

Abstract
Direct structural information obtained for many proteins supports the following conclusions. The amino acid sequences of proteins can stabilize not only the final native state but also a small set of discrete partially folded native-like intermediates. Intermediates are formed in steps that use as units the cooperative secondary structural elements of the native protein. Earlier intermediates guide the addition of subsequent units in a process of sequential stabilization mediated by native-like tertiary interactions. The resulting stepwise self-assembly process automatically constructs a folding pathway, whether linear or branched. These conclusions are drawn mainly from hydrogen exchange-based methods, which can depict the structure of infinitesimally populated folding intermediates at equilibrium and kinetic intermediates with subsecond lifetimes. Other kinetic studies show that the polypeptide chain enters the folding pathway after an initial free-energy-uphill conformational search. The search culminates by finding a native-like topology that can support forward (native-like) folding in a free-energy-downhill manner. This condition automatically defines an initial transition state, the search for which sets the maximum possible (two-state) folding rate. It also extends the sequential stabilization strategy, which depends on a native-like context, to the first step in the folding process. Thus the native structure naturally generates its own folding pathway. The same amino acid code that translates into the final equilibrium native structure—by virtue of propensities, patterning, secondary structural cueing, and tertiary context—also produces its kinetic accessibility.
The effort to understand how proteins fold has consumed the labors of generations of scientists (1–14). Unlike other fundamental problems, the folding problem cannot be solved by finding the molecules that mediate the process or by determining a molecular structure or function. Its solution will require the definition of entire sequences of structural transformations that are complete in less than 1 s and involve multiple molecular forms, none of which can be isolated for study. Given these difficulties, the present situation is that entirely opposed paradigms for the folding process are still being considered.
The classical view of protein folding is that the search for the native state through the vastness of conformational space flows through predetermined pathways defined by discrete intermediates and barriers (2–4, 15, 16). This view, formalized in the free energy diagrams in Fig. 1A, has guided the interpretation of decades of experimental observations. An important challenge arises. How is it possible for the one-dimensional sequence to code not only for the final native structure but, in addition, for a set of three-dimensional nonnative intermediate structures and their connection into a temporal folding sequence?
Figure 1.

Alternative folding paradigms. (A) Classical pathways. Following Sosnick et al. (62), the profiles shown suggest a basis for apparent two-state folding (Upper) due to an initial rate-limiting barrier, even though intermediates are present; three-state folding (Lower) due to the insertion of an error repair barrier (101, 107); and heterogeneous folding (Upper and Lower) due to the chance nature of misfolding errors. The molecular cartoons are meant to suggest a lengthy energetically uphill conformational search for a set of interactions that can pin the chain into some native-like transition-state (TS) topology, the subsequent downhill folding that marks the success of the search, and the chance incorrect placement of some group(s) (red) that can block folding and impose a time-consuming error repair process. This picture is formalized in Eq. 2. (B) A folding funnel (adapted from ref. 13) and a typical on-lattice folding model (adapted from ref. 108).
The “new view” of protein folding (13, 17–20), often represented as in Fig. 1B, judges that no coding is necessary because predefined pathways with compulsory intermediates simply do not exist. The downhill bias of the energy landscape is sufficient to drive random folding in a reasonable time period. Folding proceeds one amino acid at a time in no particular order through an unlimited continuum of undefined intermediates and paths. When discrete intermediate structures do become significantly populated, as is often observed with real proteins, this is taken to mean that the folding chain has accidentally wandered into some deep off-pathway well in the landscape, which slows rather than promotes the folding process. These conclusions derive mainly from computer simulations of simplified models, like the lattice model in Fig. 1B, which generally omit the primary, secondary, and tertiary structural characteristics that make proteins unique.
To distinguish between these opposed views and to illuminate the important mechanistic details of folding processes in real proteins, one wants to detect and characterize the intermediate forms that in fact carry unfolded proteins (U) to their native state (N). This is problematic. Folding intermediates are short lived, often heterogeneous, and cannot be studied by the usual crystallographic or NMR methods. One has had to depend on fast spectroscopic probes (21–25) or mutational analyses (26, 27). These diverse methods can provide rate information, signal the accumulation of intermediates, and give local information on the role of particular amino acids or averaged parameters of the main chain. However, they do not provide definitive structural information and have not been able to distinguish even the diametrically opposed paradigms just noted.
Structural detail for folding intermediates has come especially from hydrogen exchange (HX) measurements. HX information defines structure by identifying amino acids that have slowly exchanging, presumably hydrogen-bonded, main-chain amides. HX methods now in common use (28–32) can identify intermediates in both kinetic and equilibrium modes, track their formation on a subsecond time scale (33), and quantify their thermodynamic stability (34, 35). The purpose of this paper is to organize that information and consider its significance for the protein folding problem.
Folding Intermediates Studied by HX
It first became possible to define transiently formed submolecular structure by the use of hydrogen–deuterium (H-D) exchange labeling together with stopped-flow methods and high-resolution proton NMR analysis (36, 37), a method known as HX pulse labeling. The protein, unfolded in guanidinium chloride (GdmCl) and deuterated by exchange in D2O solvent, is diluted into H2O to initiate refolding. After various experimental folding times, a brief pulse to higher pH is used to promote fast D to H exchange and label with H the main-chain amides that are not yet protected by hydrogen bond formation. The protein folds to its native state, trapping the H-D labeling profile imposed during the labeling pulse, which can then be read out by two-dimensional NMR. To quantify the degree of protection against H exchange, the pH in the labeling period can be varied (37–39).
