

Abstract
Hereditary familial amyloidosis of Finnish type (FAF) leading to amyloid in the peripheral and central nervous systems stems from deposition of a 71 residue fragment generated from the D187N/Y variants of plasma gelsolin by two sequential endoproteolytic events. We identify the protease accomplishing the first cleavage as furin, a proprotein convertase. Endoproteolysis of plasma gelsolin occurs in the trans-Golgi network due to the inability of the FAF variants to bind and be stabilized by Ca2+. Secretion and processing of the FAF variants by furin can be uncoupled by blocking the convergence of the exocytic pathway transporting plasma gelsolin and the endocytic recycling of furin. We propose that coincidence of membrane trafficking pathways contributes to the development of proteolysis-initiated amyloid disease.
Keywords: amyloid/calcium/familial amyloidosis of Finnish type/furin/gelsolin
Introduction
Familial amyloidosis of Finnish type (FAF) appears to be caused by amyloid deposition of a 71 residue fragment of the gelsolin protein (Maury, 1991). Human gelsolin is expressed as both an intracellular (81 kDa) and a secreted protein (plasma gelsolin; PG) (83 kDa), each composed of six domains (Kwiatkowski et al., 1986; Yin, 1987; Burtnick et al., 1997; Sun et al., 1999). PG is generated from a precursor that has a 53 residue N-terminal extension, with the first 27 amino acids functioning as a cleavable leader sequence facilitating delivery to the endoplasmic reticulum (ER). PG is thought to be an important component of the actin scavenging system in serum (Lee and Galbraith, 1992). While both the intra- and extracellular forms of gelsolin could lead to the formation of the 173–243 (major) and 173–225 (minor) amyloidogenic fragments derived from domain 2, evidence strongly favors the secreted (extracellular) PG form (Ghiso et al., 1990; Haltia et al., 1990; Levy et al., 1990; Maury and Baumann, 1990; Maury et al., 1990; Paunio et al., 1994; Kangas et al., 1999). Amyloid deposits are found in numerous tissues including the peripheral and central nervous systems (Hiltunen et al., 1991).
Wild-type PG is not associated with any amyloid pathology; however, inheritance of either the D187N or D187Y mutation confers 100% penetrance of FAF. Equivalent amyloidogenicities of the 71 residue fragments of wild-type and D187N PG implicate aberrant proteolysis as the enabling event in FAF (Ratnaswamy et al., 1999). Amyloid diseases resulting from endoproteolytic processing of a large precursor protein currently are best understood in the context of Alzheimer’s disease (AD), where the transmembrane amyloid precursor protein (APP) is processed by the β- and γ-secretases producing the fragments Aβ 1–40 and 1–42, the latter being particularly amyloidogenic (Greenfield et al., 1999b). The β-cleavage is believed to occur in late Golgi and endocytic compartments, but the cellular location of the subsequent cleavage by γ-secretase remains controversial (Huse and Doms, 2001). γ-cleavage potentially is mediated by the activity of presenilins normally localized to ER and pre-Golgi compartments (Tector and Salter, 1995; Annaert et al., 1999; Greenfield et al., 1999a).
In contrast to APP, which is cleaved during transit through the secretory pathway, wild-type PG is secreted as a full-length protein; hence the proteolytic processing of the FAF variants is abnormal (Paunio et al., 1994; Kiuru, 1998). Analysis of the D187N/Y FAF-associated amyloidogenic variants of PG reveals that an initial proteolytic cleavage occurs intracellularly at position R172–A173 by a putative ‘α-gelsolinase’ producing a 68 kDa secreted C-terminal fragment of PG referred to as C68 (Paunio et al., 1994; Kiuru, 1998). Cleavage of the FAF variants is insensitive to a broad spectrum of general protease inhibitors, although EDTA inhibits proteolysis (Paunio et al., 1994). The second proteolytic cleavage at position M243–L244 in the C68 fragment by a putative ‘β-gelsolinase’ generates the 71 residue amyloidogenic fragment (Kiuru, 1998).
The identities of the putative α- and β-gelsolinases responsible for the formation of the 71 residue FAF fragment from D187N/Y PG variants currently are unknown, as is the mechanism by which the FAF mutations enable aberrant endoproteolytic processing. We now demonstrate that α-gelsolinase is furin, a member of the proprotein convertase family that recycles between the trans-Golgi network (TGN), the cell surface and endosomes (Molloy et al., 1999). Secretion and processing of PG FAF variants by furin can be separated biochemically using vesicular transport inhibitors and furin inhibitors in vivo. Thus, amyloid disease occurs in response to the dynamic trafficking pathways of PG through the Golgi that are coupled to furin recycling. Biophysical experiments demonstrate that the wild-type protein, but not the FAF variants, is stabilized by binding of Ca2+ to domain 2 under the slightly acidic conditions found in the Golgi and Golgi-derived transport containers Thus, α-gelsolinase processing of PG FAF variants occurring en route to the cell surface is a consequence of the lack of Ca2+-mediated stabilization. Our results provide key insights into the first step of FAF and suggest a prominent role for convergence of the late secretory and endocytic pathways in generating C68 PG in tissue, the precursor to the 71 residue fragment found in FAF amyloid fibrils. We propose that coincidence of membrane trafficking pathways is an important contributing factor to the cell type-specific development of amyloid diseases including FAF and, potentially, AD.
Results
Expression and processing of the wild-type and D187N FAF PG variant
To study the processing of the D187N FAF variant to the 68 kDa fragment (residues 173–755), we transfected COS cells that express an endogenous form of PG (Figure 1A) and BHK cells that also weakly express an endogenous PG (Figure 1B) with plasmids encoding the wild-type and D187N FAF variant PG. Following PG overexpression (generally 5- to 10-fold over endogenous levels in COS cells), cells were pulse-labeled in a chemically defined medium with [35S]methionine. To quantify transport, both the intracellular and extracellular (secreted) forms of PG were immunoprecipitated using a specific antibody that recognizes an epitope found in the C-terminal region of PG.

Fig. 1. Expression and processing of the wild-type and D187N FAF variant PGs. COS (A) or BHK (B) cells were transfected and radiolabeled with [35S]methionine, and PG was immunoprecipitated from cells or medium at the indicated time as described in Materials and methods. Asterisks indicate low levels (<1%) of cross-reacting protein(s). The positions of the 83 kDa (full-length) and 68 kDa fragment (C68) are indicated by arrowheads. (C) Quantitation of data presented in (A) and (B). The results are typical of three independent experiments.
Wild-type PG was secreted efficiently over a 90 min time course with a t1/2 of ∼60 min (Figure 1A and C). Strikingly, both COS and BHK cells expressing the D187N FAF variant not only secrete the full-length (83 kDa) form with kinetics similar to wild-type, but also secrete the proteolytically processed C68 fragment (Figure 1). Whereas the C68 fragment generally was not detected within COS cells, low levels (5–15%) of C68 could be detected within BHK cells. These results suggest that processing can be a late intracellular event or could occur following secretion. To test the latter possibility, a mixture of radiolabeled D187N full-length and processed (C68) PG was recovered from the medium of cells and reincubated for an additional 60 min with cells that had not been transfected. Under these conditions, <3% of the full-length D187N PG was processed further to the 68 kDa form (data not shown). These results suggest that the proteolytic processing of the D187N PG variant occurs within an intracellular compartment en route to the cell surface. Thus, both COS and BHK cells contain an intracellular protease that performs the α-gelsolinase cleavage on the D187N FAF variant, leading to the formation of the secreted C68 fragment.
