

Abstract
Fluorescence correlation spectroscopy (FCS) is now a widely used technique to measure small ensembles of labeled biomolecules with single molecule detection sensitivity (e.g., low endogenous concentrations). Fluorescence cross correlation spectroscopy (FCCS) is a derivative of this technique that detects the synchronous movement of two biomolecules with different fluorescence labels. Both methods can be applied to live cells and, therefore, can be used to address a variety of unsolved questions in cell biology. Applications of FCCS with autofluorescent proteins (AFPs) have been hampered so far by cross talk between the detector channels due to the large spectral overlap of the fluorophores. Here we present a new method that combines advantages of these techniques to analyze binding behavior of proteins in live cells. To achieve this, we have used dual color excitation of a common pair of AFPs, ECFP and EYFP, being discriminated in excitation rather than in emission. This is made possible by pulsed excitation and detection on a shorter timescale compared to the average residence time of particles in the FCS volume element. By this technique we were able to eliminate cross talk in the detector channels and obtain an undisturbed cross correlation signal. The setup was tested with ECFP/EYFP lysates as well as chimeras as negative and positive controls and demonstrated to work in live HeLa cells coexpressing the two fusion proteins ECFP-connexin and EYFP-connexin.
INTRODUCTION
The method of fluorescence correlation spectroscopy (FCS) analyzes the fluctuations of the fluorescence intensity of molecules diffusing in and out of the subfemtoliter volume of a strongly focused excitation beam in a confocal microscope. From the correlation function of the fluctuations it is possible to derive the concentration, diffusion coefficient, and intramolecular properties of the fluorophore influencing its fluorescence properties. After the introduction of FCS in the early 1970s (1,2) it took two decades before the method became popular especially due to the improvement of the signal/noise ratio and detector sensitivity down to the single molecule level (3).
Life science applications are investigated to an increasing degree: binding studies of protein subunits (4), molecular identification (5), observation of conformation changes (6,7), or microsecond protein dynamics (8).
Due to its noninvasive character, FCS studies were also rapidly expanded to live cell investigations (9–11). Applications include protein oligomerization (12), diffusion processes in the nucleus (13), or determination of the hybridization state of oligonucleotides (14). Other studies have focused on technical aspects of working in live cells such as cellular autofluorescence (15) or the confinement of the excitation volume (16).
The extension of FCS, fluorescence cross correlation spectroscopy (FCCS), deals with the problem of measuring the colocalization of two diffusing species on a molecular level. In FCCS, two species must be labeled with spectrally distinct fluorophores and the fluctuations of the fluorescence must be recorded in two separate channels. In the absence of cross talk, the cross correlation of the two channels reveals whether the two species are linked to one another or not. Hence, FCCS deals with a colocalization on a molecular level sometimes referred to as codiffusion.
FCCS has been applied to test for irreversible association kinetics of DNA renaturation (17), aggregation of prion proteins (18), vesicle fusion (19), determination of the gene expression by quantification of mRNA (20), simultaneous binding of two DNA duplexes to the NtrC-enhancer complex (21), and enzyme kinetics (22) where the method is expanded to a high throughput screening tool (23).
Though FCCS is suited for a number of unsolved questions in cell biology, there are only a few reports of in vivo studies to date. Bacia et al. have shown that subunit A and B of cholera toxin do not separate along the endocyclic pathway until reaching the Golgi apparatus (24). Kim et al. studied the binding kinetics of calmodulin and calmodulin-kinase II in HEK293 cells (25).
Particularly, the latter report suggests the possibilities of FCCS in the field of cell signaling. Most signaling cascades comprise various receptors, first and second messengers building a highly complex network where diffusion of molecular components and binding-unbinding reactions between these components are the crucial parameters for understanding the cellular response. Due to its single molecule sensitivity, FCCS studies can be performed with physiological concentrations of the molecular compounds (normally proteins) that participate in a signal cascade. It is, therefore, possible to avoid artifacts from overexpression, which might perturb the delicate balance within the cascade.
The main reason for the lack of cross correlation studies in live cells is related to the different photophysical requirements on the fluorophores used. Today, the dominant fluorophores for cell biology applications are autofluorescent proteins (AFPs; (26)). AFPs can be used for specific labeling of defined gene products; they have low cytotoxicity and do not require microinjection or other invasive techniques for entering a cell. Unfortunately, AFPs are not well suited for FCCS studies, and to our knowledge there are only two FCCS works in live cells relying exclusively on AFPs (27,28). Apart from the lower photostability of AFPs compared to that of artificial dyes, the most popular AFPs, namely, ECFP, EGFP, and EYFP, show a large spectral overlap in the fluorescence spectra (see Fig. 1 for ECFP and EYFP). This leads to cross talk in the correlation signal that is hard to correct. In live cell measurements, where the concentration of the fluorophores is not known a priori and where background fluorescence is high, cross talk usually does not allow for performing FCCS.
FIGURE 1.

Excitation and emission spectra of ECFP and EYFP (from Cubitt et al. (41)). In the shaded area are the excitation wavelengths chosen to prevent cross talk and the undetected spectral range due to the trichroic beamsplitter. The circle stresses the spectral region in which, normally, cross talk arises.
Alternative methods based on, e.g., fluorescence resonance energy transfer (FRET) proved hard to implement in cell environments due to cross talk, unknown concentrations of the fluorophores, and unknown maximal FRET efficiencies (29,30).