The initial pulse-labeling data for cytochrome c (Cyt c) (Fig. 2 A and B) showed that some amide NHs are selectively protected in milliseconds, suggesting the early concerted formation of the N- and C-terminal helices, whereas other regions form much later (36). Similar behavior for RNase A was described in a companion paper (37). A surprising result was that folding is heterogeneous (Fig. 2); different fractions of the protein population fold at different rates (33, 36, 40). Kinetic HX labeling has now been applied to many proteins. In all cases of multistate folding, one finds intermediates that appear to represent partial replicas of the native protein at a greater or lesser degree of advancement. Also, some degree of heterogeneity is often seen.
Figure 2.

HX labeling data. (A and B) The fraction of H labeling obtained at particular residue amides in Cyt c by a brief high-pH labeling pulse after increasing times of folding (36). The data indicate the early formation of an intermediate with the N- and C-terminal helices formed, the delayed formation of the rest of the protein, and heterogeneous folding in which different fractions of the protein population fold at different rates. (C) Native-like HX protection pattern for the Cyt c molten globule destabilized at acid pH (122). Protection factors are corrected (123, 124). Color coding relates to Fig. 3D. Residues 14, 15, and 18 (gray) are protected within the small covalent heme loop, even in the unfolded state (see Fig. 3A).
Structural information comes also from studies of partially destabilized molten globule forms that have been thought to represent equilibrium analogs of kinetic intermediates (41–43). Here again the usual crystallographic and NMR methods for defining structure are not applicable because molten globules are too dynamically disordered, except in the most well-structured near-native cases (44, 45). Fortunately, structure that is present can easily be defined by HX labeling. Equilibrium molten globules can be exposed to exchange by incubation in D2O for increasing periods of time and then returned to native conditions. The degree of H-to-D labeling at various amides through the molten globule can then be read out by two-dimensional NMR of the native protein (46, 47). Results for the highly ordered molten globule of Cyt c are shown in Fig. 2C. The structures found here and in other molten globules represent a partial replica of the native protein at greater or lesser degrees of advancement.
Most recently, a native-state HX method has made it possible to explore the high-free-energy folding landscape between the native and unfolded end states (48, 49). Under native conditions, proteins exist predominantly in their native form, but they must thermally unfold and refold all the time, cycling through the high-energy space pictured in Fig. 1 A and B and populating the partially folded intermediate states therein, each according to its Boltzmann factor. This cycling is invisible to most methods, which are dominated by signals from the overwhelmingly populated native state. Uniquely, the measurable exchange of structurally protected (H-bonded) hydrogens receives no contribution from the dominant native state but is wholly determined by the infinitesimally populated higher energy H-bond-broken forms.
Fig. 3 shows some of the initial native state HX results for Cyt c. H-D exchange rates were measured by two-dimensional NMR as a function of low levels of GdmCl and processed to obtain the free energy for the H-bond opening reactions that allow each NH to exchange. At low GdmCl, many adjacent main-chain NHs exchange at very different rates that are independent of denaturant, indicating exchange by way of local H-bond-breaking fluctuations. These HX pathways are interesting and not fully understood (50), but they mask the larger unfolding reactions of interest here. At higher GdmCl, but still far below the melting transition, sets of NHs merge into four common HX isotherms. This merging occurs because large unfolding reactions are selectively promoted by increasing denaturant and, when sufficiently enhanced, come to dominate the exchange of the particular hydrogens that they expose. The identity of each large unfolding unit is then revealed by the set of amino acid residues that it exposes. The free energy gap between each unfolded state and N can be computed from the HX rates. The surface exposure of each unfolding can be estimated from its denaturant sensitivity.
Figure 3.

Native-state HX results for Cyt c [pDr 7, 30°C (48)]. (A) Residues in the N-terminal helix. (B) Residues in the 60s helix. Local fluctuational HX pathways, dominant at low denaturant (m ≈ 0), are superseded by larger unfolding reactions when denaturant is increased. (C) A summary crossover curve showing how the four different unfolded states change in free energy, relative to N and to each other, with GdmCl concentration. The dashed line suggests the formation of a partially unfolded molten globule because of some selective destabilization of an unfolding unit. (D) Cyt c structure color coded in order of descending stabilization free energy (blue to green to yellow to red) to show the cooperative units indicated by native-state HX. Also shown are the peripheral histidines that can misligate to the heme iron at the Met-80-S site and trap the green loop out of place, imposing an error correction barrier that causes slow multistate folding with accumulation of the N/C helix intermediate.
The results in Fig. 3A identify the N-terminal helix of Cyt c as part of a cooperative unfolding unit, as suggested before by HX pulse labeling (Fig. 2). These data reflect a reversible unfolding reaction between the globally unfolded state (blue open) and a partially folded structure with the N and C helices formed. Fig. 3B shows the same result for all of the amide hydrogens in the 60s helix, which unfold as another unit apparently together with an Ω-loop segment. These data reflect a reversible subglobal unfolding (green open) to a state with lower free energy and surface exposure than the global unfolding, with the N and C helices still protected. Similar data (48–50), albeit with fewer probe hydrogens, depict two lower-lying partially unfolded states (yellow open with green and blue still protected; red open with yellow, green, and blue still protected). Fig. 3C summarizes these and related results, which suggest that Cyt c is composed of four separable cooperative unfolding units, color coded in Fig. 3D.
Native-state HX has been successfully applied to a number of other proteins (51–56) and has been generalized to the use of other destabilizing perturbants, including temperature (57) and pressure (58). Application of the experiment can often be limited by unfavorable protein characteristics (49, 59), but in favorable cases similar results have been obtained.
In summary, specially designed HX methods have made it possible to detect and characterize many partially folded proteins. In some cases, it has been possible to prepare, by synthesis or dissection, parts of proteins that maintain native-like structure (listed in ref. 60). These results imply that the high-free-energy landscape between N and U is characterized by a small number of discrete, partially folded, native-like intermediates rather than by an undifferentiated continuum. All other forms necessarily exist but only at higher free energy than the local minima that determine the discrete intermediates. Therefore they make no contribution to measured HX rates. The profile of the folding landscape appears to be more like that diagrammed in Fig. 1A. Evidently, amino acid sequences are able to code not only for the globally cooperative native state but also for a small number of partially unfolded intermediates in which the structured regions represent partial replicas of the native structure.
