

Abstract
The nearly neutral theory of molecular evolution predicts that slightly deleterious mutations subject to purifying selection are widespread in natural populations, particularly those of large effective population size. To test this hypothesis, the standardized difference between pairwise nucleotide difference and number of segregation sites (corrected for number of sequences) was estimated for 149 population data sets from 84 species of bacteria. This quantity (Tajima's D-statistic) was estimated separately for synonymous (Dsyn) and nonsynonymous (Dnon) polymorphisms. Dsyn was positive in 70% of data sets, and the overall median Dsyn (0.873) was positive. By contrast Dnon was negative in 68% of data sets, and the overall median Dnon (−0.656) was negative. The preponderance of negative values of Dnon is evidence that there are widespread rare nonsynonymous polymorphisms in the process of being eliminated by purifying selection, as predicted to occur in populations with large effective size by the nearly neutral theory. The major exceptions to this trend were seen among surface proteins, particularly those of bacteria parasitic on vertebrates, which included a number of cases of polymorphisms apparently maintained by balancing selection.
THE concept of purifying or negative natural selection—i.e., natural selection acting to decrease the frequency of deleterious alleles—is one of the key ideas of modern evolutionary biology (Kimura and Ohta 1974; Kimura 1983). As first pointed out by Kimura (1977), strong evidence for the widespread occurrence of purifying selection is provided by the fact that, in most comparisons between homologous genes, the number of synonymous nucleotide substitutions per synonymous site (dS) generally exceeds the number of nonsynonymous nucleotide substitutions per nonsynonymous site (dN; Li et al. 1985). However, this evidence provides no information regarding the overall strength of purifying selection, which is an important factor in the extent to which nonsynonymous polymorphisms are observed in populations.
If most nonsynonymous mutations are strongly deleterious, they will be eliminated from populations very quickly. On the other hand, the fate of slightly deleterious nonsynonymous mutations depends on effective population size (Ohta 1976). Purifying selection against slightly deleterious mutations is not efficient in populations with small effective size but is much more efficient as effective population size increases. Ohta's “nearly neutral” theory of molecular evolution emphasizes the importance of such slightly deleterious mutations in the evolutionary process (Ohta 1973, 1976, 2002). Some recent evidence in support of this theory is based on the gene diversity at single-nucleotide polymorphism (SNP) loci in the human population. Nonsynonymous SNPs—particularly those having radical effects on protein structure—tend to have lower average gene diversities than synonymous and noncoding SNPs in the same genes (Freudenberg-Hua et al. 2003; Hughes et al. 2003; Sunyaev et al. 2003; Zhao et al. 2003). This pattern suggests that slightly deleterious alleles, which had drifted to relatively high allelic frequencies when the effective size of the human population was low (Harpending et al. 1998), have decreased in frequency as a result of purifying selection after population expansion (Hughes et al. 2003).
Tajima (1989) pointed out that important inferences regarding population processes can be obtained from a sample of allelic DNA sequences by comparing the average number of pairwise nucleotide differences (k̂) with the number of segregating sites, corrected for the number of sequences compared. Specifically, if S is the number of segregating (or polymorphic) sites, we define S* = S/a1, where
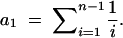 |
1 |
Both k̂ and S* are estimators of the population parameter M = 4Nu, where N is the effective population size and u is the mutation rate per generation per sequence under investigation. However, when rare variants are present in a population, S* will tend to be larger than k̂. The difference between these two quantities, divided by its standard error, is known as Tajima's D-statistic; D represents an index of the action of natural selection on a sample of allelic sequences (Tajima 1989). When D is strongly positive, there are few rare variants, a situation found under balancing selection. When D is strongly negative, rare variants are abundant, a situation indicating purifying selection (Tajima 1989).
