

Abstract
Recent work suggests that structural topology plays a key role in determining protein-folding rates and pathways. The refolding rates of small proteins that fold without intermediates are found to correlate with simple structural parameters such as relative contact order, long-range order, or the fraction of short-range contacts. To test and evaluate the role of structural topology experimentally, a set of circular permutants of the ribosomal protein S6 from Thermus thermophilus was analyzed. Despite a wide range of relative contact order, the permuted proteins all fold with similar rates. These results suggest that alternative topological parameters may better describe the role of topology in protein-folding rates.
The amino acid sequence of a protein and its chemical environment determine its native structure (1), but how this structure is determined from sequence remains one of the major unsolved problems in biology. The process of protein folding cannot be accomplished by random search through all possible conformations, because the number of structures available to an unfolded protein is too large; therefore there must be a biased or directional search in order for a protein to reach its native state.
Small Two-State Proteins Show a Wide Range in Folding Rates
The simplest models for studying the protein-folding process are those that refold without intermediates (2). To date there are more than 20 examples of such simple systems. These proteins all fold cooperatively in a mono-exponential fashion from the denatured state, but the rates at which they fold span 6 orders of magnitude. What causes this remarkable variety of refolding rates?
Folding Algorithms Based on the Topology of the Native State Have Predictive Value
Recently, several folding algorithms based on native structural information have been used to predict folding rates and nucleation sites (3–6). These efforts suggest that the topology of the final structure is an important determinant in the mechanism of protein folding.
If native topology plays a major role in protein folding, is there a simple structural parameter that will capture this feature and explain the large range of observed folding rates? Recently, several simple parameters defining topological features of the native state have been shown to correlate well with protein-refolding rates. The first and most commonly used parameter is relative contact order (RCO; ref. 7). Remarkably, RCO, which represents the normalized average sequence separation between contacting residues, was observed to correlate extremely well with the folding rates of a small set of two-state proteins. The lower the RCO, the faster the protein folds. Although it is possible to alter folding rates significantly through point mutations that do not change RCO, and there are proteins with similar RCOs and different folding rates (8), in general this correlation has improved as more two-state proteins have been characterized (9) and can explain the difference in folding rates among a family of structurally homologous proteins (10). Experimental evidence also supports the role of RCO. Loop-insertion mutants in an Src homology 3 domain (11) and chymotrypsin inhibitor 2 (12) varied in folding rate as expected by their change in RCO.
The observed correlation with RCO is purely empirical, and recently other simple structural parameters such as long-range order (13) and the percentage of short-range contacts (14) have been shown to correlate with folding rates as well. Not surprisingly, these parameters all are correlated loosely.
RCO correlates with folding rate over a range of proteins having very little in common except they are small and fold by a two-state mechanism. Across this series it is difficult to evaluate which simple topological parameter best dictates protein-folding rates. Indeed, it is unclear if a single simple parameter can describe such a complicated process. Here we have altered the RCO of a single protein, ribosomal protein S6 from Thermus thermophilus, through circular permutation, linking the N and C termini of the protein and creating alternative termini at three new positions. Such circular permutants should represent a series of proteins that do not vary in their amino acid composition, structure, or types of enthalpic interactions stabilizing the protein. Lindberg et al. (15) have also generated two other permutants of the same protein. The predicted RCO values of these permutants span ≈50% of the range observed in natural proteins. Unlike RCO, however, long-range order and the percentage of short-range contacts will be unperturbed by circular permutation, allowing us to compare these simple parameters in a single experimental system.
Several proteins have been permuted circularly and retain a similar native structure, including T4 lysozyme (16), α-spectrin Src homology 3 (SH3; ref. 17), RNase T1 (18), chymotrypsin inhibitor 2 (CI2; ref. 19), green fluorescent protein (20), and dihydrofolate reductase (21). Two of these, α-spectrin SH3 (22) and CI2 (23), fold with two-state kinetics, and both maintain a two-state folding mechanism after permutation. The permutations did not alter the RCO significantly, and as predicted the effects on the refolding rates were modest, less than 5-fold from the wild-type protein, despite the fact that the folding nuclei of the SH3 permutants were altered (24). These results suggest that circular permutation can be used to examine the role of topology in protein-folding rates.