The Building Blocks: Elements of Cooperative Secondary Structure
In most cases, the intermediate partially unfolded forms (PUFs) that have now been detected or synthesized de novo are far advanced toward the final native structure. These results demonstrate the reality of discrete and stable intermediates but they do not help to answer the question of how, in kinetic folding, the initially unfolded polypeptide efficiently reaches such advanced structures.
In some known intermediates only minimal structure is found, presumably representing more primal units. In Cyt c the N/C helical pair forms a concerted folding unit, documented by HX pulse labeling (36, 61, 62), native-state HX (48, 57), and related mutational work (63, 64). So too does the red Ω loop alone and the entire 60s helix apparently together with another loop (48, 57). In apomyoglobin the entire A, G, and H helices form a stable folding unit, seen by kinetic HX pulse labeling (65), equilibrium molten globule HX labeling (66), and direct NMR study (67). Similar results document the separable cooperative unfolding of helical segments or sets thereof in RNase H (51, 68, 69) and apo Cyt b 562 (52, 58). Analogous studies show the sequential peeling off or addition of secondary units with changing urea concentration in α-lactalbumin (70) and the intestinal fatty acid-binding protein (71). An atomic force study showed the cooperative unfolding of sequential pairs of helices in membrane-bound bacteriorhodopsin (72).
A major insight can be drawn from these various studies. Protein molecules are not monolithically cooperative all-or-none structures but can be viewed as accretions of cooperative submolecular units. When proteins are caused to unfold in the usual melting experiment, they most often appear to do so in a two-state manner, apparently because the U state in the melting region is at lower free energy than the partially unfolded forms, as shown in Fig. 3C. Unfolding and refolding of the separate units does occur, however, and can be distinguished by the native-state HX experiment under conditions of stability (Fig. 3C), by HX labeling in a kinetic mode (Fig. 2), and by other approaches.
Isolated helices (73, 74), β hairpins (75), and Ω loops (48) represent intrinsically cooperative structures in which the partially formed units are at higher free energy than the fully formed or the fully unfolded end states. When incorporated into globular proteins, these secondary structural elements are greatly stabilized by tertiary interactions but, not surprisingly, they retain their intrinsically cooperative identity and continue to act as concerted unfolding and refolding units (foldons), either individually or in larger sets. The results just reviewed indicate that these units serve as the fundamental building blocks of protein molecules. In lattice model terminology, proteins might be viewed in terms of a small number of connected beads representing cooperative folding–unfolding units rather than hundreds of beads representing independent amino acids.
Why Are Intermediates So Elusive?
Nature seems to conspire to hide folding intermediates. Many proteins fold in a kinetically two-state manner (76) so that intermediates do not populate and are not seen. In equilibrium experiments, most proteins when subjected to denaturing conditions melt in a two-state manner. Under mildly destabilizing conditions some proteins produce molten globule forms but most do not, raising the possibility that they are artifacts of the conditions used. Under native conditions, a number of proteins fail to show intermediates in native-state HX experiments. Theoretical studies generally suggest that discrete intermediates exist only as off-pathway nonnative obstructive forms.
That intermediates are not observed in given experiments has often been phrased as “intermediates do not exist.” Fortunately, as we have seen, partially unfolded forms under favorable conditions can be detected and studied by HX methods as transiently blocked species in kinetic folding (Fig. 1A Bottom; Fig. 2 A and B), as molten globules under moderately destabilizing conditions (Fig. 2C; dashed line in Fig. 3C), and in the high-free-energy landscape under native conditions (Fig. 3). These same results also show why submolecular folding behavior has been so elusive.
As illustrated in Fig. 3C, partially unfolded forms exist only at higher free energy than the native state under native conditions, and they tend to be at higher free energy than the unfolded state in the measurable melting transition zone. Thus under equilibrium conditions intermediates are minimally populated throughout and tend to escape detection. Fig. 3C also suggests the difficult criteria for populating molten globule forms (dashed line). To produce a partially unfolded form at equilibrium, it is necessary to destabilize one or more of the unfolding units selectively, for example by the protonation at low pH of some fortuitously placed ionizable group. The nonselective destabilization of all of the partially unfolded forms together, as by a solvent denaturant, is unlikely to generate a PUF or molten globule.
Fig. 3C also suggests why the native-state HX experiment often fails. Intermediates under native conditions tend to occur at high free energy levels (for reasons that are not known). The lowest-lying PUF is found at 6 kcal/mol above the native state in Cyt c and at 7.5 kcal/mol in RNase H. Thus proteins that have only 8 kcal/mol of stability or less, which is the common condition for known two-state proteins, are unlikely to reveal their intermediate forms by native-state HX. Only global and local fluctuational HX pathways will be seen. Other factors that limit the native-state HX experiment have been listed (49).
The same considerations suggest why theoretical studies most often fail to exhibit discrete folding intermediates. Intermediates are produced by the folding or unfolding of the separable cooperative elements that form naturally occurring proteins. The models usually used in theoretical studies, necessarily simplified to make them tractable for available computer capabilities, generally include only a single cooperative unit (e.g., Fig. 1B).
Finally, it appears that intermediates tend to escape detection in kinetic experiments because they often occur after the rate-limiting step. This issue is considered in a later section.
On or Off Pathway
The results noted above help to delineate the thermodynamic landscape but they do not directly define a kinetic folding–unfolding pathway. Do the PUFs observed by equilibrium methods and as trapped forms by kinetic methods in fact serve as constructive intermediates in folding pathways (Fig. 1A), or are the discrete intermediates mere curiosities in the reaction landscape, or perhaps even off-pathway trapped forms that block folding and must be completely unfolded to try again (Fig. 1B)?
