

Abstract
Cytokines play a critical role in the normal development and function of the immune system. On the other hand, many rheumatologic diseases are characterized by poorly controlled responses to or dysregulated production of these mediators. Over the past decade tremendous strides have been made in clarifying how cytokines transmit signals via pathways using the Janus kinase (Jak) protein tyrosine kinases and the Signal transducer and activator of transcription (Stat) proteins. More recently, research has focused on several distinct proteins responsible for inhibiting these pathways. It is hoped that further elucidation of cytokine signaling through these pathways will not only allow for a better comprehension of the etiopathogenesis of rheumatologic illnesses, but may also direct future treatment options.
Keywords: cytokines, signaling, Jak, Stat, immunoregulation
Introduction
Since their discovery and cloning, it has become abundantly clear that cytokines play critical roles in regulating immune and inflammatory cells. For instance, the development of lymphoid and myeloid cells is now known to be controlled to a major degree by cytokines such as interleukin (IL)-7, IL-3, granulocyte-monocyte colony-stimulating factor (GM-CSF), and granulocyte colony-stimulating factor, among others. Similarly, numerous studies have documented the role of IL-6 in promoting inflammatory responses. Other cytokines can be classified as immunoregulatory cytokines. For example, IL-2 controls lymphoid homeostasis both positively and negatively; in addition, the differentiation of CD4+ T-helper (Th) cells into Th1 and Th2 subsets has been documented to be controlled in large measure by cytokines. For instance, IL-12 promotes the differentiation of naïve Th cells to those that produce interferon (IFN)-γ and lymphotoxin (Th1 cells), whereas IL-4 drives the differentiation of T cells to those that secrete IL-4, IL-5, and IL-10 (Th2 cells).
Not only do these processes contribute to normal host defence, but also to the pathogenesis of autoimmune disease. Much research has focused on the roles that cytokines play in diseases such as rheumatoid and psoriatic arthritis, systemic lupus erythematosus, and even such disparate illnesses as scleroderma and osteoarthritis. It is clear that both the pathogenesis and clinical manifestations of these debilitating diseases are at least in part due to aberrant immune and inflammatory responses, both of which are critically dependent on cytokines. In several animal models of rheumatoid arthritis, most notably collagen-induced arthritis in mice, disease susceptibility has been shown to be highly dependent on immunoregulatory cytokines. For instance, when susceptible mouse strains are rendered genetically deficient in either IL-12 or the IL-12 receptor, they develop a much milder manifestation of arthritis when immunized with collagen compared to normal animals. Even more strikingly, mice that are incapable of producing IL-6 become totally resistant to disease in response to collagen immunization.
Obviously, then, it is of great interest to understand the molecular basis of cytokine action. Fortunately, the mechanisms by which cytokines transmit signals from the cell membrane to the nucleus have been studied extensively, and knowledge of these pathways has increased tremendously over the past several years. In particular, analysis of mice and humans with cytokine receptor mutations or mutations of signaling molecules has provided important insights into the specific functions of these molecules; this information is emphasized in the present review.
The present review focuses on signaling by receptors that are members of two structurally related families, termed type I and type II cytokine receptors. Type I cytokine receptors include those for cytokines such as erythropoietin, prolactin, growth hormone, thrombopoietin, granulocyte colony-stimulating factor, and GM-CSF. In addition, many, but not all of the receptors for different interleukins are part of this family: IL-2, IL-3, IL-4, IL-5, IL-6, IL-7, IL-9, IL-11, IL-12, IL-13, and IL-15. The type II cytokine receptors include those for the IFNs (IFN-α, IFN-β, and IFN-γ) and IL-10. Of note, the receptors for IL-1, IL-18, IL-8, transforming growth factor-β, and tumor necrosis factor are not part of this family; despite their importance to immune-mediated disease, the signaling pathways used by these cytokines are not discussed here.
Type I and II cytokine receptors lack intrinsic kinase activity and instead rely on Janus kinase (Jak) proteins to initiate signaling. Cytokine binding to these receptors can activate a variety of pathways within cells including mitogen-activated protein kinases (MAPKs) and phosphoinositide 3' kinase. However, the discovery of a new family of tyrosine phosphorylated transcription factors, the Signal transducer and activator of transcription (Stat) family, provided great insight into the action of cytokines. Recently, research has focused on molecules that attenuate cytokine signaling. Of considerable interest is the suppressor of cytokine signaling (SOCS) family of molecules [1,2,3,4,5,6,7,8,9,10,11,12].
Jaks
In contrast to other tyrosine kinase families, the Jak family is rather small. There are only four known mammalian Jaks — Jak1, Jak2, Jak3, and Tyk2 — which were identified in the early 1990s by techniques that capitalized on homology of their kinase domains to other tyrosine kinases [13,14,15,16].Since the discovery of these family members, no new mammalian members have been identified, suggesting that they may comprise the entire family. Teleost and avian Jaks have been identified, as has a single Drosophila Jak; thus, these critical signaling molecules are highly conserved throughout evolution [17,18,19]. Shortly after their discovery, their functional importance in IFN and cytokine signaling was established [1,20]. It was first shown that Jaks are essential for IFN signaling using a panel of cell lines that were resistant to IFNs [21,22,23], and subsequently, type I cytokines were also found to activate Jaks; in fact, all type I and II cytokines activate Jaks in some combination [6,24,25,26,27,28]. It was also shown that Jaks physically associate with cytokine receptors. For the IFN-α receptor, the presence of Tyk2 is required for appropriate receptor expression on the cell surface; this does not appear to be the case for other cytokines, however.
Jak function
Jak3
Jak3, in marked contrast to the relatively ubiquitous expression of Jak1, Jak2, and Tyk2, has a much more regulated and specific tissue expression. It is constitutively expressed at high levels in natural killer cells and thymocytes, and is inducible in T cells, B cells, and myeloid cells [16,29,30,31,32]. Jak3 is activated by a limited number of cytokines, only those receptors that use the common γ chain (γc) (IL-2, IL-4, IL-7, IL-9, and IL-15) [26,27,33,34,35] (Table 1). This is explained by the fact that Jak3 specifically associates with γc, and IL-2 and IL-4 signaling is markedly compromised in cells lacking Jak3 [36,37].
Table 1.
Association of Janus kinases (Jaks), Signal transducers and activators of transcription (Stats), and suppressors of cytokine signaling (SOCSs) with cytokine receptors and phenotype of knockout mice.
| Jak/Stat/SOCS | Cytokines | Deficient mice |
| Jak1 | γccytokines, gp130 cytokines, IL-10, IFN-α , IFN-β , IFN-γ, IL-13 | Neurologic defect (?), mice die perinatally |
| Jak2 | Erythropoietin, βccytokines, gp130 cytokines, IL-13, IL-12, GH, prolactin, thrombopoietin, IFN-γ | Embryonically lethal, failed to develop red blood cells |
| Jak3 | IL-2, IL-7, IL-15, γc cytokines | SCID |
| Tyk2 | gp130 cytokines, IL-13, IL-12, IFN-α, IFN-β, IL-10 | Not yet reported |
| Stat1 | IFN-α , IFN-β , IFN-γ , IL-2, IL-6, IL-10 | Susceptibility to viral and some bacterial infections |
| Defective signaling in response to IFNs | ||
| Impaired tumor rejection | ||
| Stat2 | IFN-α, IFN-β | Defective signaling in response to IFNs* |
| Stat3 | LIF, IL-10, IL-6, other gp130 cytokines, γc cytokines, GH | Embryonically lethal |
| Targeted disruption in macrophages and neutrophils showed defective | ||
| IL-6 and IL-10 signaling | ||
| Chronic enterocolitis | ||
| Conditional knockouts showed suppressed epithelial apoptosis and | ||
| delayed mammary gland involution | ||
| Stat4 | IL-12 | Defective Th1 development |
| Stat5a | Prolactin, other γc cytokines, otherβc cytokines, GH, thrombopoietin | Defective lobuloalveolar development in breast |
| Stat5b | GH, other γc cytokines,βc cytokines, prolactin, thrombopoietin | Required for sexual dimorphism |
| Stat5a/b | GH, IL-2, erythropoietin | Infertile, smaller body size |
| Impaired cell-cycle progression of peripheral T cells | ||
| Fetal anemia (?) | ||
| Stat6 | IL-4, IL-13 | Defective Th2 development |
| Stat4/6 | No Th2 cells in vitro | |
| SOCS-1 | IFN-γ, IL-6, GH, thrombopoietin, OSM, LIF | Perinatal lethality, hyper-responsiveness to IFN-γ |
| SOCS-3 | Erythropoietin, GH, IL-2, leptin, CNTF, IL-11, IL-10 | Embryonic lethality, ? due to erythrocytosis |
γ Chain (γc) cytokines: interleukin (IL)- 2, IL- 4, IL- 7, IL- 9, IL- 15. gp130 cytokines: IL- 6, IL- 11, oncostatin M (OSM), ciliary neurotropic factor (CNTF), leukemia inhibitory factor (LIF), CT- 1, IL- 12, Leptin. β chain (βc) cytokines: IL- 3, IL- 5, granulocyte- macrophage colony- stimulating factor (GM- CSF). Bold letters indicate cytokines that have been shown to be essential in knockouts. *Schindler C, personal communication. SCID, severe combined immunodeficiency; FGF, fibroblast gowth factor; GH, growth factor; IFN, interferon.
The pivotal function of Jak3 was established when a form of human severe combined immunodeficiency (SCID) was found to result from Jak3 mutations [38,39]. As was predicted on the basis of the association of Jak3 with γc, thephenotype of these patients was quite similar to that seen in patients with X-linked SCID, which results from a mutation in the γc [40]. These patients lack T cells and natural killer cells and have dysfunctional B cells. Jak3 knockout mice were subsequently generated that also have defects in the same cell lineages (T, B, and natural killer cells) [41,42,43,44].
Because Jak3 is activated by all of the γc cytokines, thequestion arises as to how the deficient signaling by these various cytokines relates to Jak3 and γc deficiency. The phenotype of Jak3 SCID and X-linked SCID is most similar to IL-7 and IL-7 receptor gene-targeted mice [45,46,47]. The lack of IL-7 signaling can clearly result in SCID in mice, and recently it was documented that a subset of patients with autosomal recessive SCID have IL-7 receptor mutations [48]. One notable feature is that these patients do have natural killer cells, indicating that IL-7 signaling is not essential for natural killer cell development. In this regard it is important to note that IL-15 receptor α chain knockout mice lack natural killer cells [49]. Thus, the SCID phenotype associated with Jak3/deficiency largely results γc from defective IL-7 and IL-15 signaling. It should be noted, however, that the phenotypes of Jak3 and deficient γc mice and humans differ somewhat. Whereas human SCID patients have dysfunctional B cells and few, if any, T cells, deficient mice lack B cells and have reduced T cell numbers. The explanation for this difference is unclear at present. It should be noted that the T cell defect in human Jak3 SCID is not absolute; some patients do develop some T cells [50].
Somewhat surprisingly, IL-2, IL-2 receptor α chain, and IL-2 receptor β chain knockout mice exhibit lymphoproliferative and autoimmune disease [51,52,53,54], an abnormality that has been attributed to defective apoptosis of activated T cells. T cells from humans with Jak3 SCID and Jak3/γc-/- mice are abnormal in that they express activation markers [41,55]; impaired negative thymic selection has been postulated as one mechanism, but an alternative explanation is that absence of IL-2 signaling results in impaired apoptosis.
Although we and others previously showed that myeloid cells express Jak3 upon stimulation with a variety of cytokines and proinflammatory stimuli, no abnormal function has been reported in this cell lineage due to the lack of Jak3 expression [56]; the function of Jak3 in this lineage, therefore, remains unclear.
Importantly, after treatment with bone marrow transplantation, no significant defects have been reported outside the immune system in Jak3 and γc deficient humans. This argues that the functions of Jak3 and γc are truly limited to the immune system, which is consistent with the relative tissue specificity of this molecule. The specificity of the defects suggests that Jak3 or the Jak3-γc interaction may represent a useful target for the development of novel immunosuppressants [33,38,39].
Jak1, Jak2, and TyK 2
As stated previously, Jak1 and Jak2 have a wide tissue expression and are activated by a variety of cytokines (Table 1). Specifically, IFN-α/β signaling requires Jak1 and Tyk2; the α subunit of the IFN-α /β receptor associates with Tyk2, and the β subunit with Jak 1 [57,58,59]. In contrast, IFN-γ requires Jak1 and Jak2 [1,21,22,23,60]. In this circumstance, the IFN-γ receptor α subunit associates solely with Jak1 and the β subunit only with Jak2 [5]. Jak1 is also activated by γc cytokines and associates with the ligand-specific receptor subunit. Hormones such as growth hormone and erythropoietin predominantly associate with and activate Jak2 [24,25], but Jak2 also associates with the common β chain, a shared subunit for IL-3, IL-5, and GM-CSF [61]. IL-6 and related cytokines can activate Jak1, Jak2, or Tyk2; gp130, the shared subunit of this family of receptors, can bind each of these Jaks [62]. Finally, the IL-12 receptor β 1 chain associates with Tyk2 and the β 2 chain associates with Jak2 [63].
As might be expected on the basis of the cytokines that activate them, mice that lack either Jak1 or Jak2 have more diverse abnormalities. Like Jak3 knockout mice, Jak1-/- mice have SCID. In contrast to Jak3 knockouts, though, Jak1 deficient mice die perinatally as a result of an incompletely defined neurologic defect [64]. In addition, they fail to manifest biologic responses to all receptors that utilize this kinase, including all type II cytokine receptors, cytokine receptors that utilize the γc subunit for signaling, and the family of cytokine receptors that depend on the gp130 subunit for signaling.
Jak2 deficiency is embryonically lethal, because these mice fail to develop erythroid cells [65,66]. Interestingly, this phenotype is more severe than that seen in erythropoietin receptor deficient mice, perhaps due to the necessity of signaling through other Jak2-requiring receptors such as IL-3 for efficient erythropoiesis. Enhanced signaling through Jak2 has also been implicated in the pathogenesis of leukemia. Chromosomal translocations in several patients with leukemia were characterized and shown to fuse the 3' portion of Jak2 to the 5' region of TEL, a gene encoding a member of the ETS transcription factor family. The TEL-Jak2 fusion protein includes the catalytic domain of Jak2 and the TEL-specific oligomerization domain. TEL-induced oligomerization of TEL-Jak2 resulted in the constitutive activation of its tyrosine kinase activity and conferred cytokine-independent proliferation to the interleukin-3-dependent Ba/F3 hematopoietic cell line [67,68]. These findings underscore the importance of Jak2 mediated signaling in driving proliferation and differentiation in both myeloid and lymphoid cells.
Tyk2 knockout mice have not been reported as of yet, and no known human disorders have been linked to a defect in this Jak, so the cytokines for which Tyk2 signaling is uniquely responsible have not been ascertained. Based on the findings with deficient cell lines, the expectation is that IFN α /β actions will be impaired in such mice.