Processing of D187N to C68 occurs in the Golgi
To gain insight into the steps in transit through the exocytic pathway that are involved in processing the D187N FAF variant to the C68 fragment, we examined the effect of dominant-negative mutants of the Sar1, Arf1 and Rab family of GTPases that interfere with the transport of cargo between different secretory compartments (Aridor and Balch, 1996; Chavrier and Goud, 1999).
Export from the ER is regulated by the coat complex II (COPII) machinery, whose assembly leading to the formation of transport vesicles is controlled by the Sar1 GTPase (Schekman and Orci, 1996). A Sar1 mutant (T39N) restricted to the GDP-bound state (Sar1-GDP) potently inhibits COPII coat recruitment in vivo and in vitro and thereby blocks ER export (Aridor et al., 1995, 2001; Rowe et al., 1996). When BHK cells were co-transfected with D187N and Sar1-GDP (5- to 10-fold overexpression of Sar1-GDP relative to endogenous wild-type Sar1), secretion of full-length and C68 PG into the medium was largely blocked, as discerned from immunoblotting (Figure 2). Importantly, no accumulation of the C68 fragment was observed in cells expressing Sar1-GDP. It is therefore unlikely that the degradative pathways associated with the ER (Ellgaard et al., 1999) participate in processing the D187N FAF variant to the C68 fragment.

Fig. 2. Processing of D187N to C68 occurs in the Golgi. BHK cells were co-transfected with wild-type (ctl) or the D187N FAF variant (ctl, a–f) and the indicated Sar1, Arf1 or Rab GTPase mutants. Following expression, cells were washed, and the intracellular or secreted full-length C68 forms and recovered after a 2 h time period were analyzed by immunoblotting (A) and quantitated (B) as described in Materials and methods. Control (ctl) is the amount of wild-type or D187N secreted or processed in the absence of mutant GTPases. The results are typical of three independent experiments.
Following exit from the ER, progress from pre-Golgi intermediates to and through the cis-, medial and trans-Golgi compartments requires the activity of a different vesicle budding apparatus, the coat complex I (COPI) machinery that is regulated by the Arf1 GTPase (Wieland and Harter, 1999). Co-transfection of D187N PG with a Q71L Arf1 mutant restricted to the GTP-bound state (Arf1-GTP) that blocks COPI coat disassembly and vesicle function (Dascher and Balch, 1994) resulted in inhibition of both transport and processing of PG (Figure 2). These results demonstrate that processing of the D187N variant to the C68 fragment is unlikely to occur prior to pre-Golgi intermediates.
Members of the Rab GTPase family control targeting and fusion of transport vesicles in both the exocytic and endocytic pathways (Zerial and McBride, 2001). In particular, Rab1 and Rab6 GTPases are important for transport of cargo to and through the Golgi (Tisdale et al., 1992; Martinez et al., 1994, 1997). A S25N Rab1 mutant restricted to the GDP-bound form (Rab1A-GDP) leads to a general disruption of trafficking from pre-Golgi intermediates to the Golgi. Co-expression of Rab1A-GDP with the D187N FAF variant reduced secretion by 90% (Figure 2). Moreover, it almost completely (>99%) blocked processing to the C68 fragment (Figure 2). Consistent with these results, the T27N Rab6 GDP-restricted mutant (Rab6-GDP), which affects transport from endosomal compartments to and through a retrograde pathway operating in the Golgi stack (Martinez et al., 1994; Girod et al., 1999; White et al., 1999; Darchen and Goud, 2000), partially reduced overall secretion, but markedly enhanced the appearance of the proteolytically processed form in the medium and in the cell (Figure 2). On the other hand, the Q72L Rab6 GTP-restricted mutant (Rab6-GTP) that leads to fragmentation of the Golgi stack (Martinez et al., 1997) completely blocked secretion and processing (Figure 2). In contrast to Golgi-specific Rab proteins, a dominant-negative T36N Rab3 mutant restricted to the GDP-bound form (Rab3A-GDP) that interferes with the targeting and fusion of post-Golgi-regulated secretory compartments to the cell surface (Jahn and Sudhof, 1999) had no detectable effect on secretion or processing of the D187N variant (Figure 2). An identical result was observed with Rab3A-GTP, a mutant restricted to the GTP-bound form (data not shown). The observed effects of Golgi-specific Rabs (1A and 6) on D187N PG export and processing are consistent with morphological studies using indirect immunofluorescence that demonstrate that the localization of either the wild type or the D187N variant PG prominently overlaps with the distribution of Golgi markers Syn5 and GM130 (cis-Golgi), Man II (medial Golgi) and TGN38 (TGN), but not endocytic markers including EEA1 (early endosome) and Lamp1/2 (late endosome) (data not shown). The combined results suggest that processing is initiated in the Golgi and that disruption of Golgi function not only interferes with movement of D187N PG to the cell surface but potently uncouples this pathway from steps involved in proteolytic processing.
α-gelsolinase is a furin-like proprotein convertase
The identification of the putative α-gelsolinase mediating the initial processing of the FAF variants in situ is crucial for understanding the origin of FAF disease. The expression of the protease appears to have some degree of cell type specificity. Whereas high levels of endogenous protease activity leading to cleavage of the D187N variant to the C68 fragment are observed in PC12 and cells of neural origin, the activity is weak or absent in a variety of other cell lines tested (Paunio et al., 1998). An examination of the sequence surrounding the cleavage site at residues 172–173 provides an important clue to the identity of α-gelsolinase. This cleavage site is reminiscent of that used by proprotein convertases (PCs), a family of related Ca2+-dependent serine endoproteases that have been studied extensively for their role in the proteolytic processing of endocrine and neuroendocrine hormone precursors (Steiner, 1998). Soluble PCs conventionally are packaged along with prohormones in secretory granules in endocrine and neuroendocrine cells. In contrast, the more ubiquitous membrane-bound forms are localized strategically to the TGN in many cell types and have an extensive recycling pathway that includes transit to the cell surface and back into the cell through endocytic compartments (Molloy et al., 1999). All PC family members are synthesized as proprotein precursors that are activated by transport to the more acidic, Ca2+-rich environment of the Golgi (Zhou et al., 1999). The putative α-gelsolinase cleavage site in PG, -169Rxx172R173A-, is a near consensus sequence for this protease family.
To provide evidence that the initial cleavage event to form the C68 fragment involves a member of the PC family, we transfected COS fibroblasts with furin, a ubiquitous membrane-bound PC that has been studied extensively (Molloy et al., 1999). If furin contributes to FAF variant processing, overexpression of furin (∼10- to 20-fold) should lead to the enhanced recovery of C68 in the medium. As predicted, co-transfection of the D187N variant PG with furin led to a striking enhancement of processing of the D187N PG mutant to the C68 fragment found in the medium (Figure 3A and B). While overexpressed furin was also capable of processing 10–15% of the wild-type PG to the C68 fragment (Figure 3A and B), the D187N variant was clearly more susceptible to proteolysis as nearly complete cleavage to the C68 secreted form was observed. Moreover, in the presence of excess furin, a significant level of intracellular C68 was detected for the first time in both COS (Figure 3A and B) and BHK cells (data not shown) expressing the D187N FAF variant, suggesting that processing is probably an intracellular event.


