

Abstract
Simian virus 40 (SV40) is a small DNA tumor virus that has been extensively characterized due to its relatively simple genetic organization and the ease with which its genome is manipulated. The large and small tumor antigens (T antigens) are the major regulatory proteins encoded by SV40. Large T antigen is responsible for both viral and cellular transcriptional regulation, virion assembly, viral DNA replication, and alteration of the cell cycle. Deciphering how a single protein can perform such numerous and diverse functions has remained elusive. Recently it was established that the SV40 T antigens, including large T antigen, are molecular chaperones, each with a functioning DnaJ domain. The molecular chaperones were originally identified as bacterial genes essential for bacteriophage growth and have since been shown to be conserved in eukaryotes, participating in an array of both viral and cellular processes. This review discusses the mechanisms of DnaJ/Hsc70 interactions and how they are used by T antigen to control viral replication and tumorigenesis. The use of the DnaJ/Hsc70 system by SV40 and other viruses suggests an important role for these molecular chaperones in the regulation of the mammalian cell cycle and sheds light on the enigmatic SV40 T antigen—a most amazing molecule.
INTRODUCTION
Simian virus 40 (SV40), a member of the Polyomaviridae family, has a rich history of discovery. Its use as a model system has led to fundamental insights into the molecular processes of genome replication, gene expression, posttranscriptional processing, and cell cycle regulation. Its simple genome structure coupled with the ease that it can be grown and manipulated in cell culture has made SV40 an ideal system for coupling genetic and biochemical approaches to complex cellular control problems. Progress in each of these areas has been aided by studies of other viral systems, particularly the adenoviruses and papillomaviruses, since they face common obstacles to successful infection. Members of each of these families can induce tumors in experimental animals or their natural host, and thus they are classified as tumor viruses. That is, they are viruses with DNA genomes that contribute to tumorigenesis.
Each of these viruses encodes a set of proteins that function to (i) take over key cellular regulatory circuits and (ii) directly contribute to viral genome replication, expression, and/or virion assembly and release. SV40 encodes a 708-amino-acid, multifunctional large tumor antigen (T antigen) that plays several of these roles in viral infection and tumorigenesis. Recently, T antigen was shown to be a DnaJ molecular chaperone, and the chaperone activity is required for T antigen to function in viral replication, transcriptional control, and virion assembly, as well as transformation. In this article we review the chaperone activity of T antigen and discuss its role in each of these viral and cellular processes.
SV40 as a Model System
SV40 was first isolated as a contaminating virus in rhesus macaque monkey cells used to grow the early versions of the active polio vaccine developed in the late 1950s (95). It was later determined that SV40 induced tumors in test animals such as baby hamsters and mice (95). Tens of millions of vaccine recipients accidentally received vaccine doses with low to high titers of the SV40 virus (reviewed in reference 22). The repercussions from this accident have led to intensive study of SV40, which has since become a model system for understanding neoplastic induction and numerous cellular activities.
More than 30 years after the discovery that SV40 induces tumors in rodents, the debate continues as to whether SV40 is an etiologic agent of human cancer. In its native host species, the rhesus macaque, SV40 forms a persistent infection in the kidneys with no apparent harmful side effects (except in circumstances when the monkeys become immunocompromised due to SIV infection [164, 214]). However, recent PCR data suggest a potential role for SV40 in human mesotheliomas, osteosarcomas, choroid plexus tumors, and other brain tumors (reviewed in references 22 and 26). Furthermore, live SV40 has been cultured from patients with neurological disease (22). Currently, however, there is no consensus as to whether SV40 is a causative agent of human cancer or whether it merely propagates well in malignant tissue. Given the potent transforming activities of SV40 proteins in multiple cellular environments, it is not difficult to envision at least a collaborative role for SV40 in tumorigenesis when it is present in human neoplasias.
SV40 is a member of the Polyomaviridae family of viruses, named after the founding member's ability to induce multiple tumorigenic lesions in newborn rodents. BK virus (BKV) and JC virus (JCV) are human polyomaviruses which form a persistent infection in 90% of the human population early in life (reviewed in reference 105). BKV has been found in human tumors (105), and PCR data suggest that JCV may be linked to neural cancers (23). However, the contribution of these viruses to cancer remains controversial. Nevertheless, it is clear that JCV is the causative agent of progressive multifocal leukoencephalopathy, a neurodegenerative disease which infects immunocompromised individuals, including approximately 5% of all AIDS patients (8). Given the high degree of sequence similarity (approximately 70%) between SV40, JCV, and BKV (65), it is not surprising that these viruses have similar life cycles. Polyomavirus family viruses replicate in differentiated cells and generally form persistent infections in their native hosts (42). Differences among the polyomavirus family members in the non-protein-coding regions are thought to account for at least some of the variations in tissue tropisms (31).
SV40 has a relatively simple genetic architecture which consists of seven gene products organized into a circular DNA genome (Fig. 1A). The late genes VP1, VP2, and VP3 are the structural proteins that form the viral capsid. Agno protein is expressed late in infection and may have a role in capsid assembly and/or release (42). The early genes—which code for large T antigen, small t antigen, and 17kT antigen—share amino acid identity in the amino-terminal 82 amino acids and are encoded by three alternatively spliced products (reviewed in reference 16). These proteins are responsible for numerous functions, including regulation of early and late gene transcription, induction of host cell transcription (to up-regulate necessary enzymes for DNA synthesis), viral DNA replication, and virion assembly (42) (see map of the T antigens [Fig. 1B]).
FIG. 1.

SV40 genomic organization and early gene products. (A) SV40 genomic organization of the alternatively spliced early (large T antigen [LT], small t antigen [St], and 17k T antigen) and late (VP1 to -3) proteins. The early (PE) and late (PL) promoters exist in opposite orientations that flank the SV40 origin (Ori) of replication. (B) T antigen is a large 708-amino-acid multidomain protein. It consists of an amino-terminal J domain (that directly contacts Hsc70), followed by an pRB family binding motif (LXCXE) which binds to all three pRB family members, pRB, p107, and p130; a nuclear localization signal (NLS); a specific DNA binding domain (ori binding); a zinc finger motif (Zn); ATPase domain; a bipartite p53 binding domain that is also essential for mediating interaction with the transcriptional adapter protein p300; and an HR specificity region. 17kT antigen is comprised of the first 131 amino acids of large T antigen (including the J domain [J]) and an pRB-binding (LXCXE) motif, plus an additional four unique amino acids. The amino terminus of small t antigen is comprised of the J domain (amino acids 1 to 82), plus an additional carboxyl-terminal domain that binds to the multimeric protein phosphatase 2A (pp2A) comprised of a catalytic (c) and a regulatory (a) peptide.
SV40 is tropic to nondividing, differentiated cells of the kidney; therefore, it has evolved mechanisms to overcome cell cycle growth arrest in order to propagate its genome. Overcoming the molecular basis of cell cycle arrest allows production of the enzymes necessary to replicate the SV40 genome and the output of progeny virus. A majority of the cell cycle-stimulating activities of SV40 are induced by T antigen (reviewed in reference 170). Consequently, expression of T antigen can lead to the transformation of a cell from a normal to a growth-deregulated state.
Expression of T antigen is sufficient to induce transformation in multiple cell types (for examples, see references 17, 36, 82, and 103). Small t antigen assists in transforming some cell types, and in some situations expression of small t antigen by itself is sufficient to induce a transformed phenotype (162, 180, 181, 192). 17kT antigen has not been studied in depth, but it is known that it is expressed during infection in small amounts relative to the other T antigens (270). It is proposed that 17kT antigen may function in fine-tuning SV40-mediated cell cycle control (270).
Study of T antigen has led to the discovery of important insights in the fields of alternative RNA splicing, nuclear localization of proteins, DNA repair, gene regulation, and tumor suppressor functions. Current research uses T antigen as a model system to understand the process of DNA replication. The advantage to using this system is that only one viral protein (T antigen) is required to initiate DNA replication, which in combination with a wealth of characterized mutants and in vitro assays has led to a better understanding of how eukaryotic DNA replication is initiated (21).
Thus, T antigen is a potent model for understanding cellular functions. A recurring theme in SV40 research is that T antigen serves as a molecular divining rod to point to the essential factors required for cellular processes, serving to navigate the researcher through the immensely complicated interactions of the cellular milieu. T antigen has played an important role in identifying and elucidating the function of tumor suppressor proteins and the following sections deal solely with this issue.
TUMOR SUPPRESSORS
Cancer is a complex, multifactorial disease that occurs when the balance between growth-promoting and growth-inhibitory signals is disrupted (reviewed in reference 84). Tumor suppressors are growth-inhibitory proteins that are often found mutated in human tumors. Two of the better-studied tumor suppressors are retinoblastoma binding protein (pRB) and p53. Both are the founding members of different families of genes related by structure and function. Why are pRB and p53 important in cancer? They are key regulatory components of cellular pathways involved in growth control. E2F can activate proliferation, and p53 inhibits abnormal cellular proliferation (Fig. 2). T antigen and other tumor virus proteins, such as the early proteins of the adenoviruses and papillomaviruses, have served as tools in elucidating the function of both p53 and pRB.
FIG. 2.

T antigen inhibits both the pRB and p53 tumor suppressor pathways. Mitogenic stimulation triggers phosphorylation of pRB by cyclin D/CDK4,6 complexes. This releases pRB-mediated repression of E2F transactivation, thus allowing the synthesis of the enzymes necessary for cell cycle progression and DNA replication. T antigen induces free E2F. p53, “the guardian of the genome,” inhibits cell cycle progression; one way this is accomplished is via p21, which inhibits phosphorylation of pRB. Additionally, p53 induces apoptosis when activated by genotoxic stresses. T antigen inhibits multiple activities of p53. Hsc70 inhibits apoptosis and may play an additional role in regulating the activities of both pRB and p53. See the text for more details.
pRB
pRB was first identified as a tumor suppressor in which homozygous null mutations in humans induce a tumor in the eye (64, 67, 133). At about the time the genetic cause of the retinoblastoma tumor was identified, it was also shown that the adenovirus transforming protein E1A complexes pRB (260). Subsequently, it was shown that T antigen also forms a stable complex with pRB (48, 53). Furthermore, some mutants of T antigen that are defective for the ability to induce transformation also fail to bind to pRB (111, 195, 223, 273). The gene encoding pRB is found mutated in many cancers, and it is likely that some component of the pRB pathway (Fig. 2) is mutated in all cancers (212).
In normal cells, pRB is negatively regulated by phosphorylation, which occurs in a cell cycle-dependent manner (254). This may be an oversimplification, because some level of phosphorylation is required to activate pRB function, with additional phosphorylation inhibiting pRB function (57). Thus, cyclin/cyclin dependent kinase (CDK) complexes negatively regulate pRB function and are, in turn, themselves negatively regulated by cell cycle inhibitors such as p16 (Fig. 2) (211). Therefore, cancers possess genetic mutations in many parts of the pRB pathway, including pRB itself and CDK inhibitors or overexpression of cyclins and E2Fs (212).
pRB is part of a family (pRB, p130, and p107) that act as transcriptional repressors but do not interact with specific DNA target sequences. The individual pRB family members share a high degree of sequence similarity in conserved regions, for example the A and B domains (Fig. 3.). The A and B domains are essential for repression of certain transcription factors (see below) (189, 206). pRB consists of multiple domains, some of which bind directly to specific transcription factors while others recruit histone deacetylases (HDACs) (reviewed in references 52, 86, 146, and 163). pRB represses transcription directly through repression domains and indirectly by recruiting HDACs and chromatin-rearranging complexes (86). An important way pRB recruits HDAC activity is through binding to RBP1 (126). In turn, RBP1 also possesses a transcriptional repression activity independent of recruiting HDAC activity (Fig. 4).
FIG. 3.

pRB binding to T antigen and chaperones. (A) The crystal structure of pRB bound to an E7 peptide (from papillomavirus) containing an LXCXE motif (PDB code 1Gux [131]). Note that pRB binds to LXCXE entirely through its B domain, even though the A domain is required for efficient complex formation with LXCXE motif-containing proteins. For comparison, the conserved amino acids of the T antigen and E7 RB binding (LXCXE) motifs are shown in black. (B) The crystal structure of pRB bound to the first 117 amino acids of T antigen (PDB code 1GH6 [122]). Notice the LXCXE motif of T antigen binds to pRB in a manner similar to the LXCXE E7 peptide. Additionally, the J domain of T antigen is depicted as four multicolored helices: helix 1 (yellow), helix 2 (dark blue), the highly conserved HPD loop in red connecting helices 2 and 3, helix 3 (green), and helix 4 (light green). (C) Domain map demonstrating that the A and B domains of pRB are highly conserved among the other pRB family members, p130 and p107. The essential regions of pRB required for various activities, such as binding to Hsp70 or T antigen, are diagrammed with black lines corresponding to particular regions of pRB (35, 106, 131).
FIG. 4.

pRB repression of the E2F transcription factors. In growth-arrested cells the pRB family of proteins (pRB, p107, and p130) can mediate transcriptional repression in at least two ways, via direct repression domains and by recruiting the activity of the protein RBP1 which directly represses transcription and indirectly represses transcription by recruiting HDAC (126). In dividing cells, pRB family members are no longer bound to E2F, thus allowing for transcriptional activation of promoters containing E2F binding sites.
Proteins of the pRB family are structurally similar and, not surprisingly, have partially overlapping functions (40, 239, 257). However, there are several key differences between the pRB family members; for example, each is active in different parts of the cell cycle to various degrees (52). Furthermore, unlike pRB, both p130 and p107 are not commonly found mutated in human tumors. Knockouts of the mouse gene encoding pRB are lethal (39, 107, 130), but in some genetic backgrounds p130 or p107 gene knockouts are viable (41, 132), demonstrating biological roles for pRB p107 or p130. However, double knockouts of both p107 and p130 genes are perinatal lethal in mice, suggesting some degree of functional compensation between p107 and p130 (41). More recently, Classon et al. have shown that pRB promotes, but p107 antagonizes, adipocyte differentiation in vitro (40), demonstrating the differing roles of the pRB family in cell fate determination. Despite such functional differences between pRB, p107, and p130, the underlying fundamental biochemical mechanism of their transcriptional repression (Fig. 4) appears to be similar (86).
pRB family members interact with multiple transcription factors (for a review, see reference 146) such as MyoD (79), Pax-3 (262), c-Abl (256), and CDP/cut (248, 253). However, the best understood, and perhaps the transcription factor most important for RB's growth-suppressive functions, is E2F (Fig. 1). E2F refers to any member of a family of transcription factors (presently there are six members; E2F-1 to -6) that are related by conserved regions of sequence similarity, as well as by the ability to bind to the same consensus DNA binding site (52).
The exact functions of the individual E2F family members are currently unclear, and this topic is the focus of ongoing research. Some E2F family members have overlapping functions in the induction of S phase, promoting expression of genes such as cyclins E and A, cdc2, CDK2, and enzymes essential for DNA synthesis such as DNA polymerase α and thymidine kinase (52). However, in certain contexts, such as when bound by pRB family members, the role of E2F is not to activate transcription, but rather the pRB-E2F complex serves to repress transcription (Fig. 4). Thus, in theory, the same E2F peptide can be a growth-promoting or growth-inhibitory agent, depending on its binding partners. Recently, it has been proposed that pRB-E2F complexes may bind near origins of DNA replication and thus play a direct inhibitory role in DNA replication (120) as well as an indirect role by preventing synthesis of the enzymes necessary to replicate DNA and drive cells to cycle.
All known E2Fs function by associating with a heterodimeric DNA binding partner called DP. There are two known DPs, and both display sequence similarity to each other as well as to the E2F family members (52). It is thought that both DP1 and DP2 can bind to all six E2Fs, enhancing their DNA binding function (271, 272).
The E2F family is commonly grouped into three subclasses based on functional and structural similarities. One class includes E2F-1 to -3, which encode their own nuclear localization signal and most frequently bind pRB (52). E2F-1, -2, and -3 are commonly thought to activate cellular division. In support of this notion, E2F-1 knockout mouse embryo fibroblasts (MEFs) are defective for exit from G0 (252), and E2F-3 is required for cell proliferation induced by loss of pRB (276). A second class includes E2F-4 and E2F-5, which do not contain a nuclear localization signal and are most commonly found associated with p130 and p107. E2F-4 and E2F-5 are not required for exit from G0 in MEFs, and unlike E2F-1 to -3, at least in some cell types, their role is not to activate cellular division but rather to mediate cell cycle inhibitory signals transduced by p16 (71). The third class includes E2F-6, which does not bind to pRB family members and lacks a transcriptional activation domain. Therefore, it has been proposed to play a predominantly inhibitory role in transcription (27, 72, 245).
The fact that the pRB and E2F families are the focus of intensive study is a testament to their essential function in cellular growth control. Their central role in the molecular mechanisms underlying human cancer have led them to be a focal point for genetic and drug therapeutic approaches (37, 93). The depth of our present understanding of these proteins is just a fraction of what is to come; however, much of what we do know has been enhanced either directly or indirectly by the study of DNA tumor viruses.
p53
p53 is one of the most-studied molecules, with over 21,000 reports found when “p53” is the subject of a Medline search on the Internet. As with pRB, the scope of the data encompassing p53 is enormous, and many reviews are available (for example see volume 18, issue 53, of Oncogene [1999]). Therefore, this review discusses only the very rudimentary aspects of p53 biology.
As is the case with the pRB family, the field of p53 research finds its roots in the study of DNA tumor viruses. p53 was initially identified as a coprecipitating protein in immunoprecipitation assays of T antigen (30, 125, 127, 145).
p53 is a specific transcription factor, referred to as the “guardian of the genome,” whose function can prevent DNA synthesis, cause G2 and G1 growth arrest, and induce apoptosis (reviewed in reference 135). The transcriptional adapter protein referred to as p300 (also known as CREB-binding protein [CBP]) is sometimes found associated with p53, and it is thought to assist in numerous cell cycle regulatory functions, including those of p53 (see below and references 2, 80, 142, and 217). p53 is ubiquitously expressed but is not stable; however, upon genotoxic stresses such as irradiation, chemotoxin exposure, or virus-induced unscheduled DNA synthesis, p53 steady-state levels increase. An increase in p53 levels leads to a cascade of events including the transcriptional activation of the CDK/cyclin kinase inhibitor p21 and the ubiquitin E3 ligase shuttling protein MDM2 (reviewed in reference 157). p21 inhibits the cell cycle-promoting functions of the CDKs (156), and MDM2 down-regulates p53 function, inducing the degradation of p53 (Fig. 2) (157). Thus, down-regulation of p53 function by MDM2 provides a feedback loop mechanism that allows the restoration of normal cell function when the genotoxic stress is diminished.
Recently several p53-related proteins have been identified, such as p63 and p73 (reviewed in reference 150). These proteins share regions of sequence similarity with p53 and can bind to the same consensus DNA binding site. Several different proteins are encoded by alternatively spliced gene products of p63 and p73. The exact biological functions of these polypeptides are still to be defined. Some p53 family members have functions similar to, and overlapping with, those of p53, including transactivation, while other members are antagonistic to p53 function (150).
Given the central roles of pRB and p53 in cellular growth control, it is not surprising that both pathways cross talk with numerous cellular signaling pathways such as Ras, MYC, and Egf (13, 136, 215). Shown in Fig. 2 is a bare bones map of the pRB and p53 signaling pathways. Note that each pathway “communicates” with the other; for example, E2F induces S phase but also stimulates p53 activity, which can inhibit cellular division. The overlapping nature of these pathways is an important issue to keep in mind when addressing the transforming activites of T antigen (see sections below).
p300
Proteins encoded by members of both the adenovirus and polyomavirus families associate with p300. p300 and the related protein CBP are transcriptional adapter (or coactivator) proteins that coordinate the formation of specific transcription factor complexes (reviewed in reference 227). p300 and CBP can enhance the transcriptional activity of many transactivators including p53. This occurs by direct interaction of p300 or CBP with the transcription factor or alternatively by modulating the chromatin structure through recruitment of histone acetyltransferase activity (reviewed in reference 98). The adenovirus E1A protein associates directly with p300 and CBP, requiring its amino-terminal conserved region 1 (CR1) motif (55, 226, 261). The p300 and CBP binding site for SV40 T antigen has been mapped to both the amino-terminal and carboxyl-terminal domains of T antigen (54, 143). Currently, it is unclear if this binding is direct, but expression of T antigen inactivates p300 transcriptional activation and changes the phosphorylation state of p300 (1, 54). One consequence of inactivation of p300 by tumor viruses is the inhibition of p53 transactivation activity, thus further fostering a cellular environment conducive to viral replication (98).