THEORY
In FCS the concentration fluctuations of fluorescing particles in an effective volume element are used to determine the diffusion constants and concentrations of the dissolved species. In practice this is achieved by focusing an excitation beam into a solution or a live cell specimen containing the particles of interest. The focus constitutes an open volume through which particles can diffuse in and out, leading to concentration fluctuations and in turn to fluctuations of the fluorescence intensity signal F. These fluctuations are correlated in time yielding a correlation function
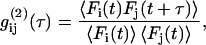 |
(1) |
where 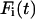 is the fluorescence intensity of the species i (
is the fluorescence intensity of the species i (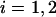 for ECFP and EYFP) at time t and the brackets denote time averaging. In the case of
for ECFP and EYFP) at time t and the brackets denote time averaging. In the case of 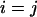 this is the autocorrelation function.
this is the autocorrelation function.
By modeling the focal volume with a three-dimensional (3D) Gaussian profile, it is possible to give an analytical model for the correlation function of free translational 3D diffusion (31):
 . .
|
(2) |
In this equation, terms 4 and 5 describe the diffusion, term 3 describes fluctuations due to protonation/deprotonation processes observed in some AFP fluorophores (32), and term 2 is a correction for uncorrelated background light. In detail, 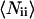 is the average number of particles within the focus,
is the average number of particles within the focus, 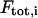 is the total fluorescence intensity,
is the total fluorescence intensity, 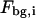 is the uncorrelated background intensity,
is the uncorrelated background intensity, 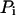 the fraction of molecules being in the protonated state,
the fraction of molecules being in the protonated state, 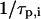 the protonation rate,
the protonation rate, 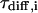 is the diffusion time, and
is the diffusion time, and 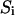 is the structural factor of the focal volume, which is defined as
is the structural factor of the focal volume, which is defined as 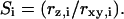 The diffusion time is inversely proportional to the diffusion constant
The diffusion time is inversely proportional to the diffusion constant 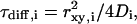 with
with 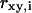 the width of the Gaussian profile and
the width of the Gaussian profile and 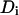 the diffusion constant.
the diffusion constant.
If the diffusion of the fluorescing particles is restricted to a planar sheet (e.g., a cell membrane), Eq. 2 can be simplified to a two-dimensional (2D) model:
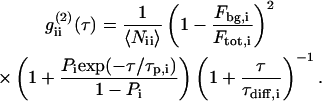 |
(3) |
In the case of different fluorophores (i.e., 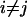 ), Eq. 1 holds for cross correlation analysis. If the excitation volumes of the two exciting laser beams overlap perfectly and the
), Eq. 1 holds for cross correlation analysis. If the excitation volumes of the two exciting laser beams overlap perfectly and the 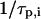 rates of the two dyes do not correlate, the cross correlation function in 3D is written as
rates of the two dyes do not correlate, the cross correlation function in 3D is written as
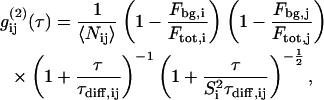 |
(4) |
and for 2D diffusion the last term can be omitted.
One of the protein complexes used for our FCCS experiments was a gap-junction hemichannel or connexon. A single connexon comprises six protein subunits called connexins. In our experiments, we have used cells that coexpress connexins labeled with ECFP and EYFP. In this case the hexamers consist of a mixture of ECFP and EYFP labeled monomers. The distribution of labels is assumed to be binomial:
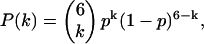 |
(5) |
with 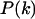 the probability of having a connexon consisting of k ECFP-connexins and
the probability of having a connexon consisting of k ECFP-connexins and 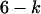 EYFP-connexins.
EYFP-connexins.
For the sake of clarity, we will first consider the simpler case of a dimer. Here, the auto- and cross correlation amplitude measure two different concentrations of the dimer because the binomial distribution gives two homolabeled dimers which do not contribute to the cross correlation amplitude and only one homolabeled dimer that does not contribute to the autocorrelation amplitude. Therefore, we introduce factors 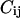 correcting this systematic error. In the dimer case, e.g., the amplitude of the cross correlation function is reduced by a factor of 2 and hence the correction factor has to be
correcting this systematic error. In the dimer case, e.g., the amplitude of the cross correlation function is reduced by a factor of 2 and hence the correction factor has to be  as it is also calculated by Eq. 6 (see below).
as it is also calculated by Eq. 6 (see below).
To derive the correction factors, Eqs. 2 and 4 are examined at 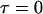 and the additional effect of different brightnesses (distribution of heterolabeled connexons) of the hexamers has to be taken into account (24). Then the cross correlation amplitude correction factor is given by
and the additional effect of different brightnesses (distribution of heterolabeled connexons) of the hexamers has to be taken into account (24). Then the cross correlation amplitude correction factor is given by
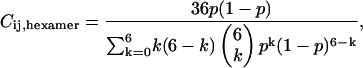 |
(6) |
for the probability of having an ECFP or a EYFP molecule assumed to be the same (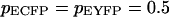 ),
), 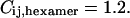 A similar calculation gives amplitude correction factors for the autocorrelation amplitudes
A similar calculation gives amplitude correction factors for the autocorrelation amplitudes 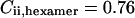 (24).
(24).
MATERIALS AND METHODS
ECFP-EYFP lysates
For the preparation of ECFP and EYFP from cell lysates we have used HEK 293 cells that were transiently transfected with the plasmids pECFP or pEYFP. In brief, cells (800 μl; 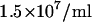 ) were harvested and 15 μg of plasmid was added to the suspension. The cells were then electroporated at 250 V and 1800 μF in a 0.4 cm cuvette (Peqbio Easyject Plus, Peqlab, Erlangen, Germany). After electroporation, the cells were immediately transferred into 15 cm cell culture dishes and grown overnight. For lysis, the cells were harvested in 500 μl buffer (10 mM HEPES pH 7.9, 10 mM KCl, 0.1 mM EDTA, 0.1 mM EGTA, 1 mM DTT, 1 mM phenylmethylsulfonyl fluoride), followed by incubation on ice for 20 min. Then 30 μl 10% Nonidet-40 was added and incubated for 2 min at 4°C while shaking. Nuclei were pelleted at 16,000 × g for 3 min at 4°C, and supernatant was used for FCCS experiments.
