

Abstract
Prion protein (PrP) is usually attached to membranes by a glycosylphosphatidylinositol-anchor that associates with detergent-resistant membranes (DRMs), or rafts. To model the molecular processes that might occur during the initial infection of cells with exogenous transmissible spongiform encephalopathy (TSE) agents, we examined the effect of membrane association on the conversion of the normal protease-sensitive PrP isoform (PrP-sen) to the protease-resistant isoform (PrP-res). A cell-free conversion reaction approximating physiological conditions was used, which contained purified DRMs as a source of PrP-sen and brain microsomes from scrapie-infected mice as a source of PrP-res. Interestingly, DRM-associated PrP-sen was not converted to PrP-res until the PrP-sen was either released from DRMs by treatment with phosphatidylinositol-specific phospholipase C (PI-PLC), or the combined membrane fractions were treated with the membrane-fusing agent polyethylene glycol (PEG). PEG-assisted conversion was optimal at pH 6–7, and acid pre-treating the DRMs was not sufficient to permit conversion without PI-PLC or PEG, arguing against late endosomes/lysosomes as primary compartments for PrP conversion. These observations raise the possibility that generation of new PrP-res during TSE infection requires (i) removal of PrP-sen from target cells; (ii) an exchange of membranes between cells; or (iii) insertion of incoming PrP-res into the raft domains of recipient cells.
Keywords: microsomes/PEG/PI-PLC/PrP/raft
Introduction
Transmissible spongiform encephalopathies (TSEs) are infectious neurodegenerative diseases naturally occurring in humans (Creutzfeldt–Jakob disease) and a variety of animals including cattle (bovine spongiform encephalopathy), sheep and goats (scrapie). The infectious agent responsible for TSEs, often called the prion, has yet to be fully defined (Chesebro, 1998, 1999). In contrast, some researchers have argued that evidence has accumulated that strongly suggests that the agent is composed of an abnormal isoform of a glycosylphosphatidylinositol (GPI)-anchored protein named PrP (reviewed in Prusiner, 1998). The two isoforms of PrP have been designated PrP-sen (or PrPC) and PrP-res (or PrPSc), based on their respective sensitivity or resistance to protease digestion. TSE agent propagation may occur through the PrP-res-directed conversion of PrP-sen to new PrP-res. This process has been modeled in cell-free reactions (Kocisko et al., 1994; Horiuchi et al., 1999; Saborio et al., 2001). However, no in vitro-generated PrP-res has been shown to be infectious (Hill et al., 1999; Caughey et al., 2001).
Although the mechanism by which PrP-sen is converted to PrP-res in TSE-infected cells and in vivo is not clear, data from cell-free reactions suggest this process is akin to autocatalytic polymerization (reviewed in Caughey et al., 2001). Some of our knowledge of the conversion process is derived from studies of neuronal cell lines persistently infected with scrapie (ScN2a cells). Based on this model, the conversion event has been proposed to occur either on the cell surface or at some point during the endocytic trafficking of PrP-sen (Caughey and Raymond, 1991; Caughey et al., 1991; Borchelt et al., 1992). PrP-res then appears to accumulate in lysosomes (Caughey et al., 1991; McKinley et al., 1991). This is consistent with some reports of disease-specific PrP localization in vivo (Laszlo et al., 1992; Arnold et al., 1995; Grigoriev et al., 1999). However, others find that in vivo most disease-specific PrP accumulates extracellularly or on neuronal plasmalemma, and have suggested the possibility that the conversion process may be primarily extracellular (Jeffrey et al., 1992, 1994, 1997). Similarly, a large proportion of PrP-res is also surface localized in ScN2a cells (Vey et al., 1996).
Like other GPI-anchored proteins, PrP is enriched in sphingolipid-cholesterol-rich membrane microdomains, or rafts (Vey et al., 1996). These membranes are also referred to as detergent-resistant membranes (DRMs) based on their insolubility in cold non-ionic detergents (e.g. Triton X-100) (Brown and Rose, 1992). Several lines of evidence suggest a role for rafts in the formation of PrP-res in scrapie-infected cultured cells: (i) PrP-sen and PrP-res co-localize to DRM fractions (Vey et al., 1996; Naslavsky et al., 1997); (ii) cholesterol depletion renders PrP-sen soluble in cold Triton X-100 and decreases PrP-res formation in ScN2a cells (Taraboulos et al., 1995); (iii) recombinant transmembrane forms of PrP-sen that are not localized to rafts do not serve as substrates for PrP-res formation in ScN2a cells (Taraboulos et al., 1995; Kaneko et al., 1997); (iv) depletion of cellular sphingolipids increases PrP-res biosynthesis (Naslavsky et al., 1999); and (v) treatment with the sterol-binding anti-scrapie drug amphotericin B modifies the floatation properties of DRMs and inhibits PrP-res synthesis (Mangé et al., 2000). Thus, rafts have attracted attention as a candidate site for the generation of PrP-res in cells.
Interactions of PrP-res with its protease-sensitive precursor, PrP-sen, are of central importance in TSE diseases. Extensive structural analyses of PrP-sen, and to a lesser extent PrP-res, have provided valuable information regarding the conformational differences between these two molecules. However, few studies have considered the GPI-anchored nature of PrP and the possible effects of membrane attachment on PrP-sen structure (DeArmond et al., 1999; Morillas et al., 1999; Zuegg and Gready, 2000) and its binding interactions with PrP-res. Although cell-free studies have provided valuable insight into the binding and conversion of PrP-sen by PrP-res, all of these studies used purified PrP-sen, often in a GPI-anchor-deficient form, and most contained denaturant and/or detergent (Kocisko et al., 1994, 1995; Bessen et al., 1997; Horiuchi et al., 1999, 2000; Wong et al., 2001).
Here, we have directly assessed the ability of raft-associated PrP-sen to interact with exogenous PrP-res molecules in an attempt to model the molecular processes that might occur during the initial infection of cells with exogenous TSE agents. Interactions between PrP-sen and PrP-res were assayed using a cell-free conversion reaction (Kocisko et al., 1994; Horiuchi et al., 1999) under near-physiological conditions in the absence of denaturants and detergents. Our data suggest that raft-bound PrP-sen is not a substrate for conversion to the protease-resistant state by exogenous PrP-res molecules unless the PrP-res is itself inserted into contiguous membranes.
Results
PrP-sen is enriched in DRMs
The Neuro2a mouse neuroblastoma cell line is widely used for TSE research. However, this cell line also expresses relatively low levels of PrP-sen compared with transfected cell lines. To facilitate our studies, we used Neuro2a cells that overexpress wild-type mouse PrP-sen (Wong et al., 2001). By fluorescence-activated cell-sorting analysis, the new cell line, designated 5E4E, expressed ∼10-fold higher levels of PrP-sen as compared with Neuro2a cells (K.Wehrly and B.Chesebro, unpublished data).
Others have shown that in neuroblastoma cells, PrP-sen is localized to low density DRMs (Gorodinsky and Harris, 1995; Vey et al., 1996; Naslavsky et al., 1997). To ensure that PrP-sen remained localized in DRMs, 5E4E cells were solubilized in cold Triton X-100 and subjected to floatation on Optiprep gradients using a protocol we modified from Naslavsky et al. (1997). Gradient fractions were collected and analyzed for the presence of PrP-sen as well as the DRM marker ganglioside GM1 (Naslavsky et al., 1997). As shown in Figure 1A, virtually all of the detectable PrP-sen was localized in the low density fractions of the Optiprep gradient. Estimates of total protein content in the gradient fractions by liquid scintillation counting of fractions from metabolically labeled cells indicated that the DRMs contained ∼3% of the total label incorporated (data not shown), consistent with the levels reported by others (Vey et al., 1996). Ganglioside GM1 was found to co-localize with PrP-sen in these low density fractions (Figure 1C). However, when 5E4E cells were solubilized at 37°C, conditions that solubilize DRM-associated molecules, both PrP-sen and ganglioside GM1 were shifted to higher density fractions (Figure 1B and D). PrP-sen was similarly shifted to higher density fractions when lysed at 4°C with 1% N-octyl β-d-glucopyranoside (data not shown), a raft-disrupting detergent (Brown and Rose, 1992). The data shown in Figure 1 indicate that PrP-sen in 5E4E cells is enriched in DRMs.