Fig. 3. α-gelsolinase is a furin-like proprotein convertase. (A) COS cells were transfected with either wild-type PG alone (a) and with furin (b) or the D187N variant alone (c) and with furin (d), labeled with [35S]methionine, chased for the indicated time, and intracellular and secreted forms of PG immunoprecipitated. Asterisks indicate low levels (<1%) of cross-reacting protein(s). (B) Quantitation of data presented in (A). The results are typical of at least four independent experiments. (C) Expression of PG in the LoVo cell line lacking furin. Cells were mock transfected (a), transfected with D187N alone (b) or co-transfected with furin and D187N PG (c). The secreted forms reflecting the level of intracellular processing are shown. (D) Distribution of furin and PG in COS cells using indirect immunofluorescence as described in Materials and methods. Arrows indicate overlap in the TGN. Arrowheads denote furin-containing punctate endosomal compartments.
To determine if processing of the D187N variant to the C68 fragment by overexpressed furin reflected the requirement for endogenous furin, we examined processing of the D187N FAF variant in the LoVo cell line that lacks furin (Takahashi et al., 1993). In contrast to the processing observed in both BHK and COS cells in the absence or presence of overexpressed furin, the C68 fragment was not formed by the LoVo cell line (Figure 3C). However, co-expression of the D187N variant and furin in the LoVo cell line restored processing (Figure 3C). Thus, the endogenous protease responsible for PG cleavage is furin and potentially related members of the PC family (Nakayama, 1997; Zhou et al., 1999). While the distribution of the D187N PG variant and furin overlaps in the TGN, it does not overlap in punctate endosomal compartments containing recycling furin (Figure 3D). The combined results are consistent with the possibility that processing by furin is a Golgi-related event in the secretory pathway (Figure 2).
FAF variant processing is inhibited by α1-PDX and peptide inhibitors
To assess the specificity of furin-dependent processing in vivo, we examined the effect of a membrane-permeant furin inhibitor (decanoyl-Arg-Val-Lys-Arg-chloromethyl ketone; dRVKRck) on D187N processing. This ketone potently inhibited processing of the D187N FAF variant in transfected BHK cells, with half-maximal inhibition observed in the range of 1–10 nM (Figure 4A), consistent with its high affinity for furin-related PCs (Steiner, 1998).
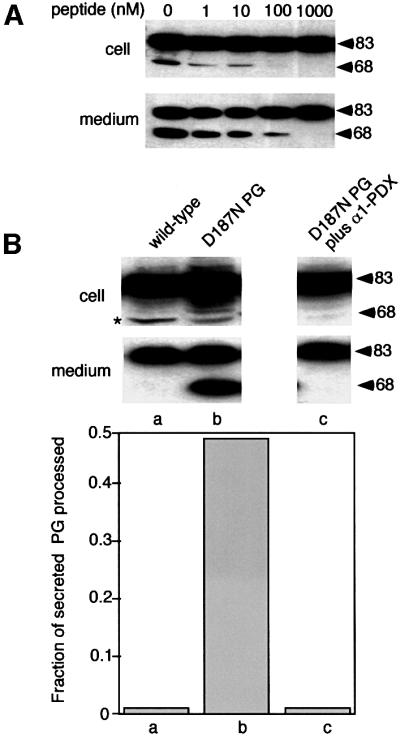
Fig. 4. α1-PDX inhibits D187N variant processing. (A) BHK cells were transfected with the D187N variant. Following 5 h of expression, the indicated amount of peptide inhibitor (dRVKRck) was added to the medium for 1 h, cells were washed, and the intracellular or secreted full-length and C68 forms recovered after a 2 h time period were analyzed by SDS–PAGE and immunoblotting. (B) Upper panel: BHK cells were co-transfected with the D187N variant and α1-PDX. Following 6 h of expression, cells were washed, and the intracellular or secreted full-length and C68 forms recovered after a 2 h time period were analyzed by immunoblotting. Lower panel: quantitation of data presented in the upper panel. Asterisks indicate low levels (<1%) of cross-reacting protein(s). The results are typical of two independent experiments.
We next examined the ability of α1-anti-trypsin Portland (α1-PDX), a bioengineered serpin highly specific for furin (Jean et al., 1998), to block the processing of the D187N FAF variant by inhibiting the endogenous α-gelsolinase activity. As shown in Figure 4B, co-expression of the D187N PG variant with α1-PDX quantitatively blocked the appearance of C68 in the cell or the medium. An identical result was observed in a triple transfection in which the D187N FAF variant, furin and the α1-PDX inhibitor were co-expressed (data not shown), demonstrating that the elevated processing observed when furin and the FAF variants were co-expressed was a direct response to excess furin. We also observed that pre-incubation of cells with recombinant α1-PDX added to the medium for 2 h, a condition expected to inactivate furin that normally is recycling between the TGN and the cell surface (Molloy et al., 1999), potently inhibited processing (data not shown). These results are consistent with a requirement for the D187N secretion pathway and the endocytic furin recycling pathway to intersect to promote efficient proteolytic processing. Furthermore, because inhibition of processing by α1-PDX did not interfere with secretion (Figure 4B), these results demonstrate our ability to uncouple secretion from processing in the Golgi (Figure 2).
Furin-like PCs constitute the major pathway of FAF variant proteolytic processing
Given that furin belongs to a large family of PCs, we examined the efficiency with which other members of the family promote the cleavage of the D187N FAF variant. We co-expressed the D187N FAF variant with representative members of various PC subgroups including PACE4, PC2, PC1/3, PC5/6B and PC7/8. PC5/6B, a furin-like protein that is inhibited potently by α1-PDX (Ki = 1–2 nM), showed significantly elevated levels of processing of the D187N variant compared with control levels (Figure 5). PC3, a PC of neuroendocrine origin that is also sensitive to α1-PDX (Ki = 60 nM), exhibits similar behavior. In contrast, processing was not stimulated by PC2, PC7 or PACE 4 when expressed at similar levels. These PCs have considerably reduced sensitivity to α1-PDX inhibition (Ki >1 µM). Because gelsolin is unlikely to be packaged in secretory vesicles in neuroendocrine cells, PC3 and the splice variant of PC5/PC6B that lacks a transmembrane domain (PC5/6A) are unlikely candidates for gelsolin proteolysis in situ. Thus, two criteria, recycling through the TGN and high sensitivity to α1-PDX (Ki = 1–2 nM), suggest that furin and functionally related PC5/6B, that also cycles through the TGN, are the primary candidates for α-gelsolinase activity in different cell types in situ. The ubiquitous distribution of these two PCs is consistent with the diversity of tissue types involved in gelsolin FAF leading to slowly progressing systemic and neurological disease.

Fig. 5. Furin-like PCs constitute the major pathway of FAF variant processing in different tissues. (A) BHK cells were co-transfected with the D187N variant and the indicated PCs. Following 6 h of expression, cells were washed, and the intracellular or secreted full-length and C68 forms recovered after a 2 h time period were analyzed by immunoblotting. (B) Quantitation of the data presented in (A).
