

Abstract
The functional properties and cellular localization of the human neuronal α7 nicotinic acetylcholine (AcCho) receptor (α7 AcChoR) and its L248T mutated (mut) form were investigated by expressing them alone or as gene fusions with the enhanced version of the green fluorescent protein (GFP). Xenopus oocytes injected with wild-type (wt), mutα7, or the chimeric subunit cDNAs expressed receptors that gated membrane currents when exposed to AcCho. As already known, AcCho currents generated by wtα7 receptors decay much faster than those elicited by the mutα7 receptors. Unexpectedly, the fusion of GFP to the wt and mutated α7 receptors led to opposite results: the AcCho-current decay of the wt receptors became slower, whereas that of the mutated receptors was accelerated. Furthermore, repetitive applications of AcCho led to a considerable “run-down” of the AcCho currents generated by mutα7-GFP receptors, whereas those of the wtα7-GFP receptors remained stable or increased in amplitude. The AcCho-current run-down of mutα7-GFP oocytes was accompanied by a marked decrease of α-bungarotoxin binding activity. Fluorescence, caused by the chimeric receptors expressed, was seen over the whole oocyte surface but was more intense and abundant in the animal hemisphere, whereas it was much weaker in the vegetal hemisphere. We conclude that fusion of GFP to wtα7 and mutα7 receptors provides powerful tools to study the distribution and function of α7 receptors. We also conclude that fused genes do not necessarily recapitulate all of the properties of the original receptors. This fact must be borne close in mind whenever reporter genes are attached to proteins.
Fluorescent tagging of cellular proteins is a very useful, and aesthetically pleasant, method for studying the intracellular traffic and distribution of many proteins, including neurotransmitter receptors. It is commonly assumed that the tagged proteins function in exactly the same way as the native nontagged proteins. To determine whether that is the case for neurotransmitter receptors, we chose to study two human receptors that are homomeric: the GABAρ1 (1) and now the α7 nicotinic receptors.
The neuronal α7 nicotinic acetylcholine receptor (α7-AcChoR) is widely expressed in the central nervous system and, being located mainly at nerve terminals, it is believed to be involved in regulating the neurotransmitter release that mediates fast cholinergic neurotransmission (2, 3). In heterologous expression systems, including the Xenopus oocyte, the α7-subunit forms functional homomeric AcChoRs that show an unusually fast desensitization and high permeability to Ca2+ (4–8). Even though it is not yet clear to what extent the α7 homomeric composition also applies in vivo, we and others have found it of interest to investigate the functional profile of homomeric α7 receptors, because they may be involved in the pathogenesis of many neurological disorders (9–16).
For this study we constructed a chimera in which the enhanced form of the green fluorescent protein (GFP) was fused to the C terminus of the wild-type (wt) human α7 subunit (wtα7-GFP). A similar construct (mutα7-GFP) was made with the Leu-to-Thr 248 (L248T) mutant α7 subunit. This mutation is known to decrease the rate of desensitization of the receptors and, very surprisingly, converts some antagonists of wtα7 AcChoRs into agonists (5, 7, 17–21). Both constructs led to the expression of functional receptors in oocytes, and the following is a study of the functional properties and distribution of the receptors expressed. We show that both wild-type and mutant α7 AcChoRs fused with GFP are grouped in clusters at or near the oocyte plasma membrane and that fusion of GFP to the α7 subunit importantly alters the desensitization of the receptors expressed, suggesting complex interactions between the ionic channel and the C terminus domains, interactions that substantially influence the receptor gating kinetics.
Materials and Methods
Chimera Constructions.