Fig. 1. PrP-sen is enriched in DRMs. Mouse neuroblastoma cells (5E4E) overexpressing wild-type mouse PrP were lysed in CBS (pH 6.0) with 1% Triton X-100 at either 4 or 37°C. Lysates were subjected to floatation on Optiprep gradients. Gradient fractions were collected and examined by immunoblotting for the presence of PrP (A and B) and the DRM marker ganglioside GM1 (C and D). Fraction numbers are indicated above each lane. Molecular mass markers are indicated in kDa on the left. The data are representative of two independent experiments.
PrP-sen is surface localized in purified DRMs
Since we planned to use DRMs as a means to assay the interaction of PrP-res with DRM-associated PrP-sen, it was important to have a measure of the amount of surface-accessible PrP-sen as purified DRMs are known to contain a large proportion of vesicle-like structures (Brown and Rose, 1992; Sargiacomo et al., 1993; Vey et al., 1996). Purified DRMs were treated with proteinase K (PK) in the absence or presence of 1% Triton X-100 at 37°C and assayed for the presence of protease-resistant PrP by immunoblotting. In the absence of detergent, the amount of PK-resistant protein would be an indicator of the amount of PrP-sen protected from digestion as a consequence of being localized within the lumen of the DRMs. As shown in Figure 2, the vast majority of the PrP-sen in purified DRMs was susceptible to PK digestion. Quantitation of the bands indicated that ∼8% of the total PrP in purified DRMs was PK resistant and thus localized to the lumen of DRMs. This was confirmed by the absence of PK-resistant PrP in parallel reactions containing Triton X-100 (at 37°C), where the DRMs would be disrupted and both luminal and surface-localized PrP-sen would be exposed to PK. Similar results were obtained by labeling purified DRMs with a membrane-impermeable biotinylation reagent (sulfo-NHS-LC-biotin) and measuring the amount of PrP precipitated by treatment with neutravidin–agarose (data not shown). Thus, the above data suggest that ∼92% of the PrP-sen in purified DRMs was surface localized.
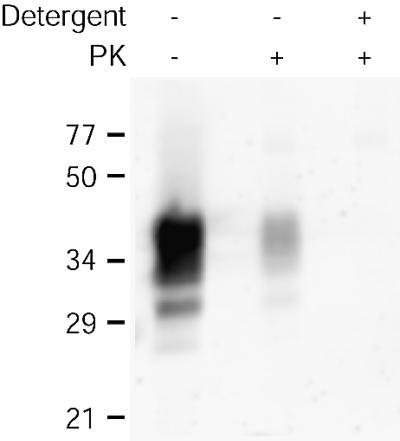
Fig. 2. PrP-sen is surface localized in purified DRMs. DRMs were digested with PK in the presence or absence of detergent (1% Triton X-100) at 37°C. Samples were assayed for PrP by immunoblotting. Results are representative of two independent experiments, each performed in duplicate.
Microsomes from scrapie-infected mouse brain contain PrP-res
To permit the use of DRMs as a source of PrP-sen in cell-free conversion reactions, a detergent-free fraction of PrP-res was required. The PrP-res used in the reactions was contained in a crude brain membrane fraction (see Materials and methods) from a mouse infected with the mouse-adapted scrapie strain 87V. This fraction contains membrane vesicles that may be derived from all types of cellular membranes including plasma, synaptosomal and endoplasmic reticulum membranes, for example. For convenience, we refer to this fraction as ‘microsomal’. We confirmed that the 87V microsomal fraction contained PrP-res by immunoblotting. As shown in Figure 3, although microsomes from both normal and scrapie-infected mouse brains contained PrP, only microsomes from infected animals (Sc+) contained PrP-res. After PK digestion, the microsome-associated PrP-res closely resembled purified, PK-digested 87V PrP-res (Figure 3), including the ∼6–8 kDa truncation corresponding to the removal of the N-terminal ∼67 amino acids (Prusiner et al., 1984; Oesch et al., 1985; Hope et al., 1986).
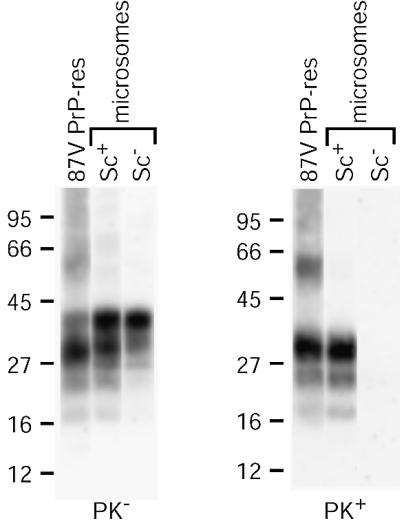
Fig. 3. Immunoblot analysis of PrP in mouse brain microsomes. Crude microsomes were prepared from the brains of normal (Sc–) or 87V scrapie-infected (Sc+) mice and assayed for total PrP (PK–) or PrP-res (PK+) by immunoblotting of untreated or PK-digested samples. Samples of PrP-res purified from 87V-infected mice (87V PrP-res) without (PK–) and with (PK+) PK digestion are shown for comparison.
Membrane-associated PrP-sen resists conversion to PrP-res
To directly assess whether raft-associated PrP-sen can serve as a substrate for the generation of new PrP-res, we purified DRMs from metabolically labeled 5E4E cells ([35S]DRMs) and used these membranes in cell-free conversion reactions. When [35S]DRMs were mixed with PrP-res-containing microsomes, we were unable to detect the generation of any PrP-res-like PK-resistant bands (Figure 4, lane 5). We observed the same results using PrP-sen contained in microsomes from neuroblastoma cells, in rafts prepared from multiple cell lines and using rafts prepared by detergent-based and detergent-free methods (data not shown). Similarly, purified PrP-sen reconstituted into raft-like liposomes (Schroeder et al., 1994) was not converted to new PrP-res when incubated with PrP-res-containing microsomes (data not shown). Collectively, our data suggest that membrane-associated PrP-sen resists conversion to PrP-res when simply incubated in the presence of exogenous PrP-res molecules.
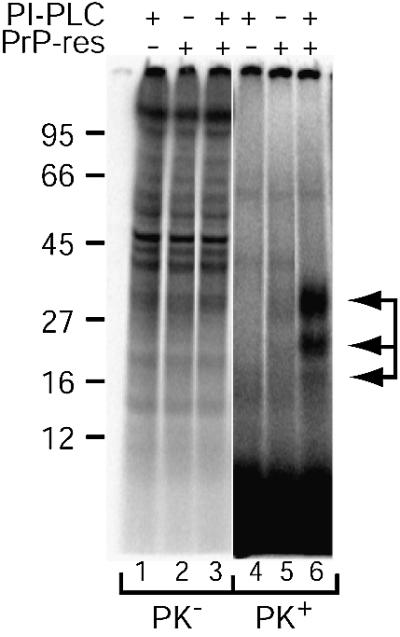
Fig. 4. PI-PLC assists cell-free conversion of DRM-associated PrP-sen. Cell-free conversion reactions were performed using [35S]DRMs and crude microsomes from the brains of normal (PrP-res negative lanes) or scrapie-infected (PrP-res positive lanes) mice. PI-PLC was added to the reactions where indicated. PK– lanes contain a one-tenth aliquot of each reaction mixture before PK digestion and are shown at reduced contrast to allow visualization of individual bands. PK+ lanes contain a nine-tenths aliquot of each reaction mixture after PK digestion. Arrows indicate PK-resistant [35S]PrP-res bands (lane 6). The data are representative of several independent experiments.
PI-PLC treatment assists cell-free conversion of DRM-associated PrP-sen
To determine whether release of PrP-sen from DRMs would permit its conversion to PrP-res, conversion reactions were performed in the presence of phosphatidylinositol-specific phospholipase C (PI-PLC), an enzyme that cleaves off the diacylglycerol moiety of the GPI anchor and releases the remainder of PrP-sen, but not PrP-res, from the surface of intact cells (Stahl et al., 1987; Caughey et al., 1989, 1990). The addition of PI-PLC to conversion reactions with [35S]DRMs produced three protease-resistant bands after PK treatment (Figure 4, lane 6). The apparent molecular mass and pattern of the bands were similar to those of the input PrP-res after PK digestion. DRM conversion reactions conducted using purified 87V PrP-res exhibited the same requirement for PI-PLC digestion, although the conversion efficiency was apparently reduced when compared with reactions with 87V microsomes (data not shown). Due to the reduced conversion efficiency, we did not pursue further DRM conversion studies with purified 87V PrP-res. When PI-PLC was added to reactions with normal brain microsomes, no protease-resistant bands were detected (Figure 4, lane 4), indicating that the protease-resistant bands observed with 87V microsomes were PrP-res dependent. Subsequent experiments discussed below showed that the PK-resistant bands were glycoforms of PrP. Thus, DRM-associated PrP-sen serves as a substrate for PrP-res synthesis after PI-PLC cleavage.