Jaks and noncytokine receptors
Since the discovery of the Jaks, occupancy of a vast array of receptors other than type I and II receptors have been shown to induce Jak phosphorylation and/or activation. To date, however, it has not been proved that Jaks are essential, nonredundant components of the signaling pathways of noncytokine receptors. For instance, although CD40 was found to associate with Jak3, no defect in CD40 signaling was apparent in Jak3 deficient cells [69,70]. At present, the only receptors documented to be absolutely dependent on Jaks for signaling are type I and II cytokine receptors. Whether this small family of tyrosine kinases is dedicated to signaling by this class of receptors or whether wider functions exist remains to be determined; this is one of the most interesting remaining questions that pertain to the biology of the Jaks.
Jak structure
The three-dimensional structure of the Jaks is presently unknown. This is no doubt partly because they are relatively large kinases of more than 1100 amino acids with apparent molecular weights of 120–130 kDa. Their messenger RNA transcripts range from 4.4 to 5.4 kilobases in length. Multiple spliced forms of Jak3 have been identified, including a variant that lacks a segment of the catalytic domain [29,71,72]. It is intriguing to speculate that a naturally occurring dominant negative form of Jak3 may have regulatory function, but this has yet to be proved.
Jaks have seven regions of homology termed Janus homology (JH) domains 1-7 (Fig. 1), and the carboxy-terminal tyrosine kinase, or JH1 domain, shares the features of other tyrosine kinase domains. For example, phosphorylation of tyrosine residues in the activation loop of kinases such as the insulin receptor play an important role in regulating phosphotransferase activity [73]. A number of autophosphorylated sites are being identified in Jaks, two of which reside within the putative activation loop. Depending upon the specfic Jak, however, mutations at these sites appear to have slightly different functional consequences. That is, mutation of tyrosine 1007 abrogated any signaling capacity of Jak2, whereas mutations in both the corresponding tyrosine residues and the adenosine triphosphate binding site were required to abolish activity of Tyk2 completely [74,75].In contrast, mutations of Y981 in Jak3 actually increased activity [76]. Thus, there may be subtle differences in the regulation of catalytic activity of each Jak. The molecule src homology (SH)2Bβ, an SH-2 domain containing protein, associates with Jak2 and increases its catalytic activity in response to growth hormone, but the mechanism of this regulation has not been determined [77,78].
Figure 1.
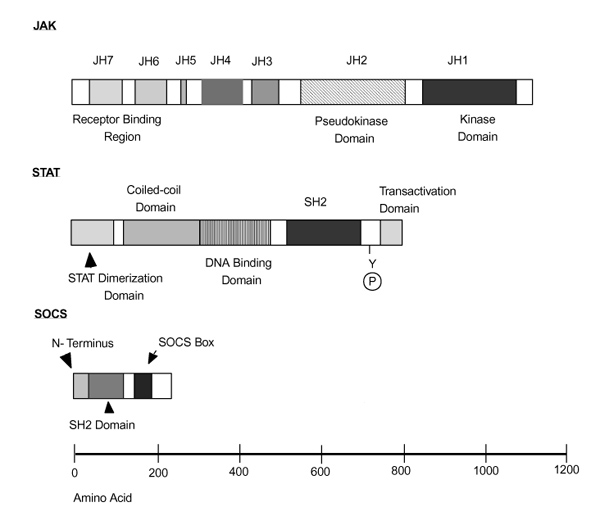
Structure of Janus kinases (Jaks), signal transducers and activators of transcription (Stats), and suppressors of cytokine signaling (SOCS). Regions of homology shared by Jaks have been termed Jak homology (JH) domains. JH1 is a kinase domain and JH2 is a pseudo-kinase domain. The amino-terminus of the Jaks appears to be important for association with cytokine receptors subunits. Stats have a conserved tyrosine residue, phosphorylation of which allows Stat dimerization; a src homology (SH2) domain that mediates the dimerization; and an amino-terminal region that is known to play a role in the dimerization of Stats dimer. The amino-terminal, carboxy-terminal and coiled-coil regions of Stats can interact with other transcription factors. SOCS proteins share a similar strucuture with a central SH2 domain, a region at the amino-terminus that is variable in both length and in amino acid sequence, and a region of homology at the carboxy-terminus termed the 'SOCS box'.
The hallmark of the Jak family of protein tyrosine kinases is the existence of tandem kinase and pseudokinase domains; it is this feature that gives the Jaks their name, and among mammalian tyrosine kinases only the Jaks have this domain. Like the Roman god of gates and doorways, the Jaks are 'two-faced'. The pseudokinase domain is also termed the JH2 domain. Although it has overall similarity to kinase domains, the JH2 domain lacks critical residues that are required for phosphotransferase activity; rather, the function of this domain appears to be to regulate catalytic activity. Mutations or deletions of this region have complicated effects that either inhibit or enhance catalytic function, depending upon the exact mutation generated [79,80] (Chen M, et al, unpublished data). Importantly, Jak3 SCID patients have been identified with mutations in this region, underscoring its critical function [50]. Another function suggested for the JH2 domain is as a docking site for Stats [81].
Although it has not been well characterized for all of the Jaks, the amino-terminus appears to confer binding to the appropriate cytokine receptor [79,82,83,84]. For Jak3, at least, the amino-terminal JH6 and JH7 domains are sufficient to confer binding specificity to γc [85,86]. For otherJaks, the amino-terminus is clearly involved in receptor interactions, but may extend beyond the JH6 and JH7 domains [87,88]. The region of the cytokine receptor to which Jaks bind has been much better characterized and is found in the membrane proximal region (reviewed in Ihle, 1995 [28]). Signal transducing adaptor molecule (STAM), a 70-kDa adaptor molecule that is phosphorylated in response to IL-2, IL-4, GM-CSF, epidermal growth factor and platelet-derived growth factor, binds to both Jak3 and Jak2, and couples cytokine stimulation to DNA synthesis [89]. STAM is a molecule that associates with Jaks and may enhance the formation of Jak-receptor complexes (authors' unpublished observations). Two STAM-associated molecules, Hrs and AMSH, have been hypothesized to act downstream of Jaks in cytokine signaling pathways [90,91].
Many tyrosine kinases have intrinsic SH2 and SH3 domains that mediate protein-protein interactions. Although the JH4 domain has overall homology to SH2 domains, mutation of the critical Arg residue, which would be expected to bind phosphotyrosine, had no effect on signaling. Thus, the function of this segment remains unclear at present [15,84].
Binding of cytokines to type I and II receptors has been suggested to initiate signaling by effecting homodimerization or heterodimerization of the receptor subunits, which in turn leads to the apposition of Jaks. This may allow transphosphorylation of the Jaks at sites within their activation loops, thus enhancing catalytic activity. A more recently proposed mechanism [92,93] is that ligand-induced allosteric alteration of the receptor itself leads to Jak activation. For receptors that heterodimerize (most of the IL and IFN receptors), heterodimerization of different Jaks also occurs, and the Jaks are interdependent for activation. For instance, in cells that lack Jak1, no phosphorylation of Tyk2 or Jak2 was observed upon stimulation with IFN [60], and conversely no phosphorylation of Jak1 was seen in cells lacking Jak2 or Tyk2. In Jak3 deficient cells, no phosphorylation of Jak1 occurs in response to IL-2 [36].
Regardless of the precise mechanism, after activation the Jaks phosphorylate receptor subunits on tyrosine residues, enabling the recruitment of proteins with SH2 or phosphotyrosine binding domains. These proteins are then phosphorylated by Jaks. A number of signaling pathways, such as the Ras-Raf-MAPK pathway and the phosphoinositide 3' kinase pathways are activated in response to cytokines; the significance of these pathways in immune function has been extensively reviewed elsewhere and is not discussed here [94,95,96,97,98,99]. However, the function of another class of SH2-containing molecules, the Stats, is discussed in detail.
Stats
By purifying factors bound to promoters of IFN-inducible genes, Darnell and coworkers [100] cloned the first members of the Stat family. The IFN-α induced complex comprised a 91-kDa polypeptide, which later became known as Stat1; a 113-kDa protein (Stat2); and p48, a member of the IFN regulatory factor family. An IFN-γ induced complex, γ activated factor, turned out to be composed of Stat1 only [101].
Following the discovery of Stat1 and Stat2, the cloning of the remaining family members, Stat3, Stat4, Stat5a, Stat5b, and Stat6, quickly ensued [9,102,103,104,105,106,107,108,109,110]. Most Stats are approximately 750 amino acids long, but Stat2 and Stat 6 are larger (850 amino acids). These transcription factors were immediately recognized as a novel family, in that they had SH2 domains and were themselves tyrosine phosphorylated. Thus, a new signaling paradigm emerged (Fig. 2). Stats are latent cytosolic transcription factors that are recruited to phosphorylated cytokine receptors via their SH2 domains [111,112,113,114]. The Stats are then phosphorylated themselves by Jaks, they heterodimerize or homodimerize via reciprocal SH2-phosphotyrosine interactions, and translocate to the nucleus to regulate gene transcription.
Figure 2.
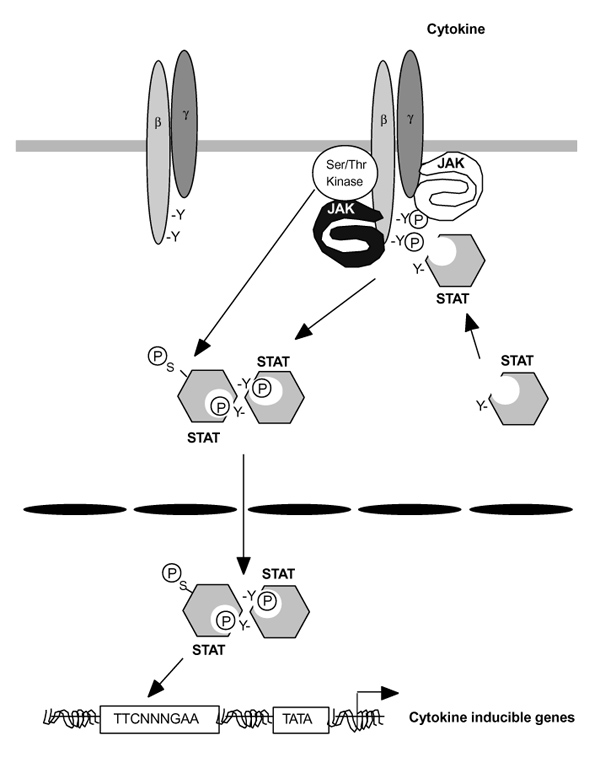
Model for cytokine signal transduction. Cytokines associate with cytokine receptor subunits and activate janus kinases (Jaks). The Jaks in turn phosphorylate tyrosine-based docking sites on the receptor. signal transducers and activators of transcription (Stats) then bind via their src homology (SH)2 domains. The STATs are then phosphorylated by the JAKs, form homo-hetero-dimers and then translocate into the nucleus, where they bind target sequences like γ activated sequence (GAS) motif. Transcriptional activation of genes typically requires the coordinated function of multiple accessory transcription factors. Additionally, serine phosphorylation of some Stats may be important for maximal transcription of target genes.
Stat structure
In contrast to the Jaks, the structure of the Stat molecules has been reasonably well characterized [115,116,117] (Figs 1 and 3). Overall, the structure of the Stats is similar to that of other transcription factors such as nuclear factor-κ B and p53. The dimeric molecule forms a C-clamp structure around the DNA, but, unlike nuclear factor-κ B and p53, there are fewer direct contact sites with the DNA backbone. Rather, the nutcracker-like structure of the Stats is largely dependent upon SH2-phosphotyrosine interactions. Stats have a conserved amino-terminal protein-protein interaction domain, followed by a segment (the coiled-coil domain) with multiple protruding α -helices. This is followed by the actual DNA binding domain, a linker domain, the SH2 domain, a conserved site of tyrosine phosphorylation, and a variable carboxy-termini transcriptional activation domain.
Figure 3.
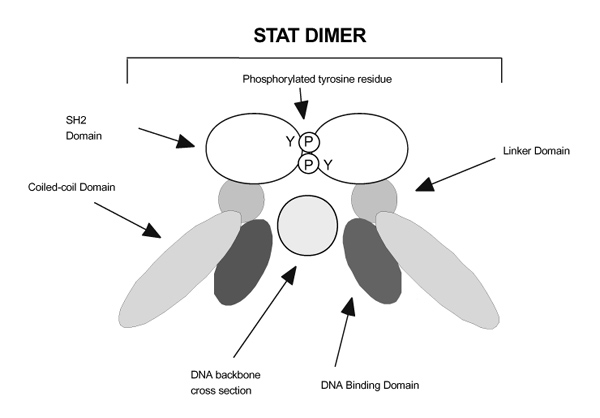
Signal transducer and activator of transcription (Stat) binding to DNA. In this two-dimensional model, the heterodimerized or homodimerized Stat molecule binds to the appropriate DNA sequence via a 'C clamp' structure. Note that in addition to the specificity controlled by the DNA binding domain, other regions of the Stat molecule may also be important in DNA interactions. Tetramerization of the Stats may allow for an even greater degree of stability in DNA binding to adjacent imperfect Stat binding sites; this is mediated by the Stat amino-terminus (not shown). Also not shown in this illustration is the Stat transcriptional activation domain.
Amino-terminal dimer-dimer interaction domain
With the exception of Stat6, Stats bind somewhat indiscriminately to the same consensus sequences; it is notable that clustered imperfect Stat binding sites are found in a number of relevant cytokine inducible promoters. Even though Stats bind poorly to these sites, cooperative dimer–dimer interactions can occur [118]. This is mediated by the conserved amino-termini of Stats, which consists of eight helices that form hook-like structures, facilitating these interactions. Perhaps unimportant for Stat binding to a single consensus binding site [119], these domains appear critical for binding to imperfect sites [120]. Combinatorial binding might be one mechanism of achieving more specificity in signaling.
Coiled-coil domain and association with other transcription factors
Consisting of four α -helices, the coiled-coil domain from amino acids 136-317 provides a structure suitable for many protein-protein interactions. The coactivator proteins p48 and p300/CBP have been shown to interact with Stats through this region [121,122,123,124,125], as well as another protein, Nmi [126]. Other transcription factors have been shown to associate with Stats such as the glucocorticoid receptor (with Stat5a and Stat5b), Sp-1, c-Jun, and nuclear factor-κ B, but the exact domain interactions have yet to be mapped [127,128,129,130,131,132].
DNA binding domain
The DNA binding region of Stats resides within the central 171 amino acids, but relatively few direct contacts exist. Rather, the clamp-like structure is imparted by phosphotyrosine-SH2 interactions [115,116]. Stats bind two types of DNA motif: IFN-stimulated response elements (consensus: AGTTTNCNTTTCC) and γ -activated sequence elements (consensus: TTCNNNGAA). Stat1, Stat2, and p48 bind to IFN-stimulated response elements, whereas Stat1, Stat3, Stat4, Stat5a, and Stat5b bind to γ -activated sequence element sites. Stat6 binds a similar but distinct site: TTCNNNNGAA.
