

Abstract
Fluorescence resonance energy transfer (FRET) between a donor (D) and an acceptor (A) at the single-molecule level currently provides qualitative information about distance, and quantitative information about kinetics of distance changes. Here, we used the sorting ability of confocal microscopy equipped with alternating-laser excitation (ALEX) to measure accurate FRET efficiencies and distances from single molecules, using corrections that account for cross-talk terms that contaminate the FRET-induced signal, and for differences in the detection efficiency and quantum yield of the probes. ALEX yields accurate FRET independent of instrumental factors, such as excitation intensity or detector alignment. Using DNA fragments, we showed that ALEX-based distances agree well with predictions from a cylindrical model of DNA; ALEX-based distances fit better to theory than distances obtained at the ensemble level. Distance measurements within transcription complexes agreed well with ensemble-FRET measurements, and with structural models based on ensemble-FRET and x-ray crystallography. ALEX can benefit structural analysis of biomolecules, especially when such molecules are inaccessible to conventional structural methods due to heterogeneity or transient nature.
INTRODUCTION
Analysis of structure, dynamics, and interactions of biomolecules is fundamental for understanding molecular mechanisms; a powerful method that can perform such an analysis is fluorescence resonance energy transfer (FRET) (Förster, 1948; Heyduk, 2002; Hillisch et al., 2001; Lilley and Wilson, 2000; Mekler et al., 2002; Selvin, 2000). FRET is the nonradiative process whereby the excitation energy of a donor fluorophore (D) is transferred to an acceptor fluorophore (A), resulting in the excitation of the latter. The FRET efficiency (E) is a function of the D-A distance R, because E = 1/[1 + (R/Ro)6], where Ro (the Förster radius) is the distance for which E equals 50%. Because E is a sensitive function of R, FRET has been used as a “spectroscopic ruler” for the 1–10-nm scale, a scale comparable to the size of most biomolecules (Stryer and Haugland, 1967). Observation of FRET at the single-molecule level (single-pair FRET or spFRET; Ha et al., 1996; Weiss, 1999) extended FRET to biomolecules with static and dynamic heterogeneity, and allowed real-time observations of biomolecular dynamics (Ha, 2004; Weiss, 2000; Zhuang and Rief, 2003). spFRET can be applied to molecules immobilized on surfaces, or diffusing in solution.
Diffusion-based spFRET combines high sensitivity, absence of surface-induced perturbations (Talaga et al., 2000), and ability to identify subpopulations; therefore, it is suitable for structural analysis of biomolecules (Deniz et al., 1999, 2000; Schuler et al., 2002; Talaga et al., 2000). Using the diffusion format, Deniz et al. demonstrated that spFRET can recover distance information within DNA (Dahan et al., 1999; Deniz et al., 1999). However, spFRET has been confined to qualitative studies of structure and structural changes (Deniz et al., 2000; Schuler et al., 2002; Talaga et al., 2000), and to a range of FRET efficiencies between ∼40% and ∼100% (Deniz et al., 1999, 2000); this is due to incomplete labeling, complex photophysics, photobleaching, and need for determining instrument-correction factors and cross-talk terms (Deniz et al., 2000; Schuler et al., 2002).
There are two general methods that measure E at the single-molecule level: a method based on the sensitized-acceptor emission (ratiometric-E method (Dahan et al., 1999; Deniz et al., 1999)), and a method based on donor-lifetime changes (Rothwell et al., 2003). A fluorescence correlation spectroscopy method specific to fluorescent acceptors that exhibit cis-trans isomerization has also been described (Widengren et al., 2001). In the ratiometric-E case, accurate determination of E is possible only after accounting for two important cross-talk terms, D-emission into the A-detection channel, and A-emission due to A-direct excitation at the D-excitation wavelength; although the first term can be easily accounted for, the second term is more difficult to obtain and is routinely omitted from expressions of E in spFRET studies. The ratiometric-E method also requires knowledge of detection-correction factor-γ (Deniz et al., 1999; Ha et al., 1999). Because γ involves quantum yields and detection efficiencies of donor and acceptor, it varies with conditions (such as solution pH, solution temperature, optical alignment, and properties of optics/filters). Ha et al. measured γ for surface-immobilized molecules using acceptor photobleaching (Ha et al., 1999); however, γ for surface-immobilized molecules is only an approximation when used for diffusing molecules, due to surface-induced differences between immobilized and diffusing molecules (Talaga et al., 2000), and to possible chromatic differences between the detection volumes on surface and in solution (Michalet et al., 2001). The donor-lifetime method bypasses the need for measuring γ, but requires sophisticated and costly instrumentation, complex data analysis, and a set of quadratic correction factors (Rothwell et al., 2003); it also requires a correction factor when the fluorophores are spectrally close to each other.
Recently, we introduced alternating-laser excitation (ALEX) to perform fluorescence-aided molecule sorting (FAMS) (Kapanidis et al., 2004). ALEX-FAMS (hereafter ALEX) employs two lasers exciting the donor and acceptor in an alternating fashion. The results are presented in two-dimensional histograms of ratiometric observables: the ratio E that reports on D-A distance, and the ratio S that reports on D-A stoichiometry. Using ALEX, we were able to sort and quantify species with different D-A stoichiometries and D-A distances, to extend the usable FRET range to 0–100%, to detect differences in molecular brightness, to detect oligomerization, and to monitor macromolecule-ligand interactions.
Here, we describe an ALEX-based method for accurate measurements of E (defined as background-, cross-talk-, and γ-corrected ratiometric-E; hereafter “accurate-E”) from single diffusing molecules. We show that one or few measurements recover all factors needed for corrections, achieved after analyzing D-only and A-only species (intrinsic to the sample of interest or added exogenously), along with D-A species featuring a wide range of E-values. Such unique capabilities result from the ability of ALEX to recover D-A stoichiometry; this new dimension enables sorting of D-only, A-only, and D-A species, and because it is γ-dependent, leads to facile calculation of γ. Because the correction factors are measured in parallel with uncorrected E for the sample of interest, accurate-E is independent of instrumental factors, such as excitation intensity or detector alignment. In the case of DNA, ALEX-based distances were in good agreement with theoretical predictions. In the case of transcription complexes, ALEX-based distances were in good agreement with ensemble-FRET measurements, and with structural models based on ensemble FRET and x-ray crystallography. ALEX-based structural analysis is well suited for biomolecules and their complexes, especially for species inaccessible to conventional structural-biology methods, such as x-ray crystallography, and NMR spectroscopy.
THEORY
Definitions
ALEX results in four distinct photon-emission streams (Fig. 1), corresponding to four photon counts for every single burst of fluorescence (i.e., the observable for a single diffusing fluorescent molecule):  and
and 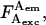 where
where 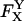 is the photon count for a single molecule upon excitation at wavelength X (where Dexc, Aexc are wavelengths of substantial excitation of donor or acceptor, respectively; in this work, Dexc = 514 nm and Aexc = 638 nm) and detection in emission wavelength range Y (where Dem, Aem are wavelengths of substantial emission of donor or acceptor, respectively, in the absence of FRET; in this work, Dem = 550–620 nm and Aem = 660–750 nm). For typical FRET pairs, photon counts for D-A species may contain photons emitted by the donor as well as photons emitted by the acceptor. For example, A-emitted photons due to FRET are detected along photons due to D-emission into the A-detection channel (hereafter “D-leakage”), and A-emission due to A-direct excitation at the D-excitation wavelength (hereafter “A-direct-excitation”); such terms are present both in ensemble-FRET (Clegg, 1992) and spFRET (Deniz et al., 1999). To define the emitting fluorophore and the wavelengths of excitation and emission associated with a photon count, we use the notation
is the photon count for a single molecule upon excitation at wavelength X (where Dexc, Aexc are wavelengths of substantial excitation of donor or acceptor, respectively; in this work, Dexc = 514 nm and Aexc = 638 nm) and detection in emission wavelength range Y (where Dem, Aem are wavelengths of substantial emission of donor or acceptor, respectively, in the absence of FRET; in this work, Dem = 550–620 nm and Aem = 660–750 nm). For typical FRET pairs, photon counts for D-A species may contain photons emitted by the donor as well as photons emitted by the acceptor. For example, A-emitted photons due to FRET are detected along photons due to D-emission into the A-detection channel (hereafter “D-leakage”), and A-emission due to A-direct excitation at the D-excitation wavelength (hereafter “A-direct-excitation”); such terms are present both in ensemble-FRET (Clegg, 1992) and spFRET (Deniz et al., 1999). To define the emitting fluorophore and the wavelengths of excitation and emission associated with a photon count, we use the notation  which represents the number of photons emitted by fluorophore Z upon excitation at wavelength X and detection in emission wavelength range Y; in the case of acceptor photons emitted due to FRET, we denote Z = D → A to signify their FRET-induced origin. Thus, the four possible photon counts from a donor are
which represents the number of photons emitted by fluorophore Z upon excitation at wavelength X and detection in emission wavelength range Y; in the case of acceptor photons emitted due to FRET, we denote Z = D → A to signify their FRET-induced origin. Thus, the four possible photon counts from a donor are 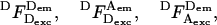 and
and  the four possible photon counts from an acceptor are
the four possible photon counts from an acceptor are  and
and  and the four possible photon counts from an acceptor due to FRET are
and the four possible photon counts from an acceptor due to FRET are 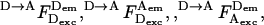 and
and  In practice, some photon counts are substantial, some are small cross-talk terms that have to be subtracted for accurate FRET measurements, and most are negligible:
In practice, some photon counts are substantial, some are small cross-talk terms that have to be subtracted for accurate FRET measurements, and most are negligible:
 |
(1) |
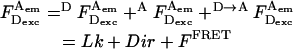 |
(2) |
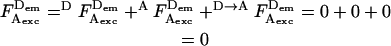 |
(3) |
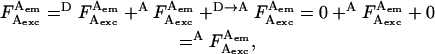 |
(4) |
where 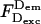 is the photon count for D-excitation-based D-emission;
is the photon count for D-excitation-based D-emission; 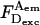 is the photon count for D-excitation-based A-emission;
is the photon count for D-excitation-based A-emission;  is the photon count for A-excitation-based D-emission;
is the photon count for A-excitation-based D-emission; 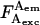 is the photon count for A-excitation-based A-emission; Lk is the photon count for D-emission into A-detection channel (D-leakage); Dir is the photon count for A-emission caused by A-direct excitation at the D-excitation wavelength (A-direct excitation); and
is the photon count for A-excitation-based A-emission; Lk is the photon count for D-emission into A-detection channel (D-leakage); Dir is the photon count for A-emission caused by A-direct excitation at the D-excitation wavelength (A-direct excitation); and  is the photon count for A-emission exclusively due to FRET.
is the photon count for A-emission exclusively due to FRET.