The DRM cell-free conversion product is PrP
To confirm that the PK-resistant bands observed above corresponded to conversion of PrP-sen to the protease-resistant state, we analyzed the immunoreactivity of the conversion products by immunoprecipitation with PrP-specific antiserum (R30) against a synthetic PrP peptide. The R30 antibody precipitated the three PK-resistant bands from a [35S]DRM conversion reaction with PI-PLC (Figure 5, lane 3) but did not precipitate any proteins from a negative control reaction lacking PrP-res (Figure 5, lane 5). Precipitation of the [35S]DRM bands was competed in the presence of the cognate peptide for the R30 antibody, demonstrating the specificity of the immunoprecipitation. Similar results were obtained from conversion reactions using purified, PI-PLC-digested [35S]PrP-sen (Figure 5, lanes 10–12). Therefore, the PK-resistant bands produced in the [35S]DRM conversion reaction are composed of protease-resistant PrP.
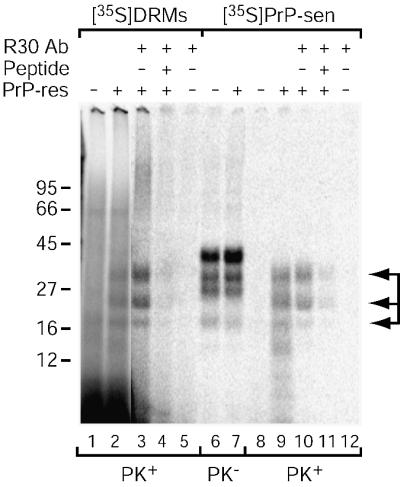
Fig. 5. The [35S]DRM conversion product is PrP. Conversion reactions were performed as in Figure 4 using microsomes and [35S]DRMs (with PI-PLC; lanes 1–5) or PI-PLC-digested [35S]PrP-sen (lanes 6–12). Conversion reaction products were immunoprecipitated using either R30 antiserum against a mouse PrP synthetic peptide (89–103; lanes 3, 5, 10 and 12) or R30 pre-absorbed with peptide 89–103 (lanes 4 and 11). Conversion reactions without immunoprecipitation (lanes 1, 2 and 6–9) are shown for comparison. PK– and PK+ indicate lanes without and with PK digestion, respectively. PK+ lanes contain nine times the reaction equivalents loaded in PK– lanes, except for the immunoprecipitated samples where the entire reaction was PK digested. Arrows indicate PK-resistant [35S]PrP-res bands (lanes 2, 3, 9 and 10). The data are representative of a single experiment performed in duplicate.
PEG treatment assists cell-free conversion of DRM-associated PrP-sen
It is possible that conversion of membrane-associated PrP-sen to PrP-res may require insertion of PrP-res seeds into PrP-sen-containing membranes. To test this idea, we attempted to induce fusion of DRMs and microsomes using a protocol commonly employed to generate hybridomas involving a brief treatment with polyethylene glycol (PEG) (Harlow and Lane, 1988). As shown in Figure 6A (lane 4), treatment with a high concentration of PEG (30%) allowed conversion of DRM-bound PrP-sen. In contrast, treatment with an intermediate PEG concentration (15%) allowed at best a very low level of conversion (Figure 6A, lane 3). Based on the input amount of [35S]PrP-sen derived by immunoprecipitation of PrP from [35S]DRMs, we estimated the conversion efficiency of PI-PLC and 30% PEG-treated reactions to be 17 and 25%, respectively. This corresponds to about half the efficiency observed with reactions using purified components under related conditions (Wong et al., 2001). A requirement for 30% PEG or PI-PLC treatment to assist PrP conversion was also observed using rafts from two other Neuro2a cell lines and prepared by detergent-based and detergent-free methods (Smart et al., 1995), suggesting the results are neither cell clone specific nor artifacts of the raft preparation procedure (data not shown). Occasionally, small amounts of two lower molecular mass species (∼21 kDa) were observed in all lanes, likely resulting from incomplete PK digestion. We also verified that PEG pre-treatment of 87V microsomes alone did not enhance their inherent converting activity as measured by conversion of purified PrP-sen (data not shown). Electron microscope (EM) analysis of DRM/normal microsome mixtures incubated with or without PEG has shown an increase in vesicle size only after treatment with 30% PEG, suggesting membrane fusion had occurred (Figure 7). Biosafety considerations prohibited us from EM analysis of scrapie brain microsomes. Although we cannot entirely rule out the possibility that a PEG-dependent effect other than membrane fusion is responsible for assisting conversion of DRM-associated PrP-sen, control reactions using purified PrP-sen suggest that the PEG treatment we have used does not have a significant direct effect on the conversion reaction (data not shown). Together, these data provide evidence that introduction of PrP-res, either alone or along with another microsome component, into DRMs or contiguous membrane domains is critical for conversion of DRM-associated PrP-sen to PrP-res.
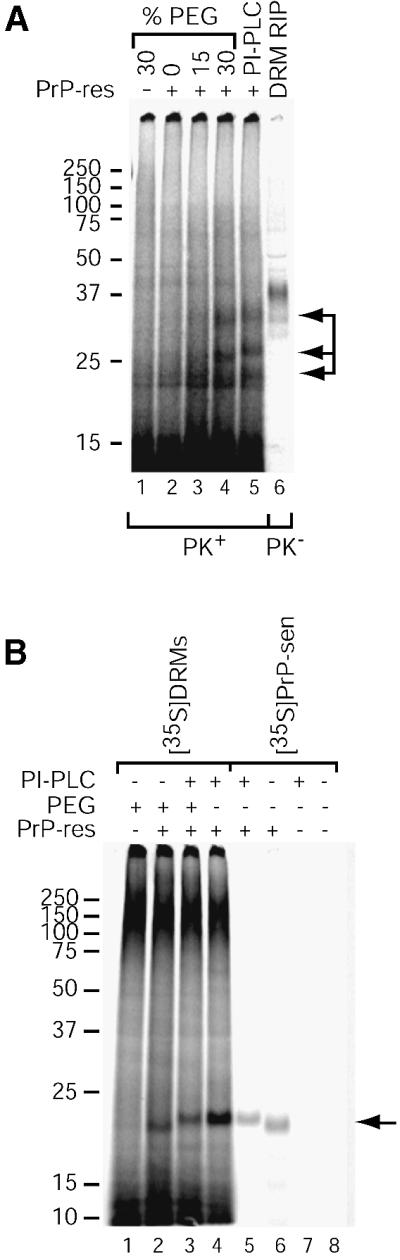
Fig. 6. PEG assists cell-free conversion of DRM-associated PrP-sen. (A) PEG-assisted [35S]DRM conversion reactions. Samples were treated with various concentrations of PEG or PI-PLC as indicated and cell-free conversion was performed as in Figure 4. PrP-sen was immunoprecipitated from one-fifth equivalents of the [35S]DRMs added to the reaction for comparison (DRM RIP, lane 6). The data are representative of several independent experiments, each performed in duplicate. Arrows indicate the PK-resistant [35S]PrP-res bands (lanes 4 and 5). (B) Effect of PNGase F and PI-PLC treatment. Cell-free conversions were performed using [35S]DRMs treated with 30% PEG or PI-PLC (lanes 1–4). Where indicated, PEG-treated [35S]DRM reactions were also digested with PI-PLC after conversion to demonstrate the PEG-assisted conversion product is GPI anchored (lane 3). For comparison, conversion reactions using purified 87V PrP-res and purified PI-PLC-digested or GPI-anchored [35S]PrP-sen are shown (lanes 5–8). All reactions were deglycosylated with PNGase F after conversion. An arrow indicates the [35S]PrP-res bands (lanes 2–6).