Several authors have extended Tajima's (1989) logic to examine synonymous and nonsynonymous polymorphisms separately (e.g., Rand and Kann 1996; Wise et al. 1998; Navarro-Sabaté et al. 2003). Here I applied this method to 149 nucleotide sequence data sets from 84 species of bacteria. To avoid ambiguity, the analysis was restricted to codons at which only a single polymorphic site was observed. Computing the D-statistic separately for synonymous (Dsyn) and nonsynonymous (Dnon) polymorphisms makes it possible to test the hypothesis that rare variants are particularly common at nonsynonymous sites, indicating purifying selection against slightly deleterious alleles.
On the nearly neutral theory, purifying selection against slightly deleterious alleles is expected in bacteria, because most bacterial species are believed to have very large effective population sizes (Lynch and Conery 2003). Feil et al. (2003) recently reported a high level of nonsynonymous nucleotide polymorphisms in Staphylococcus aureus and suggested that many of these may be deleterious mutations in the process of elimination by purifying selection. On the other hand, the bacterial species analyzed include several that are parasitic on vertebrates; certain proteins, particularly surface proteins exposed to the host immune system in these species, are known to be subject to positive selection (Reid et al. 1999). Thus, these data make it possible to compare the effects on sequence polymorphism of both positive and purifying selection.
METHODS
Data sets of protein-coding genes from bacteria (eubacteria) were chosen from the NCBI Popset database. No sets that represented laboratory isolates of a single strain were included, but only sets that had been collected as patient or environmental isolates or reference strains (e.g., American Type Culture Collection). I included only data sets for which functional information about the encoded protein was available, data sets containing at least four allelic sequences, and data sets including both synonymous and nonsynonymous polymorphisms at codons with only a single polymorphic site. Using these criteria, it was possible to find usable data sets for 7 of 21 bacterial phyla. The mean number of sequences per data set was 13.11 (±0.86 SE); the median was 11.00; the range was 4–64.
The sequences were aligned at the amino acid level using the CLUSTALW program (Thompson et al. 1994), and the resulting alignment was imposed on the DNA. Portions of any alignment that appeared unreliable on account of large numbers of gaps or poor sequence conservation and codons including undetermined nucleotides were removed. The numbers of synonymous substitutions per synonymous site (dS) and of nonsynonymous substitutions per nonsynonymous site (dN) were estimated by Nei and Gojobori's (1986) method. In preliminary analyses, more complex methods of estimating dS and dN (Li 1993; Zhang et al. 1998; Yang and Neilsen 2000) yielded essentially identical results, as is expected in a case like the present one where numbers of substitutions per site are low (Nei and Kumar 2000). dS and dN were estimated for each pairwise comparison between homologous sequences in two ways: (1) for the entire sequence and (2) for the sequence excluding codons with two or more polymorphic sites. From these values, πS (the mean of all pairwise dS values) and πN (the mean of all pairwise dN values) were estimated for each data set.
After excluding codons with two or more polymorphic sites, the following quantities were computed: the average number of pairwise synonymous differences (k̂S) and the average number of nonsynonymous pairwise differences (k̂N), the number of synonymous segregating sites (SS), and the number of nonsynonymous segregating sites (SN). Then, I computed 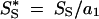 and
and 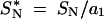 , where a1 is as in Equation 1 above. Dsyn was defined as kS − S*S, divided by the standard error of that difference (computed as described by Tajima 1989). Likewise, Dnon was defined as kN − S*N, divided by the standard error of that difference (computed as described by Tajima 1989).
, where a1 is as in Equation 1 above. Dsyn was defined as kS − S*S, divided by the standard error of that difference (computed as described by Tajima 1989). Likewise, Dnon was defined as kN − S*N, divided by the standard error of that difference (computed as described by Tajima 1989).
Bacterial species were characterized as primarily parasitic on vertebrates if a vertebrate host constitutes a major portion of the species niche. Thus species that can be vertebrate pathogens but whose primary niche is not parasitism on vertebrates (e.g., Bacillus anthracis and Vibrio cholerae) were not included in the group of vertebrate parasites. Because surface proteins may be particularly important in interactions with the immune system of vertebrates, all proteins were categorized on the basis of surface or nonsurface expression.