Furin endocytic trafficking pathways are critical for processing of the FAF variant
PCs undergo extensive recycling between the TGN and the cell surface, a pathway that involves both early and late endosomal compartments (Molloy et al., 1999). To test the hypothesis that efficient processing of the D187N FAF PG variant by furin-like PCs is a consequence of their co-localization in late Golgi or post-Golgi carriers, we examined the effect of dominant-negative Rab5, 7 and 11, GTPases that control the movement of furin and other recycling proteins through endocytic compartments (Zerial and McBride, 2001). No effect on the efficiency of secretion or processing of PG to the C68 fragment was observed with a S34N Rab5A GDP-restricted mutant (Rab5A-GDP) (Rybin et al., 1996) that alters cell surface recycling through the early, sorting endosome (Figure 6). In contrast, both the T22N Rab7 dominant-negative GDP-restricted mutant (Rab7-GDP) and the dominant-negative S25N Rab11 (Rab11-GDP) mutant that interfere with the trafficking of cargo through late endosomal compartments to the TGN (Mills et al., 1999; Mohrmann and van der Sluijs, 1999; Somsel Rodman and Wandinger-Ness, 2000) significantly decreased the proteolytic processing of D187N PG progressing through the secretory pathway (Figure 6). Importantly, co-transfection of D187N with these GTPase variants had only a modest effect on the total level of secretion (Figure 6). These results, taken together with the inhibition of proteolysis caused by administration of extracellular α1-PDX, emphasize that D187N FAF variant processing by α-gelsolinase during secretion is a step that is closely coupled to recycling of furin-like PCs through the endocytic pathway to the TGN.

Fig. 6. Furin endocytic trafficking pathways leading to processing of variant PG to C68. BHK cells were co-transfected with the wild-type (ctl) or the D187N FAF variant (ctl, a–c) and the indicated Rab GTPase mutants. Following 6 h of expression, cells were washed and the intracellular or secreted full-length and C68 forms recovered after a 2 h time period were analyzed by immunoblotting (A) and quantitated (B). The results are typical of two independent experiments.
Ca2+ binding stabilizes wild-type PG but not the FAF variants
The above results suggest that furin processing of the FAF PG variants occurs in Golgi compartments (or potentially in Golgi-derived transport vesicles) that are thought to have a high Ca2+ concentration (0.1–1 mM) (Chandra et al., 1991) and a slightly acidic pH (Wu et al., 2000). Studies carried out prior to our insight into the physiological pathway of PG processing demonstrate that the thermodynamic stabilities of the Ca2+-free wild-type, D187N and D187Y are very similar in the context of a domain 2 fragment (residues 134–266) that is structurally representative of domain 2 in full-length PG (Ratnaswamy et al., 2001). Given that the environment of the Golgi might lead to differences in stability between wild-type PG and the FAF variants that could account for the differential sensitivity to furin cleavage, the stability of the wild-type and FAF variant domain 2 constructs was reassessed in the presence of CaCl2 (100 µM) at a slightly acidic pH (6.2). Far-UV circular dichroism (CD) spectroscopy was employed to record temperature- and urea-induced denaturation curves to facilitate stability comparisons (Figure 7).

Fig. 7. Ca2+ binding stabilizes wild-type PG but not the FAF variants. The stability of gelsolin towards thermal (A) and chaotrope (urea) denaturation (B) as described in Materials and methods in the presence and absence of Ca2+ reveals that wild-type gelsolin experiences a +11°C shift in Tm in the presence of 100 µM Ca2+, whereas the FAF variants display identical Tms in both the presence and absence of Ca2+. The urea concentration required to half-denature the wild-type protein increases from 1.4 to 2.3 M in the presence of Ca2+, whereas the D187N/Y FAF variants display values that do not change significantly in the presence of Ca2+. See Table I for a summary of the thermo dynamic data. (C) The isothermal titration calorimetry binding isotherm resulting from the addition of 5 µl aliquots of 520 µM CaCl2 to the wild-type gelsolin domain 2 construct (50 µM) is depicted (raw data presented as the insert). Analogous experiments with D187N and D187Y reveal no heat evolution and hence no binding of Ca2+.
In the presence of 100 µM Ca2+ (in the absence of EDTA), the Tm of the wild-type domain 2 fold exceeded that of D187Y by 12°C and that of D187N by 8°C (Figure 7A; Table I). However, in the absence of Ca2+ (in the presence of 1 mM EDTA), the wild-type and FAF variants have very similar Tms (Table I), because removal of Ca2+ caused reduction of the wild-type Tm by 11°C while the Tms of the FAF variants remained identical to those observed in the presence of Ca2+. Consistent with these results, urea denaturation curves also demonstrate that the wild-type protein, but not the FAF variants, was stabilized by added Ca2+. The denaturant concentration where half of the protein is unfolded (D1/2) for the wild-type protein decreased from 2.3 to 1.4 M urea when Ca2+ was removed by EDTA, whereas the FAF variants exhibit a D1/2 of ∼1.4 M whether or not Ca2+ was present (Figure 7B; Table I). These combined results support the hypothesis (Zapun et al., 2000) that the D187N and D187Y mutations remove a key ligand required for the Ca2+ binding that stabilizes the wild-type PG structure in the Golgi.
Table I. Thermodynamic parameters from thermal (Tm) and urea (m, D1/2, ΔGo) denaturation of domain 2 (134–266).
| Wild type |
D187N |
D187Y |
||||
|---|---|---|---|---|---|---|
| 100 µM CaCl2 | 100 µM CaCl2 1 mM EDTA | 100 µM CaCl2 | 100 µM CaCl2 1 mM EDTA | 100 µM CaCl2 | 100 µM CaCl2 1 mM EDTA | |
| Tm (°C) | 52 | 41 | 44 | 44 | 40 | 40 |
| ΔTm (°C) | 11 | 0 | 0 | |||
| m (cal/mol/M) | 1139 | 1919 | 1705 | 1777 | 2106 | 2614 |
| ΔGo (cal/mol) | 2620 | 2687 | 2387 | 2488 | 3159 | 3398 |
| D1/2 (M) | 2.3 | 1.4 | 1.4 | 1.4 | 1.5 | 1.3 |
| ΔD1/2 (M) | 0.9 | 0 | 0.2 | |||
The hypothesis that the wild-type domain 2 construct, but not the FAF variants, binds Ca2+ was tested using isothermal titration calorimetry. Divalent cations were removed from the buffers using Chelex-100 resin and from the protein using addition and subsequent removal of EDTA. Figure 7C reveals that the wild-type protein binds Ca2+ with a dissociation constant of 650 nM, whereas the D187N and D187Y variants exhibit no binding in the micromolar concentration range, supporting the conclusion that stabilization induced by Ca2+ binding differentiates the wild-type protein from the variants in its ability to avoid proteolysis.
Furin cleaves domain 2 FAF variants but not wild-type
If the lack of Ca2+ binding and stabilization (Figure 7) is responsible for the proteolytic susceptibility of the FAF variants to furin, then the removal of Ca2+ from the wild-type domain 2 PG should result in cleavage susceptibility. However, this is not possible to test with furin given that PCs are Ca2+-dependent serine proteases. A testable prediction is that wild-type gelsolin domain 2, but not the FAF variants, should be protected from 172–173 cleavage in the presence of 1 mM Ca2+. To test this prediction, soluble furin with its membrane-spanning region deleted was incubated with the wild-type, D187N and D187Y 134–266 constructs in the presence of Ca2+. While no proteolysis was observed in the case of the wild-type domain 2, both FAF variants were 40–60% cleaved to the expected fragments of 10.4 and 4.3 kDa after 40 min (Figure 8). Analogous in vitro experiments with full-length PG (isolated from the medium of gelsolin-secreting BHK cells) revealed that wild-type PG was insensitive to furin treatment, whereas 10–15% of the D187N FAF variant exhibited furin sensitivity.