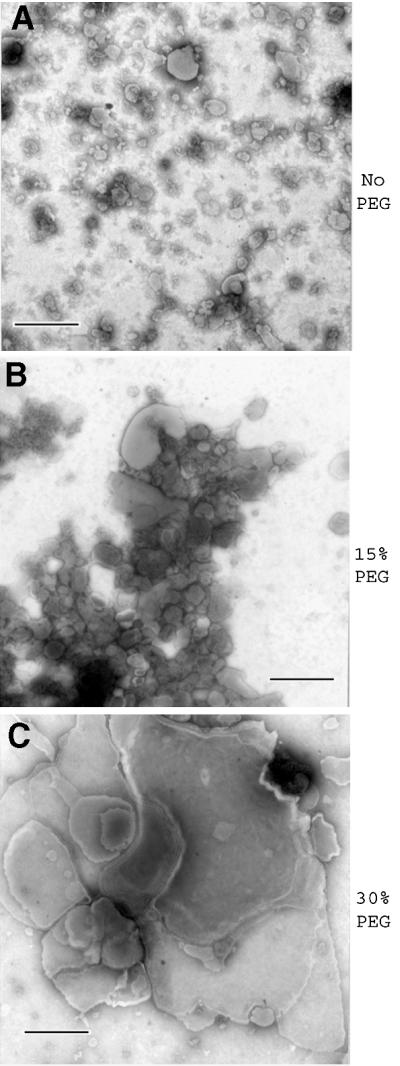
Fig. 7. PEG induces membrane fusion in DRM/microsome mixtures. Mixtures of DRMs and normal brain microsomes were subjected to the designated PEG treatment protocol used in the conversion reactions and analyzed by negative staining EM. Bar = 500 nm.
To further characterize the DRM cell-free conversion products, we treated the reactions with PNGase F after conversion to remove N-linked glycans and compared them to products generated in reactions using purified [35S]PrP-sen and PrP-res. The exact co-migration of the PI-PLC-assisted [35S]DRM conversion product with the product from a reaction using purified [35S]PrP-sen lacking a GPI anchor (Figure 6B, compare lanes 4 and 5) suggested that the PI-PLC-assisted converting species from [35S]DRMs lacked a GPI anchor, as GPI-anchored PrP can be distinguished from PI-PLC-cleaved PrP by slightly reduced electrophoretic mobility of the latter on SDS–PAGE (Caughey et al., 1989; Narwa and Harris, 1999). Conversely, the increased electrophoretic mobility of the PEG-assisted DRM conversion products (Figure 6B, lane 2) suggested that these conversion products were GPI anchored. By digestion with PI-PLC after conversion, we confirmed that the PEG-assisted DRM conversion products were indeed GPI anchored (Figure 6B, lanes 2 and 3), consistent with the model that these products were derived from the fusion of DRMs and microsomes.
pH dependence of PEG-assisted DRM conversions
The nature of the cellular compartment in which PrP conversion occurs has been a subject of debate in the TSE field. Locations from the extracellular milieu through the entire endocytic pathway to lysosomes have all been suggested. We used our DRM conversion system in an attempt to reconstitute these various compartments with respect to containing membrane-associated PrP-sen, PrP-res and a range of pH to identify conditions compatible with conversion in this system. As shown in Figure 8, conversion occurred most efficiently at pH 6–7 and was poor below pH 6.0.
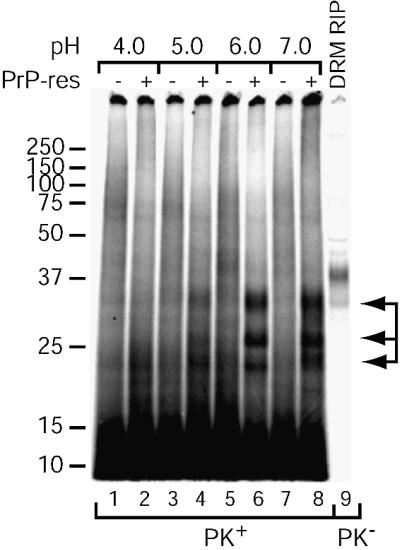
Fig. 8. pH dependence of PEG-assisted [35S]DRM conversion reactions. Conversion reactions with microsomes and [35S]DRMs and 30% PEG treatment were performed as in Figure 6. Following PEG treatment, the reactions were incubated in buffers of decreasing pH and assayed for PrP-res. PrP-sen was immunoprecipitated from one-fifth equivalents of the [35S]DRMs added to the reaction for comparison (DRM RIP, lane 9). Arrows indicate the PK-resistant [35S]PrP-res bands (lanes 4, 6 and 8). The data are representative of two independent experiments, each performed in duplicate.
Acid pre-treatment does not allow cell-free conversion of DRM-associated PrP-sen
Recent studies have suggested acidification of PrP may facilitate its conversion to PrP-res (Jackson et al., 1999; Swietnicki et al., 2000). To determine whether an acid pre-treatment might permit conversion of DRM-bound PrP-sen, DRMs were treated with various acidic buffers on ice, washed with citrate buffer (pH 6.0) and used in conversion reactions. As shown in Figure 9, conversion with acid pre-treated DRMs still required PI-PLC, indicating that acid pre-treatment alone was insufficient to allow conversion of DRM-associated PrP-sen.
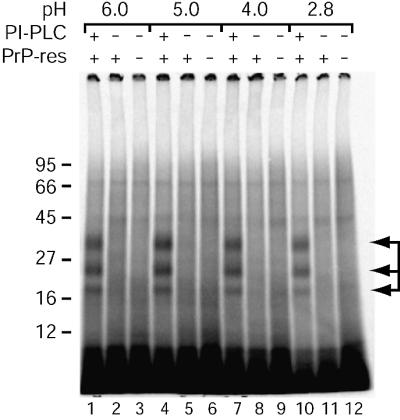
Fig. 9. Acid pre-treatment does not facilitate cell-free conversion of DRM-associated PrP-sen. [35S]DRMs were incubated in buffers of decreasing pH, washed in CBS and used in cell-free conversion reactions as in Figure 4. Arrows indicate the PK-resistant [35S]PrP-res bands (lanes 1, 4, 7 and 10). The data are representative of a single experiment performed in duplicate.
Discussion
In this study, we used DRMs purified from neuroblastoma cells as a source of membrane-bound PrP-sen and tested the ability of this PrP-sen to interact with PrP-res using a cell-free conversion assay in the absence of denaturant and detergent. Since the majority of PrP-sen in cells is associated with DRMs (Figure 1), the PrP-sen used in our experiments is provided in a context approximating that of native PrP-sen with respect to both membrane association as well as other DRM-bound molecules, some of which may influence the conversion process. Thus, these studies serve to bring the cell-free reaction closer to reconstituting the conditions under which the reaction might occur in vivo as cells are initially infected or as TSE agents are transmitted between cells.
Using these near-physiological cell-free conversion conditions, we found that simply incubating DRM-associated PrP-sen with PrP-res did not allow conversion to occur. This raises the possibility that DRM-associated PrP-sen may be incapable of binding to exogenous PrP-res, although binding can occur without conversion (Horiuchi et al., 2000, 2001). Since PrP-res is likely a component of TSE infectivity and PrP-res binds to PrP-sen in solution with high specificity (Horiuchi et al., 1999), this interaction is a good candidate for a ligand–receptor binding event that some have suggested may explain the requirement for PrP in TSE diseases (Chesebro, 1998, 1999). However, our data might challenge the idea that PrP-sen could act as a conventional receptor as DRM-bound PrP-sen is not converted to PrP-res without insertion of PrP-res into the same membrane. Previous studies of the sites of interaction between PrP-sen and PrP-res indicated that binding occurs via sites on PrP-sen that are close in space to the C-terminus where the GPI anchor is normally attached (Horiuchi et al., 1999, 2001). Thus, the association of PrP-sen with DRMs might sterically hinder its binding to exogenous PrP-res.