Materials and Methods
Structural Calculations.
RCOs were calculated according to the method described by Plaxco et al. (7). To predict the RCO of circular permutations, residues in the native structure were renumbered with the new N-terminal residue as one and the previous amino acid as the C terminus. RCO then was recalculated based on the native structure (PDB ID code ).
For long-range order and percentage of short-range calculations, residues are defined as being in contact if they have two nonhydrogen atoms within 4 Å and are not adjacent in sequence. Residues with a separation of 2 to 4 residues are defined as short-range because i, i + 4 contacts occur regularly in α-helical structure. Residues with a separation of five residues or more are considered long-range. In the S6 structure, all contacts of separation five or greater require loop closure for formation.
Gene Synthesis.
A synthetic gene encoding S6 was created by using four DNA oligomers of ≈90 base pairs, two from the sense strand and two from the antisense strand. These oligomers overlap by 18 base pairs and were annealed and extended by PCR. The synthetic gene was inserted into the pAED4 vector (pEM109; ref. 25).
A DNA template for creating circular permutants was created by using PCR to amplify the coding region of pEM109, removing the stop codon, and adding an Ala-Gly-Ala linker containing an NaeI site at both ends. After digestion with NaeI, the reaction products were ligated with T4 ligase. Permutants were created by using PCR to add an NdeI site and methionine codon before the desired N terminus and a stop codon and an EcoRI site after the desired C terminus. The single-copy product was gel-purified and ligated into pAED4 between the NdeI and EcoRI restriction sites. Plasmids pEM111, pEM112, and pEM113 contain S6cp14, S6cp55, and S6cp36, respectively. Plasmids and sequence details are available on request.
Protein Expression and Purification.
All variants were purified as described (26) with the addition of a final reverse-phase HPLC purification step (C18 column proteins eluted with a gradient from 20% acetonitrile/0.1% trifluoroacetic acid to 90% acetonitrile/0.1% trifluoroacetic acid). Final products were confirmed by electrospray ionization mass spectrometry.
A significant fraction of the S6cp14 protein was found in the insoluble fraction (inclusion bodies). Protein was recovered by extracting the sonication pellet with 50% acetonitrile/0.2% trifluoroacetic acid overnight at 25°C. The solution was centrifuged and filtered, and S6cp14 was purified by reverse-phase HPLC from the supernatant.
Stability and Kinetic Measurements.
Circular dichroism (CD) data were collected on an Aviv 62DS spectropolarimeter (25°C, 1-cm cuvette) by using 150 μg⋅ml−1 protein in 20 mM potassium phosphate/50 mM potassium chloride, pH 7.0.
Protein stabilities were determined by monitoring the CD signal at 222 nm as a function of guanidinium chloride (Gdm⋅Cl) concentration. Stabilities in aqueous solution were fit assuming a two-state transition with a linear extrapolation (27). Gdm⋅Cl was from Pierce, and concentrations were determined by refractive index (28).
Protein-folding kinetics were monitored by using the CD signal at 222 nm on an Aviv 202 stopped-flow CD spectropolarimeter. Refolding and unfolding were initiated by diluting a protein stock 1:10 into the final [Gdm⋅Cl]. Rate constants were fit to the equations S = a × e−kt + C, where S is the signal, a is the amplitude, and C is the final signal for single exponentials or when a minor refolding phase was present, and S = a1 × 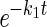 + a2 ×
+ a2 × 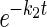 + C, where a2 is the amplitude of the minor phase, and k2 is the rate constant of the minor phase.
+ C, where a2 is the amplitude of the minor phase, and k2 is the rate constant of the minor phase.
Results
Selecting a Model Protein System.
To test the role of RCO in protein folding by circular permutation, we need a model protein with the following characteristics: (i) the structure needs to have its N and C termini close in space to be permutable, (ii) the protein must fold with two-state kinetics, and (iii) permutation needs to result in a significant change in the RCO. A representative library of protein structures previously assembled for use in the rapid autonomous fragment test library (29) was scanned for suitable candidates.