Evidence for the on-pathway nature of individual partially folded forms has been found for a number of proteins. Laurents et al. (77) extended the length of the HX labeling pulse to show that the native-like structure present in an RNase A intermediate does not unfold on its way to N. Bai obtained the same conclusion for kinetic intermediates of Cyt c (78) and hen lysozyme (79) by showing that they reach N faster than they can unfold to U. Various experiments support the on-pathway nature of kinetic intermediates in other proteins [apomyoglobin (80, 81), interleukin-1β (82), pancreatic trypsin inhibitor (83), dihydrofolate reductase (84), fatty acid-binding protein (71)]. The same partially folded intermediates found in equilibrium molten globule or native-state HX experiments have also been found in kinetic HX pulse-labeling experiments for Cyt c (36, 48), apomyoglobin (65, 66), and RNase H (51, 68), adding weight to the conclusion that these forms are intrinsic to the protein and serve in kinetic folding.
A more general observation can be made. If kinetic intermediates accumulate because they accidentally fall into some off-pathway energy well (Fig. 1B), then one might expect only small fractions of a refolding population to be involved in any trapped form. However, kinetic folding experiments often show that most or even all of the protein population moves transiently through some given intermediate form, consistent with compulsory intermediates in a defined pathway. In some cases, this behavior is obvious at the beginning of the pathway (Cyt c, apomyoglobin, RNase H) and not just at the bottom of the funnel where diversity converges to a few alternatives.
An Entire Pathway
The native-state HX experiment demonstrates protein segments that unfold and refold cooperatively (foldons). Does each foldon unfold and refold sequentially together with others in a pathway manner or are they independent? For example, in the PUF with the green unit of Cyt c unfolded, it is clear that the higher lying blue unit is still folded, but one cannot distinguish whether the lower lying red and yellow foldons unfold together with the green unit because the probe NHs that define them have already exchanged (see Fig. 3). To address the pathway issue, Xu et al. (85) used the native-state HX experiment in a “stability labeling” mode, as follows.
Binding studies show that the Met-80-S to heme iron ligation in Cyt c is stabilized by an additional 3.2 kcal/mol when the oxidized heme iron is reduced (85). Met-80 is in the red loop (Fig. 3D). When oxidized Cyt c is reduced, HX of the structurally protected red loop hydrogens is slowed by an additional 200-fold, indicating that the unfolding that exposes them is suppressed by the same 3.2 kcal/mol (85). Evidently the red loop unfolds as an independent unit and must break the S to Fe ligation (as in RYGB ↔ rYGB; uppercase refers to the color-coded segments in Fig. 3 and lowercase to the same segments unfolded).
If the other unfolding reactions seen by native-state HX are independent, they should be unaffected by the change in stability of the red loop. However, HX results show that unfolding of the yellow loop is also suppressed, again by just the same 3.2 kcal/mol, showing that the yellow unfolding represents both the red and yellow segments open together (ryGB). The green unfolding, which includes the 60s helix and the green loop, is similarly suppressed (3.3 kcal/mol), showing that in the measured green-open state the red, yellow, and green segments are open together (rygB). The blue unfolding, known to represent the final global unfolding to U (rygb) (48, 86, 87), is suppressed by 3.2 kcal plus an additional 2-kcal increment, caused apparently by neutralization of the destabilizing buried charge when Cyt c is reduced (88).
Independently, the increasing m values of the partially unfolded forms (slopes in Fig. 3C), which relate to the surface newly exposed in each unfolding reaction, are quantitatively consistent with the same identifications (48).
The Cyt c intermediates identified in this way (rYGB, ryGB, rygB) are separated from each other by one cooperative unit, and they represent just the intermediate forms necessary to construct the sequential unfolding pathway in Eq. 1.
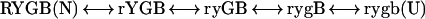 |
1 |
If this is so, then the same series in reverse order must form the major refolding sequence. This conclusion follows because the native-state HX experiment is done under equilibrium conditions where each unfolding reaction must be matched by an equal and opposite refolding reaction, as suggested by the reaction arrows.
These considerations, for Cyt c and other proteins, support the conclusion that partially folded native-like structures serve as discrete on-pathway intermediates in distinct kinetic folding pathways.
Pathway Construction by Sequential Stabilization
How is it possible for a one-dimensional amino acid sequence to guide three-dimensional structure formation through a number of partially folded intermediates in some predetermined temporal order? A surprising insight comes from comparing the suggested pathway for Cyt c with the contacts that connect its secondary structural elements (Figs. 3D and 4).
Figure 4.

Native contact diagram for the blue, green, yellow, and red folding units in equine Cyt c. Connected residues have atom–atom contacts within 4 Å in the native structure (125) without distinction between polar or apolar character. Extensive contacts connect the two blue segments (Top), the blue segments with the rest of the protein (Middle), and the green segments with the rest of the protein (excluding blue) (Bottom). Folding by sequential stabilization in the native context would explain the concerted initial formation of the N- and C-terminal helices, the subsequent apparently concerted formation of both green elements, and the later folding of the yellow and red elements.
The N and C helices of Cyt c form the first stable structure in folding and are the last to unfold (36, 48, 86). In the folding protein the initial N/C helix intermediate is more stable than U by about 3 kcal/mol (Fig. 3C). In the native protein these two helices are in extensive mutually stabilizing contact, as shown by the map of native N-to-C contacts in Fig. 4 (Top). This organization suggests that stabilization of the first intermediate in the initial folding step depends on the native-like context.
Native-state HX results suggest that the second unit to form, the green unit, consists of the 60s helix and an Ω loop on the other side of the protein (Fig. 3). Why should the green elements form next? Why do the two separate green elements, which have almost no contact with each other in the native protein, appear to form concertedly? The native contact map provides an answer.
