as selective inhibitors of steroid receptor-coactivator interactions. Proc. Natl. Acad. Sci. U.S.A 2003, 100, 11273–11278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (341).Pellegrini M; Royo M; Chorev M; Mierke DF Conformational consequences of i, i + 3 cystine linkages: nucleation for α-helicity? J. Pept. Res 1997, 49, 404–414. [DOI] [PubMed] [Google Scholar]
- (342).Wang D; Li W; Zhao R; Chen L; Liu N; Tian Y; Zhao H; Xie M; Lu F; Fang Q; et al. Stabilized Peptide HDAC Inhibitors Derived from HDAC1 Substrate H3K56 for the Treatment of Cancer Stem-Like Cells In Vivo. Cancer Res. 2019, 79, 1769–1783. [DOI] [PubMed] [Google Scholar]
- (343).Jiang Y; Deng Q; Zhao H; Xie M; Chen L; Yin F; Qin X; Zheng W; Zhao Y; Li Z Development of Stabilized Peptide-Based PROTACs against Estrogen Receptor alpha. ACS Chem. Biol 2018, 13, 628–635. [DOI] [PubMed] [Google Scholar]
- (344).Shepherd NE; Hoang HN; Desai VS; Letouze E; Young PR; Fairlie DP Modular α-helical mimetics with antiviral activity against respiratory syncitial virus. J. Am. Chem. Soc 2006, 128, 13284–13289. [DOI] [PubMed] [Google Scholar]
- (345).Walker MJ; Varani G Chapter Fifteen - Design of RNA-targeting macrocyclic peptides. In Methods Enzymol.; Hargrove AE, Ed.; Vol. 623; Academic Press, 2019; pp 339–372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (346).Athanassiou Z; Dias RLA; Moehle K; Dobson N; Varani G; Robinson JA Structural Mimicry of Retroviral Tat Proteins by Constrained β-Hairpin Peptidomimetics: Ligands with High Affinity and Selectivity for Viral TAR RNA Regulatory Elements. J. Am. Chem. Soc 2004, 126, 6906–6913. [DOI] [PubMed] [Google Scholar]
- (347).Gunasekaran K; Ramakrishnan C; Balaram P Beta-hairpins in proteins revisited: lessons for de novo design. Protein Eng. Des. Sel 1997, 10, 1131–1141. [DOI] [PubMed] [Google Scholar]
- (348).Blanco F; Ramírez-Alvarado M; Serrano L Formation and stability of β-hairpin structures in polypeptides. Curr. Opin. Struct. Biol 1998, 8, 107–111. [DOI] [PubMed] [Google Scholar]
- (349).Park JH; Waters ML Positional effects of click cyclization on β-hairpin structure, stability, and function. Org. Biomol. Chem 2013, 11, 69–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (350).Blosser SL; Sawyer N; Maksimovic I; Ghosh B; Arora PS Covalent and Noncovalent Targeting of the Tcf4/beta-Catenin Strand Interface with beta-Hairpin Mimics. ACS Chem. Biol 2021, 16, 1518–1525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (351).Pace JR; Lampkin BJ; Abakah C; Moyer A; Miao J; Deprey K; Cerulli RA; Lin YS; Baleja JD; Baker D; et al. Stapled beta-Hairpins Featuring 4-Mercaptoproline. J. Am. Chem. Soc 2021, 143, 15039–15044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (352).Nazzaro A; Lu B; Sawyer N; Watkins AM; Arora PS Macrocyclic β-Sheets Stabilized by Hydrogen Bond Surrogates. Angew. Chem., Int. Ed 2023, 62, e202303943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (353).Jagtap PK; Garg D; Kapp TG; Will CL; Demmer O; Luhrmann R; Kessler H; Sattler M Rational Design of Cyclic Peptide Inhibitors of U2AF Homology Motif (UHM) Domains To Modulate Pre-mRNA Splicing. J. Med. Chem 2016, 59, 10190–10197. [DOI] [PubMed] [Google Scholar]
- (354).Wendt M; Bellavita R; Gerber A; Efrem NL; van Ramshorst T; Pearce NM; Davey PRJ; Everard I; Vazquez-Chantada M; Chiarparin E; et al. Bicyclic beta-Sheet Mimetics that Target the Transcriptional Coactivator beta-Catenin and Inhibit Wnt Signaling. Angew. Chem., Int. Ed 2021, 60, 13937–13944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (355).Balboa JR; Essig DJ; Ma S; Karer N; Clemmensen LS; Pedersen SW; Joerger AC; Knapp S; Ostergaard S; Stromgaard K Development of a Potent Cyclic Peptide Inhibitor of the nNOS/PSD-95 Interaction. J. Med. Chem 2023, 66, 976–990. [DOI] [PubMed] [Google Scholar]
- (356).Hill MD; Goyal M; Menon BK; Nogueira RG; McTaggart RA; Demchuk AM; Poppe AY; Buck BH; Field TS; Dowlatshahi D; et al. Efficacy and safety of nerinetide for the treatment of acute ischaemic stroke (ESCAPE-NA1): a multicentre, double-blind, randomised controlled trial. Lancet 2020, 395, 878–887. [DOI] [PubMed] [Google Scholar]
- (357).Lo SC; Li X; Henzl MT; Beamer LJ; Hannink M Structure of the Keap1:Nrf2 interface provides mechanistic insight into Nrf2 signaling. EMBO J. 2006, 25, 3605–3617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (358).Lu M-C; Chen Z-Y; Wang Y-L; Jiang Y-L; Yuan Z-W; You Q-D; Jiang Z-Y Binding thermodynamics and kinetics guided optimization of potent Keap1–Nrf2 peptide inhibitors. RSC Adv. 2015, 5, 85983–85987. [Google Scholar]
- (359).Lu M-C; Jiao Q; Liu T; Tan S-J; Zhou H-S; You QD; Jiang Z-Y Discovery of a head-to-tail cyclic peptide as the Keap1-Nrf2 protein-protein interaction inhibitor with high cell potency. Eur. J. Med. Chem 2018, 143, 1578–1589. [DOI] [PubMed] [Google Scholar]
- (360).Steel RJ; O’Connell MA; Searcey M Perfluoroarene-based peptide macrocycles that inhibit the Nrf2/Keap1 interaction. Bioorg. Med. Chem. Lett 2018, 28, 2728–2731. [DOI] [PubMed] [Google Scholar]
- (361).Ortet PC; Muellers SN; Viarengo-Baker LA; Streu K; Szymczyna BR; Beeler AB; Allen KN; Whitty A Recapitulating the Binding Affinity of Nrf2 for KEAP1 in a Cyclic Heptapeptide, Guided by NMR, X-ray Crystallography, and Machine Learning. J. Am. Chem. Soc 2021, 143, 3779–3793. [DOI] [PubMed] [Google Scholar]
- (362).Fonseca Lopez F; Miao J; Damjanovic J; Bischof L; Braun MB; Ling Y; Hartmann MD; Lin Y-S; Kritzer JA Computational Prediction of Cyclic Peptide Structural Ensembles and Application to the Design of Keap1 Binders. J. Chem. Inf. Model 2023, 63, 6925–6937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (363).Iegre J; Krajcovicova S; Gunnarsson A; Wissler L; Käck H; Luchniak A; Tångefjord S; Narjes F; Spring DR A cell-active cyclic peptide targeting the Nrf2/Keap1 protein–protein interaction. Chem. Sci 2023, 14, 10800–10805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (364).Ott M; Geyer M; Zhou Q The Control of HIV Transcription: Keeping RNA Polymerase II on Track. Cell Host Microbe 2011, 10, 426–435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (365).Puglisi JD; Chen L; Blanchard S; Frankel AD Solution Structure of a Bovine Immunodeficiency Virus Tat-TAR Peptide-RNA Complex. Science 1995, 270, 1200–1203. [DOI] [PubMed] [Google Scholar]
- (366).Tok JBH; Des Jean RC; Fenker J Binding of a cyclic BIV β-tat peptide with its TAR RNA construct. Bioorg. Med. Chem. Lett 2001, 11, 43–46. [DOI] [PubMed] [Google Scholar]
- (367).Davidson A; Leeper TC; Athanassiou Z; Patora-Komisarska K; Karn J; Robinson JA; Varani G Simultaneous recognition of HIV-1 TAR RNA bulge and loop sequences by cyclic peptide mimics of Tat protein. Proc. Natl. Acad. Sci. U.S.A 2009, 106, 11931–11936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (368).Davidson A; Patora-Komisarska K; Robinson JA; Varani G Essential structural requirements for specific recognition of HIV TAR RNA by peptide mimetics of Tat protein. Nucleic Acids Res. 2011, 39, 248–256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (369).Shortridge MD; Wille PT; Jones AN; Davidson A; Bogdanovic J; Arts E; Karn J; Robinson JA; Varani G An ultra-high affinity ligand of HIV-1 TAR reveals the RNA structure recognized by P-TEFb. Nucleic Acids Res. 2019, 47, 1523–1531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (370).Leeper TC; Athanassiou Z; Dias RLA; Robinson JA; Varani G TAR RNA Recognition by a Cyclic Peptidomimetic of Tat Protein. Biochemistry 2005, 44, 12362–12372. [DOI] [PubMed] [Google Scholar]
- (371).Crawford DW; Blakeley BD; Chen P-H; Sherpa C; Le Grice SFJ; Laird-Offringa IA; McNaughton BR An Evolved RNA Recognition Motif That Suppresses HIV-1 Tat/TAR-Dependent Transcription. ACS Chem. Biol 2016, 11, 2206–2215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (372).Benitex Y; Baranger AM Recognition of essential purines by the U1A protein. BMC Biochem. 2007, 8, 22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (373).Belashov IA; Crawford DW; Cavender CE; Dai P; Beardslee PC; Mathews DH; Pentelute BL; McNaughton BR; Wedekind JE Structure of HIV TAR in complex with a Lab-Evolved RRM provides insight into duplex RNA recognition and synthesis of a constrained peptide that impairs transcription. Nucleic Acids Res. 2018, 46, 6401–6415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (374).Chavali SS; Mali SM; Jenkins JL; Fasan R; Wedekind JE Co-crystal structures of HIV TAR RNA bound to lab-evolved proteins show key roles for arginine relevant to the design of cyclic peptide TAR inhibitors. J. Biol. Chem 2020, 295, 16470–16486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (375).Chavali SS; Mali SM; Bonn R; Saseendran Anitha A; Bennett RP; Smith HC; Fasan R; Wedekind JE Cyclic peptides with a distinct arginine-fork motif recognize the HIV transactivation response RNA in vitro and in cells. J. Biol. Chem 2021, 297, 101390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (376).Shang R; Lee S; Senavirathne G; Lai EC microRNAs in action: biogenesis, function and regulation. Nat. Rev. Genet 2023, 24, 816–833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (377).Shortridge MD; Walker MJ; Pavelitz T; Chen Y; Yang W; Varani G A Macrocyclic Peptide Ligand Binds the Oncogenic MicroRNA-21 Precursor and Suppresses Dicer Processing. ACS Chem. Biol 2017, 12, 1611–1620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (378).Sun Y-T; Shortridge MD; Varani G A Small Cyclic β-Hairpin Peptide Mimics the Rbfox2 RNA Recognition Motif and Binds to the Precursor miRNA 20b. ChemBioChem. 2019, 20, 931–939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (379).Pal S; ‘t Hart P RNA-Binding Macrocyclic Peptides. Front. Mol. Biosci 2022, 9, 883060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (380).Kim YW; Verdine GL Stereochemical effects of allhydrocarbon tethers in i,i+4 stapled peptides. Bioorg. Med. Chem. Lett 2009, 19, 2533–2536. [DOI] [PubMed] [Google Scholar]
- (381).Song JM; Gallagher EE; Menon A; Mishra LD; Garner AL The role of olefin geometry in the activity of hydrocarbon stapled peptides targeting eukaryotic translation initiation factor 4E (eIF4E). Org. Biomol. Chem 2019, 17, 6414–6419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (382).Danial NN; Korsmeyer SJ Cell Death: Critical Control Points. Cell 2004, 116, 205–219. [DOI] [PubMed] [Google Scholar]
- (383).Walensky LD; Pitter K; Morash J; Oh KJ; Barbuto S; Fisher J; Smith E; Verdine GL; Korsmeyer SJ A stapled BID BH3 helix directly binds and activates BAX. Mol. Cell 2006, 24, 199–210. [DOI] [PubMed] [Google Scholar]