Fig. 8. Variant processing is a consequence of the defect in Ca2+-mediated stabilization. The susceptibility of wild-type and the FAF variants (D187N/Y) of folded gelsolin domain 2 (134–266) (2 µM) to cleavage by soluble furin (2 U) under conditions likely to be similar to those found in the TGN (pH 6.2, 37°C, 1 mM CaCl2). Proteolysis or lack thereof was evaluated as a function of time where the initial and 40 min time point are displayed in the SDS–polyacrylamide gel. Numerous experiments reveal that the wild-type domain is not susceptible to furin-mediated proteolysis whereas the FAF variants are proteolyzed into the expected 10.4 and 4.3 kDa fragments.
Discussion
Our results provide key insight into the first and probably critical step initiating FAF systemic and neurological amyloid disease, demonstrating a prominent role for the late secretory pathway in the aberrant processing of the D187N/Y variants by the PC furin. In addition, both in vivo and in vitro experiments provide a molecular explanation for the protease sensitivity of the FAF variants. We found that the D187N and D187Y variants are sensitive to furin due to a deficiency in Ca2+ binding-induced stabilization that protects the wild-type protein from proteolysis in the Golgi, rather than the previously proposed more general folding defects (Isaacson et al., 1999). These results emphasize the importance of the location of intracellular processing. Efficient processing by the furin-like PCs occurs at the convergence of the biosynthetic (exocytic) and recycling (endocytic) pathways. Thus, the presence of both furin and its recycling pathway in different tissues ultimately will dictate the efficiency of processing of the FAF variants and will probably influence the tissue specificity and extent of amyloid deposition.
FAF variant processing is a consequence of a defect in Ca2+-mediated stabilization
Using thermal and chaotrope denaturation, we found that unlike wild-type gelsolin (our data and Zapun et al., 2000), the D187N/Y variants are not stabilized by Ca2+ binding under the slightly acidic conditions found in the Golgi. The lack of Ca2+-induced stabilization appears to be correlated with furin protease susceptibility, as a soluble version of furin readily cleaved the FAF variants of domain 2 PG, but not wild-type domain 2 PG in the presence of Ca2+ (Figure 8). It is now apparent that the FAF variants are proteolyzed not because they are inherently less stable than wild-type PG (Isaacson et al., 1999), but because they lack the wild-type ability to bind Ca2+ and be stabilized against proteolysis by furin in vitro. The fact that overexpression of furin in vivo led to weak but significant processing of wild-type PG highlights the necessity for Ca2+ binding to the domain 2 region to overcome the inherent proteolytic susceptibility of secreted wild-type PG.
Consistent with this conclusion, the NMR structure of severin, the gelsolin homolog from Dictyostelium discoideum, reveals that the NHs of residues in the vicinity of D188 (equivalent to D187 in gelsolin) exhibit chemical shift changes upon addition of Ca2+, strongly suggesting that the D187 residue in human gelsolin domain 2 also participates in Ca2+ binding (Schnuchel et al., 1995). The crystal structure of severin in the absence of Ca2+ reveals that D188 is bound to a sodium ion and is thus a likely ligand for Ca2+ (Way and Weeds, 1988; Puius et al., 2000). The high homology between severin and gelsolin domain 2 considered in concert with the structural data on severin and the gelsolin domain 2 Ca2+-binding data within lead us to suggest that the FAF mutations convert an established ligand for Ca2+ into a poor ligand, rendering this region unable to bind Ca2+, unable to experience Ca2+-mediated stabilization and unable to resist furin proteolysis.
The D187N/Y mutations have no apparent effect on the function of the full-length cytosolic gelsolin (Kangas et al., 1996). While these mutations have been reported to render the secreted form unable to cap actin and promote actin severing (Weeds et al., 1993), these effects are most likely to be due to proteolysis, as the vast majority of PG in the homozygotes was fragmented. Moreover, it is unlikely that the structures of the FAF variants are dramatically altered at neutral pH (Ratnaswamy et al., 2001) given that they are exported efficiently from the ER to the cell surface as shown herein and in previous studies where the steady-state serum levels of the D187N mutant are reported to be even higher than those of wild-type PG (Paunio et al., 1997, 1998). Thus, the variants are not altered in their trafficking to the cell surface. Because gross folding defects generally are detected by the ER quality control machinery (Ellgaard et al., 1999) via ubiquitylation and proteosome degradation (Lord et al., 2000), the endoproteolytic event involved in initiation of FAF almost certainly results from a subtle structural defect that is revealed as a result of the environment found in the secretory compartments and the co-localization of PG with furin.
The trafficking and proteolytic processing pathway of the D187N FAF variant
The identification of furin as α-gelsolinase is consistent with the earlier observations that incubation with EDTA, a Ca2+ chelator, inhibits aberrant FAF proteolysis, as furin is a Ca2+-dependent serine endoprotease (Molloy et al., 1992). Furin is membrane bound, has a broad tissue distribution and functions in the constitutive exocytic/endocytic pathways to process a large variety of proprotein precursors that are targeted to the cell surface or to the endosomal/lysosomal pathways (Molloy et al., 1999). Interestingly, furin mediates proteolysis of a C-terminally extended familial British dementia (BRI) variant creating the peptide found in BRI amyloid fibrils (Kim et al., 1999).
Furin is ubiquitous; however, processing of the FAF variants is particularly efficient in cell lines of neural lineage such as PC12 cells (Paunio et al., 1997, 1998). This may reflect the contribution of additional PCs (such as PC3) present in the Golgi compartments to aberrant cleavage or be due to cell-specific recycling issues. Given that the recyling pathway of furin-like proteases is highly regulated (Molloy et al., 1999) and our observation that perturbation of this recycling pathway uncouples processing of the D187N variant from secretion, it is now apparent that PCs with furin-related cleavage specificity will contribute significantly to cell type-specific processing of PGs, particularly cells of the nervous system, leading to cranial and peripheral neuropathies. Such specialized trafficking pathways in select cell types may lead ultimately to the build-up of gelsolin amyloid deposits in response to the trafficking of α- and a putative β-gelsolinase participating in the second cleavage.
In addition to cells of neuroendocrine origin, human fibroblast, skeletal, cardiac and smooth muscle cells have large quantities of PG mRNA, in some cases devoting up to 0.5–3% of their biosynthetic capacity to making PG (Kwiatkowski et al., 1988). Whether these cells lead to furin processing of the PG variants will also probably depend on the tissue-specific transport pathways in operation. Although it remains possible that processing of the secreted full-length FAF variants can occur in situ by surface-associated or serum proteases, our results suggest that the vast majority of the extracellular C68 fragment found in plasma is likely to be generated by furin processing in the TGN.
β-secretase (BACE) is a membrane-bound aspartyl protease (de Bono and de Bono, 2001) that generates a polypeptide precursor (C99). C99 is cleaved subsequently by γ-secretase into Aβ peptides that deposit as amyloid fibrils (Selkoe, 1998, 2000) and appear to cause AD. Interestingly, β-secretase is activated by a furin-like PC (Bennett et al., 2000; Creemers et al., 2001), suggesting that it is delivered to the TGN where it subsequently is targeted to endosomal compartments and recycles between these compartments and the cell surface or the TGN (Huse et al., 2000). Given that the initial cleavage of gelsolin FAF variants is critically sensitive to the recycling pathway of furin, exposure of APP to BACE and, in turn, the C99 fragment to γ-secretase may involve a similar convergence of biosynthetic and recycling pathways in specific cell types that may be altered by mutation (Petanceska et al., 2000), a possibility supported by the observation that C99 needs to exit the ER for cleavage to occur (Maltese et al., 2001). Thus, the need for the convergence of biosynthetic and endocytic recycling pathways observed for FAF and AD (Huse and Doms, 2001) may play a more general role in the generation of amyloid disease than previously appreciated and could contribute significantly to cell type-specific amyloid deposition that has been difficult to understand. Our insight into both the physical and cellular basis for FAF disease now provides a compelling model that extends our current understanding of the key events mediating the onset of amyloid disease.