) were harvested and 15 μg of plasmid was added to the suspension. The cells were then electroporated at 250 V and 1800 μF in a 0.4 cm cuvette (Peqbio Easyject Plus, Peqlab, Erlangen, Germany). After electroporation, the cells were immediately transferred into 15 cm cell culture dishes and grown overnight. For lysis, the cells were harvested in 500 μl buffer (10 mM HEPES pH 7.9, 10 mM KCl, 0.1 mM EDTA, 0.1 mM EGTA, 1 mM DTT, 1 mM phenylmethylsulfonyl fluoride), followed by incubation on ice for 20 min. Then 30 μl 10% Nonidet-40 was added and incubated for 2 min at 4°C while shaking. Nuclei were pelleted at 16,000 × g for 3 min at 4°C, and supernatant was used for FCCS experiments.
Cell culture and transient transfection
For transient expression of an AFP labeled membrane protein, we used C-terminal fusion constructs of ECFP and EYFP to the gap-junction protein connexin46. HeLa cells (ECACC No. 96112022) were cultivated in Dulbecco's modified Eagle's medium (DMEM) containing 10% newborn calf serum (NCS) in a humidified CO2-incubator at 37°C. For transient transfection, we followed the protocol provided for the lipofectin reagent (Life Technologies, Karlsruhe, Germany). Briefly, the cells were grown in 35 mm glass bottom petri dishes (MatTek, Ashwell, MA) to 50–75% confluency. We used 35 mm glass bottom petri dishes because they allow a better observation of the cell monolayers in an inverted microscope. The cells were cotransfected in Opti-MEM1 medium (Life Technologies) containing 7–10 μl lipofectin and 1–2 μg of plasmid DNA (pECFP-N1-rCx46 and pEYFP-N1-rCx46 either alone or in 1:1 mixtures) for 6 h at 37°C. After this time the DNA/lipofectin suspension was removed and replaced with normal DMEM.
The cells were then transferred into an incubator set to a temperature of 27°C for 24–48 h for a better insertion of the protein in the plasma membrane.
Transfection efficiencies >30% were routinely obtained with this method for HeLa cells.
Experimental setup for cross talk free FCCS
FCCS experiments were performed using a modified Olympus IX-70 inverted microscope with a high numerical aperture objective (UPLAPO 60× 1.2 W, Olympus Europe, Hamburg, Germany). For confocal excitation a trichroic beamsplitter (Z425/514R at 10°, Chroma Technology, Rockingham, VT) reflects the excitation beams into the side port of the microscope. The collected photons pass the beamsplitter and a pinhole (diameter 50 μm) and are detected by an actively quenched avalanche photo diode (APD, SPCM-AQR 14, PerkinElmer Optoelectronics, Fremont, CA). The excitation wavelengths were supplied by an argon-ion laser (at 515 nm, Innova 90C, Coherent, Palo Alto, CA) and a frequency doubled (at 425 nm, BBO crystal, Fujian Castech Crystals, Fujian, China) passively mode-locked titan sapphire oscillator (at 850 nm, Mira 900 F, Coherent) with a pulse width of 100 fs and a repetition rate of 76 MHz, which was pumped by a frequency doubled Nd:YAG laser (Verdi V5, Coherent). The pulse repetition rate was reduced to 7.6 MHz by an acousto-optic modulator (AOM)-based pulse picker system (PulseSelect, APE, Berlin, Germany). It also provided the delayed pulse signal for the second AOM (350 MHz – 5 ns rise time, Crystal Technology, Palo Alto, CA), generating a 10 ns green laser pulse 50 ns after the blue one. The excitation power was adjusted to 50 μW for the blue and 5 μW for the green laser (measured continuous wave). The detector signal was recorded with a time correlated single photon counter card (TCSPC; SPC630 Fifo Mode, Becker & Hickl, Berlin, Germany) and analyzed by custom software including a software correlator. Confocal images were recorded by scanning the sample through the focus using a three-axis piezo actor (PiMars P-527.3CL, Physik Instrumente, Karlsruhe, Germany) mounted on the microscope stage. Fitting was performed with nonlinear least-square Levenberg-Marquardt routines.
The adjustment procedure followed this scheme: first, the blue and green excitation beams were brought into congruence at the beam combiner and right before the microscope. Then the detection path of the setup was adjusted with blue excitation light only and a thick solution of rhodamine 6G solvated in ethylenglycol as a sample. Alternating the fine adjustment of the incoming excitation beam and the detection pathway maximized the countrate at the detector. Only the green excitation beam was fine adjusted for maximum intensity at the same pinhole position as for the blue beam alone. A last adjusting step was to gain maximum parfocality of both excitation colors. This was achieved by changing the divergence of the blue beam with the last curved mirror of the pulse picker. Inspection of the two laser spots at the ceiling of the room (no objective in the revolver) showed a perfect overlap.