- (384).Stewart ML; Fire E; Keating AE; Walensky LD The MCL-1 BH3 helix is an exclusive MCL-1 inhibitor and apoptosis sensitizer. Nat. Chem. Biol 2010, 6, 595–601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (385).Bird GH; Gavathiotis E; LaBelle JL; Katz SG; Walensky LD Distinct BimBH3 (BimSAHB) stapled peptides for structural and cellular studies. ACS Chem. Biol 2014, 9, 831–837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (386).Braun CR; Mintseris J; Gavathiotis E; Bird GH; Gygi SP; Walensky LD Photoreactive Stapled BH3 Peptides to Dissect the BCL-2 Family Interactome. Chem. Biol 2010, 17, 1325–1333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (387).Moldoveanu T; Grace CR; Llambi F; Nourse A; Fitzgerald P; Gehring K; Kriwacki RW; Green DR BID-induced structural changes in BAK promote apoptosis. Nat. Struct. Mol. Biol 2013, 20, 589–597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (388).Rezaei Araghi R; Bird GH; Ryan JA; Jenson JM; Godes M; Pritz JR; Grant RA; Letai A; Walensky LD; Keating AE Iterative optimization yields Mcl-1-targeting stapled peptides with selective cytotoxicity to Mcl-1-dependent cancer cells. Proc. Natl. Acad. Sci. U.S.A 2018, 115, E886–E895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (389).Walensky LD; Kung AL; Escher I; Malia TJ; Barbuto S; Wright RD; Wagner G; Verdine GL; Korsmeyer SJ Activation of apoptosis in vivo by a hydrocarbon-stapled BH3 helix. Science 2004, 305, 1466–1470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (390).Yang B; Liu D; Huang Z Synthesis and helical structure of lactam bridged BH3 peptides derived from pro-apoptotic Bcl-2 family proteins. Bioorg. Med. Chem. Lett 2004, 14, 1403–1406. [DOI] [PubMed] [Google Scholar]
- (391).Verdine GL; Walensky LD The challenge of drugging undruggable targets in cancer: lessons learned from targeting BCL-2 family members. Clin. Cancer Res 2007, 13, 7264–7270. [DOI] [PubMed] [Google Scholar]
- (392).Leshchiner ES; Braun CR; Bird GH; Walensky LD Direct activation of full-length proapoptotic BAK. Proc. Natl. Acad. Sci. U.S.A 2013, 110, E986–995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (393).Kutchukian PS; Yang JS; Verdine GL; Shakhnovich EI All-Atom Model for Stabilization of α-Helical Structure in Peptides by Hydrocarbon Staples. J. Am. Chem. Soc 2009, 131, 4622–4627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (394).De Strooper B; Annaert W; Cupers P; Saftig P; Craessaerts K; Mumm JS; Schroeter EH; Schrijvers V; Wolfe MS; Ray WJ; et al. A presenilin-1-dependent γ-secretase-like protease mediates release of Notch intracellular domain. Nature 1999, 398, 518–522. [DOI] [PubMed] [Google Scholar]
- (395).Wu L; Aster JC; Blacklow SC; Lake R; Artavanis-Tsakonas S; Griffin JD MAML1, a human homologue of Drosophila Mastermind, is a transcriptional co-activator for NOTCH receptors. Nat. Genet 2000, 26, 484–489. [DOI] [PubMed] [Google Scholar]
- (396).Moellering RE; Cornejo M; Davis TN; Del Bianco C; Aster JC; Blacklow SC; Kung AL; Gilliland DG; Verdine GL; Bradner JE Direct inhibition of the NOTCH transcription factor complex. Nature 2009, 462, 182–188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (397).Maillard I; Weng AP; Carpenter AC; Rodriguez CG; Sai H; Xu L; Allman D; Aster JC; Pear WS Mastermind critically regulates Notch-mediated lymphoid cell fate decisions. Blood 2004, 104, 1696–1702. [DOI] [PubMed] [Google Scholar]
- (398).Nam Y; Sliz P; Song L; Aster JC; Blacklow SC Structural basis for cooperativity in recruitment of MAML coactivators to Notch transcription complexes. Cell 2006, 124, 973–983. [DOI] [PubMed] [Google Scholar]
- (399).Grossmann TN; Yeh JT; Bowman BR; Chu Q; Moellering RE; Verdine GL Inhibition of oncogenic Wnt signaling through direct targeting of beta-catenin. Proc. Natl. Acad. Sci. U.S.A 2012, 109, 17942–17947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (400).Takada Y; Zhu D; Bird GH; Sukhdeo K; Zhao J-J; Mani M; Lemieux M; Carrasco DE; Ryan J; Horst D; et al. Targeted disruption of the BCL9β-catenin complex inhibits oncogenic Wnt signaling. Sci. Transl. Med 2012, 4, 1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (401).Bernal F; Tyler AF; Korsmeyer SJ; Walensky LD; Verdine GL Reactivation of the p53 tumor suppressor pathway by a stapled p53 peptide. J. Am. Chem. Soc 2007, 129, 2456–2457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (402).Bernal F; Wade M; Godes M; Davis TN; Whitehead DG; Kung AL; Wahl GM; Walensky LD A stapled p53 helix overcomes HDMX-mediated suppression of p53. Cancer Cell 2010, 18, 411–422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (403).Baek S; Kutchukian PS; Verdine GL; Huber R; Holak TA; Lee KW; Popowicz GM Structure of the stapled p53 peptide bound to Mdm2. J. Am. Chem. Soc 2012, 134, 103–106. [DOI] [PubMed] [Google Scholar]
- (404).Hu B; Gilkes DM; Chen J Efficient p53 activation and apoptosis by simultaneous disruption of binding to MDM2 and MDMX. Cancer Res. 2007, 67, 8810–8817. [DOI] [PubMed] [Google Scholar]
- (405).Pazgier M; Liu M; Zou G; Yuan W; Li C; Li C; Li J; Monbo J; Zella D; Tarasov SG; et al. Structural basis for high-affinity peptide inhibition of p53 interactions with MDM2 and MDMX. Proc. Natl. Acad. Sci. U.S.A 2009, 106, 4665–4670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (406).Phan J; Li Z; Kasprzak A; Li B; Sebti S; Guida W; Schonbrunn E; Chen J Structure-based design of high affinity peptides inhibiting the interaction of p53 with MDM2 and MDMX. J. Biol. Chem 2010, 285, 2174–2183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (407).Brown CJ; Quah ST; Jong J; Goh AM; Chiam PC; Khoo KH; Choong ML; Lee MA; Yurlova L; Zolghadr K; et al. Stapled peptides with improved potency and specificity that activate p53. ACS Chem. Biol 2013, 8, 506–512. [DOI] [PubMed] [Google Scholar]
- (408).Spiegelberg D; Mortensen AC; Lundsten S; Brown CJ; Lane DP; Nestor M The MDM2/MDMX-p53 Antagonist PM2 Radiosensitizes Wild-Type p53 Tumors. Cancer Res. 2018, 78, 5084–5093. [DOI] [PubMed] [Google Scholar]
- (409).Guerlavais V; Sawyer TK; Carvajal L; Chang YS; Graves B; Ren JG; Sutton D; Olson KA; Packman K; Darlak K; et al. Discovery of Sulanemadlin (ALRN-6924), the First Cell-Permeating, Stabilized alpha-Helical Peptide in Clinical Development. J. Med. Chem 2023, 66, 9401–9417. [DOI] [PubMed] [Google Scholar]
- (410).Chang YS; Graves B; Guerlavais V; Tovar C; Packman K; To KH; Olson KA; Kesavan K; Gangurde P; Mukherjee A; et al. Stapled alpha-helical peptide drug development: a potent dual inhibitor of MDM2 and MDMX for p53-dependent cancer therapy. Proc. Natl. Acad. Sci. U.S.A 2013, 110, E3445–3454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (411).Chandramohan A; Josien H; Yuen TY; Duggal R; Spiegelberg D; Yan L; Juang YA; Ge L; Aronica PG; Kaan HYK; et al. Design-rules for stapled peptides with in vivo activity and their application to Mdm2/X antagonists. Nat. Commun 2024, 15, 489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (412).Liu M; Pazgier M; Li C; Yuan W; Li C; Lu W A left-handed solution to peptide inhibition of the p53-MDM2 interaction. Angew. Chem., Int. Ed 2010, 49, 3649–3652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (413).Kannan S; Aronica PGA; Ng S; Gek Lian DT; Frosi Y; Chee S; Shimin J; Yuen TY; Sadruddin A; Kaan HYK; et al. Macrocyclization of an all-d linear alpha-helical peptide imparts cellular permeability. Chem. Sci 2020, 11, 5577–5591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (414).Chapman RN; Dimartino G; Arora PS A Highly Stable Short α-Helix Constrained by a Main-Chain Hydrogen-Bond Surrogate. J. Am. Chem. Soc 2004, 126, 12252–12253. [DOI] [PubMed] [Google Scholar]
- (415).Henchey LK; Kushal S; Dubey R; Chapman RN; Olenyuk BZ; Arora PS Inhibition of Hypoxia Inducible Factor 1 Transcription Coactivator Interaction by a Hydrogen Bond Surrogate α-Helix. J. Am. Chem. Soc 2010, 132, 941–943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (416).Kushal S; Lao BB; Henchey LK; Dubey R; Mesallati H; Traaseth NJ; Olenyuk BZ; Arora PS Protein domain mimetics as in vivo modulators of hypoxia-inducible factor signaling. Proc. Natl. Acad. Sci. U.S.A 2013, 110, 15602–15607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (417).Henchey LK; Porter JR; Ghosh I; Arora PS High specificity in protein recognition by hydrogen-bond-surrogate alphahelices: selective inhibition of the p53/MDM2 complex. ChemBioChem. 2010, 11, 2104–2107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (418).Patgiri A; Yadav KK; Arora PS; Bar-Sagi D An orthosteric inhibitor of the Ras-Sos interaction. Nat. Chem. Biol 2011, 7, 585–587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (419).Leshchiner ES; Parkhitko A; Bird GH; Luccarelli J; Bellairs JA; Escudero S; Opoku-Nsiah K; Godes M; Perrimon N; Walensky LD Direct inhibition of oncogenic KRAS by hydrocarbon-stapled SOS1 helices. Proc. Natl. Acad. Sci. U.S.A 2015, 112, 1761–1766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (420).Spiegel J; Cromm PM; Itzen A; Goody RS; Grossmann TN; Waldmann H Direct targeting of Rab-GTPase-effector interactions. Angew. Chem., Int. Ed 2014, 53, 2498–2503. [DOI] [PubMed] [Google Scholar]
- (421).Cromm PM; Spiegel J; Kuchler P; Dietrich L; Kriegesmann J; Wendt M; Goody RS; Waldmann H; Grossmann TN Protease-Resistant and Cell-Permeable Double-Stapled Peptides Targeting the Rab8a GTPase. ACS Chem. Biol 2016, 11, 2375–2382. [DOI] [PubMed] [Google Scholar]
- (422).Mitra S; Montgomery JE; Kolar MJ; Li G; Jeong KJ; Peng B; Verdine GL; Mills GB; Moellering RE Stapled peptide inhibitors of RAB25 target context-specific phenotypes in cancer. Nat. Commun 2017, 8, 660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (423).Cheng KW; Lahad JP; Kuo W.-l.; Lapuk A; Yamada K; Auersperg N; Liu J; Smith-McCune K; Lu KH; Fishman D; et al. The RAB25 small GTPase determines aggressiveness of ovarian and breast cancers. Nat. Med 2004, 10, 1251–1256. [DOI] [PubMed] [Google Scholar]
- (424).Cheng J-M; Ding M; Aribi A; Shah P; Rao K Loss of RAB25 expression in breast cancer. Int. J. Cancer 2006, 118, 2957–2964. [DOI] [PubMed] [Google Scholar]
- (425).Sinclair JKL; Denton EV; Schepartz A Inhibiting Epidermal Growth Factor Receptor at a Distance. J. Am. Chem. Soc 2014, 136, 11232–11235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (426).Cowell JK; Teng Y; Bendzunas NG; Ara R; Arbab AS; Kennedy EJ Suppression of Breast Cancer Metastasis Using Stapled Peptides Targeting the WASF Regulatory Complex. Cancer Growth Metastasis 2017, 10, 1179064417713197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (427).Teng Y; Bahassan A; Dong D; Hanold LE; Ren X; Kennedy EJ; Cowell JK Targeting the WASF3-CYFIP1 Complex Using Stapled Peptides Suppresses Cancer Cell Invasion. Cancer Res. 2016, 76, 965–973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (428).Kim W; Bird GH; Neff T; Guo G; Kerenyi MA; Walensky LD; Orkin SH Targeted disruption of the EZH2-EED complex inhibits EZH2-dependent cancer. Nat. Chem. Biol 2013, 9, 643–650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (429).Zhang H; Zhao Q; Bhattacharya S; Waheed AA; Tong X; Hong A; Heck S; Curreli F; Goger M; Cowburn D; et al. A cell-penetrating helical peptide as a potential HIV-1 inhibitor. J. Mol. Biol 2008, 378, 565–580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (430).Zhang H; Curreli F; Waheed AA; Mercredi PY; Mehta M; Bhargava P; Scacalossi D; Tong X; Lee S; Cooper A; et al. Dual-acting stapled peptides target both HIV-1 entry and assembly. Retrovirology 2013, 10, 136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (431).Zhang H; Curreli F; Zhang X; Bhattacharya S; Waheed AA; Cooper A; Cowburn D; Freed EO; Debnath AK Antiviral activity of α-helical stapled peptides designed from the HIV-1 capsid dimerization domain. Retrovirology 2011, 8, 28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (432).Paquette AR; Boddy CN Double Stranded DNA Binding Stapled Peptides: An Emerging Tool for Transcriptional Regulation. ChemBioChem. 2023, 24, e202300594. [DOI] [PubMed] [Google Scholar]