Materials and methods
PG transport analysis
Materials were obtained and PG plasmids prepared for transport analysis using a vaccinia transient expression system as described previously (Dascher et al., 1995). Immunoblotting and indirect immunofluorescence were performed as described (Aridor et al., 1995; Rowe et al., 1996).
Domain 2 134–266 protein expression, purification and characterization
Gelsolin domain 2 (wild-type, D187N and D187Y) was prepared, and thermal and urea denaturation studies were carried out as described (Ratnaswamy et al., 2001).
Isothermal titration calorimetry
Ca2+ binding was evaluated on Ca2+-free 134–266 (wild-type and D187N/Y) using a Microcal MCS Titration Calorimeter (Microcal, Northampton, MA). Injections of 520 µM CaCl2 were made into the cell containing a domain 2 (50 µM) at 25°C. The binding isotherm resulting from integration of the thermogram was fit to several models including that of a single binding site (Microcal ORIGIN 5.0).
Furin sensitivity of PG domain 2
The susceptibility of gelsolin to cleavage by soluble furin under conditions simulating the TGN was evaluated. Wild-type or D187N/Y 134–266 (2 µM) was incubated with 2 U of furin in 20 mM sodium phosphate, 100 mM NaCl, 1 mM CaCl2 pH 6.2, 37°C. Aliquots removed as a function of time were quenched (100 mM EDTA on ice) and evaluated by SDS–PAGE.
Supplementary data
Detailed experimental procedures can be found in Supplementary data available at The EMBO Journal Online.
Acknowledgments
Acknowledgements
This work was supported by grants from the National Institutes of Health (GM42336 and GM33301-WEB; DK46335-JK), the Skaggs Institute of Chemical Biology and the Lita Annenberg Hazen Foundation.
References
- Annaert W.G. et al. (1999) Presenilin 1 controls γ-secretase processing of amyloid precursor protein in pre-Golgi compartments of hippocampal neurons. J. Cell Biol., 147, 277–294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aridor M. and Balch,W.E. (1996) Principles of selective transport: coat complexes hold the key. Trends Cell Biol., 6, 315–320. [DOI] [PubMed] [Google Scholar]
- Aridor M., Bannykh,S.I., Rowe,T. and Balch,W.E. (1995) Sequential coupling between COPII and COPI vesicle coats in endoplasmic reticulum to Golgi transport. J. Cell Biol., 131, 875–893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aridor M., Fish,K.N., Bannykh,S., Weissman,J., Roberts,T.H., Lippincott-Schwartz,J. and Balch,W.E. (2001) The Sar1 GTPase coordinates biosynthetic cargo selection with endoplasmic reticulum export site assembly. J. Cell Biol., 152, 213–229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bennett B.D., Denis,P., Haniu,M., Teplow,D.B., Kahn,S., Louis,J.C., Citron,M. and Vassar,R. (2000) A furin-like convertase mediates propeptide cleavage of BACE, the Alzheimer’s β-secretase. J. Biol. Chem., 275, 37712–37717. [DOI] [PubMed] [Google Scholar]
- Burtnick L.D., Koepf,E.K., Grimes,J., Jones,E.Y., Stuart,D.I., McLaughlin,P.J. and Robinson,R.C. (1997) The crystal structure of plasma gelsolin: implications for actin severing, capping and nucleation. Cell, 90, 661–670. [DOI] [PubMed] [Google Scholar]
- Chandra S., Kable,E.P., Morrison,G.H. and Webb,W.W. (1991) Calcium sequestration in the Golgi apparatus of cultured mammalian cells revealed by laser scanning confocal microscopy and ion microscopy. J. Cell Sci., 100, 747–752. [DOI] [PubMed] [Google Scholar]
- Chavrier P. and Goud,B. (1999) The role of ARF and Rab GTPases in membrane transport. Curr. Opin. Cell Biol., 11, 466–475. [DOI] [PubMed] [Google Scholar]
- Creemers J.W., Ines Dominguez,D., Plets,E., Serneels,L., Taylor,N.A., Multhaup,G., Craessaerts,K., Annaert,W. and De Strooper,B. (2001) Processing of β-secretase by furin and other members of the proprotein convertase family. J. Biol. Chem., 276, 4211–4217. [DOI] [PubMed] [Google Scholar]
- Darchen F. and Goud,B. (2000) Multiple aspects of Rab protein action in the secretory pathway: focus on Rab3 and Rab6. Biochimie, 82, 375–384. [DOI] [PubMed] [Google Scholar]
- Dascher C. and Balch,W.E. (1994) Dominant inhibitory mutants of ARF1 inhibit ER to Golgi transport and trigger the disassembly of the Golgi apparatus. J. Biol. Chem., 269, 1437–1448. [PubMed] [Google Scholar]
- Dascher C., Tisdale,E.J. and Balch,W.E. (1995) Transient expression of small GTPases to study protein transport along secretory pathway in vivo using recombinant T7 vaccinia virus system. Methods Enzymol., 257, 165–173. [DOI] [PubMed] [Google Scholar]
- de Bono S. and de Bono,B. (2001) β-secretase. Trends Biochem. Sci., 26, 14. [Google Scholar]
- Ellgaard L., Molinari,M. and Helenius,A. (1999) Setting the standards: quality control in the secretory pathway. Science, 286, 1882–1888. [DOI] [PubMed] [Google Scholar]
- Ghiso J., Haltia,M., Prelli,F., Novello,J. and Frangione,B. (1990) Gelsolin variant (Asn-187) in familial amyloidosis, Finnish type. Biochem. J., 272, 827–830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Girod A., Storrie,B., Simpson,J.C., Johannes,L., Goud,B., Roberts,L.M., Lord,J.M., Nilsson,T. and Pepperkok,R. (1999) Evidence for a COP-I-independent transport route from the Golgi complex to the endoplasmic reticulum. Nature Cell Biol., 1, 423–430. [DOI] [PubMed] [Google Scholar]
- Greenfield J.P., Tsai,J., Gouras,G.K., Hai,B., Thinakaran,G., Checler,F., Sisodia,S.S., Greengard,P. and Xu,H. (1999a) Endoplasmic reticulum and trans-Golgi network generate distinct populations of Alzheimer β-amyloid peptides. Proc. Natl Acad. Sci. USA, 96, 742–747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greenfield J.P., Xu,H., Greengard,P., Gandy,S. and Seeger,M. (1999b) Generation of the amyloid-β peptide N terminus in Saccharomyces cerevisiae expressing human Alzheimer’s amyloid-β precursor protein. J. Biol. Chem., 274, 33843–33846. [DOI] [PubMed] [Google Scholar]
- Haltia M., Ghiso,J., Prelli,F., Gallo,G., Kiuru,S., Somer,H., Palo,J. and Frangione,B. (1990) Amyloid in familial amyloidosis, Finnish type, is antigenically and structurally related to gelsolin. Am. J. Pathol., 136, 1223–1228. [PMC free article] [PubMed] [Google Scholar]