T-Antigen Interactions with pRB and p53
As noted in the previous sections, T antigen binds to both pRB and p53 in a stable manner (48, 121). The regions of T antigen required for these binding activities are mapped in Fig. 1. A bipartite domain in the carboxyl terminus of T antigen, from amino acids 351 to 450 and amino acids 533 to 650, is required for binding to p53 (121). T antigen does not bind to the p53 family member p63 or p73 (94, 190). The LXCXE motif at amino acids 103 to 107 is required for stable association with all three members of the pRB family of proteins and is commonly found in many cellular pRB-binding proteins. The crystal structure of the highly conserved pRB A and B domains bound to an LXCXE peptide has been solved (Fig. 3A) (131). The conserved residues of the LXCXE motif (L, C, and E [black] in Fig. 3A) are buried in a pocket that is entirely contained in the B domain (depicted in red in Fig. 3A). Note: even though E2F binds to this region of pRB, E2F does not contain an LXCXE motif (91) and can coexist in a complex of pRB bound to LXCXE-containing proteins (46, 233).
Until recently, the mechanism of how T antigen disrupts the function of pRB and p53 was thought to be analogous to that of an absorbent sponge; T antigen binds to the tumor suppressor protein and “soaks up” the available pools of pRB and p53. This hypothesis has been referred to in the literature as a “sequestration model.” It is now understood that the mechanisms by which T antigen inhibits pRB and p53 function are more complicated. For example, T antigen down-regulates p53 function, but at least part of this activity does not require the p53 binding domain of T antigen. In fact, an amino-terminal fragment of T antigen that lacks the p53 binding domain can still inhibit p53 function in some assays (74, 185, 194). This argues that sequestration alone cannot account for the effects of T antigen on p53. In addition to the pRB binding motif, T antigen requires a functional chaperone J domain to down-regulate some activities of the pRB family members (16, 47). Additionally, a transforming activity(ies) in the carboxyl terminus of T antigen also requires the function of the J domain (223). Several lines of new evidence demonstrate that T antigen is not a static sponge, but rather a dynamic machine, with many of its activities depending on its chaperone function. The rest of this review focuses on the role of the chaperone functions of T antigen in SV40 biology.
WHAT IS A CHAPERONE?
Chaperone proteins promote the proper folding of proteins and prevent protein aggregation during periods of cellular stress (90). Historically, some of the first chaperones identified were the heat shock class of proteins identified by the work of Polissi et al. as proteins that are required for the ability of phage λ to infect Escherichia coli (179). This work identified two structurally unrelated classes of chaperones: the chaperonins (including the Hsp60 family of proteins) and the DnaK/DnaJ families of cochaperones. Both families are involved in promoting the proper folding of protein substrates and are especially important under conditions of cellular stress, such as exposure to extreme temperatures or chemotoxic agents that may lead to denaturation of protein tertiary structure (20). Homologues of both the chaperonins and the DnaK/DnaJ families of chaperones are found in a broad range of species, including all known eukaryotes (186).
Another class of chaperones is that of the Hsp90-related proteins. Hsp90-like proteins are found in prokaryotes and eukaryotes and are involved in activating specific protein signaling molecules, including kinases and steroid hormone binding receptors (19). Hsp90 proteins are the most abundant cytosolic chaperones and are involved in the general stress response functioning to assist in proper protein folding and prevention of aggregation. Perhaps not surprisingly, Hsp90s interact at multiple levels with the Hsp70 chaperone machine (19, 66). For the purposes of this review, discussion is limited to the DnaK/DnaJ families of proteins and their homologues. (For in-depth reviews of the different classes of chaperones see references 19, 20, and 90).
DnaK/Hsc70 Families of Proteins
DnaK is a member of the Hsc70 family of chaperones. All Hsc70 family members have a conserved domain structure consisting of a large amino-terminal ATPase domain, a peptide (substrate) binding domain, and an extreme carboxyl-terminal region that is sometimes designated as the variable or “lid” domain (Fig. 5D) (12, 20). Hsc70 binds to polypeptide substrates, usually with hydrophobic or basic side chains of at least 7 amino acids (10, 62, 75, 191). Hsc70 hydrolyzes ATP, which induces changes in the conformation of Hsc70, which in turn is transmitted to substrates bound by Hsc70. Through this mechanism, Hsc70 performs various kinds of work, including protein membrane transport, prevention of aggregation of denatured proteins, refolding of denatured proteins, and disruption of multiprotein complexes such as the replication machinery of phage λ (14, 90, 179). Thus, Hsc70 and its homologues can be thought of as molecular motors that, when present in the proper cellular context, are able to drive a multitude of different tasks.
FIG. 5.

Domain structure of Hsc70. (A) The peptide binding domain of the E. coli Hsc70 protein DnaK (PDB code 1DKZ [275]). The substrate binding domain is shown in purple, the lid (or “variable”) domain is shown in blue, and the bound peptide substrate is shown in yellow. (B) NMR structure of the J domain of E. coli DnaJ (PDB code 1BQZ [100]). The D35 residue is modeled with space filling in red to highlight its important role in directly contacting the ATPase domain of DnaK (see the text for more details). (C) The crystal structure of the ATPase domain of DnaK is shown as four alpha-helical regions in purple (PDB code 1DKG [89]). Residue R167 is modeled with red space filling to underscore the importance of the surrounding region in directly binding to residue D35 of the DnaJ J domain. (D) Domain map of Hsc70, the ATPase (amino acids 1 to 386), and the substrate binding (P [for peptide]) domains are shown in purple. The lid (L) domain is shown in blue.
Hsc70 by itself has only a weak intrinsic ATPase activity. In the presence of cochaperones and peptide substrates, the ATPase activity of Hsc70 increases dramatically (113, 155). It has been posited that this dual mechanism of stimulation required for maximal ATPase activity requires both a cochaperone and peptide substrate to prevent wasteful unproductive “misfirings” of the Hsc70 ATPase cycle (117).
Crystallographic studies show that the DnaK ATPase domain consists of four alpha-helical domains (Fig. 5C) (89). The crystal structure for the carboxyl-terminal substrate binding and lid domain has also been solved for DnaK (275). This structure, shown in Fig. 5A, reveals that the first half (depicted in purple in Fig. 5A) has a β-sandwich structure that forms the channel which interacts with peptide substrates (depicted in yellow), followed by an alpha-helical region (depicted in blue) that closes over the channel. This alpha-helical region, therefore, has been proposed to be a lid that, when in the appropriate conformation, traps bound substrates in the β-channel (275). The crystal structures for mammalian Hsc70 ATPase domain (60, 225) and the nuclear magnetic resonance (NMR) structure for the substrate binding domain (159) reveal extensive similarities with the prokaryotic DnaK, suggesting a conservation of the mechanism of function between diverse species.
The ATP-bound form of Hsc70 is in a state of rapid flux between binding and release of substrate (61, 62, 152, 172, 176, 201, 243). Structural evidence suggests that this is accomplished through the action of the extreme carboxyl-terminal lid domain of Hsc70, which clamps shut or open, trapping and untrapping substrates bound by the substrate binding domain in response to completion of the ATPase cycle (Fig. 5A and 6) (275). Upon J-protein stimulation of Hsc70-mediated ATP hydrolysis, a global conformational change takes place in Hsc70 (5, 6, 18, 112, 140), causing the lid domain to clamp shut, thus trapping the substrate in a bound conformation (Fig. 5A and 6).
FIG. 6.

The ATPase cycle of Hsc70. When bound to ADP, Hsc70 has a high substrate affinity; conversely when bound to ATP, Hsc70 displays a weak affinity for peptide substrates. BAG-1 and GrpE are nucleotide exchange factors that promote the exchange of ATP for ADP, increasing the steady-state ATPase activity of Hsc70. J domain-containing proteins (J proteins) stimulate the ATPase activity of Hsc70, which is inhibited by CHIP. Hip promotes the stabilization of the ADP-bound form of Hsc70.
DnaJ and its homologous proteins (J proteins) are cochaperone regulators that stimulate the ATPase activity of Hsc70 and promote substrate interactions with Hsc70s (reviewed in references 25, 34, 45, 116, 117, and 213). Additionally, J proteins have been implicated in altering the phosphorylation state and inducing the degradation of substrates (229, 266). J proteins have been implicated as components of the Hsp90 chaperone machine (34, 124, 204). All J proteins contain a domain of approximately 70 amino acids that directly binds to Hsc70, known as the J domain. NMR and crystal structural analysis of the E. coli DnaJ, human HDJ1, polyomavirus large-T-antigen, and SV40 T-antigen J domains reveals that the structure of a J domain is composed of three or four alpha-helices in which helices II and III form an antiparallel finger-like projection held together by extensive hydrophobic interactions (Fig. 7A) (9, 100, 108, 122). Helices II and III (depicted in blue and green, respectively, in Fig. 7A) are linked by a solvent-exposed loop consisting of amino acids HPD (depicted in red in Fig. 7A and B). The HPD tripeptide motif directly contacts Hsc70 and is universally conserved in all J proteins.
FIG. 7.

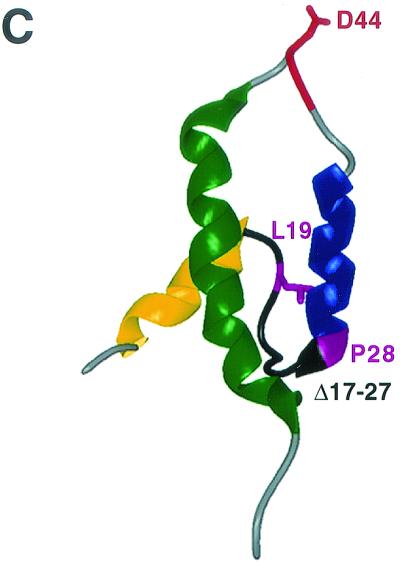
J domain structure. (A) NMR structures of the J domains of E. coli DnaJ (PDB code 1BQZ [100]), human HDJ1 (PDB code 1HDJ [184]), polyomavirus T antigen (PYV) (PDB code 1Faf [9]) and crystal structure of SV40 T antigen (SV40) (PDB code 1GH6 [122]). Alpha-helix I is shown in yellow, alpha-helix II is shown in blue, alpha-helix III is shown in dark green, and alpha-helix IV (DnaJ and HDJ1 only) is shown in light green. The absolutely conserved HPD tripeptide comprising the loop between helices II and II is shown in red. (B) Amino acid alignment of DnaJ, HDJ1, PYV, and SV40 is shown. The amino acids that make up the particular helices are indicated by colored boxes. The absolutely conserved HPD tripeptide is shown in red. (C) Amino acids of key SV40 T antigen mutants. Alpha-helix 4 of the SV40 J domain is omitted to better show the location of three distinct mutants of SV40 T antigen, representing three different phenotypes (Table 1). The locations of the D44N point mutant and the L19F,P28S double point mutant are indicated by highlighting these residues and their side chains in cyan. The region that corresponds to the small deletion mutant, Δ17-27, is shown in black.
There are three broad classes of J proteins (34). (i) Type 1 proteins are those that contain a J domain, a glycine-phenylalanine-rich region, and a zinc finger-like domain, for example, E. coli DnaJ and Saccharomyces cerevisiae Ydj1p. (ii) Type 2 proteins are those that contain a J domain and a zinc finger-like domain, for example, human HDJ1 and E. coli CbpA. (iii) Type 3 proteins are those that contain the J domain with neither a glycine-phenylalanine-rich region nor a zinc finger-like domain, such as T antigen, yeast Sec63, and mammalian P58IPK. Within this subset, the J domain can be found at the amino terminus, in the middle, or at the carboxyl terminus of the protein. The functions of the glycine-phenylalanine and zinc finger-like regions are not known, but it is possible that they facilitate interaction with Hsc70 (34).
J proteins may regulate Hsc70 function by binding to substrates of Hsc70 and recruiting and/or stabilizing the substrates into a complex with Hsc70 (116). Tertiary complex formation between Hsc70, a peptide substrate, and a J protein induces maximal stimulation of the ATPase activity of Hsc70 (113, 129, 155). Hsc70-mediated hydrolysis of ATP “powers” the desired “work” being performed, by changing the conformation of the bound substrate and Hsc70 itself. Mutational analysis has revealed that the J domain of J proteins is essential for the induction of increased ATPase stimulation by Hsc70 (223, 247, 249). Structure and function as well as NMR perturbation analysis demonstrated that the J domain directly contacts the ATPase domain of Hsc70 through alpha-helix 2 and the HPD motif (70, 77, 231). J proteins may also contact the carboxyl-terminal region of Hsc70 since mutations of Hsc70 in the substrate binding or the EEVD motif of the lid domain impair productive DnaJ/Hsc70 interactions in various species (63, 113, 232).
One can visualize this basic Hsc70 ATPase mechanism (Fig. 6) accounting for multiple Hsc70 activities. For example, transport of newly synthesized proteins into the lumen of the ER requires a luminal Hsc70 homologue, BiP (153). Various models depict BiP as either anchoring peptides that pass through the membrane pore via Brownian motion, or alternatively, BiP may act as a motor that actively pulls peptides through the pore (14). Either model requires the binding and release of peptide substrates by BiP. Another example of the versatility of the Hsc70 ATPase cycle is the disassembly of λO-λP-DnaB multiprotein complex at the origin of phage λ replication (73, 139). Phage λ uses the E. coli host DnaK and DnaJ proteins to assemble and disassemble the components of the replication machinery necessary for phage DNA replication. Thus, the role of Hsc70 in multiple contexts is to use the force generated by ATP hydrolysis to bind, alter, and then release the substrate so that Hsc70 is recycled to perform additional functions. Clearly, disparate results can occur by the same basic Hsc70 chaperone mechanism—depending on the context in which the Hsc70 is found.
There are several modulators of Hsc70 function that regulate different parts of the Hsc70 ATPase cycle (Fig. 6). For example, in prokaryotes there is a nucleotide exchange factor referred to as GrpE that promotes release of ADP and binding of ATP by DnaK (171). The result is a GrpE-induced enhancement of the steady-state ATPase activity of Hsc70. Thus far, except in the mitochondria and chloroplasts, no structural homologues to GrpE have been identified in eukaryotes. However, functional homologues that foster the nucleotide exchange of Hsc70 from the ADP-bound to the ATP-bound form have been found. These include isoforms of the Bag-1 protein. Like GrpE, Bag-1 binds to Hsc70 and stimulates the exchange of ADP for ATP, thus enhancing the steady-state ATPase activity of Hsc70 (96, 228, 240); however, Bag-1 also serves to negatively regulate some Hsc70 functions in cultured cells (166). Additionally, the multiple isoforms of Bag-1 are involved in different elements of hormone receptor regulation (81).
Several regulators of Hsc70 function contain tetratricopeptide motifs that (in part) foster binding to Hsc70. These include Hip, CHIP, and HOP (4, 49, 66). Hip stabilizes Hsc70 into the ADP-bound high substrate affinity form and facilitates the activation of hormone receptors (97, 183). CHIP performs the opposite function by promoting the stabilization of the low substrate affinity ATP-bound form of Hsc70 (4). Additionally, CHIP targets some proteins for proteasome-mediated degradation (reviewed in reference 154). HOP is a bridging molecule that fosters association of Hsc70 with Hsp90 (205, 216). The roles of some of the regulators of Hsc70 function are depicted in an Hsc70 ATPase cycle mechanistic model in Fig. 6. Note that these added points of regulation can allow for a subtle fine-tuning of the Hsc70 chaperone motor machine.
There are many DnaJ and Hsc70 homologues in the cell. In yeast there are more than 14 different Hsc70-like or DnaJ-like proteins, some of which are localized to the same cellular compartment (186). How then does a particular Hsc70 mediate interaction with its proper binding J-protein partner? Experiments in yeast have demonstrated that there is specificity to the interaction between DnaJ-like proteins and Hsc70 family members. The endoplasmic reticulum (ER) luminal DnaK homologue BiP but not Ssa1p (a cytosolic Hsc70) can associate with the J domain of the ER luminal chaperone Sec63p (153). Ydj1p, a cytosolic J protein, stimulates the ATPase activity of Ssa1p by 10-fold, but does so only 2-fold for BiP (153). When the J domain of the ER luminal chaperone Sec63p was replaced with the J domain of either Sis1p or Mdj1p, these cytosolic and mitochondrial J domains could not substitute for the J domain of Sec63p. However, changing only three amino acids in Sis1p restored function, presumably by fostering interaction with an ER luminal DnaJ homologue (200). Other experiments indicate that the J domains from E. coli DnaJ and other nonmitochondrial J proteins can substitute (to various degrees) for the J domain of the mitochondrial luminal J protein Mdj1p when their expression is targeted to the mitochondria (147). DnaJ restored wild-type function; Xdj1p, Ydj1p, and Sis1p restored function to an intermediate level; and Scj1p lacked the ability to restore viability and respiration. These results suggest that while the basic mechanism of Hsc70-J-protein function is conserved in the various orthologues, elements within and surrounding the J domain contribute the specificity of interaction of particular chaperone partners. Similar approaches, when applied to other Hsc70/DnaJ-like proteins, may provide an understanding of the determinants of specificity in chaperone interactions.
T Antigen Is a J-Protein Chaperone
Many different type of viruses, including bacteriophages, utilize molecular chaperones for some part of their viral life cycle (reviewed in reference 235). Some viruses such as bacteriophage λ utilize only host-encoded chaperones, while others, like SV40, encode virus-specific chaperones. Multiple lines of evidence confirm that SV40 T antigen is a functional molecular chaperone J protein.
First, there is sequence similarity between the amino-terminal region of the SV40 T antigens and the conserved residues of the type 3 DnaJ-like proteins including the absolutely conserved HPD loop of the J domain (Fig. 7) (33; W. L. Kelley and S. J. Landry, Letter, Trends Biochem. Sci. 19:277-278, 1994). Recently, the crystal structure of SV40 T antigen bound to the A and B domains of the pRB protein has been reported (122). SV40 T antigen shares 44% identity in the J-domain region with polyomavirus T antigen, and the NMR structural determination of the first 79 amino acids of polyomavirus T antigen demonstrates that it bears extreme similarity to the T-antigen J domain (Fig. 7A) (9). Consistent with the NMR structures of DnaJ, HDJ1, and polyomavirus T antigen, the SV40 T antigen is composed of a finger-like projection of two antiparallel helices (Fig. 3). Like the polyomavirus T antigen, the first alpha-helix of SV40 T antigen is longer than the nonpolyomavirus J domains, suggesting the possibility of additional functions in the extreme amino-terminal region. Interestingly, alpha-helix 4 curls back in the opposite direction of other known nonpolyomavirus J domains and makes contacts with alpha-helix 2 and the HPD loop. The extended long loop connecting alpha-helices 3 and 4 is also novel to T antigen. Not surprisingly, the conserved residues of the LXCXE motif of T antigen directly contact the B domain of pRB in a manner analogous to the papillomavirus E7 peptide (Fig. 3). Interestingly, helices 3 and 4 of T antigen make additional hydrogen bond contacts with pRB. Because regions of T antigen that contact Hsc70 (HPD motif) are proximal to the regions of T antigen that bind to pRB, this structure supports the model that T antigen serves as a bridge to direct the action of Hsc70 to RB-E2F complexes (122).
Second, functional studies involving domain-swapping experiments show that the J domain of T antigen can functionally substitute for the J domains of E. coli DnaJ in a phage λ growth assay (118) and for Ydj1p in a yeast viability assay (S. Fewell and J. L. Brodsky, unpublished observation). Mutation in the amino-terminal region of T antigen in residues conserved with other J proteins renders T antigen defective for SV40 replication, transformation, and assembly. These mutants fail to substitute for the DnaJ J domain in the E. coli complementation assay (J. V. Vartikar and W. L. Kelley, unpublished observations). Furthermore, the J domain of human J proteins Hsj1 and DnaJ2 can functionally substitute for the T-antigen J domain in DNA replication, albeit DnaJ2 is less efficient than Hsj1 in this activity (24).
