

Abstract
The identification of genes contributing to the adaptation of local populations is of great biological interest. In an attempt to characterize functionally important differences among African and non-African Drosophila melanogaster populations, we surveyed neutral microsatellite variation in an 850-kb genomic sequence. Three genomic regions were identified that putatively bear an adaptive mutation associated with the habitat expansion of D. melanogaster. A further inspection of two regions by sequence analysis of multiple fragments confirmed the presence of a recent beneficial mutation in the non-African populations. Our study suggests that hitchhiking mapping is a universal approach for the identification of ecologically important mutations.
One of the major challenges in biology is the elucidation of the functions encoded by the genomic sequence. It is only recently that the key role of natural variation has been recognized as an important tool for the functional characterization of genomic regions (1–3). Although most research efforts have been directed at epidemiological questions, we used an approach that aims to identify and characterize beneficial mutations.
Given that directional selection is much more common than predicted by the neutral theory of molecular evolution (4–6), appropriate experiments should allow the detection of individual selective events and provide insight into their molecular basis and phenotypic consequence.
Starting from Africa, Drosophila melanogaster colonized the rest of the world ≈10,000 years ago (7). This habitat shift presumably required several genetic adaptations to counter the biotic and abiotic changes. We used a mapping strategy (hitchhiking mapping) to identify adaptive mutations associated with the colonization event. The underlying principle of hitchhiking mapping is that a beneficial mutation is either lost or increases in frequency until it eventually becomes fixed. This spread of a positively selected mutation through a population (selective sweep) removes variability at the selected site and its flanking region, a process that has been called “hitchhiking” (8). Thus, regions that recently experienced an episode of positive selection can be detected by a local reduction in variability (8–10).
Microsatellites, short tandem repetitions of 1–5-bp motifs, are generally considered to evolve neutrally (11) and are highly abundant components of eukaryotic genomes. Several recent studies used microsatellites as markers to detect putatively selected regions in the genome (12–16). Here, we extend this approach and use a combination of a high-density microsatellite screen and sequence-polymorphism analysis along the chromosome to map regions in the D. melanogaster genome that recently have experienced positive selection.
Materials and Methods
Population Samples.
Microsatellites were typed in six European, two North-American, and two African populations. A summary of populations and sample sizes can be found at http://i122server.vu-wien.ac.at (Table 4, which is published as supporting information on the PNAS web site, www.pnas.org). For the sequencing analysis, only one European (Austria) population was used for all fragments apart from the cramped (crm) locus, for which Austrian and Italian samples were analyzed jointly. North-American populations were not considered in the sequencing analysis, because they are believed to have colonized only very recently and most likely represent a subsample of European populations (7). For one genome region (partial coding region of crm), where a strong deviation from neutrality was observed in the European sample, we included one additional population from Germany (14 lines). African flies were represented by samples from multiple Kenyan localities and one population collected in Zimbabwe (Harare). For the cramped locus, only individuals from Kenya were sequenced.
Microsatellite Loci.
The X-chromosomal region was selected based on a previous study (17), which showed one locus (DS06335b) with strongly reduced variability in a German D. melanogaster population. To describe the pattern of variation around this locus we selected flanking loci covering a region of 274.4 kb of the D. melanogaster X chromosome (polytene bands 3B6–3C3; Fig. 1). A set of 15 autosomal loci covering a chromosomal segment of 577.5 kb (polytene band 62B6–62E5; Fig. 1) was selected as a reference. This second segment was chosen randomly with respect to chromosomal location, but care was taken that the X and autosomal segments have similar recombination rates (when adjusted for the lack of recombination in males). Both investigated segments are located in chromosomal regions of intermediate recombination rate. Location and spacing between the loci are shown in Fig. 1A. Only dinucleotide microsatellites with at least six uninterrupted repeats in the database sequence (release 2) were included in the survey. Details of primer sequences and amplification conditions are given at http://i122server.vu-wien.ac.at (Table 5, which is published as supporting information on the PNAS web site). Typing protocols were essentially as described (18).
Figure 1.

Relative variability estimates in regions affected by a selective sweep. (A) Location of microsatellite loci in polytene band 3B-C (X chromosome) and 62E (left arm of chromosome 3) and their respective variability expressed as standardized lnRV values. Both chromosomal segments have approximately the same distance from the tip of the chromosome and exhibit comparable and intermediate levels of recombination. The 95% confidence interval of the lnRV statistic is indicated by dashed lines. The gray line marks the extension of the conservative larger mapping interval (see Results for explanation). Black lines above gray lines indicate the mapping interval using a deterministic hitchhiking model (hitchhiking-mapping interval; see Materials and Methods). Small boxes below mapping intervals indicate the position of the sequenced DNA pieces. Positions are given relative to the start of the chromosome of according to release 2 of the complete D. melanogaster genomic sequence. (B) Ratio of nucleotide diversities (π, average number of pairwise differences) in European and African populations in X-chromosomal sweep regions 1 (Left) and 2 (Right).