Expression plasmids for human α7-GFP and mutα7-GFP chimeras were constructed by inserting the α7 coding region into the SalI/BamHI restriction sites of vector pEGFP-N2 (CLONTECH) which encodes an enhanced (E)GFP variant. The α7 ORFs were amplified from original α7pcDNA3 constructs (22) by PCR, using the Platinum Pfx High Fidelity DNA polymerase (Invitrogen). The primer pair used for amplification corresponds to the 5′ and the 3′ α7 cDNA ends including restriction sites for SalI and BamHI (sense oligonucleotide, 5′-CAGTCGACAAGCATCCGCCACCATGCGCTGCTCGCCGGGA-3′; antisense oligonucleotide, 5′-CAGGGATCCTCGCAAAGTCTTTGGCGGCCTCCACGAAG-3′). By using the pEGFP-N2 vector, α7 ORFs were placed upstream of the EGFP gene to produce fluorescent fusion proteins.
Oocyte Injection.
Recombinant plasmids encoding human wt α7, mut α7, wt α7-GFP, or mut α7-GFP neuronal nicotinic subunits in the pcDNA3 vector were intranuclearly injected into stage V-VI oocytes (2 ng of cDNA in 10 nl of buffer). Preparation of oocytes and nuclear injection procedures were as detailed elsewhere (4, 7).
Electrophysiology.
Two to four days after injection, whole cell membrane currents were recorded in voltage-clamped oocytes by using two microelectrodes filled with 3 M KCl (23). The oocytes were placed in a recording chamber (volume, about 0.1 ml) and perfused continuously, 9–11 ml/min, with oocyte Ringer's solution (82.5 mM NaCl/2.5 mM KCl/2.5 mM CaCl2/1 mM MgCl2/5 mM Hepes/adjusted to pH 7.4 with NaOH) or normal frog Ringer's solution (23) at room temperature (20–22°C). To obtain dose/response relations, AcCho was applied to the oocytes at 3-min intervals. The half dissociation constants (EC50) of AcCho were estimated by fitting the data to the Hill equation:
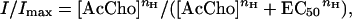 |
1 |
where [AcCho] is the dose of AcCho, nH is the Hill coefficient, and Imax is the maximum AcCho current response. The times for 10% or 50% current decay (T0.1 or T0.5), that is, the time taken for the current to decay from its peak to 90% or 50%, respectively, were used to estimate the rate of receptor desensitization. Drugs were applied by adding them to the superfusing fluid. All drugs were purchased from Sigma, except methyllycaconitine (MLA; Research Biochemicals, Natick, MA).
Binding Assay on Oocytes.
Oocytes that had been injected with mutα7 or mutα7-GFP cDNAs were first tested electrophysiologically and then selected to estimate toxin binding. Some oocytes were exposed to 1 mM AcCho for 90 min, after which plasma-membrane binding of α-bungarotoxin (α-BuTX) was determined (20). To measure toxin binding, nonstimulated control and AcCho-stimulated oocytes were incubated for 2 h in oocyte Ringer's medium containing 40 nM [α-125I]BuTX (Amersham Biosciences). Some oocytes were preincubated for 1 h with 10 μM MLA before applying [α-125I]BuTX plus MLA. After incubation, the labeled medium was removed, and the oocytes were washed rapidly 5 times with Ringer's solution (or Ringer's solution plus MLA) and counted in a Beckman Coulter γ counter. The nonspecific binding was measured in noninjected oocytes.
Confocal Imaging.
Two laser confocal microscopes were used to examine the oocytes: a Bio-Rad MRC600 with an argon-ion laser and filters set to observe GFP fluorescence and a homemade confocal microscope built by I. Parker and N. Callamaras (Univ. of California, Irvine), which allowed for a more detailed z-scan imaging.
Results
Functional Expression of Human wtα7-GFP Receptors.