Addition of PI-PLC to conversion reactions with DRMs to release PrP-sen from DRMs dramatically enhanced its conversion to PrP-res, indicating that the PrP-sen molecules that co-purify with DRMs are indeed capable of being converted to PrP-res. These observations were somewhat unexpected given the current biochemical data indicating the apparent importance of PrP-sen association with DRMs to PrP-res biosynthesis in ScN2a cells (Taraboulos et al., 1995; Kaneko et al., 1997) and that PrP-res produced in cell culture (Vey et al., 1996) or in vivo (Stahl et al., 1987) likely possesses a GPI anchor. In addition, PI-PLC treatment can inhibit PrP-res formation in ScN2a cells (Caughey and Raymond, 1991; Enari et al., 2001), presumably by release of cell-surface PrP-sen and dilution of the released PrP into the culture medium. Thus, in cell culture and in vivo, PrP-sen release by GPI-anchor cleavage is not likely to be a predominant mechanism mediating conversion of PrP-sen to PrP-res.
We theorized that the PrP-res-binding site might be occluded on DRM-bound PrP-sen, thereby preventing membrane-bound PrP-sen from binding to exogenous PrP-res molecules, at least in a manner leading to conversion. Assuming that at least some of the microsomal PrP-res was membrane associated, as suggested by EM studies (Jeffrey et al., 1992, 1994), we reasoned that addition of a membrane fusogen to a mixture of DRMs and microsomes could induce fusion of the two sets of membranes and create larger vesicles containing both PrP-res and PrP-sen. In these new vesicles, PrP-res and PrP-sen, now in cis (i.e. in the same membrane), might then be able to interact in a manner leading to conversion. We tested this theory in our system by inducing membrane fusion using PEG. Studies of PEG-induced liposome fusion have shown that although intermediate (10–15%) and high (>20%) concentrations of PEG both induce vesicle aggregation, only the latter are capable of inducing membrane fusion (Massenburg and Lentz, 1993; Viguera et al., 1993). Consistent with our model, only treatment with a high, fusogenic concentration of PEG allowed conversion of DRM-bound, GPI-anchored PrP-sen. The small amount of conversion observed with 15% PEG might be attributed to the low level lipid mixing that can occur at intermediate PEG concentrations (Viguera et al., 1993).
Bearing in mind that our system models the initial interaction of cell-surface PrP-sen with exogenous PrP-res molecules, our data suggest two scenarios for initiation and propagation of PrP-res synthesis. First, if conversion is a cell-associated process, this would imply a requirement for insertion of PrP-res molecules into host cell membranes prior to induction of PrP-sen conversion. Insertion of PrP-res could be mediated via uptake of membrane microparticles (Mack et al., 2000) or exchange of membrane components between closely apposed cells (Batista et al., 2001), as might conceivably occur at nerve synapses (Figure 10A). Alternatively, insertion could occur by GPI-anchor-directed introduction of PrP-res molecules into target membranes, a process known as ‘painting’ (Medof et al., 1996) (Figure 10B). GPI-anchor-dependent painting has been shown to occur in vivo (Kooyman et al., 1995). The concept of a role for membrane fusion in initiation of scrapie infection of cells has been proposed previously (Clarke and Millson, 1976), although a subsequent attempt to enhance infection of cultured fibroblasts using Sendai virus as a fusogen was unsuccessful (Clarke, 1979). On the other hand, if conversion occurs extracellularly, as proposed by Jeffrey et al. (1997), release of PrP-sen from cell membranes would be necessary to provide substrate for the synthesis of new PrP-res.

Fig. 10. Possible mechanisms for transmission of PrP-res (squares) from an infected cell (thick membrane) to an uninfected cell (thin membrane). (A) Exchange of PrP-res-containing membrane microparticles. (B) GPI-anchor-dependent ‘painting’. PrP-sen (shaded circles).
At this point in our studies, we cannot rule out some alternative explanations for our observations. It is possible that our reactions lack soluble factors present in the extracellular matrix, such as glycosaminoglycans, which assist the conversion of membrane-bound PrP-sen although our reactions were supplemented with heparan sulfate, a candidate accessory factor (Snow et al., 1990; Caughey and Raymond 1993; Gabizon et al., 1993; Caughey et al., 1994; McBride et al., 1998; Wong et al., 2001). Similarly, it could be that the 5E4E cell line used in our studies expresses reduced levels of a conversion accessory factor(s) or that such a hypothetical factor was removed from the raft membranes during the raft preps.
Another possibility was that acidification may be a prerequisite for membrane-bound PrP-sen to adopt a conversion-competent conformation and/or to disrupt a hypothetical association with an inhibitory molecule. Studies using recombinant PrP-sen and GPI-anchor-deficient PrP-sen expressed in the culture supernatant of fibroblast cells have suggested a role for acidification in PrP fibrillization and acquisition of a PK-resistant conformation (Jackson et al., 1999; Swietnicki et al., 2000; M.Horiuchi and B.Caughey, unpublished data). This is consistent with the concept that in neuroblastoma cells, the conversion event may occur in lysosomes or at some point along the endocytic pathway (Caughey and Raymond, 1991; Caughey et al., 1991; Borchelt et al., 1992) in acidic cellular compartments. As observed previously using purified components (Horiuchi et al., 1999), we found that conversion was most efficient at pH 6–7. This led us to consider the possibility that PrP-sen may need to transiently cycle through an acidic compartment to acquire a conversion-competent state prior to conversion in a neutral or near-neutral compartment. However, we found acid pre-treatment followed by incubation at pH 6.0 insufficient to allow the conversion of DRM-bound PrP-sen (Figure 9). Thus, our data support early endosomes and/or the cell surface/extracellular space as primary locations for the conversion reaction.
Materials and methods
Neuroblastoma cells
The construction of the 5E4E mouse neuroblastoma cell line is described elsewhere (Wong et al., 2001). The cells were maintained at 37°C in a humidified atmosphere of 5% CO2 in Dulbecco’s modified Eagle’s medium (DMEM; Gibco) supplemented with 10% fetal bovine serum (FBS; Gibco).
Antibodies
The R30 monospecific rabbit antiserum was raised against a mouse PrP synthetic peptide corresponding to residues 89–103 (Caughey et al., 1991). D13 is a mouse/human recombinant monoclonal antibody Fab that binds mouse PrP between residues 96 and 104 (Williamson et al., 1998) and was a generous gift from Drs Anthony Williamson and Dennis Burton (The Scripps Research Institute, CA). Alkaline phosphatase-conjugated goat anti-rabbit antiserum was obtained from Zymed (62-6122). Alkaline phosphatase-conjugated goat anti-human IgG antibody (AP-GAHIgG) was purchased from Sigma (A8542).
Purification of 35S-labeled PrP-sen
Mouse neuroblastoma cells (5E4E) were metabolically labeled with [35S]methionine as described previously (Caughey et al., 1999a) in methionine-deficient DMEM. To isolate GPI-anchor-deficient PrP-sen, cells were chased for 30 min with complete DMEM (with 10% FBS), washed twice with phosphate-buffered balanced salts solution (PBBS) and incubated at 37°C for 30 min with ∼0.4 U/ml PI-PLC from Bacillus thuringiensis (ICN) in PBBS. After centrifugation at 1000 g for 2 min, the PI-PLC medium was adjusted to contain 0.5% NP-40, 1 mM EDTA, 20 mM Tris–HCl pH 7.4 and a cocktail of protease inhibitors. Radiolabeled PrP-sen was then immunoprecipitated from the PI-PLC media with R30 antibody (Caughey et al., 1999a). For GPI-anchored PrP-sen, PrP was immunoprecipitated with R30 antibody directly from a radiolabeled 5E4E cell lysate without a chase as described previously (Caughey et al., 1999a).
Preparation of DRMs
DRMs were prepared from neuroblastoma cells as described by Naslavsky et al. (1997) with some modifications. Cells (one confluent 75 cm2 plate) were lysed on ice in 225 µl of either citrate-buffered saline (CBS, 10 mM citrate, 137 mM NaCl pH 6.0) or phosphate-buffered saline (PBS, pH 7.0) containing 1% Triton X-100, 1 mM EDTA, 0.1 mM phenylmethylsulfonyl fluoride (PMSF), 0.7 µg/ml pepstatin A and 1 µg/ml aprotinin. To 200 µl of lysate, 216.5 µl of 50% Optiprep (in CBS or PBS) were added to give a final concentration of ∼26% Optiprep. The sample was then overlaid with an Optiprep step gradient (in CBS or PBS) with 200 µl/step of 23, 20, 16, 12, 8, 4 and 2.5% Optiprep. The tubes were spun in a Beckman TLS-55 rotor at 25 000 r.p.m. for 90 min at 4°C. Nine 200 µl fractions were collected from the top of the tubes and stored on ice in a cold room until use. To prepare [35S]DRMs, cells were labeled as described above, chased for 30 min with complete medium, washed with PBBS (three times) then CBS or PBS (three times) and lysed as above. PrP-enriched DRM fractions (1–6) were combined prior to use. We confirmed that the labeling procedure had no effect on the fractionation of PrP and that similar results were obtained using either CBS or PBS (data not shown).