FIGURE 1.

Alternating-laser excitation microscopy. (A) Microscope setup for ALEX. EOM, electrooptical modulator; P, polarizer; DM, dichroic mirror; OBJ, objective; PH, pinhole; F, filter; APD, avalanche photodiode. Modulators combined with polarizers result in alternating-laser excitation. After spatial and spectral filtering, emitted fluorescence photons were detected on APDs. (B) Time traces for a high-E DNA. The emission streams are 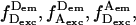 and
and  where
where  represents the emission rate in the Y emission detection channel while X-excitation is on. Burst a is due to a high-E D-A species (low
represents the emission rate in the Y emission detection channel while X-excitation is on. Burst a is due to a high-E D-A species (low  and high
and high  ). Burst b is due to a D-only species (high
). Burst b is due to a D-only species (high  and very low
and very low 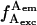 ). D-only species (stars); D-A species (solid circles).
). D-only species (stars); D-A species (solid circles).
The nonzero photon counts in Eqs. 1, 2, and 4 can be written as a function of the excitation and emission properties, and of FRET efficiency:
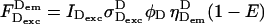 |
(5) |
 |
(6) |
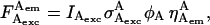 |
(7) |
where IDexc, IAexc are D- and A-excitation laser intensities, respectively; σDexcD, σDexcA, and σAexcA are absorption crosss sections of D upon D-excitation, of A upon D-excitation, and of A upon A-excitation, respectively; φD and φA are quantum yields of D and A, respectively; ηDemD, ηAemD, and ηAemA are detection efficiencies of D-emission in the D-detection channel, of D-emission in the A-detection channel, and of A-emission in the A-detection channel, respectively; and E is FRET efficiency.
Defining the Lk contribution
The Lk contribution can be defined on the basis of 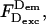 because:
because:
 |
(8) |
where 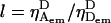 is the D-leakage coefficient; l can be easily determined using the ratio
is the D-leakage coefficient; l can be easily determined using the ratio 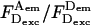 for D-only species (Fig. 2, A and B). After correcting for Lk, the E of D-only species should be zero.
for D-only species (Fig. 2, A and B). After correcting for Lk, the E of D-only species should be zero.
FIGURE 2.

Sorting single molecules using ALEX-based EPR-S histograms. (A) Expected location of labeled molecules depending on D-A stoichiometry and D-A distance. (B) Species required for recovering all corrections factor needed for accurate-E measurements using the method that depends on laser-alternation characteristics. D-only species provides the D-leakage factor l, A-only species provides the A-direct-excitation factor d, and two D-A species with large difference in E provide the γ-factor. (C) Species required for recovering all corrections needed for accurate-E measurements using the method that is independent of laser-alternation characteristics. D-only species provides the D-leakage factor l, a D-A species with E ∼ 0 (“simple-coincidence” control) provides the modified A-direct-excitation factor d′, and a D-A species with appreciable E provides the γ-factor. Use of A-only species is not necessary.
Defining the Dir contribution
One way of defining Dir is based on 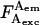 :
:
 |
(9) |
where 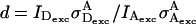 is an A-direct excitation coefficient determined using the ratio
is an A-direct excitation coefficient determined using the ratio  for A-only species (Fig. 2B). Coefficient d requires use of an A-excitation laser, and thus is available using ALEX. It is important to note that although
for A-only species (Fig. 2B). Coefficient d requires use of an A-excitation laser, and thus is available using ALEX. It is important to note that although 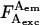 can be used to measure Dir, the Dir photon count is independent of the A-excitation intensity.
can be used to measure Dir, the Dir photon count is independent of the A-excitation intensity.
Ratiometric expressions E and S
When the detection-correction factor 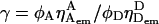 is known, the FRET efficiency E for a single burst is defined (Dahan et al., 1999; Deniz et al., 1999) as:
is known, the FRET efficiency E for a single burst is defined (Dahan et al., 1999; Deniz et al., 1999) as:
 |
(10) |
where
 |
(11) |
Recently, we defined E-independent ratio S, that reports on the D-A stoichiometry of diffusing species (Kapanidis et al., 2004). Here, we redefine S as Sγ, to point out that calculation of E-independent stoichiometry ratios requires the knowledge of γ-factor:
 |
(12) |
(Note: to standardize the notation used for ALEX-based analysis, we recommend that future ALEX-based studies use the E and S definitions described in this article). The presence of γ in the formula renders Sγ independent of E, because replacing 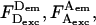 and
and  by their definitions (Eqs. 5, 7, and 11) yields:
by their definitions (Eqs. 5, 7, and 11) yields:
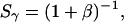 |
(13) |
where
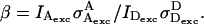 |
(14) |
Equations 13 and 14 show that Sγ depends on the excitation power ratio 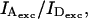 and on the D-A stoichiometry (as reflected by ratio
and on the D-A stoichiometry (as reflected by ratio 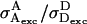 ). This relation can be extended to species with many
). This relation can be extended to species with many 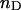 donor fluorophores and
donor fluorophores and 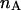 acceptor fluorophores, and a common, average FRET efficiency
acceptor fluorophores, and a common, average FRET efficiency 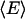 from each donor to the ensemble of acceptors (see Supplementary Material), resulting in Eq. 15:
from each donor to the ensemble of acceptors (see Supplementary Material), resulting in Eq. 15:
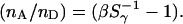 |
(15) |
Calculation of γ
When γ is unknown, E (Eq. 10) and Sγ (Eq. 12) are not experimental values. Thus, we define simplified E and S expressions that eliminate γ from Eqs. 10 and 12. This simplification results in the cross-talk-corrected proximity ratio EPR (Dahan et al., 1999), and cross-talk-corrected stoichiometry ratio S, which are directly calculated for single bursts without the knowledge of γ.
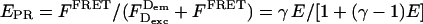 |
(16) |
 |
(17) |
(Note: EPR is different from Ec (Kapanidis et al., 2004), because the latter expression of E involves only D-leakage correction.) By substituting Eqs. 5, 7, and 11 into Eq. 17, a new expression for S is obtained:
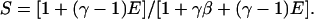 |
(18) |
From Eq. 18 and the reciprocal definition of E in terms of EPR:
 |
(19) |
one can obtain a linear relation between 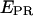 and 1/S:
and 1/S:
 |
(20) |
Therefore, by plotting 1/S against different 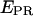 for two or more samples, the intercept Ω = 1 + γβ and slope Σ = β(1 − γ) of the best linear fit yield the values of β and γ.
for two or more samples, the intercept Ω = 1 + γβ and slope Σ = β(1 − γ) of the best linear fit yield the values of β and γ.
 |
(21) |
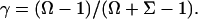 |
(22) |
The calculated γ-factor is used to determine E from Eq. 19. The distances were obtained from the E-values and the Förster radius Ro using Eq. 23 (assuming κ2 = 2/3):
 |
(23) |
Determination of γ for various D-A pairs using a standard pair
Factor-γ for a D-A pair subject to a specific local/global fluorophore environment and measured using a given instrument alignment can be used as a “standard” to calculate “unknown” γ for D-A pairs that feature identical probes and alignment, but different fluorophore environment (which mainly affects the quantum yield of the fluorophores). The change in fluorophore environment might be due to a change in the incorporation site of the fluorophore, or in buffer conditions. Defining 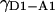 as the γ of the standard pair, and
as the γ of the standard pair, and  as the γ of the unknown pair, we have:
as the γ of the unknown pair, we have:
 |
(24) |
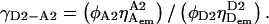 |
(25) |
In the usual case of minor shift (<2–3 nm) in the emission spectra of the fluorophores upon change in the environment, the convolution of emission spectra with transmission spectra of optics and detection-efficiency spectra of the detectors will not change:  and
and  Therefore,
Therefore, 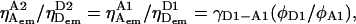 and
and
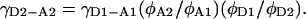 |
(26) |
In this case, 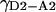 is determined from
is determined from 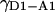 by simply measuring
by simply measuring 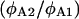 and
and 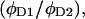 the differences in quantum yield due to change in fluorophore environment. Such measurements require simple ensemble measurements of absorbance and fluorescence, also used to calculate the Ro for the D2-A2 pair from the Ro of the “standard” pair. The case is simpler when only the donor or only the acceptor experience a different environment.
the differences in quantum yield due to change in fluorophore environment. Such measurements require simple ensemble measurements of absorbance and fluorescence, also used to calculate the Ro for the D2-A2 pair from the Ro of the “standard” pair. The case is simpler when only the donor or only the acceptor experience a different environment.