Third, biochemical evidence confirms that T antigen functions as a J domain in multiple assays, including stimulating the ATPase activity of bovine Hsc70 and Ssa1p (cytosolic yeast Hsc70) (223), promoting the release of a bound substrate from Ssa1p (223), and binding to Hsc70 (24, 198, 209, 234, 236). Thus, structural, functional, and biochemical assays demonstrate with clarity that T antigen contains a functional J domain.
FUNCTIONS OF T-ANTIGEN CHAPERONE ACTIVITY
Mutational analysis has demonstrated that the T-antigen J domain is essential for multiple viral activities, including viral DNA replication, transformation, transcriptional activation, and virion assembly (Table 1). Each of these topics is discussed in detail below.
TABLE 1.
Large-T-antigen J-domain mutantsa
| Mutant type | Mutant name | Ability to transformb
|
Growth
|
Induction of:
|
Ability to:
|
||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| REF52 | 10T1/2 | REF/CREF | Soft agarc | Advant. MEFsd | Low serumd | Choroid plexuse | Lymphomae | Hepatocarcinomaf | Bind pRBg | Over. pRB G1 arresth | Alter p130i | Disrupt p130/E2Fj | Transactivate E2Fk | Incorp. BrdUl | Enhance Tst1m | Override p53n | Replicate in cello | Replicate in vitroo | Bind Hsc70p | Stimulate 70 ATPaseq | Bacterial/viabilityr | ||
| SV40 | WT | + | + | + | + | + | + | + | + | + | + | + | + | + | + | + | + | + | + | + | + | + | + |
| H42Q | ND | ND | ND | + | − | − | ND | ND | ND | + | − | − | − | − | ND | ND | + | ND | ND | − | ND | − | |
| D44N | +/− | + | ND | + | − | − | ND | ND | ND | + | − | − | − | − | ND | − | ND | − | + | − | +/− | − | |
| L17K | ND | ND | ND | ND | ND | ND | ND | ND | ND | + | ND | − | ND | ND | ND | ND | ND | ND | ND | ND | ND | ND | |
| L19F,P28S | +/− | +/− | ND | ND | ND | ND | + | + | ND | ND | ND | ND | ND | ND | ND | ND | ND | +/− | ND | ND | ND | − | |
| P43F | ND | ND | ND | ND | ND | ND | ND | ND | ND | ND | ND | − | ND | ND | ND | ND | ND | − | ND | − | ND | ND | |
| P43F,T57 | ND | ND | ND | ND | ND | ND | ND | ND | ND | ND | ND | − | ND | ND | ND | ND | ND | − | ND | − | ND | ND | |
| K45Q | ND | ND | ND | ND | ND | ND | ND | ND | ND | ND | ND | − | ND | ND | ND | ND | ND | − | ND | − | ND | ND | |
| G47E | ND | ND | ND | ND | ND | ND | ND | ND | ND | ND | ND | ND | ND | ND | ND | ND | ND | − | ND | − | ND | ND | |
| K67P | ND | ND | ND | ND | ND | ND | ND | ND | ND | ND | ND | ND | ND | ND | ND | − | ND | ND | ND | ND | ND | ND | |
| pRB mutants | E107KK1 | ND | ND | ND | − | − | − | ND | ND | − | − | − | − | − | − | ND | ND | − | + | ND | + | ND | + |
| E107,108K3213 | − | − | ND | ND | ND | ND | ND | ND | ND | − | ND | ND | − | ND | ND | + | ND | − | + | + | ND | + | |
| Deletions | Δ2-82 | ND | ND | ND | − | ND | ND | ND | ND | ND | + | ND | − | ND | ND | ND | ND | +/− | − | + | − | − | ND |
| Δ17-27 | − | − | − | ND | ND | ND | − | + | − | + | ND | ND | ND | ND | ND | − | − | − | + | ND | ND | − | |
| Δ46-74 | ND | ND | ND | ND | ND | ND | ND | ND | ND | + | ND | − | ND | ND | ND | ND | ND | ND | ND | ND | ND | ND | |
| Δsb59-64 | ND | ND | − | ND | ND | ND | ND | ND | ND | + | ND | − | ND | ND | ND | ND | ND | − | ND | ND | ND | ND | |
| Δ65-70 | ND | ND | +/− | ND | ND | ND | ND | ND | ND | + | ND | + | ND | ND | ND | ND | ND | + | ND | ND | ND | ND | |
| N fragments | N121 | − | +/− | ND | ND | ND | ND | + | − | + | + | ND | ND | ND | ND | ND | ND | +/− | ND | ND | ND | ND | + |
| N136 | − | +/− | ND | ND | ND | ND | ND | ND | ND | + | ND | ND | ND | ND | ND | ND | ND | − | − | − | + | + | |
| N147 | ND | +/− | ND | + | ND | ND | ND | ND | ND | ND | ND | + | ND | ND | ND | ND | ND | ND | ND | ND | ND | + | |
| N136/D44N | − | − | ND | ND | ND | ND | ND | ND | ND | ND | ND | ND | ND | ND | ND | ND | ND | ND | ND | − | − | − | |
| pRB mutants | N136/K | − | − | ND | ND | ND | ND | ND | ND | ND | − | ND | ND | ND | ND | ND | ND | ND | ND | ND | − | ND | + |
| N136/3213 | − | − | ND | ND | ND | ND | ND | ND | ND | − | ND | ND | ND | ND | ND | ND | ND | ND | ND | − | ND | + | |
| Chimeras | DnaJ2-T | ND | ND | ND | ND | ND | ND | ND | ND | ND | + | ND | + | ND | ND | ND | ND | ND | +/− | ND | + | ND | ND |
| DnaJ2-T HQ | ND | ND | ND | ND | ND | ND | ND | ND | ND | + | ND | − | ND | ND | ND | ND | ND | − | ND | − | ND | ND | |
| Hsj1-T | ND | ND | ND | + | +/− | + | ND | ND | ND | + | + | + | + | + | ND | +/− | ND | + | ND | + | ND | ND | |
| Hsj1-T HQ | ND | ND | ND | + | − | − | ND | ND | ND | + | − | − | − | − | ND | − | ND | − | ND | − | ND | ND | |
| SV-JCV | + | ND | ND | ND | ND | ND | ND | ND | ND | ND | ND | ND | + | ND | ND | ND | ND | + | ND | ND | ND | ND | |
| SV-Ydj | − | ND | ND | ND | ND | ND | ND | ND | ND | ND | ND | ND | − | ND | ND | ND | ND | − | ND | ND | ND | ND | |
| SV-DnaJ | − | ND | ND | ND | ND | ND | ND | ND | ND | ND | ND | ND | − | ND | ND | ND | ND | − | ND | + | +/− | ND | |
| Polyomavirus | WT | ND | ND | ND | ND | ND | ND | ND | ND | ND | + | ND | ND | ND | + | + | ND | ND | + | ND | + | ND | + |
| V4D | ND | ND | ND | ND | ND | ND | ND | ND | ND | ND | ND | ND | ND | ND | ND | ND | ND | ND | ND | ND | ND | +/− | |
| L13V | ND | ND | ND | ND | ND | ND | ND | ND | ND | ND | ND | ND | ND | − | ND | ND | ND | +/− | ND | ND | ND | − | |
| 32E | ND | ND | ND | ND | ND | ND | ND | ND | ND | ND | ND | ND | ND | ND | ND | ND | ND | ND | ND | ND | ND | − | |
| A33S | ND | ND | ND | ND | ND | ND | ND | ND | ND | ND | ND | ND | ND | − | ND | ND | ND | ND | ND | ND | ND | ND | |
| Y34N | ND | ND | ND | ND | ND | ND | ND | ND | ND | ND | ND | ND | ND | − | ND | ND | ND | ND | ND | ND | ND | ND | |
| H42 | ND | ND | ND | ND | ND | ND | ND | ND | ND | ND | ND | ND | ND | − | ND | ND | ND | ND | ND | ND | ND | ND | |
| P43S | ND | ND | ND | ND | ND | ND | ND | ND | ND | + | ND | ND | ND | − | − | ND | ND | ND | − | − | ND | ND | |
| 44N | ND | ND | ND | ND | ND | ND | ND | ND | ND | ND | ND | ND | ND | − | ND | ND | ND | ND | ND | ND | ND | ND | |
| RB mutant | 142VE146Q | ND | ND | ND | ND | ND | ND | ND | ND | ND | − | ND | ND | ND | − | − | ND | ND | ND | ND | + | ND | + |
| BKV | WT | ND | ND | ND | ND | ND | + | ND | ND | ND | + | ND | + | ND | + | ND | ND | ND | ND | ND | ND | ND | + |
| H42Q | ND | ND | ND | ND | ND | − | ND | ND | ND | + | ND | ND | ND | − | ND | ND | ND | ND | ND | ND | ND | ND | |
| pRB mutant | E109K | ND | ND | ND | ND | ND | + | ND | ND | ND | − | ND | ND | ND | − | ND | ND | ND | ND | ND | ND | ND | + |
Mutants in the large T antigens of SV40, polyomavirus and BKV as well as their phenotypes in various assays. See individual footnotes for specifics of each assay. Mutants in the J domain that possess levels of activity near those of the wild-type T antigen are not listed. For comparison purposes, mutant T antigens defective for binding to pRB family members are included.
The ability to transform REF52, C3H10T1/2, REF, or CREF cells was scored on the basis of the appearance of dense foci. Symbols: +, >50% of the foci induced by wild-type T antigen; +/−, ≤50% of the foci induced by wild-type T antigen; −, <3% of the foci induced by wild-type T antigen (175, 178, 219, 223, 224, 236).
The ability to convey a growth advantage to primary MEFs in 10% or low serum was scored. The increased number of cells induced by wild-type T antigen was scored as +, mutants that induced an advantage but less than the wild type until late in the experiment (day 10) were scored as +/−, and mutants that induced little advantage throughout the entire experiment were scored as − (88, 229).
Induction of tumors in mice when expression was driven by the lymphotropic polyomavirus promoter was scored. Wild-type T antigen induced choroid plexus papillomas which induced death early in the mice due to hydroencephalopy. Some mice survived and later developed lymphomas (36, 237, 238).
Induction of hepatocarcinomas in mice when expression is driven by the antithrombin III promoter (7).
The ability to bind to pRB family members as determined through coimmunoprecipitation assays (56, 87, 88, 210, 223, 229, 230, 268).
Alleviation of a G1 arrest mediated by transfection with pRB, p107, and p130 in Saos-2 (RB−/−) cells (269).
The ability to alter the phosphorylation state of p107 and p130 correlates with a reduction in the overall p130 steady-state levels (24, 87, 229, 230).
Disruption of p130/E2F complexes as assayed by gel shift analysis of cell culture lysate or immunoprecipitation release assays in vitro (233, 236, 269).
Bromodeoxyuridine incorporation (209; M. Saenz-Robles and J. M. Pipas, unpublished observation).
Ability to enhance the transcriptional activity of the POU protein transcription factor Tst1/Oct6/SCIP. Symbols: −, <30% of the activity of wild type; +/−, ∼50% of the activity of wild type (218).
Ability to override p53-mediated growth arrest induced by a temperature-sensitive allele of p53 in a rat embryo fibroblast cell line. Symbols: −, no activity; +/−, 20 to 50% of the activity of wild-type T antigen (74, 185).
Replication of viral DNA in cell culture or in a cellular lysate in vitro system. Symbols:+/−, between approximately 20 and 60% that of the wild type: −, <5% of that of wild type (24, 111, 134, 137, 151, 175, 195; P. G. Cantalupo and J. M. Pipas, unpublished results).
Ability to coimmunoprecipitate Hsc70 in the context of a cellular lysate or directly as assayed with purified components (24, 198, 209, 234, 236).
Stimulation of the ATPase activity of Hsc70 measured using either steady-state or single-turnover assays. Symbols: − and +/−, <5% and 30 to 60% of wild type, respectively (223, 236).
Ability to substitute as chimeric proteins for the J domain of DnaJ in a hypersensitive E. coli strain at extreme temperatures (9, 118). Note that the pRB-binding mutants and amino-terminal fragments contain a wild-type J domain. Because this is the same domain used in wild-type T-antigen chimeric DnaJ proteins, these mutants were scored as +.
Replication
Early mutational analysis demonstrated the essential role for an intact amino terminus of T antigen for SV40 replication in tissue cell culture models (178). Small-deletion mutants and point mutants of T antigen in residues highly conserved among J domains are defective for replication by 20-fold (174). Furthermore, a chimeric T antigen, HsjT, in which the T-antigen J domain is replaced with the human Hsj1 J domain, is functional for replication (24). Thus, it is evident that J-domain function is required for SV40 DNA replication in a cellular context.
Currently the role of the J domain of T antigen in DNA replication is unknown, and two different explanations are possible. First, the J-domain requirement may be direct. In such a model, the T-antigen J domain directly recruits the activity of an Hsc70 homologue to rearrange the necessary replication machinery to drive DNA replication in a manner analogous to phage λ DNA replication. Second, the J domain may be indirectly required. There are several ways to envision this hypothesis. The J domain may be required to recruit a cellular Hsc70 homologue activity to disrupt chromatin complexes which are inhibitory to DNA replication in the cellular context but absent in the noncellular in vitro assays. Recent evidence demonstrating a possible inhibitory role for pRB-E2F complexes at origins of replication (120) lends credence to this hypothesis. Alternatively, the J domain may be indirectly required for replication to drive the production of a necessary cellular enzyme(s) required for DNA synthesis such as DNA polymerase α or thymidine kinase. In support of these indirect hypotheses, in vitro assays in which the J domain of T antigen is mutated (43; P. G. Cantalupo and J. M. Pipas, unpublished observations), or even completely deleted (255), still replicate SV40 DNA in noncellular replication assays (albeit at a partially reduced level) (255). Furthermore, Hsc70 (or homologues) is not a major required component in a reconstituted noncellular in vitro replication assay (263). Finally, addition of purified Hsc70 to an in vitro replication reaction mixture does not enhance the replication efficiency (Cantalupo and Pipas, unpublished observations). Surely there are other ways to envision an indirect role for the J domain in replication; however, the actual mechanism remains undetermined, and only future experimentation will elucidate whether the J-domain requirement is direct, indirect, or some combination of both.
Role of J Domain in pRB Complexes
Using the powerful technique of expressing wild-type or mutant T antigens in MEFs null for various pRB family members, it was demonstrated that the J domain and pRB-binding motif are required to effect p130 and p107 to elicit cellular growth to a high density (38, 229). Furthermore, T antigen alters the phosphorylation state and decreases the half-life of p130 in established rodent cells (229). Down-regulating p130 function is essential for T-antigen-induced tumorigenesis, since overexpression of p130 inhibits the tumorigenic effects induced by JCV T antigen (99).
Because the J domain is required in cis with the pRB-binding motif to elicit transformation (223), a model in which the chaperone activity of the J domain directly acts on pRB-transcription factor complexes has developed. In this model (Fig. 8A) the J domain acts to recruit Hsc70 to the pRB-transcription factor complex (in this case E2F, but could easily apply to others such as MyoD or c-Abl) (16). The action of Hsc70 ATP hydrolysis induced by the J domain of T antigen induces the disruption of pRB-E2F, either directly by altering the conformation of pRB or E2F (Fig. 8A) or by recruiting cellular factors to the complex (Fig. 8B). Thus, free E2F is liberated and transcription of the genes necessary for viral replication proceeds.
FIG. 8.

Chaperone models for disruption of pRB-E2F family complexes. The multiple pRB and E2F family members are represented by “RB” and “E2F.” (A) In this model, T antigen recruits Hsc70 to pRB to directly act as a molecular machine that pries apart the pRB-E2F multiprotein complexes. (B) In this model, T antigen recruits Hsc70 to a multiprotein complex that requires the action of an additional unknown cellular protein (C [for cellular factor]) to disrupt pRB-E2F complexes (see the text for details). Factor C may posttranslationally modify pRB or E2F (denoted with asterisks) after they are separated from each other by the action of Hsc70. An alternative explanation is that factor C may enhance the activity of Hsc70 directly.
In support of this model, it has been shown that the J domain is required in cis with the pRB-binding motif to up-regulate exogenous promoters containing multiple E2F binding sites (209, 269). J-domain function is conserved among other polyomavirus family viruses, because both polyomavirus and BKV require a functional J domain to stimulate E2F-dependent transcription (88, 209). Furthermore, lysates made from cells not expressing T antigen or those expressing J-domain mutants of T antigen contain a p130-E2F-4 DNA binding complex that is not present in cellular lysates from cells expressing wild-type T antigen (88, 236, 269). These data are consistent with the chaperone-induced disruption of p130 from E2F.
However, there exists a caveat to interpreting the above data. Since T antigen is a powerful mitogen with multiple growth-inducing activities, it is possible that the J domain is required to drive the cells to cycle in a manner that indirectly disrupts pRB-E2F complexes. For example, several different mitogens will induce free E2F and transcriptional activity even though they are not known to directly bind to the pRB family (83, 92, 169, 258). Biochemical evidence, however, clearly demonstrates that T antigen has the capability to disrupt pRB-E2F family complexes in vitro (233). When a lysate from cells that overexpressed p130-E2F complexes was incubated with T antigen, p130 remained bound to E2F-4. However, inclusion of exogenous Hsc70 and an ATP regeneration system in the reaction released a portion of the E2F from p130. These results indicate that disruption of p130-E2F requires a functional J domain. The released E2F is capable of binding DNA containing an E2F consensus-binding site, consistent with its role as a transcription factor. Interestingly, the chaperone-mediated release of pRB family members from E2F is more efficient (approximately sixfold) in the presence of an unknown proteinaceous factor (designated factor C, for cellular protein) (233; C. S. Sullivan and J. M. Pipas, unpublished observation). Because the release reaction is enhanced by Hsc70 and ATP, it is possible that factor C is a cochaperone such as a Hip or a Bag. Alternatively, it is known that the phosphorylation state of pRB and E2F family members changes as cells proceed through the cell cycle and divide (52, 148). Phosphorylation of pRB prevents its association with E2F, and phosphorylation of E2F prevents its ability to bind to DNA in vitro (50, 51). Therefore, another plausible possibility is that factor C is a phosphatase or a kinase that requires Hsc70 to potentiate the disruption of the pRB-E2F family complex. In this model, T antigen acts as a switch that uses its chaperone activity to propagate some secondary effector such as a kinase to p130 or E2F-4 (Fig. 8B). In support of this notion, the J domain of T antigen is required to alter the phosphorylation state of p130 in cultured cells (229). An in vitro system of defined components will greatly enhance the testing of these models.
Non-J-domain-mediated T-antigen effects on the pRB family.
pRB has multiple functions including repressing the activities of E2F and other transcription factors (see section “pRB” of this review). The role of the J domain on non-E2F transcription factors remains to be determined. Consistent with the multifaceted growth regulation mechanisms that pRB possesses, T antigen can alter pRB cellular growth regulation in a J-domain-dependent (see above) or J-domain-independent manner. M. J. Tevethia and coworkers demonstrated that fusing the pRB-binding motif to a heterologous site on a carboxyl-terminal T-antigen fragment (amino acids 128 to 708) enabled this fragment to induce cells to grow to a high density (242). Therefore, the pRB-binding motif can contribute some growth-enhancing function(s) without the presence of a J domain. In an established rat embryo fibroblast cell line, the T-antigen J-domain mutant Δ17-27 induces expression of B-myb mRNA as well as wild-type T antigen, which the authors interpret as a productive interaction with p130 (182). Sheng and coworkers (210) have shown that the polyomavirus T antigen induces apoptosis in C2C12 myoblast cells and that this depends on an intact pRB-binding motif (LXCXE), but not a J domain, since mutant H42Q induces apoptosis nearly as well as the wild type. In contrast, SV40 T antigen prevents apoptosis in a neural astrocyte precursor cell line upon growth factor withdrawal, and this activity requires both the pRB-binding motif as well as the J domain of T antigen (215). Therefore, pRB possesses multiple activities intimately associated with the balance of cell growth and death which T antigen can disrupt—some in a J-domain-independent manner.