Preparation of crude brain microsomes
Brains were removed from either normal or terminally ill VM/Dk mice infected with 87V scrapie, flash frozen in liquid nitrogen and stored at –80°C until use. Two brains were used per prep. A 10% (w/v) brain homogenate was made in PBS (pH 6.9) containing 1 mM EDTA, 0.1 mM Pefabloc (Roche Molecular Biochemicals), 0.7 µg/ml pepstatin A, 1 µg/ml aprotinin, 0.5 µg/ml leupeptin and 1 mM dithiothreitol. Brains were homogenized by Dounce homogenization (12 strokes with a loose fitting pestle, 20 strokes with a tight fitting pestle). The homogenate was sonicated for 3 min in a cuphorn sonicator and centrifuged at 3000 g for 10 min. The supernatant was removed and the pellet was resuspended to the original volume in PBS and homogenized again (30 strokes with a tight fitting pestle). After centrifugation, the two supernatants were combined, diluted with PBS and spun in a Beckman Type 50.2 rotor at 34 000 r.p.m. for 1 h at 4°C. The microsome pellet was resuspended in PBS with sonication in a cuphorn sonicator and stored on ice in a cold room until use. Total protein content in the microsome fractions was measured by BCA assay (Pierce). PrP-sen and PrP-res in the microsome fractions were analyzed by immunoblotting of untreated samples, or PK-treated (25 µg/ml, 37°C, 1 h) samples after the addition of sarkosyl to 1% to disrupt the membranes. Untreated or PK-treated purified 87V PrP-res standards were used to quantify total PrP or PrP-res, respectively.
Cell-free conversion reactions
Cell-free conversion reactions were performed in 50 mM citrate pH 6.0, 137 mM NaCl and 5 mM MgCl2 (conversion buffer) in a 20 µl final volume similar to that described by Horiuchi et al. (1999) but without sarkosyl. Heparan sulfate (Sigma; H 5393) was added to all reactions at a concentration of 100 µg/ml as in Wong et al. (2001). For some reactions, protease inhibitors (0.1 mM PMSF, 0.7 µg/ml pepstatin A, 1 µg/ml aprotinin) were also included. Where indicated, PI-PLC was added to a final concentration of 0.71 U/ml. Reactions without PI-PLC received an equal volume of PI-PLC enzyme buffer (10 mM Tris–HCl, 50% glycerol, 0.1 M NaCl) instead. Reactions using [35S]PrP-sen contained ∼15 000–20 000 c.p.m./reaction. For reactions with [35S]DRMs, usually ∼1/50 equivalents (24 µl) of the prep was used per reaction. Prior to a reaction, [35S]DRMs were washed by 10-fold dilution with CBS and pelleting at 100 000 g for 2.5 h at 4°C. [35S]DRM pellets were resuspended in CBS and ∼5–7 × 105 c.p.m. were added per reaction. To prepare acid pre-treated DRMs, pellets were resuspended in saline with 10 mM citrate (pH 6.0), 10 mM acetate (pH 5.0 or 4.0) or 0.1 M acetic acid (pH 2.8) and incubated on ice for 30 min. These samples were then diluted 10-fold with CBS, pelleted and resuspended in CBS as above, normalizing for total c.p.m. added to each reaction. [35S]PrP-sen or [35S]DRMs was mixed with brain microsomes containing 100 ng of PrP-res. Control reactions contained brain microsomes from uninfected mice normalized for total protein. For [35S]DRM reactions, DRMs and microsomes were first mixed and then pelleted at 21 000 g for 20 min. The pellets were resuspended with stirring and gentle pipetting in 20 µl of either conversion buffer or PEG (15 or 30%) in conversion buffer and incubated at room temperature for 5 min. The samples were pelleted again, washed once with 40 µl of CBS (again with stirring and gentle pipetting) and finally resuspended in conversion buffer supplemented with heparan sulfate and PI-PLC (where indicated). To examine the pH dependence of PEG-induced conversions, samples treated with 30% PEG in conversion buffer were washed in saline buffers of decreasing pH [10 mM HEPES pH 7.2 (at room temperature); 10 mM citrate pH 6.0; 10 mM acetate pH 5.0 or 4.0] and conversion was performed in conversion buffer substituted with 50 mM of each buffer. Reactions were incubated at 37°C for 3 days. After conversion, [35S]DRM reactions were adjusted to 50 mM Tris–HCl pH 8.0, 0.5% Triton X-100, 0.5% sodium deoxycholate and 137 mM NaCl (1× extraction buffer) and incubated at 37°C for 20 min to disrupt DRMs. Reaction mixtures were processed as described by Horiuchi et al. (1999) with PK digestion for 1 h at 37°C with either 20 µg/ml ([35S]PrP-sen reactions) or 50 µg/ml ([35S]DRM reactions) PK. In some cases, the samples were deglycosylated before methanol precipitation by treatment with PNGase F (New England Biolabs) as per the manufacturer’s instructions. Samples treated with PI-PLC after conversion were digested with PNGase F and PI-PLC simultaneously. PrP-sen was immunoprecipitated from one-fifth the amount of [35S]DRMs added to the reactions as described above after solubilization with 1× extraction buffer, and eluted by boiling in SDS–PAGE sample buffer.
To immunoprecipitate the conversion products, all reactions were adjusted to 1× extraction buffer prior to PK digestion. After PK digestion, the samples were pelleted at 10 000 g for 15 min and washed once with 1× extraction buffer (containing 0.1 mM Pefabloc). Pellets were resuspended in conversion buffer with 1× extraction buffer, 20 µg thyroglobulin and 2 mM Pefabloc. SDS was added to a final concentration of 0.5% and the samples were boiled for 10 min. The samples were then adjusted to 1% NP-40 and 50 mM sodium phosphate pH 7.5. Untreated or pre-absorbed (with the mouse PrP peptide 89–103, 100 µg/ml, 2 h, 37°C) R30 antibody was added, and the samples were treated as described previously (Caughey et al., 1999a) with elution by boiling in SDS–PAGE sample buffer.
Immunoblotting
Immunoblot detection of PrP was performed as described by Horiuchi et al. (1999) using the D13 antibody Fab at 0.7 µg/ml and AP-GAHIgG secondary antibody at 1:10 000 dilution. Detection of ganglioside GM1 in Optiprep gradient fractions was performed by dot blotting on nitrocellulose membranes (Bio-Rad) using horseradish peroxidase-conjugated cholera toxin B subunit (Calbiochem) (Stulnig et al., 1998). Purified 87V PrP-res was prepared from infected VM/Dk mice as described previously (Caughey et al., 1999b).
Electron microscopy
Mixtures of DRMs and normal brain microsomes were treated with or without PEG as described for the cell-free conversion reactions. The samples were analyzed by electron microscopy (60 kV) essentially as described (Xiong et al., 2001), with the exception that the grids were washed with 150 mM ammonium acetate.
Acknowledgments
Acknowledgements
The authors thank Gregory and Lynne Raymond for technical assistance, Gary Hettrick for graphics assistance, and Sue Priola and John Portis for critical reading of the manuscript. G.S.B. was supported in part by a post-doctoral fellowship from the Natural Sciences and Engineering Research Council of Canada.