Microsatellite Data Analysis.
We calculated the variability at each locus for African and non-African samples independently according to the formula
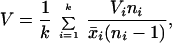 |
where k is the number of African or non-African populations, respectively, and ni is the number of chromosomes analyzed in the ith population. Variances in repeat number (Vi) were divided by the mean repeat number (x̄) at the locus to account for length dependence of microsatellite mutation rates (19). For inbred lines, one allele was discarded randomly to account for drift during the propagation of the isofemale lines.
Because microsatellite mutation rates differ widely among loci, we compared for each locus the variance in repeat number (V) of several non-African (Europe and North America) populations to African populations. Under neutrality, the distribution of the standardized measurement of variability, ln RV = ln〚VnAfr/VAfr〛, can be approximated by a Gaussian distribution (13). To estimate the mean and variance of the Gaussian distribution, we used polymorphism data from 31 X-chromosomal and 45 autosomal microsatellite loci from a similar recombinational environment but located outside the two analyzed contiguous DNA segments. For all microsatellite loci in the two chromosomal segments we determined the ln RV values and asked whether they fall significantly outside the Gaussian distribution.
Sequencing of Microsatellite Alleles.
Recently it was shown that microsatellite loci with a base substitution in the repeat unit have a lower mutation rate (20–22). To rule out the possibility that a base substitution in the microsatellite is responsible for the reduced variability in non-African populations, we sequenced the entire allele spectrum of the loci MSX-2, -3, -4, -12, and MS3L-11. No new base substitution was detected interrupting the microsatellite structure in European individuals.
Microsatellite-Based Mapping of the Selected Site.
A deterministic hitchhiking framework (23) was used to obtain the expected position of candidate genes from the reduction in microsatellite variability assuming a selective sweep at linked loci. The model requires knowledge of the distance to the selected site, the recombination rate between the two loci, the mutation rate at the microsatellite locus, and the fixation time of the advantageous mutation. Most of these parameters can be estimated, but the fixation time and distance to the selected locus are unknown. We solved this problem by assuming that two adjacent microsatellite loci were affected by the same selective sweep. Hence, if the distance of the first microsatellite locus to the selected site is x, the distance to the second microsatellite locus is x + b, where b is the distance between the two microsatellite loci. If the variability of both microsatellite loci is affected by the same selective sweep, the fixation time is the same for both sets of equations, allowing one to determine x. The variability before the selective sweep was approximated by the variance in repeat number in African populations (VAfr) and the variability after the fixation of the beneficial mutation was estimated by the variance in repeat number in non-African populations (VnAfr). For sweep regions 2 and 3 (see Fig. 1), we used the two microsatellite loci, which showed the strongest reduction in variability. For sweep region 1, all three microsatellite loci with strongly reduced variability were used. Based on equation 18.3 in ref. 24, we derived Eq. 1 to calculate the distance x between the microsatellite locus with the strongest reduction in variability (i.e., locus 1) and the selected site. A slight modification of Eq. 1 (not shown) was used for the case in which the selected site was assumed to be between the two characterized microsatellite loci.
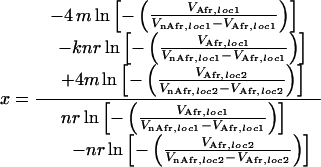 |
1 |
The following parameter values also were used: average microsatellite mutation rate m = 8.3 × 10−6 per generation (19, 25, 26); number of neutral microsatellite alleles n = 7 [estimated from 35 X-chromosomal loci analyzed in African populations (27)]; distance between the flanking microsatellite loci k (in bp); and the recombination rate r per bp (r = 1.1 × 10−8 for X-chromosomal region, r = 1.2 × 10−8 for autosomal region) (28). Recombination rates were adjusted for the lack of recombination in males. It should be noted that the distance obtained between the microsatellite and the selected site is based on the assumption that no novel microsatellite mutations occurred after the fixation of the beneficial mutation. Confidence intervals were obtained by resampling microsatellite alleles within populations of both geographic origins separately. The expected positions of candidate genes were defined as the 95% confidence interval of all resamples after applying Eq. 1 to each individual resample.
Sequence Polymorphism Analysis.
In sweep region 1, five fragments were sequenced, including two candidate genes [cramped and syntaxin4 (syx4)] and three flanking fragments (denoted frag.1–frag.3). Although the complete coding region has been determined for the cramped gene (3.5 kb), only exons 2, 3, and 4, and parts of exon 5 and intervening introns were sequenced for the syx4 gene. Intron 2 of the syx4 gene was only sequenced partially, because it contained several long poly(A)/T-rich regions, which could not be determined unambiguously. In sweep region 2, four fragments were sequenced, including exon 2 and 3 of the candidate gene CG3588 and three flanking fragments (denoted frag.4–frag.7). Locations of fragments were chosen according to two criteria: (i) fragments should cover the “conservative” mapping interval determined by the microsatellite data (see Results), (ii) and all identified candidate genes (Table 1) should be sequenced at least partially.