In another example, wild-type T antigen and J-domain mutants of T antigen restore growth to a cell line that is growth inhibited by the conditional expression of p53 (74, 185). The pRB-binding motif is required for this activity, but the J-domain mutant H42Q functions as well as the wild type. Interestingly, a deletion mutant of the entire J domain (Δ2-82) compromises the ability of T antigen to inhibit growth arrest by 80%, which the authors attribute to poor expression levels of Δ2-82 (74). The small-deletion mutant Δ17-27 is not functional in this assay, reflecting a more severe phenotype than point mutants in the J domain (see “Transformation” section below). Thus, while it is clear that the J domain is required to disrupt E2F from pRB, it has yet to be determined what activity the LXCXE motif possesses independent of the J domain. One interesting hypothesis is that the LXCXE motif by itself is able to disrupt HDACs from pRB (74, 210), thereby reducing the ability of pRB to inhibit transcription. There is support for such an idea since peptides corresponding to the LXCXE motif of T antigen inhibit or disrupt complex formation between HDAC-1 and pRB (149), but whether or not this occurs in the context of the cell remains to be determined. Another possibility is that the LXCXE motif or amino acid sequences surrounding it interact with some other cellular target that affects growth control independent of T antigen's actions on pRB.
Role of J domain in transactivating E2F promoters.
Multiple studies of polyomavirus, BKV, and SV40 T antigen demonstrate that the J domain and pRB-binding motif of T antigen contribute to alleviating repression of E2F transactivation (32, 74, 88, 134, 209, 210, 269). These studies are performed by transiently transfecting reporter constructs containing an E2F site(s) upstream of a reporter gene in the context of a minimal (E2F site upstream of a TATA box) or a portion of a physiological promoter such as the E2F-1 promoter. In five of the six reports, a J-domain point mutant is diminished in inducing the transcriptional activity of E2F relative to wild type (32, 74, 88, 134, 209, 210, 269). Additionally, J-domain mutants are consistently less defective than pRB-binding mutants in these assays. This suggests that the LXCXE pRB-binding motif contains additional activities that alleviate pRB-mediated repression of E2F family members in a J-domain-independent fashion. An alternative interpretation is that the J-domain point mutants assayed are “leaky,” thus retaining partial J-domain function. The J domain and LXCXE pRB-binding motif also act to alleviate the trans-repression activity of pRB that is independent of binding to E2F. This was demonstrated by experiments in which the repression activity of a fragment of pRB (containing the trans-repression domain) which cannot bind to E2F, is alleviated by T antigen (74). This activity was dependent on an intact LXCXE motif and J domain. These data suggest that in addition to alleviating pRB-mediated repression of E2F, both the J domain and LXCXE motif combine to alleviate pRB transcriptional repression that is independent of disrupting pRB-E2F complexes. Using a physiological cyclin A promoter with the E2F binding sites mutated, Sheng et al. demonstrated that LXCXE mutants transactivate cyclin A as well as wild-type T antigen (210). In this assay, a J-domain mutant is unable to transactivate cyclin A. These results suggests that the J domain has the ability to regulate some promoters independent of liberating E2F from pRB family repression.
The question arises as to why the J domain is required to transactivate promoters containing E2F to various degrees, ranging from no requirement with J-domain mutants transactivating reporter constructs as well as wild-type T antigen (32) up to J-domain mutants that are 10-fold defective relative to wild type (134). There are several experimental parameters that may account for at least some of these differences. Because many of these studies depend on transient transfections, proper normalization to account for discrepancies in transfection efficiencies must occur. Additionally, in the six studies (32, 74, 88, 134, 209, 210, 269), four different cell types were used, making it plausible that each may have differing pRB/E2F or chaperone levels. Finally, there is some suggestion that the stage of cell cycle division of the cells affects the degree to which the various domains of T antigen are required to transactivate E2F (210).
Transformation
There are at least three regions of T antigen required for cellular transformation, including the carboxyl-terminal region, the LXCXE pRB-binding motif, and the J domain (28, 111, 196, 223, 224, 244, 268, 273). The J domain is required in cis with both the pRB-binding (LXCXE) motif and the carboxyl terminus to transform REF52 cells (223). Furthermore, as noted in previous sections, the J domain is required in cis with the pRB-binding motif to transactivate E2F-responsive promoters (209). These findings suggest that the J-domain function must act on pRB complexes bound by the same molecule of T antigen. In addition, some other binding target of the carboxyl terminus is affected by J-domain activity (223). A likely possibility for the carboxyl-terminal transforming activity is binding to p53; however, studies suggest that other transforming activities in addition to p53 binding reside in the carboxyl terminus of T antigen (28, 196). Therefore, the J domain may also function on an as-yet-unidentified T-antigen carboxyl-terminal activity.
Different J-domain mutants displayed various degrees of penetrance for transformation ability (Table 1). The small-deletion mutant Δ17-27 (dl1135) (diagrammed in Fig. 7C) is 100% defective for transformation, while the D44N mutant (Fig. 7C), which contains a mutation in the highly conserved HPD motif, is capable of inducing transformation, albeit in a partially defective manner, inducing only 50% of the foci that wild-type T antigen does.
What can account for this discrepancy? There are at least three possible explanations. First, it is possible that the D44N mutation only partially disrupts J-domain function while the Δ17-27 mutation completely abolishes activity. The D44N mutation is analogous to the E. coli DnaJ mutant D35N, which is defective for J-domain function. A suppressor mutation in DnaK of the E. coli D35N mutant is R167H (Fig. 5B) (231). Suh et al. have shown that the cleft region surrounding R167 is a likely binding site for the HPD and helix 2 of the J domain (shown in Fig. 5C). It is reasonable to infer that the J domain of T antigen contacts an analogous cleft domain in Hsc70 and possibly other mammalian DnaK homologues. Extending this inference, the D44N mutation likely diminishes stimulation of Hsc70 activity in a manner analogous to that of the phenotypic null E. coli D35N mutant. Furthermore, studies using D44N confirm that it is defective for several chaperone-related activities, including binding to Hsc70 (234), stimulating Ssa1p ATP hydrolysis in a single turnover assay (although D44N still has approximately twofold more activity than a control mutant lacking the entire J domain) (233), and functioning for the E. coli DnaJ J domain in a phage λ replication assay (Vartikar and Kelley, unpublished observation). These data suggest that D44N retains little, if any, chaperone activity.
Second, it is possible that Δ17-27 causes a gross structural abnormality in the T-antigen molecule, such that it would interfere with the other more carboxyl-terminal transforming functions of T antigen including binding to the pRB or p53 family. This seems unlikely, because purified Δ17-27 is functional in numerous biochemistry assays such as stimulation of DNA replication, double hexamer assembly, ATPase activity, helicase activity, and ori unwinding, suggesting that it has structural integrity (43). Furthermore, Δ17-27 can bind to both p53 and pRB (56, 151, 178) and induce lymphomas when expressed in mice (238). However, it has been suggested that the Δ17-27 may be less stable than wild-type T antigen (151). Thus, while maintaining global structural integrity, lower steady-state levels in some cells could account for the more severe phenotype of Δ17-27, independent of its J-domain activity.
Third, it is possible that Δ17-27 abolishes another transforming function in the amino terminus in addition to the J-domain activity. In support of this idea, Cavender et al. have shown that the amino-terminal first 82 amino acids convey an essential, independent function that does not require binding to pRB to transactivate a polI-dependent promoter (29). Additionally, mutants in the polyomavirus J domain in the extreme amino-terminal region are defective for transformation induced by middle T antigen (44), even though the NMR structure does not support a role for these residues in contacting Hsc70 (9). The amino terminus of T antigen has a stretch of amino acids with weak amino acid similarity to the conserved CR1 of adenovirus E1A (177). The CR1 of E1A is required to bind to p300 (250) and to inhibit pRB activities in vitro (58, 104). Because both E1A and T antigen bind to pRB and induce transformation, it has been proposed that the CR1-like region of T antigen may share functionality with the corresponding sequence in E1A (265). The Δ17-27 mutation deletes portions of both the CR1-like region as well as a part of alpha-helix 2 of the J domain. Thus, it is possible that this mutant targets a CR1-like function as well as a J-domain function of T antigen. However, the crystal structure of the SV40 large T antigen predicts that the CR1-like region of SV40 is buried in the hydrophobic core of alpha-helices 2 and 3 of the J domain (Fig. 7C), which makes it difficult to envision a separate transforming activity for this exact region (amino acids 17 to 27). The Δ17-27 mutant, however, could abolish additional transforming activities in the extreme amino terminus of SV40 T antigen, similar to that of polyomavirus middle T antigen. Notice in Fig. 7 that the J domains of polyomavirus and SV40 T antigens contain extra conserved sequences amino terminal to the start of alpha-helix 1 of the nonpolyomavirus J domains. Therefore, it is possible that the Δ17-27 mutant alters 2 independent functions: the J domain and a transforming function amino terminal to amino acid 17. Construction of mutants in the first 17 amino acids of SV40 T antigen should be informative regarding this possibility.
Further complicating matters, the degree of the J-domain transforming contribution is dependent on the assay used to measure cellular growth deregulation. The cis requirement for the J domain and pRB-binding motif to transform REF52 cells was determined using crystal violet staining for dense focus formation. In this assay, cells are allowed to grow for approximately 6 weeks. As mentioned above, Δ17-27 is totally defective for focus formation, but D44N is only partially defective, forming approximately 50% the number of foci. However, when D44N is examined in an assay with growth to high density, it is completely defective (229). This assay is performed on a much shorter time scale (approximately 2 weeks) than the dense-focus assay and therefore likely measures different aspects of cellular growth control. Importantly, another mutant in the conserved loop of the J domain, H42Q, is also defective at inducing cellular growth to high density (229). However, the D44N mutant induces growth in soft agar as well as wild-type T antigen (229). Furthermore, TN136, a fragment of T antigen consisting of the J domain and the pRB-binding (LXCXE) motif, is capable of inducing foci in C3H10T1/2 cells (at a much reduced level relative to the wild type) but is unable to induce foci in REF52 cells (223). Interestingly, unlike the full-length situation, a D44N mutant in the context of TN136 is completely defective at inducing foci in C3H10T1/2 cells. The above data point out the varied requirements for J-domain function depending on cell type and the transformation assay employed. Unlike the J domain, a functional pRB-binding motif of T antigen is required to induce transformation in most assays, including induction of foci in REF52 and growth in soft agar. Interestingly, the fact that the J domain is required for transformation of some cell types in the context of TN136, but not full-length T antigen, suggests that the carboxyl-terminal domain of T antigen may encode transforming activities redundant with the J domain. Work from Cavender et al. supports such a notion (28).
Role of J domain in carboxyl-terminal transforming functions.
The J domain is required in cis with some as-yet-unknown carboxyl-terminal function for transformation. This was established using the TN136 amino-terminal fragment of T antigen and Δ17-27 which were unable to complement each other for full transformation of REF52 or C3H10T1/2 cells (223). As mentioned previously, the carboxyl-terminal function of T antigen that is required is unknown, but p53 is a likely candidate. However, J-domain mutants of T antigen function as well as the wild type in blocking p53 DNA binding in vitro (P. A. Carroll and J. M. Pipas, unpublished data), suggesting that the J domain has no role in this activity of T antigen. Another possible target of the J-domain function is the p300 protein, whose binding has been mapped to the carboxyl terminus of T antigen (143). Mutants in the amino terminus of T antigen are defective for altering the phosphorylation state of p300 and inhibiting its transactivation activity (54). This suggests that a carboxyl-terminally bound p300 substrate is a potential target for the J-domain activity. Alternatively, since p53 and p300 form a complex, it is possible that the J domain may be required to somehow alter this complex. Finally, it is possible that there are still undiscovered transforming functions in the carboxyl-terminal regions. Consistent with this idea, coexpression of TN136 and DD53, a dominant negative mutant of p53, is transformation defective compared to the p53 binding-competent full-length T antigen (196). Therefore, it is formally possible that the J domain acts independently of p53 and p300 on some other carboxyl-terminal function.
It should be noted that the J-domain function can be complemented in trans in several assays including induction of hepatic tumors and transactivation of a ribosomal promoter (7, 29). Why the J domain is required in cis for some activities (such as transformation of REF52 cells and transactivation of E2F responsive promoters) (209, 223) and not others is unknown. One possible determinant may be whether J-domain mutants of T antigen can functionally oligomerize with mutants of T antigen that contain a wild-type J domain. However, Cavender et al. argue that while oligomerization may be required for trans complementation, it is not sufficient for transactivation, since mutants that contain the regions of T antigen sufficient for oligomerization fail to complement in trans (29). Future experimentation is required to understand the differing cis and trans J-domain requirements in these multiple assays.
J Domain and Virion Assembly
SV40 virions are composed of three viral proteins, VP1, VP2, and VP3, which form an icosahedral structure of 72 pentamers of VP1, with each pentamer associated with one polypeptide of either VP2 or VP3 (141). The mature virion encapsulates the viral DNA molecule and has a sedimentation coefficient of 240S in a sucrose density gradient (42). Developing virions composed of chromatin and some VP polypeptides have less-dense sedimentation coefficients ranging between 100 and 150S (221, 222). Both genetic and biochemical data indicate that a wild-type T antigen is required for virion assembly for at least two independent steps of virion maturation. Some T-antigen mutants in either the extreme carboxyl-terminal host range (HR) specificity function or the J domain (L19F,P28S) replicate DNA and produce VP1 to -3 but fail to assemble mature virions (221, 222). Both mutants produce immature capsid intermediates that migrate at unique lower densities in a sucrose gradient. When these mutants are expressed in the same cell, the HR and L19F,P28S mutants complement each other to infect cells as well as wild-type virus, suggesting that they code for two separate functions (175).
The L19F,P28S double point mutant contains substitutions in both the second and third alpha-helices of the J domain (Fig. 7C). L19 has a functional group that protrudes into the hydrophobic core of the J domain, making contacts directly with the functional group of M30 of the third alpha-helix (Fig. 7C). Thus, the SV40 L19F,P28S mutant alters the stability of the interactions between the second and third helices of the J domain. Unlike other J-domain mutants, L19F,P28S replicates DNA to near wild-type levels (175) but is defective for virion assembly. The ability of other J-domain mutants that do not replicate their DNA or express the VP virion proteins has not been determined.
The virion assembly defect of the T-antigen HR or L19F,P28S mutants may be indirect, for example, due to the failed induction of a cellular peptide that is necessary for proper virion assembly. Alternatively, these mutants may implicate a direct role of T antigen in virion assembly due to the inability of T antigen to physically function in the assembly process. For L19F,P28S there is some evidence that the defect may be direct, since Hsc70 binds to VP1 (197). This links, at least circumstantially, the J-protein/Hsc70 chaperone machinery to the process of virion assembly.
J Domain and Transactivation
T antigen is both a transactivator of the SV40 structural late genes and a repressor of the SV40 early genes (42, 85, 114, 115). Furthermore, T antigen is a promiscuous transactivator of polymerase I (PolI), PolII, and PolIII (29, 274). Multiple domains of T antigen are required to induce transactivation, including the J domain (29, 274). Cavender et al. have shown that a J-domain mutant (Δ2-82) is defective for transactivation of a PolI ribosomal promoter but can be complemented in trans by a transactivation-defective carboxyl-terminal mutant (dl400). In these assays, an intact pRB-binding (LXCXE) motif is not required for T-antigen-induced transactivation (29, 274). This suggests that the J-domain activity required for transactivation is different from the J-domain activity required to disrupt pRB/E2F complexes which requires a functional pRB-binding site. T antigen associates with multiprotein complexes at promoters; perhaps the J domain is required to rearrange such complexes.
J Domain and Tumorigenesis
Ectopic expression of T antigen results in tumors in numerous rodent models—including brain, breast, bladder, choroid plexus, pancreas, intestinal, lymphatic, and liver (7, 36, 76, 78, 99, 138, 187, 241)—and may be linked to human tumors as well (see Introduction). The function of the T-antigen J domain during in vivo tumorigenesis has not been studied in detail; however some conclusions can be made.
A fragment of T antigen that expresses the J domain and pRB-binding motif (N1 to 121) is sufficient to induce pancreatic and hepatic tumors as well as full-length T antigen, suggesting that binding to p53 is not required to induce tumors in these systems (7, 241). N121 induces slow-growing tumors in the choroid plexus (36). These tumors become fast growing (similar to ones induced by expression of full-length T antigen) if the mice are crossed into a p53-null mouse (237), suggesting that in these cells, the role of the T-antigen J domain and pRB-binding motif is to drive the cells into a state of hyperplasia but the role of the p53 binding domain is to inhibit p53-dependent apoptosis. Furthermore, if N121-expressing mice are crossed to an E2F-1−/− background then apoptosis induction is greatly reduced (173). This suggests that a fragment of T antigen containing the J domain and pRB-binding motif liberates E2F from pRB family members in vivo which induces p53-dependent apoptosis. In another transgenic murine model system, full-length T antigen induces dysplasia (onset at approximately 6 weeks of age) when expressed with the oncoprotein-activated K-ras in the villi of the intestine, but expression of N121 with activated K-ras induces only hyperplasia (123). If the N121 mice are crossed into a p53-null background, they do not progress to a further growth deregulated state (C. M. Coopersmith and J. Gordon, unpublished observation) as is the case in the choroid plexus model. This suggests that T antigen possesses transforming functions in its carboxyl terminus other than inactivating p53 (see above). The differing requirements for inactivation of p53 in the various model systems underscore the tissue specific effects of T-antigen expression.
The J-domain mutant Δ17-27 is defective at inducing tumors in the choroid plexus, liver, intestine, and pancreas but does induce T-cell lymphomas (7, 187, 238). In contrast, the L19F,P28S J-domain mutant is able to induce choroid plexus tumors (36). The basis for the differing abilities of Δ17-27 and L19F,P28S to induce choroid plexus tumors is not known; one possibility is that L19F,P28S only partially inhibits J-domain function (see discussion on Δ17-27 and L19F,P28S in sections above). To our knowledge, other J-domain mutants have not been studied in vivo; therefore a comprehensive understanding of the J-domain function during in vivo tumorigenesis will require the construction of additional transgenic animals.
Transplantation of ovarian carcinoma cells that overexpress the Her2 gene into mice results in tumors (144). Expression of J-domain-containing amino-terminal fragments of T antigen in these cells inhibits tumor growth (144, 264). This effect is observed if small t antigen, the N121 amino-terminal fragment of large T antigen, or the N1-82 fragment consisting solely of the J domain is expressed. The mechanism of how T antigen inhibits tumor growth is unknown; however, the expression of J-domain-containing fragments of T antigen induces transcriptional repression of the Her2 and other promoters (144, 251). Because expression of the J domain by itself has dominant negative effects on the transactivation of an E2F reporter construct (209), it is possible that the J domain may be acting as a dominant negative for some chaperone requirement for Her2 transactivation. Further experiments that use point mutations in key J-domain residues would demonstrate if this effect is truly J domain dependent.
Role of the J Domain in Other T Antigens
There are three SV40 splice variants that contain the J domain in their amino terminus, which compose the T antigens: large T antigen, small t antigen, and 17kT antigen (Fig. 1). Several other alternatively spliced T antigens are made by the various polyomaviruses; however, the role of the J domain in the function of these proteins has not been studied in detail. Small t antigen is made by most polyomaviruses, can stimulate the ATPase activity of Hsc70, and is involved in down-regulating the phosphatase activity of protein phosphatase 2A (193, 223). Middle T antigen is not encoded by SV40 but is made by a subset of the polyomaviruses including polyomavirus itself. Middle T antigen is a transforming protein that activates cytoplasmic signal transduction proteins (168). Mutations in the middle-T-antigen J domain render it defective for inducing transformation (44).