A second method for calculating accurate-E
It is often desirable to change the ratio of excitation powers, the duty cycle of alternation, or the laser source exciting the acceptor in ALEX. This requires repeating the A-only measurements that recover the A-direct excitation factor d, because  An alternative way to obtain Dir independent of the details of alternation uses a “simple-coincidence” control, a molecule or complex that contains a single donor and a single acceptor at distances where E ∼ 0, with a local environment for the fluorophores identical to the environment in the sample of interest (Fig. 2 C, D-A species with E ∼ 0, and with 1/S-value matching the intercept of the E − 1/S plot); for this control sample, Eq. 6 becomes
An alternative way to obtain Dir independent of the details of alternation uses a “simple-coincidence” control, a molecule or complex that contains a single donor and a single acceptor at distances where E ∼ 0, with a local environment for the fluorophores identical to the environment in the sample of interest (Fig. 2 C, D-A species with E ∼ 0, and with 1/S-value matching the intercept of the E − 1/S plot); for this control sample, Eq. 6 becomes 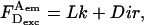 and Dir is obtained after subtraction of Lk (Fig. 2 C). This can be done by expressing Dir based on
and Dir is obtained after subtraction of Lk (Fig. 2 C). This can be done by expressing Dir based on 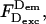 after defining a modified A-direct excitation correction factor
after defining a modified A-direct excitation correction factor 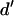 :
:
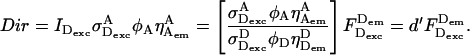 |
(27) |
Factor  is obtained using the D-A species of the simple-coincidence control (after removing D-leakage; Fig. 2 C). To recover E using factor
is obtained using the D-A species of the simple-coincidence control (after removing D-leakage; Fig. 2 C). To recover E using factor  we define the γ-uncorrected cross-talk-uncorrected proximity ratio
we define the γ-uncorrected cross-talk-uncorrected proximity ratio  :
:
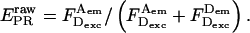 |
(28) |
Accounting for cross-talk contributions (using l and d′) and detection-efficiency differences (using γ), we can convert 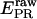 to E (see Supplementary Material):
to E (see Supplementary Material):
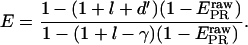 |
(29) |
It is important to note that the “simple-coincidence” control relies on the ability of ALEX to separate D-only species from D-A species with E ∼ 0.
The availability of two ways to account for direct excitation allows for flexibility in addressing different biomolecules. For instance, when the preparation of a “simple-coincidence” control is facile (as in the case of DNA fragments or well-characterized protein-DNA complexes), one can use the 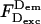 -based expression for A-direct excitation, and analyze an additional sample with appreciable E to obtain γ (thus bypassing the need for accounting for the alternation properties; Fig. 2 C) and then calculate accurate-E. In contrast, when the “simple-coincidence” control is not readily available (as in the case of small proteins, or poorly characterized complexes), A-direct excitation can be based on the
-based expression for A-direct excitation, and analyze an additional sample with appreciable E to obtain γ (thus bypassing the need for accounting for the alternation properties; Fig. 2 C) and then calculate accurate-E. In contrast, when the “simple-coincidence” control is not readily available (as in the case of small proteins, or poorly characterized complexes), A-direct excitation can be based on the  -based expression, and γ can be obtained from a standard D-A pair (Fig. 2 B).
-based expression, and γ can be obtained from a standard D-A pair (Fig. 2 B).
MATERIALS AND METHODS
DNA
Oligodeoxyribonucleotides were prepared by automated synthesis (Kapanidis et al., 2004), labeled, and hybridized to form D-only, A-only, and D-A double-stranded DNA (dsDNA) fragments (Fig. S1 in Supplementary Material). For the set of five DNA fragments used for the determination of γ and accurate-E values, the top-strand sequence was 5′-TAAATCTAAAGTAACATAAGGTAACATAACGGTAAGTCCA-3′, with amino-C6-dT residues (Glen Research, Sterling, VA) at position 1 of the top strand, and at each of positions 8, 13, 18, 23, or 28 of the bottom strands (positions underlined in the top-strand sequence shown above). Oligodeoxyribonucleotides were high-performance liquid chromatography (HPLC)-purified, labeled with N-hydroxy-succinimidyl esters of carboxytetramethylrhodamine (TMR) or Alexa 647 (Molecular Probes, Eugene, OR) using manufacturer's instructions, and HPLC-purified. We incorporated TMR (FRET donor) at position 1 of the top strand, and Alexa 647 (FRET acceptor) at each of five distinct positions in the bottom strand; the acceptor was incorporated within the same 3-bp sequence (TAA) to eliminate any changes in fluorescence properties due to change in local environment. dsDNA was formed by hybridization of top and bottom strands in 40 mM Tris-HCl, pH 8, 500 mM NaCl after heating for 2 min at 95°C and cooling to 25°C overnight; we used 50% molar excess of D-labeled top strand to ensure complete hybridization of A-labeled bottom strands. We denote dsDNA fragments as T1Bx, with T1 representing position 1 of top strand labeled by TMR, and Bx representing position x of bottom strand labeled by Alexa 647. To form D-only and A-only dsDNA, 10-fold molar excess of unlabeled strand was used for hybridization. For the comparison of accurate-E values to values predicted from DNA models, we added three more DNA fragments (T1B15, T1B20, and T1B25; Fig. S1B in Supplementary Material), by making a single 2-bp insertion (in gray boxes, Fig. S1B in Supplementary Material) between the D-A pairs of T1B13, T1B18, and T1B23 sequences; the insertion does not change the local environment of the fluorophores, allowing the eight DNA fragments to be used as a consistent set. The DNA fragments used for recovery of cross-talk terms and correction factor-γ within transcription complexes were lacUV5-11(Cy5,+25), lacUV5-14(Cy5,+28), lacUV5-15(Cy5,+29), and lacUV5-50(Cy5,+64) (Mukhopadhyay et al., 2001). Fragments lacUV5-11(Cy5,+25) and lacUV5-11(Cy5,−40) (Mukhopadhyay et al., 2001) were used to analyze distances within transcription complexes.
RNA polymerase derivatives
Escherichia coli core RNAP was purchased from Epicentre (Madison, WI). Derivatives of σ70 (σTMR,366, σTMR,396, σTMR,569, and σTMR,596; Mukhopadhyay et al., 2001) were prepared by labeling single-Cys derivatives of σ70 with tetramethylrhodamine-5-maleimide (TMR; Molecular Probes). Labeling, purification, and storage of labeled σ-derivatives, and formation of RNAP holoenzymes was performed as described (Mekler et al., 2002; Mukhopadhyay et al., 2001, 2003).
Sample preparation: DNA
Stock solutions of 20 nM for each DNA were prepared in SM buffer (10 mM HEPES-NaOH, pH 7, 500 mM NaCl, 100 μg/ml BSA, 1 mM mercaptoethylamine, and 5% glycerol). Final DNA concentration was 50 pM, resulting in <0.5% probability of simultaneous presence of two molecules in the detection volume (Deniz et al., 1999). For the ensemble study, we used 0.1 μM DNA in SM buffer.
Sample preparation: transcription complexes
For the series of transcription complexes studied to measure γ, the donor (TMR) was incorporated on residue Cys366 of σ70. The acceptor (Cy5) was incorporated at the downstream end of a series of DNA fragments with increasing length (positions +25, +28, +29, or +66 of DNA; the numbering refers to the location of the labeling site relative to transcription start site (Mukhopadhyay et al., 2001)). The local environment of Cy5 was kept identical to eliminate differences in local interactions of the acceptor with DNA. RNAP-DNA open complexes were prepared as described (Mukhopadhyay et al., 2001). Before data acquisition, the complexes were diluted to 50–100 pM in KG7 buffer (20 mM HEPES-NaOH, pH 7, 100 mM potassium glutamate, 10 mM MgCl2, 1 mM DTT, 100 μg/ml BSA, 1 mM mercaptoethylamine, and 5% glycerol), and 15 μL were transferred in eight-well chambered coverglass (Grace Biolabs, Bend, OR). All incubations and measurements were performed at 37°C.
Ensemble fluorescence spectroscopy
Measurements were performed on a T-format steady-state spectrofluorometer (QM-6/2003SE, PTI, Lawrenceville, NJ), equipped with polarization optics. The quantum yields for TMR and Alexa 647 were measured as described (Kapanidis et al., 2001) using D-only and A-only dsDNA in SM buffer; the values were 0.56 and 0.32 for TMR and Alexa 647, respectively. The steady-state fluorescence anisotropy values of TMR and Alexa 647 in dsDNA were measured to be 0.21 and 0.20, respectively; these values are low compared to the fundamental anisotropies of the probes (0.36–0.4; corresponding to immobile fluorophores), indicating substantial rotational freedom of the probes, and justifying the assumption that the orientation factor κ2 equals 2/3. Using the κ2 approximation, the Förster radius Ro for the TMR-Alexa 647 pair was ∼69 Å. Ensemble FRET efficiencies were measured using the method of sensitized A-emission (Clegg, 1992; Mekler et al., 2002) (excitation wavelengths = 530 nm for D and 620 nm for A, detection wavelengths = 588 nm for D and 665 nm for A, slit width = 10 nm) using extinction coefficients of 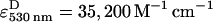 (for TMR) and
(for TMR) and 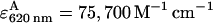 (for Alexa 647) (Haugland, 2002).
(for Alexa 647) (Haugland, 2002).
ALEX-based microscopy
The instrumentation, data acquisition, and data analysis for ALEX have been described (Kapanidis et al., 2004); a schematic of the setup is in Fig.1. For DNA, the alternation period was 50 μs, the excitation duty cycle was 44%, and the excitation intensities were 200 μW at 514 nm, and 20–60 μW at 638 nm (measured at the continuous-wave mode). An 100× objective with 1.3 numerical aperture was used to place the focal point in solution (20 μm from the surface), and a 100-μm pinhole was placed at the image plane of the tube lens to reject out-of-focus light. For transcription complexes, the same setup was used, but the alternation period was 100 μs, the duty cycle was 47%, and the excitation intensities were 250 μW at 514 nm, and 50 μW at 638 nm. When necessary, the ratio of detection efficiencies of the two emission channels  was calculated by convoluting the fluorophore emission spectra with transmission spectra of dichroic mirrors and filters of the detection path, and with the detection-efficiency spectra of the avalanche photodiode detectors (APDs), followed by integrating the resulting transmitted signal; the calculation does not consider any wavelength-dependent transmittance of the microscope objective, lenses, and pinhole, or differences between APD sensitivities.
was calculated by convoluting the fluorophore emission spectra with transmission spectra of dichroic mirrors and filters of the detection path, and with the detection-efficiency spectra of the avalanche photodiode detectors (APDs), followed by integrating the resulting transmitted signal; the calculation does not consider any wavelength-dependent transmittance of the microscope objective, lenses, and pinhole, or differences between APD sensitivities.