The src homology 2 domain and tyrosine phosphorylation site
The SH2 domain (amino acids 600-700) serves two critical functions: to allow Stats to bind phosphorylated receptor subunits and be phosphorylated themselves by Jaks on a conserved tyrosine residue; and to enable Stat dimerization and DNA binding. The crystal structure of a Stat-DNA complex underscores the importance of the SH2 domain, as the SH2-phosphotyrosine interaction forms the hinge of the clamp that is largely responsible for DNA binding (Fig. 3).
Transcriptional activation domain
Stat1, Stat2, and Stat5 have been documented to have carboxy-terminal transcriptional activation domains [119,133,134,135,136,137]. In addition to tyrosine phosphorylation, it has been shown that Stat1, Stat3, Stat4, and Stat5 are also phosphorylated on serine residues in response to cytokine stimulation [133,138,139]. For these proteins, a conserved site of serine phosphorylation, residing in a consensus sequence for MAPK-mediated phosphorylation has been mapped within the carboxy-terminal transcriptional activation domain [135,140] (Visconti et al, unpublished data). However, the functional significance of Stat serine phosphorylation and the identity of the kinase(s) responsible for this event remain deeply controversial. Recently, a large number of reports have been published that link STAT serine phosphorylation to the activation of various MAPKs. Notably though, they provide significantly divergent results, perhaps due to the differences in the Stat proteins investigated and in the systems utilized [141,142,143,144,145,146].
Thus, it has been reported that p38, which is activated in response to IFNs, is indispensable for Stat1 serine 727 phosphorylation and transcriptional activity [145]. Accordingly, we have found that IL-12-induced Stat4 serine 721 phosphorylation and transcriptional activity requires p38 activity (Visconti et al, unpublished data). Other findings indicate that JNKs, but not p38, mediate Stat3 serine 727 phosphorylation in response to various stress treatments and that this event results in the inhibition of Stat3 activity [146]. In contrast, it has been shown that both JNKs and p38 are required for STAT3 transcriptional activity induced by the Src oncoprotein [143]. Other MAPKs, including extracellular signal related kinase family members, can also phosphorylate serine residues in Stat proteins and activate them [147,148]. Others have found, however, that signaling through the extracellular signal related kinase pathway can also downregulate Stat activity [149]. The most plausible hypothesis at the moment is that the effect of serine phosphorylation of Stat proteins depends on the cell type and on the class of serine kinases activated in response to different extracellular stimuli.
Nuclear translocation
Stats lack a classic nuclear localization signal, and in general dimerization of the Stats is believed to be essential for nuclear localization. This appears not to be sufficient, however, and sequences in the amino-terminus contribute to nuclear translocation (and perhaps deactivation, as well) [150]. For Stat1 at least, nuclear import has been shown to be dependent upon the activity of the small guanosine triphosphatase Ran and may involve the importin receptor [151,152,153,154]. One group has hypothesized that Stats may translocate to the nucleus via the nuclear localization signal of the ligands or receptors themselves, but the importance of this possibility remains to be established [155,156]. Finally, regulation of nuclear export of the Stats may also be an important means of nuclear localization. In any case, it is very clear that much needs to be learned about the regulation of Stat intracellular trafficking.
Stat function
Stat1
Stat1 has been shown to be activated by the IFNs, cytokines such as IL-2, IL-6, and IL-10, and noncytokine signals such as epidermal growth factor (Table 1). Not surprisingly, Stat1 knockout mice were found to be highly susceptible to viral and some bacterial infections, reminiscent of defects observed in IFN-α receptor and IFN-γ receptor knockout mice and IFN-γ receptor deficient humans [157,158,159,160]. Interestingly, defects in signaling by cytokines other than IFNs have not been reported, but it does seem to be important for the fibroblast growth factor-mediated growth inhibition of chondrocytes [161].
A role for IFN-γ signaling through Stat1 for tumor surveillance has recently been underscored. Mice deficient in Stat1 or the IFN-γ receptor were much more susceptible to tumor development when challenged with a chemical carcinogen, and when bred with mice deficient in the tumor suppressor p53 gene, these mice developed a broader spectrum of tumors compared with mice lacking p53 alone [162]. A recent study [163] has shown that Stat1 deficient mice are unable to clear immunogenic tumors that their wild-type littermates easily controlled, and that they are unable to reject poorly immunogenic tumors when immunized with an IL-12 based vaccine. Severe defects in lytic activity in both T and natural killer cells were also noted.
Stat1 has additional functions in regulating apoptosis. That is, tumor necrosis factor-α dependent apoptosis is impaired in Stat1 deficient mice because of reduced expression of the caspases Ice, CPP32, and Ich-1 [164]. Unlike its role in mediating cytokine dependent gene expression, however, Stat1 dimerization does not appear to be necessary for the regulation of these genes, because an SH2 mutant Stat1 supports expression. The mechanism by which this occurs is obscure, particularly in view of the structural information present. The importance of Stat1 in apoptosis mediated by type I cytokines was further underscored when it was found that Stat1-/- mice were resistant to virus-induced apoptosis [165].
Stat2
Like Stat1, Stat2 is also activated by interferons; indeed, only IFN-α/β has been reported to activate Stat2 (Table 1). Unlike other Stats, it requires Stat1 and p48 for interaction with DNA [166]. As of yet, Stat2 knockout mice have not been reported, but evidently they are severely deficient in IFN-α/β signaling, as might be expected (Schindler C, personal communication).
Stat3
Stat3 was first identified as a factor activated by cytokines signaling through gp130 (IL-6, leukemia inhibitory factor, and ciliary neurotropic factor). Stat3 deficiency is embryonically lethal, perhaps due to the absence of leukocyte inhibiting factor function, as well as its role in maintaining stem cell pluripotency [167,168]. In contrast, gene targeting of Stat3 only in myeloid cells produced an exaggerated inflammatory response, resulting in premature death largely due to impaired IL-10 function [169]. These animals became highly susceptible to endotoxic shock with increased production of inflammatory cytokines such as tumor necrosis factor-α, IL-1, IFN-γ, and IL-6. The suppressive effects of IL-10 on the production of inflammatory cytokines by macrophages and neutrophils was completely abolished, and these mice developed chronic enterocolitis with age. Additionally, these mice manifested an exaggerated Th1 response, which may also help to explain the inflammatory bowel disease seen. These results might suggest a role for abnormal Stat3 signaling in other autoimmune processes, but no studies have been published to date. Although it is clear that Stat3 is essential for appropriate IL-10 signaling, its function for other cytokines remains unclear because of the embryonic lethality seen in Stat3 knockout embryos.
Stat4
Stat4 is activated by a limited number of cytokines; IL-12 is the predominant cytokine that activates Stat4 in mice, whereas in humans both IFN-α/β and IL-12 activate it. More recently it has been shown that IL-2 is capable of activating Stat4 in natural killer cells [170]. Of note, though, Stat4 deficient mice only demonstrate defects of impaired IL-12 responses (ie defective Th1 development and impaired cell-mediated immune responses), a phenotype similar to that seen in IL-12 and IL-12 receptor knockout mice and IL-12 receptor deficient humans [171,172,173,174]. Although predominantly expressed in lymphoid cells, Stat4 has recently been found to be inducibly expressed in activated macrophages, most notably those found in synovium from rheumatoid arthritis patients (Frucht et al, submitted). The target genes of Stat4 in macrophages are currently unknown, but it is interesting to speculate that macrophages may provide some functions of cellular immunity previously assigned only to lymphoid cells.
Stat5
Encoded by two genes, Stat5a and Stat5b share 93% identity at the protein level [109,175], and are both activated by a plethora of cytokines, including prolactin, growth hormone, erythropoietin, thrombopoeitin, and IL-2. The development of knockout mice, however, underscores the very different biologic functions they each serve; Stat5a knockout mice have impaired mammary gland development [176], whereas Stat5b deficient mice are defective in both sexually dimorphic growth as well as in growth hormone dependent regulation of liver gene expression [177].
To assess potential redundancy in function, Stat5a/Stat5b double knockouts were created [178]; one-third of these mice died within 48 h of birth, with the surviving mice developing a smaller than normal body size, which was apparently due to aberrant growth hormone signaling. Despite the fact that lymphoid development is normal, T cells are hyporesponsive to IL-2, and these animals develop lymphoproliferative disease, similar to that in IL-2, IL-2 receptor α chain and IL-2 receptor β chain deficient mice [179]. These results underscore an essential role for Stat5 for IL-2 signaling; whether Stat5a or Stat5b individually are critical is somewhat controversial at present, because several studies have clearly shown that IL-2 responsiveness is impaired in either Stat5a or Stat5b deficient mice [180,181]. Clinically, adult females are infertile, but unlike Jak2 knockout mice, which have no blood, these animals are only moderately anemic; the severity of anemia, however, is also a point of contention [182] (Ihle J, personal communication).
Of additional interest is the current controversy regarding the role of Stat5 in T-cell receptor-mediated signaling. Initially found not to be involved with T-cell receptor signaling [183], a more recent study has shown that Stat5 phosphorylation induced by T-cell receptor crosslinking is abolished in lck deficient mice [184].Other studies [185] have suggested a role for Stat3 but not Stat5 in T-cell receptor-mediated signaling; the different results may reflect the different model systems used. Nonetheless, the potential significance of antigen-mediated signaling through the Stats remains intriguing.
Stat6
Stat6 was originally identified as an IL-4 inducible transcription factor [106]. It was therefore not surprising to find that Stat6 deficient mice failed to develop Th2 immunity in response to IL-4 or IL-13, were unable to upregulate cell surface expression of major histocompatibility complex class II, CD23, or IL-4 receptor α chain in response to IL-4, and failed to produce immunoglobulin E in response to cross-linking of surface immunoglobulin D [186,187,188,189]. Accordingly, lack of Stat6 dramatically attenuates allergic and asthmatic diseases in several animal models [190,191,192,193]. Stat6 deficient animals are also unable to clear parasites [194]. Remarkably, a recent study of Stat6 knockout mice in a murine acquired immune deficiency syndrome model revealed normal serum immunoglobulin E levels and lymphoproliferation, indicating that B cells from mice with murine acquired immune deficiency syndrome activate unique IL-4-independent and STAT6-independent signaling pathways for B-cell activation and differentiation [195].
The generation of Stat4/Stat6 double knockout mice has provided an interesting model system for the study of Th1/Th2 differentiation, a complex and exciting topic in immunology today. Interestingly, these mice develop Th1 responses, suggesting that Stat4 may be dispensable for Th1 differentiation in the absence of Th2 responses [196].
Activation of signal transducers and activators of transcription by noncytokine receptors
As with the Jaks, ligation of many receptors has been reported to activate various Stats. As discussed above, T-cell receptor crosslinking, for instance, has been reported to activate Stats, but this remains controversial [179,184, 197]. Less controversial are the findings that epidermal growth factor, angiotensin II, and other ligands activate Stats [198,199,200,201,202,203]. Through the use of knockout mice or deficient cells, however, the essential function of a specific Stat for a noncytokine stimulus has yet to be established; this is clearly the major challenge in this area. It should be noted, though, that Stats are evolutionarily very old. Several Dictyostelium Stats have been identifed, but this organism has not been reported to have Jaks [204, 205]. If this is the case, it would add credence to the notion that Stats have broader functions beyond type I and II cytokine signaling mediated by Jaks. Interestingly, v-src and other oncogenes have been reported to activate Stats and, importantly, a dominant-negative Stat3 construct has been shown to block transformation [206]. Conversely, a gain-of-function Stat3 construct is transforming [207].The importance of Stats in malignant transformation will need to be established using knockout animals, but this is clearly an exciting area.
Attenuation of cytokine signaling
Equally as important as the ability to initiate cytokine signaling is the ability to terminate it. Indeed, one might speculate that this will be more important in terms of the pathogenesis of human autoimmune diseases. The ability to regulate cytokine signaling occurs by multiple proposed mechanisms. These include the following: phosphatases, cytokine-inducible inhibitor molecules, transcriptional repressors, and Stat degradation (Fig. 4).
Figure 4.
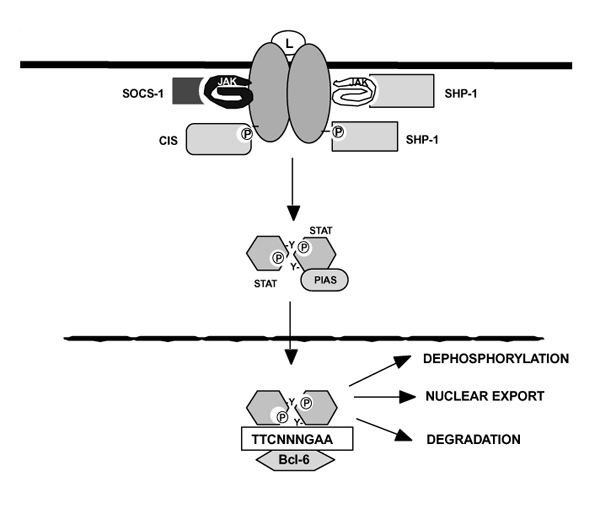
Attenuation of cytokine signaling. Suppressor of cytokine signaling (SOCS) proteins are induced in response to cytokines and suppress signal transduction in two ways. Some SOCS proteins bind directly to janus kinases (Jaks) and inhibit their catalytic activity, whereas others like cytokine inducible src homology-2 protein (CIS) can bind to activated receptors and prevent docking by signaling intermediates such as the Stats. SHP-1 can either dephosphorylate Jaks or activated receptor subunits, depending upon the pathway activated. Protein inhibitors of activated Stats (PIAS) family members inactivate Stat dimers by an as yet unknown mechanism. Stat dimers are also probably downregulated by degradation and dephosphorylated by unknown mechanisms. The accumulation of STATs in the nucleus could be regulated at the level of nuclear import, nuclear export, or a combination of the two; the mechanisms that control these processes are not well characterized. Finally, molecules like Bcl-6 can bind to consensus Stat binding sites and function as repressors.
Phosphatases
It is well recognized that cytokine-induced phosphorylation of various substrates, including the Jaks, cytokine receptors, and the Stats, is transient. The tyrosine phosphatase SHP-1 has been hypothesized as one regulator that can interact with cytokine receptors and downregulate their function [208,209,210]. Interestingly, motheaten and viable motheaten mice have a mutation in the gene that encodes SHP-1 and exhibit many characteristics of systemic autoimmunity [211,212,213]. Motheaten mice are characterized by increased levels of plasma cells in secondary lymphoid organs with abnormal levels of immunoglobulins and elevated serum anti-double-stranded DNA antibodies, a peripheral neutrophilia and monocytosis, decreased erythropoiesis, neutrophilic skin lesions, and a severe pneumonitis that is induced by activated macrophages. These mice rarely live beyond 8 weeks [214]. This striking presentation of autoimmune phenomena suggests that some manifestations of human disease might also be a result of aberrant downregulation of cytokine signaling. The regulation of cytokine signaling via SHP-1 is also important in bone remodeling, because motheaten mice have recently been shown to develop osteopenia due to a lack of SHP-1 mediated control of bone resorption [215].