References
- Arnold J.E., Tipler,C., Laszlo,L., Hope,J., Landon,M. and Mayer,R.J. (1995) The abnormal isoform of the prion protein accumulates in late-endosome-like organelles in scrapie-infected mouse brain. J. Pathol., 176, 403–411. [DOI] [PubMed] [Google Scholar]
- Batista F.D., Iber,D. and Neuberger,M.S. (2001) B cells acquire antigen from target cells after synapse formation. Nature, 411, 489–494. [DOI] [PubMed] [Google Scholar]
- Bessen R.A., Raymond,G.J. and Caughey,B. (1997) In situ formation of protease-resistant prion protein in transmissible spongiform encephalopathy-infected brain slices. J. Biol. Chem., 272, 15227–15231. [DOI] [PubMed] [Google Scholar]
- Borchelt D.R., Taraboulos,A. and Prusiner,S.B. (1992) Evidence for synthesis of scrapie prion proteins in the endocytic pathway. J. Biol. Chem., 267, 16188–16199. [PubMed] [Google Scholar]
- Brown D.A. and Rose,J.K. (1992) Sorting of GPI-anchored proteins to glycolipid-enriched membrane subdomains during transport to the apical cell surface. Cell, 68, 533–544. [DOI] [PubMed] [Google Scholar]
- Caughey B. and Raymond,G.J. (1991) The scrapie-associated form of PrP is made from a cell surface precursor that is both protease- and phospholipase-sensitive. J. Biol. Chem., 266, 18217–18223. [PubMed] [Google Scholar]
- Caughey B. and Raymond,G.J. (1993) Sulfated polyanion inhibition of scrapie-associated PrP accumulation in cultured cells. J. Virol., 67, 643–650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caughey B., Race,R.E., Ernst,D., Buchmeier,M.J. and Chesebro,B. (1989) Prion protein biosynthesis in scrapie-infected and uninfected neuroblastoma cells. J. Virol., 63, 175–181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caughey B., Neary,K., Buller,R., Ernst,D., Perry,L.L., Chesebro,B. and Race,R.E. (1990) Normal and scrapie-associated forms of prion protein differ in their sensitivities to phospholipase and proteases in intact neuroblastoma cells. J. Virol., 64, 1093–1101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caughey B., Raymond,G.J., Ernst,D. and Race,R.E. (1991) N-terminal truncation of the scrapie-associated form of PrP by lysosomal protease(s): implications regarding the site of conversion of PrP to the protease-resistant state. J. Virol., 65, 6597–6603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caughey B., Brown,K., Raymond,G.J., Katzenstein,G.E. and Thresher,W. (1994) Binding of the protease-sensitive form of prion protein PrP to sulfated glycosaminoglycan and Congo red. J. Virol., 68, 2135–2141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caughey B., Horiuchi,M., Demaimay,R. and Raymond,G.J. (1999a) Assays of protease-resistant prion protein and its formation. Methods Enzymol., 309, 122–133. [DOI] [PubMed] [Google Scholar]
- Caughey B., Raymond,G.J., Priola,S.A., Kocisko,D.A., Race,R.E., Bessen,R.A., Lansbury,P.T.,Jr and Chesebro,B. (1999b) Methods for studying prion protein (PrP) metabolism and the formation of protease-resistant PrP in cell culture and cell-free systems. Mol. Biotechnol., 13, 45–55. [DOI] [PubMed] [Google Scholar]
- Caughey B., Raymond,G.J., Callahan,M.A., Wong,C., Baron,G.S. and Xiong,L.W. (2001) Interactions and conversions of prion protein isoforms. Adv. Protein Chem., 57, 139–169. [DOI] [PubMed] [Google Scholar]
- Chesebro B. (1995) Prion protein and the transmissible spongiform encephalopathy diseases. Neuron, 24, 503–506. [DOI] [PubMed] [Google Scholar]
- Chesebro B. (1998) BSE and prions: uncertainties about the agent. Science, 279, 42–43. [DOI] [PubMed] [Google Scholar]
- Clarke M.C. (1979) Infection of cell cultures with scrapie agent. In Prusiner,S.B. and Hadlow,W.J. (eds), Slow Transmissible Diseases of the Nervous System: Volume 2. Academic Press, New York, NY, pp. 225–233.
- Clarke M.C. and Millson,G.C. (1976) Infection of a cell line of mouse L fibroblasts with scrapie agent. Nature, 261, 144–145. [DOI] [PubMed] [Google Scholar]
- DeArmond S.J., Qiu,Y., Sanchez,H., Spilman,P.R., Ninchak-Casey,A., Alonso,D. and Daggett,V. (1999) PrPC glycoform heterogeneity as a function of brain region: implications for selective targeting of neurons by prion strains. J. Neuropathol. Exp. Neurol., 58, 1000–1009. [DOI] [PubMed] [Google Scholar]
- Enari M., Flechsig,E. and Weissmann,C. (2001) Scrapie prion protein accumulation by scrapie-infected neuroblastoma cells abrogated by exposure to a prion protein antibody. Proc. Natl Acad. Sci. USA, 98, 9295–9299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gabizon R., Meiner,Z., Halimi,M. and Ben-Sasson,S.A. (1993) Heparin-like molecules bind differentially to prion-proteins and change their intracellular metabolic fate. J. Cell. Physiol., 157, 319–325. [DOI] [PubMed] [Google Scholar]
- Gorodinsky A. and Harris,D.A. (1995) Glycolipid-anchored proteins in neuroblastoma cells form detergent-resistant complexes without caveolin. J. Cell Biol., 129, 619–627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grigoriev V., Escaig-Haye,F., Streichenberger,N., Kopp,N., Langeveld,J., Brown,P. and Fournier,J.-G. (1999) Submicroscopic immunodetection of PrP in the brain of a patient with a new-variant of Creutzfeldt–Jakob disease. Neurosci. Lett., 264, 57–60. [DOI] [PubMed] [Google Scholar]
- Harlow E. and Lane,D. (1988) Antibodies: A Laboratory Manual. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY.
- Hill A.F., Antoniou,M. and Collinge,J. (1999) Protease-resistant prion protein produced in vitro lacks detectable infectivity. J. Gen. Virol., 80, 11–14. [DOI] [PubMed] [Google Scholar]
- Hope J., Morton,L.J.D., Farquhar,C.F., Multhaup,G., Beyreuther,K. and Kimberlin,R.H. (1986) The major polypeptide of scrapie-associated fibrils (SAF) has the same size, charge distribution and N-terminal protein sequence as predicted for the normal brain protein (PrP). EMBO J., 5, 2591–2597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horiuchi M., Chabry,J. and Caughey,B. (1999) Specific binding of normal prion protein to the scrapie form via a localized domain initiates its conversion to the protease-resistant state. EMBO J., 18, 3193–3203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horiuchi M., Priola,S.A., Chabry,J. and Caughey,B. (2000) Interactions between heterologous forms of prion protein: binding, inhibition of conversion and species barriers. Proc. Natl Acad. Sci. USA, 97, 5836–5841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horiuchi M., Baron,G.S., Xiong,L.-W. and Caughey,B. (2001) Inhibition of interactions and interconversions of prion protein isoforms by peptide fragments from the C-terminal folded domain. J. Biol. Chem., 276, 15489–15497. [DOI] [PubMed] [Google Scholar]
- Jackson G.S. et al. (1999) Reversible conversion of monomeric human prion protein between native and fibrilogenic conformations. Science, 283, 1935–1937. [DOI] [PubMed] [Google Scholar]
- Jeffrey M., Goodsir,C.M., Bruce,M.E., McBride,P.A., Scott,J.R. and Halliday,W.G. (1992) Infection specific prion protein (PrP) accumulates on neuronal plasmalemma in scrapie infected mice. Neurosci. Lett., 147, 106–109. [DOI] [PubMed] [Google Scholar]
- Jeffrey M., Goodsir,C.M., Bruce,M., McBride,P.A., Scott,J.R. and Halliday,W.G. (1994) Correlative light and electron microscopy studies of PrP localisation in 87V scrapie. Brain Res., 656, 329–343. [DOI] [PubMed] [Google Scholar]
- Jeffrey M., Goodsir,C.M., Bruce,M.E., McBride,P.A. and Fraser,J.R. (1997) In vivo toxicity of prion protein in murine scrapie: ultrastructural and immunogold studies. Neuropathol. Appl. Neurobiol., 23, 93–101. [PubMed] [Google Scholar]