Because the distributions of πS, πN, Dsyn, and Dnon across the 149 data sets deviated significantly from normality (Kolmogorov-Smironov test), nonparametric methods were used for testing hypotheses regarding differences among data sets.
RESULTS
The occurrence of polymorphic sites per codon was compared with that expected, assuming a Poisson distribution with μ equal to the observed frequency of polymorphic sites per codon (Figure 1). The hypothesis that the median differerence between observed and expected proportions of codons having a given number of polymorphic sites equaled zero was tested by Wilcoxon signed-rank tests (Figure 1). There were significantly fewer codons than expected with zero or two substitutions, but significantly more codons than expected with one substitution (Figure 1). Thus, the distribution of polymorphic sites in these data showed overall a more dispersed pattern than did a Poisson distribution.
Figure 1.—
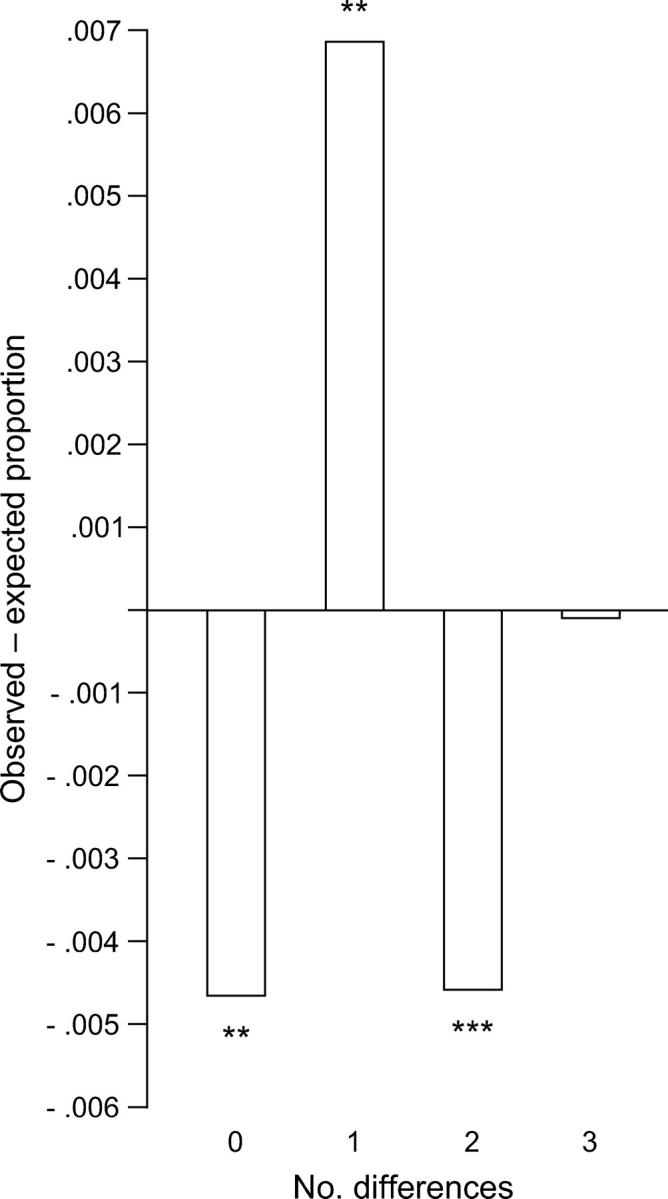
Median pairwise difference between observed and expected (assuming a Poisson distribution) proportions of codons with zero to three polymorphic nucleotides in the 149 data sets. Tests of the equality of the median proportion observed and the median proportion expected (Wilcoxon signed-rank test) are **P < 0.01 and ***P < 0.001.