Oocytes injected with wtα7 subunit cDNA (wtα7 oocytes) and held at −60 to −100 mV responded to AcCho (100 μM ≈ EC50; refs. 4, 7, and 8) with inward currents (AcCho currents), whose amplitude depended on the donors and varied greatly from oocyte to oocyte, ranging from about 50 nA to over 50 μA. Independently of their amplitude, AcCho currents decayed rapidly with a T0.5 of 0.29 ± 0.08 s (mean ± SE 15 oocytes held at −100 mV, 2 donors; e.g., Fig. 1A). Repetitive applications of AcCho did not substantially modify the peak amplitude or decay of the AcCho currents, indicating a fast recovery from receptor desensitization (Fig. 1B). The T0.5 values remained substantially the same after raising the AcCho concentration to 1 mM (0.20 ± 0.05 s; n = 5) or after depolarizing the oocytes from −100 to −60 mV (0.23 ± 0.03 s; n = 5; e.g., Fig. 1A). Thus, under our experimental conditions, the current desensitization of wtα7 receptors seems to be maximal at −60 mV and 100 μM AcCho.
Figure 1.

Properties of wtα7 and wtα7-GFP AcChoRs expressed in Xenopus oocytes. (A) AcCho currents in an oocyte injected with wtα7 subunit cDNA and held at the indicated potentials. Horizontal bars indicate the timing of AcCho applications. (B) AcCho-current amplitudes during consecutive 4-s applications of AcCho (1 mM) to wtα7 or wtα7-GFP oocytes held at −100 mV. Points represent means ± SE (4 oocytes; 1 donor). The peak currents were normalized to 2.7 μA (○) and to 0.5 μA (●). (C) Currents evoked in an oocyte injected with wtα7-GFP cDNA. Note slower decay at −60 mV compared with A. (D) Dose-response relationships in oocytes held at −60 mV expressing wtα7 or wtα7-GFP receptors. Each point represents mean (± SE) from 5 oocytes (2 donors). Peak currents were normalized to 1.5 μA (○) and 0.36 μA (●), with AcCho 3 mM. (○) EC50 = 75 μM; nH = 2.7. (●) EC50 = 79 μM; nH = 2.3.
Oocytes injected with wtα7-GFP cDNA also expressed functional receptors, but the AcCho currents generated were generally smaller than those of the wtα7 oocytes (cf. Fig. 1 A and C). Nevertheless, many “wtα7-GFP oocytes” generated very large currents (up to 50 μA at −60 mV), indicating a strong expression of functional wtα7-GFP chimeric receptors. The AcCho currents had again a fast desensitization and recovered promptly after AcCho washout. During repetitive AcCho applications, the AcCho-current amplitude was well maintained in some experiments (Fig. 1B), but in others with longer applications of AcCho, the amplitude of successive responses increased and reached a value that was 2–3 times that of the first AcCho response (Fig. 2). The current decay was not greatly altered by raising the AcCho concentration to 1 mM (e.g., cf. Fig. 1 C and A). However, with 100 μM AcCho, the T0.5 values at −100 mV matched (0.29 ± 0.09 s; 11/2) and at −60 mV were significantly larger (T0.5 = 0.53 ± 0.09 s; Student's t test, P < 0.001; e.g., Fig. 1 A and C) than those of wtα7 oocytes, indicating both a different voltage-dependence and a slowing of the AcCho-current desensitization at −60 mV. The decreased rate of wtα7-GFP receptor desensitization was not caused by a change in receptor affinity, because the EC50 values of wtα7 and wtα7-GFP oocytes were similar (Fig. 1D).
Figure 2.

Increase of consecutive AcCho currents in an oocyte expressing wtα7-GFP. The oocyte was held at −60 mV and repetitively exposed to AcCho 100 μM for ≈30 s.
Functional Expression of Human mutα7-GFP Receptors.
It is known that mutation of Leu-to-Thr 248, within the AcChoR-channel domain, considerably slows down the process of α7 receptor desensitization (8), offering a model of AcChoR dysfunction possibly associated with neurodegenerative diseases (24). To gain further insights on the behavior of homomeric α7 receptors fused to GFP, experiments were also performed in oocytes injected with the mutα7 subunit or with the mutα7-GFP chimera.