- Hiltunen T., Kiuru,S., Hongell,V., Helio,T., Palo,J. and Peltonen,L. (1991) Finnish type of familial amyloidosis: cosegregation of Asp187: Asn mutation of gelsolin with the disease in three large families. Am. J. Hum. Genet., 49, 522–528. [PMC free article] [PubMed] [Google Scholar]
- Huse J.T. and Doms,R.W. (2001) Neurotoxic traffic: uncovering the mechanics of amyloid production in Alzheimer’s disease. Traffic, 2, 75–81. [DOI] [PubMed] [Google Scholar]
- Huse J.T., Pijak,D.S., Leslie,G.J., Lee,V.M. and Doms,R.W. (2000) Maturation and endosomal targeting of β-site amyloid precursor protein-cleaving enzyme. The Alzheimer’s disease β-secretase. J. Biol. Chem., 275, 33729–33737. [DOI] [PubMed] [Google Scholar]
- Isaacson R.L., Weeds,A.G. and Fersht,A.R. (1999) Equilibria and kinetics of folding of gelsolin domain 2 and mutants involved in familial amyloidosis-Finnish type. Proc. Natl Acad. Sci. USA, 96, 11247–11252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jahn R. and Sudhof,T.C. (1999) Membrane fusion and exocytosis. Annu. Rev. Biochem., 68, 863–911. [DOI] [PubMed] [Google Scholar]
- Jean F., Stella,K., Thomas,L., Liu,G., Xiang,Y., Reason,A.J. and Thomas,G. (1998) α1-Antitrypsin Portland, a bioengineered serpin highly selective for furin: application as an antipathogenic agent. Proc. Natl Acad. Sci. USA, 95, 7293–7298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kangas H., Paunio,T., Kalkkinen,N., Jalanko,A. and Peltonen,L. (1996) In vitro expression analysis shows that the secretory form of gelsolin is the sole source of amyloid in gelsolin-related amyloidosis. Hum. Mol. Genet., 5, 1237–1243. [DOI] [PubMed] [Google Scholar]
- Kangas H., Ulmanen,I., Paunio,T., Kwiatkowski,D.J., Lehtovirta,M., Jalanko,A. and Peltonen,L. (1999) Functional consequences of amyloidosis mutation for gelsolin polypeptide—analysis of gelsolin–actin interaction and gelsolin processing in gelsolin knock-out fibroblasts. FEBS Lett., 454, 233–239. [DOI] [PubMed] [Google Scholar]
- Kim S.H., Wang,R., Gordon,D.J., Bass,J., Steiner,D.F., Lynn,D.G., Thinakaran,G., Meredith,S.C. and Sisodia,S.S. (1999) Furin mediates enhanced production of fibrillogenic ABri peptides in familial British dementia. Nature Neurosci., 2, 984–988. [DOI] [PubMed] [Google Scholar]
- Kiuru S. (1998) Gelsolin-related familial amyloidosis, Finnish type (FAF) and its variants found worldwide. Amyloid, 5, 55–66. [DOI] [PubMed] [Google Scholar]
- Kwiatkowski D.J., Stossel,T.P., Orkin,S.H., Mole,J.E., Colten,H.R. and Yin,H.L. (1986) Plasma and cytoplasmic gelsolins are encoded by a single gene and contain a duplicated actin-binding domain. Nature, 323, 455–458. [DOI] [PubMed] [Google Scholar]
- Kwiatkowski D.J., Mehl,R., Izumo,S., Nadal-Ginard,B. and Yin,H.L. (1988) Muscle is the major source of plasma gelsolin. J. Biol. Chem., 263, 8239–8243. [PubMed] [Google Scholar]
- Lee W.M. and Galbraith,R.M. (1992) The extracellular actin-scavenger system and actin toxicity. N. Engl. J. Med., 326, 1335–1341. [DOI] [PubMed] [Google Scholar]
- Levy E., Haltia,M., Fernandez-Madrid,I., Koivunen,O., Ghiso,J., Prelli,F. and Frangione,B. (1990) Mutation in gelsolin gene in Finnish hereditary amyloidosis. J. Exp. Med., 172, 1865–1867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lord J.M., Davey,J., Frigerio,L. and Roberts,L.M. (2000) Endoplasmic reticulum-associated protein degradation. Semin. Cell. Dev. Biol., 11, 159–164. [DOI] [PubMed] [Google Scholar]
- Maltese W.A., Wilson,S., Tan,Y., Suomensaari,S., Sinha,S., Barbour,R. and McConlogue,L. (2001) Retention of the Alzheimer’s amyloid precursor fragment C99 in the endoplasmic reticulum prevents formation of amyloid β-peptide. J. Biol. Chem., 276, 20267–20279. [DOI] [PubMed] [Google Scholar]
- Martinez O., Schmidt,A., Salamero,J., Hoflack,B., Roa,M. and Goud,B. (1994) The small GTP-binding protein rab6 functions in intra-Golgi transport. J. Cell Biol., 127, 1575–1588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martinez O., Antony,C., Pehau-Arnaudet,G., Berger,E.G., Salamero,J. and Goud,B. (1997) GTP-bound forms of rab6 induce the redistribution of Golgi proteins into the endoplasmic reticulum. Proc. Natl Acad. Sci. USA, 94, 1828–1833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maury C.P. (1991) Gelsolin-related amyloidosis. Identification of the amyloid protein in Finnish hereditary amyloidosis as a fragment of variant gelsolin. J. Clin. Invest., 87, 1195–1199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maury C.P. and Baumann,M. (1990) Isolation and characterization of cardiac amyloid in familial amyloid polyneuropathy type IV (Finnish): relation of the amyloid protein to variant gelsolin. Biochim. Biophys. Acta, 1096, 84–86. [DOI] [PubMed] [Google Scholar]
- Maury C.P., Alli,K. and Baumann,M. (1990) Finnish hereditary amyloidosis. Amino acid sequence homology between the amyloid fibril protein and human plasma gelsolin. FEBS Lett., 260, 85–87. [DOI] [PubMed] [Google Scholar]
- Mills I.G., Jones,A.T. and Clague,M.J. (1999) Regulation of endosome fusion. Mol. Membr. Biol., 16, 73–79. [DOI] [PubMed] [Google Scholar]
- Mohrmann K. and van der Sluijs,P. (1999) Regulation of membrane transport through the endocytic pathway by Rab GTPases. Mol. Membr. Biol., 16, 81–87. [DOI] [PubMed] [Google Scholar]
- Molloy S.S., Bresnahan,P.A., Leppla,S.H., Klimpel,K.R. and Thomas,G. (1992) Human furin is a calcium-dependent serine endoprotease that recognizes the sequence Arg-X-X-Arg and efficiently cleaves anthrax toxin protective antigen. J. Biol. Chem., 267, 16396–16402. [PubMed] [Google Scholar]
- Molloy S.S., Anderson,E.D., Jean,F. and Thomas,G. (1999) Bi-cycling the furin pathway: from TGN localization to pathogen activation and embryogenesis. Trends Cell Biol., 9, 28–35. [DOI] [PubMed] [Google Scholar]