For each data set, similar results were obtained when πS and πN were estimated for the entire sequence and for the sequence excluding codons with two or three polymorphic sites. In both cases, median πS (0.0836 and 0.0835, respectively) was significantly greater than median πN (0.0070 and 0.0044, respectively). In seven data sets πN exceeded πS by both methods of computation, but in none of these was there a significant difference between πN and πS (z-test).
Dsyn was positive in 104 (69.8%) of the 149 data sets, and median Dsyn (0.8726) was positive (Figure 2). On the other hand, Dnon was negative in 102 (69.4%) of data sets, and median Dnon (−0.6555) was negative (Figure 2). Median Dsyn exceeded Dnon in 123 (82.6%) of 149 data sets, and the two medians differed significantly from each other (Wilcoxon signed-rank test; P < 0.001; Figure 2). Also both median Dsyn and median Dnon were significantly different from zero (Wilcoxon signed-rank test; P < 0.001 in each case).
Figure 2.—

Plot of Dnon vs. Dsyn for the 149 data sets. The line is a 45° line. Median Dsyn was significantly greater than median Dnon (Wilcoxon signed-rank test, P < 0.001).
Overall, Dsyn and Dnon were positively correlated (Spearman's rank correlation coefficient, rS = 0.236; P = 0.004; Figure 2). An examination of the data showed six data sets with high positive values of both Dnon and Dsyn. These unusual data sets were the following (with Dsyn and Dnon values, respectively, in parentheses): Borrelia burgdoerferi flagellin (8.8576, 2.4235), Clostridium difficile adhesin (4.2776, 2.9993) and S-layer protein (3.5102, 1.9122), Streptococcus pneumoniae cspB (4.9233, 1.6862), Vibrio cholerae toxin coregulated pilus subunit (10.1966, 2.0389), and Wolbachia pipientis surface protein wsp (4.2608, 3.1798). When these six data sets were excluded, there was no longer a significant correlation between Dsyn and Dnon (rS = 0.147; NS).
Bacteria were categorized in terms of parasitism on vertebrates and in terms of surface expression of the proteins, and median πS and πN were computed for each category (Figure 3). Figure 3 illustrates median πS and πN computed excluding codons with two or three polymorphic sites; similar results were seen when the latter codons were included (data not shown). There was no significant difference with respect to median πS among categories (Figure 3). However, there was a highly significant difference among categories with respect to median πN (Kruskal-Wallis test; P < 0.001). Median πN was much higher for surface proteins from bacteria parasitic on vertebrates than for the other three categories (Figure 3B).
Figure 3.—

Median values of (A) πS and (B) πN for data sets categorized by parasitism on vertebrates and surface expression of the protein. There was no significant difference among categories with respect to median πS (Kruskal-Wallis test), but median πN differed significantly among categories (Kruskal-Wallis test, P < 0.001).
Similar patterns were seen when median Dsyn and Dnon were compared across categories (Figure 4). No significant differences among categories were observed with respect to median Dsyn, but median Dnon differed significantly among categories (Kruskal-Wallis test; P = 0.035). Median Dnon was higher for surface proteins from bacteria parasitic on vertebrates than for the other three categories (Figure 4B). Four of the six data sets identified as having unusually high values of both Dsyn and Dnon were surface proteins of bacteria primarily parasitic on vertebrates, namely B. burgdoerferi flagellin, C. difficile adhesin and S-layer protein, and S. pneumoniae cspB. Yet even when these six data sets were excluded from the analysis, there was still a significant difference among categories with respect to median Dnon (Kruskal-Wallis test; P = 0.04), and the highest median Dnon (−0.1683) was still seen in surface proteins of bacteria primarily parasitic on vertebrates.
Figure 4.—

Median values of (A) Dsyn and (B) Dnon for data sets categorized by parasitism on vertebrates and surface expression of the protein. There was no significant difference among categories with respect to median Dsyn (Kruskal-Wallis test), but median Dnon differed significantly among categories (Kruskal-Wallis test, P < 0.001).