Table 1.
Candidate genes located in sweep regions
| Loci used | Hitchhiking mapping interval* | Position of the closest gene(s) | Distance from predicted interval†, kb | Identified gene‡ | |
|---|---|---|---|---|---|
| Sweep 1 | MSX-2/-3/-4 | 2503839–2512339 | 2488732–2492958 | 17.2 | syx4 |
| 2483719–2488249 | 22.1 | cramped | |||
| Sweep 2 | MSX-11/-12 | 2669739–2670739 | 2671827–2677737 | 4.5 | CG3588 |
| Sweep 3 | MS3L-10/-11 | 2299310–2299770 | 2272095–2274956 | 26 | CG13803 |
| CG13802 | |||||
| 2313216–2318779 | 16.5 | CG8985 |
Based on a deterministic hitchhiking framework (23).
Distance is calculated from the center of the coding region of the gene to the center of the mapping interval.
Fragment containing predicted genes CG13803 and CG13802. This region was newly annotated by using GENESCAN, yielding only a single ORF containing both genes.
All PCR products were obtained from male flies and sequenced in both directions. PCR and sequencing primers are available from the authors on request. Whenever possible, we used Drosophila simulans as an outgroup. For the cramped locus, which could be amplified only partially in D. simulans, we used Drosophila sechellia as outgroup. Sequences were aligned by using CLUSALX (29) and adjusted manually. Silent polymorphism, divergence, Fu and Li's D statistic (30), and Hudson–Kreitman–Aguade (HKA) tests (31) were calculated by using the program DNASP 3.53 (32). For Fu and Li's D statistic, ancestral and derived sites were distinguished by using the outgroup. The HKA test contrasts within-species polymorphism and between-species divergence at one test locus and one reference locus. Locus frag.1 served as a reference locus against which all other fragments were tested, because frag.1 showed no deviation from neutrality in our non-African population. The H statistics (33) were calculated separately for the African and European populations. To incorporate sites that are fixed between European and African populations, we restricted the maximum frequency of the derived allele to (n − 1)/n (n = number of individuals sequenced). Significance levels were obtained online (http://crimp.lbl.gov/htest.html) for each of the sequenced fragments by 10,000 iterations. The simulations were conditioned on the number of segregating sites in the respective populations. The outgroup species was used to infer the derived and ancestral states. P values were determined for the conservative assumption of no recombination.
Estimation of the Selection Coefficient and the Position of the Selected Site.
We used a composite maximum-likelihood method (10) to detect the signature of genetic hitchhiking along a recombining chromosome. The test calculates the ratio of the likelihood (LR) of the data under a neutral model and a hitchhiking model. The P value represents the fraction of LR values obtained from simulated neutral data that are larger than the observed LR. The method was applied to the European samples for sweep regions 1 and 2 separately by using a range of values for the mutation and recombination rate. The method also can be used to obtain a composite maximum-likelihood estimate for the position of the selected site and the selection coefficient. Initial guesses for X extended over the whole sequenced fragment and were moved in 1-kb steps along the chromosome. The selection coefficient (s) and the position of the selected site (X) are the values that maximize the likelihood of the data under a hitchhiking model. The effective population size (Ne) of non-African D. melanogaster was assumed to be 106 (34).
Results
The power of a microsatellite-based hitchhiking-mapping approach depends on the size of the window of reduced variation around the selected site. This window size is determined by the recombination rate (r) and selection intensity (s). For the identification of a selective sweep, at least one microsatellite locus must be located in the window of reduced variability. Using the deterministic hitchhiking framework (23), we calculated the expected size of the window in which variability would be reduced by 50% because of a selective sweep. Assuming a typical recombination rate of 1 × 10−8 (28, 35), a reasonable estimate for the effective population size of D. melanogaster [Ne = 106 (34)] and a selection intensity (s) of 0.005 variability is reduced over a 28-kb region (see Fig. 2, which is published as supporting information on the PNAS web site, for a broader range of parameters). Although this is a rough estimate and microsatellites are not distributed randomly (36), the typical microsatellite density of D. melanogaster [four microsatellites per 100 kb (36, 37)] seems appropriate to detect a substantial number of selective sweeps.
Evidence for Nonneutral Evolution in Non-African D. melanogaster.