Mutα7 oocytes, held at −60 or −100 mV and exposed to 0.3 μM AcCho (≈EC50) (25), generated AcCho currents that decayed slowly (T0.1 > 10 s), had peak amplitudes that ranged from 1 to 40 μA, and recovered fully after a few minutes (Fig. 3). The peak amplitude increased by ≈1.9-fold as the AcCho concentration was raised to 100 μM (“plateau” concentration) and even with this high concentration the T0.1 was always greater than 4 s. In parallel experiments, mutα7-GFP oocytes generated AcCho currents that were again of variable amplitude but generally smaller then those of mutα7 oocytes, rarely as high as 40 μA. Similarly to mutα7 oocytes, the AcCho-current amplitude increased ≈1.9-fold as the AcCho concentration was raised from 0.3 to 100 μM (Fig. 3C), suggesting that mutα7 and mutα7-GFP oocytes had similar AcCho sensitivities. However, and in sharp contrast to mut oocytes, mut-GFP oocytes always generated AcCho currents that had a faster decay: T0.1 values averaged 1.9 ± 0.2 s (15 oocytes, 3 donors) at −100 mV membrane potential and 0.3 μM AcCho and 0.8 ± 0.1 s after raising the AcCho concentration to 100 μM (Fig. 3C), and 3.6 ± 1.2 s with 100 μM AcCho at −60 mV (data not shown). Furthermore, the AcCho currents generated by mutα7 receptors decreased only less than 10% during repetitive activation (Fig. 3D). In contrast, repetitive activation of mutα7-GFP receptors led to a drastic decrease of the AcCho-current amplitude. Specifically, at −60 mV and 100 μM AcCho, the AcCho current decreased to ≈30% of the control in the experiments illustrated in Fig. 3D; and to 3% in that of Fig. 4. After the AcCho-current run-down, some oocytes were allowed to rest at 16°C for 18–24 h, after which they again generated large AcCho currents with a similar run-down (Fig. 4). It seems that the AcCho-current run-down is not restricted to applications of high concentrations of AcCho, because the currents elicited by 0.1 μM AcCho also decreased progressively in amplitude (e.g., in Fig. 4, first current = 142 nA, third = 105 nA).
Figure 3.

Fast decay and run-down of AcCho currents elicited by mutα7-GFP receptors. AcCho currents evoked in oocytes expressing mutα7 (A) or mutα7-GFP (C) receptors. Note the faster AcCho-current decay compared with A. (B) Superimposed traces of AcCho currents in two other oocytes. AcCho-current amplitudes: 7.3 μA (mutα7); 1.4 μA (mutα7-GFP). Holding potential, −100 mV. (D) AcCho-current amplitudes of consecutive responses in oocytes injected with mutα7 or mutα7-GFP subunit cDNAs. Points represent means (± SE) of 7 oocytes (2 donors). AcCho (100 μM) applied at −100 mV as in A. Peak currents normalized to 5.1 μA (mutα7) or to 1.9 μA (mutα7-GFP).
Figure 4.

Run-down of AcCho currents in an oocyte expressing mutα7-GFP receptors. Oocyte held at −60 mV and repetitively exposed to AcCho for ≈60 s at indicated concentrations. Open symbols show the first series of application, which was repeated 24 h later (closed symbols). Note the partial recovery and the similar run-down after 24 h.
Because serotonin (5HT) is a competitive agonist of mutα7 receptors (7), it was of interest to determine whether the chimeric mutα7-GFP receptors were also activated by 5HT. Mutα7-GFP oocytes exposed to 5HT (10 μM ≈ EC50; 25) generated inward currents with profiles similar to those elicited by 0.3 μM AcCho (not shown). The ratio of 5HT currents elicited by 10 μM vs. 1 mM (plateau concentration, 25) was 59% in mutα7 vs. 56% in mutα7-GFP oocytes, indicating that mutα7 and mutα7-GFP receptors have a similar sensitivity to 5HT. All this evidence suggests that the agonist-binding sites of mutα7 receptors are not greatly modified by the fusion to GFP. Moreover, the amplitude of the 5HT current was also reduced during the run-down caused by repetitive AcCho applications.