RESULTS
Main principle of cross talk free measurements using FCCS
The aim of this study is to measure the colocalization of proteins in live cells using the fluorophore pair ECFP and EYFP. This is not possible by standard FCCS setups due to cross talk caused by the overlap of the emission spectra of these fluorophores (Fig. 1). Indeed, using our setup with a standard filter set for ECFP and EYFP, we always see fluorescence of ECFP in the EYFP detection channel leading to cross correlation without EYFP fluorescence at all. This problem is addressed by using a pulsed excitation pattern of alternating blue and green laser pulses, which exclusively excite the cyan fluorescent protein (ECFP) with a wavelength of 425 nm and the yellow fluorescent protein (EYFP) with 515 nm (shaded bars in Fig. 1). We use a single detector to count the emitted photons of both fluorophores, which are afterwards rearranged by the software to form time traces of the two labels. For this purpose a TCSPC stores each photon with an additional time tag giving the arrival time of this photon relative to a synchronization pulse (see Fig. 2 A). The pulse repetition rate is optimized to allow for fluorescence decay after excitation to prevent cross talk in the time domain. In fact, the delay between succeeding pulses (50 ns) is much larger than the fluorescence lifetime of the AFPs (∼3 ns). The length of the blue pulse of 100 fs is given by the TiSa laser and would in principle allow additional analysis of the fluorescence lifetime of ECFP although this is not required for the colocalization studies. The cross correlation setup would also work with a continuous wave (cw) laser around 425 nm together with a fast AOM and a pulse generator to synchronize the AOMs for blue and green light.
FIGURE 2.

(A) Pulsed excitation scheme for cross talk free FCCS. Fluorescence time intervals for data analysis can be selected by software. (B) Schematic overview of the FCCS setup.
For testing the setup we have used samples of ECFP and EYFP and an ECFP/EYFP chimera (both proteins connected by a calcium sensitive calmodulin linker) in solution. In addition we have used live HeLa cells cotransfected with ECFP-connexin and EYFP-connexin fusion proteins. These connexin-AFP fusion proteins form hexameric membrane channels that are inserted into the membrane in the endoplasmatic reticulum. The channels are then transported into the plasma membrane via the Golgi apparatus. These channels form a pool of free diffusing particles in the plasma membrane. The particles of this pool show a distribution of different numbers of ECFP and EYFP labels that is assumed to be binomial. Therefore the majority of the channels should contain both labels (see Theory).
Signal enhancement by filtering in the time domain
To separate fluorescence of ECFP and EYFP, we determine the arrival time of all fluorescence photons with respect to the blue laser pulses. The blue pulses were chosen for synchronization due to the much better time resolution of these pulses (100 fs) compared to the green pulses generated by switching an AOM (10 ns).
Fig. 3 A shows the histogram of the arrival times. It exhibits two main peaks, a sharp one at 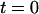 ns and a broader peak around
ns and a broader peak around 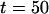 ns, corresponding to the fluorescence excited by the blue laser and the green laser, respectively. The smaller peak at
ns, corresponding to the fluorescence excited by the blue laser and the green laser, respectively. The smaller peak at 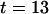 ns corresponds to an after pulse, which could not be totally suppressed by the pulse picker. The delay between main pulse and after pulse reflects the inverse repetition rate of the Titan Sapphire oscillator. Due to the time resolution of 100 fs of the blue laser, it is possible to determine the fluorescence lifetime of the fluorescence component excited by the blue laser. Here, we were able to determine the lifetime of ECFP to 3.1 ns, which is in good agreement with published values for this fluorophore (33). The fluorescence peak of the EYFP fluorescence is asymmetric and can be described as a convolution of a Gaussian and an exponential decay curve. Considering only the right wing, it is possible to observe a decay in the range of 3 ns corresponding to the lifetime of EYFP (34).
ns corresponds to an after pulse, which could not be totally suppressed by the pulse picker. The delay between main pulse and after pulse reflects the inverse repetition rate of the Titan Sapphire oscillator. Due to the time resolution of 100 fs of the blue laser, it is possible to determine the fluorescence lifetime of the fluorescence component excited by the blue laser. Here, we were able to determine the lifetime of ECFP to 3.1 ns, which is in good agreement with published values for this fluorophore (33). The fluorescence peak of the EYFP fluorescence is asymmetric and can be described as a convolution of a Gaussian and an exponential decay curve. Considering only the right wing, it is possible to observe a decay in the range of 3 ns corresponding to the lifetime of EYFP (34).
FIGURE 3.

(A) Histogram of the photon arrival times. Two main peaks corresponding to photons originating from ECFP (left) and EYFP (right) are observable. (B) Background corrected FCS curves of ECFP, EYFP, and the FCCS curve for a mixture of both fluorophores in solution (cyan, CFP autocorrelation; yellow, YFP autocorrelation; black, cross correlation; red, fit functions). (C and D) FCS curves of ECFP, EYFP, and the FCCS curve for the ECFP/EYFP chimera in solution. (C) Measured curves. (D) Background corrected curves. (E and F) FCS curves of ECFP, EYFP, and the FCCS curve for coexpressed ECFP/Cx46 and EYFP/Cx46 in live HeLa cells. (E) Measured curves. (F) Background and permutation corrected curves. (Inset) Schematic top view of six connexins fused to three ECFPs' respective EYFPs.
The histogram was measured from a solution containing exactly the same amount of ECFP and EYFP. Taking into account the absorption cross sections and quantum yields of ECFP and EYFP, the excitation power, and detection efficiencies of ECFP and EYFP, one would expect two times more photons in the EYFP channel than in the ECFP channel. Instead we observed 1.6 times more counts in the ECFP channel. These additional background photons in the ECFP channel could arise from autofluorescence or stray light from within the cell. Stray processes are fast and should appear in the histogram at 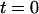 ns. For fluorescence processes from components other than ECFP, we would expect longer or shorter fluorescence lifetimes than for ECFP. In any case, it is possible to enhance the signal/noise ratio by disregarding time channels with high background fluorescence. Unfortunately, the main part of autofluorescence detected in the spectral range of ECFP also has a lifetime comparable to ECFP, resulting in a poor enhancement of the signal/noise ratio for the ECFP channel.
ns. For fluorescence processes from components other than ECFP, we would expect longer or shorter fluorescence lifetimes than for ECFP. In any case, it is possible to enhance the signal/noise ratio by disregarding time channels with high background fluorescence. Unfortunately, the main part of autofluorescence detected in the spectral range of ECFP also has a lifetime comparable to ECFP, resulting in a poor enhancement of the signal/noise ratio for the ECFP channel.