In addition, there are several different T antigens made that demonstrate similarity to amino-terminal fragments of large T antigen. These include tiny t antigen (polyomavirus), 17kT antigen (SV40), and the T′ T antigens of JCV: N135, N136, and N165. Polyomavirus tiny t is comprised of the J domain plus an additional unique six amino acids, has a short half-life during infection, and stimulates the ATPase activity of Hsc70 (188). Its function is unknown. SV40 17kT antigen is comprised of the first 131 amino acids of large T antigens plus an additional 4 unique amino acids. 17kT antigen is expressed at low levels during infection but is able to transform rat fibroblast cells upon plasmid-mediated expression (270). The JCV T′ molecules are unique in that they are expressed at higher levels with a longer half-life than tiny t or 17kT. All three T′ proteins have the same first 132 amino acids as JCV large T antigen and contribute to infectivity (246). The carboxyl terminus of N165 contains the HR specificity domain in common with the extreme carboxyl terminus of large T antigen. In addition to the common 132 amino acids, N136 and N135 each have unique amino acids at their carboxyl terminus (four and three, respectively). A similar construct engineered in SV40, TN136, can transform cells (223); therefore it is likely that JCV N136 and N135 proteins alter cellular growth properties during infection. Consistent with this, the three T′ proteins can bind to the pRB family members (11). The exact role of the J domain in the function in these peptides resembling the amino terminus of SV40 large T antigen awaits future functional characterizations and mutational analyses.
SPECIFICITY OF T-ANTIGEN J-DOMAIN INTERACTIONS
The T-antigen J domain can functionally replace the endogenous J domain of both E. coli and yeast J proteins in viability assays (118; Fewell and Brodsky, unpublished observation). Furthermore, heterologous T-antigen chimeras containing J domains from human proteins (Hsj1, DnaJ2) are functional for viral activities, including DNA replication and inducing cellular growth to a high density (24, 229). Interestingly, chimeric T antigens containing the J domains of E. coli DnaJ or yeast Ydj1p are defective for several SV40 activities, including the ability to liberate E2F from p130, induce transformation, and replicate viral DNA (Table 1) (236). Thus, mammalian but neither the yeast nor E. coli J domains contain structural elements that allow for productive association with the appropriate mammalian Hsc70. Conversely, the T-antigen J domain can functionally interact with the yeast and E. coli Hsc70s.
The first alpha-helix of T antigen is longer than all other known J domains (Fig. 7); therefore, it may contain specific structural determinants for T-antigen function that are not encoded by other J domains. However, one must still account for the functionality of T-antigen chimeras containing human J domains. One possibility is that the human J domains possess similar specificity elements (for example similar charged residues or length) with the T-antigen J domain. In support of this, the NMR structure of the polyomavirus J domain shares greater similarity with the structure of HDJ1 than E. coli DnaJ (9). However, sequence gazing fails to produce any sequence similarities among Hsj1, DnaJ2, and SV40 that are not present in yeast or E. coli J domains. It is possible that the human J domains have only some of the T-antigen-specific elements, rendering T-antigen chimeras containing these J domains only partly functional. In support of this notion, the chimeras containing human J domains are not as fully active as wild-type T antigen for induction of cellular growth to a high density, DNA replication, and the synergistic transactivation of the Oct1/scip transcriptional complex (24, 218, 229). These observations suggest that, for certain T-antigen functions, human J domains provide only some of the specific elements that the T-antigen J domain possesses. Thus, the key residues that mediate interaction of J proteins with Hsc70 are evolutionarily conserved, but additional functions may be present in the T-antigen J domain. In support of this notion, Berjanskii and coworkers have suggested that the NMR structural analysis of the polyomavirus J domain supports the existence of polyomavirus family virus-specific structural elements in the extreme amino terminus of T antigen (9).
CANCER AND CHAPERONES
What is the role of chaperones in nonvirally mediated cellular transformation? Hsc70 has the hallmarks of a tumor suppressor (it is mutated in some breast cancers [3]), and Hsp90 overexpression enhances apoptosis in at least some cell types (69). On the other hand, it is well established that some chaperones, including Hsc70 and Hsp90, are overexpressed or display changes in their subcellular localization in many tumor types (109, 110, 220). This suggests that chaperones may contribute to tumorigenesis; however, it is not clear whether chaperone overexpression is a cause or effect or the transformed phenotype.
Overexpression of Hsc70 in transgenic mice induces T-cell lymphomas (late in development at age 8 to 10 months), which suggests that Hsc70 can be a cause of transformation (207). However, in these studies, it is unclear why only some of the founder mice (three of nine) develop lymphomas and why they occur later in development. Not surprisingly, the cellular stress and death pathways are linked, and consequently Hsc70 expression protects against apoptosis in multiple model systems (68, 160, 161, 203). This occurs through at least two activities, one upstream of caspase activation involving JNK and another involving inhibition of caspases (161). Furthermore, inhibition of Hsc70 synthesis results in tumor cell-specific apoptosis (167). Hsc70-mediated inhibition of apoptosis could explain how overexpression of Hsc70 induces the lymphomas described above (207).
Chaperones have been implicated in tumorigenesis by directly binding to and modulating the function of tumor suppressors. Hsp90 binds to several kinases and is thought to modulate their oncogenicity (19, 109). An Hsp90 homologue, Hsp75, binds to pRB through an LXCXE motif during mitosis (Fig. 3B) (35). In addition, Hsc70 binds to pRB in the context of some cellular lysates (165). Hsc70 preferentially associates with hypophosphorylated pRB in vitro, with the smallest fragment of pRB that binds to Hsc70 being mapped to the amino-terminal region including a small portion of the A box (Fig. 3B) (106). E1A, an LXCXE motif-containing protein, cannot compete with Hsc70 for binding to pRB, suggesting that Hsc70 and LXCXE motif-containing proteins can coexist in the same complex with pRB. Hsc70 binds to both wild-type and mutant p53s (109, 128), and overexpression of Hsc70 can overcome transformation induced by coexpression of a mutant p53 and ras (267). Finally, DnaK, a prokaryotic homologue to Hsc70, can activate the DNA binding activity of p53 in vitro (101, 102). The above data imply that in many circumstances chaperones are involved in promoting the function of tumor suppressors. Paradoxically, chaperones are often found overexpressed in tumors, which could be specific or alternatively due to the stress induced by an increase in protein synthesis that occurs in rapidly dividing cancer cells. At least in the case of viral infection, chaperone induction is specific, since sometimes only a subset of chaperones are induced upon infection (235). Whether the same can be concluded for nonvirally induced tumorigenesis awaits further study. The above data suggest the existence of a delicate balance between chaperone activity and maintenance of proper cellular growth control.
Are chaperones useful targets for cancer therapy? Thus far, the best chance for chaperone-based cancer therapy comes from small molecule inhibitors such as geldanamycin and radicol. Both target the ATP binding activity of Hsp90 and both have shown positive results as cancer therapeutic agents (109, 199, 202, 208, 259). Two promising compounds that alter Hsc70 function,15-deoxyspergualin and R/1, are currently being tested for therapeutic applications (15, 59). The immunoreactivity of Hsc70-bound antigens has led some to develop therapeutic applications by generating a chaperone-induced immune response to cancer cells (109, 220). These and other approaches are in still in their infancy, and future research is required to determine their efficacy.
THE FUTURE
While in recent years much progress has been made, several important questions regarding SV40 chaperone function remain. Much of our understanding of the J domain comes from mutational studies conducted before it was known that T antigen contains a J domain. Critically, the construction of more surgical mutants within and surrounding the J domain should afford us a more precise view into the role of the J domain, its cellular targets, and the factors that determine its specific interactions. For example, what is the carboxyl-terminal function that interacts with the J domain to cause transformation? Does T antigen alter p300 function or some other novel as-yet-unidentified transformation function between amino acids 137 and 708? Why is it that the J-domain function is required in cis with the rest of T antigen in some assays but may be complemented in trans in others? Is Hsc70 the only biologically relevant DnaK that participates in SV40 function? There are many Hsc70 homologues in mammalian cells, so it is entirely possible that other Hsc70 homologues interact with T antigen. For example, mouse beta islet cells expressing T antigen via a tetracycline-induced promoter express sevenfold-higher mRNA levels of the Hsc70-related protein NST-1 (277). Does NST-1 contribute to T-antigen activities, or is its overexpression simply an artifact of stressed cells? What cellular factor(s) is required in addition to Hsc70 to efficiently drive the disruption of pRB/E2F family complexes? One could envision a cochaperone such as a functional homologue to the eukaryotic nucleotide exchange factor GrpE. Alternatively, a posttranslational modification such as phosphorylation of pRB or E2F may be required to modulate separation of pRB and E2F proteins. As more of the components are purified (for example, p130 and E2F-4), biochemical analysis to identify the cellular proteins required for the release reaction can be performed. Is it possible to inhibit viral life cycles by targeting chaperone activities? A better understanding of what determines chaperone specificity is required before such therapies can be efficiently designed. Finally, like the SV40-mediated disruption of pRB-E2F complexes, is there a similar role for the J-protein/Hsc70 chaperone machine in the nonviral context?
Throughout this review we have tried to present not only the progress that has been made with understanding the functions of the T-antigen J domain but also the interesting, unresolved questions as well. The take-home message from this review is that the two distinct fields of chaperone research and cell cycle regulation intersect at the T-antigen J domain.
Acknowledgments
We thank Lisa Engler for her input and valuable assistance with the Molecular Modeling program Midas. We thank Sheara Fewell and Steve Van Doren for helpful comments regarding the manuscript. We thank Amy E. Baker and Paul Cantalupo for formatting assistance.
This work was supported by grant CA40586 from the NIH.
REFERENCES
- 1.Avantaggiati, M. L., M. Carbone, A. Graessmann, Y. Nakatani, B. Howard, and A. S. Levine. 1996. The SV40 large T antigen and adenovirus E1a oncoproteins interact with distinct isoforms of the transcriptional co-activator, p300. EMBO J. 15:2236-2248. [PMC free article] [PubMed] [Google Scholar]
- 2.Avantaggiati, M. L., V. Ogryzko, K. Gardner, A. Giordano, A. S. Levine, and K. Kelly. 1997. Recruitment of p300/CBP in p53-dependent signal pathways. Cell 89:1175-1184. [DOI] [PubMed] [Google Scholar]
- 3.Bakkenist, C. J., J. Koreth, C. S. Williams, N. C. Hunt, and J. O. McGee. 1999. Heat shock cognate 70 mutations in sporadic breast carcinoma. Cancer Res. 59:4219-4221. [PubMed] [Google Scholar]
- 4.Ballinger, C. A., P. Connell, Y. Wu, Z. Hu, L. J. Thompson, L. Y. Yin, and C. Patterson. 1999. Identification of CHIP, a novel tetratricopeptide repeat-containing protein that interacts with heat shock proteins and negatively regulates chaperone functions. Mol. Cell. Biol. 19:4535-4545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Banecki, B., and M. Zylicz. 1996. Real time kinetics of the DnaK/DnaJ/GrpE molecular chaperone machine action. J. Biol. Chem. 271:6137-6143. [DOI] [PubMed] [Google Scholar]
- 6.Banecki, B., M. Zylicz, E. Bertoli, and F. Tanfani. 1992. Structural and functional relationships in DnaK and DnaK756 heat-shock proteins from Escherichia coli. J. Biol. Chem. 267:25051-25058. [PubMed] [Google Scholar]
- 7.Bennoun, M., G. Grimber, D. Couton, A. Seye, T. Molina, P. Briand, and V. Joulin. 1998. The amino-terminal region of SV40 large T antigen is sufficient to induce hepatic tumours in mice. Oncogene 17:1253-1259. [DOI] [PubMed] [Google Scholar]
- 8.Berger, J. R., and E. O. Major. 1999. Progressive multifocal leukoencephalopathy. Semin. Neurol. 19:193-200. [DOI] [PubMed] [Google Scholar]
- 9.Berjanskii, M. V., M. I. Riley, A. Xie, V. Semenchenko, W. R. Folk, and S. R. Van Doren. 2000. NMR structure of the N-terminal J domain of murine polyomavirus T antigens: implications for DnaJ-like domains and for mutations of T antigens. J. Biol. Chem. 275:36094-36103. [DOI] [PubMed]
- 10.Blond-Elguindi, S., S. E. Cwirla, W. J. Dower, R. J. Lipshutz, S. R. Sprang, J. F. Sambrook, and M. J. Gething. 1993. Affinity panning of a library of peptides displayed on bacteriophages reveals the binding specificity of BiP. Cell 75:717-728. [DOI] [PubMed] [Google Scholar]
- 11.Bollag, B., C. Prins, E. L. Snyder, and R. J. Frisque. 2000. Purified JC virus T and T′ proteins differentially interact with the retinoblastoma family of tumor suppressor proteins. Virology 274:165-178. [DOI] [PubMed] [Google Scholar]
- 12.Boorstein, W. R., T. Ziegelhoffer, and E. A. Craig. 1994. Molecular evolution of the HSP70 multigene family. J. Mol. Evol. 38:1-17. [DOI] [PubMed] [Google Scholar]
- 13.Bringold, F., and M. Serrano. 2000. Tumor suppressors and oncogenes in cellular senescence. Exp. Gerontol. 35:317-329. [DOI] [PubMed] [Google Scholar]
- 14.Brodsky, J. L. 1996. Post-translational protein translocation: not all hsc70s are created equal. Trends Biochem. Sci. 21:122-126. [PubMed] [Google Scholar]
- 15.Brodsky, J. L. 1999. Selectivity of the molecular chaperone-specific immunosuppressive agent 15-deoxyspergualin: modulation of Hsc70 ATPase activity without compromising DnaJ chaperone interactions. Biochem. Pharmacol. 57:877-880. [DOI] [PubMed] [Google Scholar]
- 16.Brodsky, J. L., and J. M. Pipas. 1998. Polyomavirus T antigens: molecular chaperones for multiprotein complexes. J. Virol. 72:5329-5334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bryan, T. M., and R. R. Reddel. 1994. SV40-induced immortalization of human cells. Crit. Rev. Oncog. 5:331-357. [DOI] [PubMed] [Google Scholar]
- 18.Buchberger, A., H. Theyssen, H. Schroder, J. S. McCarty, G. Virgallita, P. Milkereit, J. Reinstein, and B. Bukau. 1995. Nucleotide-induced conformational changes in the ATPase and substrate binding domains of the DnaK chaperone provide evidence for interdomain communication. J. Biol. Chem. 270:16903-16910. [DOI] [PubMed] [Google Scholar]
- 19.Buchner, J. 1999. Hsp90 & Co.—a holding for folding. Trends Biochem. Sci. 24:136-141. [DOI] [PubMed] [Google Scholar]
- 20.Bukau, B., and A. L. Horwich. 1998. The Hsp70 and Hsp60 chaperone machines. Cell 92:351-366. [DOI] [PubMed] [Google Scholar]
- 21.Bullock, P. A. 1997. The initiation of simian virus 40 DNA replication in vitro. Crit. Rev. Biochem. Mol. Biol. 32:503-568. [DOI] [PubMed] [Google Scholar]
- 22.Butel, J. S., and J. A. Lednicky. 1999. Cell and molecular biology of simian virus 40: implications for human infections and disease. J. Natl. Cancer Inst. 91:119-134. [DOI] [PubMed] [Google Scholar]
- 23.Caldarelli-Stefano, R., R. Boldorini, G. Monga, E. Meraviglia, E. O. Zorini, and P. Ferrante. 2000. JC virus in human glial-derived tumors. Hum. Pathol. 31:394-395. [DOI] [PubMed] [Google Scholar]
- 24.Campbell, K. S., K. P. Mullane, I. A. Aksoy, H. Stubdal, J. Zalvide, J. M. Pipas, P. A. Silver, T. M. Roberts, B. S. Schaffhausen, and J. A. DeCaprio. 1997. DnaJ/hsp40 chaperone domain of SV40 large T antigen promotes efficient viral DNA replication. Genes Dev. 11:1098-1110. [DOI] [PubMed] [Google Scholar]
- 25.Caplan, A. J., D. M. Cyr, and M. G. Douglas. 1993. Eukaryotic homologues of Escherichia coli dnaJ: a diverse protein family that functions with hsp70 stress proteins. Mol. Biol. Cell 4:555-563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Carbone, M. 1999. Simian virus 40 and human tumors: it is time to study mechanisms. J. Cell. Biochem. 76:189-193. [DOI] [PubMed] [Google Scholar]
- 27.Cartwright, P., H. Muller, C. Wagener, K. Holm, and K. Helin. 1998. E2F-6: a novel member of the E2F family is an inhibitor of E2F-dependent transcription. Oncogene 17:611-623. [DOI] [PubMed] [Google Scholar]
- 28.Cavender, J. F., A. Conn, M. Epler, H. Lacko, and M. J. Tevethia. 1995. Simian virus 40 large T antigen contains two independent activities that cooperate with a ras oncogene to transform rat embryo fibroblasts. J. Virol. 69:923-934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Cavender, J. F., C. Mummert, and M. J. Tevethia. 1999. Transactivation of a ribosomal gene by simian virus 40 large-T antigen requires at least three activities of the protein. J. Virol. 73:214-224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Chang, C., D. T. Simmons, M. A. Martin, and P. T. Mora. 1979. Identification and partial characterization of new antigens from simian virus 40-transformed mouse cells. J. Virol. 31:463-471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Chang, C. F., H. Tada, and K. Khalili. 1994. The role of a pentanucleotide repeat sequence, AGGGAAGGGA, in the regulation of JC virus DNA replication. Gene 148:309-314. [DOI] [PubMed] [Google Scholar]
- 32.Chao, H. H., A. M. Buchmann, and J. A. DeCaprio. 2000. Loss of p19(ARF) eliminates the requirement for the pRB-binding motif in simian virus 40 large T antigen-mediated transformation. Mol. Cell. Biol. 20:7624-7633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Cheetham, M. E., J. P. Brion, and B. H. Anderton. 1992. Human homologues of the bacterial heat-shock protein DnaJ are preferentially expressed in neurons. Biochem. J. 284:469-476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Cheetham, M. E., and A. J. Caplan. 1998. Structure, function and evolution of DnaJ: conservation and adaptation of chaperone function. Cell Stress Chaperones 3:28-36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Chen, C. F., Y. Chen, K. Dai, P. L. Chen, D. J. Riley, and W. H. Lee. 1996. A new member of the hsp90 family of molecular chaperones interacts with the retinoblastoma protein during mitosis and after heat shock. Mol. Cell. Biol. 16:4691-4699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Chen, J., G. J. Tobin, J. M. Pipas, and T. Van Dyke. 1992. T-antigen mutant activities in vivo: roles of p53 and pRB binding in tumorigenesis of the choroid plexus. Oncogene 7:1167-1175. [PubMed] [Google Scholar]
- 37.Chen, Y. N., S. K. Sharma, T. M. Ramsey, L. Jiang, M. S. Martin, K. Baker, P. D. Adams, K. W. Bair, and W. G. Kaelin, Jr. 1999. Selective killing of transformed cells by cyclin/cyclin-dependent kinase 2 antagonists. Proc. Natl. Acad. Sci. USA 96:4325-4329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Christensen, J. B., and M. J. Imperiale. 1995. Inactivation of the retinoblastoma susceptibility protein is not sufficient for the transforming function of the conserved region 2-like domain of simian virus 40 large T antigen. J. Virol. 69:3945-3948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Clarke, A. R., E. R. Maandag, M. van Roon, N. M. van der Lugt, M. van der Valk, M. L. Hooper, A. Berns, and H. te Riele. 1992. Requirement for a functional Rb-1 gene in murine development. Nature 359:328-330. [DOI] [PubMed] [Google Scholar]
- 40.Classon, M., B. K. Kennedy, R. Mulloy, and E. Harlow. 2000. Opposing roles of pRB and p107 in adipocyte differentiation. Proc. Natl. Acad. Sci. USA 97:10826-10831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Cobrinik, D., M. H. Lee, G. Hannon, G. Mulligan, R. T. Bronson, N. Dyson, E. Harlow, D. Beach, R. A. Weinberg, and T. Jacks. 1996. Shared role of the pRB-related p130 and p107 proteins in limb development. Genes Dev. 10:1633-1644. [DOI] [PubMed] [Google Scholar]
- 42.Cole, C. N. 1996. Polyomaviridae: the viruses and their replication, p. 1997-2043. In B. N. Fields, D. M. Knipe, and P. M. Howley (ed.), Fields virology, 3rd ed. Lippincott-Raven Publishers, Philadelphia, Pa.