Twenty-nine microsatellite loci distributed over a genomic region of 850 kb were scanned for variability in African and non-African D. melanogaster. Visual inspection of a plot of relative variability (Fig. 1A) suggests that three genomic regions have unusually low levels of variability in the non-African D. melanogaster populations. Based on a 95% confidence interval of the lnRV test statistic (13), which accounts for stochastic fluctuations in relative microsatellite variability among loci, four loci were found to deviate significantly from neutral expectations (Table 2). Although two of the loci with a significant reduction in variability fall into one region of reduced variability, the remaining loci define separate regions of reduced variability (Fig. 1A), suggesting that two regions of significantly reduced variability are located on the X chromosome and one is located on the third chromosome.
Table 2.
lnRV values of microsatellite loci deviating from neutral expectations
| Locus | ln RV | Std ln RV | Two-tailed, P |
|---|---|---|---|
| MSX-2 | −2.95 | −1.83 | 0.0673 |
| MSX-3 | −4.00 | −2.79 | 0.0053 |
| MSX-4 | −3.18 | −2.04 | 0.0414 |
| MSX-12 | −4.25 | −3.02 | 0.0025 |
| MS3L-11 | −4.71 | −3.44 | 0.0006 |
Strong positive selection is expected to leave its footprint in a wider genomic region than just a single microsatellite locus. Under such a hitchhiking scenario, the pattern of reduced microsatellite variability should extend over several loci. We observe this pattern in its most extreme in the case of sweep region 1, where three adjacent loci have a strong reduction in variability in non-African populations (see Fig. 1A for the nomenclature of sweep regions). A similar but less extreme trend of reduced variation at loci flanking the significantly reduced microsatellite can be observed in sweep regions 2 and 3.
For sweep regions 1 and 2, we used sequence polymorphism to further verify the deviation from neutrality. Several DNA fragments (between 0.8 and 3.5 kb) around the regions of reduced microsatellite variability were analyzed (Table 3 and Table 6, which is published as supporting information on the PNAS web site). Location and spacing between the sequenced fragments on the X chromosome is shown in Fig. 1B. Corresponding to the microsatellite analysis we compared levels of variability in African and non-African flies. The ratio of the average number of pairwise differences (π) for each of the sequenced fragments (Fig. 1B) is in good agreement with the one obtained with microsatellites. In sweep region 1, two fragments containing the genes crm and syx4 showed the strongest reduction in variability in non-African populations (7.8- and 8.1-fold). Twenty kilobases further upstream and downstream of the crm and syx4 genes sequence variability has recovered substantially but still remains 3-fold lower in non-African flies (Fig. 1B). Complete recovery of neutral variability in non-African flies was observed in locus frag.1. This fragment showed only 2-fold lower variability in non-African populations, a value that is well within the range observed for other X-chromosomal genes (38). Similarly, in sweep region 2, the predicted gene CG3588 (i.e., frag.6) and frag.5 had a 10.6- and 12.5-fold reduction in variability, respectively. The remaining two sequence stretches were only slightly less variable in comparison with the African samples (Fig. 1B, Table 3).
Table 3.
Diversity estimates and results of tests statistics for selective sweeps
| Sequenced fragment | Gene | No. of silent sites | No. of individuals | Nucleotide diversity, π | Tajima's D* | Fu and Li's D | Divergence | P value, H test | P value, HKA test† |
|---|---|---|---|---|---|---|---|---|---|
| Africa | |||||||||
| frag.1 | period/CG2650 | 861 | 32 | 0.0178 | −0.569 | −0.446 | 0.058 | 0.452 | — |
| frag.2 | 100G10.2 | 781 | 28 | 0.0169 | −0.677 | −1.007 | 0.074 | 0.368 | 0.66 |
| crm‡ | cramped | 1210 | 7 | 0.0233 | −0.199 | 0.411 | 0.117 | 0.234 | 0.305 |
| syx4 | syxntaxin4 | 657 | 25 | 0.0072 | −0.750 | −1.352 | 0.071 | 0.241 | 0.101 |
| frag.3 | Noncoding | 1279 | 16 | 0.0122 | −0.596 | −1.077 | 0.063 | 0.38 | 0.414 |
| frag.4 | Noncoding | 824 | 11 | 0.0076 | −0.741 | −1.146 | 0.045 | 0.551 | 0.392 |
| frag.5 | Noncoding | 992 | 14 | 0.0127 | −0.765 | −1.041 | 0.074 | 0.2462 | 0.372 |
| frag.6 | CG3588 | 459 | 14 | 0.0325 | −0.191 | −0.039 | 0.135 | 0.1752 | 0.693 |
| frag.7 | CG3592 | 431 | 18 | 0.0278 | 0.587 | −0.769 | 0.155 | 0.6409 | 0.343 |
| Europe | |||||||||
| frag.1 | period/CG2650 | 861 | 30 | 0.0090 | 0.122 | −0.161 | 0.059 | 0.099 | — |
| frag.2 | 100G10.2 | 781 | 28 | 0.0062 | −0.135 | 0.699 | 0.073 | 0.070 | 0.295 |
| crm | cramped | 1210 | 27 | 0.0030 | −0.730 | 1.824§ | 0.120 | 0.003 | 0.001 |
| syx4 | syxntaxin4 | 657 | 29 | 0.0009 | −0.338 | −0.296 | 0.074 | 0.064 | 0.002 |
| frag.3 | Noncoding | 1279 | 28 | 0.0036 | 1.270 | 0.651 | 0.066 | 0.100 | 0.015 |
| frag.4 | Noncoding | 824 | 13 | 0.0052 | 1.229 | 0.549 | 0.048 | 0.440 | 0.322 |
| frag.5 | Noncoding | 992 | 10 | 0.0012 | 0.131 | 0.915 | 0.073 | 0.050 | 0.003 |
| frag.6 | CG3588 | 459 | 9 | 0.0026 | −0.788 | −0.806 | 0.147 | 0.093 | 0.005 |
| frag.7 | CG3592 | 431 | 12 | 0.016 | 1.01 | 1.634¶ | 0.159 | 0.114 | 0.212 |
Tajima's D was in all loci nonsignificant (P > 0.05).