Whether SHP-1 or another nuclear tyrosine phosphatase is responsible for Stat dephosphorylation is not clear. Indeed, one might speculate that a nuclear phosphatase would be required. Because some Stats are also serine phosphorylated, it is also reasonable to expect that a serine phosphatase might also regulate Stat function. Alternatively, it has been suggested that degradation of Stats via ubiquitination is also a means of terminating Stat induced signaling [216].
Suppressor of cytokine signaling/Jak binding/Stat-induced Stat inhibitor/cytokine inducible SH2 protein family of inhibitors
A recently described family of SH2-containing molecules [alternatively named Jak binding, SOCS, Stat-induced Stat inhibitor and cytokine inducible SH2 protein (CIS)] comprises several molecules that are induced rapidly upon cytokine stimulation and serve as classic feedback inhibitors of signaling [11,217,218,219,220]. There are at least eight members of this family characterized by a central SH2 domain and a carboxy-terminal region of homology, termed the 'SOCS box' (Fig. 1); for the purpose of this paper, this family will be referred to as the 'SOCS' family. The exact function of this region remains unclear at present, but a recent study [221] has shown that the SOCS box mediates interactions with elongins B and C. In this manner, they may couple SOCS proteins and their activated substrates to the proteasomal protein degradation pathway. The proteins SOCS-1, SOCS-3, and CIS-1 have been the most carefully studied in terms of regulating cytokine signaling, and this is discussed further.
The first member of this family, CIS, was discovered in 1995 and was shown to associate with the IL-3 and ery-thropoietin receptors [217]. Subsequently, three different groups identified the second family member (named SOCS-1, Jak binding, or Stat-induced Stat inhibitor-1) based on its ability to interact with Jaks and/or inhibit Jak mediated Stat activation [218,219]. Via their SH2 domains, some SOCS members bind the phosphorylated activation loop tyrosine residue in Jaks, thereby inhibiting Jak activity [219,222].
The importance of this downregulation is highlighted in SOCS-1 knockout mice, which have marked growth retardation, display increased lymphocyte apoptosis, and perish within 3 weeks of birth [223,224]. Two very recent studies of these animals have shown that this lethality is due almost entirely to systemic hyper-responsiveness to IFN-γ and that aberrant T lymphocytes may be the source of the IFN-γ [225,226]. Although these mice have enhanced IFN-γ dependent ability to kill Leishmania major parasites, they exhibit exaggerated and lethal responses to viral infections. Deficiency of SOCS-1 also results in impaired lymphopoiesis, as thymi from these animals undergo a loss of cellularity and a switch from predominantly CD4+CD8+ to single positive cells. Additionally, peripheral T cells express activation markers and respond to IL-2 in the absence of T-cell receptor cross-linking. The relative specificity of SOCS-1 for IFN-γ signaling in this model is underscored by experiments that demonstrate that all pathology can be prevented by administering anti-IFN-γ antibodies or by crossing the mice with IFN-γ knockout mice. Clearly, the main role of SOCS-1, therefore, is to prevent uncontrolled and lethal IFN-γ signaling. It is interesting to speculate that SOCS-1 mutations or polymorphisms could potentially underlie immunologic diseases in humans.
SOCS-3 is another family member that has been shown in vitro to interact with the Jaks to regulate Stat activation [222,227,228]. However, a recent study in SOCS-3 deficient mice reveals the critical role for this protein in down-regulating fetal, but not adult, hematopoiesis [229]. This suggests a more specific role for SOCS-3 in Jak2 regulation, because Jak2 knockout mice have hematopoietic disorders as well [66]. CIS-1, another member of this family, may downregulate cytokine signaling by binding directly to receptors, rather than Jaks. In this regard, CIS-1 has been shown to interact with the IL-2 receptor β chain and inhibit IL-2 dependent signaling [230]. Recently, CIS-1 transgenic mice have been created, and their phenotype is remarkably similar to those of Stat5a and Stat5b knockout mice, indicating the critical role of CIS-1 as a negative regulator of Stat5 function [231]. It is becoming more evident with time that in vivo models are necessary to dissect the specificity of SOCS interactions. Also, we are learning that SOCS members may play a role in noncytokine signaling, including the leptin, growth hormone, and prolactin signaling pathways [232,233,234], but findings in SOCS knockout mice do not necessarily support a critical function.
Protein inhibitors of activated Stats
Recently, a family of proteins that interact with Stats, termed protein inhibitors of activated Stats (PIAS), have been identified [235,236]. PIAS1 and PIAS3 bind to Stat1 and Stat3, respectively. They inhibit transcriptional activity of the Stats, but do not affect phosphorylation. Just how specific they are in terms of regulating cytokine signaling has not been determined; no knockouts have yet been reported. In addition, these molecules were cloned by yeast two-hybrid screens using baits other than Stat molecules, and therefore may affect proteins other than Stats. Thus, it will be important to characterize the physiologic function of this family of molecules.
Bcl-6
Bcl-6, a zinc-finger protein expressed in B cells and CD4+ T cells and frequently associated with non-Hodgkin's lymphoma, has also been recently shown to regulate Stat function negatively. Bcl-6 deficient mice develop a severe systemic inflammatory disease typical of a Th2-mediated hyperimmune response, which is characterized by infiltrates of immunoglobulin E-bearing B cells and eosinophils [237]. Because the Bcl-6 DNA recognition motif resembles sites bound by Stat6, it was surprising to find that when Bcl-6 mice were bred to either Stat6 or IL-4 deficient mice, the animals still manifested the same hyperinflammatory process. In vitro, however, Stat6 was required for the differentiation of Bcl-6 deficient T cells into Th2 cells. These findings indicate that this transcriptional repressor can regulate Th2 responses by pathways both dependent on and independent of IL-4 and Stat6 [238].
Conclusion
The Jaks are a small family of tyrosine kinases with very specific functions; these are best illustrated by humans with mutations and gene-targeted mice. The present data indicate that they have critical functions in transmitting cytokine-dependent signals. The Stats, too, appear to be a small, but conserved family of transcription factors that serve to further transmit signals initiated by receptor-Jak interactions, also with highly specific functions. Indeed, at least four of the six Stats have major functions in regulating host defense and immune responses. Although we have learned a great deal about the cytokine signaling pathways, relatively few cytokine-inducible genes have been identified. This, of course, will rapidly change with the advent of microarray and gene chip technologies. The challenge will remain to dissect how Stats interact with the growing list of other transcription factors to regulate the expression of these genes and how signals emanating from cytokine receptors affect transcriptional activation. Another important issue that needs to be resolved is to what extent Jaks and Stats function as essential intermediates for noncytokine receptors. Equally exciting as the discovery of the Jaks and Stats is the discovery of families of molecules that serve to attenuate cytokine signalling; it is exciting to consider that these molecules might be mutated or polymorphic in human autoimmune diseases.
A knowledge of these signaling pathways is of particular importance to rheumatologists, because cytokines clearly regulate the inflammatory and immune responses. One significant lesson gleaned from these investigations is that cytokines may act as a double-edged sword: That is, although cytokines play important physiologic roles in promoting immune development and fighting off infections, maladapted cytokine responses can lead to autoimmunity. Perhaps a clearer understanding of how members of the Jak and Stat families, as well as the more recently discovered SOCS family members, interact and function may allow us to target specific pathways, such as those involving proinflammatory cytokines including IFN-γ or IL-6, for therapeutic intervention. Knowledge in cytokine signaling pathways has grown exponentially over the past decade and, conceivably, manipulation of these pathways through pharmaceutical intervention may provide the rheumatologist with a unique way to treat autoimmune diseases.
References
- Darnell JEJ, Kerr IM, Stark GR. Jak-STAT pathways and transcriptional activation in response to IFNs and other extracellular signaling proteins. Science. 1994;264:1415–1421. doi: 10.1126/science.8197455. [DOI] [PubMed] [Google Scholar]
- Pellegrini S, Dusanter-Fourt I. The structure, regulation and function of the Janus kinases (JAKs) and the signal transducers and activators of transcription (STATs). Eur J Biochem. 1997;248:615–633. doi: 10.1111/j.1432-1033.1997.00615.x. [DOI] [PubMed] [Google Scholar]
- Ihle JN, Thierfelder W, Teglund S, et al. Signaling by the cytokine receptor superfamily. Ann N Y Acad Sci. 1998;865:1–9. doi: 10.1111/j.1749-6632.1998.tb11157.x. [DOI] [PubMed] [Google Scholar]
- O'Shea JJ. Jaks, STATs, cytokine signal transduction, and immunoregulation: are we there yet? Immunity. 1997;7:1–11. doi: 10.1016/s1074-7613(00)80505-1. [DOI] [PubMed] [Google Scholar]
- Bach EA, Aguet M, Schreiber RD. The IFN-gamma receptor: a paradigm for cytokine receptor signaling. Annu Rev Immunol. 1997;15:563–593. doi: 10.1146/annurev.immunol.15.1.563. [DOI] [PubMed] [Google Scholar]
- Leonard WJ, O'Shea JJ. Jaks and STATs: biological implications. Annu Rev Immunol. 1998;16:293–322. doi: 10.1146/annurev.immunol.16.1.293. [DOI] [PubMed] [Google Scholar]
- Carter-Su C, Smit LS. Signaling via JAK tyrosine kinases: growth hormone receptor as a model system. Recent Prog Horm Res. 1998;53:61–82; discussion 82-83. [PubMed] [Google Scholar]
- Aringer M, Cheng A, Nelson JW, et al. Janus kinases and their role in growth and disease. Life Sci. 1999;64:2173–2186. doi: 10.1016/s0024-3205(98)00538-4. [DOI] [PubMed] [Google Scholar]
- Hoey T, Grusby MJ. STATs as mediators of cytokine-induced responses. Adv Immunol. 1999;71:145–162. doi: 10.1016/s0065-2776(08)60401-0. [DOI] [PubMed] [Google Scholar]
- Starr R, Hilton DJ. Negative regulation of the JAK/STAT pathway. Bioessays. 1999;21:47–52. doi: 10.1002/(SICI)1521-1878(199901)21:1<47::AID-BIES6>3.3.CO;2-E. [DOI] [PubMed] [Google Scholar]
- Hilton DJ. Negative regulators of cytokine signal transduction. . Cell Mol Life Sci. 1999;55:1568–1577. doi: 10.1007/s000180050396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baird AM, Gerstein RM, Berg LJ. The role of cytokine receptor signaling in lymphocyte development. Curr Opin Immunol. 1999;11:157–166. doi: 10.1016/s0952-7915(99)80027-2. [DOI] [PubMed] [Google Scholar]
- Krolewski JJ, Lee R, Eddy R, Shows TB, Dalla-Favera R. Identification and chromosomal mapping of new human tyrosine kinase genes. Oncogene. 1990;5:277–282. [PubMed] [Google Scholar]
- Wilks AF, Harpur AG, Kurban RR, et al. Two novel protein-tyrosine kinases, each with a second phosphotransferase-related catalytic domain, define a new class of protein kinase. Mol Cell Biol. 1991;11:2057–2065. doi: 10.1128/mcb.11.4.2057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harpur AG, Andres AC, Ziemiecki A, Aston RR, Wilks AF. JAK2, a third member of the JAK family of protein tyrosine kinases. . Oncogene. 1992;7:1347–1353. [PubMed] [Google Scholar]
- Kawamura M, McVicar DW, Johnston JA, et al. Molecular cloning of L-JAK, a Janus family protein-tyrosine kinase expressed in natural killer cells and activated leukocytes. Proc Natl Acad Sci USA. 1994;91:6374–6378. doi: 10.1073/pnas.91.14.6374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sofer L, Kampa D, Burnside J. Molecular cloning of a chicken JAK homolog from activated T cells. Gene. 1998;215:29–36. doi: 10.1016/S0378-1119(98)00284-4. [DOI] [PubMed] [Google Scholar]
- Harrison DA, Binari R, Nahreini TS, Gilman M, Perrimon N. Activation of a Drosophila Janus kinase (JAK) causes hematopoietic neoplasia and developmental defects. . EMBO J. 1995;14:2857–2865. doi: 10.1002/j.1460-2075.1995.tb07285.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Binari R, Perrimon N. Stripe-specific regulation of pair-rule genes by hopscotch, a putative Jak family tyrosine kinase in Drosophila. . Genes Dev. 1994;8:300–312. doi: 10.1101/gad.8.3.300. [DOI] [PubMed] [Google Scholar]
- Ihle JN, Witthuhn BA, Quelle FW, Yamamoto K, Silvennoinen O. Signaling through the hematopoietic cytokine receptors. . Annu Rev Immunol. 1995;13:369–398. doi: 10.1146/annurev.iy.13.040195.002101. [DOI] [PubMed] [Google Scholar]