Data analysis: DNA
All data analysis was performed using homebuilt LabVIEW software (National Instruments, Austin, TX). As described (Kapanidis et al., 2004), ALEX-based microscopy generates streams of four emission rates: 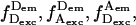 and
and 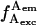 (Fig. 1 B; f-values represent emission rates, as opposed to intensities F, which represent photon counts per diffusing molecule). For time traces generated using D-A DNA featuring short or long interprobe distances, all D-A species show high E (Fig. 1 B), or low E (not shown), respectively. In such traces, high
(Fig. 1 B; f-values represent emission rates, as opposed to intensities F, which represent photon counts per diffusing molecule). For time traces generated using D-A DNA featuring short or long interprobe distances, all D-A species show high E (Fig. 1 B), or low E (not shown), respectively. In such traces, high  signals the presence of D, high
signals the presence of D, high 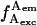 signals the presence of A, and high
signals the presence of A, and high 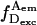 signals the occurrence of FRET. Background levels were <1 kHz for all emission streams. From the 500-μs binned photon time trace, we selected bursts using a start/stop criterion on the sum of all channels, and a threshold of 15–30 total photon counts removed any false positives due to background. At the end of the analysis, each burst is characterized by photon counts
signals the occurrence of FRET. Background levels were <1 kHz for all emission streams. From the 500-μs binned photon time trace, we selected bursts using a start/stop criterion on the sum of all channels, and a threshold of 15–30 total photon counts removed any false positives due to background. At the end of the analysis, each burst is characterized by photon counts 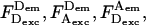 and
and  (Eqs. 1–4); any expression including these four quantities can be calculated during burst analysis.
(Eqs. 1–4); any expression including these four quantities can be calculated during burst analysis.
After burst identification, we: i), calculate cross-talk-uncorrected proximity ratio 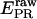 (Eq. 28), and cross-talk-uncorrected stoichiometry ratio
(Eq. 28), and cross-talk-uncorrected stoichiometry ratio 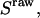 defined as:
defined as:
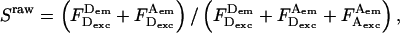 |
(30) |
for all species, and plot a two-dimensional histogram of 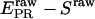 ; ii), select D-only species (species with
; ii), select D-only species (species with 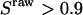 ); plot and fit the distribution of ratio
); plot and fit the distribution of ratio  to recover D-leakage factor l; iii), select A-only species (species with
to recover D-leakage factor l; iii), select A-only species (species with  ); plot and fit the distribution of ratio
); plot and fit the distribution of ratio 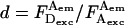 to recover A-direct excitation factor d; iv), reprocess data to subtract Lk and Dir contributions (Eqs. 8 and 9) from a D-A species and calculate
to recover A-direct excitation factor d; iv), reprocess data to subtract Lk and Dir contributions (Eqs. 8 and 9) from a D-A species and calculate 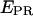 and S (Eq. 16 and 17); v), plot the two-dimensional
and S (Eq. 16 and 17); v), plot the two-dimensional 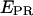 -S histogram for the D-A species, and fit to a Gaussian distribution to recover the mean (
-S histogram for the D-A species, and fit to a Gaussian distribution to recover the mean (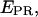 S) values for the D-A species; vi), repeat steps iv–v for more samples with D-A species that feature a wide range of E; vii), plot mean (
S) values for the D-A species; vi), repeat steps iv–v for more samples with D-A species that feature a wide range of E; vii), plot mean ( 1/S) values on a scatter plot and fit a straight line to recover factors β and γ (using Eq. 20); and viii), use γ-factor to calculate accurate-E values for all D-A species (using Eq. 19).
1/S) values on a scatter plot and fit a straight line to recover factors β and γ (using Eq. 20); and viii), use γ-factor to calculate accurate-E values for all D-A species (using Eq. 19).
Several of the processing steps are manual, leading to processing time of a few hours for a complete data set. By using a single-measurement approach to recover all correction factors (see Results and Discussion), and by straightforward automation of all steps, processing time will be reduced to ∼10 min, allowing the procedure to be used daily for characterizing both the optical system and the D-A pair.
Data analysis: transcription complexes
Data analysis was performed as above, apart from the use of the modified A-direct excitation factor d′, obtained using the open complex of RNAP σTMR,366 with lacUV50(Cy5,+64) as the “simple-coincidence” control sample (because R ≫ 120 Å; see Theory).
RESULTS AND DISCUSSION
ALEX-based determination of accurate-E
Recently, we showed that ALEX sorts fluorescent species based on D-A stoichiometry (measured as S or Sγ) and D-A distance (measured as E) (Fig. 2 A) (Kapanidis et al., 2004). D-only species show E ∼ 0 and S ∼ 1, A-only species show high apparent E (provided that the photon count Dir is significantly higher than the corresponding background counts) and low S (0 < S < 0.25), and D-A species show intermediate S and variable E (0–1), depending on D-A distance. Sorting does not require corrections; however, accurate-E measurements require three main corrections: a), separation of D-leakage from FRET-induced A-emission; b), separation of A-direct excitation from FRET-induced A-emission; and c), correction for differences in the quantum yield and detection efficiency of the fluorophores (compounded in factor-γ).
To obtain accurate-E from single molecules, we performed ALEX on each of five DNA fragments labeled with D and A separated by 27, 22, 17, 12, and 7 bp, along with control experiments with D-only and A-only DNA (not shown). To determine EPR in Fig. 3, A–E, we subtracted D-leakage and A-direct excitation contributions from the 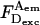 signal of every burst (Eqs. 2 and 6), using measured correction factors l ∼ 0.20, and d ∼ 0.06 (see Materials and Methods).
signal of every burst (Eqs. 2 and 6), using measured correction factors l ∼ 0.20, and d ∼ 0.06 (see Materials and Methods).
FIGURE 3.

ALEX-based EPR-S histograms for DNA fragments used for the determination of accurate-E. Light and dark gray curves in EPR and S histograms: individual and sum of Gaussian fits to the one-dimensional histograms. (A–E) Histogram for T1B28, T1B23, T1B18, T1B13, and T1B8 DNA, respectively. The thick solid lines correspond to EPR, S-values as predicted for D-A species using γ = 0.71 and β = 1.25.
All EPR-S histograms show two main fluorescent species: D-only species (EPR ∼ 0; S ∼ 1), and D-A species (0.1 < EPR < 1.0; S ∼ 0.5). When the D-A distance is short, the EPR-values of D-only and D-A species are well separated (Fig. 3, C–E), allowing accurate extraction of mean EPR using a two-Gaussian fit of the collapsed one-dimensional EPR histogram. However, for long D-A distances (Fig. 3, A and B), the large overlap of D-only and D-A species along the EPR axis hinders accurate determination of EPR; this is the main reason why conventional single-laser excitation spFRET underperforms on FRET species with E < 0.4 (Kapanidis et al., 2004). On the other hand, because ALEX can examine species with specific stoichiometry, we can use the window of 0.25 < S < 0.8 to construct EPR distributions solely for D-A species (not shown).
To determine detection-correction factor-γ, we measured EPR and S for the D-A species of each sample (Table 1), and generated an EPR versus 1/S plot (Fig. 4 A, solid line). As expected from Eq. 20, a linear relation exists between the two parameters (correlation coefficient R ∼ 0.992). From the slope and intercept of the plot, we obtained γ = 0.71 ± 0.03, and β = 1.25 ± 0.05. To our knowledge, this is the first report of measuring γ within single diffusing molecules. Moreover, excitation factor-β is a new observable that reports on two important (and difficult to obtain) properties of biomolecules: the D-A stoichiometry, and the absorbance spectra of the donor and acceptor. Using γ = 0.71 and Eq. 19, we obtained accurate-E values (Table 1; solid circles in Fig. 4 B).
TABLE 1.
EPR- and E-values measured using different detector alignments for DNA fragments
| D-A separation (bp) | EPR (optimal alignment) | E (optimal alignment) | EPR (suboptimal alignment) | E (suboptimal alignment) |
|---|---|---|---|---|
| 7 | 0.95 | 0.96 | 0.96 | 0.96 |
| 12 | 0.77 | 0.82 | 0.84 | 0.83 |
| 17 | 0.50 | 0.58 | 0.62 | 0.60 |
| 22 | 0.29 | 0.36 | 0.38 | 0.37 |
| 27 | 0.14 | 0.18 | 0.19 | 0.18 |
FIGURE 4.

ALEX-based distance measurement and its dependence on detection-correction factor-γ. (A) EPR-1/S plot for the DNA of Fig. 3 and its dependence on alignment. Mean EPR-1/S values and linear fit for optimal alignment (solid circles and solid line); mean EPR-1/S values and linear fit for suboptimal alignment (open circles and dotted line). Error bars are the standard deviations of three measurements; error bars for EPR are not visible (<0.01). With optimal alignment, the linear fit yields γ = 0.71, and β = 1.25. Suboptimal alignment changes the correction factors (γ = 1.05; β = 1.30), leading to changes in EPR and 1/S; however, corrected values of E are identical to the one obtained by optimal alignment (Table 1; Fig. 4 B). (B) Relation between EPR and E, and its dependence on γ. Optimal alignment (•); suboptimal alignment (○). (Gray lines) EPR − E correction curves for γ = 0.71, and γ = 1.05. (Dotted lines) EPR − E correction curves for 0.25 < γ < 4. Differences between EPR and E are maximal for intermediate values of E, and for γ ≫ 1 and γ ≪ 1. (C) Relation between RPR and R, and its dependence on factor-γ . The differences between RPR and R increase linearly with increasing R.