If folding occurs in a native context, the initial formation of the N/C helix intermediate will naturally prepare a docking surface, with a native-like patterning of interactions, that can guide and stabilize formation of both the 60s helix and the green loop (Fig. 4 Middle). The green segments by themselves in solution have no stability, yet the intermediate with the blue-plus-green units formed (ryGB) is more stable than U by 5 kcal/mol (Fig. 3C). This difference in stability can be explained by the extensive native-like interactions that bind both green segments to the blue unit. The native contact map further indicates that the yellow and red elements receive no support from the initially formed blue unit but do require both green elements to be formed (Fig. 4 Bottom). Thus the two green elements are coupled only indirectly by the fact that, within the native context, both must follow formation of the blue unit and both must precede formation of the yellow and red loops.
In this sequence the heme group undoubtedly plays a role. Indeed, because of its covalently bound heme Cyt c might be considered a questionable model for protein folding behavior. Several points can be noted here. The heme is present during both in vivo and in vitro Cyt c folding. It supports a small loop structure (residues 14–18) that may stabilize the early formation of the N-terminal helix, but other proteins also show indications of residual native-like structure in the U state, possibly designed to aid in folding (89). The single five-residue loop held by the Cyt c heme compares not unfavorably with other folding models (three major disulfide loops in bovine pancreatic trypsin inhibitor, four in RNase A, hen lysozyme, and α-lactalbumin). Three methyl groups on the inner edge of the Cyt c heme participate in some core contacts, as do side-chain methyls in other proteins, but the large heme surface does not provide a hydrophobic organizing center (unlike the globins), because most heme interactions in native Cyt c are polar. Most pertinently, Cyt c possesses all of the natural attributes of other proteins (primary structural stereochemistry, cooperative secondary elements, specific tertiary interactions and topology, exact potential functions, solvent interaction), and these characteristics appear to dominate its folding behavior, as for other proteins, despite their postfolding modifications. This can be compared with simplified models widely used in theoretical studies, which often have none of these unique protein characteristics.
In summary, the native Cyt c structure provides a simple but compelling rationale for its experimentally inferred folding sequence. It appears that the folding pathway can naturally construct itself through a process of sequential stabilization in which, at each step, earlier formed native-like structure provides a template that guides and stabilizes the next native-like unit in turn through native-like tertiary interactions. It is difficult to think why other proteins would behave very differently.
These results support and make more specific the concept of hierarchic condensation, drawn originally from the observation that protein structures consist of distinguishable units that interact but do not intertwine, suggesting a process in which structural modules, each laid down in a distinct step, pyramid upward toward the native state (89–94). However, these results are silent on the more recently entrained issue of pre-organized structure in the U state (89).
Plasticity
Various observations indicate that the ordering of pathway intermediates is likely to be malleable, depending on protein structure and the relative stability of the folding units. The Cyt c contact map suggests that the red loop may be able to form before or after the yellow loop (Fig. 4). Contact maps for other proteins studied by native-state HX also suggest that given units may form in alternative orders (53, 58, 95). A forked pathway is suggested by native-state HX results for apo Cyt b-562 (52, 58), consistent with its structure. In the λ repressor, mutational stabilization of helix 3 appears to add it to the initial transition-state structure (96). In apomyoglobin, destabilization of helix H appears to remove it from the early AGH intermediate (97), whereas stabilization of helix E adds it in (98). Apparently for similar stability reasons, the GH helix pair is found together with either helix A (+B) or helix E in early populated kinetic intermediates of the homologous apomyoglobin and leghemoglobin proteins (99). A similar mixing of alternative elements is seen among three RNase H homologs (56). In the same way, the detailed identities of secondary elements that act as folding units in an intermediate are likely to be similarly malleable rather than wholly identical to their native counterparts because they depend on the existing tertiary context, which is not fully in place during folding.
These considerations suggest that the precise folding trajectory, although rather strictly predetermined, will be somewhat flexible, depending on the participating secondary structural elements, their relative stability, and detailed interactions within the developing tertiary context. The unifying principle seems to be that the use of a limited number of cooperative native-like elements, shaped by the propensities and patterning (100) of the primary amino acid sequence and cues embedded therein, profoundly limits unfolding and refolding to a small number of alternative pathway sequences in which each step is guided by previously formed native-like structure.
The Kinetic Barriers: Misfolding and Error Repair in Cyt c
Kinetic folding studies raise some challenges to the conclusions reached here. Small proteins tend to fold in an apparently two-state (monoexponential) manner without the accumulation of intermediates (76). This manner of folding is often interpreted to mean that intermediates do not exist. Larger proteins tend to fold in a slower multistate (multiexponential) way in which intermediate forms accumulate transiently before the native state is reached. It is often conceived that these intermediates are off-pathway nonnative forms that act to slow rather than to promote folding.
Particularly relevant evidence comes from studies of Cyt c, which can be induced to fold in either a two-state or a multistate manner, even under the very same folding conditions. Other proteins can do the same. It is commonly conceived that two- and three-state folders are fundamentally different; this difference is often phrased in terms of the roughness of their folding landscapes. Clearly not.
When equine Cyt c is unfolded at low pH and then mixed into folding conditions at pH 5 or below, it folds largely in a two-state manner. When it is unfolded at higher pH and then mixed into the very same pH 5 folding conditions as before, most of the population reaches N much more slowly, in a multistate manner, with the transient accumulation of the N/C helix intermediate (101, 102). The reason is that, in the unfolded protein, the Met-80-S deligates from the heme-Fe and can be replaced by a peripheral histidine in its neutral (high pH) form. This forces the segment that carries the histidine, the green loop in Fig. 3, to the “wrong side” of the heme. When folding is initiated, the chain collapses and traps the green loop out of place. Initial N/C helix formation can proceed unhindered, but the next step requires the green loop to fold. Therefore, forward folding halts until the heme-bound histidine and its entrained segment are freed and folding can resume.