Except for this autofluorescence, which obviously can be excited by 425 nm, other sources of background have been systematically eliminated by time resolved measurements of the buffer, several different coverslips (BK7, SUPRASIL I and II), and the setup without any specimens (data not shown).
Signal/noise ratio
In this section we want to discuss the signal/noise ratio of the correlation curve using cw and pulsed excitation, respectively. First assume time intervals of the inverse repetition rate (Δt = 130 ns) and cw excitation, i.e., pulse lengths of 130 ns, too. Using a typical count rate of 10 kcps and a detection efficiency of 10% of the microscope, the probability for emitting a photon in the focus per inverse repetition rate Δt is 1.3%. Provided that the number of photons follows a Poisson distribution, the probability that two photons are emitted is <0.01%, i.e., per 150 intervals containing one photon there is one interval containing two photons. Because of the missing photon pairs with short time distance, marginally lower signal/noise values for short correlation times τ would be expected in pulsed excitation. To check this, we recorded 50 correlation curves with cw and pulsed (pulse width 10 ns) excitation, respectively, using the same count rate of 4 kcps. The signal/noise ratio as a function of time is 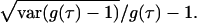 For short times, the signal/noise ratio is slightly better for cw excitation (factor of 1.4), for times around the diffusion time there is no difference, and for times longer then the diffusion time the signal/noise ratio is slightly better for pulsed excitation (factor 1.4). The differences become smaller for longer pulse widths.
For short times, the signal/noise ratio is slightly better for cw excitation (factor of 1.4), for times around the diffusion time there is no difference, and for times longer then the diffusion time the signal/noise ratio is slightly better for pulsed excitation (factor 1.4). The differences become smaller for longer pulse widths.
Deviation of the diffusion time of ECFP and EYFP
All ECFP autocorrelation curves showed a systematic error toward apparently longer diffusion times (see Fig. 3 B, D, and F). Two effects can account for this enlargement of the confocal volume: i), the short excitation pulses (∼100 fs) with a high peak power caused saturation of the fluorescence emission in the Gaussian excitation profile, effectively broadening the focus (35); and ii), due to chromatic aberrations of the objective for the emission wavelength of ECFP, the detected focal volume for ECFP is shifted in the z direction with respect to the effective detection volume for EYFP (spanned by 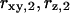 ). This results in an effective detection volume for ECFP that is bigger than the focal volume for EYFP.
). This results in an effective detection volume for ECFP that is bigger than the focal volume for EYFP.
To estimate the impact of the first effect, we measured the diffusion times of a diluted ECFP lysate excited with 100 fs pulses as a function of the mean excitation power. The diffusion time started to increase for excitation powers >10 μW and rose until a maximum of a factor of 1.9 was reached at 70 μW, comparable to data published before (35). A further increase of the excitation power yielded a decrease of the diffusion time due to bleaching of the fluorophore in the excitation volume. The correlation time measured at lowest power is close to the value obtained with cw excitation. We want to compare the ECFP diffusion with the EYFP diffusion excited with 10 ns pulses. For such long pulses the saturation effect is relatively small (35), i.e., the correlation times measured for EYFP are close to values of cw measurements, too.
The correlation curves measured in live cells (see Fig. 3 F) were excited with 80 μW and show diffusion times of τYFP = 7.5 ms and τCFP = 16.4 ms, i.e., the diffusion time observed with short pulses is a factor of 2.2 longer than with long pulses or cw. Compared to this result, a factor of 1.8 was found for the diffusion of ECFP excited with 5 μW (comparable to cw) and 70 μW in the lysate. For the correlation curves of the chimera in lysate, which was excited with 50μW/blue, the difference is smaller. Here, an increase of the diffusion time of a factor of 1.2 (τYFP, = 0.18 ms and τCFP = 0.22 ms) was observed compared to 1.3 for ECFP excited with 5 μW and 50 μW. Hence, most of the increase of the diffusion time can be explained by saturation effects due to short pulsed excitation.
ECFP-EYFP in lysate
The first step to test our experimental approach is to check whether there is any cross correlation between unbound ECFP and EYFP in solution. For this we have used either single or mixed solutions of free ECFP and EYFP from cell lysates. The lysates were diluted with phosphate-buffered saline to concentrations of 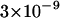 mol/l, i.e., to a concentration where on average a single molecule is found in the focal volume.
mol/l, i.e., to a concentration where on average a single molecule is found in the focal volume.
Experiments that test for channel cross talk with solutions containing only ECFP or EYFP showed no emission of ECFP with excitation by 515 nm laser light (EYFP excitation) and vice versa (data not shown).
Fig. 3 B shows the corresponding background corrected autocorrelation and cross correlation curves for freely diffusing ECFP and EYFP in mixed solution. The FCS curves are obtained by taking all photons of the arrival time histogram into account. The curves were least square fitted with the diffusion term of Eq. 2 (all errors given originate from the fitting procedure). The diffusion constants have similar values of 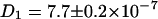 cm2/s and
cm2/s and 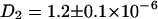 cm2/s, which are reasonable values for small proteins in solution (13,36). A deviation of the fit function compared to the measured curve for the EYFP at longer times τ most probably arises from small aggregates with other proteins in the lysate. No cross correlation is observable (noisy black line in Fig. 3 B) as expected for this solution of unbound ECFP and EYFP labels.
cm2/s, which are reasonable values for small proteins in solution (13,36). A deviation of the fit function compared to the measured curve for the EYFP at longer times τ most probably arises from small aggregates with other proteins in the lysate. No cross correlation is observable (noisy black line in Fig. 3 B) as expected for this solution of unbound ECFP and EYFP labels.