The ribosomal protein S6 from T. thermophilus was selected as the best fit for all these criteria. The structure of S6 has been solved by x-ray crystallography (30), and the N and C termini are close in space. The folding and unfolding kinetics apparently are two-state (26). Finally, S6 satisfies the most difficult criterion, permutations can cause large changes in RCO. Permutations of S6 are predicted to change the RCO from 14 to 22% spanning half the observed range (8% of a total of 15%; ref. 9). Three specific permutants of varying RCO were selected for comparison to the wild-type protein. Residues 14, 36, and 55 were selected as new N termini, because they are in loop regions (Fig. 1A) and correspond to the lowest (residues 14 and 36) and highest (residue 55) RCOs accessible by permutation of S6 (Fig. 1B).
Fig 1.

Effect of circular permutation on RCO. (A) Permutation sites are marked on a ribbon diagram of S6 generated from PDB ID code . (B) The predicted RCO of every permutant of S6 is shown, with closed circles showing the permutations selected for this study.
Structure and Stability.
Fig. 2A shows the far-UV CD spectra of all four variants (S6, S6cp14, S6cp36, and S6cp55). The spectra are virtually identical. This result, together with the cooperative denaturant-induced transitions (see below), indicates that, similar to circular permutants of many other proteins, all three permutants fold into a native-like protein.
Fig 2.

Characterization of the permutants. Far-UV CD spectra (A), equilibrium denaturation with Gdm⋅Cl (B), and relaxation kinetics as a function of Gdm⋅Cl (C) are shown. Closed circles are data for S6, open squares for S6cp14, closed triangles for S6cp36, and open diamonds for S6cp55.
To determine the energetic effects of permuting the protein, guanidinium-induced denaturation was monitored by CD (Fig. 2B). No equilibrium folding intermediates were observed for any of the variants. Measurements on the wild-type protein stability (ΔGunf = 8.5 kcal/mol) are within error of those determined previously (26). The stabilities of S6cp14, S6cp36, and S6cp55 were determined to be 4.1, 5.5, and 9.1 kcal/mol, respectively (Table 1). The m values of all four proteins were similar, once again suggesting a similar fold and that all four have a similar amount of surface area exposed after unfolding (31).
Table 1.
Equilibrium and kinetic parameters for S6 and permutants
| RCO, % | Long-range order | % short-range contacts | ΔGu, kcal⋅mol−1 | m, kcal⋅mol−1⋅M−1 | kf, s−1 | mf, kcal⋅M−1 | ku, s−1 | mu, kcal⋅M−1 | |
|---|---|---|---|---|---|---|---|---|---|
| WTS6 | 19.0 | 1.33 | 50 | 8.5 ± 0.5 | 2.5 ± 0.1 | 400 ± 70 | 1.68 ± 0.04 | 3 × 10−4 ± 1 × 10−4 | −0.71 ± 0.03 |
| S6cp14 | 14.1 | 1.31 | 49 | 4.1 ± 0.1 | 2.7 ± 0.2 | 27 ± 8 | 1.2 ± 0.2 | 0.02 ± 0.01 | −1.4 ± 0.1 |
| S6cp36 | 14.4 | 1.30 | 49 | 5.4 ± 0.3 | 2.0 ± 0.2 | 350 ± 50 | 1.65 ± 0.05 | 0.021 ± 0.004 | −0.61 ± 0.03 |
| S6cp55 | 21.6 | 1.32 | 48 | 9.1 ± 0.5 | 2.6 ± 0.1 | 470 ± 70 | 1.56 ± 0.04 | 1 × 10−4 ± 5 × 10−5 | −1.01 ± 0.03 |
Folding Kinetics.