- Velazquez L, Fellous M, Stark GR, Pellegrini S. A protein tyrosine kinase in the interferon alpha/beta signaling pathway. . Cell. 1992;70:313–322. doi: 10.1016/0092-8674(92)90105-l. [DOI] [PubMed] [Google Scholar]
- Watling D, Guschin D, Muller M, et al. Complementation by the protein tyrosine kinase JAK2 of a mutant cell line defective in the interferon-gamma signal transduction pathway. Nature. 1993;366:166–170. doi: 10.1038/366166a0. [DOI] [PubMed] [Google Scholar]
- Silvennoinen O, Ihle JN, Schlessinger J, Levy DE. Interferon-induced nuclear signalling by Jak protein tyrosine kinases. Nature. 1993;366:583–585. doi: 10.1038/366583a0. [DOI] [PubMed] [Google Scholar]
- Witthuhn BA, Quelle FW, Silvennoinen O, et al. JAK2 associates with the erythropoietin receptor and is tyrosine phosphorylated and activated following stimulation with erythropoietin. Cell. 1993;74:227–236. doi: 10.1016/0092-8674(93)90414-l. [DOI] [PubMed] [Google Scholar]
- Argetsinger LS, Campbell GS, Yang X, et al. Identification of JAK2 as a growth hormone receptor-associated tyrosine kinase. Cell. 1993;74:237–244. doi: 10.1016/0092-8674(93)90415-m. [DOI] [PubMed] [Google Scholar]
- Witthuhn BA, Silvennoinen O, Miura O, et al. Involvement of the Jak-3 Janus kinase in signalling by interleukins 2 and 4 in lymphoid and myeloid cells. Nature. 1994;370:153–157. doi: 10.1038/370153a0. [DOI] [PubMed] [Google Scholar]
- Johnston JA, Kawamura M, Kirken RA, et al. Phosphorylation and activation of the Jak-3 Janus kinase in response to interleukin-2. Nature. 1994;370:151–153. doi: 10.1038/370151a0. [DOI] [PubMed] [Google Scholar]
- Ihle JN. The Janus protein tyrosine kinase family and its role in cytokine signaling. Adv Immunol. 1995;60:1–35. doi: 10.1016/s0065-2776(08)60582-9. [DOI] [PubMed] [Google Scholar]
- Gurniak CB, Berg LJ. Murine JAK3 is preferentially expressed in hematopoietic tissues and lymphocyte precursor cells. Blood. 1996;87:3151–3160. [PubMed] [Google Scholar]
- Sharfe N, Dadi HK, Shahar M, Roifman CM. Human immune disorder arising from mutation of the alpha chain of the interleukin-2 receptor. . Proc Natl Acad Sci USA. 1997;94:3168–3171. doi: 10.1073/pnas.94.7.3168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tortolani PJ, Lal BK, Riva A, et al. Regulation of JAK3 expression and activation in human B cells and B cell malignancies. . J Immunol. 1995;155:5220–5226. [PubMed] [Google Scholar]
- Musso T, Johnston JA, Linnekin D, et al. Regulation of JAK3 expression in human monocytes: phosphorylation in response to interleukins 2, 4, and 7. J Exp Med. 1995;181:1425–1431. doi: 10.1084/jem.181.4.1425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Russell SM, Johnston JA, Noguchi M, et al. Interaction of IL-2R beta and gamma c chains with Jak1 and Jak3: implications for XSCID and XCID. Science. 1994;266:1042–1045. doi: 10.1126/science.7973658. [DOI] [PubMed] [Google Scholar]
- Miyazaki T, Kawahara A, Fujii H, et al. Functional activation of Jak1 and Jak3 by selective association with IL-2 receptor subunits. Science. 1994;266:1045–1047. doi: 10.1126/science.7973659. [DOI] [PubMed] [Google Scholar]
- Boussiotis VA, Barber DL, Nakarai T, et al. Prevention of T cell anergy by signaling through the gamma c chain of the IL-2 receptor. Science. 1994;266:1039–1042. doi: 10.1126/science.7973657. [DOI] [PubMed] [Google Scholar]
- Oakes SA, Candotti F, Johnston JA, et al. Signaling via IL-2 and IL-4 in JAK3-deficient severe combined immunodeficiency lymphocytes: JAK3-dependent and independent pathways. Immunity. 1996;5:605–615. doi: 10.1016/s1074-7613(00)80274-5. [DOI] [PubMed] [Google Scholar]
- Candotti F, Oakes SA, Johnston JA, et al. In vitro correction of JAK3-deficient severe combined immunodeficiency by retroviral-mediated gene transduction. J Exp Med. 1996;183:2687–2692. doi: 10.1084/jem.183.6.2687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Macchi P, Villa A, Gillani S, et al. Mutations of Jak-3 gene in patients with autosomal severe combined immune deficiency (SCID). Nature. 1995;377:65–68. doi: 10.1038/377065a0. [DOI] [PubMed] [Google Scholar]
- Russell SM, Tayebi N, Nakajima H, et al. Mutation of Jak3 in a patient with SCID: essential role of Jak3 in lymphoid development. Science. 1995;270:797–800. doi: 10.1126/science.270.5237.797. [DOI] [PubMed] [Google Scholar]
- Noguchi M, Yi H, Rosenblatt HM, et al. Interleukin-2 receptor gamma chain mutation results in X-linked severe combined immunodeficiency in humans. Cell. 1993;73:147–157. doi: 10.1016/0092-8674(93)90167-o. [DOI] [PubMed] [Google Scholar]
- Nosaka T, van Deursen JM, Tripp RA, et al. Defective lymphoid development in mice lacking Jak3. Science. 1995;270:800–802. doi: 10.1126/science.270.5237.800. [DOI] [PubMed] [Google Scholar]
- Thomis DC, Gurniak CB, Tivol E, Sharpe AH, Berg LJ. Defects in B lymphocyte maturation and T lymphocyte activation in mice lacking Jak3. Science. 1995;270:794–797. doi: 10.1126/science.270.5237.794. [DOI] [PubMed] [Google Scholar]
- Park SY, Saijo K, Takahashi T, et al. Developmental defects of lymphoid cells in Jak3 kinase-deficient mice. Immunity . 1995;3:771–782. doi: 10.1016/1074-7613(95)90066-7. [DOI] [PubMed] [Google Scholar]
- Thomis DC, Berg LJ. The role of Jak3 in lymphoid development, activation, and signaling. Curr Opin Immunol. 1997;9:541–547. doi: 10.1016/s0952-7915(97)80108-2. [DOI] [PubMed] [Google Scholar]
- Maeurer MJ, Lotze MT. Interleukin-7 (IL-7) knockout mice. Implications for lymphopoiesis and organ-specific immunity. Int Rev Immunol. 1998;16:309–322. doi: 10.3109/08830189809042999. [DOI] [PubMed] [Google Scholar]
- Maraskovsky E, Teepe M, Morrissey PJ, et al. Impaired survival and proliferation in IL-7 receptor-deficient peripheral T cells. . J Immunol. 1996;157:5315–5323. [PubMed] [Google Scholar]
- von Freeden-Jeffry U, Vieira P, Lucian LA, et al. Lymphopenia in interleukin (IL)-7 gene-deleted mice identifies IL-7 as a nonredundant cytokine. J Exp Med. 1995;181:1519–1526. doi: 10.1084/jem.181.4.1519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Puel A, Ziegler SF, Buckley RH, Leonard WJ. Defective IL7R expression in T(-)B(+)NK(+) severe combined immunodeficiency. . Nature Genet. 1998;20:394–397. doi: 10.1038/3877. [DOI] [PubMed] [Google Scholar]
- Lodolce JP, Boone DL, Chai S, et al. IL-15 receptor maintains lymphoid homeostasis by supporting lymphocyte homing and proliferation. Immunity. 1998;9:669–676. doi: 10.1016/s1074-7613(00)80664-0. [DOI] [PubMed] [Google Scholar]
- Candotti F, Oakes SA, Johnston JA, et al. Structural and functional basis for JAK3-deficient severe combined immunodeficiency. Blood. 1997;90:3996–4003. [PubMed] [Google Scholar]
- Suzuki H, Kundig TM, Furlonger C, et al. Deregulated T cell activation and autoimmunity in mice lacking interleukin-2 receptor beta. Science. 1995;268:1472–1476. doi: 10.1126/science.7770771. [DOI] [PubMed] [Google Scholar]
- Willerford DM, Chen J, Ferry JA, et al. Interleukin-2 receptor alpha chain regulates the size and content of the peripheral lymphoid compartment. Immunity. 1995;3:521–530. doi: 10.1016/1074-7613(95)90180-9. [DOI] [PubMed] [Google Scholar]
- Fujii H, Ogasawara K, Otsuka H, et al. Functional dissection of the cytoplasmic subregions of the IL-2 receptor betac chain in primary lymphocyte populations. EMBO J. 1998;17:6551–6557. doi: 10.1093/emboj/17.22.6551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saijo K, Park SY, Ishida Y, Arase H, Saito T. Crucial role of Jak3 in negative selection of self-reactive T cells. J Exp Med . 1997;185:351–356. doi: 10.1084/jem.185.2.351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brugnoni D, Notarangelo LD, Sottini A, et al. Development of autologous, oligoclonal, poorly functioning T lymphocytes in a patient with autosomal recessive severe combined immunodeficiency caused by defects of the Jak3 tyrosine kinase. . Blood. 1998;91:949–955. [PubMed] [Google Scholar]
- Villa A, Sironi M, Macchi P, et al. Monocyte function in a severe combined immunodeficient patient with a donor splice site mutation in the Jak3 gene. Blood. 1996;88:817–823. [PubMed] [Google Scholar]
- Colamonici O, Yan H, Domanski P, et al. Direct binding to and tyrosine phosphorylation of the alpha subunit of the type I interferon receptor by p135tyk2 tyrosine kinase. Mol Cell Biol . 1994;14:8133–8142. doi: 10.1128/mcb.14.12.8133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Novick D, Cohen B, Rubinstein M. The human interferon alpha/beta receptor: characterization and molecular cloning. Cell. 1994;77:391–400. doi: 10.1016/0092-8674(94)90154-6. [DOI] [PubMed] [Google Scholar]
- Abramovich C, Shulman LM, Ratovitski E, et al. Differential tyrosine phosphorylation of the IFNAR chain of the type I interferon receptor and of an associated surface protein in response to IFN-alpha and IFN-beta. EMBO J. 1994;13:5871–5877. doi: 10.1002/j.1460-2075.1994.tb06932.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muller M, Briscoe J, Laxton C, et al. The protein tyrosine kinase JAK1 complements defects in interferon-alpha/beta and -gamma signal transduction. Nature. 1993;366:129–135. doi: 10.1038/366129a0. [DOI] [PubMed] [Google Scholar]
- Quelle FW, Sato N, Witthuhn BA, et al. JAK2 associates with the beta c chain of the receptor for granulocyte-macrophage colony-stimulating factor, and its activation requires the membrane-proximal region. Mol Cell Biol. 1994;14:4335–4341. doi: 10.1128/mcb.14.7.4335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stahl N, Boulton TG, Farruggella T, et al. Association and activation of Jak-Tyk kinases by CNTF-LIF-OSM-IL-6 beta receptor components. Science. 1994;263:92–95. doi: 10.1126/science.8272873. [DOI] [PubMed] [Google Scholar]
- Zou J, Presky DH, Wu CY, Gubler U. Differential associations between the cytoplasmic regions of the interleukin-12 receptor subunits beta1 and beta2 and JAK kinases. J Biol Chem. 1997;272:6073–6077. doi: 10.1074/jbc.272.9.6073. [DOI] [PubMed] [Google Scholar]
- Rodig SJ, Meraz MA, White JM, et al. Disruption of the Jak1 gene demonstrates obligatory and nonredundant roles of the Jaks in cytokine-induced biologic responses. Cell. 1998;93:373–383. doi: 10.1016/s0092-8674(00)81166-6. [DOI] [PubMed] [Google Scholar]
- Neubauer H, Cumano A, Muller M, et al. Jak2 deficiency defines an essential developmental checkpoint in definitive hematopoiesis. Cell. 1998;93:397–409. doi: 10.1016/s0092-8674(00)81168-x. [DOI] [PubMed] [Google Scholar]
- Parganas E, Wang D, Stravopodis D, et al. Jak2 is essential for signaling through a variety of cytokine receptors. . Cell. 1998;93:385–395. doi: 10.1016/s0092-8674(00)81167-8. [DOI] [PubMed] [Google Scholar]
- Lacronique V, Boureux A, Valle VD, et al. A TEL-JAK2 fusion protein with constitutive kinase activity in human leukemia. . Science. 1997;278:1309–1312. doi: 10.1126/science.278.5341.1309. [DOI] [PubMed] [Google Scholar]
- Peeters P, Raynaud SD, Cools J, et al. Fusion of TEL, the ETS-variant gene 6 (ETV6), to the receptor-associated kinase JAK2 as a result of t(9;12) in a lymphoid and t(9;15;12) in a myeloid leukemia. . Blood. 1997;90:2535–2540. [PubMed] [Google Scholar]
- Hanissian SH, Geha RS. Jak3 is associated with CD40 and is critical for CD40 induction of gene expression in B cells. . Immunity. 1997;6:379–387. doi: 10.1016/s1074-7613(00)80281-2. [DOI] [PubMed] [Google Scholar]
- Jabara HH, Buckley RH, Roberts JL, et al. Role of JAK3 in CD40-mediated signaling. Blood. 1998;92:2435–2440. [PubMed] [Google Scholar]
- Rane SG, Reddy EP. JAK3: a novel JAK kinase associated with terminal differentiation of hematopoietic cells. Oncogene. 1994;9:2415–2423. [PubMed] [Google Scholar]
- Lai KS, Jin Y, Graham DK, et al. A kinase-deficient splice variant of the human JAK3 is expressed in hematopoietic and epithelial cancer cells. J Biol Chem. 1995;270:25028–25036. doi: 10.1074/jbc.270.42.25028. [DOI] [PubMed] [Google Scholar]
- Hubbard SR, Wei L, Ellis L, Hendrickson WA. Crystal structure of the tyrosine kinase domain of the human insulin receptor. Nature. 1994;372:746–754. doi: 10.1038/372746a0. [DOI] [PubMed] [Google Scholar]
- Gauzzi MC, Velazquez L, McKendry R, et al. Interferon-alpha-dependent activation of Tyk2 requires phosphorylation of positive regulatory tyrosines by another kinase. . J Biol Chem. 1996;271:20494–20500. doi: 10.1074/jbc.271.34.20494. [DOI] [PubMed] [Google Scholar]
- Feng J, Witthuhn BA, Matsuda T, et al. Activation of Jak2 catalytic activity requires phosphorylation of Y1007 in the kinase activation loop. Mol Cell Biol. 1997;17:2497–2501. doi: 10.1128/mcb.17.5.2497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou YJ, Hanson EP, Chen YQ, et al. Distinct tyrosine phosphorylation sites in JAK3 kinase domain positively and negatively regulate its enzymatic activity. Proc Natl Acad Sci USA. 1997;94:13850–13855. doi: 10.1073/pnas.94.25.13850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rui L, Mathews LS, Hotta K, Gustafson TA, Carter-Su C. Identification of SH2-Bbeta as a substrate of the tyrosine kinase JAK2 involved in growth hormone signaling. Mol Cell Biol. 1997;17:6633–6644. doi: 10.1128/mcb.17.11.6633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rui L, Carter-Su C. Identification of SH2-bbeta as a potent cytoplasmic activator of the tyrosine kinase Janus kinase 2. Proc Natl Acad Sci USA. 1999;96:7172–7177. doi: 10.1073/pnas.96.13.7172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frank SJ, Gilliland G, Kraft AS, Arnold CS. Interaction of the growth hormone receptor cytoplasmic domain with the JAK2 tyrosine kinase. . Endocrinology. 1994;135:2228–2239. doi: 10.1210/endo.135.5.7956946. [DOI] [PubMed] [Google Scholar]