- (433).Bird GH; Irimia A; Ofek G; Kwong PD; Wilson IA; Walensky LD Stapled HIV-1 peptides recapitulate antigenic structures and engage broadly neutralizing antibodies. Nat. Struct. Mol. Biol 2014, 21, 1058–1067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (434).Varambally S; Dhanasekaran SM; Zhou M; Barrette TR; Kumar-Sinha C; Sanda MG; Ghosh D; Pienta KJ; Sewalt RGAB; Otte AP; et al. The polycomb group protein EZH2 is involved in progression of prostate cancer. Nature 2002, 419, 624–629. [DOI] [PubMed] [Google Scholar]
- (435).Sinclair JKL; Schepartz A Influence of Macrocyclization on Allosteric, Juxtamembrane-Derived, Stapled Peptide Inhibitors of the Epidermal Growth Factor Receptor (EGFR). Org. Lett 2014, 16, 4916–4919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (436).Sinclair JKL; Robertson WE; Mozumdar D; Quach K; Schepartz A Allosteric Inhibition of the Epidermal Growth Factor Receptor. Biochemistry 2021, 60, 500–512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (437).Dema A; Perets E; Schulz MS; Deák VA; Klussmann E Pharmacological targeting of AKAP-directed compartmentalized cAMP signalling. Cell. Signal 2015, 27, 2474–2487. [DOI] [PubMed] [Google Scholar]
- (438).Carlson CR; Lygren B; Berge T; Hoshi N; Wong W; Taskén K; Scott JD Delineation of Type I Protein Kinase A-selective Signaling Events Using an RI Anchoring Disruptor*. J. Biol. Chem 2006, 281, 21535–21545. [DOI] [PubMed] [Google Scholar]
- (439).Carr DW; Hausken ZE; Fraser ID; Stofko-Hahn RE; Scott JD Association of the type II cAMP-dependent protein kinase with a human thyroid RII-anchoring protein. Cloning and characterization of the RII-binding domain. J. Biol. Chem 1992, 267, 1337613382. [PubMed] [Google Scholar]
- (440).Gold MG; Lygren B; Dokurno P; Hoshi N; McConnachie G; Taskén K; Carlson CR; Scott JD; Barford D Molecular Basis of AKAP Specificity for PKA Regulatory Subunits. Mol. Cell 2006, 24, 383–395. [DOI] [PubMed] [Google Scholar]
- (441).Wang Y; Ho TG; Bertinetti D; Neddermann M; Franz E; Mo GCH; Schendowich LP; Sukhu A; Spelts RC; Zhang J; et al. Isoform-Selective Disruption of AKAP-Localized PKA Using Hydrocarbon Stapled Peptides. ACS Chem. Biol 2014, 9, 635–642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (442).Flaherty BR; Wang Y; Trope EC; Ho TG; Muralidharan V; Kennedy EJ; Peterson DS The Stapled AKAP Disruptor Peptide STAD-2 Displays Antimalarial Activity through a PKA-Independent Mechanism. PLoS One 2015, 10, No. e0129239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (443).Wang Y; Ho TG; Franz E; Hermann JS; Smith FD; Hehnly H; Esseltine JL; Hanold LE; Murph MM; Bertinetti D; et al. PKA-Type I Selective Constrained Peptide Disruptors of AKAP Complexes. ACS Chem. Biol 2015, 10, 1502–1510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (444).Limaye AJ; Bendzunas GN; Kennedy EJ Targeted disruption of PKC from AKAP signaling complexes. RSC Chem. Biol 2021, 2, 1227–1231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (445).Monty OBC; Nyshadham P; Bohren KM; Palaniappan M; Matzuk MM; Young DW; Simmons N Homogeneous and Functional Group Tolerant Ring-Closing Metathesis for DNA-Encoded Chemical Libraries. ACS Comb. Sci 2020, 22, 80–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (446).Nair DP; Podgórski M; Chatani S; Gong T; Xi W; Fenoli CR; Bowman CN The Thiol-Michael Addition Click Reaction: A Powerful and Widely Used Tool in Materials Chemistry. Chem. Mater 2014, 26, 724–744. [Google Scholar]
- (447).Lindley H The reaction of thiol compounds and chloroacetamide. 2. The reaction between chloroacetamide and cysteine peptides. Biochem. J 1962, 82, 418–425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (448).Jo H; Meinhardt N; Wu Y; Kulkarni S; Hu X; Low KE; Davies PL; DeGrado WF; Greenbaum DC Development of alpha-helical calpain probes by mimicking a natural protein-protein interaction. J. Am. Chem. Soc 2012, 134, 17704–17713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (449).Diderich P; Bertoldo D; Dessen P; Khan MM; Pizzitola I; Held W; Huelsken J; Heinis C Phage Selection of Chemically Stabilized alpha-Helical Peptide Ligands. ACS Chem. Biol 2016, 11, 1422–1427. [DOI] [PubMed] [Google Scholar]
- (450).Timmerman P; Beld J; Puijk WC; Meloen RH Rapid and Quantitative Cyclization of Multiple Peptide Loops onto Synthetic Scaffolds for Structural Mimicry of Protein Surfaces. ChemBioChem. 2005, 6, 821–824. [DOI] [PubMed] [Google Scholar]
- (451).Iqbal ES; Richardson SL; Abrigo NA; Dods KK; Osorio Franco HE; Gerrish HS; Kotapati HK; Morgan IM; Masterson DS; Hartman MCT A new strategy for the in vitro selection of stapled peptide inhibitors by mRNA display. Chem. Commun 2019, 55, 8959–8962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (452).Peraro L; Zou Z; Makwana KM; Cummings AE; Ball HL; Yu H; Lin Y-S; Levine B; Kritzer JA Diversity-Oriented Stapling Yields Intrinsically Cell-Penetrant Inducers of Autophagy. J. Am. Chem. Soc 2017, 139, 7792–7802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (453).Tennett JC; Epstein SR; Sawyer N Enhanced EphB2-Specific Peptide Inhibitors through Stabilization of Polyproline II Helical Structure. ACS Chem. Biol 2024, 19, 1214–1221. [DOI] [PubMed] [Google Scholar]
- (454).Shim SY; Kim YW; Verdine GL A new i, i + 3 peptide stapling system for alpha-helix stabilization. Chem. Biol. Drug Des 2013, 82, 635–642. [DOI] [PubMed] [Google Scholar]
- (455).Li K; Tokareva OS; Thomson TM; Wahl SCT; Travaline TL; Ramirez JD; Choudary SK; Agarwal S; Walkup WG; Olsen TJ; et al. De novo mapping of α-helix recognition sites on protein surfaces using unbiased libraries. Proc. Natl. Acad. Sci. U.S.A 2022, 119, e2210435119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (456).Tokareva OS; Li K; Travaline TL; Thomson TM; Swiecicki J-M; Moussa M; Ramirez JD; Litchman S; Verdine GL; McGee JH Recognition and reprogramming of E3 ubiquitin ligase surfaces by α-helical peptides. Nat. Commun 2023, 14, 6992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (457).Weiergräber OH; Stangler T; Thielmann Y; Mohrluüder J; Wiesehan K; Willbold D Ligand Binding Mode of GABAA Receptor-Associated Protein. J. Mol. Biol 2008, 381, 1320–1331. [DOI] [PubMed] [Google Scholar]
- (458).Brown H; Chung M; Uffing A; Batistatou N; Tsang T; Doskocil S; Mao W; Willbold D; Bast RC Jr.; Lu Z; et al. Structure-Based Design of Stapled Peptides That Bind GABARAP and Inhibit Autophagy. J. Am. Chem. Soc 2022, 144, 14687–14697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (459).Assem N; Ferreira DJ; Wolan DW; Dawson PE Acetone-Linked Peptides: A Convergent Approach for Peptide Macrocyclization and Labeling. Angew. Chem., Int. Ed 2015, 54, 8665–8668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (460).Wang Y; Chou DH A Thiol-Ene Coupling Approach to Native Peptide Stapling and Macrocyclization. Angew. Chem., Int. Ed 2015, 54, 10931–10934. [DOI] [PubMed] [Google Scholar]
- (461).Muppidi A; Wang Z; Li X; Chen J; Lin Q Achieving cell penetration with distance-matching cysteine cross-linkers: a facile route to cell-permeable peptide dual inhibitors of Mdm2/Mdmx. Chem. Commun 2011, 47, 9396–9398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (462).Islam MS; Junod SL; Zhang S; Buuh ZY; Guan Y; Zhao M; Kaneria KH; Kafley P; Cohen C; Maloney R; et al. Unprotected peptide macrocyclization and stapling via a fluorine-thiol displacement reaction. Nat. Commun 2022, 13, 350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (463).Spokoyny AM; Zou Y; Ling JJ; Yu H; Lin Y-S; Pentelute BL A Perfluoroaryl-Cysteine SNAr Chemistry Approach to Unprotected Peptide Stapling. J. Am. Chem. Soc 2013, 135, 5946–5949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (464).Jimenez-Macias JL; Lee YC; Miller E; Finkelberg T; Zdioruk M; Berger G; Farquhar CE; Nowicki MO; Cho CF; Fedeles BI; et al. A Pt(IV)-conjugated brain penetrant macrocyclic peptide shows pre-clinical efficacy in glioblastoma. J. Controlled Release 2022, 352, 623–636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (465).Hu K; Geng H; Zhang Q; Liu Q; Xie M; Sun C; Li W; Lin H; Jiang F; Wang T; et al. An In-tether Chiral Center Modulates the Helicity, Cell Permeability, and Target Binding Affinity of a Peptide. Angew. Chem., Int. Ed 2016, 55, 8013–8017. [DOI] [PubMed] [Google Scholar]
- (466).Hu K; Li W; Yu M; Sun C; Li Z Investigation of Cellular Uptakes of the In-Tether Chiral-Center-Induced Helical Pentapeptides. Bioconjugate Chem. 2016, 27, 2824–2827. [DOI] [PubMed] [Google Scholar]
- (467).Hu K; Sun C; Yu M; Li W; Lin H; Guo J; Jiang Y; Lei C; Li Z Dual In-Tether Chiral Centers Modulate Peptide Helicity. Bioconjugate Chem. 2017, 28, 1537–1543. [DOI] [PubMed] [Google Scholar]
- (468).Jiang Y; Hu K; Shi X; Tang Q; Wang Z; Ye X; Li Z Switching substitution groups on the in-tether chiral centre influences backbone peptides’ permeability and target binding affinity. Org. Biomol. Chem 2017, 15, 541–544. [DOI] [PubMed] [Google Scholar]
- (469).Speltz TE; Fanning SW; Mayne CG; Fowler C; Tajkhorshid E; Greene GL; Moore TW Stapled Peptides with gamma-Methylated Hydrocarbon Chains for the Estrogen Receptor/Coactivator Interaction. Angew. Chem., Int. Ed 2016, 55, 4252–4255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (470).Hart PT; Hommen P; Noisier A; Krzyzanowski A; Schuüler D; Porfetye AT; Akbarzadeh M; Vetter IR; Adihou H; Waldmann H Structure Based Design of Bicyclic Peptide Inhibitors of RbAp48. Angew. Chem., Int. Ed 2021, 60, 1813–1820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (471).Adzhubei AA; Sternberg MJE; Makarov AA Polyproline-II Helix in Proteins: Structure and Function. J. Mol. Biol 2013, 425, 2100–2132. [DOI] [PubMed] [Google Scholar]
- (472).Johansson JR; Beke-Somfai T; Said Stålsmeden A; Kann N Ruthenium-Catalyzed Azide Alkyne Cycloaddition Reaction: Scope, Mechanism, and Applications. Chem. Rev 2016, 116, 14726–14768. [DOI] [PubMed] [Google Scholar]