It is well known that bacterial species have characteristic patterns of codon usage, and it has been proposed that bacterial codon usage is selectively maintained (Grantham et al. 1980). To test whether the trends in πN and Dnon (Figures 3B and 4B) were correlated with codon usage, the median percentage of G + C at third-codon positions (GC3) was compared across data sets (Figure 5). The data in Figure 5 were computed excluding codons with two or three polymorphic sites; similar results were seen when the latter codons were included (data not shown). Although there were significant differences among categories (Figure 5), there was no obvious relationship between the pattern of GC3 variation and that of πN and Dnon.
Figure 5.—

Median percentage of G + C at third-codon positions (GC3) for data sets categorized by parasitism on vertebrates and surface expression of the protein. There was a significant difference among categories with respect to median GC3 (Kruskal-Wallis test, P = 0.004).
Tajima (1989) provides a formula for estimating the total number of deleterious mutants per DNA sequence when the D-statistic is negative. For codons with a single polymorphic site in the present data set, this method yielded the estimates that 25 of 5248 synonymous polymorphisms (0.48%) are deleterious and 58 of 1840 nonsynonymous polymorphisms (3.15%) are deleterious. Thus the ratio of the proportions of deleterious mutations and nonsynonymous sites to that at synonymous sites is ∼6.6:1. This ratio is over twice the ratio of nonsynonymous to synonymous sites in the data set (79,806:24,621 or ∼3.24:1).
DISCUSSION
An examination of DNA sequence of polymorphism in 149 data sets from 84 bacterial species showed a significant preponderance of negative Dnon values but not of Dsyn values. This pattern indicates the widespread occurrence of relatively rare nonsynonymous polymorphisms but not of synonymous polymorphisms. The abundance of such rare polymorphisms at nonsynonymous but not synonymous sites in the same genes is in turn evidence that bacterial populations harbor abundant slightly deleterious nonsynonymous allelic substitutions, which are subject to ongoing purifying selection.
It is believed that, in many bacterial species, synonymous codon usage is subject to selection relating to tRNA abundance or other factors (Grantham et al. 1980). Thus, purifying selection at synonymous as well as nonsynonymous sites might be expected to occur in bacteria. The present results suggested that such selection on synonymous sites is a minor factor in comparison to that on nonsynonymous sites. Deleterious mutations were estimated to be present at only 0.48% of synonymous sites, in comparison to 3.15% of nonsynonymous sites. These results are consistent with earlier estimates that the selection coefficients at synonymous sites in bacteria are quite low (Bulmer 1991).
Deleterious mutations present in the bacterial populations examined here presumably largely represent nearly neutral mutations, i.e., those whose selection coefficients are close to the reciprocal of the effective population size (Ohta and Gillespie 1996). Deleterious synonymous mutations are expected to have very low selection coefficients (Bulmer 1991). By contrast, at least some nonsynonymous mutations are expected to be strongly deleterious because they damage protein structure and thereby render the cell nonviable. These mutations are likely to be eliminated very quickly and thus not appear as polymorphisms within populations. The low dN/dS ratios reported for bacteria largely reflect the elimination of such strongly deleterious mutations. For example, in a comparison of >14,000 phylogenetically independent pairs of genes between closely related pairs of bacterial species, Friedman et al. (2004) found a mean dN/dS ratio of ∼0.14; and Jordan et al. (2002) reported similar values. On the other hand, the present results indicate that there are abundant nearly neutral nonsynonymous mutations present in bacterial populations, where they are subject to ongoing purifying selection.