- 43.Collins, B. S., and J. M. Pipas. 1995. T antigens encoded by replication-defective simian virus 40 mutants dl1135 and 5080. J. Biol. Chem. 270:15377-15384. [DOI] [PubMed] [Google Scholar]
- 44.Cook, D. N., and J. A. Hassell. 1990. The amino terminus of polyomavirus middle T antigen is required for transformation. J. Virol. 64:1879-1887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Cyr, D. M., T. Langer, and M. G. Douglas. 1994. DnaJ-like proteins: molecular chaperones and specific regulators of Hsp70. Trends Biochem. Sci. 19:176-181. [DOI] [PubMed] [Google Scholar]
- 46.Dahiya, A., M. R. Gavin, R. X. Luo, and D. C. Dean. 2000. Role of the LXCXE binding site in Rb function. Mol. Cell. Biol. 20:6799-6805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.DeCaprio, J. A. 1999. The role of the J domain of SV40 large T in cellular transformation. Biologicals 27:23-28. [DOI] [PubMed] [Google Scholar]
- 48.DeCaprio, J. A., J. W. Ludlow, J. Figge, J. Y. Shew, C. M. Huang, W. H. Lee, E. Marsilio, E. Paucha, and D. M. Livingston. 1988. SV40 large tumor antigen forms a specific complex with the product of the retinoblastoma susceptibility gene. Cell 54:275-283. [DOI] [PubMed] [Google Scholar]
- 49.Demand, J., J. Luders, and J. Hohfeld. 1998. The carboxy-terminal domain of Hsc70 provides binding sites for a distinct set of chaperone cofactors. Mol. Cell. Biol. 18:2023-2028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Dynlacht, B. D., O. Flores, J. A. Lees, and E. Harlow. 1994. Differential regulation of E2F transactivation by cyclin/cdk2 complexes. Genes Dev. 8:1772-1786. [DOI] [PubMed] [Google Scholar]
- 51.Dynlacht, B. D., K. Moberg, J. A. Lees, E. Harlow, and L. Zhu. 1997. Specific regulation of E2F family members by cyclin-dependent kinases. Mol. Cell. Biol. 17:3867-3875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Dyson, N. 1998. The regulation of E2F by pRB-family proteins. Genes Dev. 12:2245-2262. [DOI] [PubMed] [Google Scholar]
- 53.Dyson, N., R. Bernards, S. H. Friend, L. R. Gooding, J. A. Hassell, E. O. Major, J. M. Pipas, T. Vandyke, and E. Harlow. 1990. Large T antigens of many polyomaviruses are able to form complexes with the retinoblastoma protein. J. Virol. 64:1353-1356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Eckner, R., J. W. Ludlow, N. L. Lill, E. Oldread, Z. Arany, N. Modjtahedi, J. A. DeCaprio, D. M. Livingston, and J. A. Morgan. 1996. Association of p300 and CBP with simian virus 40 large T antigen. Mol. Cell. Biol. 16:3454-3464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Egan, C., T. N. Jelsma, J. A. Howe, S. T. Bayley, B. Ferguson, and P. E. Branton. 1988. Mapping of cellular protein-binding sites on the products of early region 1A of human adenovirus type 5. Mol. Cell. Biol. 8:3955-3959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Ewen, M. E., J. W. Ludlow, E. Marsilio, J. A. DeCaprio, R. C. Millikan, S. H. Cheng, E. Paucha, and D. M. Livingston. 1989. An N-terminal transformation-governing sequence of SV40 large T antigen contributes to the binding of both p110Rb and a second cellular protein, p120. Cell 58:257-267. [DOI] [PubMed] [Google Scholar]
- 57.Ezhevsky, S. A., H. Nagahara, A. M. Vocero-Akbani, D. R. Gius, M. C. Wei, and S. F. Dowdy. 1997. Hypo-phosphorylation of the retinoblastoma protein (pRb) by cyclin D:Cdk4/6 complexes results in active pRb. Proc. Natl. Acad. Sci. USA 94:10699-10704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Fattaey, A. R., E. Harlow, and K. Helin. 1993. Independent regions of adenovirus E1A are required for binding to and dissociation of E2F-protein complexes. Mol. Cell. Biol. 13:7267-7277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Fewell, S. W., B. W. Day, and J. L. Brodsky. 2001. Identification of an inhibitor of hsc70-mediated protein translocation and ATP hydrolysis. J. Biol. Chem. 276:910-914. [DOI] [PubMed] [Google Scholar]
- 60.Flaherty, K. M., C. DeLuca-Flaherty, and D. B. McKay. 1990. Three-dimensional structure of the ATPase fragment of a 70K heat-shock cognate protein. Nature 346:623-628. [DOI] [PubMed] [Google Scholar]
- 61.Flynn, G. C., T. G. Chappell, and J. E. Rothman. 1989. Peptide binding and release by proteins implicated as catalysts of protein assembly. Science 245:385-390. [DOI] [PubMed] [Google Scholar]
- 62.Flynn, G. C., J. Pohl, M. T. Flocco, and J. E. Rothman. 1991. Peptide-binding specificity of the molecular chaperone BiP. Nature 353:726-730. [DOI] [PubMed] [Google Scholar]
- 63.Freeman, B. C., M. P. Myers, R. Schumacher, and R. I. Morimoto. 1995. Identification of a regulatory motif in Hsp70 that affects ATPase activity, substrate binding and interaction with HDJ-1. EMBO J. 14:2281-2292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Friend, S. H., R. Bernards, S. Rogelj, R. A. Weinberg, J. M. Rapaport, D. M. Albert, and T. P. Dryja. 1986. A human DNA segment with properties of the gene that predisposes to retinoblastoma and osteosarcoma. Nature 323:643-646. [DOI] [PubMed] [Google Scholar]
- 65.Frisque, R. J., G. L. Bream, and M. T. Cannella. 1984. Human polyomavirus JC virus genome. J. Virol. 51:458-469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Frydman, J., and J. Hohfeld. 1997. Chaperones get in touch: the Hip-Hop connection. Trends Biochem. Sci. 22:87-92. [DOI] [PubMed] [Google Scholar]
- 67.Fung, Y. K., A. L. Murphree, A. T'Ang, J. Qian, S. H. Hinrichs, and W. F. Benedict. 1987. Structural evidence for the authenticity of the human retinoblastoma gene. Science 236:1657-1661. [DOI] [PubMed] [Google Scholar]
- 68.Gabai, V. L., A. B. Meriin, J. A. Yaglom, V. Z. Volloch, and M. Y. Sherman. 1998. Role of Hsp70 in regulation of stress-kinase JNK: implications in apoptosis and aging. FEBS Lett. 438:1-4. [DOI] [PubMed] [Google Scholar]
- 69.Galea-Lauri, J., A. J. Richardson, D. S. Latchman, and D. R. Katz. 1996. Increased heat shock protein 90 (hsp90) expression leads to increased apoptosis in the monoblastoid cell line U937 following induction with TNF-alpha and cycloheximide: a possible role in immunopathology. J. Immunol. 157:4109-4118. [PubMed] [Google Scholar]
- 70.Gassler, C. S., A. Buchberger, T. Laufen, M. P. Mayer, H. Schroder, A. Valencia, and B. Bukau. 1998. Mutations in the DnaK chaperone affecting interaction with the DnaJ cochaperone. Proc. Natl. Acad. Sci. USA 95:15229-15234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Gaubatz, S., G. J. Lindeman, S. Ishida, L. Jakoi, J. R. Nevins, D. M. Livingston, and R. E. Rempel. 2000. E2F4 and E2F5 play an essential role in pocket protein-mediated G1 control. Mol. Cell 6:729-735. [DOI] [PubMed] [Google Scholar]
- 72.Gaubatz, S., J. G. Wood, and D. M. Livingston. 1998. Unusual proliferation arrest and transcriptional control properties of a newly discovered E2F family member, E2F-6. Proc. Natl. Acad. Sci. USA 95:9190-9195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Georgopoulos, C., and W. J. Welch. 1993. Role of the major heat shock proteins as molecular chaperones. Annu. Rev. Cell Biol. 9:601-634. [DOI] [PubMed] [Google Scholar]
- 74.Gjoerup, O., H. Chao, J. A. DeCaprio, and T. M. Roberts. 2000. pRB-dependent, J domain-independent function of simian virus 40 large T antigen in override of p53 growth suppression. J. Virol. 74:864-874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Gragerov, A., L. Zeng, X. Zhao, W. Burkholder, and M. E. Gottesman. 1994. Specificity of DnaK-peptide binding. J. Mol. Biol. 235:848-854. [DOI] [PubMed] [Google Scholar]
- 76.Green, J. E., M. A. Shibata, K. Yoshidome, M. L. Liu, C. Jorcyk, M. R. Anver, J. Wigginton, R. Wiltrout, E. Shibata, S. Kaczmarczyk, W. Wang, Z. Y. Liu, A. Calvo, and C. Couldrey. 2000. The C3(1)/SV40 T-antigen transgenic mouse model of mammary cancer: ductal epithelial cell targeting with multistage progression to carcinoma. Oncogene 19:1020-1027. [DOI] [PubMed] [Google Scholar]
- 77.Greene, M. K., K. Maskos, and S. J. Landry. 1998. Role of the J-domain in the cooperation of Hsp40 with Hsp70. Proc. Natl. Acad. Sci. USA 95:6108-6113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Grippo, P. J., and E. P. Sandgren. 2000. Highly invasive transitional cell carcinoma of the bladder in a simian virus 40 T-antigen transgenic mouse model. Am. J. Pathol. 157:805-813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Gu, W., J. W. Schneider, G. Condorelli, S. Kaushal, V. Mahdavi, and B. Nadal-Ginard. 1993. Interaction of myogenic factors and the retinoblastoma protein mediates muscle cell commitment and differentiation. Cell 72:309-324. [DOI] [PubMed] [Google Scholar]
- 80.Gu, W., X. L. Shi, and R. G. Roeder. 1997. Synergistic activation of transcription by CBP and p53. Nature 387:819-823. [DOI] [PubMed] [Google Scholar]
- 81.Guzey, M., S. Takayama, and J. C. Reed. 2000. BAG1L enhances trans-activation function of the vitamin D receptor. J. Biol. Chem. 275:40749-40756. [DOI] [PubMed] [Google Scholar]
- 82.Hahn, W. C., C. M. Counter, A. S. Lundberg, R. L. Beijersbergen, M. W. Brooks, and R. A. Weinberg. 1999. Creation of human tumour cells with defined genetic elements. Nature 400:464-468. [DOI] [PubMed] [Google Scholar]
- 83.Halaban, R., E. Cheng, Y. Smicun, and J. Germino. 2000. Deregulated E2F transcriptional activity in autonomously growing melanoma cells. J. Exp. Med. 191:1005-1016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Hanahan, D., and R. A. Weinberg. 2000. The hallmarks of cancer. Cell 100:57-70. [DOI] [PubMed] [Google Scholar]
- 85.Hansen, U., D. G. Tenen, D. M. Livingston, and P. A. Sharp. 1981. T antigen repression of SV40 early transcription from two promoters. Cell 27:603-613. [DOI] [PubMed] [Google Scholar]
- 86.Harbour, J. W., and D. C. Dean. 2000. The Rb/E2F pathway: expanding roles and emerging paradigms. Genes Dev. 14:2393-2409. [DOI] [PubMed] [Google Scholar]
- 87.Harris, K. F., J. B. Christensen, and M. J. Imperiale. 1996. BK virus large T antigen: interactions with the retinoblastoma family of tumor suppressor proteins and effects on cellular growth control. J. Virol. 70:2378-2386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Harris, K. F., J. B. Christensen, E. H. Radany, and M. J. Imperiale. 1998. Novel mechanisms of E2F induction by BK virus large-T antigen: requirement of both the pRb-binding and the J domains. Mol. Cell. Biol. 18:1746-1756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Harrison, C. J., M. Hayer-Hartl, M. Di Liberto, F. Hartl, and J. Kuriyan. 1997. Crystal structure of the nucleotide exchange factor GrpE bound to the ATPase domain of the molecular chaperone DnaK. Science 276:431-435. [DOI] [PubMed] [Google Scholar]
- 90.Hartl, F. U. 1996. Molecular chaperones in cellular protein folding. Nature 381:571-579. [DOI] [PubMed] [Google Scholar]
- 91.Helin, K., J. A. Lees, M. Vidal, N. Dyson, E. Harlow, and A. Fattaey. 1992. A cDNA encoding a pRB-binding protein with properties of the transcription factor E2F. Cell 70:337-350. [DOI] [PubMed] [Google Scholar]
- 92.Hengstschlager, M., E. Hengstschlager-Ottnad, O. Pusch, and E. Wawra. 1996. The role of p16 in the E2F-dependent thymidine kinase regulation. Oncogene 12:1635-1643. [PubMed] [Google Scholar]
- 93.Hermiston, T. 2000. Gene delivery from replication-selective viruses: arming guided missiles in the war against cancer. J. Clin. Investig. 105:1169-1172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Higashino, F., J. M. Pipas, and T. Shenk. 1998. Adenovirus E4orf6 oncoprotein modulates the function of the p53-related protein, p73. Proc. Natl. Acad. Sci. USA 95:15683-15687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Hilleman, M. R. 1998. Discovery of simian virus 40 (SV40) and its relationship to poliomyelitis virus vaccines. Dev. Biol. Stand. 94:183-190. [PubMed] [Google Scholar]
- 96.Hohfeld, J., and S. Jentsch. 1997. GrpE-like regulation of the hsc70 chaperone by the anti-apoptotic protein BAG-1. EMBO J. 16:6209-6216. (Erratum, 17:847, 1998.) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Hohfeld, J., Y. Minami, and F. U. Hartl. 1995. Hip, a novel cochaperone involved in the eukaryotic Hsc70/Hsp40 reaction cycle. Cell 83:589-598. [DOI] [PubMed] [Google Scholar]
- 98.Hottiger, M. O., and G. J. Nabel. 2000. Viral replication and the coactivators p300 and CBP. Trends Microbiol. 8:560-565. [DOI] [PubMed] [Google Scholar]
- 99.Howard, C. M., P. P. Claudio, G. L. Gallia, J. Gordon, G. G. Giordano, W. W. Hauck, K. Khalili, and A. Giordano. 1998. Retinoblastoma-related protein pRb2/p130 and suppression of tumor growth in vivo. J. Natl. Cancer Inst. 90:1451-1460. [DOI] [PubMed] [Google Scholar]
- 100.Huang, K., J. M. Flanagan, and J. H. Prestegard. 1999. The influence of C-terminal extension on the structure of the “J-domain” in E. coli DnaJ. Protein Sci. 8:203-214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Hupp, T. R., D. W. Meek, C. A. Midgley, and D. P. Lane. 1993. Activation of the cryptic DNA binding function of mutant forms of p53. Nucleic Acids Res. 21:3167-3174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Hupp, T. R., D. W. Meek, C. A. Midgley, and D. P. Lane. 1992. Regulation of the specific DNA binding function of p53. Cell 71:875-886. [DOI] [PubMed] [Google Scholar]
- 103.Ibaraki, N., S. C. Chen, L. R. Lin, H. Okamoto, J. M. Pipas, and V. N. Reddy. 1998. Human lens epithelial cell line. Exp. Eye Res. 67:577-585. [DOI] [PubMed] [Google Scholar]
- 104.Ikeda, M. A., and J. R. Nevins. 1993. Identification of distinct roles for separate E1A domains in disruption of E2F complexes. Mol. Cell. Biol. 13:7029-7035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Imperiale, M. J. 2000. The human polyomaviruses, BKV and JCV: molecular pathogenesis of acute disease and potential role in cancer. Virology 267:1-7. [DOI] [PubMed] [Google Scholar]
- 106.Inoue, A., T. Torigoe, K. Sogahata, K. Kamiguchi, S. Takahashi, Y. Sawada, M. Saijo, Y. Taya, S. Ishii, N. Sato, et al. 1995. 70-kDa heat shock cognate protein interacts directly with the N-terminal region of the retinoblastoma gene product pRb. Identification of a novel region of pRb-mediating protein interaction. J. Biol. Chem. 270:22571-22576. [DOI] [PubMed] [Google Scholar]
- 107.Jacks, T., A. Fazeli, E. M. Schmitt, R. T. Bronson, M. A. Goodell, and R. A. Weinberg. 1992. Effects of an Rb mutation in the mouse. Nature 359:295-300. [DOI] [PubMed] [Google Scholar]
- 108.Jiang, R. F., T. Greener, W. Barouch, L. Greene, and E. Eisenberg. 1997. Interaction of auxilin with the molecular chaperone, Hsc70. J. Biol. Chem. 272:6141-6145. [DOI] [PubMed] [Google Scholar]
- 109.Jolly, C., and R. I. Morimoto. 2000. Role of the heat shock response and molecular chaperones in oncogenesis and cell death. J. Natl. Cancer Inst. 92:1564-1572. [DOI] [PubMed] [Google Scholar]
- 110.Jolly, C., and R. I. Morimoto. 1999. Stress and the cell nucleus: dynamics of gene expression and structural reorganization. Gene Expr. 7:261-270. [PMC free article] [PubMed] [Google Scholar]
- 111.Kalderon, D., and A. E. Smith. 1984. In vitro mutagenesis of a putative DNA binding domain of SV40 large-T. Virology 139:109-137. [DOI] [PubMed] [Google Scholar]
- 112.Kamath-Loeb, A. S., C. Z. Lu, W. C. Suh, M. A. Lonetto, and C. A. Gross. 1995. Analysis of three DnaK mutant proteins suggests that progression through the ATPase cycle requires conformational changes. J. Biol. Chem. 270:30051-30059. [DOI] [PubMed] [Google Scholar]
- 113.Karzai, A. W., and R. McMacken. 1996. A bipartite signaling mechanism involved in DnaJ-mediated activation of the Escherichia coli DnaK protein. J. Biol. Chem. 271:11236-11246. [DOI] [PubMed] [Google Scholar]
- 114.Keller, J. M., and J. C. Alwine. 1984. Activation of the SV40 late promoter: direct effects of T antigen in the absence of viral DNA replication. Cell 36:381-389. [DOI] [PubMed] [Google Scholar]
- 115.Keller, J. M., and J. C. Alwine. 1985. Analysis of an activatable promoter: sequences in the simian virus 40 late promoter required for T-antigen-mediated trans activation. Mol. Cell. Biol. 5:1859-1869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Kelley, W. L. 1998. The J-domain family and the recruitment of chaperone power. Trends Biochem. Sci. 23:222-227. [DOI] [PubMed] [Google Scholar]
- 117.Kelley, W. L. 1999. Molecular chaperones: how J domains turn on Hsp70s. Curr. Biol. 9:R305-R308. [DOI] [PubMed] [Google Scholar]
- 118.Kelley, W. L., and C. Georgopoulos. 1997. The T/t common exon of simian virus 40, JC, and BK polyomavirus T antigens can functionally replace the J-domain of the Escherichia coli DnaJ molecular chaperone. Proc. Natl. Acad. Sci. USA 94:3679-3684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Reference deleted
- 120.Kennedy, B. K., D. A. Barbie, M. Classon, N. Dyson, and E. Harlow. 2000. Nuclear organization of DNA replication in primary mammalian cells. Genes Dev. 14:2855-2868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Kierstead, T. D., and M. J. Tevethia. 1993. Association of p53 binding and immortalization of primary C57BL/6 mouse embryo fibroblasts by using simian virus 40 T-antigen mutants bearing internal overlapping deletion mutations. J. Virol. 67:1817-1829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Kim, H. Y., B. Y. Ahn, and Y. Cho. 2001. Structural basis for the inactivation of retinoblastoma tumor suppressor by SV40 large T antigen. EMBO J. 20:295-304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Kim, S. H., K. A. Roth, C. M. Coopersmith, J. M. Pipas, and J. I. Gordon. 1994. Expression of wild-type and mutant simian virus 40 large tumor antigens in villus-associated enterocytes of transgenic mice. Proc. Natl. Acad. Sci. USA 91:6914-6918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Kimura, Y., I. Yahara, and S. Lindquist. 1995. Role of the protein chaperone YDJ1 in establishing Hsp90-mediated signal transduction pathways. Science 268:1362-1365. [DOI] [PubMed] [Google Scholar]