The folding and unfolding kinetics were evaluated by stopped-flow CD. Unfolding for all proteins fit well to a single exponential. Refolding of S6 has been reported to have two phases (26), with the major phase accounting for 90% of the signal change and the minor phase attributed to proline isomerization. Similar phases described the refolding of S6cp14, S6cp36, and S6cp55. As in the previous study of S6, we used the major phase of refolding in our analysis of the mutants. “Chevron” curves, the denaturant dependence of the ln(kobs), were fit to a two-state process to determine the extrapolated refolding-rate constant in the absence of denaturant (Fig. 2C). These folding-rate constants, 400, 27, 350, and 470 s−1 for S6, S6cp14, S6cp36, and S6cp55, respectively (Table1), all are similar and certainly do not correlate with RCO in the predicted manner. The extrapolated unfolding rate constants are 3 × 10−4, 0.02, 0.021, and 1 × 10−4 s−1, respectively (Table 1). When fit to a two-state kinetic model, the free energies of unfolding calculated from the equation ΔG = −RT ln(kuk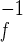 ) are consistent with the two-state equilibrium model.
) are consistent with the two-state equilibrium model.
Discussion
Current literature suggests proteins with lower RCOs will fold more rapidly, but this was not observed in our experimental system (Fig. 3A). All the variants refold with similar rates. Although other factors such as stability will affect folding rates, in natural proteins RCO seems to be the most important determinant (9). Peptide insertions into loop regions of proteins have been found also to follow the expected trend, decelerating the folding process exponentially with respect to RCO (11, 12, 32). Why does changing RCO through circular permutation not result in a similar effect?
Fig 3.

Parameters for predicting refolding kinetics. (A) RCO correlates to protein-refolding rates for naturally occurring proteins (open circles) but not for circular permutants of S6 (closed circle for S6, closed square for S6cp14, closed triangle for S6cp36, and closed diamond for S6cp55). Folding rates for S6 and permutants were also corrected to the average stability of the data set, 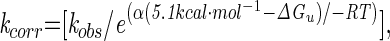 where α is the solvent exposure of the transition state (bold symbols). (B) Long-range order correlates with protein-refolding rates including circular permutants of S6. (C) The percentage of short-range contacts correlates with protein-refolding rates including circular permutants of S6. Fits and correlation coefficients were calculated excluding S6 and permutants by using SIGMAPLOT 2000 (SPSS, Chicago).
where α is the solvent exposure of the transition state (bold symbols). (B) Long-range order correlates with protein-refolding rates including circular permutants of S6. (C) The percentage of short-range contacts correlates with protein-refolding rates including circular permutants of S6. Fits and correlation coefficients were calculated excluding S6 and permutants by using SIGMAPLOT 2000 (SPSS, Chicago).
Unlike RCO, topological parameters such as long-range order (13) or the percentage of short-range contacts (14) predict that permutants should have little effect on folding rates (Fig. 3 B and C). Both of these parameters divide contacts into short-range and long-range classes and are insensitive to the absolute sequence separation of contacting residues. Although permuting a protein will change the absolute sequence separation of contacting residues significantly, these two parameters will remain nearly constant for any circular permutation of any protein, assuming the native structure remains unchanged. Recently two circular permutants of S6 studied by Lindberg et al. (15) also were noted to fold at approximately the same rate. Indeed previous studies on circular permutants in other systems show little change in folding rate; however, the RCO order of these proteins remained unchanged. Our results suggest that despite the changes in RCO, circular permutation does not alter folding rates significantly and that dividing contacts into shortrange and long-range classes may better reflect the role of topology in protein folding.
The Effect of Changes in Protein Stability.
The circular permutants studied here do vary in stability, and changes in stability will be reflected in the folding and/or unfolding rates. Topological features such as loop-closure entropy that drive folding pathways will contribute to protein stability also (33). In some families of proteins with similar folds, stability has been observed to correlate with folding rates (8, 34). One possible explanation for our results is that the destabilization of the permutants results in a deceleration of the refolding process that opposes the acceleration of refolding caused by the decrease in RCO. Lindberg et al. (15) examined two circular permutants of S6, the folding rates of which in water were similar also, varying by a factor of 1.7. They analyzed their data by comparing folding rates at the denaturant midpoints rather than under the same conditions; when adjusted for stability, the folding rates do vary and are in closer agreement with the expectations from the RCO correlation. However, a similar comparison of the folding rates of the naturally occurring examples of two-state proteins at the denaturant midpoints results in a weaker correlation with RCO than comparing the rates extrapolated to water (R = 0.79 vs. 0.89 from table 2 of ref. 15). The poorer correlation at iso-energetic conditions is consistent with the observation that protein stability does not correlate with folding rates among the set of two-state proteins. The best correlation between RCO and folding rates is in aqueous conditions, and therefore to test the dependence of folding rate on RCO we have compared these permutants in aqueous conditions as well.