The eventual fate of nearly neutral mutations is predicted by the nearly neutral theory of molecular evolution to be determined by a combination of selection and drift (Ohta 1973, 1976, 2002). The present results support the prediction of this theory that selection against slightly deleterious mutations is likely to be efficient in species with large effective population sizes, since most bacterial species have very large effective population sizes (Lynch and Conery 2003). Thus, the present results are consistent with Jordan et al.'s (2002) observation of a lower dN/dS ratio in Escherichia coli than in three other bacterial species. Since E. coli was believed to have a larger effective population size than the other species examined, Jordan et al. (2002) interpreted their results as support for the prediction of the nearly neutral theory that purifying selection will be most effective in species with large effective population sizes.
In addition, the fact that evidence for purifying selection was found in the case of nonsynonymous but not synonymous sites supports the hypothesis that slightly deleterious mutations are more likely to be nonsynonymous than synonymous because of the effect of the former on protein structure. In the present data, deleterious nonsynonymous mutations were estimated to exceed deleterious synonymous mutations by a ratio over twice the ratio of nonsynonymous to synonymous sites.
The fact that a majority of data sets analyzed had positive Dsyn is most likely to be due to a certain degree of population subdivision in the bacterial species studied (Kreitman 2000). In the case of bacteria, limits on the extent of recombination are expected to impose limits on gene flow among clonal lineages and thus to create population subdivision. However, it is important to recognize that the present results are inconsistent with an entirely clonal population structure in the bacterial species examined. The evidence of purifying selection reducing the frequency of slightly nonsynonymous variants implies recombination, since without recombination such variants cannot be purged from a given genetic background. The present results are thus consistent with numerous other studies showing evidence of both small-scale and large-scale recombination events among bacterial genomes (e.g., Nelson et al. 1997; McGraw et al. 1999; Hughes and Friedman 2004).
A small number of the data sets analyzed here showed high positive values of both Dnon and Dsyn, suggestive of balancing selection. Of the six data sets with the highest Dnon and Dsyn, all encoded surface proteins, and four encoded surface proteins of bacterial species classified as parasitic on vertebrates. Because vertebrates possess an immune system capable of specific recognition of foreign proteins, it is expected that the parasitic organisms exposed to vertebrate immune mechanisms will be under selective pressure to evade this recognition (Frank 2002). As a result, organisms parasitic on vertebrates are expected to be subject to balancing selection favoring amino acid sequence diversity of immunogenic proteins, including cell-surface proteins (e.g., Hughes and Hughes 1995).
The data sets with high positive values of both Dnon and Dsyn that were not classified among surface proteins of vertebrate parasites were the toxin coregulated pilus subunit of V. cholerae and the W. pipientis surface protein wps. As mentioned previously (see materials and methods), V. cholerae was not classified as a vertebrate parasite for the purpose of analyses because the vast majority of known V. cholerae strains belong to the natural flora of the aquatic environment (Faruque and Mekalanos 2003). However, strains pathogenic on humans have acquired by horizontal gene transfer plasmids, phages, and chromosomal gene clusters (“pathogenicity islands”). The toxin coregulated pilus subunit gene forms part of such a pathogenicity island and the protein it encodes is thus exposed to vertebrate immune recognition (Kotetishvili et al. 2003).
On the other hand W. pipientis is a maternally transmitted obligate parasite of insects that influences its host's life history (Tsutsui et al. 2003). The present results provide evidence for balancing selection on a surface protein (wsp) of this species. This in turn suggests that polymorphism at the locus encoding this protein may play a role in the evolutionary dynamics of the host-parasite relationship.
Acknowledgments
This research was supported by grant GM43940 from the National Institutes of Health.