- 125.Kress, M., E. May, R. Cassingena, and P. May. 1979. Simian virus 40-transformed cells express new species of proteins precipitable by anti-simian virus 40 tumor serum. J. Virol. 31:472-483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Lai, A., J. M. Lee, W. M. Yang, J. A. DeCaprio, W. G. Kaelin, Jr., E. Seto, and P. E. Branton. 1999. RBP1 recruits both histone deacetylase-dependent and -independent repression activities to retinoblastoma family proteins. Mol. Cell. Biol. 19:6632-6641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Lane, D. P., and L. V. Crawford. 1979. T antigen is bound to a host protein in SV40-transformed cells. Nature 278:261-263. [DOI] [PubMed] [Google Scholar]
- 128.Lane, D. P., C. Midgley, and T. Hupp. 1993. Tumour suppressor genes and molecular chaperones. Philos. Trans. R. Soc. Lond. B 339:369-373. [DOI] [PubMed] [Google Scholar]
- 129.Laufen, T., M. P. Mayer, C. Beisel, D. Klostermeier, A. Mogk, J. Reinstein, and B. Bukau. 1999. Mechanism of regulation of hsp70 chaperones by DnaJ cochaperones. Proc. Natl. Acad. Sci. USA 96:5452-5457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Lee, E. Y., C. Y. Chang, N. Hu, Y. C. Wang, C. C. Lai, K. Herrup, W. H. Lee, and A. Bradley. 1992. Mice deficient for Rb are nonviable and show defects in neurogenesis and haematopoiesis. Nature 359:288-294. [DOI] [PubMed] [Google Scholar]
- 131.Lee, J. O., A. A. Russo, and N. P. Pavletich. 1998. Structure of the retinoblastoma tumour-suppressor pocket domain bound to a peptide from HPV E7. Nature 391:859-865. [DOI] [PubMed] [Google Scholar]
- 132.Lee, M. H., B. O. Williams, G. Mulligan, S. Mukai, R. T. Bronson, N. Dyson, E. Harlow, and T. Jacks. 1996. Targeted disruption of p107: functional overlap between p107 and Rb. Genes Dev. 10:1621-1632. [DOI] [PubMed] [Google Scholar]
- 133.Lee, W. H., J. Y. Shew, F. D. Hong, T. W. Sery, L. A. Donoso, L. J. Young, R. Bookstein, and E. Y. Lee. 1987. The retinoblastoma susceptibility gene encodes a nuclear phosphoprotein associated with DNA binding activity. Nature 329:642-645. [DOI] [PubMed] [Google Scholar]
- 134.Lemieux, B., and M. Bastin. 2000. Polyomavirus large T antigen mutants affected in viral DNA replication. Virology 269:370-376. [DOI] [PubMed] [Google Scholar]
- 135.Levine, A. J. 1997. p53, the cellular gatekeeper for growth and division. Cell 88:323-331. [DOI] [PubMed] [Google Scholar]
- 136.Levy, R., S. D. Smith, K. Chandler, Y. Sadovsky, and D. M. Nelson. 2000. Apoptosis in human cultured trophoblasts is enhanced by hypoxia and diminished by epidermal growth factor. Am. J. Physiol. Cell Physiol. 278:C982-C988. [DOI] [PubMed] [Google Scholar]
- 137.Li, J. J., and T. J. Kelly. 1984. Simian virus 40 DNA replication in vitro. Proc. Natl. Acad. Sci. USA 81:6973-6977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Li, M., B. Lewis, A. V. Capuco, R. Laucirica, and P. A. Furth. 2000. WAP-TAg transgenic mice and the study of dysregulated cell survival, proliferation, and mutation during breast carcinogenesis. Oncogene 19:1010-1019. [DOI] [PubMed] [Google Scholar]
- 139.Liberek, K., C. Georgopoulos, and M. Zylicz. 1988. Role of the Escherichia coli DnaK and DnaJ heat shock proteins in the initiation of bacteriophage lambda DNA replication. Proc. Natl. Acad. Sci. USA 85:6632-6636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Liberek, K., D. Skowyra, M. Zylicz, C. Johnson, and C. Georgopoulos. 1991. The Escherichia coli DnaK chaperone, the 70-kDa heat shock protein eukaryotic equivalent, changes conformation upon ATP hydrolysis, thus triggering its dissociation from a bound target protein. J. Biol. Chem. 266:14491-14496. [PubMed] [Google Scholar]
- 141.Liddington, R. C., Y. Yan, J. Moulai, R. Sahli, T. L. Benjamin, and S. C. Harrison. 1991. Structure of simian virus 40 at 3.8-A resolution. Nature 354:278-284. [DOI] [PubMed] [Google Scholar]
- 142.Lill, N. L., S. R. Grossman, D. Ginsberg, J. DeCaprio, and D. M. Livingston. 1997. Binding and modulation of p53 by p300/CBP coactivators. Nature 387:823-827. [DOI] [PubMed] [Google Scholar]
- 143.Lill, N. L., M. J. Tevethia, R. Eckner, D. M. Livingston, and N. Modjtahedi. 1997. p300 family members associate with the carboxyl terminus of simian virus 40 large tumor antigen. J. Virol. 71:129-137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Lin, Y. C., J. M. Peng, and W. B. Wang. 2000. The N-terminal common domain of simian virus 40 large T and small t antigens acts as a transformation suppressor of the HER-2/neu oncogene. Oncogene 19:2704-2713. [DOI] [PubMed] [Google Scholar]
- 145.Linzer, D. I., and A. J. Levine. 1979. Characterization of a 54K dalton cellular SV40 tumor antigen present in SV40-transformed cells and uninfected embryonal carcinoma cells. Cell 17:43-52. [DOI] [PubMed] [Google Scholar]
- 146.Lipinski, M. M., and T. Jacks. 1999. The retinoblastoma gene family in differentiation and development. Oncogene 18:7873-7882. [DOI] [PubMed] [Google Scholar]
- 147.Lisse, T., and E. Schwarz. 2000. Functional specificity of the mitochondrial DnaJ protein, Mdj1p, in Saccharomyces cerevisiae. Mol. Gen. Genet. 263:527-534. [DOI] [PubMed] [Google Scholar]
- 148.Ludlow, J. W., J. Shon, J. M. Pipas, D. M. Livingston, and J. A. DeCaprio. 1990. The retinoblastoma susceptibility gene product undergoes cell cycle-dependent dephosphorylation and binding to and release from SV40 large T. Cell 60:387-396. [DOI] [PubMed] [Google Scholar]
- 149.Magnaghi-Jaulin, L., R. Groisman, I. Naguibneva, P. Robin, S. Lorain, J. P. Le Villain, F. Troalen, D. Trouche, and A. Harel-Bellan. 1998. Retinoblastoma protein represses transcription by recruiting a histone deacetylase. Nature 391:601-605. [DOI] [PubMed] [Google Scholar]
- 150.Marin, M. C., and W. G. Kaelin, Jr. 2000. p63 and p73: old members of a new family. Biochim. Biophys. Acta 1470:M93-M100. [DOI] [PubMed] [Google Scholar]
- 151.Marsilio, E., S. H. Cheng, B. Schaffhausen, E. Paucha, and D. M. Livingston. 1991. The T/t common region of simian virus 40 large T antigen contains a distinct transformation-governing sequence. J. Virol. 65:5647-5652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.McCarty, J. S., A. Buchberger, J. Reinstein, and B. Bukau. 1995. The role of ATP in the functional cycle of the DnaK chaperone system. J. Mol. Biol. 249:126-137. [DOI] [PubMed] [Google Scholar]
- 153.McClellan, A. J., J. B. Endres, J. P. Vogel, D. Palazzi, M. D. Rose, and J. L. Brodsky. 1998. Specific molecular chaperone interactions and an ATP-dependent conformational change are required during posttranslational protein translocation into the yeast ER. Mol. Biol. Cell 9:3533-3545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.McClellan, A. J., and J. Frydman. 2001. Molecular chaperones and the art of recognizing a lost cause. Nat. Cell Biol. 3:E51-E53. [DOI] [PubMed] [Google Scholar]
- 155.Misselwitz, B., O. Staeck, and T. A. Rapoport. 1998. J proteins catalytically activate Hsp70 molecules to trap a wide range of peptide sequences. Mol. Cell 2:593-603. [DOI] [PubMed] [Google Scholar]
- 156.Mittnacht, S. 1998. Control of pRB phosphorylation. Curr. Opin. Genet. Dev. 8:21-27. [DOI] [PubMed] [Google Scholar]
- 157.Momand, J., H. H. Wu, and G. Dasgupta. 2000. MDM2: master regulator of the p53 tumor suppressor protein. Gene 242:15-29. [DOI] [PubMed] [Google Scholar]
- 158.Montano, X., R. Millikan, J. M. Milhaven, D. A. Newsom, J. W. Ludlow, A. K. Arthur, E. Fanning, I. Bikel, and D. M. Livingston. 1990. Simian virus 40 small tumor antigen and an amino-terminal domain of large tumor antigen share a common transforming function. Proc. Natl. Acad. Sci. USA 87:7448-7452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Morshauser, R. C., W. Hu, H. Wang, Y. Pang, G. C. Flynn, and E. R. Zuiderweg. 1999. High-resolution solution structure of the 18 kDa substrate-binding domain of the mammalian chaperone protein Hsc70. J. Mol. Biol. 289:1387-1403. [DOI] [PubMed] [Google Scholar]
- 160.Mosser, D. D., A. W. Caron, L. Bourget, C. Denis-Larose, and B. Massie. 1997. Role of the human heat shock protein hsp70 in protection against stress-induced apoptosis. Mol. Cell. Biol. 17:5317-5327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Mosser, D. D., A. W. Caron, L. Bourget, A. B. Meriin, M. Y. Sherman, R. I. Morimoto, and B. Massie. 2000. The chaperone function of hsp70 is required for protection against stress-induced apoptosis. Mol. Cell. Biol. 20:7146-7159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Mungre, S., K. Enderle, B. Turk, A. Porras, Y. Q. Wu, M. C. Mumby, and K. Rundell. 1994. Mutations which affect the inhibition of protein phosphatase 2A by simian virus 40 small-t antigen in vitro decrease viral transformation. J. Virol. 68:1675-1681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Nevins, J. R. 1998. Toward an understanding of the functional complexity of the E2F and retinoblastoma families. Cell Growth Differ. 9:585-593. [PubMed] [Google Scholar]
- 164.Newman, J. S., G. B. Baskin, and R. J. Frisque. 1998. Identification of SV40 in brain, kidney and urine of healthy and SIV-infected rhesus monkeys. J. Neurovirol. 4:394-406. [DOI] [PubMed] [Google Scholar]
- 165.Nihei, T., S. Takahashi, S. Sagae, N. Sato, and K. Kikuchi. 1993. Protein interaction of retinoblastoma gene product pRb110 with Mr 73,000 heat shock cognate protein. Cancer Res. 53:1702-1705. [PubMed] [Google Scholar]
- 166.Nollen, E. A., J. F. Brunsting, J. Song, H. H. Kampinga, and R. I. Morimoto. 2000. Bag1 functions in vivo as a negative regulator of Hsp70 chaperone activity. Mol. Cell. Biol. 20:1083-1088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Nylandsted, J., M. Rohde, K. Brand, L. Bastholm, F. Elling, and M. Jaattela. 2000. Selective depletion of heat shock protein 70 (Hsp70) activates a tumor-specific death program that is independent of caspases and bypasses Bcl-2. Proc. Natl. Acad. Sci. USA 97:7871-7876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Oliveira, M. L., S. M. Brochado, and M. C. Sogayar. 1999. Mechanisms of cell transformation induced by polyomavirus. Braz. J. Med. Biol. Res. 32:861-865. [DOI] [PubMed] [Google Scholar]
- 169.Onishi, T., W. Zhang, X. Cao, and K. Hruska. 1997. The mitogenic effect of parathyroid hormone is associated with E2F-dependent activation of cyclin-dependent kinase 1 (cdc2) in osteoblast precursors. J. Bone Miner. Res. 12:1596-1605. [DOI] [PubMed] [Google Scholar]
- 170.Ozer, H. L. 2000. SV40-mediated immortalization. Prog. Mol. Subcell. Biol. 24:121-153. [DOI] [PubMed] [Google Scholar]
- 171.Packschies, L., H. Theyssen, A. Buchberger, B. Bukau, R. S. Goody, and J. Reinstein. 1997. GrpE accelerates nucleotide exchange of the molecular chaperone DnaK with an associative displacement mechanism. Biochemistry 36:3417-3422. [DOI] [PubMed] [Google Scholar]
- 172.Palleros, D. R., K. L. Reid, L. Shi, W. J. Welch, and A. L. Fink. 1993. ATP-induced protein-Hsp70 complex dissociation requires K+ but not ATP hydrolysis. Nature 365:664-666. [DOI] [PubMed] [Google Scholar]
- 173.Pan, H., C. Yin, N. J. Dyson, E. Harlow, L. Yamasaki, and T. Van Dyke. 1998. Key roles for E2F1 in signaling p53-dependent apoptosis and in cell division within developing tumors. Mol. Cell 2:283-292. [DOI] [PubMed] [Google Scholar]
- 174.Peden, K. W., and J. M. Pipas. 1992. Simian virus 40 mutants with amino-acid substitutions near the amino terminus of large T antigen. Virus Genes 6:107-118. [DOI] [PubMed] [Google Scholar]
- 175.Peden, K. W., S. L. Spence, L. C. Tack, C. A. Cartwright, A. Srinivasan, and J. M. Pipas. 1990. A DNA replication-positive mutant of simian virus 40 that is defective for transformation and the production of infectious virions. J. Virol. 64:2912-2921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Pierpaoli, E. V., E. Sandmeier, A. Baici, H. J. Schonfeld, S. Gisler, and P. Christen. 1997. The power stroke of the DnaK/DnaJ/GrpE molecular chaperone system. J. Mol. Biol. 269:757-768. [DOI] [PubMed] [Google Scholar]
- 177.Pipas, J. M. 1992. Common and unique features of T antigens encoded by the polyomavirus group. J. Virol. 66:3979-3985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Pipas, J. M., K. W. Peden, and D. Nathans. 1983. Mutational analysis of simian virus 40 T antigen: isolation and characterization of mutants with deletions in the T-antigen gene. Mol. Cell. Biol. 3:203-213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Polissi, A., L. Goffin, and C. Georgopoulos. 1995. The Escherichia coli heat shock response and bacteriophage lambda development. FEMS Microbiol. Rev. 17:159-169. [DOI] [PubMed] [Google Scholar]
- 180.Porràs, A., J. Bennett, A. Howe, K. Tokos, N. Bouck, B. Henglein, S. Sathyamangalam, B. Thimmapaya, and K. Rundell. 1996. A novel simian virus 40 early-region domain mediates transactivation of the cyclin A promoter by small-t antigen and is required for transformation in small-t antigen-dependent assays. J. Virol. 70:6902-6908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Porrás, A., S. Gaillard, and K. Rundell. 1999. The simian virus 40 small-t and large-T antigens jointly regulate cell cycle reentry in human fibroblasts. J. Virol. 73:3102-3107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182.Powell, A. J., A. J. Darmon, E. S. Gonos, E. W. Lam, K. W. Peden, and P. S. Jat. 1999. Different functions are required for initiation and maintenance of immortalization of rat embryo fibroblasts by SV40 large T antigen. Oncogene 18:7343-7350. [DOI] [PubMed] [Google Scholar]
- 183.Prapapanich, V., S. Chen, S. C. Nair, R. A. Rimerman, and D. F. Smith. 1996. Molecular cloning of human p48, a transient component of progesterone receptor complexes and an Hsp70-binding protein. Mol. Endocrinol. 10:420-431. [DOI] [PubMed] [Google Scholar]
- 184.Qian, Y. Q., D. Patel, F. U. Hartl, and D. J. McColl. 1996. Nuclear magnetic resonance solution structure of the human Hsp40 (HDJ-1) J-domain. J. Mol. Biol. 260:224-235. [DOI] [PubMed] [Google Scholar]
- 185.Quartin, R. S., C. N. Cole, J. M. Pipas, and A. J. Levine. 1994. The amino-terminal functions of the simian virus 40 large T antigen are required to overcome wild-type p53-mediated growth arrest of cells. J. Virol. 68:1334-1341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186.Rassow, J., O. von Ahesen, U. Bomer, and N. Pfanner. 1997. Molecular chaperones: towards a characterization of the heat-shock protein 70 family. Trends Cell Biol. 7:129-133. [DOI] [PubMed] [Google Scholar]
- 187.Ratineau, C., A. Ronco, and A. B. Leiter. 2000. Role of the amino-terminal domain of simian virus 40 early region in inducing tumors in secretin-expressing cells in transgenic mice. Gastroenterology 119:1305-1311. [DOI] [PubMed] [Google Scholar]
- 188.Riley, M. I., W. Yoo, N. Y. Mda, and W. R. Folk. 1997. Tiny T antigen: an autonomous polyomavirus T antigen amino-terminal domain. J. Virol. 71:6068-6074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189.Ross, J. F., X. Liu, and B. D. Dynlacht. 1999. Mechanism of transcriptional repression of E2F by the retinoblastoma tumor suppressor protein. Mol. Cell 3:195-205. [DOI] [PubMed] [Google Scholar]
- 190.Roth, J., and M. Dobbelstein. 1999. Failure of viral oncoproteins to target the p53-homologue p51A. J. Gen. Virol. 80:3251-3255. [DOI] [PubMed] [Google Scholar]
- 191.Rudiger, S., L. Germeroth, J. Schneider-Mergener, and B. Bukau. 1997. Substrate specificity of the DnaK chaperone determined by screening cellulose-bound peptide libraries. EMBO J. 16:1501-1507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192.Rundell, K., S. Gaillard, and A. Porras. 1998. Small-t and large-T antigens cooperate to drive cell proliferation. Dev. Biol. Stand. 94:289-295. [PubMed] [Google Scholar]
- 193.Rundell, K., and R. Parakati. 2001. The role of the SV40 ST antigen in cell growth promotion and transformation. Semin. Cancer Biol. 11:5-13. [DOI] [PubMed] [Google Scholar]
- 194.Rushton, J. J., D. Jiang, A. Srinivasan, J. M. Pipas, and P. D. Robbins. 1997. Simian virus 40 T antigen can regulate p53-mediated transcription independent of binding p53. J. Virol. 71:5620-5623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 195.Rutila, J. E., M. J. Imperiale, and W. W. Brockman. 1986. Replication and transformation functions of in vitro-generated simian virus 40 large T antigen mutants. J. Virol. 58:526-535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 196.Sachsenmeier, K. F., and J. M. Pipas. 2001. Inhibition of Rb and p53 is insufficient for SV40 T-antigen transformation. Virology 283:40-48. [DOI] [PubMed] [Google Scholar]
- 197.Sainis, I., C. Angelidis, G. N. Pagoulatos, and I. Lazaridis. 2000. HSC70 interactions with SV40 viral proteins differ between permissive and nonpermissive mammalian cells. Cell Stress Chaperones 5:132-138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 198.Sawai, E. T., and J. S. Butel. 1989. Association of a cellular heat shock protein with simian virus 40 large T antigen in transformed cells. J. Virol. 63:3961-3973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 199.Scheibel, T., and J. Buchner. 1998. The Hsp90 complex—a super-chaperone machine as a novel drug target. Biochem. Pharmacol. 56:675-682. [DOI] [PubMed] [Google Scholar]
- 200.Schlenstedt, G., S. Harris, B. Risse, R. Lill, and P. A. Silver. 1995. A yeast DnaJ homologue, Scj1p, can function in the endoplasmic reticulum with BiP/Kar2p via a conserved domain that specifies interactions with Hsp70s. J. Cell Biol. 129:979-988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 201.Schmid, D., A. Baici, H. Gehring, and P. Christen. 1994. Kinetics of molecular chaperone action. Science 263:971-973. [DOI] [PubMed] [Google Scholar]
- 202.Schulte, T. W., S. Akinaga, T. Murakata, T. Agatsuma, S. Sugimoto, H. Nakano, Y. S. Lee, B. B. Simen, Y. Argon, S. Felts, D. O. Toft, L. M. Neckers, and S. V. Sharma. 1999. Interaction of radicicol with members of the heat shock protein 90 family of molecular chaperones. Mol. Endocrinol. 13:1435-1448. [DOI] [PubMed] [Google Scholar]
- 203.Schulz, J. B., D. Bremen, J. C. Reed, J. Lommatzsch, S. Takayama, U. Wullner, P. A. Loschmann, T. Klockgether, and M. Weller. 1997. Cooperative interception of neuronal apoptosis by BCL-2 and BAG-1 expression: prevention of caspase activation and reduced production of reactive oxygen species. J. Neurochem. 69:2075-2086. [DOI] [PubMed] [Google Scholar]