One of the circular permutants studied here (S6cp14) is very similar to a permutant studied by Lindberg et al. (15), P13-14. Both of these variants of S6 begin with residue 14 at the N terminus. S6cp14 folds slightly slower than the other permutants (Fig. 2C), whereas P13-14 folds at the wild-type rate. The folding nucleus of S6 is centered around strand 1 and helix 1 (35). By examining site-specific mutations, Lindberg et al. demonstrated that permuting the protein at residue 14 causes folding to nucleate near the newly linked N and C termini (15). The linker used to create S6cp14 and all the permutants in this study simply connected the unstructured residues at the C terminus of S6 to the N terminus, whereas P13-14 has these residues truncated with a designed linker between the termini. This subtle difference in the linker may account for the slower refolding of S6cp14 compared with the others. The other permutants leave the S6 folding nucleus intact. In a lattice model system, circular permutation in the nucleation site had significant effects on the folding rate (36). Remarkably, in P13-14 where the linker has been optimized by design, changing the folding nucleus of S6 has almost no effect on the folding rate in the P13-14 variant with the designed linker. Loop design has been demonstrated previously to play a significant role in determining protein-folding pathways and rates in protein G and protein L (37, 38). Similar loop effects may account for the modestly decelerated refolding of S6cp14.
Local Structure Affects Folding Rates.
Recent folding prediction algorithms based on the structure of the native state are consistent with the observation that the topology plays a fundamental role in determining folding rates. Our results suggest that if there is a single structural parameter to describe the effect of topology, it should be related to the ratio of long- and short-range contacts, which for most proteins is directly related to RCO.
One popular model of folding, the diffusion-collision model, however, seems to be related directly to the relationship between short- and long-range contacts (39). This model has been used successfully to model the refolding of apomyoglobin (40), monomeric λ repressor (41), and the B domain of protein A (42). The diffusion-collision model considers the protein as a collection of microdomains connected by the polypeptide backbone. During the folding process, these microdomains diffuse and collide in solution. If two microdomains are sampling a native-like structure when they collide, they will coalesce and remain native-like. In this model, two major variables determine the refolding rate of a protein. First, the length of unstructured polypeptide chain between microdomains limits the volume over which they can diffuse. Second, the likelihood of a successful collision between two microdomains varies as the product of the structural propensities of the microdomains. Both of these variables depend on the topology of the native structure.
Several experiments have demonstrated the importance of the first variable: microdomain diffusion. Protein-refolding rates depend on solution viscosity (43, 44). Increasing the diffusion space by extending unstructured loops has also been shown to decelerate refolding (11, 12).
The second variable, structural propensities of microdomains, also plays an important role in determining refolding rates. Because peptide helices are well characterized (45) and helical propensities can be predicted computationally (46), proteins that are predominantly α-helical can be modeled with diffusion collision (40–42), and increasing helical propensities through mutation can accelerate refolding (47). Determining the native propensities of regions that are not α-helical is more difficult. These propensities should be correlated with the number of short-range contacts in a region of structure. Thus, proteins with more short-range contacts will have higher local structural propensities than proteins with more long-range contacts.
Conclusions
We evaluated the hypothesis that lowering the RCO of a protein structure through circular permutation should result in acceleration of the refolding process. Our set of permutants spans half the range of RCOs seen in proteins that fold without intermediates, yet no significant change in folding rates was observed.
In these experiments, we altered the RCO of S6 by changing the average contact separation. The distribution of short- and long-range contacts, however, remained the same. RCO is correlated with long-range order and the percentage of short-range contacts for previously characterized proteins (data not shown), making it difficult to determine which factors are influencing protein-refolding rates. With the circular permutants, varying RCO without changing the distribution of long- and short-range contacts has no significant effect on folding rates, suggesting that this distribution is the feature normally captured by RCO that gives rise to the correlation.