- (473).Lau YH; Wu Y; Rossmann M; Tan BX; de Andrade P; Tan YS; Verma C; McKenzie GJ; Venkitaraman AR; Hyvonen M; et al. Double Strain-Promoted Macrocyclization for the Rapid Selection of Cell-Active Stapled Peptides. Angew. Chem., Int. Ed 2015, 54, 15410–15413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (474).Kawamoto SA; Coleska A; Ran X; Yi H; Yang CY; Wang S Design of triazole-stapled BCL9 alpha-helical peptides to target the beta-catenin/B-cell CLL/lymphoma 9 (BCL9) protein-protein interaction. J. Med. Chem 2012, 55, 1137–1146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (475).Sharma K; Strizhak AV; Fowler E; Xu W; Chappell B; Sore HF; Galloway W; Grayson MN; Lau YH; Itzhaki LS; et al. Functionalized Double Strain-Promoted Stapled Peptides for Inhibiting the p53-MDM2 Interaction. ACS Omega 2020, 5, 1157–1169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (476).Lau YH; de Andrade P; Quah S-T; Rossmann M; Laraia L; Sköld N; Sum TJ; Rowling PJE; Joseph TL; Verma C; et al. Functionalised staple linkages for modulating the cellular activity of stapled peptides. Chem. Sci 2014, 5, 1804–1809. [Google Scholar]
- (477).Wu Y; Kaur A; Fowler E; Wiedmann MM; Young R; Galloway W; Olsen L; Sore HF; Chattopadhyay A; Kwan TT; et al. Toolbox of Diverse Linkers for Navigating the Cellular Efficacy Landscape of Stapled Peptides. ACS Chem. Biol 2019, 14, 526–533. [DOI] [PubMed] [Google Scholar]
- (478).Charoenpattarapreeda J; Tan YS; Iegre J; Walsh SJ; Fowler E; Eapen RS; Wu Y; Sore HF; Verma CS; Itzhaki L; et al. Targeted covalent inhibitors of MDM2 using electrophilebearing stapled peptides. Chem. Commun 2019, 55, 7914–7917. [DOI] [PubMed] [Google Scholar]
- (479).St Amant AH; Lemen D; Florinas S; Mao S; Fazenbaker C; Zhong H; Wu H; Gao C; Christie RJ; Read de Alaniz J Tuning the Diels-Alder Reaction for Bioconjugation to Maleimide Drug-Linkers. Bioconjugate Chem. 2018, 29, 2406–2414. [DOI] [PubMed] [Google Scholar]
- (480).St Amant AH; Huang F; Lin J; Rickert K; Oganesyan V; Lemen D; Mao S; Harper J; Marelli M; Wu H; et al. A Diene-Containing Noncanonical Amino Acid Enables Dual Functionality in Proteins: Rapid Diels–Alder Reaction with Maleimide or Proximity-Based Dimerization. Angew. Chem., Int. Ed 2019, 58, 8489–8493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (481).Montgomery JE; Donnelly JA; Fanning SW; Speltz TE; Shangguan X; Coukos JS; Greene GL; Moellering RE Versatile Peptide Macrocyclization with Diels-Alder Cycloadditions. J. Am. Chem. Soc 2019, 141, 16374–16381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (482).Riggs BL; Hartmann LC Selective Estrogen-Receptor Modulators — Mechanisms of Action and Application to Clinical Practice. New Engl. J. Med 2003, 348, 618–629. [DOI] [PubMed] [Google Scholar]
- (483).Fanning SW; Mayne CG; Dharmarajan V; Carlson KE; Martin TA; Novick SJ; Toy W; Green B; Panchamukhi S; Katzenellenbogen BS; et al. Estrogen receptor alpha somatic mutations Y537S and D538G confer breast cancer endocrine resistance by stabilizing the activating function-2 binding conformation. eLife 2016, 5, e12792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (484).Phillips C; Roberts LR; Schade M; Bazin R; Bent A; Davies NL; Moore R; Pannifer AD; Pickford AR; Prior SH; et al. Design and structure of stapled peptides binding to estrogen receptors. J. Am. Chem. Soc 2011, 133, 9696–9699. [DOI] [PubMed] [Google Scholar]
- (485).Speltz TE; Danes JM; Stender JD; Frasor J; Moore TW A Cell-Permeable Stapled Peptide Inhibitor of the Estrogen Receptor/Coactivator Interaction. ACS Chem. Biol 2018, 13, 676684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (486).Speltz TE; Mayne CG; Fanning SW; Siddiqui Z; Tajkhorshid E; Greene GL; Moore TWA ″cross-stitched″ peptide with improved helicity and proteolytic stability. Org. Biomol. Chem 2018, 16, 3702–3706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (487).Xie M; Zhao H; Liu Q; Zhu Y; Yin F; Liang Y; Jiang Y; Wang D; Hu K; Qin X; et al. Structural Basis of Inhibition of ERalpha-Coactivator Interaction by High-Affinity N-Terminus Isoaspartic Acid Tethered Helical Peptides. J. Med. Chem 2017, 60, 8731–8740. [DOI] [PubMed] [Google Scholar]
- (488).Oliveira BL; Guo Z; Bernardes GJL Inverse electron demand Diels-Alder reactions in chemical biology. Chem. Soc. Rev 2017, 46, 4895–4950. [DOI] [PubMed] [Google Scholar]
- (489).Pagel M Inverse electron demand Diels-Alder (IEDDA) reactions in peptide chemistry. J. Pept. Sci 2019, 25, e3141. [DOI] [PubMed] [Google Scholar]
- (490).Chen F-J; Lin W; Chen F-E Non-symmetric stapling of native peptides. Nat. Rev. Chem 2024, 8, 304–318. [DOI] [PubMed] [Google Scholar]
- (491).de Araujo AD; Hoang HN; Kok WM; Diness F; Gupta P; Hill TA; Driver RW; Price DA; Liras S; Fairlie DP Comparative alpha-helicity of cyclic pentapeptides in water. Angew. Chem., Int. Ed 2014, 53, 6965–6969. [DOI] [PubMed] [Google Scholar]
- (492).Bartling CRO; Alexopoulou F; Kuschert S; Chin YKY; Jia X; Sereikaite V; Ozcelik D; Jensen TM; Jain P; Nygaard MM; et al. Comprehensive Peptide Cyclization Examination Yields Optimized APP Scaffolds with Improved Affinity toward Mint2. J. Med. Chem 2023, 66, 3045–3057. [DOI] [PubMed] [Google Scholar]
- (493).de Araujo AD; Lim J; Wu K-C; Hoang HN; Nguyen HT; Fairlie DP Landscaping macrocyclic peptides: stapling hDM2-binding peptides for helicity, protein affinity, proteolytic stability and cell uptake. RSC Chem. Biol 2022, 3, 895–904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (494).Esteban JJ; Mason JR; Kaminski J; Ramachandran R; Luyt LG A survey of stapling methods to increase affinity, activity, and stability of ghrelin analogues. RSC Med. Chem 2024, 15, 254–266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (495).Hilinski GJ; Kim YW; Hong J; Kutchukian PS; Crenshaw CM; Berkovitch SS; Chang A; Ham S; Verdine GL Stitched alpha-helical peptides via bis ring-closing metathesis. J. Am. Chem. Soc 2014, 136, 12314–12322. [DOI] [PubMed] [Google Scholar]
- (496).de Araujo AD; Lim J; Wu K-C; Xiang Y; Good AC; Skerlj R; Fairlie DP Bicyclic Helical Peptides as Dual Inhibitors Selective for Bcl2A1 and Mcl-1 Proteins. J. Med. Chem 2018, 61, 2962–2972. [DOI] [PubMed] [Google Scholar]
- (497).Anananuchatkul T; Chang IV; Miki T; Tsutsumi H; Mihara H Construction of a Stapled α-Helix Peptide Library Displayed on Phage for the Screening of Galectin-3-Binding Peptide Ligands. ACS Omega 2020, 5, 5666–5674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (498).Miki T; Namii K; Seko K; Kakehi S; Moro G; Mihara H Pattern enrichment analysis for phage selection of stapled peptide ligands. Chem. Sci 2022, 13, 12634–12642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (499).Yokoo H; Ohoka N; Takyo M; Ito T; Tsuchiya K; Kurohara T; Fukuhara K; Inoue T; Naito M; Demizu Y Peptide Stapling Improves the Sustainability of a Peptide-Based Chimeric Molecule That Induces Targeted Protein Degradation. Int. J. Mol. Sci 2021, 22, 8772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (500).Ma B; Liu D; Zheng M; Wang Z; Zhang D; Jian Y; Ma J; Fan Y; Chen Y; Gao Y; et al. Development of a Double-Stapled Peptide Stabilizing Both alpha-Helix and beta-Sheet Structures for Degrading Transcription Factor AR-V7. JACS Au 2024, 4, 816–827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (501).Frackenpohl J; Arvidsson PI; Schreiber JV; Seebach D The outstanding biological stability of beta- and gamma-peptides toward proteolytic enzymes: an in vitro investigation with fifteen peptidases. ChemBioChem. 2001, 2, 445–455. [DOI] [PubMed] [Google Scholar]
- (502).Al Musaimi O; Lombardi L; Williams DR; Albericio F Strategies for Improving Peptide Stability and Delivery. Pharmaceuticals 2022, 15, 1283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (503).Gellman SH Foldamers: A Manifesto. Acc. Chem. Res 1998, 31, 173–180. [Google Scholar]
- (504).Ko E; Liu J; Burgess K Minimalist and universal peptidomimetics. Chem. Soc. Rev 2011, 40, 4411–4421. [DOI] [PubMed] [Google Scholar]
- (505).Ahn JM; Boyle NA; MacDonald MT; Janda KD Peptidomimetics and peptide backbone modifications. Mini Rev. Med. Chem 2002, 2, 463–473. [DOI] [PubMed] [Google Scholar]
- (506).Cheng RP; Gellman SH; DeGrado WF β-Peptides: From Structure to Function. Chem. Rev 2001, 101, 3219–3232. [DOI] [PubMed] [Google Scholar]
- (507).Seebach D; Abele S; Gademann K; Guichard G; Hintermann T; Jaun B; Matthews JL; Schreiber JV; Oberer L; Hommel U; et al. β2- and β3-Peptides with Proteinaceous Side Chains: Synthesis and solution structures of constitutional isomers, a novel helical secondary structure and the influence of solvation and hydrophobic interactions on folding. Helv. Chim. Acta 1998, 81, 932982. [Google Scholar]
- (508).Seebach D; Matthews JL β-Peptides: a surprise at every turn. Chem. Commun 1997, 2015–2022. [Google Scholar]
- (509).Seebach D; Beck AK; Bierbaum DJ The world of beta- and gamma-peptides comprised of homologated proteinogenic amino acids and other components. Chem. Biodivers 2004, 1, 1111–1239. [DOI] [PubMed] [Google Scholar]
- (510).Seebach D; Gardiner J β-Peptidic Peptidomimetics. Acc. Chem. Res 2008, 41, 1366–1375. [DOI] [PubMed] [Google Scholar]
- (511).Cabrele C; Martinek TA; Reiser O; Berlicki Ł Peptides Containing β-Amino Acid Patterns: Challenges and Successes in Medicinal Chemistry. J. Med. Chem 2014, 57, 9718–9739. [DOI] [PubMed] [Google Scholar]
- (512).Koyack MJ; Cheng RP Design and Synthesis of β-Peptides With Biological Activity. In Protein Design: Methods and Applications; Guerois R, de la Paz ML, Eds.; Humana Press, 2006; pp 95–109. [DOI] [PubMed] [Google Scholar]
- (513).Horne WS Peptide and peptoid foldamers in medicinal chemistry. Expert Opin. Drug Discovery 2011, 6, 1247–1262. [DOI] [PubMed] [Google Scholar]
- (514).Horne WS; Gellman SH Foldamers with Heterogeneous Backbones. Acc. Chem. Res 2008, 41, 1399–1408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (515).Johnson LM; Mortenson DE; Yun HG; Horne WS; Ketas TJ; Lu M; Moore JP; Gellman SH Enhancement of α-Helix Mimicry by an α/β-Peptide Foldamer via Incorporation of a Dense Ionic Side-Chain Array. J. Am. Chem. Soc 2012, 134, 7317–7320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (516).Martinek TA; Fuülöp F Peptidic foldamers: ramping up diversity. Chem. Soc. Rev 2012, 41, 687–702. [DOI] [PubMed] [Google Scholar]
- (517).Cheloha RW; Maeda A; Dean T; Gardella TJ; Gellman SH Backbone modification of a polypeptide drug alters duration of action in vivo. Nat. Biotechnol 2014, 32, 653–655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (518).Johnson LM; Barrick S; Hager MV; McFedries A; Homan EA; Rabaglia ME; Keller MP; Attie AD; Saghatelian A; Bisello A; et al. A Potent α/β-Peptide Analogue of GLP-1 with Prolonged Action in Vivo. J. Am. Chem. Soc 2014, 136, 12848–12851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (519).Kritzer JA; Lear JD; Hodsdon ME; Schepartz A Helical β-Peptide Inhibitors of the p53-hDM2 Interaction. J. Am. Chem. Soc 2004, 126, 9468–9469. [DOI] [PubMed] [Google Scholar]
- (520).Murray JK; Gellman SH Targeting protein–protein interactions: Lessons from p53/MDM2. Pept. Sci 2007, 88, 657–686. [DOI] [PubMed] [Google Scholar]