References
- Bulmer, M., 1991. The selection-mutation-drift theory of synonymous codon usage. Genetics 129: 897–907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Faruque, S. M., and J. J. Mekalanos, 2003. Pathogenicity islands and phages in Vibrio cholerae evolution. Trends Microbiol. 11: 505–510. [DOI] [PubMed] [Google Scholar]
- Feil, E. J., J. E. Cooper, H. Grundmann, D. A. Robinson, M. C. Enright et al., 2003. How clonal is Staphylococcus aureus? J. Bacteriol. 185: 3307–3316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frank, S. A., 2002 Immunology and Evolution of Infectious Disease. Princeton University Press, Princeton, NJ. [PubMed]
- Freudenberg-Hua, Y., J. Freudenberg, N. Kluck, S. Cichon, P. Propping et al., 2003. Single nucleotide variation analysis in 65 candidate genes for CNS disorders in a representative sample of the European population. Genome Res. 13: 2271–2276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Friedman, R., J. Drake and A. L. Hughes, 2004. Genome-wide patterns of nucleotide substitution reveal stringent functional constraints on the protein sequences of thermophiles. Genetics 167: 1507–1512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grantham, R., C. Gautier, M. Gouy, R. Mercier and A. Pave, 1980. Codon catalog usage and the genome hypothesis. Nucleic Acids Res. 8: r49–r62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harpending, H. C., M. A. Batzer, M. Gurven, L. B. Jorde, A. R. Rogers et al., 1998. Genetic traces of ancient demography. Proc. Natl. Acad. Sci. USA 95: 1961–1967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hughes, A. L., and R. Friedman, 2004. Patterns of sequence divergence in 5′ intergenic spacers and linked coding regions in 10 species of pathogenic bacteria reveal distinct recombinational histories. Genetics 168: 1795–1803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hughes, A. L., B. Packer, R. Welch, A. W. Bergen, S. J. Chanock et al., 2003. Widespread purifying selection at polymorphic sites in human protein-coding loci. Proc. Natl. Acad. Sci. USA 100: 15754–15757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hughes, M. K., and A. L. Hughes, 1995. Natural selection on Plasmodium surface proteins. Mol. Biochem. Parasitol. 71: 99–113. [DOI] [PubMed] [Google Scholar]
- Jordan, I. K., I. B. Rogozin, Y. I. Wolf and E. V. Koonin, 2002. Microevolutionary genomics of bacteria. Theor. Popul. Biol. 61: 435–447. [DOI] [PubMed] [Google Scholar]
- Kimura, M., 1977. Preponderance of synonymous changes as evidence for the neutral theory of molecular evolution. Nature 267: 275–276. [DOI] [PubMed] [Google Scholar]
- Kimura, M., 1983 The Neutral Theory of Molecular Evolution. Cambridge University Press, Cambridge, UK.
- Kimura, M., and T. Ohta, 1974. On some principles governing molecular evolution. Proc. Natl. Acad. Sci. USA 71: 2848–2852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kotetishvili, M., O. C. Stine, Y. Chen, A. Kreger, A. Sulakvelidze et al., 2003. Multilocus sequence typing has better discriminatory ability for typing Vibrio cholerae than does pulsed-field gel electrophoresis and provides a measure of phylogenetic relatedness. J. Clin. Microbiol. 41: 2191–2196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kreitman, M., 2000. Methods to detect selection in populations with applications to the human. Annu. Rev. Genomics Hum. Genet. 1: 539–559. [DOI] [PubMed] [Google Scholar]
- Li, W.-H., 1993. Unbiased estimates of the rates of synonymous and nonsynonymous substitution. J. Mol. Evol. 36: 96–99. [DOI] [PubMed] [Google Scholar]
- Li, W.-H., C.-C. Luo and C.-I Wu, 1985 Evolution of DNA sequences, pp. 1–94 in Molecular Evolutionary Genetics, edited by R. J. MacIntyre. Plenum Press, New York.