To verify the accuracy of the measured γ, we compared it to a value calculated using the spectral properties of the probes and the transmission properties of microscope components. Using the quantum yields of TMR and Alexa 647 (0.56 and 0.32, respectively), and the ratio of detection efficiencies of the two channels ( ; Materials and Methods), we obtained γ = 0.81, in a reasonable agreement with the experimentally determined factor.
; Materials and Methods), we obtained γ = 0.81, in a reasonable agreement with the experimentally determined factor.
Dependence of β and γ on excitation power and detector alignment
Because γ depends on the ratio of quantum yields and the ratio of detection efficiencies, it should be independent of the excitation-power ratio 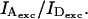 In contrast, β is by definition (Eq. 14) proportional to
In contrast, β is by definition (Eq. 14) proportional to  To test such dependences, we studied the effects of changing excitation power ratio and detector alignment on β and γ.
To test such dependences, we studied the effects of changing excitation power ratio and detector alignment on β and γ.
Increasing 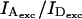 decreased S for D-A species (Kapanidis et al., 2004), but did not change EPR (Fig. S2A in Supplementary Material); using Eq. 20, β and γ were extracted for each power-ratio measurement (Fig. S2B in Supplementary Material). As expected, β was directly proportional to the power ratio (1:2.1:3.2 vs. 1:2:3), whereas γ was independent of power ratio (values 0.75, 0.71 and 0.72).
decreased S for D-A species (Kapanidis et al., 2004), but did not change EPR (Fig. S2A in Supplementary Material); using Eq. 20, β and γ were extracted for each power-ratio measurement (Fig. S2B in Supplementary Material). As expected, β was directly proportional to the power ratio (1:2.1:3.2 vs. 1:2:3), whereas γ was independent of power ratio (values 0.75, 0.71 and 0.72).
We tested the effect of detector alignment on γ and β by increasing the detection-efficiency ratio 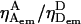 using suboptimal alignment of the D-emission detection channel. Because γ is proportional to
using suboptimal alignment of the D-emission detection channel. Because γ is proportional to  such misalignment increases γ, but does not affect β. Upon D-detection misalignment, the detected D-emission decreased, increasing EPR for all species (Fig. 4 A, open circles). However, the linearity of 1/S and EPR is maintained (Fig. 4 A, dotted line; cf. with aligned detectors), allowing calculation of γ = 1.05 and β = 1.30. As expected, γ increased by ∼50% compared to the values for aligned detectors, whereas β was unchanged. Significantly, using γ obtained for misaligned detectors to convert EPR to E yields values identical to the ones recovered for the optimally aligned detectors (Table 1; cf. columns 3 and 5; Fig. 4 B), showing that the ALEX-based accurate-E is independent of alignment.
such misalignment increases γ, but does not affect β. Upon D-detection misalignment, the detected D-emission decreased, increasing EPR for all species (Fig. 4 A, open circles). However, the linearity of 1/S and EPR is maintained (Fig. 4 A, dotted line; cf. with aligned detectors), allowing calculation of γ = 1.05 and β = 1.30. As expected, γ increased by ∼50% compared to the values for aligned detectors, whereas β was unchanged. Significantly, using γ obtained for misaligned detectors to convert EPR to E yields values identical to the ones recovered for the optimally aligned detectors (Table 1; cf. columns 3 and 5; Fig. 4 B), showing that the ALEX-based accurate-E is independent of alignment.
Differences between EPR and E as a function of γ are more pronounced for intermediate-E species, and minimal at either large or small values of E (Fig. 4 B; dotted lines represent theoretical values for a range of γ). However, because the relation between apparent distances RPR obtained using EPR, and distances R obtained using accurate-E values is given by the simple relation R = γ1/6RPR, distance deviations due to incorrect γ become significant at long D-A distances (Fig. 4 C). For distances close to the midpoint of the dynamic range (∼70 Å), a twofold difference in γ results in only ∼8 Å difference in the recovered distance.
Effect of cross-talk signals to apparent FRET efficiencies
To determine the error in E associated with absence of D-leakage and A-direct excitation corrections, we compared E-values with and without corrections for these two cross-talk terms.
If D-leakage is not subtracted from 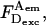 it increases the extracted E (Table 2). The error due to D-leakage is larger for low-E species: for DNA fragments with 7- and 12-bp D-A separation, the error in E is ∼0.01, but for 27-bp separation, the error amounts to 0.16, i.e., half of the uncorrected value. As with D-leakage, the effect of A-direct excitation is small for high-E species and increases for low-E species (Table 2); e.g., the deviation of E for 27-bp D-A separation is ∼0.07. For intermediate E, where most high-resolution distance measurements are performed, the deviation is significant (∼0.03.) Single-molecule-based measurements of E that exceed the theoretical E for DNA fragments with wide D-A separation have been reported (Deniz et al., 1999; Dietrich et al., 2002); absence of Dir subtraction might have contributed to such a discrepancy. We conclude that accurate-E measurements require rigorous corrections for both Lk and Dir.
it increases the extracted E (Table 2). The error due to D-leakage is larger for low-E species: for DNA fragments with 7- and 12-bp D-A separation, the error in E is ∼0.01, but for 27-bp separation, the error amounts to 0.16, i.e., half of the uncorrected value. As with D-leakage, the effect of A-direct excitation is small for high-E species and increases for low-E species (Table 2); e.g., the deviation of E for 27-bp D-A separation is ∼0.07. For intermediate E, where most high-resolution distance measurements are performed, the deviation is significant (∼0.03.) Single-molecule-based measurements of E that exceed the theoretical E for DNA fragments with wide D-A separation have been reported (Deniz et al., 1999; Dietrich et al., 2002); absence of Dir subtraction might have contributed to such a discrepancy. We conclude that accurate-E measurements require rigorous corrections for both Lk and Dir.
TABLE 2.
The effect of D-leakage and A-direct excitation on the measured values of E for DNA fragments
| D-A separation (bp) | E (all corrections) | Elk (no D-leakage correction) | EDir (no A-direct-excitation correction) | Eraw (no D-leakage or A-direct-excitation correction) |
|---|---|---|---|---|
| 7 | 0.96 | 0.97 | 0.97 | 0.97 |
| 12 | 0.82 | 0.84 | 0.84 | 0.85 |
| 17 | 0.58 | 0.63 | 0.62 | 0.65 |
| 22 | 0.36 | 0.46 | 0.41 | 0.49 |
| 27 | 0.18 | 0.34 | 0.25 | 0.38 |
Accurate measurements of low-E values can extend the upper limit of single-molecule FRET range beyond 100 Å. This is due to the fact that Lk and Dir cross-talk terms are substantial for pairs with high Ro (such as Alexa 594→Alexa 647, a pair with expected Ro > 80 Å; Haugland, 2002), because such pairs are spectrally close (to maximize their spectral overlap), and the cross-talk terms are significant. Use of such pairs combined with ALEX-based corrections will allow reliable distance measurements up to 120 Å (equivalent to E ∼ 0.08 for Ro ∼ 80 Å), making single-molecule FRET compatible with large, multicomponent complexes.
Determination of all correction factors and accurate-E from a single ALEX measurement
Because ALEX can sort D-only, A-only, low-E D-A, and high-E D-A species present in the same solution, it can obtain all information needed for accurate-E from a single measurement (where all the species found in Fig. 2 B are present). The EPR-S histogram of a minimal mixture (D-only, A-only, T1B28, and T1B13) is shown in Fig. 5; D-only species provide the D-leakage correction, A-only species provide for A-direct excitation correction, and the T1B28/T1B13 pair provides the γ-correction. The two EPR-1/S points defined by the T1B28/T1B13 pair (0.14 and 1.95 for T1B28, and 0.77 and 2.17 for T1B13) are identical to measurements for the individual DNA fragments of Fig. 3, and define a linear relation between EPR and 1/S identical to the one defined by the full set of five measurements (Fig. 4 A). This results in γ- and β-factors that are identical for the pair and the full set (γ = 0.72 ± 0.06 and β = 1.26 ± 0.05 for the pair; cf. with γ = 0.71 ± 0.03 and β = 1.25 ± 0.05 for the full set). Accurate-E values obtained using the T1B28 (0.18) and T1B13 (0.82) are identical to the ones obtained by the full set. We conclude that a minimal set of two D-A species featuring different D-A distances is necessary and sufficient for accurate measurements of E. The single-measurement concept for γ-determination can be extended with ensemble measurements that determine γ for various D-A pairs (see Theory). Overall, the γ for the standard pair evaluates the alignment and the wavelength dependence of the emission path, whereas the ensemble measurements of the standard and unknown account for differences in fluorophore properties. Similar results can be obtained using the alternation-independent method for extracting d′ and γ (Fig. 2 C).
FIGURE 5.

A single ALEX measurement can recover accurate-E. EPR-S histogram for a mixture of D-only, A-only, T1B28, and T1B13 DNA. (Dotted rectangle) Area of histogram shown in the EPR histogram. D-only species were used for D-leakage correction, A-only species for A-direct-excitation correction, and the T1B28/T1B13 pair for determination of γ.