- Luo H, Rose P, Barber D, et al. Mutation in the Jak kinase JH2 domain hyperactivates Drosophila and mammalian Jak-Stat pathways. . Mol Cell Biol. 1997;17:1562–1571. doi: 10.1128/mcb.17.3.1562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fujitani Y, Hibi M, Fukada T, et al. An alternative pathway for STAT activation that is mediated by the direct interaction between JAK and STAT. Oncogene. 1997;14:751–761. doi: 10.1038/sj/onc/1200907. [DOI] [PubMed] [Google Scholar]
- Frank SJ, Yi W, Zhao Y, et al. Regions of the JAK2 tyrosine kinase required for coupling to the growth hormone receptor. . J Biol Chem. 1995;270:14776–14785. doi: 10.1074/jbc.270.24.14776. [DOI] [PubMed] [Google Scholar]
- Zhao Y, Wagner F, Frank SJ, Kraft AS. The amino-terminal portion of the JAK2 protein kinase is necessary for binding and phosphorylation of the granulocyte-macrophage colony-stimulating factor receptor beta c chain. . J Biol Chem. 1995;270:13814–13818. doi: 10.1074/jbc.270.23.13814. [DOI] [PubMed] [Google Scholar]
- Kohlhuber F, Rogers NC, Watling D, et al. A JAK1/JAK2 chimera can sustain alpha and gamma interferon responses. Mol Cell Biol. 1997;17:695–706. doi: 10.1128/mcb.17.2.695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen M, Cheng A, Chen YQ, et al. The amino terminus of JAK3 is necessary and sufficient for binding to the common gamma chain and confers the ability to transmit interleukin 2-mediated signals. Proc Natl Acad Sci USA. 1997;94:6910–6915. doi: 10.1073/pnas.94.13.6910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cacalano NA, Migone TS, Bazan F, et al. Autosomal SCID caused by a point mutation in the N-terminus of Jak3: mapping of the Jak3-receptor interaction domain. EMBO J. 1999;18:1549–1558. doi: 10.1093/emboj/18.6.1549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan H, Piazza F, Krishnan K, Pine R, Krolewski JJ. Definition of the interferon-alpha receptor-binding domain on the TYK2 kinase. JBiol Chem. 1998;273:4046–4051. doi: 10.1074/jbc.273.7.4046. [DOI] [PubMed] [Google Scholar]
- Richter MF, Dumenil G, Uze G, Fellous M, Pellegrini S. Specific contribution of tyk2 JH regions to the binding and the expression of the interferon alpha/beta receptor component IFNAR1. J Biol Chem. 1998;273:24723–24729. doi: 10.1074/jbc.273.38.24723. [DOI] [PubMed] [Google Scholar]
- Takeshita T, Arita T, Higuchi M, et al. STAM, signal transducing adaptor molecule, is associated with Janus kinases and involved in signaling for cell growth and c-myc induction. Immunity. 1997;6:449–457. doi: 10.1016/s1074-7613(00)80288-5. [DOI] [PubMed] [Google Scholar]
- Asao H, Sasaki Y, Arita T, et al. Hrs is associated with STAM, a signal-transducing adaptor molecule. Its suppressive effect on cytokine-induced cell growth. J Biol Chem. 1997;272:32785–32791. doi: 10.1074/jbc.272.52.32785. [DOI] [PubMed] [Google Scholar]
- Tanaka N, Kaneko K, Asao H, et al. Possible involvement of a novel STAM-associated molecule "AMSH" in intracellular signal transduction mediated by cytokines. J Biol Chem. 1999;274:19129–19135. doi: 10.1074/jbc.274.27.19129. [DOI] [PubMed] [Google Scholar]
- Livnah O, Stura EA, Middleton SA, et al. Crystallographic evidence for preformed dimers of erythropoietin receptor before ligand activation. Science. 1999;283:987–990. doi: 10.1126/science.283.5404.987. [DOI] [PubMed] [Google Scholar]
- Remy I, Wilson IA, Michnick SW. Erythropoietin receptor activation by a ligand-induced conformation change. Science. 1999;283:990–993. doi: 10.1126/science.283.5404.990. [DOI] [PubMed] [Google Scholar]
- Tibbles LA, Woodgett JR. The stress-activated protein kinase pathways. Cell Mol Life Sci. 1999;55:1230–1254. doi: 10.1007/s000180050369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campbell KS. Signal transduction from the B cell antigen-receptor. Curr Opin Immunol. 1999;11:256–264. doi: 10.1016/s0952-7915(99)80042-9. [DOI] [PubMed] [Google Scholar]
- Nelms K, Keegan AD, Zamorano J, Ryan JJ, Paul WE. The IL-4 receptor: signaling mechanisms and biologic functions. Annu Rev Immunol. 1999;17:701–738. doi: 10.1146/annurev.immunol.17.1.701. [DOI] [PubMed] [Google Scholar]
- Kurosaki T. Genetic analysis of B cell antigen receptor signaling. Annu Rev Immunol. 1999;17:555–592. doi: 10.1146/annurev.immunol.17.1.555. [DOI] [PubMed] [Google Scholar]
- Hardy K, Chaudhri G. Activation and signal transduction via mitogen-activated protein (MAP) kinases in T lymphocytes. Immunol Cell Biol. 1997;75:528–545. doi: 10.1038/icb.1997.84. [DOI] [PubMed] [Google Scholar]
- Su B, Karin M. Mitogen-activated protein kinase cascades and regulation of gene expression. Curr Opin Immunol. 1996;8:402–411. doi: 10.1016/s0952-7915(96)80131-2. [DOI] [PubMed] [Google Scholar]
- Fu XY, Schindler C, Improta T, Aebersold R, Darnell JEJ. The proteins of ISGF-3, the interferon alpha-induced transcriptional activator, define a gene family involved in signal transduction. Proc Natl Acad Sci USA. 1992;89:7840–7843. doi: 10.1073/pnas.89.16.7840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shuai K, Schindler C, Prezioso VR, Darnell JEJ. Activation of transcription by IFN-gamma: tyrosine phosphorylation of a 91-kD DNA binding protein. Science. 1992;258:1808–1812. doi: 10.1126/science.1281555. [DOI] [PubMed] [Google Scholar]
- Zhong Z, Wen Z, Darnell JEJ. Stat3 and Stat4: members of the family of signal transducers and activators of transcription. Proc Natl Acad Sci USA. 1004;91:4806–4810. doi: 10.1073/pnas.91.11.4806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Akira S, Nishio Y, Inoue M, et al. Molecular cloning of APRF, a novel IFN-stimulated gene factor 3 p91-related transcription factor involved in the gp130-mediated signaling pathway. Cell . 1994;77:63–71. doi: 10.1016/0092-8674(94)90235-6. [DOI] [PubMed] [Google Scholar]
- Yamamoto K, Quelle FW, Thierfelder WE, et al. Stat4, a novel gamma interferon activation site-binding protein expressed in early myeloid differentiation. Mol Cell Biol. 1994;14:4342–4349. doi: 10.1128/mcb.14.7.4342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quelle FW, Shimoda K, Thierfelder W, et al. Cloning of murine Stat6 and human Stat6, Stat proteins that are tyrosine phosphorylated in responses to IL-4 and IL-3 but are not required for mitogenesis. Mol Cell Biol. 1995;15:3336–3343. doi: 10.1128/mcb.15.6.3336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hou J, Schindler U, Henzel WJ, et al. An interleukin-4-induced transcription factor: IL-4 Stat. Science . 1994;265:1701–1706. doi: 10.1126/science.8085155. [DOI] [PubMed] [Google Scholar]
- Wakao H, Gouilleux F, Groner B. Mammary gland factor (MGF) is a novel member of the cytokine regulated transcription factor gene family and confers the prolactin response. EMBO J. 1994;13:2182–2191. doi: 10.1002/j.1460-2075.1994.tb06495.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hou J, Schindler U, Henzel WJ, Wong SC, McKnight SL. Identification and purification of human Stat proteins activated in response to interleukin-2. Immunity. 1995;2:321–329. doi: 10.1016/1074-7613(95)90140-x. [DOI] [PubMed] [Google Scholar]
- Liu X, Robinson GW, Gouilleux F, Groner B, Hennighausen L. Cloning and expression of Stat5 and an additional homologue (Stat5b) involved in prolactin signal transduction in mouse mammary tissue. . Proc Natl Acad Sci USA. 1995;92:8831–8835. doi: 10.1073/pnas.92.19.8831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Darnell JEJ. STATs and gene regulation. Science. 1997;277:1630–1635. doi: 10.1126/science.277.5332.1630. [DOI] [PubMed] [Google Scholar]
- Schindler C, Fu XY, Improta T, Aebersold R, Darnell JEJ. Proteins of transcription factor ISGF-3: one gene encodes the 91-and 84-kDa ISGF-3 proteins that are activated by interferon alpha. Proc Natl Acad Sci USA. 1992;89:7836–7839. doi: 10.1073/pnas.89.16.7836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shuai K, Stark GR, Kerr IM, Darnell JEJ. A single phosphotyrosine residue of Stat91 required for gene activation by interferon-gamma. Science. 1993;261:1744–1746. doi: 10.1126/science.7690989. [DOI] [PubMed] [Google Scholar]
- Greenlund AC, Morales MO, Viviano BL, et al. Stat recruitment by tyrosine-phosphorylated cytokine receptors: an ordered reversible affinity-driven process. Immunity. 1995;2:677–687. doi: 10.1016/1074-7613(95)90012-8. [DOI] [PubMed] [Google Scholar]
- Schindler U, Wu P, Rothe M, Brasseur M, McKnight SL. Components of a Stat recognition code: evidence for two layers of molecular selectivity. Immunity. 1995;2:689–697. doi: 10.1016/1074-7613(95)90013-6. [DOI] [PubMed] [Google Scholar]
- Becker S, Groner B, Muller CW. Three-dimensional structure of the Stat3beta homodimer bound to DNA. Nature. 1998;394:145–151. doi: 10.1038/28101. [DOI] [PubMed] [Google Scholar]
- Chen X, Vinkemeier U, Zhao Y, et al. Crystal structure of a tyrosine phosphorylated STAT-1 dimer bound to DNA. Cell. 1998;93:827–839. doi: 10.1016/s0092-8674(00)81443-9. [DOI] [PubMed] [Google Scholar]
- Vinkemeier U, Moarefi I, Darnell JEJ, Kuriyan J. Structure of the amino-terminal protein interaction domain of STAT-4. Science. 1998;279:1048–1052. doi: 10.1126/science.279.5353.1048. [DOI] [PubMed] [Google Scholar]
- Xu X, Sun YL, Hoey T. Cooperative DNA binding and sequence-selective recognition conferred by the STAT amino-terminal domain. . Science. 1996;273:794–797. doi: 10.1126/science.273.5276.794. [DOI] [PubMed] [Google Scholar]
- Mikita T, Campbell D, Wu P, Williamson K, Schindler U. Requirements for interleukin-4-induced gene expression and functional characterization of Stat6. Mol Cell Biol. 1996;16:5811–5820. doi: 10.1128/mcb.16.10.5811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- John S, Vinkemeier U, Soldaini E, Darnell JEJ, Leonard WJ. The significance of tetramerization in promoter recruitment by Stat5. . Mol Cell Biol. 1999;19:1910–1918. doi: 10.1128/mcb.19.3.1910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhattacharya S, Eckner R, Grossman S, et al. Cooperation of Stat2 and p300/CBP in signalling induced by interferon-alpha. Nature. 1996;383:344–347. doi: 10.1038/383344a0. [DOI] [PubMed] [Google Scholar]
- Zhang JJ, Vinkemeier U, Gu W, et al. Two contact regions between Stat1 and CBP/p300 in interferon gamma signaling. . Proc Natl Acad Sci USA. 1996;93:15092–15096. doi: 10.1073/pnas.93.26.15092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horvai AE, Xu L, Korzus E, et al. Nuclear integration of JAK/STAT and Ras/AP-1 signaling by CBP and p300. Proc Natl Acad Sci USA. 1997;94:1074–1079. doi: 10.1073/pnas.94.4.1074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Korzus E, Torchia J, Rose DW, et al. Transcription factor-specific requirements for coactivators and their acetyltransferase functions. Science. 1998;279:703–707. doi: 10.1126/science.279.5351.703. [DOI] [PubMed] [Google Scholar]
- Pfitzner E, Jahne R, Wissler M, Stoecklin E, Groner B. p300/CREB-binding protein enhances the prolactin-mediated transcriptional induction through direct interaction with the transactivation domain of Stat5, but does not participate in the Stat5-mediated suppression of the glucocorticoid response. Mol Endocrinol . 1998;12:1582–1593. doi: 10.1210/mend.12.10.0180. [DOI] [PubMed] [Google Scholar]
- Zhu M, John S, Berg M, Leonard WJ. Functional association of Nmi with Stat5 and Stat1 in IL-2- and IFNgamma-mediated signaling. . Cell. 1999;96:121–130. doi: 10.1016/s0092-8674(00)80965-4. [DOI] [PubMed] [Google Scholar]
- Stocklin E, Wissler M, Gouilleux F, Groner B. Functional interactions between Stat5 and the glucocorticoid receptor. Nature . 1996;383:726–728. doi: 10.1038/383726a0. [DOI] [PubMed] [Google Scholar]
- Moriggl R, Berchtold S, Friedrich K, et al. Comparison of the trans-activation domains of Stat5 and Stat6 in lymphoid cells and mammary epithelial cells. Mol Cell Biol. 1997;17:3663–3678. doi: 10.1128/mcb.17.7.3663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cella N, Groner B, Hynes NE. Characterization of Stat5a and Stat5b homodimers and heterodimers and their association with the glucocortiocoid receptor in mammary cells. Mol Cell Biol. 1998;18:1783–1792. doi: 10.1128/mcb.18.4.1783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Look DC, Pelletier MR, Tidwell RM, Roswit WT, Holtzman MJ. Stat1 depends on transcriptional synergy with Sp1. J Biol Chem. 1995;270:30264–30267. doi: 10.1074/jbc.270.51.30264. [DOI] [PubMed] [Google Scholar]
- Schaefer TS, Sanders LK, Nathans D. Cooperative transcriptional activity of Jun and Stat3 beta, a short form of Stat3. Proc Natl Acad Sci USA. 1995;92:9097–9101. doi: 10.1073/pnas.92.20.9097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen CH, Stavnezer J. Interaction of stat6 and NF-kappaB: direct association and synergistic activation of interleukin-4-induced transcription. Mol Cell Biol. 1998;18:3395–3404. doi: 10.1128/mcb.18.6.3395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wen Z, Zhong Z, Darnell JEJ. Maximal activation of transcription by Stat1 and Stat3 requires both tyrosine and serine phosphorylation. Cell. 1995;82:241–250. doi: 10.1016/0092-8674(95)90311-9. [DOI] [PubMed] [Google Scholar]