- Lynch, M., and J. S. Conery, 2003. The origins of genome complexity. Science 302: 1401–1404. [DOI] [PubMed] [Google Scholar]
- McGraw, E. A., J. Li, R. K. Selander and T. S. Whittam, 1999. Molecular evolution and mosaic structure of α, β, and γ intimins of pathogenic Escherichia coli. Mol. Biol. Evol. 16: 12–22. [DOI] [PubMed] [Google Scholar]
- Navarro-Sabaté, À., M. Aguadé and C. Segarra, 2003. Excess of nonsynonymous polymorphism at Acph-1 in different gene arrangements of Drosophila subobscura. Mol. Biol. Evol. 20: 1833–1843. [DOI] [PubMed] [Google Scholar]
- Nei, M., and T. Gojobori, 1986. Simple methods for estimating the numbers of synonymous and nonsynonymous nucleotide substitutions. Mol. Biol. Evol. 3: 418–426. [DOI] [PubMed] [Google Scholar]
- Nei, M., and S. Kumar, 2000 Molecular Evolution and Phylogenetics. Oxford University Press, New York.
- Nelson, K., F.-S. Wang, E. F. Boyd and R. K. Selander, 1997. Size and sequence polymorphism in the isocitrate dehydrogenase kinase/phosphatase gene (aceK) and flanking regions in Salmonella enterica and Escherichia coli. Genetics 147: 1509–1520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohta, T., 1973. Slightly deleterious mutations in evolution. Nature 246: 96–98. [DOI] [PubMed] [Google Scholar]
- Ohta, T., 1976. Role of very slightly deleterious mutations in molecular evolution and polymorphism. Theor. Popul. Biol. 10: 254–275. [DOI] [PubMed] [Google Scholar]
- Ohta, T., 2002. Near-neutrality in evolution of genes and gene regulation. Proc. Natl. Acad. Sci. USA 99: 16134–16137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohta, T., and J. H. Gillespie, 1996. Development of neutral and nearly neutral theories. Theor. Popul. Biol. 49: 128–142. [DOI] [PubMed] [Google Scholar]
- Rand, D. M., and L. M. Kann, 1996. Excess amino acid polymorphism in mitochondrial DNA: contrasts among Drosophila, mice, and humans. Mol. Biol. Evol. 13: 735–748. [DOI] [PubMed] [Google Scholar]
- Reid, S. D., R. K. Selander and T. S. Whittam, 1999. Sequence diversity of flagellin (fliC) alleles in pathogenic Escherichia coli. J. Bacteriol. 181: 153–160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sunyaev, S., F. A. Kondrashov, P. Bork and V. Ramensky, 2003. Impact of selection, mutation rate and genetic drift on human genetic variation. Hum. Mol. Genet. 15: 3325–3330. [DOI] [PubMed] [Google Scholar]
- Tajima, F., 1989. Statistical method for testing the neutral mutation hypothesis by DNA polymorphism. Genetics 123: 585–595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thompson, J. D., D. G. Higgins and T. Gibson, 1994. CLUSTALW: improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice. Nucleic Acids Res. 22: 4673–4680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsutsui, N. D., S. N. Kauppinen, A. F. Ofayuso and R. K. Grosberg, 2003. The distribution and evolutionary history of Wolbachia infection in native and introduced populations of the invasive argentine ant (Linepithema humile). Mol. Ecol. 12: 3057–3068. [DOI] [PubMed] [Google Scholar]
- Wise, C. A., M. Sraml and S. Easteal, 1998. Departure from neutrality at the mitochondrial NADH dehydrogenase subunit 2 gene in humans, but not in chimpanzees. Genetics 148: 409–421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang, Z., and R. Nielsen, 2000. Estimating synonymous and nonsynonymous substitution rates under realistic evolutionary models. Mol. Biol. Evol. 17: 32–43. [DOI] [PubMed] [Google Scholar]
- Zhang, J., H. F. Rosenberg and M. Nei, 1998. Positive Darwinian selection after gene duplication in primate ribonuclease genes. Proc. Natl. Acad. Sci. USA 98: 3708–3713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao, Z., Y.-X. Fu, D. Hewett-Emmett and E. Boerwinkle, 2003. Investigating single nucleotide polymorphism (SNP) density in the human genome and its implications for molecular evolution. Gene 312: 207–213. [DOI] [PubMed] [Google Scholar]