Acknowledgments
We thank E. Nicholson and J. Myers for insightful discussion about the diffusion-collision model and N. Pokala and the Marqusee lab for discussion and critical reading of the manuscript. This work was supported by the National Institutes of Health. E.J.M. is supported by a Howard Hughes predoctoral fellowship.
Abbreviations
RCO, relative contact order
CD, circular dichroism
Gdm⋅Cl, guanidinium chloride
This paper was submitted directly (Track II) to the PNAS office.
References
- 1.Anfinsen C. B. (1973) Science 181, 223-230. [DOI] [PubMed] [Google Scholar]
- 2.Jackson S. E. (1998) Folding Des. 3, R81-R91. [DOI] [PubMed] [Google Scholar]
- 3.Clementi C., Nymeyer, H. & Onuchic, J. N. (2000) J. Mol. Biol. 298, 937-953. [DOI] [PubMed] [Google Scholar]
- 4.Alm E. & Baker, D. (1999) Proc. Natl. Acad. Sci. USA 96, 11305-11310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Galzitskaya O. V. & Finkelstein, A. V. (1999) Proc. Natl. Acad. Sci. USA 96, 11299-11304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Muñoz V. & Eaton, W. A. (1999) Proc. Natl. Acad. Sci. USA 96, 11311-11316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Plaxco K. W., Simons, K. T. & Baker, D. (1998) J. Mol. Biol. 277, 985-994. [DOI] [PubMed] [Google Scholar]
- 8.Clarke J., Cota, E., Fowler, S. B. & Hamill, S. J. (1999) Structure (London) 7, 1145-1153. [DOI] [PubMed] [Google Scholar]
- 9.Plaxco K. W., Simons, K. T., Ruczinski, I. & Baker, D. (2000) Biochemistry 39, 11177-11183. [DOI] [PubMed] [Google Scholar]
- 10.Chiti F., Taddei, N., White, P. M., Bucciantini, M., Magherini, F., Stefani, M. & Dobson, C. M. (1999) Nat. Struct. Biol. 6, 1005-1009. [DOI] [PubMed] [Google Scholar]
- 11.Viguera A.-R. & Serrano, L. (1997) Nat. Struct. Biol. 4, 939-946. [DOI] [PubMed] [Google Scholar]
- 12.Ladurner A. G. & Fersht, A. R. (1997) J. Mol. Biol. 273, 330-337. [DOI] [PubMed] [Google Scholar]
- 13.Gromiha M. M. & Selvaraj, S. (2001) J. Mol. Biol. 310, 27-32. [DOI] [PubMed] [Google Scholar]
- 14.Mirny L. & Shakhnovich, E. (2001) Annu. Rev. Biophys. Biomol. Struct. 30, 361-396. [DOI] [PubMed] [Google Scholar]
- 15.Lindberg M. O., Tangrot, J., Otzen, D. E., Dolgikh, D. A., Finkelstein, A. V. & Oliveberg, M. (2001) J. Mol. Biol. 314, 891-900. [DOI] [PubMed] [Google Scholar]
- 16.Zhang T., Bertelsen, E., Benvegnu, D. & Alber, T. (1993) Biochemistry 32, 12311-12318. [DOI] [PubMed] [Google Scholar]
- 17.Viguera A. R., Blanco, F. J. & Serrano, L. (1995) J. Mol. Biol. 247, 670-681. [DOI] [PubMed] [Google Scholar]
- 18.Garrett J. B., Mullins, L. S. & Raushel, F. M. (1996) Protein Sci. 5, 204-211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Otzen D. E. & Fersht, A. R. (1998) Biochemistry 37, 8139-8146. [DOI] [PubMed] [Google Scholar]
- 20.Baird G. S., Zacharias, D. A. & Tsien, R. Y. (1999) Proc. Natl. Acad. Sci. USA 96, 11241-11246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Iwakura M., Nakamura, T., Yamane, C. & Maki, K. (2000) Nat. Struct. Biol. 7, 580-585. [DOI] [PubMed] [Google Scholar]