HKA test for all fragments was performed with frag.1 as the control locus.
Two fragments covering the fixed differences between African and non-African flies (≈800 bp each) were sequenced in an additional 22 African individuals (Kenya and Zimbabwe) to confirm that the intronic and the replacement substitutions were fixed between African and non-African flies.
P (Fu and Li's D) < 0.02.
P (Fu and Li's D) < 0.05.
To evaluate the statistical significance of the observed reduction in sequence variability, we performed the Hudson–Kreitman–Aguade test (31), which compares the level of polymorphism within populations to between-species divergence. Although none of the sequenced fragments deviated from neutral expectations in African D. melanogaster, crm and syx4 in sweep region 1 and frag.5 and frag.6 in sweep region 2 had the most significant deviation from neutral expectations in non-African flies (Table 3). Further support for recent selective sweeps in non-African D. melanogaster is provided by the H test, which uses the frequency spectrum of derived sites to detect deviation from neutral expectations (33). Although no significant deviation from neutrality was observed in African flies, crm had a highly significant H test (P = 0.003), and frag.5 was marginally significant (P = 0.05) in non-African D. melanogaster. The remaining sequenced fragments showed no significant deviation from neutral expectations. Tajima's D statistic (39), a standard neutrality test, failed to provide evidence for nonneutral evolution in all tests. Similarly, Fu and Li's D statistic did not reject neutrality. Only for two fragments (crm and frag.7) was Fu and Li's D significant in European populations. In both cases, a significant deficiency of external and/or surplus of internal mutations was observed. Two reasons could be responsible for the low power of Tajima's and Fu and Li's tests in detecting the selective sweep. Because non-African flies show a strong reduction in variability, only a few segregating sites can be analyzed, which in turn results in a low power of the tests. Furthermore, the beneficial mutation may have been fixed too recently for new variation to recover by mutation, resulting in a deficiency of singletons.
Although the neutrality tests based on sequence variation seem to confirm the microsatellite-based result of recent selective sweeps, it should be noted that they are single-locus tests. For unlinked sequences, the problem of multiple testing can be taken into account by adjusting the significance level. In this study, however, the sequence stretches are located in close proximity and are not independent. When independent estimates about the effective population size, mutation, and recombination rates are available, it is possible to construct a likelihood ratio test, which considers the sequenced fragments jointly (10). Using a realistic range of parameters, we demonstrated that the partitioning of sequence variability in both sweep regions fits a hitchhiking model significantly better than a neutral model (P < 0.05; Table 7, which is published as supporting information on the PNAS web site). Moreover, the choice of parameters used had only a limited impact on the significance of the test statistic.
Mapping of Beneficial Mutations.
The shape of the standardized variability plot in Fig. 1A provides a rough idea of the location of the selected gene. An intuitive estimate of the size of the selected regions can be obtained from the distance between those two microsatellites, which are located 5′ and 3′ of the most strongly reduced loci, but the lnRV of which are within 1 standard deviation of the mean lnRV value. Using this criterion, we defined a mapping interval (later referred to as conservative mapping interval) for the location of the beneficial mutation to be 99 kb wide for sweep region 1, 31 kb for sweep region 2, and 79 kb for sweep region 3 (Fig. 1A, gray line). An alternative approach is based on the deterministic hitchhiking model (23). This model provides the expected reduction in variability in relation to the distance from the selected site. Based on the reduction in variability of two adjacent microsatellite loci, we calculated the expected position of the selected site. To account for the sampling variance in our data, we constructed a confidence interval for the position of the selected site by bootstrapping of genotypes. Note, however, that this procedure does not account for the variation among independent realizations of the selective sweep; in the absence of analytical methods to estimate this variance, we restrict our analysis to the confidence interval, which we refer to as the “hitchhiking-mapping interval” (Fig. 1A). For each sweep region, one or two candidate genes are located closest to the predicted hitchhiking-mapping interval (Table 1) with CG3588 being the gene closest to the predicted interval (4.5-kb distance) and CG13803 the farthest (26-kb distance). Some functional properties of the identified candidates are given in Table 8, which is published as supporting information on the PNAS web site.