α-BuTX Binding Activity.
To determine whether long-term exposure to AcCho alters the number of receptors in the plasma membrane, oocytes injected with either mutα7 or mutα7-GFP cDNAs were assayed for α-BuTX binding activity before and after a prolonged application of AcCho. The binding of α-BuTX to mutα7 oocytes decreased only by ≈11% after the oocytes had been exposed to AcCho (1 mM) for 90 min. In sharp contrast, the binding of α-BuTX to mutα7-GFP oocytes decreased by ≈64% after the same treatment (Fig. 5). It should be noted that the binding of α-BuTX to both mutα7 and mutα7-GFP receptors was blocked well by the α7 antagonist MLA, because in both cases the binding was reduced to nearly that seen in noninjected oocytes (Fig. 5).
Figure 5.

α-BuTX binding activity in oocytes injected with either mutα7 or mutα7-GFP subunit cDNAs. Columns indicate α-BuTX binding to untreated oocytes (black columns) or oocytes pretreated for 90 min with 1 mM AcCho (open columns). MLA effectively blocked the binding of α-BuTX to both receptors (striped columns). Each column represents data from 27 oocytes (one experiment of two). The oocytes, tested for AcCho sensitivity (100 μM; −100 mV) before the binding experiment, generated AcCho-current amplitudes ranging from 1.5 to 2.3 μA (mutα7 oocytes) and 0.3 to 2 μA (mutα7-GFP oocytes). (−), noninjected oocytes.
Mutα7 oocytes tested 30 min after the AcCho treatment generated AcCho currents whose amplitudes were the same, or only slightly smaller, than the control currents obtained before AcCho treatment. On the other hand, after a similar treatment, mutα7-GFP oocytes generated little or no AcCho current.
Fluorescent α7 Receptors.
Most of the noninjected oocytes, as well as oocytes injected with the wtα7 and mutα7 subunits, had a marked native fluorescence. Nevertheless, oocytes expressing the α7-GFP chimeras generated a fluorescence that was clearly above the background at or near the cell surface (Fig. 6 A–E), especially in oocytes that generated AcCho currents larger than 2 μA (1 μM AcCho). Oocytes injected with either of the α7-GFP chimeras displayed similar fluorescence patterns (Fig. 6). It is worth noting that many α7 chimera-injected oocytes did not display clear additional GFP fluorescence above the general background, even though they generated large AcCho currents (>3 μA with 1 μM AcCho). The oocytes with low native fluorescent background showed a strong green fluorescence in the animal hemisphere (Fig. 6A), whereas the vegetal hemisphere displayed very low if any fluorescence. Z-scans showed that the fluorescence was concentrated mainly over a region that extended from the surface of the oocyte to a depth of about 5–10 μm (Fig. 6 B and C). The z-scans as well as the xy-scans (Fig. 6 D and E) of the surface of several oocytes indicate that the receptors are distributed in clusters (Fig. 6 D and E), with highly fluorescent “patches” mingling with nonfluorescent areas. These patches were abundant in the animal hemisphere (Fig. 6 D and E) and were very sparse, and less bright, in the vegetal one (Fig. 6 F–H).
Figure 6.

Confocal microscope fluorescent images of oocytes expressing α7-GFP receptors. (A) An oocyte expressing mutα7-GFP. Notice that fluorescence is strongly polarized. Z-scans of animal hemisphere sections: wtα7-GFP (B), mutα7-GFP (C). Surface scans of the animal hemispheres of oocytes expressing wtα7-GFP (D) or mutα7-GFP (E). Surface scan of the vegetal hemisphere of an oocyte expressing wtα7-GFP (F). G and H show a detail (×8 magnification) of the fluorescent clusters sparsely distributed in the vegetal hemisphere of the same oocyte. [Bars = 200 μm (in A ), 5 μm (in B and C), and 20 μm (in D–F).]