- (521).Michel J; Harker EA; Tirado-Rives J; Jorgensen WL; Schepartz A In Silico Improvement of β3-Peptide Inhibitors of p53•hDM2 and p53•hDMX. J. Am. Chem. Soc 2009, 131, 63566357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (522).Harker EA; Daniels DS; Guarracino DA; Schepartz A β-Peptides with improved affinity for hDM2 and hDMX. Biorg. Med. Chem 2009, 17, 2038–2046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (523).Harker EA; Schepartz A Cell-Permeable β-Peptide Inhibitors of p53/hDM2 Complexation. ChemBioChem. 2009, 10, 990–993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (524).Bautista AD; Appelbaum JS; Craig CJ; Michel J; Schepartz A Bridged β3-Peptide Inhibitors of p53-hDM2 Complexation: Correlation between Affinity and Cell Permeability. J. Am. Chem. Soc 2010, 132, 2904–2906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (525).Sadowsky JD; Fairlie WD; Hadley EB; Lee H-S; Umezawa N; Nikolovska-Coleska Z; Wang S; Huang DCS; Tomita Y; Gellman SH (α/β+α)-Peptide Antagonists of BH3 Domain/Bcl-xL Recognition: Toward General Strategies for Foldamer-Based Inhibition of Protein-Protein Interactions. J. Am. Chem. Soc 2007, 129, 139–154. [DOI] [PubMed] [Google Scholar]
- (526).Sadowsky JD; Schmitt MA; Lee H-S; Umezawa N; Wang S; Tomita Y; Gellman SH Chimeric (α/β + α)-Peptide Ligands for the BH3-Recognition Cleft of Bcl-xL: Critical Role of the Molecular Scaffold in Protein Surface Recognition. J. Am. Chem. Soc 2005, 127, 11966–11968. [DOI] [PubMed] [Google Scholar]
- (527).Lee EF; Sadowsky JD; Smith BJ; Czabotar PE; Peterson-Kaufman KJ; Colman PM; Gellman SH; Fairlie WD High-Resolution Structural Characterization of a Helical α/β-Peptide Foldamer Bound to the Anti-Apoptotic Protein Bcl-xL. Angew. Chem., Int. Ed 2009, 48, 4318–4322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (528).Horne WS; Boersma MD; Windsor MA; Gellman SH Sequence-Based Design of α/β-Peptide Foldamers That Mimic BH3 Domains. Angew. Chem., Int. Ed 2008, 47, 2853–2856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (529).Checco JW; Lee EF; Evangelista M; Sleebs NJ; Rogers K; Pettikiriarachchi A; Kershaw NJ; Eddinger GA; Belair DG; Wilson JL; et al. α/β-Peptide Foldamers Targeting Intracellular Protein-Protein Interactions with Activity in Living Cells. J. Am. Chem. Soc 2015, 137, 11365–11375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (530).Gelman MA; Richter S; Cao H; Umezawa N; Gellman SH; Rana TM Selective Binding of TAR RNA by a Tat-Derived β-Peptide. Org. Lett 2003, 5, 3563–3565. [DOI] [PubMed] [Google Scholar]
- (531).Horne WS; Johnson LM; Ketas TJ; Klasse PJ; Lu M; Moore JP; Gellman SH Structural and biological mimicry of protein surface recognition by α/β-peptide foldamers. Proc. Natl. Acad. Sci. U.S.A 2009, 106, 14751–14756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (532).Stephens OM; Kim S; Welch BD; Hodsdon ME; Kay MS; Schepartz A Inhibiting HIV Fusion with a β-Peptide Foldamer. J. Am. Chem. Soc 2005, 127, 13126–13127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (533).Outlaw VK; Cheloha RW; Jurgens EM; Bovier FT; Zhu Y; Kreitler DF; Harder O; Niewiesk S; Porotto M; Gellman SH; et al. Engineering Protease-Resistant Peptides to Inhibit Human Parainfluenza Viral Respiratory Infection. J. Am. Chem. Soc 2021, 143, 5958–5966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (534).Liu S; Daley EJ; Tran LM-L; Yu Z; Reyes M; Dean T; Khatri A; Levine PM; Balana AT; Pratt MR; et al. Backbone Modification Provides a Long-Acting Inverse Agonist of Pathogenic, Constitutively Active PTH1R Variants. J. Am. Chem. Soc 2024, 146, 6522–6529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (535).Hanessian S; Luo X; Schaum R; Michnick S Design of Secondary Structures in Unnatural Peptides: Stable Helical γ-Tetra-, Hexa-, and Octapeptides and Consequences of α-Substitution. J. Am. Chem. Soc 1998, 120, 8569–8570. [Google Scholar]
- (536).Legrand B; Maillard LT α,β-Unsaturated γ-Peptide Foldamers. ChemPlusChem. 2021, 86, 629–645. [DOI] [PubMed] [Google Scholar]
- (537).Hintermann T; Gademann K; Jaun B; Seebach D γ-Peptides Forming More Stable Secondary Structures than α-Peptides: Synthesis and helical NMR-solution structure of the γ-hexapeptide analog of H-(Val-Ala-Leu)2-OH. Helv. Chim. Acta 1998, 81, 983–1002. [Google Scholar]
- (538).Bouillerèe F; Feytens D; Gori D; Guillot R; Kouklovsky C; Miclet E; Alezra V Constrained α/γ-peptides: a new stable extended structure in solution without any hydrogen bond and characterized by a four-fold symmetry. Chem. Commun 2012, 48, 1982–1984. [DOI] [PubMed] [Google Scholar]
- (539).Zhang Y; Zhong Y; Connor AL; Miller DP; Cao R; Shen J; Song B; Baker ES; Tang Q; Pulavarti SVSRK; et al. Folding and Assembly of Short α, β, γ-Hybrid Peptides: Minor Variations in Sequence and Drastic Differences in Higher-Level Structures. J. Am. Chem. Soc 2019, 141, 14239–14248. [DOI] [PubMed] [Google Scholar]
- (540).Grison CM; Miles JA; Robin S; Wilson AJ; Aitken DJ An alpha-Helix-Mimicking 12,13-Helix: Designed alpha/beta/gamma-Foldamers as Selective Inhibitors of Protein-Protein Interactions. Angew. Chem., Int. Ed 2016, 55, 11096–11100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (541).Baldauf C; Gunther R; Hofmann HJ Side-chain control of folding of the homologous alpha-, beta-, and gamma-peptides into ″mixed″ helices (beta-helices). Biopolymers 2005, 80, 675–687. [DOI] [PubMed] [Google Scholar]
- (542).Guo L; Almeida AM; Zhang W; Reidenbach AG; Choi SH; Guzei IA; Gellman SH Helix Formation in Preorganized β/ γ-Peptide Foldamers: Hydrogen-Bond Analogy to the α-Helix without α-Amino Acid Residues. J. Am. Chem. Soc 2010, 132, 7868–7869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (543).Sawada T; Gellman SH Structural Mimicry of the α-Helix in Aqueous Solution with an Isoatomic α/β/γ-Peptide Backbone. J. Am. Chem. Soc 2011, 133, 7336–7339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (544).Shah SKH; Modi U; Patel K; James A; N S; De S; Vasita R; Prabhakaran P Site-selective post-modification of short α/γ hybrid foldamers: a powerful approach for molecular diversification towards biomedical applications. Biomater. Sci 2023, 11, 6210–6222. [DOI] [PubMed] [Google Scholar]
- (545).Kaffy J; Berardet C; Mathieu L; Legrand B; Taverna M; Halgand F; Van Der Rest G; Maillard LT; Ongeri S Helical γ-Peptide Foldamers as Dual Inhibitors of Amyloid-β Peptide and Islet Amyloid Polypeptide Oligomerization and Fibrillization. Chem. Eur. J 2020, 26, 14612–14622. [DOI] [PubMed] [Google Scholar]
- (546).Mathieu L; Legrand B; Deng C; Vezenkov L; Wenger E; Didierjean C; Amblard M; Averlant-Petit M-C; Masurier N; Lisowski V; et al. Helical Oligomers of Thiazole-Based γ-Amino Acids: Synthesis and Structural Studies. Angew. Chem., Int. Ed 2013, 52, 6006–6010. [DOI] [PubMed] [Google Scholar]
- (547).Burgess K; Shin H; Linthicum DS Solid-Phase Syntheses of Unnatural Biopolymers Containing Repeating Urea Units. Angew. Chem., Int. Ed 1995, 34, 907–909. [Google Scholar]
- (548).Yoo SH; Li B; Dolain C; Pasco M; Guichard G Chapter Three - Urea based foldamers. In Methods Enzymol.; Petersson EJ, Ed.; Vol. 656; Academic Press, 2021; pp 59–92. [DOI] [PubMed] [Google Scholar]
- (549).Guichard G; Semetey V; Didierjean C; Aubry A; Briand J-P; Rodriguez M Effective Preparation of O-Succinimidyl-2- (tert-butoxycarbonylamino)ethylcarbamate Derivatives from β-Amino Acids. Application to the Synthesis of Urea-Containing Pseudopeptides and Oligoureas. J. Org. Chem 1999, 64, 8702–8705. [Google Scholar]
- (550).Burgess K; Ibarzo J; Linthicum DS; Russell DH; Shin H; Shitangkoon A; Totani R; Zhang AJ Solid Phase Syntheses of Oligoureas. J. Am. Chem. Soc 1997, 119, 1556–1564. [Google Scholar]
- (551).Antunes S; Douat C; Guichard G Solid-Phase Synthesis of Hybrid Urea Oligomers Containing Conservative Thiourea Mutations. Eur. J. Org. Chem 2016, 2016, 2131–2138. [Google Scholar]
- (552).Douat-Casassus C; Pulka K; Claudon P; Guichard G Microwave-Enhanced Solid-Phase Synthesis of N,N′-Linked Aliphatic Oligoureas and Related Hybrids. Org. Lett 2012, 14, 3130–3133. [DOI] [PubMed] [Google Scholar]
- (553).Fischer L; Claudon P; Pendem N; Miclet E; Didierjean C; Ennifar E; Guichard G The Canonical Helix of Urea Oligomers at Atomic Resolution: Insights Into Folding-Induced Axial Organization. Angew. Chem., Int. Ed 2010, 49, 1067–1070. [DOI] [PubMed] [Google Scholar]
- (554).Tallet L; Frisch E; Bornerie M; Medemblik C; Frisch B; Lavalle P; Guichard G; Douat C; Kichler A Design of Oligourea-Based Foldamers with Antibacterial and Antifungal Activities. Molecules 2022, 27, 1749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (555).Semetey V; Rognan D; Hemmerlin C; Graff R; Briand JP; Marraud M; Guichard G Stable Helical Secondary Structure in Short-Chain N,N′-Linked Oligoureas Bearing Proteinogenic Side Chains. Angew. Chem., Int. Ed 2002, 41, 1893–1895. [PubMed] [Google Scholar]
- (556).Cussol L; Mauran-Ambrosino L; Buratto J; Belorusova AY; Neuville M; Osz J; Fribourg S; Fremaux J; Dolain C; Goudreau SR; et al. Structural Basis for α-Helix Mimicry and Inhibition of Protein-Protein Interactions with Oligourea Foldamers. Angew. Chem., Int. Ed 2021, 60, 2296–2303. [DOI] [PubMed] [Google Scholar]
- (557).Mbianda J; Bakail M; André C; Moal G; Perrin ME; Pinna G; Guerois R; Becher F; Legrand P; Traoré S; et al. Optimal anchoring of a foldamer inhibitor of ASF1 histone chaperone through backbone plasticity. Sci. Adv 2021, 7, eabd9153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (558).Hu X; Zhu H; Zhang X; He X; Xu X Comprehensive analysis of pan-cancer reveals potential of ASF1B as a prognostic and immunological biomarker. Cancer Med. 2021, 10, 6897–6916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (559).Im JS; Keaton M; Lee KY; Kumar P; Park J; Dutta A ATR checkpoint kinase and CRL1βTRCP collaborate to degrade ASF1a and thus repress genes overlapping with clusters of stalled replication forks. Genes Dev. 2014, 28, 875–887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (560).English CM; Adkins MW; Carson JJ; Churchill MEA; Tyler JK Structural Basis for the Histone Chaperone Activity of Asf1. Cell 2006, 127, 495–508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (561).Bakail M; Gaubert A; Andreani J; Moal G; Pinna G; Boyarchuk E; Gaillard M-C; Courbeyrette R; Mann C; Thuret J-Y; et al. Design on a Rational Basis of High-Affinity Peptides Inhibiting the Histone Chaperone ASF1. Cell Chem. Biol 2019, 26, 1573–1585.e1510. [DOI] [PubMed] [Google Scholar]
- (562).Teyssières E; Corre J-P; Antunes S; Rougeot C; Dugave C; Jouvion G; Claudon P; Mikaty G; Douat C; Goossens PL; et al. Proteolytically Stable Foldamer Mimics of Host-Defense Peptides with Protective Activities in a Murine Model of Bacterial Infection. J. Med. Chem 2016, 59, 8221–8232. [DOI] [PubMed] [Google Scholar]
- (563).Fremaux J; Venin C; Mauran L; Zimmer RH; Guichard G; Goudreau SR Peptide-oligourea hybrids analogue of GLP-1 with improved action in vivo. Nat. Commun 2019, 10, 924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (564).Simon RJ; Kania RS; Zuckermann RN; Huebner VD; Jewell DA; Banville S; Ng S; Wang L; Rosenberg S; Marlowe CK Peptoids: a modular approach to drug discovery. Proc. Natl. Acad. Sci. U.S.A 1992, 89, 9367–9371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (565).Kessler H Peptoids—A New Approach to the Development of Pharmaceuticals. Angew. Chem., Int. Ed 1993, 32, 543–544. [Google Scholar]