- Nakayama K. (1997) Furin: a mammalian subtilisin/Kex2p-like endoprotease involved in processing of a wide variety of precursor proteins. Biochem. J., 327, 625–635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paunio T., Kangas,H., Kalkkinen,N., Haltia,M., Palo,J. and Peltonen,L. (1994) Toward understanding the pathogenic mechanisms in gelsolin-related amyloidosis: in vitro expression reveals an abnormal gelsolin fragment. Hum. Mol. Genet., 3, 2223–2229. [DOI] [PubMed] [Google Scholar]
- Paunio T., Kangas,H., Kiuru,S., Palo,J., Peltonen,L. and Syvanen,A.C. (1997) Tissue distribution and levels of gelsolin mRNA in normal individuals and patients with gelsolin-related amyloidosis. FEBS Lett., 406, 49–55. [DOI] [PubMed] [Google Scholar]
- Paunio T., Kangas,H., Heinonen,O., Buc-Caron,M.H., Robert,J.J., Kaasinen,S., Julkunen,I., Mallet,J. and Peltonen,L. (1998) Cells of the neuronal lineage play a major role in the generation of amyloid precursor fragments in gelsolin-related amyloidosis. J. Biol. Chem., 273, 16319–16324. [DOI] [PubMed] [Google Scholar]
- Petanceska S.S., Seeger,M., Checler,F. and Gandy,S. (2000) Mutant presenilin 1 increases the levels of Alzheimer amyloid β-peptide Aβ42 in late compartments of the constitutive secretory pathway. J. Neurochem., 74, 1878–1884. [DOI] [PubMed] [Google Scholar]
- Puius Y.A., Fedorov,E.V., Eichinger,L., Schleicher,M. and Almo,S.C. (2000) Mapping the functional surface of domain 2 in the gelsolin superfamily. Biochemistry, 39, 5322–5331. [DOI] [PubMed] [Google Scholar]
- Ratnaswamy G., Koepf,E., Bekele,H., Yin,H. and Kelly,J.W. (1999) The amyloidogenicity of gelsolin is controlled by proteolysis and pH. Chem. Biol., 6, 293–304. [DOI] [PubMed] [Google Scholar]
- Ratnaswamy G., Huff,M.E., Su,A.I., Rion,S. and Kelly,J.W. (2001) Destabilization of Ca2+-free gelsolin may not be responsible for proteolysis in familial amyloidosis of Finnish type. Proc. Natl Acad. Sci. USA, 98, 2334–2339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rowe T., Aridor,M., McCaffery,J.M., Plutner,H. and Balch,W.E. (1996) COPII vesicles derived from mammalian endoplasmic reticulum (ER) microsomes recruit COPI. J. Cell Biol., 135, 895–911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rybin V., Ullrich,O., Rubino,M., Alexandrov,K., Simon,I., Seabra,M.C., Goody,R. and Zerial,M. (1996) GTPase activity of Rab5 acts as a timer for endocytic membrane fusion. Nature, 383, 266–269. [DOI] [PubMed] [Google Scholar]
- Schekman R. and Orci,L. (1996) Coat proteins and vesicle budding. Science, 271, 1526–1533. [DOI] [PubMed] [Google Scholar]
- Schnuchel A., Wiltscheck,R., Eichinger,L., Schleicher,M. and Holak,T.A. (1995) Structure of severin domain 2 in solution. J. Mol. Biol., 247, 21–27. [DOI] [PubMed] [Google Scholar]
- Selkoe D.J. (1998) The cell biology of β-amyloid precursor protein and presenilin in Alzheimer’s disease. Trends Cell Biol., 8, 447–453. [DOI] [PubMed] [Google Scholar]
- Selkoe D.J. (2000) The genetics and molecular pathology of Alzheimer’s disease: roles of amyloid and the presenilins. Neurol. Clin., 18, 903–922. [DOI] [PubMed] [Google Scholar]
- Somsel Rodman J. and Wandinger-Ness,A. (2000) Rab GTPases coordinate endocytosis. J. Cell Sci., 113, 183–192. [DOI] [PubMed] [Google Scholar]
- Steiner D.F. (1998) The proprotein convertases. Curr. Opin. Chem. Biol., 2, 31–39. [DOI] [PubMed] [Google Scholar]
- Sun H.Q., Yamamoto,M., Mejillano,M. and Yin,H.L. (1999) Gelsolin, a multifunctional actin regulatory protein. J. Biol. Chem., 274, 33179–33182. [DOI] [PubMed] [Google Scholar]
- Takahashi S., Kasai,K., Hatsuzawa,K., Kitamura,N., Misumi,Y., Ikehara,Y., Murakami,K. and Nakayama,K. (1993) A mutation of furin causes the lack of precursor-processing activity in human colon carcinoma LoVo cells. Biochem. Biophys. Res. Commun., 195, 1019–1026. [DOI] [PubMed] [Google Scholar]
- Tector M. and Salter,R.D. (1995) Calnexin influences folding of human class I histocompatibility proteins but not their assembly with β2-microglobulin. J. Biol. Chem., 270, 19638–19642. [DOI] [PubMed] [Google Scholar]
- Tisdale E.J., Bourne,J.R., Khosravi-Far,R., Der,C.J. and Balch,W.E. (1992) GTP-binding mutants of Rab1 and Rab2 are potent inhibitors of vesicular transport from the endoplasmic reticulum to the Golgi complex. J. Cell Biol., 119, 749–761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Way M. and Weeds,A. (1988) Nucleotide sequence of pig plasma gelsolin. Comparison of protein sequence with human gelsolin and other actin-severing proteins shows strong homologies and evidence for large internal repeats. J. Mol. Biol., 203, 1127–1133. [DOI] [PubMed] [Google Scholar]
- Weeds A.G., Gooch,J., McLaughlin,P. and Maury,C.P. (1993) Variant plasma gelsolin responsible for familial amyloidosis (Finnish type) has defective actin severing activity. FEBS Lett., 335, 119–123. [DOI] [PubMed] [Google Scholar]
- White J. et al. (1999) Rab6 coordinates a novel Golgi to ER retrograde transport pathway in live cells. J. Cell Biol., 147, 743–760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wieland F. and Harter,C. (1999) Mechanisms of vesicle formation: insights from the COP system. Curr. Opin. Cell Biol., 11, 440–446. [DOI] [PubMed] [Google Scholar]
- Wu M.M., Llopis,J., Adams,S., McCaffery,J.M., Kulomaa,M.S., Machen,T.E., Moore,H.P. and Tsien,R.Y. (2000) Organelle pH studies using targeted avidin and fluorescein–biotin. Chem. Biol., 7, 197–209. [DOI] [PubMed] [Google Scholar]
- Yin H.L. (1987) Gelsolin: calcium- and polyphosphoinositide-regulated actin-modulating protein. BioEssays, 7, 176–179. [DOI] [PubMed] [Google Scholar]
- Zapun A., Grammatyka,S., Deral,G. and Vernet,T. (2000) Calcium dependent conformational stability of modules 1 and 2 of gelsolin. Biochem. J., 350, 873–881. [PMC free article] [PubMed] [Google Scholar]
- Zerial M. and McBride,M. (2001) Rab proteins as membrane organizers. Nature Rev., 2, 107–119. [DOI] [PubMed] [Google Scholar]
- Zhou A., Webb,G., Zhu,X. and Steiner,D.F. (1999) Proteolytic processing in the secretory pathway. J. Biol. Chem., 274, 20745–20748. [DOI] [PubMed] [Google Scholar]