Discussion
GFP has been used widely to construct chimeric proteins to study their distribution and function in various expression systems. In this work, we fused GFP to the human α7 subunit in its wild-type and mutated (L248T) forms and expressed them in the Xenopus oocyte, with the main following results. First, the chimeric α7 homomeric receptors are functionally expressed in clusters in the oocyte surface and mainly in the animal hemisphere, as evidenced by robust fluorescence signals and AcCho-activated currents. Second, the chimeric wtα7-GFP receptors, activated by AcCho, generate currents that have a slower decay than those of the wtα7 receptors. In contrast, the chimeric mutα7-GFP receptors generate AcCho currents that have a faster decay and show a considerable run-down after repetitive AcCho stimulation.
Although the “patchy” distribution of fluorescence in the oocyte membrane reported here for the human α7 receptor-GFP chimeras has been observed with other receptor GFP constructs (1, 26), some of our findings were unexpected, and are in marked contrast to observations on other ligand-gated receptors fused to GFP. For example, such fusion does not seem to alter greatly the properties of N-methyl-d-aspartate (NMDA), glycine, GABAA, and GABAρ1 receptors (1, 26–28). Instead, in this work we show that the kinetics of AcCho-current inactivation change appreciably after receptor fusion with GFP, in such a way that desensitization of the chimeric wtα7-GFP receptor becomes slower, whereas that of the chimeric mutα7-GFP receptor becomes faster than their respective non-GFP controls. These findings show that the functional properties of α7 receptors are considerably modified after tagging them with GFP in their carboxyl ends. We have also found important differences in the single-channel kinetics of α7 receptors and their GFP-chimeric counterparts (29).
Receptor desensitization and recovery from desensitization are particularly fast events for wtα7 receptors that, when stimulated by AcCho, generate currents exhibiting a rapid decay in the presence of the transmitter and a maintained amplitude after brief repetitive applications of AcCho. Moreover, the L248T mutation leads to a marked decrease of receptor desensitization. Interestingly, after fusion of GFP to the mutα7 receptor C terminus, the AcCho-current decay becomes faster, and the peak current amplitude is greatly reduced during repetitive AcCho applications. A similar run-down occurs after a prolonged application of AcCho, after which the AcCho current becomes very small or undetectable.
Several mechanisms may account for the AcCho-current run-down. For example, after a prolonged application of AcCho the receptors might be internalized or they could be structurally altered in such a way that they become fully refractory to gating by AcCho and partially to binding of α-BuTX. The slow recovery from the run-down (cf. Fig. 4) could be the result of reactivation of the receptors or to membrane insertion of new receptors derived from intracellular pools. Unfortunately, the results of the α-BuTX binding experiments alone do not discriminate well between these possibilities. But, whatever the explanation turns out to be, the present work already suggests complex molecular interactions between the C terminus domain fused with GFP and the channel domain, interactions that considerably influence receptor desensitization.
Altogether, our results show that the implicit assumption that fusion of ligand-gated receptors with GFP does not alter receptor function is not generally correct and suggests more extensive investigations of chimeric receptors. Moreover, our findings show that fusion of GFP to human neuronal nicotinic α7 receptors provides potent tools to study the synthesis, traffic, and localization of α7 receptors, and it would be interesting to extend this study to neuronal cells in various stages of development. Finally, the functional changes that we found after fusing GFP to the wtα7 and the mutα7 receptors may offer experimental models to study receptor run-down and neurodegeneration associated with receptor desensitization.