- (566).Yu P; Liu B; Kodadek T A high-throughput assay for assessing the cell permeability of combinatorial libraries. Nat. Biotechnol 2005, 23, 746–751. [DOI] [PubMed] [Google Scholar]
- (567).Miller SM; Simon RJ; Ng S; Zuckermann RN; Kerr JM; Moos WH Proteolytic studies of homologous peptide and N-substituted glycine peptoid oligomers. Bioorg. Med. Chem. Lett 1994, 4, 2657–2662. [Google Scholar]
- (568).Zuckermann RN Peptoid origins. Biopolymers 2011, 96, 545–555. [DOI] [PubMed] [Google Scholar]
- (569).Sanborn TJ; Wu CW; Zuckermann RN; Barron AE Extreme stability of helices formed by water-soluble poly-N-substituted glycines (polypeptoids) with α-chiral side chains. Biopolymers 2002, 63, 12–20. [DOI] [PubMed] [Google Scholar]
- (570).Armand P; Kirshenbaum K; Falicov A; Dunbrack RL; Dill KA; Zuckermann RN; Cohen FE Chiral N-substituted glycines can form stable helical conformations. Fold. Des 1997, 2, 369–375. [DOI] [PubMed] [Google Scholar]
- (571).Kirshenbaum K; Barron AE; Goldsmith RA; Armand P; Bradley EK; Truong KT; Dill KA; Cohen FE; Zuckermann RN Sequence-specific polypeptoids: a diverse family of heteropolymers with stable secondary structure. Proc. Natl. Acad. Sci. U.S.A 1998, 95, 4303–4308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (572).Oh M; Lee JH; Moon H; Hyun Y-J; Lim H-S A Chemical Inhibitor of the Skp2/p300 Interaction that Promotes p53-Mediated Apoptosis. Angew. Chem., Int. Ed 2016, 55, 602–606. [DOI] [PubMed] [Google Scholar]
- (573).Eastwood JRB; Weisberg EI; Katz D; Zuckermann RN; Kirshenbaum K Guidelines for designing peptoid structures: Insights from the Peptoid Data Bank. Pept. Sci 2023, 115, e24307. [Google Scholar]
- (574).Zuckermann RN; Kerr JM; Kent SBH; Moos WH Efficient method for the preparation of peptoids [oligo(N-substituted glycines)] by submonomer solid-phase synthesis. J. Am. Chem. Soc 1992, 114, 10646–10647. [Google Scholar]
- (575).Clapperton AM; Babi J; Tran H A Field Guide to Optimizing Peptoid Synthesis. ACS Polym. Au 2022, 2, 417–429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (576).Figliozzi GM; Goldsmith R; Ng SC; Banville SC; Zuckermann RN [25] Synthesis of N-substituted glycine peptoid libraries. In Methods Enzymol., Vol. 267; Academic Press, 1996; pp 437–447. [DOI] [PubMed] [Google Scholar]
- (577).Alluri PG; Reddy MM; Bachhawat-Sikder K; Olivos HJ; Kodadek T Isolation of Protein Ligands from Large Peptoid Libraries. J. Am. Chem. Soc 2003, 125, 13995–14004. [DOI] [PubMed] [Google Scholar]
- (578).Schneider JA; Craven TW; Kasper AC; Yun C; Haugbro M; Briggs EM; Svetlov V; Nudler E; Knaut H; Bonneau R; et al. Design of Peptoid-peptide Macrocycles to Inhibit the β-catenin TCF Interaction in Prostate Cancer. Nat. Commun 2018, 9, 4396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (579).Zuckermann RN; Martin EJ; Spellmeyer DC; Stauber GB; Shoemaker KR; Kerr JM; Figliozzi GM; Goff DA; Siani MA; Simon RJ; et al. Discovery of nanomolar ligands for 7-transmembrane G-protein-coupled receptors from a diverse N-(substituted)glycine peptoid library. J. Med. Chem 1994, 37, 2678–2685. [DOI] [PubMed] [Google Scholar]
- (580).García-Martinez C; Humet M; Planells-Cases R; Gomis A; Caprini M; Viana F; De La Pena E; Sanchez-Baeza F; Carbonell T; De Felipe C; et al. Attenuation of thermal nociception and hyperalgesia by VR1 blockers. Proc. Natl. Acad. Sci. U.S.A 2002, 99, 2374–2379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (581).Sun J; Zuckermann RN Peptoid Polymers: A Highly Designable Bioinspired Material. ACS Nano 2013, 7, 4715–4732. [DOI] [PubMed] [Google Scholar]
- (582).Morimoto J; Fukuda Y; Kuroda D; Watanabe T; Yoshida F; Asada M; Nakamura T; Senoo A; Nagatoishi S; Tsumoto K; et al. A Peptoid with Extended Shape in Water. J. Am. Chem. Soc 2019, 141, 14612–14623. [DOI] [PubMed] [Google Scholar]
- (583).Fukuda Y; Yokomine M; Kuroda D; Tsumoto K; Morimoto J; Sando S Peptoid-based reprogrammable template for cell-permeable inhibitors of protein-protein interactions. Chem. Sci 2021, 12, 13292–13300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (584).Hahne G; Grossmann TN Direct targeting of β-catenin: Inhibition of protein-protein interactions for the inactivation of Wnt signaling. Biorg. Med. Chem 2013, 21, 4020–4026. [DOI] [PubMed] [Google Scholar]
- (585).Boehm M; Beaumont K; Jones R; Kalgutkar AS; Zhang L; Atkinson K; Bai G; Brown JA; Eng H; Goetz GH; et al. Discovery of Potent and Orally Bioavailable Macrocyclic Peptide–Peptoid Hybrid CXCR7Modulators. J. Med. Chem 2017, 60, 9653–9663. [DOI] [PubMed] [Google Scholar]
- (586).Niu Y; Hu Y; Li X; Chen J; Cai J γ-AApeptides: design, synthesis and evaluation. New J. Chem 2011, 35, 542–545. [Google Scholar]
- (587).Dragulescu-Andrasi A; Rapireddy S; Frezza BM; Gayathri C; Gil RR; Ly DH A Simple γ-Backbone Modification Preorganizes Peptide Nucleic Acid into a Helical Structure. J. Am. Chem. Soc 2006, 128, 10258–10267. [DOI] [PubMed] [Google Scholar]
- (588).Swenson CS; Velusamy A; Argueta-Gonzalez HS; Heemstra JM Bilingual Peptide Nucleic Acids: Encoding the Languages of Nucleic Acids and Proteins in a Single Self-Assembling Biopolymer. J. Am. Chem. Soc 2019, 141, 19038–19047. [DOI] [PubMed] [Google Scholar]
- (589).Argueta-Gonzalez HS; Swenson CS; Song G; Heemstra JM Stimuli-responsive assembly of bilingual peptide nucleic acids. RSC Chem. Biol 2022, 3, 1035–1043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (590).Wu H; Teng P; Cai J Rapid Access to Multiple Classes of Peptidomimetics from Common γ-AApeptide Building Blocks. Eur. J. Org. Chem 2014, 2014, 1760–1765. [Google Scholar]
- (591).Teng P; Zheng M; Cerrato DC; Shi Y; Zhou M; Xue S; Jiang W; Wojtas L; Ming L-J; Hu Y; et al. The folding propensity of α/sulfono-γ-AA peptidic foldamers with both left- and right-handedness. Commun. Chem 2021, 4, 58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (592).Teng P; Ma N; Cerrato DC; She F; Odom T; Wang X; Ming L-J; van der Vaart A; Wojtas L; Xu H; et al. Right-Handed Helical Foldamers Consisting of De Novo d-AApeptides. J. Am. Chem. Soc 2017, 139, 7363–7369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (593).She F; Teng P; Peguero-Tejada A; Wang M; Ma N; Odom T; Zhou M; Gjonaj E; Wojtas L; van der Vaart A; et al. De Novo Left-Handed Synthetic Peptidomimetic Foldamers. Angew. Chem., Int. Ed 2018, 57, 9916–9920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (594).Sang P; Shi Y; Lu J; Chen L; Yang L; Borcherds W; Abdulkadir S; Li Q; Daughdrill G; Chen J; et al. α-Helix-Mimicking Sulfono-γ-AApeptide Inhibitors for p53-MDM2/MDMX Protein-Protein Interactions. J. Med. Chem 2020, 63, 975–986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (595).Sang P; Zhang M; Shi Y; Li C; Abdulkadir S; Li Q; Ji H; Cai J Inhibition of β-catenin/B cell lymphoma 9 protein-protein interaction using α-helix–mimicking sulfono-γ-AApeptide inhibitors. Proc. Natl. Acad. Sci. U.S.A 2019, 116, 10757–10762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (596).Liu H; Cui Y; Zhao X; Wei L; Wang X; Shen N; Odom T; Li X; Lawless W; Karunarathne K; et al. Helical sulfonyl-γ-AApeptides modulating Aβ oligomerization and cytotoxicity by recognizing Aβ helix. Proc. Natl. Acad. Sci. U.S.A 2024, 121, e2311733121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (597).Yang Y; Niu Y; Hong H; Wu H; Zhang Y; Engle JW; Barnhart TE; Cai J; Cai W Radiolabeled γ-AApeptides: a new class of tracers for positron emission tomography. Chem. Commun 2012, 48, 7850–7852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (598).Wu H; Li Y; Bai G; Niu Y; Qiao Q; Tipton JD; Cao C; Cai J γ-AApeptide-based small-molecule ligands that inhibit Aβ aggregation. Chem. Commun 2014, 50, 5206–5208. [DOI] [PubMed] [Google Scholar]
- (599).Teng P; Zhang X; Wu H; Qiao Q; Sebti SM; Cai J Identification of novel inhibitors that disrupt STAT3–DNA interaction from a γ-AApeptide OBOC combinatorial library. Chem. Commun 2014, 50, 8739–8742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (600).Teng P; Shi Y; Sang P; Cai J γ-AApeptides as a New Class of Peptidomimetics. Chem. Eur. J 2016, 22, 5458–5466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (601).Yin H; Hamilton AD Strategies for Targeting Protein–Protein Interactions With Synthetic Agents. Angew. Chem., Int. Ed 2005, 44, 4130–4163. [DOI] [PubMed] [Google Scholar]
- (602).Orner BP; Ernst JT; Hamilton AD Toward Proteomimetics: Terphenyl Derivatives as Structural and Functional Mimics of Extended Regions of an α-Helix. J. Am. Chem. Soc 2001, 123, 5382–5383. [DOI] [PubMed] [Google Scholar]
- (603).Yin H; Lee G. -i.; Park HS; Payne GA; Rodriguez JM; Sebti SM; Hamilton AD Terphenyl-Based Helical Mimetics That Disrupt the p53/HDM2 Interaction. Angew. Chem., Int. Ed 2005, 44, 2704–2707. [DOI] [PubMed] [Google Scholar]
- (604).Davis JM; Tsou LK; Hamilton AD Synthetic non-peptide mimetics of α-helices. Chem. Soc. Rev 2007, 36, 326–334. [DOI] [PubMed] [Google Scholar]
- (605).Jayatunga MKP; Thompson S; Hamilton AD α-Helix mimetics: Outwards and upwards. Bioorg. Med. Chem. Lett 2014, 24, 717–724. [DOI] [PubMed] [Google Scholar]
- (606).Saraogi I; Hamilton AD α-Helix mimetics as inhibitors of protein–protein interactions. Biochem. Soc. Trans 2008, 36, 1414–1417. [DOI] [PubMed] [Google Scholar]
- (607).Algar S; Martín-Martínez M; González-Munñiz R Evolution in non-peptide α-helix mimetics on the road to effective protein-protein interaction modulators. Eur. J. Med. Chem 2021, 211, 113015. [DOI] [PubMed] [Google Scholar]
- (608).Yin H; Lee G.-i.; Sedey KA; Rodriguez JM; Wang H-G; Sebti SM; Hamilton AD Terephthalamide Derivatives as Mimetics of Helical Peptides: Disruption of the Bcl-xL/Bak Interaction. J. Am. Chem. Soc 2005, 127, 5463–5468. [DOI] [PubMed] [Google Scholar]
- (609).Kutzki O; Park HS; Ernst JT; Orner BP; Yin H; Hamilton AD Development of a Potent Bcl-xL Antagonist Based on α-Helix Mimicry. J. Am. Chem. Soc 2002, 124, 11838–11839. [DOI] [PubMed] [Google Scholar]
- (610).Chen L; Yin H; Farooqi B; Sebti S; Hamilton AD; Chen J p53 α-Helix mimetics antagonize p53/MDM2 interaction and activate p53. Mol. Cancer Ther 2005, 4, 1019–1025. [DOI] [PubMed] [Google Scholar]
- (611).Jung K-Y; Wang H; Teriete P; Yap JL; Chen L; Lanning ME; Hu A; Lambert LJ; Holien T; Sundan A; et al. Perturbation of the c-Myc–Max Protein–Protein Interaction via Synthetic α-Helix Mimetics. J. Med. Chem 2015, 58, 3002–3024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (612).Burslem GM; Kyle HF; Breeze AL; Edwards TA; Nelson A; Warriner SL; Wilson AJ Small-Molecule Proteomimetic Inhibitors of the HIF-1α–p300 Protein–Protein Interaction. ChemBioChem. 2014, 15, 1083–1087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (613).Campbell F; Plante JP; Edwards TA; Warriner SL; Wilson AJ N-alkylated oligoamide α-helical proteomimetics. Org. Biomol. Chem 2010, 8, 2344–2351. [DOI] [PubMed] [Google Scholar]