For the two sweep regions for which sequence polymorphism data were also generated, we also used a recently published likelihood ratio test (10) to estimate the position of the selected site and the selection coefficient for each of the two regions independently. For sweep region 1, the location of the selected site was mapped to the syx4 gene, which is located 17 kb away from the selected site predicted by the hitchhiking framework. The only case for which the selected site mapped to the crm gene was if a low mutation rate was assumed (Table 7). In sweep region 2, the predicted selected site mapped to the fragment showing the lowest variability (frag.5). No gene could be predicted for this chromosomal region, which is located 21 kb away from the selected site predicted by the hitchhiking framework.
To evaluate the reliability of the sequence polymorphism mapping approach further, we repeated the likelihood ratio test for sweep region 2 but omitted in each test one of the sequenced fragments with strongly reduced variability. In these tests, the predicted selected site coincided with the corresponding fragment with a strong reduction in variability (data not shown). These analyses suggest that the mapping accuracy could be improved further by more sequence data. However, both sequence polymorphism and microsatellite data provided similar estimates for the position of the selected site. The maximum discrepancy between the two approaches was found to be less than 10 kb.
In addition to an estimate of the position of the selected site, the likelihood ratio test also provided an estimate for the selection coefficient. Depending on the assumed parameters of recombination and mutation rate, slightly different estimates for the selection coefficient were obtained. The estimated selection coefficient for sweep region 1 ranged from 0.002 to 0.01 and for sweep region 2 from 0.0001 to 0.002 (Table 7).
Discussion
A genome-wide, systematic departure from the neutral expectation for a panmictic population at equilibrium has been described for non-African D. melanogaster (34), which most likely is attributable to complex demographic scenarios. It has been shown that standard tests of neutrality using single-locus sequence polymorphism data are sensitive to past demographic events (40, 41). In contrast, the microsatellite-based lnRV test uses information from many unlinked microsatellites. Thus, the distribution of lnRV values captures the demographic history, leaving the lnRV test statistic rather insensitive to a range of demographic scenarios (13). Because neither single-locus sequence data nor the multilocus sequence-based likelihood ratio test of Kim and Stephan (10) incorporate demography, they are not sufficient to exclude the possibility that a demographic event could explain the reduction in sequence variability. However, the combination with the microsatellite-based test statistic, which is affected only marginally by demography, makes a hitchhiking scenario the favorite hypothesis.
Given that ecological adaptations probably are very difficult to reproduce in the laboratory, an emerging question is whether this hitchhiking-mapping approach could lead to the identification of the actual mutation conferring the selective advantage. Assuming that the selective sweep has been completed, we would expect the beneficial mutations to be fixed in non-African populations, whereas the variant would be absent or segregating in African populations. In sweep region 1, two genes with reduced variability were identified. Interestingly, in both genes we found fixed amino acid replacements in non-African D. melanogaster (one in the cramped gene and two in the syntaxin4 gene). Moreover, one fixed difference was detected in intron 1 of the cramped gene. Based on low Grantham's distances, the fixed amino acid replacements are predicted to cause minor changes in the protein (42). Given the short time for the fixation of beneficial mutations, it is nearly impossible that the combination of four mutations (three fixed replacements and one intronic site) caused the selective sweep, because the waiting time is too long for subsequent mutations to have occurred after the habitat expansion of D. melanogaster 10,000 years ago. Most likely, some or all of the fixed amino acid replacements are slightly deleterious mutations, which have been hitchhiking with the beneficial mutation. The consequence of such a large number of fixed differences is that the identification of the beneficial mutation, which originally caused the selective sweep in this region, is difficult without further functional tests of each of the differentially fixed sites independently. For sweep region 2, the situation is fundamentally different. We did not detect a single fixed difference between African and non-African populations in any of the sequenced fragments. Thus, sequence analysis of the whole window of reduced polymorphism potentially could identify a single fixed difference, which would then represent a candidate single-nucleotide polymorphism.
A potential strategy to confirm beneficial mutations associated with the out-of-Africa habitat expansion would be to analyze fixed substitutions in multiple non-African populations. To evaluate this strategy, we used a 2-kb fragment in the crm gene region covering one fixed amino acid replacement. Sequence analysis of this fragment in three non-African populations revealed similar variability estimates (π and θ, data not shown), and the same amino acid replacement was fixed in all populations. This result suggests that the selective sweep extends over a wider geographic range but also indicates that the comparison of different European populations may provide only limited information to discriminate between selected and hitchhiked replacements.