RESULTS: VALIDATION
Single-molecule versus ensemble FRET measurements: DNA
To compare ALEX-based E-values (Esm) to E-values measured in ensembles (Eens), we studied the DNA fragments of Fig. 3 using the ensemble method of sensitized A-emission (which measures E by comparing FRET-induced A-emission with A-emission due to direct A-excitation at the A-excitation wavelength; Table 3 and Fig. 6, open circles). For intermediate- and low-E species, Esm agrees well with Eens. As interprobe distances decrease (high-E species), Eens becomes significantly lower than Esm (0.80 vs. 0.96 for T1B8; 0.74 vs. 0.82 for T1B13). Moreover, Eens-values (Fig. 6) deviate significantly from predictions derived from DNA models (Fig. 6, curves; next section), regardless of the exact model parameters. Discrepancies between Eens and Esm were reported (Dietrich et al., 2002), and attributed to direct ground- and excited-state interactions between D and A fluorophores at short interprobe distances (Marras et al., 2002). Such interactions quench D-emission and sensitized A-emission (thus lowering 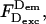 and
and 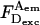 ) (Vamosi et al., 1996), but do not significantly change the A-emission upon A-excitation
) (Vamosi et al., 1996), but do not significantly change the A-emission upon A-excitation 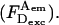 The combined changes reduce apparent Eens; however, because Esm is defined using solely
The combined changes reduce apparent Eens; however, because Esm is defined using solely 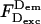 and
and 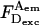 (reduced comparably due to D-quenching), Esm is not affected by D-quenching. The timescale of short-range D-quenching is faster than the transit (diffusion) time of the D-A species (∼1 ms), because A-only species are rare regardless of Esm (Fig. 3) (Edman et al., 1996; Eggeling et al., 1998; Wennmalm et al., 1997). The decreased Eens-values observed for short interprobe distances call for caution in the interpretation of high (Eens > 0.8) ensemble-FRET values measured using the sensitized A-emission method. Because short-range D-A interactions depend on probe properties (e.g., absorbance and emission spectra, charge complementarity; Marras et al., 2002), the extent of the discrepancy will vary. Presence of any A-only species in the ensemble experiment will also contribute to the differences between Esm and Eens.
(reduced comparably due to D-quenching), Esm is not affected by D-quenching. The timescale of short-range D-quenching is faster than the transit (diffusion) time of the D-A species (∼1 ms), because A-only species are rare regardless of Esm (Fig. 3) (Edman et al., 1996; Eggeling et al., 1998; Wennmalm et al., 1997). The decreased Eens-values observed for short interprobe distances call for caution in the interpretation of high (Eens > 0.8) ensemble-FRET values measured using the sensitized A-emission method. Because short-range D-A interactions depend on probe properties (e.g., absorbance and emission spectra, charge complementarity; Marras et al., 2002), the extent of the discrepancy will vary. Presence of any A-only species in the ensemble experiment will also contribute to the differences between Esm and Eens.
TABLE 3.
Comparison of ensemble and single-molecule measurements of E for DNA fragments
| D-A separation (bp) | Eens | Rens (Å) | Esm | Rens (Å) |
|---|---|---|---|---|
| 7 | 0.80 | 55 | 0.96 | 41 |
| 12 | 0.74 | 58 | 0.82 | 54 |
| 17 | 0.59 | 65 | 0.58 | 65 |
| 22 | 0.39 | 74 | 0.36 | 76 |
| 27 | 0.24 | 84 | 0.18 | 89 |
FIGURE 6.

Comparison of E-values measured for DNA fragments with values predicted from cylindrical models of DNA. ALEX-based E (Esm) (•). Ensemble E (Eens) (○). Theoretical E (Ethe) was calculated (Clegg et al., 1993; Norman et al., 2000) using  where n is the interprobe separation (in bp), L is the rise of the terminal probe along the helix axis, d is the radial distance of the center of the donor probe from the helix axis (in Å), a is the radial distance of the center of the acceptor probe from the helix axis (in Å), θ is the rotation angle for fluorophores separated by n bp (calculated using θ = 34 (n − 1)), ϕ is the cylindrical angle between radially extended donor and acceptor when spaced by 1 bp, and Ro is the Förster radius (in Å). The solid curve represents Ethe for a DNA model with the donor probe proximal to the DNA helical axis (L = 4 Å, a = 25 Å, d = 0 Å, ϕ = 232°, and Ro = 69 Å), whereas the dotted curve represents Ethe for a DNA model with the donor probe distal from the DNA helical axis (L = 4 Å, a = 25 Å, d = 15 Å, ϕ = 232°, and Ro = 69 Å). Error bars are the standard deviations of three measurements. In all cases, Esm-values fit better to theoretical values than Eens.
where n is the interprobe separation (in bp), L is the rise of the terminal probe along the helix axis, d is the radial distance of the center of the donor probe from the helix axis (in Å), a is the radial distance of the center of the acceptor probe from the helix axis (in Å), θ is the rotation angle for fluorophores separated by n bp (calculated using θ = 34 (n − 1)), ϕ is the cylindrical angle between radially extended donor and acceptor when spaced by 1 bp, and Ro is the Förster radius (in Å). The solid curve represents Ethe for a DNA model with the donor probe proximal to the DNA helical axis (L = 4 Å, a = 25 Å, d = 0 Å, ϕ = 232°, and Ro = 69 Å), whereas the dotted curve represents Ethe for a DNA model with the donor probe distal from the DNA helical axis (L = 4 Å, a = 25 Å, d = 15 Å, ϕ = 232°, and Ro = 69 Å). Error bars are the standard deviations of three measurements. In all cases, Esm-values fit better to theoretical values than Eens.
Single-molecule measurements versus structural model predictions: DNA
To determine whether E-values measured within DNA are consistent with DNA structure and the local disposition of the probes, we compared Esm with values of E calculated using simple DNA models (Ethe). To increase the confidence in the analysis, we added three DNA fragments to the original set of five (Fig. S1B in Supplementary Material) and analyzed the set of eight DNA fragments; it is remarkable that the accurate-E values for the original set of five DNA fragments were identical to the ones obtained for the same fragments during the analysis of the eight-fragment set, despite the fact that the measurements were taken a year apart, by a different experimenter, and using different detection filters.
Simple inspection of the dependence of Esm to the D-A separation (Fig. 6) shows a monotonic FRET increase as the separation decreases, without pronounced FRET modulation caused by helical DNA geometry. This dependence was observed before (Norman et al., 2000), and was attributed to the fact that one of the probes assumes an effective position proximal to the helix axis; this is consistent with the tendency of the TMR (donor) to stack at DNA-helix ends (Hillisch et al., 2001). It is likely that observation of helix-dependent modulation of E will require use of internal fluorophores, which cannot stack to the helix ends, and show minimal or negligible groove binding (C. Seidel, personal communication).
Using a cylindrical model for DNA (Clegg et al., 1993; Deniz et al., 1999; Norman et al., 2000), we calculated E-values (Ethe, calculated using equation in the legend of Fig. 6) for a model that includes donor stacking on the end of the DNA helix (d = 0), a donor rise similar to the one obtained for terminal Cy3 (L = 4 Å) (Norman et al., 2000), an extended acceptor conformation (a = 25 Å), a cylindrical angle that places probes on opposites sides of the helix (ϕ = 232°), and experimentally determined Ro (69 Å). The Ethe-values derived from this model (Fig. 6, solid line) agrees with the experimental data (rmsdE ∼ 0.057; Fig. 6, solid circles). If we translate deviations of E into deviations of RD-A, a fit with rmsdE of 0.057 for a FRET pair with Ro = 69 Å corresponds to rmsdR ≤ 5 Å for distances within the 50–85 Å range (rmsdR calculated from the extremes of the distance range). (An alternative model (Fig. 6, dotted line), with identical parameters except of an effective donor position distal from the helical axis (d = 15), fits significantly worst to the experimental data; rmsdE ∼ 0.089.) When the Ro is fitted with the rest of the parameters fixed, the best fit recovers an Ro-value that matches (within 1 Å) the experimentally determined Ro-value (see Materials and Methods). This agreement represents a marked improvement over prior work, including ours (Deniz et al., 1999; Dietrich et al., 2002), without the use of scaling factors (necessary in Widengren et al., 2001), and thus validates ALEX-based quantitative distance measurements.
Single-molecule versus ensemble FRET measurements: transcription complexes
To test the ability of ALEX to measure accurate distances within large, multicomponent complexes, we determined distances within RNAP-DNA open transcription complexes (RPo) and compared them to ensemble-FRET data (Mukhopadhyay et al., 2001, 2003) and structural models (Lawson et al., 2004).
We first used the EPR-S histograms for the D-A species of a series of transcription complexes with different D-A distances (Fig. 7, A–D) to calculate mean EPR- and S-values for the EPR-1/S plot (Fig. 7 E); the four points were fitted with a straight line (R ∼ 0.91), resulting in β = 1.44 and γ = 0.33; the cross-talk coefficients were l ∼ 0.10, and d′ ∼ 0.10. If only three points are fitted (the points with extreme values of E plus one of the remaining two points), we obtain γ′ = 0.29 or γ″= 0.35, showing that the small scatter of the points with E ∼ 0.05 has a minimal effect on γ.
FIGURE 7.

EPR-S histograms for RNAPσTMR,366 complexed with DNA carrying an acceptor at various positions, and γ-determination. Histograms display only the D-A species; D-only and A-only species were removed using 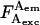 > 20 photons and
> 20 photons and  > 20 photons, respectively. (A–D) Complexes with acceptor at +64, +29, +28, and +25, respectively. (E) Linear relation between EPR and 1/S allows extraction of β- and γ-factors; each point reflects the averages and standard deviations of three experiments.
> 20 photons, respectively. (A–D) Complexes with acceptor at +64, +29, +28, and +25, respectively. (E) Linear relation between EPR and 1/S allows extraction of β- and γ-factors; each point reflects the averages and standard deviations of three experiments.
We also calculated the γ for the 366/+25 TMR-Cy5 D-A pair (based on the complex of RNAPσTMR,366 with lacUV11(Cy5,+25)) using Eq. 26; the “standard” γ was the one obtained for the TMR-Alexa647 D-A pair of the DNA series (0.71), whereas the ratio  was found to be 0.43. The final result of γ = 0.31 agrees well with the γ measured using the graphical method, validating both methods.
was found to be 0.43. The final result of γ = 0.31 agrees well with the γ measured using the graphical method, validating both methods.
Using the γ and the Ro of the 366/+25 D-A pair as a standard to calculate the γ- and Ro-values of additional D-A pairs we obtained (γ, Ro) values of (0.50, 57.6 Å), (0.27, 65.4 Å), and (0.29, 64.4 Å) for 396/+25, 569/−40, and 596/−40 D-A pairs, respectively. We subsequently calculated accurate-E values and corresponding D-A distances for several open complexes (Table 4, column 3).
TABLE 4.