In the presence of heme misligation, the reaction sequence for Cyt c in Eq. 1 can be rewritten as in Eq. 2, which formalizes the upper and lower diagrams in Fig. 1A taken together:
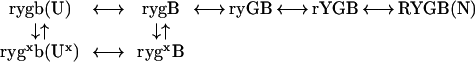 |
2 |
In misligated Cyt c, a misfolding error blocks the folding of the green segment (gx). The error can already be present in U (Ux) or can develop as folding proceeds. The blue (N/C) intermediate forms normally, from Ux as from U (see below). When gx is present, further folding halts until a time-consuming thermal search succeeds in correcting the error, freeing the green loop and returning the molecule to the fast track, either directly or round about by way of U. The time-consuming error repair process might be viewed as an optionally inserted “misfold–reorganization” barrier (Fig. 1A Bottom; Eq. 2).
Misfolding and Error Repair in Other Proteins
Two-state to three-state switching and the inserted error repair behavior that this switching implies are not peculiar to Cyt c. Many proteins can be switched from two-state to multistate folding simply by allowing a proline misisomerization to develop in the initially unfolded protein before the refolding experiment is initiated (103). In known cases, the intermediates that then become kinetically populated are in part strikingly native like but they have evidently been corrupted by some misfolding error. For example, a T1 ribonuclease intermediate trapped by proline misisomerization was shown by NMR to be impressively native-like in part (104). In bovine pancreatic trypsin inhibitor, a near-native intermediate accumulates when folding is slowed by the premature burial of two cysteines before they can be oxidized to the native disulfide bond by reaction with external glutathione (83). For Cyt c, folding past the misligation error is accelerated by mild denaturant, indicating that error repair involves some partial unfolding of the native-like intermediate (101), but the native-like structure is carried forward, here and in other proteins (77–84).
An emphasis on the nonnative nature of observed folding intermediates has confused discussions of their role. Intermediates that are blocked and accumulate in folding are nonnative only in part because of some added misfolding error. Error repair can slow the rate of final native-state acquisition. Slow folding and intermediate accumulation then occur together, not because intermediate accumulation causes slow folding but because both are caused by the insertion of a kinetic barrier, a misfolding-dependent error correction process. Only the misfolding error is optional, nonnative, and obstructive, and must be removed. The native-like part of the intermediate is retained and carried forward.
The very same circumstances explain the often observed heterogeneity of folding. One can expect that different molecules will by chance encounter different error-dependent barriers, or none at all, and thus will appear to populate different intermediates and fold at different rates. This variety is precisely what is observed. In such cases, one often observes some fraction of the protein population that folds all the way to N in a fast two-state manner. This manner of folding clearly shows that the late barrier that slows native-state acquisition in three-state folding does not represent an obligatory on-pathway folding step. The usual interpretation of heterogeneous folding behavior in terms of nonobligatory nonnative intermediates, or in terms of independent parallel pathways, must be reconsidered.
In summary, it appears that slow folding, intermediate accumulation, and folding heterogeneity are all caused by the chance insertion of a time-consuming error-repair process, which may be viewed in terms of a misfold–reorganization barrier (Fig. 1A, Eq. 2). The concept that slow folding is limited by a partial back-unfolding to release random misfolding errors fits well with current views of the forceful unfolding function of the GroEL chaperonin (105).
Two-State Folding: The Initial Kinetic Barrier
When an optional error-dependent blockage is not encountered, folding can appear to be a two-state process without the accumulation of intermediates. Does this finding mean that intermediates do not exist? An alternative is that intermediates are present but escape detection because the rate-limiting step occurs as the first step in folding, before any intermediates are formed, as in Fig. 1A Top. In fact, Cyt c demonstrates just this result. The ability to cause Cyt c to fold in either a two- or three-state manner allowed Sosnick et al. (62, 106, 107) to characterize the transition state in two-state folding in some detail. The picture that emerges indicates that the very first step in folding leads the protein into its sequential stabilization sequence, as follows.
The rate-limiting barrier in two-state Cyt c folding has the same thermodynamic (ΔG‡, ΔH‡, ΔS‡) and structural (large m value) parameters as the initial barrier in three-state folding (compare Fig. 1A Upper vs. Lower). Evidently, they both represent the very same process. In three-state folding, this barrier occurs before (or during) the formation of the initial N/C helix intermediate and well before the folding of the rest of the protein. Therefore, the equivalent barrier in two-state folding must also occur before (or during) formation of the initial N/C intermediate and so must also represent the initial folding step. When later barriers are smaller, the initial barrier becomes rate limiting and folding appears to be a two-state process (Fig. 1A Upper). Subsequent kinetic intermediates then escape detection, not because they do not exist but because they do not accumulate to a visible level.
The rate-limiting step in two-state folding is accompanied by a massive chain collapse, shown by the strong dependence of the folding rate on denaturant concentration (high m value). Equally large surface occlusion is seen even in very small proteins. The kinetic transition that leads to the native state is thought to involve a nucleation process (6, 62, 106–110). Nucleation is often suggested to occur late in folding for two reasons: (i) A large fraction of the surface that is buried in the native state becomes buried in the initial rate-limiting step (m‡/m° is 0.5 or more). When one takes the degree of surface burial as the reaction coordinate, the rate-limiting step therefore appears to be late in folding. In fact it is early, before any intermediates are formed. (ii) In three-state folding, the rate-limiting event occurs after the observable intermediate, which is often well advanced. This too is misleading. It appears that large late barriers are not intrinsic to the folding process but are caused by error repair. These are not nucleation events.