ECFP/EYFP chimera in lysate
Fig. 3 C shows the correlation curves for the ECFP/EYFP chimera in lysate, which is diluted with phosphate-buffered saline (no Ca2+ ions included to prevent FRET in the chimera). Here, the diffusion constants are slightly smaller (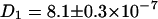 cm2/s and
cm2/s and 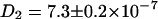 cm2/s) than in the mixture sample of ECFP-EYFP due to the bigger hydrodynamic radius of the fusion protein. As expected this system shows a cross correlation amplitude. The value for the diffusion constant of the cross correlation curve is slightly lower (
cm2/s) than in the mixture sample of ECFP-EYFP due to the bigger hydrodynamic radius of the fusion protein. As expected this system shows a cross correlation amplitude. The value for the diffusion constant of the cross correlation curve is slightly lower (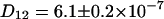 cm2/s) than for the autocorrelation. Applying the fluorescence background correction term of Eq. 2 (Eq. 4 for the cross correlation) results in a cross correlation amplitude of half the theoretical maximum value, assuming the green channel is free of background fluorescence (see Fig. 3 D). Possible reasons are a nonideal overlap of the excitation volumes and nonfunctional AFPs in the construct.
cm2/s) than for the autocorrelation. Applying the fluorescence background correction term of Eq. 2 (Eq. 4 for the cross correlation) results in a cross correlation amplitude of half the theoretical maximum value, assuming the green channel is free of background fluorescence (see Fig. 3 D). Possible reasons are a nonideal overlap of the excitation volumes and nonfunctional AFPs in the construct.
ECFP-connexin and EYFP-connexin in live cells
Fig. 3 E shows the autocorrelation and cross correlation curves for ECFP- and EYFP-connexin fusion proteins in the plasma membrane of live HeLa cells. Connexins form a hexameric complex called connexon, schematically shown in Fig. 3 F (inset). For the correlation curve of EYFP, the protonation term has to be taken into account (Eqs. 3 and 4 for cross correlation without the last term) to achieve satisfactory fitting. The values derived for 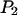 and
and 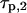 are in good agreement with previously reported values (32). In the measurement results shown in Fig. 3, B–D, this blinking was obscured because it is in the same time region as the diffusion of the fast chromophores in solution. The ECFP curve, on the other hand, does not show this protonation behavior.
are in good agreement with previously reported values (32). In the measurement results shown in Fig. 3, B–D, this blinking was obscured because it is in the same time region as the diffusion of the fast chromophores in solution. The ECFP curve, on the other hand, does not show this protonation behavior.
The diffusion constants of 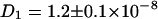 cm2/s for ECFP and
cm2/s for ECFP and 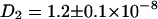 cm2/s for EYFP correspond well with membrane protein diffusion in live cells (37,38). The cross correlation diffusion constant is again close to the autocorrelation values (
cm2/s for EYFP correspond well with membrane protein diffusion in live cells (37,38). The cross correlation diffusion constant is again close to the autocorrelation values (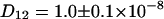 cm2/s), as expected. After fluorescence background correction of the data and taking into account the various compositions of a connexon (homo- and several heterolabeled hexamers, see Theory), an amplitude of the cross correlation curve (see Fig. 3 F) of 1.86, compared to a maximum of 2.18, shows the spectral overlap of the excitation volumes to be ∼86%.
cm2/s), as expected. After fluorescence background correction of the data and taking into account the various compositions of a connexon (homo- and several heterolabeled hexamers, see Theory), an amplitude of the cross correlation curve (see Fig. 3 F) of 1.86, compared to a maximum of 2.18, shows the spectral overlap of the excitation volumes to be ∼86%.
CONCLUSIONS
We were able to design and test a new setup with pulsed excitation lasers to prevent cross talk in the detection pathway to measure the colocalization of proteins on a single molecule level. With this setup we have successfully demonstrated colocalization of membrane proteins labeled with the living colors ECFP and EYFP at molecular level in live cells. We were also able to prove the concept with positive and negative control experiments using unbound ECFP and EYFP from cell lysates in buffer. The major problem in all measurements was autofluorescence in the ECFP channel. Our method provides the advantage of filtering in the time regime, i.e., suppressing all fluorescence photons arriving from faster (e.g., Raman scattering) and slower decay processes (e.g., fluorescence of color centers in glass). Unfortunately, in the biological specimens investigated in this study, most of the autofluorescence shows the same decay time as the ECFP molecules. However, working with other cell lines may result in other fluorescing components and, hence, in less autofluorescence from fluorophores exactly matching the photophysical properties of ECFP.
For measurements in the presence of high autofluorescence, multiple ECFP labels (2–3) per protein proved to be useful to detect this protein in the ECFP channel above background.
Future developments could improve the investigations of colocalizations in live cells. Obviously, the development of AFPs with properly separated emission spectra would allow the application of the standard FCCS method using two detectors. Nowadays available monomeric red fluorescent proteins show weak fluorescence (39) that is difficult to detect effectively above the background in cells. Nevertheless, Saito et al. (27) and Baudendistel et al. (28) succeeded recently in this task by using mRFP1 fused to the sample protein and measured FCCS curves in live cells. A very promising technique for all live cell studies would be the application of fusion proteins in combination with artificial dyes that have much better photophysical properties than AFPs. First results on this technique were shown recently (40).