Conclusion
Scanning 850-kb genomic DNA, we identified three genomic regions that were the target of selection associated with the out-of-Africa colonization in D. melanogaster. Although our data are currently too limited to extrapolate to a genome-wide frequency of selective sweeps in non-African populations, they suggest that selective sweeps may be common in these populations. The contrasting pattern of X-chromosomal vs. autosomal variation previously observed for African and non-African populations of D. melanogaster (27) supports the notion of a high density of selective sweeps in colonizing populations of D. melanogaster.
Supplementary Material
Acknowledgments
We are grateful to T. Wiehe, M. Kohn, B. Charlesworth, C.-I. Wu, J. Fay, G. Muir, and A. Greenberg for helpful discussion and comments on various versions of the manuscript. J. Fay and Y. Kim provided C codes. D. Dieringer shared unpublished data. This work was supported by Fonds zur Förderung der Wissenschaftlichen Forschung grants (to C.S.). B.H. was partially supported by an Emmy-Noether Fellowship by the Deutsche Forschungsgemeinschaft.
Note Added in Proof.
Two other research groups independently identified some of the sweep regions described here [W. Stephan and D. de Lorenzo (sweep region 1) and C. Aquadro and J. Pool (sweep region 2), personal communications].
Footnotes
This paper was submitted directly (Track II) to the PNAS office.
Data deposition: The sequences reported in this paper have been deposited in the GenBank database (accession nos: cramped, AF365364–AF365399; syntaxin4, AY093819–AY093873; fragment 1, AY047980–AY048043; fragment 2, AY047924–AY047979; fragment 3, AY047879–AY047923; fragment 4, AF495973–AF495997; fragment 5, AF 495998–AF496022; fragment 6, AF495949–AF495972; and fragment 7, AF495918–AF495948).
References
- 1.Chakravarti A. Nature (London) 2001;409:822–823. doi: 10.1038/35057281. [DOI] [PubMed] [Google Scholar]
- 2.Collins F S, Guyer M S, Charkravarti A. Science. 1997;278:1580–1581. doi: 10.1126/science.278.5343.1580. [DOI] [PubMed] [Google Scholar]
- 3.Fullerton S M, Clark A G, Weiss K M, Nickerson D A, Taylor S L, Stengard J H, Salomaa V, Vartiainen E, Perola M, Boerwinkle E, Sing C F. Am J Hum Genet. 2000;67:881–900. doi: 10.1086/303070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Fay J C, Wyckoff G J, Wu C I. Nature (London) 2002;415:1024–1026. doi: 10.1038/4151024a. [DOI] [PubMed] [Google Scholar]
- 5.Fay J C, Wu C I. Curr Opin Genet Dev. 2001;11:642–646. doi: 10.1016/s0959-437x(00)00247-1. [DOI] [PubMed] [Google Scholar]
- 6.Smith N G, Eyre-Walker A. Nature (London) 2002;415:1022–1024. doi: 10.1038/4151022a. [DOI] [PubMed] [Google Scholar]
- 7.David J R, Capy P. Trends Genet. 1988;4:106–111. doi: 10.1016/0168-9525(88)90098-4. [DOI] [PubMed] [Google Scholar]
- 8.Maynard Smith J, Haigh J. Genet Res. 1974;23:23–35. [PubMed] [Google Scholar]
- 9.Wiehe T H, Stephan W. Mol Biol Evol. 1993;10:842–854. doi: 10.1093/oxfordjournals.molbev.a040046. [DOI] [PubMed] [Google Scholar]
- 10.Kim Y, Stephan W. Genetics. 2002;160:765–777. doi: 10.1093/genetics/160.2.765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Schlötterer C. Chromosoma. 2000;109:365–371. doi: 10.1007/s004120000089. [DOI] [PubMed] [Google Scholar]
- 12.Kohn M H, Pelz H J, Wayne R K. Proc Natl Acad Sci USA. 2000;97:7911–7915. doi: 10.1073/pnas.97.14.7911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Schlötterer C. Genetics. 2002;160:753–763. doi: 10.1093/genetics/160.2.753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wootton J C, Feng X, Ferdig M T, Cooper R A, Mu J, Baruch D I, Magill A J, Su X Z. Nature (London) 2002;418:320–323. doi: 10.1038/nature00813. [DOI] [PubMed] [Google Scholar]
- 15.Vigouroux Y, McMullen M, Hittinger C T, Houchins K, Schulz L, Kresovich S, Matsuoka Y, Doebley J. Proc Natl Acad Sci USA. 2002;99:9650–9655. doi: 10.1073/pnas.112324299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Payseur B A, Cutter A D, Nachman M W. Mol Biol Evol. 2002;19:1143–1153. doi: 10.1093/oxfordjournals.molbev.a004172. [DOI] [PubMed] [Google Scholar]