Acknowledgments
We thank Drs. F. Grassi and S. Fucile for critical reading of the manuscript. The wtα7 was a gift from Janssen (Belgium). We also thank Drs. I. Parker and J. S. Marchant and the UCI Image facility for the use of confocal microscopes. This work was supported in part by Ministero Università Ricerca Scientifica e Tecnologica (to F.E.) and the National Science Foundation, Neuronal and Glial Mechanisms Grant 9982856 (to R.M.).
Abbreviations
- GFP
green fluorescent protein
- AcCho
acetylcholine
- AcChoR
nicotinic AcCho receptor
- wtα7 AcChoR
wild-type α7 AcChoR
- mutα7 nAcChoR
Leu-to-Thr 248 mutant α7 AcChoR
- wtα7-GFP AcChoR
wtα7 AcChoR fused to GFP
- mutα7-GFP AcChoR
Leu-to-Thr 248 mutant α7 AcChoR fused to GFP
- MLA
methyllycaconitine
- α-BuTX
α-bungarotoxin
- 5HT
serotonin
References
- 1.Martinez-Torres A, Miledi R. Proc Natl Acad Sci USA. 2001;98:1947–1951. doi: 10.1073/pnas.031584898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Gray R, Rajan A S, Radcliffe K A, Yakehiro M, Dani J A. Nature (London) 1996;383:713–716. doi: 10.1038/383713a0. [DOI] [PubMed] [Google Scholar]
- 3.Chang K, Berg D K. J Neurosci. 1999;19:3701–3710. doi: 10.1523/JNEUROSCI.19-10-03701.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Couturier S, Bertrand D, Matter J M, Hernandez M C, Bertrand S, Millar N, Valera S, Barkas T, Ballivet M. Neuron. 1990;5:847–856. doi: 10.1016/0896-6273(90)90344-f. [DOI] [PubMed] [Google Scholar]
- 5.Bertrand D, Devillers-Thiery A, Revah F, Galzi J L, Hussy N, Mulle C, Bertrand S, Ballivet M, Changeux J P. Proc Natl Acad Sci USA. 1992;89:1261–1265. doi: 10.1073/pnas.89.4.1261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Seguela P, Wadiche J, Dineley-Miller K, Dani J A, Patrick J W. J Neurosci. 1993;13:596–604. doi: 10.1523/JNEUROSCI.13-02-00596.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Palma E, Mileo A M, Eusebi F, Miledi R. Proc Natl Acad Sci USA. 1996;93:11231–11235. doi: 10.1073/pnas.93.20.11231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Fucile S, Palma E, Mileo A M, Miledi R, Eusebi F. Proc Natl Acad Sci USA. 2000;97:3643–3648. doi: 10.1073/pnas.050582497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kihara T, Shimohama S, Sawada H, Kimura J, Kume T, Kochiyama H, Maeda T, Akaike A. Ann Neurol. 1997;42:159–163. doi: 10.1002/ana.410420205. [DOI] [PubMed] [Google Scholar]
- 10.Kihara T, Shimohama S, Sawada H, Honda K, Nakamizo T, Shibasaki H, Kume T, Akaike A. J Biol Chem. 2001;276:13541–13546. doi: 10.1074/jbc.M008035200. [DOI] [PubMed] [Google Scholar]
- 11.Liu Q, Berg D K. Proc Natl Acad Sci USA. 2001;98:4734–4739. doi: 10.1073/pnas.081553598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Stassen H H, Bridler R, Hagele S, Hergersberg M, Mehman B, Schinzel A, Weisbrod M, Scharfetter C. Am J Med Genet. 2000;96:173–177. [PubMed] [Google Scholar]