Today, AFPs (and among them ECFP and EYFP) are the dominant fluorophores in cell biology applications and are widely used in many laboratories. Our new technique of FCCS might support the investigations of protein-protein interactions on a single molecule level within a live cell. For this an easier to use and cheaper setup can be used utilizing two pulsed laser diodes or two AOMs and suitable laser sources for excitation. The relatively short data acquisition time of 30 s allows time resolved observation of protein interactions, e.g., in a signaling cascade. This will shed new light on cell functions, which now can be monitored under physiological conditions in live cells with resolution below diffraction limit. This should work in cellular membranes and in the cytosol as well as in any other larger compartment of cells.
Acknowledgments
We thank Anja Krippner-Heidenreich from the Institute of Cell Biology and Immunology IZI of the University of Stuttgart for preparing the ECFP and EYFP lysates, and Hans Rudolph from the Institute of Biochemistry of the University of Stuttgart for the supply of the chimera.
The Deutsche Forschungsgemeinschaft (DFG; grants SFB 495 and Ec141/6-1) provided financial support.
References
- 1.Ehrenberg, M., and R. Rigler. 1974. Rotational Brownian-motion and fluorescence intensity fluctuations. Chem. Phys. 4:390–401. [Google Scholar]
- 2.Magde, D., W. W. Webb, and E. Elson. 1972. Thermodynamic fluctuations in a reacting system—measurement by fluorescence correlation spectroscopy. Phys. Rev. Lett. 29:705–708. [Google Scholar]
- 3.Rigler, R., and J. Widengren. 1990. Ultrasensitive detection of single molecules as observed by fluorescence correlation spectroscopy. In Bioscience. B. Klinge and C. Owman, editors. Lund University Press, Lund, Sweden. 180–183.
- 4.Diez, M., M. Börsch, B. Zimmermann, P. Turina, S. D. Dunn, and P. Gräber. 2004. Binding of the b-subunit in the ATP synthase from Escherichia coli. Biochemistry. 43:1054–1064. [DOI] [PubMed] [Google Scholar]
- 5.Földes-Papp, Z., U. Demel, and G. P. Tilz. 1998. Ultrasensitive detection and identification of fluorescent molecules by FCS: impact for immunobiology (in biological sciences). Proc. Natl. Acad. Sci. USA. 98:11509–11514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Börsch, M., P. Turina, C. Eggeling, J. R. Fries, C. A. M. Seidel, A. Labahn, and P. Gräber. 1998. Conformational changes of the H+-ATPase from Escherichia coli upon nucleotide binding detected by single molecule fluorescence. FEBS Lett. 437:251–254. [DOI] [PubMed] [Google Scholar]
- 7.Kim, H. D., G. U. Nienhaus, T. Ha, J. W. Orr, J. R. Williamson, and S. Chu. 2002. Mg2+-dependent conformational change of RNA studied by fluorescence correlation and FRET on immobilized single molecules. Proc. Natl. Acad. Sci. USA. 99:4284–4289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Chattopadhyay, K., S. Saffarian, E. L. Elson, and C. Frieden. 2002. Measurement of microsecond dynamic motion in the intestinal fatty acid binding protein by using fluorescence correlation spectroscopy. Proc. Natl. Acad. Sci. USA. 99:14171–14176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Berland, K. M., P. T. C. So, and E. Gratton. 1995. 2-Photon fluorescence correlation spectroscopy—method and application to the intracellular environment. Biophys. J. 68:694–701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Maiti, S., U. Haupts, and W. W. Webb. 1997. Fluorescence correlation spectroscopy: diagnostics for sparse molecules. Proc. Natl. Acad. Sci. USA. 94:11753–11757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Schwille, P., U. Haupts, S. Maiti, and W. W. Webb. 1999. Molecular dynamics in living cells observed by fluorescence correlation spectroscopy with one- and two-photon excitation. Biophys. J. 77:2251–2265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Chen, Y., L. N. Wei, and J. D. Muller. 2003. Probing protein oligomerization in living cells with fluorescence fluctuation spectroscopy. Proc. Natl. Acad. Sci. USA. 100:15492–15497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wachsmuth, M., W. Waldeck, and J. Langowski. 2000. Anomalous diffusion of fluorescent probes inside living cell nuclei investigated by spatially-resolved fluorescence correlation spectroscopy. J. Mol. Biol. 298:677–689. [DOI] [PubMed] [Google Scholar]
- 14.Politz, J. C., E. S. Browne, D. E. Wolf, and T. Pederson. 1998. Intranuclear diffusion and hybridization state of oligonucleotides measured by fluorescence correlation spectroscopy in living cells. Proc. Natl. Acad. Sci. USA. 95:6043–6048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Brock, R., M. A. Hink, and T. M. Jovin. 1998. Fluorescence correlation microscopy of cells in the presence of autofluorescence. Biophys. J. 75:2547–2557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Gennerich, A., and D. Schild. 2000. Fluorescence correlation spectroscopy in small cytosolic compartments depends critically on the diffusion model used. Biophys. J. 79:3294–3306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Schwille, P., F. J. Meyer-Almes, and R. Rigler. 1997. Dual-color fluorescence cross-correlation spectroscopy for multicomponent diffusional analysis in solution. Biophys. J. 72:1878–1886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Bieschke, J., A. Giese, W. Schulz-Schaeffer, I. Zerr, S. Poser, M. Eigen, and H. Kretzschmar. 2000. Ultrasensitive detection of pathological prion protein aggregates by dual-color scanning for intensely fluorescent targets. Proc. Natl. Acad. Sci. USA. 97:5468–5473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Swift, J. L., A. Carnini, T. E. S. Dahms, and D. T. Cramb. 2004. Anesthetic-enhanced membrane fusion examined using two-photon fluorescence correlation spectroscopy. J. Phys. Chem. B. 108:11133–11138. [Google Scholar]