The rate-limiting step in the two-state folding of Cyt c and other small proteins most often requires a time scale of milliseconds, orders of magnitude longer than the time required for simple pairwise residue encounters (111, 112). This time scale implies that the free-energy-uphill process before successful nucleation involves a lengthy conformational search to find some relatively special combination of interacting residues. The sequence-dispersed interactions that come together must provide sufficient drive to condense the chain in competition with unfavorable loop closure entropy. Nevertheless, the overall entropic search process represents an energetically uphill barrier crossing rather than a simple downhill collapse, and the uphill kinetic barrier is the same one that separates the equilibrium U and N states. This conclusion is supported by the fact that, for many proteins in two-state kinetic folding, the folding and unfolding ΔG‡ values sum to equal the equilibrium ΔG°, and the same is true for m‡ and m°.
Given these results, Sosnick et al. (62, 106, 107) proposed that the successful interaction set reached in the initial rate-limiting search nucleation step is one that manages to pin the chain into a native-like topology and thus establishes some initial native-like structural context (see the molecular diagrams in Fig. 1A). The successful topology is “recognized” by the fact that it discontinuously supports forward (native-like) folding in an energetically downhill manner. Therefore it naturally defines the operative folding transition state. The idea that the rate-determining transition state sets up some native topology (113–116) and that the time-consuming search for it limits the folding rate (117, 118) has recently gained considerable support.
Sosnick et al. (62, 106, 107) focused their studies on the folding limb of the folding–unfolding chevron at moderate denaturant concentration where interactions are weakened. The character of the rate-limiting process could then be studied in isolation because chain collapse is postponed until a concerted set of native-like interactions is found that can together overcome loop closure entropy and condense the chain. At lower denaturant, interactions are stronger and smaller more random sets of local sequence interactions that are found rapidly are able to condense the chain in a more random way. This kind of off-pathway solvent-dependent chain collapse produces fast “burst-phase” structural and spectroscopic signals that are easily mistaken for specific intermediate formation (107, 119–121). The burst phase collapse also obscures the important on-pathway search-nucleation process, which must then proceed unseen within the condensed milieu.
In summary, kinetic folding can appear to be two-state, even when discrete intermediates are present because the rate-limiting process is the first step in the pathway. The initial barrier represents an uphill whole-molecule search for a state that can, by creating a native-like context, support forward downhill folding. This behavior automatically defines the transition state, and it extends to the very first step in the pathway the sequential stabilization strategy discussed here, in which earlier formed native-like structure guides and stabilizes processive native structure formation.
Conclusions
How does the amino acid sequence code for kinetic folding? Most simply stated, it appears to do so according to the same design principles that code for the native state. The structure-based information reviewed here consistently affirms that the folding process is dominated by native-like structure and interactions throughout. The supporting native-like structural context is laid down at the very first on-pathway step. Native-like tertiary interactions promote the progressive association of cooperative secondary structural units to form discrete folding intermediates. These factors determine a limited set of possible folding pathways. The effective kinetic barriers represent an initial large-scale conformational search for a native-like topology that ultimately limits the folding rate, subsequent smaller search-dependent barriers for putting sequential intermediates into place, and, when necessary, an additionally inserted thermal search to reverse nonnative misfolding errors.
These same principles appear to explain the folding behavior of many proteins.
Acknowledgments
We thank Joshua Wand, George Rose, Ken Dill, and Bill DeGrado for comments on the manuscript and members of the Englander laboratory, past and present, for continuing discussion of the issues. This work was supported by the National Institutes of Health and the Mathers Charitable Foundation.
Abbreviations
- HX
hydrogen exchange
- GdmCl
guanidinium chloride
- PUFs
partially unfolded forms
- Cyt
cytochrome
Footnotes
This contribution is part of the special series of Inaugural Articles by members of the National Academy of Sciences elected on April 29, 1997.
References
- 1.Anfinsen C B. Science. 1973;181:223–230. doi: 10.1126/science.181.4096.223. [DOI] [PubMed] [Google Scholar]
- 2.Creighton T E. Adv Biophys. 1984;18:1–20. doi: 10.1016/0065-227x(84)90004-2. [DOI] [PubMed] [Google Scholar]