- (614).Lao BB; Grishagin I; Mesallati H; Brewer TF; Olenyuk BZ; Arora PS In vivo modulation of hypoxia-inducible signaling by topographical helix mimetics. Proc. Natl. Acad. Sci. U.S.A 2014, 111, 7531–7536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (615).Cummings CG; Ross NT; Katt WP; Hamilton AD Synthesis and Biological Evaluation of a 5–6-5 Imidazole-Phenyl-Thiazole Based α-Helix Mimetic. Org. Lett 2009, 11, 25–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (616).Saraogi I; Hebda JA; Becerril J; Estroff LA; Miranker AD; Hamilton AD Synthetic α-Helix Mimetics as Agonists and Antagonists of Islet Amyloid Polypeptide Aggregation. Angew. Chem., Int. Ed 2010, 49, 736–739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (617).Kumar S; Schlamadinger DE; Brown MA; Dunn JM; Mercado B; Hebda JA; Saraogi I; Rhoades E; Hamilton AD; Miranker AD Islet Amyloid-Induced Cell Death and Bilayer Integrity Loss Share a Molecular Origin Targetable with Oligopyridylamide-Based α-Helical Mimetics. Chem. Biol 2015, 22, 369–378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (618).Kumar S; Hamilton AD α-Helix Mimetics as Modulators of Aβ Self-Assembly. J. Am. Chem. Soc 2017, 139, 5744–5755. [DOI] [PubMed] [Google Scholar]
- (619).Pellegrino S; Tonali N; Erba E; Kaffy J; Taverna M; Contini A; Taylor M; Allsop D; Gelmi ML; Ongeri S β-Hairpin mimics containing a piperidine–pyrrolidine scaffold modulate the β-amyloid aggregation process preserving the monomer species. Chem. Sci 2017, 8, 1295–1302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (620).Kaur A; Goyal D; Goyal B An α-helix mimetic oligopyridylamide, ADH-31, modulates Aβ42 monomer aggregation and destabilizes protofibril structures: insights from molecular dynamics simulations. Phys. Chem. Chem. Phys 2020, 22, 28055–28073. [DOI] [PubMed] [Google Scholar]
- (621).Kazi A; Sun J; Doi K; Sung S-S; Takahashi Y; Yin H; Rodriguez JM; Becerril J; Berndt N; Hamilton AD; et al. The BH3 α-Helical Mimic BH3-M6 Disrupts Bcl-XL, Bcl-2, and MCL-1 Protein-Protein Interactions with Bax, Bak, Bad, or Bim and Induces Apoptosis in a Bax- and Bim-dependent Manner*. J. Biol. Chem 2011, 286, 9382–9392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (622).Lee Y; Im H; Das S; Oh M; Lee JH; Ham S; Lim H-S Bridged α-helix mimetic small molecules. Chem. Commun 2019, 55, 13311–13314. [DOI] [PubMed] [Google Scholar]
- (623).Maity D; Kumar S; Curreli F; Debnath AK; Hamilton AD α-Helix-Mimetic Foldamers for Targeting HIV-1 TAR RNA. Chem. Eur. J 2019, 25, 7265–7269. [DOI] [PubMed] [Google Scholar]
- (624).González-Bulnes L; Ibánñez I; Bedoya LM; Beltrán M; Catalán S; Alcamí J; Fustero S; Gallego J Structure-Based Design of an RNA-Binding p-Terphenylene Scaffold that Inhibits HIV-1 Rev Protein Function. Angew. Chem., Int. Ed 2013, 52, 13405–13409. [DOI] [PubMed] [Google Scholar]
- (625).Medina-Trillo C; Sedgwick DM; Herrera L; Beltrán M; Moreno Á; Barrio P; Bedoya LM; Alcamí J; Fustero S; Gallego J Nucleic acid recognition and antiviral activity of 1,4-substituted terphenyl compounds mimicking all faces of the HIV-1 Rev protein positively-charged α-helix. Sci. Rep 2020, 10, 7190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (626).Shaginian A; Whitby LR; Hong S; Hwang I; Farooqi B; Searcey M; Chen J; Vogt PK; Boger DL Design, Synthesis, and Evaluation of an α-Helix Mimetic Library Targeting Protein-Protein Interactions. J. Am. Chem. Soc 2009, 131, 5564–5572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (627).Huc I Aromatic Oligoamide Foldamers. Eur. J. Org. Chem 2004, 2004, 17–29. [Google Scholar]
- (628).Barnard A; Long K; Martin HL; Miles JA; Edwards TA; Tomlinson DC; Macdonald A; Wilson AJ Selective and Potent Proteomimetic Inhibitors of Intracellular Protein–Protein Interactions. Angew. Chem., Int. Ed 2015, 54, 2960–2965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (629).Tošovská P; Arora PS Oligooxopiperazines as Nonpeptidic α-Helix Mimetics. Org. Lett 2010, 12, 1588–1591. [DOI] [PubMed] [Google Scholar]
- (630).Lao BB; Drew K; Guarracino DA; Brewer TF; Heindel DW; Bonneau R; Arora PS Rational Design of Topographical Helix Mimics as Potent Inhibitors of Protein–Protein Interactions. J. Am. Chem. Soc 2014, 136, 7877–7888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (631).Barnard A; Miles JA; Burslem GM; Barker AM; Wilson AJ Multivalent helix mimetics for PPI-inhibition. Org. Biomol. Chem 2015, 13, 258–264. [DOI] [PubMed] [Google Scholar]
- (632).Arrata I; Barnard A; Tomlinson DC; Wilson AJ Interfacing native and non-native peptides: using Affimers to recognise α-helix mimicking foldamers. Chem. Commun 2017, 53, 2834–2837. [DOI] [PubMed] [Google Scholar]
- (633).Hegedus Z; Grison CM; Miles JA; Rodriguez-Marin S; Warriner SL; Webb ME; Wilson AJ A catalytic protein–proteomimetic complex: using aromatic oligoamide foldamers as activators of RNase S. Chem. Sci 2019, 10, 3956–3962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (634).Barnard A; Long K; Yeo DJ; Miles JA; Azzarito V; Burslem GM; Prabhakaran P; Edwards TA; Wilson AJ Orthogonal functionalisation of α-helix mimetics. Org. Biomol. Chem 2014, 12, 6794–6799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (635).Murray JK; Farooqi B; Sadowsky JD; Scalf M; Freund WA; Smith LM; Chen J; Gellman SH Efficient Synthesis of a β-Peptide Combinatorial Library with Microwave Irradiation. J. Am. Chem. Soc 2005, 127, 13271–13280. [DOI] [PubMed] [Google Scholar]
- (636).Long K; Edwards TA; Wilson AJ Microwave assisted solid phase synthesis of highly functionalized N-alkylated oligobenzamide α-helix mimetics. Biorg. Med. Chem 2013, 21, 4034–4040. [DOI] [PubMed] [Google Scholar]
- (637).Gopalakrishnan R; Frolov AI; Knerr L; Drury WJ III.; Valeur E Therapeutic Potential of Foldamers: From Chemical Biology Tools To Drug Candidates? J. Med. Chem 2016, 59, 9599–9621. [DOI] [PubMed] [Google Scholar]
- (638).Whaby M; Khan I; O’Bryan JP Chapter Eight - Targeting the “undruggable” RAS with biologics. In Adv. Cancer Res; O’Bryan JP, Piazza GA, Eds.; Vol. 153; Academic Press, 2022; pp 237–266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (639).Sha F; Salzman G; Gupta A; Koide S Monobodies and other synthetic binding proteins for expanding protein science. Protein Sci. 2017, 26, 910–924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (640).Shilova ON; Deyev SM DARPins: Promising Scaffolds for Theranostics. Acta Naturae 2019, 11, 42–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (641).Ståhl S; Gräslund T; Eriksson Karlström A; Frejd FY; Nygren P-Å; Löfblom J Affibody Molecules in Biotechnological and Medical Applications. Trends Biotechnol. 2017, 35, 691–712. [DOI] [PubMed] [Google Scholar]
- (642).Wang X; Li F; Qiu W; Xu B; Li Y; Lian X; Yu H; Zhang Z; Wang J; Li Z; et al. SYNBIP: synthetic binding proteins for research, diagnosis and therapy. Nucleic Acids Res. 2022, 50, D560–D570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (643).Vazquez-Lombardi R; Phan TG; Zimmermann C; Lowe D; Jermutus L; Christ D Challenges and opportunities for non-antibody scaffold drugs. Drug Discovery Today 2015, 20, 1271–1283. [DOI] [PubMed] [Google Scholar]
- (644).Frejd FY; Kim K-T Affibody molecules as engineered protein drugs. Exp. Mol. Med 2017, 49, e306–e306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (645).Horne WS; Grossmann TN Proteomimetics as protein-inspired scaffolds with defined tertiary folding patterns. Nat. Chem 2020, 12, 331–337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (646).Lin SH; Konishi Y; Denton ME; Scheraga HA Influence of an extrinsic crosslink on the folding pathway of ribonuclease A. Conformational and thermodynamic analysis of crosslinked (7-lysine, 41-lysine)-ribonuclease A. Biochemistry 1984, 23, 5504–5512. [DOI] [PubMed] [Google Scholar]
- (647).Marfey PS; Nowak H; Uziel M; Yphantis DA Reaction of Bovine Pancreatic Ribonuclease A with 1,5-Difluoro-2,4-dinitrobenzene: I. PREPARATION OF MONOMERIC INTRAMOLECULARLY BRIDGED DERIVATIVES. J. Biol. Chem 1965, 240, 3264–3269. [PubMed] [Google Scholar]
- (648).Kent SBH Novel protein science enabled by total chemical synthesis. Protein Sci. 2019, 28, 313–328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (649).Xiao Q; Bécar NA; Brown NP; Smith MS; Stern KL; Draper SRE; Thompson KP; Price JL Stapling of two PEGylated side chains increases the conformational stability of the WW domain via an entropic effect. Org. Biomol. Chem 2018, 16, 8933–8939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (650).Wuo MG; Mahon AB; Arora PS An Effective Strategy for Stabilizing Minimal Coiled Coil Mimetics. J. Am. Chem. Soc 2015, 137, 11618–11621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (651).Ekblad T; Tolmachev V; Orlova A; Lendel C; Abrahmsén L; Karlström AE Synthesis and chemoselective intramolecular crosslinking of a HER2-binding affibody. Pept. Sci 2009, 92, 116–123. [DOI] [PubMed] [Google Scholar]
- (652).Sawyer N; Watkins AM; Arora PS Protein Domain Mimics as Modulators of Protein–Protein Interactions. Acc. Chem. Res 2017, 50, 1313–1322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (653).Dang B; Wu H; Mulligan VK; Mravic M; Wu Y; Lemmin T; Ford A; Silva D-A; Baker D; DeGrado WF De novo design of covalently constrained mesosize protein scaffolds with unique tertiary structures. Proc. Natl. Acad. Sci. U.S.A 2017, 114, 10852–10857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (654).Wuo MG; Hong SH; Singh A; Arora PS Synthetic Control of Tertiary Helical Structures in Short Peptides. J. Am. Chem. Soc 2018, 140, 16284–16290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (655).Lindgren J; Eriksson Karlström A Intramolecular Thioether Crosslinking of Therapeutic Proteins to Increase Proteolytic Stability. ChemBioChem. 2014, 15, 2132–2138. [DOI] [PubMed] [Google Scholar]
- (656).Paulussen FM; Schouten GK; Moertl C; Verheul J; Hoekstra I; Koningstein GM; Hutchins GH; Alkir A; Luirink RA; Geerke DP; et al. Covalent Proteomimetic Inhibitor of the Bacterial FtsQB Divisome Complex. J. Am. Chem. Soc 2022, 144, 15303–15313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (657).Wang C; Li X; Yu F; Lu L; Jiang X; Xu X; Wang H; Lai W; Zhang T; Zhang Z; et al. Site-specific Isopeptide Bridge Tethering of Chimeric gp41 N-terminal Heptad Repeat Helical Trimers for the Treatment of HIV-1 Infection. Sci. Rep 2016, 6, 32161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (658).Montgomery JE; Moellering RE Diels-Alder Cycloadditions for Peptide MacrocyclePeptidemacrocyclesFormation. In Peptide Macrocycles: Methods and Protocols; Coppock MB, Winton AJ, Eds.; Springer: US, 2022; pp 159–174. [DOI] [PubMed] [Google Scholar]
- (659).Reinert ZE; Lengyel GA; Horne WS Protein-like Tertiary Folding Behavior from Heterogeneous Backbones. J. Am. Chem. Soc 2013, 135, 12528–12531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (660).Lombardo CM; Kumar MVV; Douat C; Rosu F; Mergny J-L; Salgado GF; Guichard G Design and Structure Determination of a Composite Zinc Finger Containing a Nonpeptide Foldamer Helical Domain. J. Am. Chem. Soc 2019, 141, 2516–2525. [DOI] [PubMed] [Google Scholar]