- 204.Schumacher, R. J., W. J. Hansen, B. C. Freeman, E. Alnemri, G. Litwack, and D. O. Toft. 1996. Cooperative action of Hsp70, Hsp90, and DnaJ proteins in protein renaturation. Biochemistry 35:14889-14898. [DOI] [PubMed] [Google Scholar]
- 205.Schumacher, R. J., R. Hurst, W. P. Sullivan, N. J. McMahon, D. O. Toft, and R. L. Matts. 1994. ATP-dependent chaperoning activity of reticulocyte lysate. J. Biol. Chem. 269:9493-9499. [PubMed] [Google Scholar]
- 206.Sellers, W. R., J. W. Rodgers, and W. G. Kaelin, Jr. 1995. A potent transrepression domain in the retinoblastoma protein induces a cell cycle arrest when bound to E2F sites. Proc. Natl. Acad. Sci. USA 92:11544-11548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 207.Seo, J. S., Y. M. Park, J. I. Kim, E. H. Shim, C. W. Kim, J. J. Jang, S. H. Kim, and W. H. Lee. 1996. T cell lymphoma in transgenic mice expressing the human Hsp70 gene. Biochem. Biophys. Res. Commun. 218:582-587. [DOI] [PubMed] [Google Scholar]
- 208.Sharma, S. V., T. Agatsuma, and H. Nakano. 1998. Targeting of the protein chaperone, HSP90, by the transformation suppressing agent, radicicol. Oncogene 16:2639-2645. [DOI] [PubMed] [Google Scholar]
- 209.Sheng, Q., D. Denis, M. Ratnofsky, T. M. Roberts, J. A. DeCaprio, and B. Schaffhausen. 1997. The DnaJ domain of polyomavirus large T antigen is required to regulate Rb family tumor suppressor function. J. Virol. 71:9410-9416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 210.Sheng, Q., T. M. Love, and B. Schaffhausen. 2000. J domain-independent regulation of the Rb family by polyomavirus large T antigen. J. Virol. 74:5280-5290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 211.Sherr, C. J. 1996. Cancer cell cycles. Science 274:1672-1677. [DOI] [PubMed] [Google Scholar]
- 212.Sherr, C. J. 2000. The Pezcoller lecture: cancer cell cycles revisited. Cancer Res. 60:3689-3695. [PubMed] [Google Scholar]
- 213.Silver, P. A., and J. C. Way. 1993. Eukaryotic DnaJ homologs and the specificity of Hsp70 activity. Cell 74:5-6. [DOI] [PubMed] [Google Scholar]
- 214.Simon, M. A., P. O. Ilyinskii, G. B. Baskin, H. Y. Knight, D. R. Pauley, and A. A. Lackner. 1999. Association of simian virus 40 with a central nervous system lesion distinct from progressive multifocal leukoencephalopathy in macaques with AIDS. Am. J. Pathol. 154:437-446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 215.Slinskey, A., D. Barnes, and J. M. Pipas. 1999. Simian virus 40 large T antigen J domain and Rb-binding motif are sufficient to block apoptosis induced by growth factor withdrawal in a neural stem cell line. J. Virol. 73:6791-6799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 216.Smith, D. F., W. P. Sullivan, T. N. Marion, K. Zaitsu, B. Madden, D. J. McCormick, and D. O. Toft. 1993. Identification of a 60-kilodalton stress-related protein, p60, which interacts with hsp90 and hsp70. Mol. Cell. Biol. 13:869-876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 217.Snowden, A. W., and N. D. Perkins. 1998. Cell cycle regulation of the transcriptional coactivators p300 and CREB binding protein. Biochem. Pharmacol. 55:1947-1954. [DOI] [PubMed] [Google Scholar]
- 218.Sock, E., J. Enderich, and M. Wegner. 1999. The J domain of papovaviral large tumor antigen is required for synergistic interaction with the POU-domain protein Tst-1/Oct6/SCIP. Mol. Cell. Biol. 19:2455-2464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 219.Sompayrac, L., and K. J. Danna. 1988. A new SV40 mutant that encodes a small fragment of T antigen transforms established rat and mouse cells. Virology 163:391-396. [DOI] [PubMed] [Google Scholar]
- 220.Soti, C., and P. Csermely. 1998. Molecular chaperones in the etiology and therapy of cancer. Pathol. Oncol. Res. 4:316-321. [DOI] [PubMed] [Google Scholar]
- 221.Spence, S. L., and J. M. Pipas. 1994. Simian virus 40 large T antigen host range domain functions in virion assembly. J. Virol. 68:4227-4240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 222.Spence, S. L., and J. M. Pipas. 1994. SV40 large T antigen functions at two distinct steps in virion assembly. Virology 204:200-209. [DOI] [PubMed] [Google Scholar]
- 223.Srinivasan, A., A. J. McClellan, J. Vartikar, I. Marks, P. Cantalupo, Y. Li, P. Whyte, K. Rundell, J. L. Brodsky, and J. M. Pipas. 1997. The amino-terminal transforming region of simian virus 40 large T and small t antigens functions as a J domain. Mol. Cell. Biol. 17:4761-4773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 224.Srinivasan, A., K. W. Peden, and J. M. Pipas. 1989. The large tumor antigen of simian virus 40 encodes at least two distinct transforming functions. J. Virol. 63:5459-5463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 225.Sriram, M., J. Osipiuk, B. Freeman, R. Morimoto, and A. Joachimiak. 1997. Human Hsp70 molecular chaperone binds two calcium ions within the ATPase domain. Structure 5:403-414. [DOI] [PubMed] [Google Scholar]
- 226.Stein, R. W., M. Corrigan, P. Yaciuk, J. Whelan, and E. Moran. 1990. Analysis of E1A-mediated growth regulation functions: binding of the 300-kilodalton cellular product correlates with E1A enhancer repression function and DNA synthesis-inducing activity. J. Virol. 64:4421-4427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 227.Struhl, K. 1998. Histone acetylation and transcriptional regulatory mechanisms. Genes Dev. 12:599-606. [DOI] [PubMed] [Google Scholar]
- 228.Stuart, J. K., D. G. Myszka, L. Joss, R. S. Mitchell, S. M. McDonald, Z. Xie, S. Takayama, J. C. Reed, and K. R. Ely. 1998. Characterization of interactions between the anti-apoptotic protein BAG-1 and Hsc70 molecular chaperones. J. Biol. Chem. 273:22506-22514. [DOI] [PubMed] [Google Scholar]
- 229.Stubdal, H., J. Zalvide, K. S. Campbell, C. Schweitzer, T. M. Roberts, and J. A. DeCaprio. 1997. Inactivation of pRB-related proteins p130 and p107 mediated by the J domain of simian virus 40 large T antigen. Mol. Cell. Biol. 17:4979-4990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 230.Stubdal, H., J. Zalvide, and J. A. DeCaprio. 1996. Simian virus 40 large T antigen alters the phosphorylation state of the RB-related proteins p130 and p107. J. Virol. 70:2781-2788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 231.Suh, W. C., W. F. Burkholder, C. Z. Lu, X. Zhao, M. E. Gottesman, and C. A. Gross. 1998. Interaction of the Hsp70 molecular chaperone, DnaK, with its cochaperone DnaJ. Proc. Natl. Acad. Sci. USA 95:15223-15228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 232.Suh, W. C., C. Z. Lu, and C. A. Gross. 1999. Structural features required for the interaction of the Hsp70 molecular chaperone DnaK with its cochaperone DnaJ. J. Biol. Chem. 274:30534-30539. [DOI] [PubMed] [Google Scholar]
- 233.Sullivan, C. S., P. Cantalupo, and J. M. Pipas. 2000. The molecular chaperone activity of simian virus 40 large T antigen is required to disrupt Rb-E2F family complexes by an ATP-dependent mechanism. Mol. Cell. Biol. 20:6233-6243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 234.Sullivan, C. S., S. P. Gilbert, and J. M. Pipas. 2001. ATP-dependent simian virus 40 T-antigen-Hsc70 complex formation. J. Virol. 75:1601-1610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 235.Sullivan, C. S., and J. M. Pipas. 2001. The virus-chaperone connection. Virology 287:1-8. [DOI] [PubMed] [Google Scholar]
- 236.Sullivan, C. S., J. D. Tremblay, S. W. Fewell, J. A. Lewis, J. L. Brodsky, and J. M. Pipas. 2000. Species-specific elements in the large T-antigen J domain are required for cellular transformation and DNA replication by simian virus 40. Mol. Cell. Biol. 20:5749-5757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 237.Symonds, H., L. Krall, L. Remington, M. Saenz-Robles, S. Lowe, T. Jacks, and T. Van Dyke. 1994. p53-dependent apoptosis suppresses tumor growth and progression in vivo. Cell 78:703-711. [DOI] [PubMed] [Google Scholar]
- 238.Symonds, H. S., S. A. McCarthy, J. Chen, J. M. Pipas, and T. Van Dyke. 1993. Use of transgenic mice reveals cell-specific transformation by a simian virus 40 T-antigen amino-terminal mutant. Mol. Cell. Biol. 13:3255-3265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 239.Takahashi, Y., J. B. Rayman, and B. D. Dynlacht. 2000. Analysis of promoter binding by the E2F and pRB families in vivo: distinct E2F proteins mediate activation and repression. Genes Dev. 14:804-816. [PMC free article] [PubMed] [Google Scholar]
- 240.Terada, K., and M. Mori. 2000. Human DnaJ homologs dj2 and dj3, and bag-1 are positive cochaperones of hsc70. J. Biol. Chem. 275:24728-24734. [DOI] [PubMed] [Google Scholar]
- 241.Tevethia, M. J., R. H. Bonneau, J. W. Griffith, and L. Mylin. 1997. A simian virus 40 large T-antigen segment containing amino acids 1 to 127 and expressed under the control of the rat elastase-1 promoter produces pancreatic acinar carcinomas in transgenic mice. J. Virol. 71:8157-8166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 242.Tevethia, M. J., H. A. Lacko, T. D. Kierstead, and D. L. Thompson. 1997. Adding an Rb-binding site to an N-terminally truncated simian virus 40 T antigen restores growth to high cell density, and the T common region in trans provides anchorage-independent growth and rapid growth in low serum concentrations. J. Virol. 71:1888-1896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 243.Theyssen, H., H. P. Schuster, L. Packschies, B. Bukau, and J. Reinstein. 1996. The second step of ATP binding to DnaK induces peptide release. J. Mol. Biol. 263:657-670. [DOI] [PubMed] [Google Scholar]
- 244.Thompson, D. L., D. Kalderon, A. E. Smith, and M. J. Tevethia. 1990. Dissociation of Rb-binding and anchorage-independent growth from immortalization and tumorigenicity using SV40 mutants producing N-terminally truncated large T antigens. Virology 178:15-34. [DOI] [PubMed] [Google Scholar]
- 245.Trimarchi, J. M., B. Fairchild, R. Verona, K. Moberg, N. Andon, and J. A. Lees. 1998. E2F-6, a member of the E2F family that can behave as a transcriptional repressor. Proc. Natl. Acad. Sci. USA 95:2850-2855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 246.Trowbridge, P. W., and R. J. Frisque. 1995. Identification of three new JC virus proteins generated by alternative splicing of the early viral mRNA. J. Neurovirol. 1:195-206. [DOI] [PubMed] [Google Scholar]
- 247.Tsai, J., and M. G. Douglas. 1996. A conserved HPD sequence of the J-domain is necessary for YDJ1 stimulation of Hsp70 ATPase activity at a site distinct from substrate binding. J. Biol. Chem. 271:9347-9354. [DOI] [PubMed] [Google Scholar]
- 248.van Wijnen, A. J., M. F. van Gurp, M. C. de Ridder, C. Tufarelli, T. J. Last, M. Birnbaum, P. S. Vaughan, A. Giordano, W. Krek, E. J. Neufeld, J. L. Stein, and G. S. Stein. 1996. CDP/cut is the DNA-binding subunit of histone gene transcription factor HiNF-D: a mechanism for gene regulation at the G1/S phase cell cycle transition point independent of transcription factor E2F. Proc. Natl. Acad. Sci. USA 93:11516-11521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 249.Wall, D., M. Zylicz, and C. Georgopoulos. 1994. The NH2-terminal 108 amino acids of the Escherichia coli DnaJ protein stimulate the ATPase activity of DnaK and are sufficient for lambda replication. J. Biol. Chem. 269:5446-5451. [PubMed] [Google Scholar]
- 250.Wang, H. G., Y. Rikitake, M. C. Carter, P. Yaciuk, S. E. Abraham, B. Zerler, and E. Moran. 1993. Identification of specific adenovirus E1A N-terminal residues critical to the binding of cellular proteins and to the control of cell growth. J. Virol. 67:476-488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 251.Wang, W. B., I. Bikel, E. Marsilio, D. Newsome, and D. M. Livingston. 1994. Transrepression of RNA polymerase II promoters by the simian virus 40 small t antigen. J. Virol. 68:6180-6187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 252.Wang, Z. M., H. Yang, and D. M. Livingston. 1998. Endogenous E2F-1 promotes timely G0 exit of resting mouse embryo fibroblasts. Proc. Natl. Acad. Sci. USA 95:15583-15586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 253.Webster, M. A., N. Martin-Soudant, A. Nepveu, R. D. Cardiff, and W. J. Muller. 1998. The induction of uterine leiomyomas and mammary tumors in transgenic mice expressing polyomavirus (PyV) large T (LT) antigen is associated with the ability of PyV LT antigen to form specific complexes with retinoblastoma and CUTL1 family members. Oncogene 16:1963-1972. [DOI] [PubMed] [Google Scholar]
- 254.Weinberg, R. A. 1995. The retinoblastoma protein and cell cycle control. Cell 81:323-330. [DOI] [PubMed] [Google Scholar]
- 255.Weisshart, K., M. K. Bradley, B. M. Weiner, C. Schneider, I. Moarefi, E. Fanning, and A. K. Arthur. 1996. An N-terminal deletion mutant of simian virus 40 (SV40) large T antigen oligomerizes incorrectly on SV40 DNA but retains the ability to bind to DNA polymerase α and replicate SV40 DNA in vitro. J. Virol. 70:3509-3516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 256.Welch, P. J., and J. Y. Wang. 1993. A C-terminal protein-binding domain in the retinoblastoma protein regulates nuclear c-Abl tyrosine kinase in the cell cycle. Cell 75:779-790. [DOI] [PubMed] [Google Scholar]
- 257.Wells, J., K. E. Boyd, C. J. Fry, S. M. Bartley, and P. J. Farnham. 2000. Target gene specificity of E2F and pocket protein family members in living cells. Mol. Cell. Biol. 20:5797-5807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 258.Wharton, W., J. Savell, W. D. Cress, E. Seto, and W. J. Pledger. 2000. Inhibition of mitogenesis in Balb/c-3T3 cells by trichostatin A. Multiple alterations in the induction and activation of cyclin-cyclin-dependent kinase complexes. J. Biol. Chem. 275:33981-33987. [DOI] [PubMed] [Google Scholar]
- 259.Whitesell, L., S. D. Shifrin, G. Schwab, and L. M. Neckers. 1992. Benzoquinonoid ansamycins possess selective tumoricidal activity unrelated to src kinase inhibition. Cancer Res. 52:1721-1728. [PubMed] [Google Scholar]
- 260.Whyte, P., K. J. Buchkovich, J. M. Horowitz, S. H. Friend, M. Raybuck, R. A. Weinberg, and E. Harlow. 1988. Association between an oncogene and an anti-oncogene: the adenovirus E1A proteins bind to the retinoblastoma gene product. Nature 334:124-129. [DOI] [PubMed] [Google Scholar]
- 261.Whyte, P., N. M. Williamson, and E. Harlow. 1989. Cellular targets for transformation by the adenovirus E1A proteins. Cell 56:67-75. [DOI] [PubMed] [Google Scholar]
- 262.Wiggan, O., A. Taniguchi-Sidle, and P. A. Hamel. 1998. Interaction of the pRB-family proteins with factors containing paired-like homeodomains. Oncogene 16:227-236. [DOI] [PubMed] [Google Scholar]
- 263.Wold, M. S., D. H. Weinberg, D. M. Virshup, J. J. Li, and T. J. Kelly. 1989. Identification of cellular proteins required for simian virus 40 DNA replication. J. Biol. Chem. 264:2801-2809. [PubMed] [Google Scholar]
- 264.Xing, X., A. Matin, D. Yu, W. Xia, F. Sorgi, L. Huang, and M. C. Hung. 1996. Mutant SV40 large T antigen as a therapeutic agent for HER-2/neu-overexpressing ovarian cancer. Cancer Gene Ther. 3:168-174. [PubMed] [Google Scholar]
- 265.Yaciuk, P., M. C. Carter, J. M. Pipas, and E. Moran. 1991. Simian virus 40 large-T antigen expresses a biological activity complementary to the p300-associated transforming function of the adenovirus E1A gene products. Mol. Cell. Biol. 11:2116-2124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 266.Yaglom, J. A., A. L. Goldberg, D. Finley, and M. Y. Sherman. 1996. The molecular chaperone Ydj1 is required for the p34CDC28-dependent phosphorylation of the cyclin Cln3 that signals its degradation. Mol. Cell. Biol. 16:3679-3684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 267.Yehiely, F., and M. Oren. 1992. The gene for the rat heat-shock cognate, hsc70, can suppress oncogene-mediated transformation. Cell Growth Differ. 3:803-809. [PubMed] [Google Scholar]
- 268.Zalvide, J., and J. A. DeCaprio. 1995. Role of pRb-related proteins in simian virus 40 large-T-antigen-mediated transformation. Mol. Cell. Biol. 15:5800-5810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 269.Zalvide, J., H. Stubdal, and J. A. DeCaprio. 1998. The J domain of simian virus 40 large T antigen is required to functionally inactivate RB family proteins. Mol. Cell. Biol. 18:1408-1415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 270.Zerrahn, J., U. Knippschild, T. Winkler, and W. Deppert. 1993. Independent expression of the transforming amino-terminal domain of SV40 large I antigen from an alternatively spliced third SV40 early mRNA. EMBO J. 12:4739-4746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 271.Zhang, Y., and S. P. Chellappan. 1995. Cloning and characterization of human DP2, a novel dimerization partner of E2F. Oncogene 10:2085-2093. [PubMed] [Google Scholar]
- 272.Zheng, N., E. Fraenkel, C. O. Pabo, and N. P. Pavletich. 1999. Structural basis of DNA recognition by the heterodimeric cell cycle transcription factor E2F-DP. Genes Dev. 13:666-674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 273.Zhu, J., P. W. Rice, L. Gorsch, M. Abate, and C. N. Cole. 1992. Transformation of a continuous rat embryo fibroblast cell line requires three separate domains of simian virus 40 large T antigen. J. Virol. 66:2780-2791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 274.Zhu, J. Y., P. W. Rice, M. Chamberlain, and C. N. Cole. 1991. Mapping the transcriptional transactivation function of simian virus 40 large T antigen. J. Virol. 65:2778-2790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 275.Zhu, X., X. Zhao, W. F. Burkholder, A. Gragerov, C. M. Ogata, M. E. Gottesman, and W. A. Hendrickson. 1996. Structural analysis of substrate binding by the molecular chaperone DnaK. Science 272:1606-1614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 276.Ziebold, U., T. Reza, A. Caron, and J. A. Lees. 2001. E2F3 contributes both to the inappropriate proliferation and to the apoptosis arising in Rb mutant embryos. Genes Dev. 15:386-391. [DOI] [PMC free article] [PubMed]
- 277.Zimmer, Y., D. Milo-Landesman, A. Svetlanov, and S. Efrat. 1999. Genes induced by growth arrest in a pancreatic beta cell line: identification by analysis of cDNA arrays. FEBS Lett. 457:65-70. [DOI] [PubMed] [Google Scholar]