- Zhang X, Blenis J, Li HC, Schindler C, Chen-Kiang S. Requirement of serine phosphorylation for formation of STAT-promoter complexes. Science. 1995;267:1990–1994. doi: 10.1126/science.7701321. [DOI] [PubMed] [Google Scholar]
- Wen Z, Darnell JEJ. Mapping of Stat3 serine phosphorylation to a single residue (727) and evidence that serine phosphorylation has no influence on DNA binding of Stat1 and Stat3. Nucleic Acids Res . 1997;25:2062–2067. doi: 10.1093/nar/25.11.2062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bromberg JF, Horvath CM, Wen Z, Schreiber RD, Darnell JEJ. Transcriptionally active Stat1 is required for the antiproliferative effects of both interferon alpha and interferon gamma. . Proc Natl Acad Sci USA. 1996;93:7673–7678. doi: 10.1073/pnas.93.15.7673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moriggl R, Gouilleux-Gruart V, Jahne R, et al. Deletion of the carboxyl-terminal transactivation domain of MGF-Stat5 results in sustained DNA binding and a dominant negative phenotype. Mol Cell Biol. 1996;16:5691–5700. doi: 10.1128/mcb.16.10.5691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cho SS, Bacon CM, Sudarshan C, et al. Activation of STAT4 by IL-12 and IFN-alpha: evidence for the involvement of ligand-induced tyrosine and serine phosphorylation. J Immunol. 1996;157:4781–4789. [PubMed] [Google Scholar]
- Beadling C, Ng J, Babbage JW, Cantrell DA. Interleukin-2 activation of STAT5 requires the convergent action of tyrosine kinases and a serine/threonine kinase pathway distinct from the Raf1/ERK2 MAP kinase pathway. EMBO J. 1996;15:1902–1913. [PMC free article] [PubMed] [Google Scholar]
- Yamashita H, Xu J, Erwin RA, et al. Differential control of the phosphorylation state of proline-juxtaposed serine residues ser725 of stat5a and ser730 of stat5b in prolactin-sensitive cells. . J Biol Chem. 1998;273:30218–30224. doi: 10.1074/jbc.273.46.30218. [DOI] [PubMed] [Google Scholar]
- Woetmann A, Nielsen M, Christensen ST, et al. Inhibition of protein phosphatase 2A induces serine/threonine phosphorylation, subcellular redistribution, and functional inhibition of STAT3. Proc Natl Acad Sci USA. 1999;96:10620–10625. doi: 10.1073/pnas.96.19.10620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zauberman A, Zipori D, Krupsky M, Ben-Levy R. Stress activated protein kinase p38 is involved in IL-6 induced transcriptional activation of STAT3. Oncogene. 1999;18:3886–3893. doi: 10.1038/sj/onc/1202738. [DOI] [PubMed] [Google Scholar]
- Turkson J, Bowman T, Adnane J, et al. Requirement for Ras/Rac1-mediated p38 and c-Jun N-terminal kinase signaling in stat3 transcriptional activity induced by the src oncoprotein. Mol Cell Biol. 1999;19:7519–7528. doi: 10.1128/mcb.19.11.7519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bode JG, Gatsios P, Ludwig S, et al. The mitogen-activated protein (MAP) kinase p38 and its upstream activator MAP kinase kinase 6 are involved in the activation of signal transducer and activator of transcription by hyperosmolarity. J Biol Chem. 1999;274:30222–30227. doi: 10.1074/jbc.274.42.30222. [DOI] [PubMed] [Google Scholar]
- Goh KC, Haque SJ, Williams BR. p38 MAP kinase is required for STAT1 serine phosphorylation and transcriptional activation induced by interferons. EMBO J. 1999;18:5601–5608. doi: 10.1093/emboj/18.20.5601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lim CP, Cao X. Serine phosphorylation and negative regulation of stat3 by JNK. J Biol Chem. 1999;274:31055–31061. doi: 10.1074/jbc.274.43.31055. [DOI] [PubMed] [Google Scholar]
- David M, Petricoin ER, Benjamin C, et al. Requirement for MAP kinase (ERK2) activity in interferon alpha- and interferon beta-stimulated gene expression through STAT proteins. Science . 1995;269:1721–1723. doi: 10.1126/science.7569900. [DOI] [PubMed] [Google Scholar]
- Chung J, Uchida E, Grammer TC, Blenis J. STAT3 serine phosphorylation by ERK-dependent and -independent pathways negatively modulates its tyrosine phosphorylation. Mol Cell Biol. 1997;17:6508–6516. doi: 10.1128/mcb.17.11.6508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sengupta TK, Talbot ES, Scherle PA, Ivashkiv LB. Rapid inhibition of interleukin-6 signaling and stat3 activation mediated by mitogen-activated protein kinases. Proc Natl Acad Sci USA. 1998;95:11107–11112. doi: 10.1073/pnas.95.19.11107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strehlow I, Schindler C. Amino-terminal signal transducer and activator of transcription (STAT) domains regulate nuclear translocation and STAT deactivation. J Biol Chem. 1998;273:28049–28056. doi: 10.1074/jbc.273.43.28049. [DOI] [PubMed] [Google Scholar]
- Sekimoto T, Nakajima K, Tachibana T, Hirano T, Yoneda Y. Interferon-gamma-dependent nuclear import of Stat1 is mediated by the GTPase activity of Ran/TC4. J Biol Chem. 1996;271:31017–31020. doi: 10.1074/jbc.271.49.31017. [DOI] [PubMed] [Google Scholar]
- Sekimoto T, Yoneda Y. Nuclear import and export of proteins: the molecular basis for intracellular signaling. Cytokine Growth Factor Rev. 1998;9:205–211. doi: 10.1016/s1359-6101(98)00012-4. [DOI] [PubMed] [Google Scholar]
- Ullman KS, Powers MA, Forbes DJ. Nuclear export receptors: from importin to exportin. Cell. 1997;90:967–970. doi: 10.1016/s0092-8674(00)80361-x. [DOI] [PubMed] [Google Scholar]
- Gorlich D. Transport into and out of the cell nucleus. . EMBO J. 1998;17:2721–2727. doi: 10.1093/emboj/17.10.2721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Subramaniam PS, Mujtaba MG, Paddy MR, Johnson HM. The carboxyl terminus of interferon-gamma contains a functional polybasic nuclear localization sequence. J Biol Chem. 1999;274:403–407. doi: 10.1074/jbc.274.1.403. [DOI] [PubMed] [Google Scholar]
- Johnson HM, Torres BA, Green MM, et al. Hypothesis: ligand/receptor-assisted nuclear translocation of STATs. Proc Soc Exp Biol Med. 1998;218:149–155. doi: 10.3181/00379727-218-44282. [DOI] [PubMed] [Google Scholar]
- Car BD, Eng VM, Schnyder B, et al. Interferon gamma receptor deficient mice are resistant to endotoxic shock. J Exp Med . 1994;179:1437–1444. doi: 10.1084/jem.179.5.1437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Durbin JE, Hackenmiller R, Simon MC, Levy DE. Targeted disruption of the mouse Stat1 gene results in compromised innate immunity to viral disease. Cell. 1996;84:443–450. doi: 10.1016/s0092-8674(00)81289-1. [DOI] [PubMed] [Google Scholar]
- Meraz MA, White JM, Sheehan KC, et al. Targeted disruption of the Stat1 gene in mice reveals unexpected physiologic specificity in the JAK-STAT signaling pathway. Cell. 1996;84:431–442. doi: 10.1016/s0092-8674(00)81288-x. [DOI] [PubMed] [Google Scholar]
- Jouanguy E, Lamhamedi-Cherradi S, Lammas D, et al. A human IFNGR1 small deletion hotspot associated with dominant susceptibility to mycobacterial infection. Nature Genet. 1999;21:370–378. doi: 10.1038/7701. [DOI] [PubMed] [Google Scholar]
- Sahni M, Ambrosetti DC, Mansukhani A, et al. FGF signaling inhibits chondrocyte proliferation and regulates bone development through the STAT-1 pathway. Genes Dev. 1999;13:1361–1366. doi: 10.1101/gad.13.11.1361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaplan DH, Shankaran V, Dighe AS, et al. Demonstration of an interferon gamma-dependent tumor surveillance system in immuno-competent mice. Proc Natl Acad Sci USA. 1998;95:7556–7561. doi: 10.1073/pnas.95.13.7556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fallarino F, Gajewski TF. Cutting edge: differentiation of antitumor CTL In vivo requires host expression of stat1. J Immunol . 1999;163:4109–4113. [PubMed] [Google Scholar]
- Kumar A, Commane M, Flickinger TW, Horvath CM, Stark GR. Defective TNF-alpha-induced apoptosis in STAT1-null cells due to low constitutive levels of caspases. Science. 1997;278:1630–1632. doi: 10.1126/science.278.5343.1630. [DOI] [PubMed] [Google Scholar]
- Tanaka N, Sato M, Lamphier MS, et al. Type I interferons are essential mediators of apoptotic death in virally infected cells. Genes Cells. 1998;3:29–37. doi: 10.1046/j.1365-2443.1998.00164.x. [DOI] [PubMed] [Google Scholar]
- Bluyssen HA, Levy DE. Stat2 is a transcriptional activator that requires sequence-specific contacts provided by stat1 and p48 for stable interaction with DNA. J Biol Chem. 1997;272:4600–4605. doi: 10.1074/jbc.272.7.4600. [DOI] [PubMed] [Google Scholar]
- Takeda K, Noguchi K, Shi W, et al. Targeted disruption of the mouse Stat3 gene leads to early 'embryonic' lethality. Proc Natl Acad Sci USA. 1997;94:3801–3804. doi: 10.1073/pnas.94.8.3801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Escary JL, Perreau J, Dumenil D, Ezine S, Brulet P. Leukaemia inhibitory factor is necessary for maintenance of haematopoietic stem cells and thymocyte stimulation. Nature. 1993;363:361–364. doi: 10.1038/363361a0. [DOI] [PubMed] [Google Scholar]
- Takeda K, Clausen BE, Kaisho T, et al. Enhanced Th1 activity and development of chronic enterocolitis in mice devoid of Stat3 in macrophages and neutrophils. Immunity. 1999;10:39–49. doi: 10.1016/s1074-7613(00)80005-9. [DOI] [PubMed] [Google Scholar]
- Wang KS, Ritz J, Frank DA. IL-2 induces STAT4 activation in primary NK cells and NK cell lines, but not in T cells. J Immunol. 1999;162:299–304. [PubMed] [Google Scholar]
- Kaplan MH, Sun YL, Hoey T, Grusby MJ. Impaired IL-12 responses and enhanced development of Th2 cells in Stat4-deficient mice. Nature. 1996;382:174–177. doi: 10.1038/382174a0. [DOI] [PubMed] [Google Scholar]
- Magram J, Connaughton SE, Warrier RR, et al. IL-12-deficient mice are defective in IFN gamma production and type 1 cytokine responses. Immunity. 1996;4:471–481. doi: 10.1016/s1074-7613(00)80413-6. [DOI] [PubMed] [Google Scholar]
- Wu C, Ferrante J, Gately MK, Magram J. Characterization of IL-12 receptor beta1 chain (IL-12Rbeta1)-deficient mice: IL-12Rbeta1 is an essential component of the functional mouse IL-12 receptor. J Immunol. 1997;159:1658–1665. [PubMed] [Google Scholar]
- de Jong R, Altare F, Haagen IA, et al. Severe mycobacterial and Salmonella infections in interleukin-12 receptor-deficient patients. Science. 1998;280:1435–1438. doi: 10.1126/science.280.5368.1435. [DOI] [PubMed] [Google Scholar]
- Lin JX, Mietz J, Modi WS, John S, Leonard WJ. Cloning of human Stat5B. Reconstitution of interleukin-2-induced Stat5A and Stat5BDNA binding activity in COS-7 cells. J Biol Chem. 1996;271:10738–10744. [PubMed] [Google Scholar]
- Liu X, Robinson GW, Wagner KU, et al. Stat5a is mandatory for adult mammary gland development and lactogenesis. . Genes Dev. 1997;11:179–186. doi: 10.1101/gad.11.2.179. [DOI] [PubMed] [Google Scholar]
- Udy GB, Towers RP, Snell RG, et al. Requirement of STAT5b for sexual dimorphism of body growth rates and liver gene expression. . Proc Natl Acad Sci USA. 1997;94:7239–7244. doi: 10.1073/pnas.94.14.7239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teglund S, McKay C, Schuetz E, et al. STAT5a and STAT5b proteins have essential and nonessential, or redundant, roles in cytokine responses. Cell. 1998;93:841–850. doi: 10.1016/s0092-8674(00)81444-0. [DOI] [PubMed] [Google Scholar]
- Moriggl R, Topham DJ, Teglund S, et al. Stat5 is required for IL-2-induced cell cycle progression of peripheral T cells. . Immunity. 1999;10:249–259. doi: 10.1016/s1074-7613(00)80025-4. [DOI] [PubMed] [Google Scholar]
- Nakajima H, Liu XW, Wynshaw-Boris A, et al. An indirect effect of Stat5a in IL-2-induced proliferation: a critical role for Stat5a in IL-2-mediated IL-2 receptor alpha chain induction. . Immunity. 1997;7:691–701. doi: 10.1016/s1074-7613(00)80389-1. [DOI] [PubMed] [Google Scholar]
- Imada K, Bloom ET, Nakajima H, et al. Stat5b is essential for natural killer cell-mediated proliferation and cytolytic activity. J Exp Med. 1998;188:2067–2074. doi: 10.1084/jem.188.11.2067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Socolovsky M, Fallon AE, Wang S, Brugnara C, Lodish HF. Fetal anemia and apoptosis of red cell progenitors in Stat5a-/-5b-/- mice: a direct role for Stat5 in Bcl-X(L) induction. Cell. 1999;98:181–191. doi: 10.1016/s0092-8674(00)81013-2. [DOI] [PubMed] [Google Scholar]
- Beadling C, Guschin D, Witthuhn BA, et al. Activation of JAK kinases and STAT proteins by interleukin-2 and interferon alpha, but not the T cell antigen receptor, in human T lymphocytes. EMBO J. 1994;13:5605–5615. doi: 10.1002/j.1460-2075.1994.tb06898.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Welte T, Leitenberg D, Dittel BN, et al. STAT5 interaction with the T cell receptor complex and stimulation of T cell proliferation. Science. 1999;283:222–225. doi: 10.1006/abio.2000.4639. [DOI] [PubMed] [Google Scholar]
- Gerwien J, Nielsen M, Labuda T, et al. Cutting edge: TCR stimulation by antibody and bacterial superantigen induces stat3 activation in human T cells. J Immunol. 1999;163:1742–1745. [PubMed] [Google Scholar]