- 20.Camacho, A., K. Korn, M. Damond, J. F. Cajot, E. Litborn, B. H. Liao, P. Thyberg, H. Winter, A. Honegger, P. Gardellin, and R. Rigler. 2004. Direct quantification of mRNA expression levels using single molecule detection. J. Biotechnol. 107:107–114. [DOI] [PubMed] [Google Scholar]
- 21.Rippe, K. 2000. Simultaneous binding of two DNA duplexes to the NtrC-enhancer complex studied by two-color fluorescence cross-correlation spectroscopy. Biochemistry. 39:2131–2139. [DOI] [PubMed] [Google Scholar]
- 22.Kettling, U., A. Koltermann, P. Schwille, and M. Eigen. 1998. Real-time enzyme kinetics monitored by dual-color fluorescence cross-correlation spectroscopy. Proc. Natl. Acad. Sci. USA. 95:1416–1420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Koltermann, A., U. Kettling, J. Bieschke, T. Winkler, and M. Eigen. 1998. Rapid assay processing by integration of dual-color fluorescence cross-correlation spectroscopy: high throughput screening for enzyme activity. Proc. Natl. Acad. Sci. USA. 95:1421–1426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Bacia, K., I. V. Majoul, and P. Schwille. 2002. Probing the endocytic pathway in live cells using dual-color fluorescence cross-correlation analysis. Biophys. J. 83:1184–1193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kim, S. A., K. G. Heinze, M. N. Waxham, and P. Schwille. 2004. Intracellular calmodulin availability accessed with two-photon cross-correlation. Proc. Natl. Acad. Sci. USA. 101:105–110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Tsien, R. Y. 1998. The green fluorescent protein. Annu. Rev. Biochem. 67:509–544. [DOI] [PubMed] [Google Scholar]
- 27.Saito, K., I. Wada, M. Tamura, and M. Kinjo. 2004. Direct detection of caspase-3 activation in single live cells by cross-correlation analysis. Biochem. Biophys. Res. Commun. 324:849–854. [DOI] [PubMed] [Google Scholar]
- 28.Baudendistel, N., G. Müller, W. Waldeck, P. Angel, and J. Langowski. 2005. Two-hybrid fluorescence cross-correlation spectroscopy detects protein-protein interactions in vivo. ChemPhysChem. 6:984–990. [DOI] [PubMed] [Google Scholar]
- 29.Day, R. N., A. Periasamy, and F. Schaufele. 2001. Fluorescence resonance energy transfer microscopy of localized protein interactions in the living cell nucleus. Methods. 25:4–18. [DOI] [PubMed] [Google Scholar]
- 30.Selvin, P. R. 2000. The renaissance of fluorescence resonance energy transfer. Nat. Struct. Biol. 7:730–734. [DOI] [PubMed] [Google Scholar]
- 31.Aragón, S. R., and R. Pecora. 1975. Fluorescence correlation spectroscopy and Brownian rotational diffusion. Biopolymers. 14:119–137. [Google Scholar]
- 32.Schwille, P., S. Kummer, A. A. Heikal, W. E. Moerner, and W. W. Webb. 2000. Fluorescence correlation spectroscopy reveals fast optical excitation-driven intramolecular dynamics of yellow fluorescent proteins. Proc. Natl. Acad. Sci. USA. 97:151–156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hoppe, A., K. Christensen, and J. A. Swanson. 2002. Fluorescence resonance energy transfer-based stoichiometry in living cells. Biophys. J. 83:3652–3664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Striker, G., V. Subramaniam, C. A. M. Seidel, and A. Volkmer. 1999. Photochromicity and fluorescence lifetimes of green fluorescent protein. J. Phys. Chem. B. 103:8612–8617. [Google Scholar]
- 35.Gregor, I., D. Patra, and J. Enderlein. 2005. Optical saturation in fluorescence correlation spectroscopy under continuous-wave and pulsed excitation. ChemPhysChem. 6:164–170. [DOI] [PubMed] [Google Scholar]
- 36.Harms, G. S., L. Cognet, P. H. M. Lommerse, G. A. Blab, and T. Schmidt. 2001. Autofluorescent proteins in single-molecule research: applications to live cell imaging microscopy. Biophys. J. 80:2396–2408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Schütz, G. J., G. Kada, V. P. Pastushenko, and H. Schindler. 2000. Properties of lipid microdomains in a muscle cell membrane visualized by single molecule microscopy. EMBO J. 19:892–901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Borisenko, V., T. Lougheed, J. Hesse, E. Fureder-Kitzmuller, N. Fertig, J. C. Behrends, G. A. Woolley, and G. J. Schütz. 2003. Simultaneous optical and electrical recording of single gramicidin channels. Biophys. J. 84:612–622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Campbell, R. E., O. Tour, A. E. Palmer, P. A. Steinbach, G. S. Baird, D. A. Zacharias, and R. Y. Tsien. 2002. A monomeric red fluorescent protein. Proc. Natl. Acad. Sci. USA. 99:7877–7882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Keppler, A., H. Pick, C. Arrivoli, H. Vogel, and K. Johnsson. 2004. Labeling of fusion proteins with synthetic fluorophores in live cells. Proc. Natl. Acad. Sci. USA. 101:9955–9959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Cubitt, A. B., L. A. Woollenweber, and R. Heim. 1999. Understanding structure-function relationships in the Aequorea victoria green fluorescent protein. In Methods in Cell Biology: Green Fluorescent Proteins. K. F. Sullivan and S. A. Kay, editors. Academic Press, San Diego. 58:19–30. [DOI] [PubMed]