- 22.Viguera A. R., Martínez, J. C., Filimonov, V. V., Mateo, P. L. & Serrano, L. (1994) Biochemistry 33, 2142-2150. [DOI] [PubMed] [Google Scholar]
- 23.Jackson S. E. & Fersht, A. R. (1991) Biochemistry 30, 10428-10435. [DOI] [PubMed] [Google Scholar]
- 24.Viguera A. R., Serrano, L. & Wilmanns, M. (1996) Nat. Struct. Biol. 3, 874-880. [DOI] [PubMed] [Google Scholar]
- 25.Doering D. S. & Matsudaira, P. (1996) Biochemistry 35, 12677-12685. [DOI] [PubMed] [Google Scholar]
- 26.Otzen D. E., Kristensen, O., Proctor, M. & Oliveberg, M. (1999) Biochemistry 38, 6499-6511. [DOI] [PubMed] [Google Scholar]
- 27.Santoro M. M. & Bolen, D. W. (1988) Biochemistry 27, 8063-8068. [DOI] [PubMed] [Google Scholar]
- 28.Nozaki Y. (1972) Methods Enzymol. 26, 43-50. [DOI] [PubMed] [Google Scholar]
- 29.Fischer K. F. & Marqusee, S. (2000) J. Mol. Biol. 302, 701-712. [DOI] [PubMed] [Google Scholar]
- 30.Lindahl M., Svensson, L. A., Liljas, A., Sedelnikova, S. E., Eliseikina, I. A., Fomenkova, N. P., Nevskaya, N., Nikonov, S. V. & Garber, M. B. (1994) EMBO J. 13, 1249-1254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Myers J. K., Pace, C. N. & Scholtz, J. M. (1995) Protein Sci. 4, 2138-2148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Fersht A. R. (2000) Proc. Natl. Acad. Sci. USA 97, 1525-1529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Zhang T., Bertelsen, E. & Alber, T. (1994) Nat. Struct. Biol. 1, 434-438. [DOI] [PubMed] [Google Scholar]
- 34.Sato S., Xiang, S. & Raleigh, D. P. (2001) J. Mol. Biol. 312, 569-577. [DOI] [PubMed] [Google Scholar]
- 35.Oliveberg M. (2001) Curr. Opin. Struct. Biol. 11, 94-100. [DOI] [PubMed] [Google Scholar]
- 36.Li L. & Shakhnovich, E. I. (2001) J. Mol. Biol. 306, 121-132. [DOI] [PubMed] [Google Scholar]
- 37.Nauli S., Kuhlman, B. & Baker, D. (2001) Nat. Struct. Biol. 8, 602-605. [DOI] [PubMed] [Google Scholar]
- 38.Kuhlman B., O'Neill, J. W., Kim, D. E., Zhang, K. Y. & Baker, D. (2002) J. Mol. Biol. 315, 471-477. [DOI] [PubMed] [Google Scholar]
- 39.Karplus M. & Weaver, D. L. (1976) Nature (London) 260, 404-406. [DOI] [PubMed] [Google Scholar]
- 40.Pappu R. V. & Weaver, D. L. (1998) Protein Sci. 7, 480-490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Burton R. E., Myers, J. K. & Oas, T. G. (1998) Biochemistry 37, 5337-5343. [DOI] [PubMed] [Google Scholar]
- 42.Myers J. K. & Oas, T. G. (2001) Nat. Struct. Biol. 8, 552-558. [DOI] [PubMed] [Google Scholar]
- 43.Jacob M. & Schmid, F. X. (1999) Biochemistry 38, 13773-13779. [DOI] [PubMed] [Google Scholar]
- 44.Jacob M., Geeves, M., Holtermann, G. & Schmid, F. X. (1999) Nat. Struct. Biol. 6, 923-926. [DOI] [PubMed] [Google Scholar]
- 45.Scholtz J. M. & Baldwin, R. L. (1992) Annu. Rev. Biophys. Biomol. Struct. 21, 95-118. [DOI] [PubMed] [Google Scholar]
- 46.Munoz V. & Serrano, L. (1994) Nat. Struct. Biol. 1, 399-409. [DOI] [PubMed] [Google Scholar]
- 47.Viguera A. R., Villegas, V., Aviles, F. X. & Serrano, L. (1997) Folding Des. 2, 23-33. [DOI] [PubMed] [Google Scholar]