- (661).Lelais G; Seebach D; Jaun B; Mathad RI; Flögel O; Rossi F; Campo M; Wortmann A β-Peptidic Secondary Structures Fortified and Enforced by Zn2+ Complexation – On the Way to β-Peptidic Zinc Fingers? Helv. Chim. Acta 2006, 89, 361–403. [Google Scholar]
- (662).George KL; Horne WS Heterogeneous-Backbone Foldamer Mimics of Zinc Finger Tertiary Structure. J. Am. Chem. Soc 2017, 139, 7931–7938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (663).Cheng P-N; Liu C; Zhao M; Eisenberg D; Nowick JS Amyloid β-sheet mimics that antagonize protein aggregation and reduce amyloid toxicity. Nat. Chem 2012, 4, 927–933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (664).Checco JW; Kreitler DF; Thomas NC; Belair DG; Rettko NJ; Murphy WL; Forest KT; Gellman SH Targeting diverse protein–protein interaction interfaces with α/β-peptides derived from the Z-domain scaffold. Proc. Natl. Acad. Sci. U.S.A 2015, 112, 4552–4557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (665).Moore EJ; Zorine D; Hansen WA; Khare SD; Fasan R Enzyme stabilization via computationally guided protein stapling. Proc. Natl. Acad. Sci. U.S.A 2017, 114, 12472–12477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (666).Yin L; Yuvienco C; Montclare JK Protein based therapeutic delivery agents: Contemporary developments and challenges. Biomaterials 2017, 134, 91–116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (667).Sadek J; Wuo MG; Rooklin D; Hauenstein A; Hong SH; Gautam A; Wu H; Zhang Y; Cesarman E; Arora PS Modulation of virus-induced NF-κB signaling by NEMO coiled coil mimics. Nat. Commun 2020, 11, 1786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (668).Yoo DY; Barros SA; Brown GC; Rabot C; Bar-Sagi D; Arora PS Macropinocytosis as a Key Determinant of Peptidomimetic Uptake in Cancer Cells. J. Am. Chem. Soc 2020, 142, 14461–14471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (669).Hong SH; Yoo DY; Conway L; Richards-Corke KC; Parker CG; Arora PS A Sos proteomimetic as a pan-Ras inhibitor. Proc. Natl. Acad. Sci. U.S.A 2021, 118, e2101027118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (670).Yang G; Zhang J; You W; Zhao X; Hou P; He W; Yan J; Guo H Targeted disruption of the BCL9/β-catenin interaction by endosomal-escapable nanoparticles functionalized with an E-cadherin-derived peptide. Nanotechnology 2020, 31, 115102. [DOI] [PubMed] [Google Scholar]
- (671).Tao Z; Wu X Targeting Transcription Factors in Cancer: From “Undruggable” to “Druggable. In Transcription Factor Regulatory Networks; Song Q, Tao Z, Eds.; Humana, New York, NY, 2023; pp 107–131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (672).Lambert M; Jambon S; Depauw S; David-Cordonnier M-H Targeting Transcription Factors for Cancer Treatment. Molecules 2018, 23, 1479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (673).Adihou H; Gopalakrishnan R; Förster T; Guéret SM; Gasper R; Geschwindner S; Carrillo García C; Karatas H; Pobbati AV; Vazquez-Chantada M; et al. A protein tertiary structure mimetic modulator of the Hippo signalling pathway. Nat. Commun 2020, 11, 5425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (674).Kyle HF; Wickson KF; Stott J; Burslem GM; Breeze AL; Tiede C; Tomlinson DC; Warriner SL; Nelson A; Wilson AJ; et al. Exploration of the HIF-1α/p300 interface using peptide and Adhiron phage display technologies. Mol. Biosyst 2015, 11, 2738–2749. [DOI] [PubMed] [Google Scholar]
- (675).Burslem GM; Kyle HF; Breeze AL; Edwards TA; Nelson A; Warriner SL; Wilson AJ Towards “bionic” proteins: replacement of continuous sequences from HIF-1α with proteomimetics to create functional p300 binding HIF-1α mimics. Chem. Commun 2016, 52, 5421–5424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (676).Blancafort P; Segal DJ; Barbas CF Designing Transcription Factor Architectures for Drug Discovery. Mol. Pharmacol 2004, 66, 1361. [DOI] [PubMed] [Google Scholar]
- (677).Boga S; Bouzada D; García Penña D; Vázquez López M; Vázquez ME Sequence-Specific DNA Recognition with Designed Peptides. Eur. J. Org. Chem 2018, 2018, 249–261. [Google Scholar]
- (678).Montclare JK; Schepartz A Miniature Homeodomains: High Specificity without an N-Terminal Arm. J. Am. Chem. Soc 2003, 125, 3416–3417. [DOI] [PubMed] [Google Scholar]
- (679).Ghosh B; Boila LD; Choudhury S; Mondal P; Bhattacharjee S; Pal SK; Sengupta A; Roy S A Potent Conformation-Constrained Synthetic Peptide Mimic of a Homeodomain Selectively Regulates Target Genes in Cells. ACS Chem. Biol 2018, 13, 2003–2009. [DOI] [PubMed] [Google Scholar]
- (680).Talanian RV; McKnight CJ; Rutkowski R; Kim PS Minimum length of a sequence-specific DNA binding peptide. Biochemistry 1992, 31, 6871–6875. [DOI] [PubMed] [Google Scholar]
- (681).Canne LE; Ferre- D’Amare AR; Burley SK; Kent SBH Total Chemical Synthesis of a Unique Transcription Factor-Related Protein: cMyc-Max. J. Am. Chem. Soc 1995, 117, 2998–3007. [Google Scholar]
- (682).Madden SK; de Araujo AD; Gerhardt M; Fairlie DP; Mason JM Taking the Myc out of cancer: toward therapeutic strategies to directly inhibit c-Myc. Mol. Cancer 2021, 20, 3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (683).Soucek L; Helmer-Citterich M; Sacco A; Jucker R; Cesareni G; Nasi S Design and properties of a Myc derivative that efficiently homodimerizes. Oncogene 1998, 17, 2463–2472. [DOI] [PubMed] [Google Scholar]
- (684).Brown ZZ; Mapelli C; Farasat I; Shoultz AV; Johnson SA; Orvieto F; Santoprete A; Bianchi E; McCracken AB; Chen K; et al. Multiple Synthetic Routes to the Mini-Protein Omomyc and Coiled-Coil Domain Truncations. J. Org. Chem 2020, 85, 1466–1475. [DOI] [PubMed] [Google Scholar]
- (685).Jbara M; Pomplun S; Schissel CK; Hawken SW; Boija A; Klein I; Rodriguez J; Buchwald SL; Pentelute BL Engineering Bioactive Dimeric Transcription Factor Analogs via Palladium Rebound Reagents. J. Am. Chem. Soc 2021, 143, 11788–11798. [DOI] [PubMed] [Google Scholar]
- (686).Beaulieu M-E; Jauset T; Massó-Vallés D; Martínez-Martín S; Rahl P; Maltais L; Zacarias-Fluck MF; Casacuberta-Serra S; Serrano del Pozo E; Fiore C; et al. Intrinsic cell-penetrating activity propels Omomyc from proof of concept to viable anti-MYC therapy. Sci. Transl. Med 2019, 11, eaar5012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (687).Demma MJ; Mapelli C; Sun A; Bodea S; Ruprecht B; Javaid S; Wiswell D; Muise E; Chen S; Zelina J; et al. Omomyc Reveals New Mechanisms To Inhibit the MYC Oncogene. Mol. Cell. Biol 2019, 39, e00248–00219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (688).Beaulieu M-E; Martínez-Martín S; Kaur J; Castillo Cano V; Massó-Vallés D; Foradada Felip L; López-Estévez S; Serrano del Pozo E; Thabussot H; Soucek L Pharmacokinetic Analysis of Omomyc Shows Lasting Structural Integrity and Long Terminal HalfLife in Tumor Tissue. Cancers (Basel) 2023, 15, 826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (689).Garralda E; Beaulieu M-E; Moreno V; Casacuberta-Serra S; Martínez-Martín S; Foradada L; Alonso G; Massó-Vallés D; López-Estévez S; Jauset T; et al. MYC targeting by OMO-103 in solid tumors: a phase 1 trial. Nat. Med 2024, 30, 762–771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (690).Speltz TE; Qiao Z; Swenson CS; Shangguan X; Coukos JS; Lee CW; Thomas DM; Santana J; Fanning SW; Greene GL; et al. Targeting MYC with modular synthetic transcriptional repressors derived from bHLH DNA-binding domains. Nat. Biotechnol 2023, 41, 541–551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (691).Jones S An overview of the basic helix-loop-helix proteins. Genome Biol. 2004, 5, 226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (692).Qiao Z; Nguyen LC; Yang D; Dann C; Thomas DM; Henn M; Valdespino A; Swenson CS; Oakes SA; Rosner MR; et al. Direct inhibition of tumor hypoxia response with synthetic transcriptional repressors. Nat. Chem. Biol 2024. DOI: 10.1038/s41589-024-01716-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (693).Ebrahimi SB; Samanta D Engineering protein-based therapeutics through structural and chemical design. Nat. Commun 2023, 14, 2411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (694).Löfblom J; Feldwisch J; Tolmachev V; Carlsson J; Ståhl S; Frejd FY Affibody molecules: Engineered proteins for therapeutic, diagnostic and biotechnological applications. FEBS Lett. 2010, 584, 2670–2680. [DOI] [PubMed] [Google Scholar]
- (695).Fletcher JM; Horner KA; Bartlett GJ; Rhys GG; Wilson AJ; Woolfson DN De novo coiled-coil peptides as scaffolds for disrupting protein–protein interactions. Chem. Sci 2018, 9, 7656–7665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (696).Listov D; Goverde CA; Correia BE; Fleishman SJ Opportunities and challenges in design and optimization of protein function. Nat. Rev. Mol. Cell. Biol 2024, 25, 639–653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (697).Lau JL; Dunn MK Therapeutic peptides: Historical perspectives, current development trends, and future directions. Biorg. Med. Chem 2018, 26, 2700–2707. [DOI] [PubMed] [Google Scholar]
- (698).Peraro L; Deprey KL; Moser MK; Zou Z; Ball HL; Levine B; Kritzer JA Cell Penetration Profiling Using the Chloroalkane Penetration Assay. J. Am. Chem. Soc 2018, 140, 11360–11369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (699).Peier A; Ge L; Boyer N; Frost J; Duggal R; Biswas K; Edmondson S; Hermes JD; Yan L; Zimprich C; et al. NanoClick: A High Throughput, Target-Agnostic Peptide Cell Permeability Assay. ACS Chem. Biol 2021, 16, 293–309. [DOI] [PubMed] [Google Scholar]
- (700).Bruno BJ; Miller GD; Lim CS Basics and recent advances in peptide and protein drug delivery. Ther. Delivery 2013, 4, 1443–1467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (701).Mitchell MJ; Billingsley MM; Haley RM; Wechsler ME; Peppas NA; Langer R Engineering precision nanoparticles for drug delivery. Nat. Rev. Drug Discovery 2021, 20, 101–124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (702).Rao SR; Schettler SL; Horne WS Metal-Binding Foldamers. ChemPlusChem. 2021, 86, 137–145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (703).Metrano AJ; Chinn AJ; Shugrue CR; Stone EA; Kim B; Miller SJ Asymmetric Catalysis Mediated by Synthetic Peptides, Version 2.0: Expansion of Scope and Mechanisms. Chem. Rev 2020, 120, 11479–11615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (704).Wang Z; Ji H Characterization of Hydrophilic α-Helical Hot Spots on the Protein-Protein Interaction Interfaces for the Design of α-Helix Mimetics. J. Chem. Inf. Model 2022, 62, 1873–1890. [DOI] [PubMed] [Google Scholar]
- (705).Watson JL; Juergens D; Bennett NR; Trippe BL; Yim J; Eisenach HE; Ahern W; Borst AJ; Ragotte RJ; Milles LF; et al. De novo design of protein structure and function with RFdiffusion. Nature 2023, 620, 1089–1100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (706).Huang P-S; Boyken SE; Baker D The coming of age of de novo protein design. Nature 2016, 537, 320–327. [DOI] [PubMed] [Google Scholar]