- 13.Leonard S, Breese C, Adams C, Benhammou K, Gault J, Stevens K, Lee M, Adler L, Olincy A, Ross R, Freedman R. Eur J Pharmacol. 2000;393:237–242. doi: 10.1016/s0014-2999(00)00035-2. [DOI] [PubMed] [Google Scholar]
- 14.Freedman R, Coon H, Myles-Worsley M, Orr-Urtreger A, Olincy A, Davis A, Polymeropoulos M, Holik J, Hopkins J, Hoff M, et al. Proc Natl Acad Sci USA. 1997;94:587–592. doi: 10.1073/pnas.94.2.587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Rei R T, Sabbagh M N, Corey-Bloom J, Tirabioschi P, Thal L J. Neurobiol Aging. 2000;5:741–746. doi: 10.1016/s0197-4580(00)00168-8. [DOI] [PubMed] [Google Scholar]
- 16.Banerjee C, Nyengaard J R, Wevers A, de Vos R A, Jansen Steur E N, Lindstrom J, Pilz K, Nowacki S, Bloch W, Schroder H. Neurobiol Dis. 2000;7:666–672. doi: 10.1006/nbdi.2000.0317. [DOI] [PubMed] [Google Scholar]
- 17.Revah F, Bertrand D, Galzi J L, Devillers-Thiery A, Mulle C, Hussy N, Bertrand S, Ballivet M, Changeux J P. Nature (London) 1991;353:846–849. doi: 10.1038/353846a0. [DOI] [PubMed] [Google Scholar]
- 18.Bertrand S, Devillers-Thiery A, Palma E, Buisson B, Edelstein S J, Corringer P J, Changeux J P, Bertrand D. NeuroReport. 1997;8:3591–3596. doi: 10.1097/00001756-199711100-00034. [DOI] [PubMed] [Google Scholar]
- 19.Palma E, Eusebi F, Miledi R. Proc Natl Acad Sci USA. 1997;94:1539–1543. doi: 10.1073/pnas.94.4.1539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Palma E, Maggi L, Barabino B, Eusebi F, Ballivet M. J Biol Chem. 1999;274:18335–18340. doi: 10.1074/jbc.274.26.18335. [DOI] [PubMed] [Google Scholar]
- 21.Palma E, Maggi L, Miledi R, Eusebi F. Proc Natl Acad Sci USA. 1998;95:10246–10250. doi: 10.1073/pnas.95.17.10246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Groot Kormelink P J, Luyten W H M L. FEBS Lett. 1997;400:309–314. doi: 10.1016/s0014-5793(96)01383-x. [DOI] [PubMed] [Google Scholar]
- 23.Miledi R. Proc R Soc London B Biol Sci. 1982;215:491–497. doi: 10.1098/rspb.1982.0056. [DOI] [PubMed] [Google Scholar]
- 24.Changeux J P, Edelstein S J. Curr Opin Neurobiol. 2001;11:369–377. doi: 10.1016/s0959-4388(00)00221-x. [DOI] [PubMed] [Google Scholar]
- 25. Fucile, S., Palma, E., Eusebi, F. & Miledi, R. (2002) Neuroscience, in press. [DOI] [PubMed]
- 26.David-Watine B, Shorte S L, Fucile S, Saint Jan D, Korn H, Bregestovski P. Neuropharmacology. 1999;38:785–792. doi: 10.1016/s0028-3908(99)00015-5. [DOI] [PubMed] [Google Scholar]
- 27.Bueno O F, Robinson L C, Alvarez-Hernandez X, Leidenheimer N J. Brain Res Mol Brain Res. 1998;59:165–177. doi: 10.1016/s0169-328x(98)00129-6. [DOI] [PubMed] [Google Scholar]
- 28.Marshall J, Molloy R, Moss G W, Howe J R, Hughes T E. Neuron. 1995;14:211–215. doi: 10.1016/0896-6273(95)90279-1. [DOI] [PubMed] [Google Scholar]
- 29.Fucile S, Palma E, Martínez-Torres A, Miledi R, Eusebi F. Proc Natl Acad Sci USA. 2002;99:3956–3961. doi: 10.1073/pnas.052699599. [DOI] [PMC free article] [PubMed] [Google Scholar]