- Kaneko K., Vey,M., Scott,M., Pilkuhn,S., Cohen,F.E. and Prusiner,S.B. (1997) COOH-terminal sequence of the cellular prion protein directs subcellular trafficking and controls conversion into the scrapie isoform. Proc. Natl Acad. Sci. USA, 94, 2333–2338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kocisko D.A., Come,J.H., Priola,S.A., Chesebro,B., Raymond,G.J., Lansbury,P.T. and Caughey,B. (1994) Cell-free formation of protease-resistant prion protein. Nature, 370, 471–474. [DOI] [PubMed] [Google Scholar]
- Kocisko D.A., Priola,S.A., Raymond,G.J., Chesebro,B., Lansbury,P.T.,Jr and Caughey,B. (1995) Species specificity in the cell-free conversion of prion protein to protease-resistant forms: a model for the scrapie species barrier. Proc. Natl Acad. Sci. USA, 92, 3923–3927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kooyman D.L. et al. (1995) In vivo transfer of GPI-linked complement restriction factors from erythrocytes to the endothelium. Science, 269, 89–92. [DOI] [PubMed] [Google Scholar]
- Laszlo L. et al. (1992) Lysosomes as key organelles in the pathogenesis of prion encephalopathies. J. Pathol., 166, 333–341. [DOI] [PubMed] [Google Scholar]
- Mack M. et al. (2000) Transfer of the chemokine receptor CCR5 between cells by membrane-derived microparticles: a mechanism for cellular human immunodeficiency virus 1 infection. Nature Med., 6, 769–775. [DOI] [PubMed] [Google Scholar]
- Mangé A., Nishida,N., Milhavet,O., McMahon,H.E.M., Casanova,D. and Lehmann,S. (2000) Amphotericin B inhibits the generation of the scrapie isoform of the prion protein in infected cultures. J. Virol., 74, 3135–3140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Massenburg D. and Lentz,B.R. (1993) Poly(ethylene glycol)-induced fusion and rupture of dipalmitoylphosphatidylcholine large, unilamellar extruded vesicles. Biochemistry, 32, 9172–9180. [DOI] [PubMed] [Google Scholar]
- McBride P.A., Wilson,M.I., Eikelenboom,P., Tunstall,A. and Bruce,M.E. (1998) Heparan sulfate proteoglycan is associated with amyloid plaques and neuroanatomically targeted PrP pathology throughout the incubation period of scrapie-infected mice. Exp. Neurol., 149, 447–454. [DOI] [PubMed] [Google Scholar]
- McKinley M.P., Taraboulos,A., Kenaga,L., Serban,D., Stieber,A., DeArmond,S.J., Prusiner,S.B. and Gonatas,N. (1991) Ultrastructural localization of scrapie prion proteins in cytoplasmic vesicles of infected cultured cells. Lab. Invest., 65, 622–630. [PubMed] [Google Scholar]
- Medof M.E., Nagarajan,S. and Tykocinski,M.L. (1996) Cell-surface engineering with GPI-anchored proteins. FASEB J., 10, 574–586. [DOI] [PubMed] [Google Scholar]
- Morillas M., Swietnicki,W., Gambetti,P. and Surewicz,W.K. (1999) Membrane environment alters the conformational structure of the recombinant human prion protein. J. Biol. Chem., 274, 36859–36865. [DOI] [PubMed] [Google Scholar]
- Narwa R. and Harris,D.A. (1999) Prion proteins carrying pathogenic mutations are resistant to phospholipase cleavage of their glycolipid anchors. Biochemistry, 38, 8770–8777. [DOI] [PubMed] [Google Scholar]
- Naslavsky N., Stein,R., Yanai,A., Friedlander,G. and Taraboulos,A. (1997) Characterization of detergent-insoluble complexes containing the cellular prion protein and its scrapie isoform. J. Biol. Chem., 272, 6324–6331. [DOI] [PubMed] [Google Scholar]
- Naslavsky N., Shmeeda,H., Friedlander,G., Yanai,A., Futerman,A.H., Barenholz,Y. and Taraboulos,A. (1999) Sphingolipid depletion increases formation of the scrapie prion protein in neuroblastoma cells infected with prions. J. Biol. Chem., 274, 20763–20771. [DOI] [PubMed] [Google Scholar]
- Oesch B. et al. (1985) A cellular gene encodes scrapie PrP 27-30 protein. Cell, 40, 735–746. [DOI] [PubMed] [Google Scholar]
- Prusiner S.B. (1998) Prions. Proc. Natl Acad. Sci. USA, 95, 13363–13383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prusiner S.B., Groth,D.F., Bolton,D.C., Kent,S.B. and Hood,L.E. (1984) Purification and structural studies of a major scrapie prion protein. Cell, 38, 127–134. [DOI] [PubMed] [Google Scholar]
- Saborio G.P., Permanne,B. and Soto,C. (2001) Sensitive detection of pathological prion protein by cyclic amplification of protein misfolding. Nature, 411, 810–813. [DOI] [PubMed] [Google Scholar]
- Sargiacomo M., Sudol,M., Tang,Z. and Lisanti,M.P. (1993) Signal transducing molecules and glycosyl-phosphatidylinositol-linked proteins form a caveolin-rich insoluble complex in MDCK cells. J. Cell Biol., 122, 789–807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schroeder R., London,E. and Brown,D. (1994) Interactions between saturated acyl chains confer detergent resistance on lipids and glycosylphosphatidylinositol (GPI)-anchored proteins: GPI-anchored proteins in liposomes and cells show similar behavior. Proc. Natl Acad. Sci. USA, 91, 12130–12134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smart E.J., Ying,Y.-S., Mineo,C. and Anderson,R.G.W. (1995) A detergent-free method for purifying caveolae membrane from tissue culture cells. Proc. Natl Acad. Sci. USA, 92, 10104–10108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Snow A.D., Wight,T.N., Nochlin,D., Koike,Y., Kimata,K., DeArmond,S.J. and Prusiner,S.B. (1990) Immunolocalization of heparan sulfate proteoglycans to the prion protein amyloid plaques of Gerstmann–Straussler syndrome, Creutzfeldt–Jakob disease and scrapie. Lab. Invest., 63, 601–611. [PubMed] [Google Scholar]
- Stahl N., Borchelt,D.R., Hsiao,K. and Prusiner,S.B. (1987) Scrapie prion protein contains a phosphatidylinositol glycolipid. Cell, 51, 229–240. [DOI] [PubMed] [Google Scholar]
- Stulnig T.M., Berger,M., Sigmund,T., Raederstorff,D., Stockinger,H. and Waldhäusl,W. (1998) Polyunsaturated fatty acids inhibit T cell signal transduction by modification of detergent-insoluble membrane domains. J. Cell Biol., 143, 637–644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swietnicki W., Morillas,M., Chen,S.G., Gambetti,P. and Surewicz,W.K. (2000) Aggregation and fibrillization of the recombinant human prion protein huPrP90-231. Biochemistry, 39, 424–431. [DOI] [PubMed] [Google Scholar]
- Taraboulos A., Scott,M., Semenov,A., Avrahami,D., Laszlo,L. and Prusiner,S.B. (1995) Cholesterol depletion and modification of COOH-terminal targeting sequence of the prion protein inhibit formation of the scrapie isoform. J. Cell Biol., 129, 121–132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vey M., Pilkuhn,S., Wille,H., Nixon,R., DeArmond,S.J., Smart,E.J., Anderson,R.W.G., Taraboulos,A. and Prusiner,S.B. (1996) Subcellular colocalization of the cellular and scrapie prion proteins in caveolae-like membranous domains. Proc. Natl Acad. Sci. USA, 93, 14945–14949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Viguera A.-R., Mencia,M. and Goni,F.M. (1993) Time-resolved and equilibrium measurements of the effects of poly(ethylene glycol) on small unilamellar phospholipid vesicles. Biochemistry, 32, 3708–3713. [DOI] [PubMed] [Google Scholar]
- Williamson R.A., Peretz,D., Pinilla,C., Ball,H., Bastidas,R.B., Rozenshteyn,R., Houghten,R.A., Prusiner,S.B. and Burton,D.R. (1998) Mapping the prion protein using recombinant antibodies. J. Virol., 72, 9413–9418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wong C., Xiong,L.-W., Horiuchi,M., Raymond,L., Wehrly,K., Chesebro,B. and Caughey,B. (2001) Sulfated glycans and elevated temperature stimulate PrPSc-dependent cell-free formation of protease-resistant prion protein. EMBO J., 20, 377–386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiong L.-W., Raymond,L.D., Hayes,S.F., Raymond,G.J. and Caughey,B. (2001) Conformational change, aggregation and fibril formation induced by detergent treatments of cellular prion protein. J. Neurochem., 79, 669–678. [DOI] [PubMed] [Google Scholar]
- Zuegg J. and Gready,J.E. (2000) Molecular dynamics simulation of human prion protein including both N-linked oligosaccharides and the GPI anchor. Glycobiology, 10, 959–974. [DOI] [PubMed] [Google Scholar]