- Kaplan MH, Schindler U, Smiley ST, Grusby MJ. Stat6 is required for mediating responses to IL-4 and for development of Th2 cells. Immunity. 1996;4:313–319. doi: 10.1016/s1074-7613(00)80439-2. [DOI] [PubMed] [Google Scholar]
- Takeda K, Tanaka T, Shi W, et al. Essential role of Stat6 in IL-4 signalling. Nature. 1996;380:627–630. doi: 10.1038/380627a0. [DOI] [PubMed] [Google Scholar]
- Takeda K, Kamanaka M, Tanaka T, Kishimoto T, Akira S. Impaired IL-13-mediated functions of macrophages in STAT6-deficient mice. JImmunol. 1996;157:3220–3222. [PubMed] [Google Scholar]
- Shimoda K, van Jong J, Sangster MY, et al. Lack of IL-4-induced Th2 response and IgE class switching in mice with disrupted Stat6 gene. Nature. 1996;380:630–633. doi: 10.1038/380630a0. [DOI] [PubMed] [Google Scholar]
- Akimoto T, Numata F, Tamura M, et al. Abrogation of bronchial eosinophilic inflammation and airway hyperreactivity in signal transducers and activators of transcription (STAT)6-deficient mice. . J Exp Med. 1998;187:1537–1542. doi: 10.1084/jem.187.9.1537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuperman D, Schofield B, Wills-Karp M, Grusby MJ. Signal transducer and activator of transcription factor 6 (Stat6)- deficient mice are protected from antigen-induced airway hyperresponsiveness and mucus production. J Exp Med. 1998;187:939–948. doi: 10.1084/jem.187.6.939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller RL, Eppinger TM, McConnell D, Cunningham-Rundles C, Rothman P. Analysis of cytokine signaling in patients with extrinsic asthma and hyperimmunoglobulin E. J Allergy Clin Immunol. 1998;102:503–511. doi: 10.1016/s0091-6749(98)70141-1. [DOI] [PubMed] [Google Scholar]
- Miyata S, Matsuyama T, Kodama T, et al. STAT6 deficiency in a mouse model of allergen-induced airways inflammation abolishes eosinophilia but induces infiltration of CD8+ T cells. . Clin Exp Allergy. 1999;29:114–123. doi: 10.1046/j.1365-2222.1999.00405.x. [DOI] [PubMed] [Google Scholar]
- Kaplan MH, Whitfield JR, Boros DL, Grusby MJ. Th2 cells are required for the Schistosoma mansoni egg-induced granulomatous response. J Immunol. 1998;160:1850–1856. [PubMed] [Google Scholar]
- Morse HCR, McCarty T, Giese NA, Taddesse-Heath L, Grusby MJ. STAT6-deficient mice exhibit normal induction of murine AIDS and expression of immunoglobulin E following infection with LP-BM5 murine leukemia viruses. J Virol. 1999;73:7093–7095. doi: 10.1128/jvi.73.8.7093-7095.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaplan MH, Wurster AL, Grusby MJ. A signal transducer and activator of transcription (Stat)4-independent pathway for the development of T helper type 1 cells. J Exp Med. 1998;188:1191–1196. doi: 10.1084/jem.188.6.1191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cantrell D. T cell antigen receptor signal transduction pathways. Annu Rev Immunol. 1996;14:259–274. doi: 10.1146/annurev.immunol.14.1.259. [DOI] [PubMed] [Google Scholar]
- David M, Wong L, Flavell R, et al. STAT activation by epidermal growth factor (EGF) and amphiregulin. Requirement for the EGF receptor kinase but not for tyrosine phosphorylation sites or JAK1. J Biol Chem. 1996;271:9185–9188. doi: 10.1074/jbc.271.16.9185. [DOI] [PubMed] [Google Scholar]
- Zhong Z, Wen Z, Darnell JEJ. Stat3: a STAT family member activated by tyrosine phosphorylation in response to epidermal growth factor and interleukin-6. Science. 1994;264:95–98. doi: 10.1126/science.8140422. [DOI] [PubMed] [Google Scholar]
- Leaman DW, Pisharody S, Flickinger TW, et al. Roles of JAKs in activation of STATs and stimulation of c-fos gene expression by epidermal growth factor. Mol Cell Biol. 1996;16:369–375. doi: 10.1128/mcb.16.1.369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhat GJ, Thekkumkara TJ, Thomas WG, Conrad KM, Baker KM. Activation of the STAT pathway by angiotensin II in T3CHO/AT1A cells. Cross-talk between angiotensin II and interleukin-6 nuclear signaling. J Biol Chem. 1995;270:19059–19065. doi: 10.1074/jbc.270.32.19059. [DOI] [PubMed] [Google Scholar]
- Marrero MB, Schieffer B, Paxton WG, et al. Direct stimulation of Jak/STAT pathway by the angiotensin II AT1 receptor. . Nature. 1995;375:247–250. doi: 10.1038/375247a0. [DOI] [PubMed] [Google Scholar]
- Mellado M, Rodriguez-Frade JM, Aragay A, et al. The chemokine monocyte chemotactic protein 1 triggers Janus kinase 2 activation and tyrosine phosphorylation of the CCR2B receptor. J Immunol. 1998;161:805–813. [PubMed] [Google Scholar]
- Kawata T, Shevchenko A, Fukuzawa M, et al. SH2 signaling in a lower eukaryote: a STAT protein that regulates stalk cell differentiation in dictyostelium. Cell. 1997;89:909–916. doi: 10.1016/s0092-8674(00)80276-7. [DOI] [PubMed] [Google Scholar]
- Mohanty S, Jermyn KA, Early A, et al. Evidence that the Dictyostelium Dd-STATa protein is a repressor that regulates commitment to stalk cell differentiation and is also required for efficient chemotaxis. . Development. 1999;126:3391–3405. doi: 10.1242/dev.126.15.3391. [DOI] [PubMed] [Google Scholar]
- Bromberg JF, Horvath CM, Besser D, Lathem WW, Darnell JEJ. Stat3 activation is required for cellular transformation by v-src. . Mol Cell Biol. 1998;18:2553–2558. doi: 10.1128/mcb.18.5.2553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bromberg JF, Wrzeszczynska MH, Devgan G, et al. Stat3 as an oncogene. Cell. 1999;98:295–303. doi: 10.1016/s0092-8674(00)81959-5. [DOI] [PubMed] [Google Scholar]
- Klingmuller U, Lorenz U, Cantley LC, Neel BG, Lodish HF. Specific recruitment of SH-PTP1 to the erythropoietin receptor causes inactivation of JAK2 and termination of proliferative signals. . Cell. 1995;80:729–738. doi: 10.1016/0092-8674(95)90351-8. [DOI] [PubMed] [Google Scholar]
- Haque SJ, Harbor P, Tabrizi M, Yi T, Williams BR. Protein-tyrosine phosphatase Shp-1 is a negative regulator of IL-4- and IL-13-dependent signal transduction. J Biol Chem. 1998;273:33893–33896. doi: 10.1074/jbc.273.51.33893. [DOI] [PubMed] [Google Scholar]
- Migone TS, Cacalano NA, Taylor N, et al. Recruitment of SH2-containing protein tyrosine phosphatase SHP-1 to the interleukin 2 receptor; loss of SHP-1 expression in human T-lymphotropic virus type I-transformed T cells. Proc Natl Acad Sci USA. 1998;95:3845–3850. doi: 10.1073/pnas.95.7.3845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsui HW, Siminovitch KA, de Souza L, Tsui FW. Motheaten and viable motheaten mice have mutations in the haematopoietic cell phosphatase gene. Nature Genet. 1993;4:124–129. doi: 10.1038/ng0693-124. [DOI] [PubMed] [Google Scholar]
- Bignon JS, Siminovitch KA. Identification of PTP1C mutation as the genetic defect in motheaten and viable motheaten mice: a step toward defining the roles of protein tyrosine phosphatases in the regulation of hemopoietic cell differentiation and function. Clin Immunol Immunopathol. 1994;73:168–179. doi: 10.1006/clin.1994.1185. [DOI] [PubMed] [Google Scholar]
- Shultz LD, Schweitzer PA, Rajan TV, et al. Mutations at the murine motheaten locus are within the hematopoietic cell protein-tyrosine phosphatase (Hcph) gene. Cell. 1993;73:1445–1454. doi: 10.1016/0092-8674(93)90369-2. [DOI] [PubMed] [Google Scholar]
- Green MC, Shultz LD. Motheaten, an immunodeficient mutant of the mouse. I. Genetics and pathology. J Hered. 1975;66:250–258. doi: 10.1093/oxfordjournals.jhered.a108625. [DOI] [PubMed] [Google Scholar]
- Aoki K, Didomenico E, Sims NA, et al. The tyrosine phosphatase SHP-1 is a negative regulator of osteoclastogenesis and osteoclast resorbing activity: increased resorption and osteopenia in me(v)/me(v) mutant mice. Bone. 1999;25:261–267. doi: 10.1016/S8756-3282(99)00174-X. [DOI] [PubMed] [Google Scholar]
- Kim TK, Maniatis T. Regulation of interferon-gamma-activated STAT1 by the ubiquitin-proteasome pathway. Science. 1996;273:1717–1719. doi: 10.1126/science.273.5282.1717. [DOI] [PubMed] [Google Scholar]
- Yoshimura A, Ohkubo T, Kiguchi T, et al. A novel cytokine-inducible gene CIS encodes an SH2-containing protein that binds to tyrosine-phosphorylated interleukin 3 and erythropoietin receptors. EMBO J. 1995;14:2816–2826. doi: 10.1002/j.1460-2075.1995.tb07281.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Starr R, Willson TA, Viney EM, et al. A family of cytokine-inducible inhibitors of signalling. Nature. 1997;387:917–921. doi: 10.1038/43206. [DOI] [PubMed] [Google Scholar]
- Endo TA, Masuhara M, Yokouchi M, et al. A new protein containing an SH2 domain that inhibits JAK kinases. Nature. 1997;387:921–924. doi: 10.1038/43213. [DOI] [PubMed] [Google Scholar]
- Naka T, Narazaki M, Hirata M, et al. Structure and function of a new STAT-induced STAT inhibitor. Nature. 1997;387:924–929. doi: 10.1038/43219. [DOI] [PubMed] [Google Scholar]
- Zhang JG, Farley A, Nicholson SE, et al. The conserved SOCS box motif in suppressors of cytokine signaling binds to elongins B and C and may couple bound proteins to proteasomal degradation. Proc Natl Acad Sci USA. 1999;96:2071–2076. doi: 10.1073/pnas.96.5.2071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sasaki A, Yasukawa H, Suzuki A, et al. Cytokine-inducible SH2 protein-3 (CIS3/SOCS3) inhibits janus tyrosine kinase by binding through the N-terminal kinase inhibitory region as well as SH2 domain. Genes Cells. 1999;4:339–351. doi: 10.1046/j.1365-2443.1999.00263.x. [DOI] [PubMed] [Google Scholar]
- Naka T, Matsumoto T, Narazaki M, et al. Accelerated apoptosis of lymphocytes by augmented induction of Bax in SSI-1 (STAT-induced STAT inhibitor-1) deficient mice. Proc Natl Acad Sci USA. 1998;95:15577–15582. doi: 10.1073/pnas.95.26.15577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Starr R, Metcalf D, Elefanty AG, et al. Liver degeneration and lymphoid deficiencies in mice lacking suppressor of cytokine signaling-1. Proc Natl Acad Sci USA. 1998;95:14395–14399. doi: 10.1073/pnas.95.24.14395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander WS, Starr R, Fenner JE, et al. SOCS1 is a critical inhibitor of interferon gamma signaling and prevents the potentially fatal neonatal actions of this cytokine. Cell. 1999;98:597–608. doi: 10.1016/s0092-8674(00)80047-1. [DOI] [PubMed] [Google Scholar]
- Marine JC, Topham DJ, McKay C, et al. SOCS1 deficiency causes a lymphocyte-dependent perinatal lethality. Cell. 1999;98:609–616. doi: 10.1016/s0092-8674(00)80048-3. [DOI] [PubMed] [Google Scholar]
- Adams TE, Hansen JA, Starr R, et al. Growth hormone preferentially induces the rapid, transient expression of SOCS-3, a novel inhibitor of cytokine receptor signaling. J Biol Chem. 1998;273:1285–1287. doi: 10.1074/jbc.273.3.1285. [DOI] [PubMed] [Google Scholar]
- Cohney SJ, Sanden D, Cacalano NA, et al. SOCS-3 is tyrosine phosphorylated in response to interleukin-2 and suppresses STAT5 phosphorylation and lymphocyte proliferation. Mol Cell Biol. 1999;19:4980–4988. doi: 10.1128/mcb.19.7.4980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marine JC, McKay C, Wang D, et al. SOCS3 is essential in the regulation of fetal liver erythropoiesis. Cell. 1999;98:617–627. doi: 10.1016/s0092-8674(00)80049-5. [DOI] [PubMed] [Google Scholar]
- Aman MJ, Migone TS, Sasaki A, et al. CIS associates with the interleukin-2 receptor beta chain and inhibits interleukin-2-dependent signaling. J Biol Chem. 1999;274:30266–30272. doi: 10.1074/jbc.274.42.30266. [DOI] [PubMed] [Google Scholar]
- Matsumoto A, Seki Y, Kubo M, et al. Suppression of STAT5 functions in liver, mammary glands, and T cells in cytokine-inducible SH2-containing protein 1 transgenic mice. Mol Cell Biol. 1999;19:6396–6407. doi: 10.1128/mcb.19.9.6396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pezet A, Favre H, Kelly PA, Edery M. Inhibition and restoration of prolactin signal transduction by suppressors of cytokine signaling. J Biol Chem. 1999;274:24497–24502. doi: 10.1074/jbc.274.35.24497. [DOI] [PubMed] [Google Scholar]
- Karlsson H, Gustafsson JA, Mode A. Cis desensitizes GH induced Stat5 signaling in rat liver cells. Mol Cell Endocrinol. 1999;154:37–43. doi: 10.1016/s0303-7207(99)00101-x. [DOI] [PubMed] [Google Scholar]
- Bjorbaek C, Elmquist JK, El-Haschimi K, et al. Activation of SOCS-3 messenger ribonucleic acid in the hypothalamus by ciliary neurotrophic factor. Endocrinology. 1999;140:2035–2043. doi: 10.1210/endo.140.5.6736. [DOI] [PubMed] [Google Scholar]
- Liu B, Liao J, Rao X, et al. Inhibition of Stat1-mediated gene activation by PIAS1. Proc Natl Acad Sci USA . 1998;95:10626–10631. doi: 10.1073/pnas.95.18.10626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chung CD, Liao J, Liu B, et al. Specific inhibition of Stat3 signal transduction by PIAS3. Science. 1997;278:1803–1805. doi: 10.1126/science.278.5344.1803. [DOI] [PubMed] [Google Scholar]
- Ye BH, Cattoretti G, Shen Q, et al. The BCL-6 protooncogene controls germinal-centre formation and Th2-type inflammation. . Nature Genet. 1997;16:161–170. doi: 10.1038/ng0697-161. [DOI] [PubMed] [Google Scholar]
- Dent AL, Hu-Li J, Paul WE, Staudt LM. T helper type 2 inflammatory disease in the absence of interleukin 4 and transcription factor STAT6. Proc Natl Acad Sci USA. 1998;95:13823–13828. doi: 10.1073/pnas.95.23.13823. [DOI] [PMC free article] [PubMed] [Google Scholar]