- 17.Schlötterer C, Vogl C, Tautz D. Genetics. 1997;146:309–320. doi: 10.1093/genetics/146.1.309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Schlötterer C. In: Molecular Genetic Analysis of Populations: A Practical Approach. 2nd Ed. Hoelzel A R, editor. Oxford: Oxford Univ. Press; 1998. pp. 237–261. [Google Scholar]
- 19.Schlötterer C, Ritter R, Harr B, Brem G. Mol Biol Evol. 1998;15:1269–1274. doi: 10.1093/oxfordjournals.molbev.a025855. [DOI] [PubMed] [Google Scholar]
- 20.Jin L, Macaubas C, Hallmayer J, Kimura A, Mignot E. Proc Natl Acad Sci USA. 1996;93:15285–15288. doi: 10.1073/pnas.93.26.15285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Weber J L. Genomics. 1990;7:524–530. doi: 10.1016/0888-7543(90)90195-z. [DOI] [PubMed] [Google Scholar]
- 22.Petes T D, Greenwell P W, Dominska M. Genetics. 1997;146:491–498. doi: 10.1093/genetics/146.2.491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wiehe T. Theor Popul Biol. 1998;53:272–283. doi: 10.1006/tpbi.1997.1346. [DOI] [PubMed] [Google Scholar]
- 24.Schlötterer C, Wiehe T. In: Microsatellites—Evolution and Applications. Goldstein D, Schlötterer C, editors. Oxford: Oxford Univ. Press; 1999. pp. 238–248. [Google Scholar]
- 25.Schug M D, Mackay T F C, Aquadro C F. Nat Genet. 1997;15:99–102. doi: 10.1038/ng0197-99. [DOI] [PubMed] [Google Scholar]
- 26.Schug M D, Hutter C M, Wetterstrand K A, Gaudette M S, Mackay T F, Aquadro C F. Mol Biol Evol. 1998;15:1750–1760. doi: 10.1093/oxfordjournals.molbev.a025901. [DOI] [PubMed] [Google Scholar]
- 27.Kauer M, Zangerl B, Dieringer D, Schlötterer C. Genetics. 2002;160:247–256. doi: 10.1093/genetics/160.1.247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Comeron J M, Kreitman M, Aguade M. Genetics. 1999;151:239–249. doi: 10.1093/genetics/151.1.239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Thompson J D, Higgins D G, Gibson T J. Nucleic Acids Res. 1994;22:4673–4680. doi: 10.1093/nar/22.22.4673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Fu Y X, Li W H. Genetics. 1993;133:693–709. doi: 10.1093/genetics/133.3.693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hudson R R, Kreitman M, Aguade M. Genetics. 1987;116:153–159. doi: 10.1093/genetics/116.1.153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Rozas J, Rozas R. Bioinformatics. 1999;15:174–175. doi: 10.1093/bioinformatics/15.2.174. [DOI] [PubMed] [Google Scholar]
- 33.Fay J C, Wu C I. Genetics. 2000;155:1405–1413. doi: 10.1093/genetics/155.3.1405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Andolfatto P, Przeworski M. Genetics. 2000;156:257–268. doi: 10.1093/genetics/156.1.257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ashburner M. Drosphila: A Laboratory Handbook. Plainview, NY: Cold Spring Harbor Lab. Press; 1989. [Google Scholar]
- 36.Bachtrog D, Weiss S, Zangerl B, Brem G, Schlötterer C. Mol Biol Evol. 1999;16:602–610. doi: 10.1093/oxfordjournals.molbev.a026142. [DOI] [PubMed] [Google Scholar]
- 37.Schug M D, Wetterstrand K A, Gaudette M S, Lim R H, Hutter C M, Aquadro C F. Mol Ecol. 1998;7:57–69. doi: 10.1046/j.1365-294x.1998.00304.x. [DOI] [PubMed] [Google Scholar]
- 38.Begun D, Aquadro C F. Nature (London) 1993;365:548–550. doi: 10.1038/365548a0. [DOI] [PubMed] [Google Scholar]
- 39.Tajima F. Genetics. 1989;123:585–595. doi: 10.1093/genetics/123.3.585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Przeworski M. Genetics. 2002;160:1179–1189. doi: 10.1093/genetics/160.3.1179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Nielsen R. Heredity. 2001;86:641–647. doi: 10.1046/j.1365-2540.2001.00895.x. [DOI] [PubMed] [Google Scholar]
- 42.Grantham R. Science. 1974;185:862–864. doi: 10.1126/science.185.4154.862. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.