Comparison of ALEX-based distances within transcription complexes with distances obtained using ensemble-FRET measurements, or a model constructed combining FRET-based distance-restrained docking and x-ray crystallography
| Donor position on σ70 | Acceptor position on DNA | Rsm (Å) (this work) | Rens (Å) (Mukhophadyay et al., 2001, 2003) | Rens (Å) (Mekler et al., 2002) | Rcalc (Å) (current model, freely rotating acceptor*) |
|---|---|---|---|---|---|
| 366 | +25 | 79 | 74 | 84 | 80 |
| 396 | +25 | 88 | 85 | 84 | 96 |
| 569 | −40 | 50 | 50 | – | 51 |
| 596 | −40 | 41 | 38 | – | 27 |
Values are determined using the reference model of Lawson et al. (2004) and the methods of Mekler et al. (2002) (explicit modeling of linker and probe, identification of sterically allowed linker and probe conformations, assignment of pseudoatom for each sterically allowed linker and conformation), assuming free rotation of the +25 and −40 DNA probes.
Comparison of ALEX-based distances with distances available from ensemble-FRET measurements (Mekler et al., 2002; Mukhopadhyay et al., 2001) show that all ALEX-based distances are within <5 Å from distances obtained from ensemble-FRET measurements, for identical complexes (Mukhopadhyay et al., 2001), or essentially identical complexes (Mekler et al., 2002). Small changes in the measured γ (e.g., calculating distances using γ′ = 0.29 or γ″ = 0.35) do not change distances significantly (ΔR: 0–1 Å). The agreement between ALEX-based distances and ensemble measurements validates ALEX as a method capable of generating structural information from large biomolecules. The better agreement seen at short interprobe distances compared to the comparison for DNA fragments might be due to gel-based purification of transcription complexes in the case of ensemble-FRET measurements (Mukhopadhyay et al., 2001), or due to a relative probe orientation that prevents donor-acceptor contact and associated D-quenching.
Single-molecule measurements versus structural model predictions: transcription complexes
Comparison of ALEX-based distances with distances predicted by a structural model based on ensemble-FRET measurements and distance-restrained docking was done using the RPo model of Lawson et al. (2004), and the methods of Mekler et al. (2002) (explicit modeling of linker and probe, identification of sterically allowed linker and probe conformations, and assignment of pseudoatom for each sterically allowed linker and conformation). The model assumed the presence of a freely rotating acceptor on the DNA-helix ends (Table 4, column 6). The D-A pairs with intermediate FRET values agree well with the model-based distances (Rcalc; 79 vs. 80 Å for the 366/+25 pair; 50 vs. 51 Å for the 569/−40 pair). The discrepancy is larger for small (<0.1) and large (>0.9) values of Esm, resulting in distance differences of 8 and 14 Å. As with the DNA fragments, both single-molecule and ensemble FRET seems to underestimate long D-A distances, and overestimate short D-A distances. We conclude that ALEX-based distances serve as accurate and quantitative measures of biomolecular structure for an E-range of 0.15–0.85, and as qualitative indicators for extreme values of E (E < 0.15 or E > 0.85). Considering the Ro range of 40–70 Å for the D-A pairs compatible with single-molecule FRET (Kapanidis and Weiss, 2002), the ALEX-based method can perform reliable distance measurement in the range of 30–95 Å, a scale compatible with most biomolecules.
Possible sources of differences between ALEX-based and model-predicted distances
The small but systematic discrepancy between ALEX-based and model-predicted distances at extreme values of E is likely due to the way the models describe or average the FRET process. A likely explanation is that both DNA and RPo models do not generate the full E-distribution, because they do not explicitly model the influence of orientation factor κ2 on the computed E for a specific configuration. Underestimation of orientational and/or distance heterogeneity results in predicted distances that are shorter than the measured apparent mean distances, and predicted mean E-values that are higher that the measured mean E-values. This effect is minimized for distances longer or close to Ro (Wu and Brand, 1992).
Furthermore, a more efficient excitation (due to faster decay of fluorescence) of donor while it adopts acceptor-proximal configurations might bias the mean of the ensemble toward the distance of closest approach (Kapanidis et al., 2001), as in diffusion-enhanced fluorescence energy transfer (Stryer et al., 1982). Proper description of the contribution of such configurations to E for diffusing molecules should consider the extent and dynamics of the fluorophore linker, as well as the flexibility of DNA (recently shown to be significant even for DNA fragments much shorter than persistence length; Coultier and Widom, 2004).
Another possibility is that there are additional, non-Förster, energy-transfer mechanisms operating at the short interprobe distances, possibly due to donor-acceptor contact (Marras et al., 2002); this is coupled to low sensitivity of FRET to distance changes at R ≫ Ro and R ≪ Ro (Kapanidis and Weiss, 2002). Understanding of the deviations present at extreme values of E will increase the useful range of the measurement, increase its attractiveness as a structural tool, and help probe the dynamic properties of biomolecules.
CONCLUSION AND OUTLOOK
We have shown that ALEX allows facile and quantitative analysis of FRET within single diffusing molecules. The ability to perform all corrections using a small set of measurements minimizes the effects of sample preparation, error propagation, instrument alignment, and data-acquisition time, and leads to rapid accumulation of distances for structural analysis. The overall agreement of ALEX-based distances with distances obtained by ensemble FRET and predicted by structural models validates the method, and paves the way for analysis of biomolecules inaccessible to conventional structural-biology methods.
There are several advantages of using ALEX for structural analysis: first, ALEX can analyze samples of static heterogeneity (presence of multiple, distinct, noninterconverting species in a mixture). This capability permits accurate-E measurements by selecting the species of interest after sorting, and can uncover additional species in samples that would otherwise be treated as homogeneous. Second, ALEX can analyze samples of dynamic heterogeneity (presence of multiple, interconverting states of a given biomolecule), permitting analysis of transient states and intermediates difficult to trap and study using other methods. Third, ALEX is compatible with partially purified, partially labeled, and partially active samples (conditions that complicate ensemble FRET). The ability to work with partially purified samples enables the homogeneous nature of the assay (e.g., by obviating gel purification of the species of interest), which in turn, permits fast exchange of reagents.
This work is one of a series of steps toward increasingly accurate structural analysis on single biomolecules, as indicated by recent work on single-molecule three-color FRET (Hohng et al., 2004), on polyproline helices (Schuler et al., 2005) and immobilized DNA fragments (Sabanayagam et al., 2005) as well as on an extensive collection of dsDNA fragments (C. Seidel, personal communication). Our work provides a facile way to characterize the instrument alignment and correction factors needed to obtain accurate-E, and was used to study the stoichiometry and structure of transcription initiation and elongation complexes (A. N. Kapanidis, R. H. Ebright, and S. Weiss, unpublished data). However, the current ALEX-based method does not explicitly consider linker or DNA dynamics, or polarization information at the single-molecule level; moreover, it cannot distinguish static heterogeneity from dynamic heterogeneity. A new ALEX method, based on interlaced pulsed laser excitation (nanosecond-ALEX; T. A. Laurence, X. Kong, and S. Weiss, unpublished data), surveys the rotational freedom of fluorophores, setting limits to the effects of probe orientation (expressed as κ2) on distance measurements; moreover, the new method improves distance measurements by recovering distances that fluctuate on timescales slower than the timescale of fluorescence emission, thus providing a handle for the analysis of dynamics. Extension of the ALEX principle to single-molecule three-color FRET systems will soon lead to simultaneous extraction of two or three distances (N. K. Lee, A. N. Kapanidis, and S. Weiss, unpublished data).
Because the sensitivity of single-molecule FRET methods is high (requiring less than a femtomole of biomolecules, 6–10 orders of magnitude less material than x-ray crystallography or NMR spectroscopy), advances in ALEX-based structural analysis combined with high-throughput, rapid preparation of labeled biomolecules may allow measurements of multiple distances and construction of solution-based, low-resolution structures of biomolecules. Such an approach will be suitable for the analysis of multicomponent complexes that make up cellular machinery. Structural analysis by single-molecule FRET will benefit from the large collection of individual structures available through x-ray crystallographic or NMR analysis, as was done at the ensemble level (Mekler et al., 2002).
SUPPLEMENTARY MATERIAL
An online supplement to this article can be found by visiting BJ Online at http://www.biophysj.org.
Supplementary Material
Acknowledgments
We thank Ekaterine Kortkhonjia for technical assistance and discussions, Jennifer Knight and Ron Levy for discussions, and Joanne Tang for editorial assistance.
This work was funded by the National Institutes of Health grants GM65382 to S.W. and GM41376 to R.H.E., Department of Energy grant FG03-02ER63339 to S.W., and a Howard Hughes Medical Institute Investigatorship to R.H.E.
Nam Ki Lee and Achillefs N. Kapanidis contributed equally to this work.
Nam Ki Lee's present address is School of Chemistry, Seoul National University, Seoul, Korea 151-747.
Achillefs N. Kapanidis' present address is Dept. of Physics, Clarendon Laboratory, University of Oxford, Parks Rd., Oxford, OX1 3PU, UK.
References
- Clegg, R. M. 1992. Fluorescence resonance energy transfer and nucleic acids. Methods Enzymol. 211:353–388. [DOI] [PubMed] [Google Scholar]
- Clegg, R. M., A. I. Murchie, A. Zechel, and D. M. J. Lilley. 1993. Observing the helical geometry of double-stranded DNA in solution by fluorescence resonance energy transfer. Proc. Natl. Acad. Sci. USA. 90:2994–2998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coultier, T. E., and J. Widom. 2004. Spontaneous sharp bending of double-stranded DNA. Mol. Cell. 14:355–362. [DOI] [PubMed] [Google Scholar]
- Dahan, M., A. A. Deniz, T. J. Ha, D. S. Chemla, P. G. Schultz, and S. Weiss. 1999. Ratiometric measurement and identification of single diffusing molecules. Chem. Phys. 247:85–106. [Google Scholar]
- Deniz, A. A., M. Dahan, J. R. Grunwell, T. J. Ha, A. E. Faulhaber, D. S. Chemla, S. Weiss, and P. G. Schultz. 1999. Single-pair fluorescence resonance energy transfer on freely diffusing molecules: observation of Förster distance dependence and subpopulations. Proc. Natl. Acad. Sci. USA. 96:3670–3675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deniz, A. A., T. A. Laurence, G. S. Beligere, M. Dahan, A. B. Martin, D. S. Chemla, P. E. Dawson, P. G. Schultz, and S. Weiss. 2000. Single-molecule protein folding: diffusion fluorescence resonance energy transfer studies of the denaturation of chymotrypsin inhibitor 2. Proc. Natl. Acad. Sci. USA. 97:5179–5184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dietrich, A., V. Buschmann, C. Muller, and M. Sauer. 2002. Fluorescence resonance energy transfer (FRET) and competing processes in donor-acceptor substituted DNA strands: a comparative study of ensemble and single-molecule data. Rev. Mol. Biotechnol. 82:211–231. [DOI] [PubMed] [Google Scholar]
- Edman, L., U. Mets, and R. Rigler. 1996. Conformational transitions monitored for single molecules in solution. Proc. Natl. Acad. Sci. USA. 93:6710–6715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eggeling, C., J. R. Fries, L. Brand, R. Gunther, and C. A. M. Seidel. 1998. Monitoring conformational dynamics of a single molecule by selective fluorescence spectroscopy. Proc. Natl. Acad. Sci. USA. 95:1556–1561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Förster, T. 1948. Intermolecular energy migration and fluorescence test. Annalen der Physik. 2:55–75. [Google Scholar]
- Ha, T. 2004. Structural dynamics and processing of nucleic acids revealed by single-molecule spectroscopy. Biochemistry. 43:4055–4063. [DOI] [PubMed] [Google Scholar]
- Ha, T., T. Enderle, D. F. Ogletree, D. S. Chemla, P. R. Selvin, and S. Weiss. 1996. Probing the interaction between two single molecules: fluorescence resonance energy transfer between a single donor and a single acceptor. Proc. Natl. Acad. Sci. USA. 93:6264–6268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ha, T. J., A. Y. Ting, J. Liang, W. B. Caldwell, A. A. Deniz, D. S. Chemla, P. G. Schultz, and S. Weiss. 1999. Single-molecule fluorescence spectroscopy of enzyme conformational dynamics and cleavage mechanism. Proc. Natl. Acad. Sci. USA. 96:893–898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haugland, R. P. 2002. Handbook of Fluorescent Probes and Research Products, 9th Ed. Molecular Probes, Eugene, OR.
- Heyduk, T. 2002. Measuring protein conformational changes by FRET/LRET. Curr. Opin. Biotechnol. 13:292–296. [DOI] [PubMed] [Google Scholar]
- Hillisch, A., M. Lorenz, and S. Diekmann. 2001. Recent advances in FRET: distance determination in protein-DNA complexes. Curr. Opin. Struct. Biol. 11:201–207. [DOI] [PubMed] [Google Scholar]
- Hohng, S., C. Joo, and T. Ha. 2004. Single-molecule three-color FRET. Biophys. J. 87:1328–1337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kapanidis, A. N., Y. W. Ebright, R. D. Ludescher, S. Chan, and R. H. Ebright. 2001. Mean DNA bend angle and distribution of DNA bend angles in the CAP-DNA complex in solution. J. Mol. Biol. 312:453–468. [DOI] [PubMed] [Google Scholar]
- Kapanidis, A. N., N. K. Lee, T. A. Laurence, S. Doose, E. Margeat, and S. Weiss. 2004. Fluorescence-aided molecule sorting: analysis of structure and interactions by alternating-laser excitation of single molecules. Proc. Natl. Acad. Sci. USA. 101:8936–8941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kapanidis, A. N., and S. Weiss. 2002. Fluorescent probes and bioconjugation chemistries for single-molecule fluorescence analysis of biomolecules. J. Chem. Phys. 117:10953–10964. [Google Scholar]
- Lawson, C. L., D. Swigon, K. S. Murakami, S. A. Darst, H. M. Berman, and R. H. Ebright. 2004. Catabolite activator protein: DNA binding and transcription activation. Curr. Opin. Struct. Biol. 14:10–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lilley, D. M. J., and T. J. Wilson. 2000. Fluorescence resonance energy transfer as a structural tool for nucleic acids. Curr. Opin. Chem. Biol. 4:507–517. [DOI] [PubMed] [Google Scholar]
- Marras, S. A., F. R. Kramer, and S. Tyagi. 2002. Efficiencies of fluorescence resonance energy transfer and contact-mediated quenching in oligonucleotide probes. Nucleic Acids Res. 30:e122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mekler, V., E. Kortkhonjia, J. Mukhopadhyay, J. Knight, A. Revyakin, A. N. Kapanidis, W. Niu, Y. W. Ebright, R. Levy, and R. H. Ebright. 2002. Structural organization of bacterial RNA polymerase holoenzyme and the RNA polymerase-promoter open complex. Cell. 108:599–614. [DOI] [PubMed] [Google Scholar]
- Michalet, X., T. D. Lacoste, and S. Weiss. 2001. Ultrahigh-resolution colocalization of spectrally separable point-like fluorescent probes. Methods. 25:87–102. [DOI] [PubMed] [Google Scholar]
- Mukhopadhyay, J., A. N. Kapanidis, V. Mekler, E. Kortkhonjia, Y. W. Ebright, and R. H. Ebright. 2001. Translocation of sigma(70) with RNA polymerase during transcription: fluorescence resonance energy transfer assay for movement relative to DNA. Cell. 106:453–463. [DOI] [PubMed] [Google Scholar]
- Mukhopadhyay, J., V. Mekler, E. Kortkhonjia, A. N. Kapanidis, Y. W. Ebright, and R. H. Ebright. 2003. Fluorescence resonance energy transfer (FRET) in analysis of transcription-complex structure and function. In RNA Polymerases and Associated Factors, Part D 44–159. Academic Press, New York. [DOI] [PubMed]
- Norman, D. G., R. J. Grainger, D. Uhrin, and D. M. J. Lilley. 2000. Location of cyanine-3 on double-stranded DNA: importance for fluorescence resonance energy transfer studies. Biochemistry. 39:6317–6324. [DOI] [PubMed] [Google Scholar]
- Rothwell, P. J., S. Berger, O. Kensch, S. Felekyan, M. Antonik, B. M. Wohrl, T. Restle, R. S. Goody, and C. A. M. Seidel. 2003. Multiparameter single-molecule fluorescence spectroscopy reveals heterogeneity of HIV-1 reverse transcriptase: primer/template complexes. Proc. Natl. Acad. Sci. USA. 100:1655–1660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sabanayagam, C. R., J. S. Eid, and A. Meller. 2005. Using fluorescence resonance energy transfer to measure distances along individual DNA molecules: Corrections due to nonideal transfer. J. Chem. Phys. 122:061103. [DOI] [PubMed] [Google Scholar]
- Schuler, B., E. A. Lipman, and W. A. Eaton. 2002. Probing the free-energy surface for protein folding with single-molecule fluorescence spectroscopy. Nature. 419:743–747. [DOI] [PubMed] [Google Scholar]
- Schuler, B., E. A. Lipman, P. J. Steinbach, M. Kumke, and W. A. Eaton. 2005. Polyproline and the “spectroscopic ruler” revisited and with single-molecule fluorescence. Proc. Natl. Acad. Sci. USA. 102:2754–2759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Selvin, P. R. 2000. The renaissance of fluorescence resonance energy transfer. Nat. Struct. Biol. 7:730–734. [DOI] [PubMed] [Google Scholar]
- Stryer, L., and R. P. Haugland. 1967. Energy transfer: a spectroscopic ruler. Proc. Natl. Acad. Sci. USA. 58:719–726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stryer, L., D. D. Thomas, and C. F. Meares. 1982. Diffusion-enhanced fluorescence energy transfer. Annu. Rev. Biophys. Bioeng. 11:203–222. [DOI] [PubMed] [Google Scholar]
- Talaga, D. S., W. L. Lau, H. Roder, J. Y. Tang, Y. W. Jia, W. F. DeGrado, and R. M. Hochstrasser. 2000. Dynamics and folding of single two-stranded coiled-coil peptides studied by fluorescent energy transfer confocal microscopy. Proc. Natl. Acad. Sci. USA. 97:13021–13026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vamosi, G., C. Gohlke, and R. M. Clegg. 1996. Fluorescence characteristics of 5-carboxytetramethylrhodamine linked covalently to the 5′ end of oligonucleotides: multiple conformers of single-stranded and double-stranded dye-DNA complexes. Biophys. J. 71:972–994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weiss, S. 1999. Fluorescence spectroscopy of single biomolecules. Science. 283:1676–1683. [DOI] [PubMed] [Google Scholar]
- Weiss, S. 2000. Measuring conformational dynamics of biomolecules by single molecule fluorescence spectroscopy. Nat. Struct. Biol. 7:724–729. [DOI] [PubMed] [Google Scholar]
- Wennmalm, S., L. Edman, and R. Rigler. 1997. Conformational fluctuations in single DNA molecules. Proc. Natl. Acad. Sci. USA. 94:10641–10646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Widengren, J., E. Schweinberger, S. Berger, and C. A. M. Seidel. 2001. Two new concepts to measure fluorescence resonance energy transfer via fluorescence correlation spectroscopy: theory and experimental realizations. J. Phys. Chem. A. 105:6851–6866. [Google Scholar]
- Wu, P., and L. Brand. 1992. Orientation factor in steady-state and time-resolved resonance energy transfer measurements. Biochemistry. 31:7939–7947. [DOI] [PubMed] [Google Scholar]
- Zhuang, X. W., and M. Rief. 2003. Single-molecule folding. Curr. Opin. Struct. Biol. 13:88–97. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.