- 3.Kim P S, Baldwin R L. Annu Rev Biochem. 1990;59:631–660. doi: 10.1146/annurev.bi.59.070190.003215. [DOI] [PubMed] [Google Scholar]
- 4.Baldwin R L. Nat Struct Biol. 1999;6:814–817. doi: 10.1038/12268. [DOI] [PubMed] [Google Scholar]
- 5.Fersht A R, Serrano L. Curr Opin Struct Biol. 1993;3:75–83. [Google Scholar]
- 6.Fersht A R. Curr Opin Struct Biol. 1997;7:3–9. doi: 10.1016/s0959-440x(97)80002-4. [DOI] [PubMed] [Google Scholar]
- 7.Ptitsyn O B. Curr Opin Struct Biol. 1995;5:74–78. doi: 10.1016/0959-440x(95)80011-o. [DOI] [PubMed] [Google Scholar]
- 8.Dobson C M, Karplus M. Curr Opin Struct Biol. 1999;9:92–101. doi: 10.1016/s0959-440x(99)80012-8. [DOI] [PubMed] [Google Scholar]
- 9.Matthews C R. Curr Opin Struct Biol. 1991;1:28–35. doi: 10.1016/j.sbi.2006.01.007. [DOI] [PubMed] [Google Scholar]
- 10.Karplus M, Sali A. Curr Opin Struct Biol. 1995;5:58–73. doi: 10.1016/0959-440x(95)80010-x. [DOI] [PubMed] [Google Scholar]
- 11.Shakhnovich E I. Curr Opin Struct Biol. 1997;7:29–40. doi: 10.1016/s0959-440x(97)80005-x. [DOI] [PubMed] [Google Scholar]
- 12.Chan H S, Dill K A. Annu Rev Biophys Biomol Struct. 1997;26:425–459. doi: 10.1146/annurev.biophys.26.1.425. [DOI] [PubMed] [Google Scholar]
- 13.Bryngelson J D, Onuchic J N, Socci N D, Wolynes P G. Proteins Struct Funct Genet. 1995;21:167–195. doi: 10.1002/prot.340210302. [DOI] [PubMed] [Google Scholar]
- 14.Lazaridis T, Karplus M. Science. 1997;278:1928–1931. doi: 10.1126/science.278.5345.1928. [DOI] [PubMed] [Google Scholar]
- 15.Levinthal C. J Chim Phys. 1968;65:44–45. [Google Scholar]
- 16.Matthews C R. Annu Rev Biochem. 1993;62:653–683. doi: 10.1146/annurev.bi.62.070193.003253. [DOI] [PubMed] [Google Scholar]
- 17.Baldwin R L. J Biomol NMR. 1995;5:103–109. doi: 10.1007/BF00208801. [DOI] [PubMed] [Google Scholar]
- 18.Dill K A, Chan H S. Nat Struct Biol. 1997;4:10–19. doi: 10.1038/nsb0197-10. [DOI] [PubMed] [Google Scholar]
- 19.Pande V S, Grosberg A, Tanaka T, Rokhsar D S. Curr Opin Struct Biol. 1998;8:68–79. doi: 10.1016/s0959-440x(98)80012-2. [DOI] [PubMed] [Google Scholar]
- 20.Onuchic J N, Nymeyer H, Garcia A E, Chahine J, Socci N D. Adv Protein Chem. 2000;53:87–152. doi: 10.1016/s0065-3233(00)53003-4. [DOI] [PubMed] [Google Scholar]
- 21.Plaxco K W, Dobson C M. Curr Opin Struct Biol. 1996;6:630–636. doi: 10.1016/s0959-440x(96)80029-7. [DOI] [PubMed] [Google Scholar]
- 22.Eaton W A, Munoz V, Thompson P A, Chan C-K, Hofrichter J. Curr Opin Struct Biol. 1997;7:10–14. doi: 10.1016/s0959-440x(97)80003-6. [DOI] [PubMed] [Google Scholar]
- 23.Roder H, Colon W. Curr Opin Struct Biol. 1997;7:15–28. doi: 10.1016/s0959-440x(97)80004-8. [DOI] [PubMed] [Google Scholar]
- 24.Segel D J, Bachmann A, Hofrichter J, Hodgson K O, Doniach S, Kiefhaber T. J Mol Biol. 1999;288:489–499. doi: 10.1006/jmbi.1999.2703. [DOI] [PubMed] [Google Scholar]
- 25.Wen Y X, Chen E F, Lewis J W, Kliger D S. Rev Sci Instrum. 1996;67:3010–3016. [Google Scholar]
- 26.Fersht A R, Matouschek A, Serrano L. J Mol Biol. 1992;224:771–782. doi: 10.1016/0022-2836(92)90561-w. [DOI] [PubMed] [Google Scholar]
- 27.Fersht A R. Philos Trans R Soc London B. 1995;348:11–15. doi: 10.1098/rstb.1995.0040. [DOI] [PubMed] [Google Scholar]
- 28.Woodward C K. Curr Opin Struct Biol. 1994;4:112–116. [Google Scholar]
- 29.Scholtz J M, Robertson A D. Methods Mol Biol. 1995;40:291–311. doi: 10.1385/0-89603-301-5:291. [DOI] [PubMed] [Google Scholar]
- 30.Englander S W, Sosnick T R, Englander J J, Mayne L. Curr Opin Struct Biol. 1996;6:18–23. doi: 10.1016/s0959-440x(96)80090-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Englander S W, Mayne L, Bai Y, Sosnick T R. Protein Sci. 1997;6:1101–1109. doi: 10.1002/pro.5560060517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Raschke T M, Marqusee S. Curr Opin Biotechnol. 1998;9:80–86. doi: 10.1016/s0958-1669(98)80088-8. [DOI] [PubMed] [Google Scholar]
- 33.Englander S W, Mayne L. Annu Rev Biophys Biomol Struct. 1992;21:243–265. doi: 10.1146/annurev.bb.21.060192.001331. [DOI] [PubMed] [Google Scholar]
- 34.Linderstrøm-Lang K. In: Symposium on Protein Structure. Neuberger A, editor. London: Methuen; 1958. [Google Scholar]
- 35.Englander S W, Kallenbach N R. Q Rev Biophys. 1984;16:521–655. doi: 10.1017/s0033583500005217. [DOI] [PubMed] [Google Scholar]
- 36.Roder H, Elove G A, Englander S W. Nature (London) 1988;335:700–704. doi: 10.1038/335700a0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Udgaonkar J B, Baldwin R L. Nature (London) 1988;335:694–699. doi: 10.1038/335694a0. [DOI] [PubMed] [Google Scholar]
- 38.Roder H. Methods Enzymol. 1989;176:447–473. doi: 10.1016/0076-6879(89)76024-9. [DOI] [PubMed] [Google Scholar]
- 39.Gladwin S T, Evans P A. Folding Des. 1996;1:407–417. doi: 10.1016/S1359-0278(96)00057-0. [DOI] [PubMed] [Google Scholar]
- 40.Radford S E, Dobson C M, Evans P A. Nature (London) 1992;358:302–307. doi: 10.1038/358302a0. [DOI] [PubMed] [Google Scholar]
- 41.Ptitsyn O B, Pain R H, Semisotnov G V, Zerovnik E, Razgulyaev O I. FEBS Lett. 1990;262:20–24. doi: 10.1016/0014-5793(90)80143-7. [DOI] [PubMed] [Google Scholar]
- 42.Ptitsyn O B. J Protein Chem. 1987;6:273–293. [Google Scholar]
- 43.Kuwajima K. Proteins Struct Funct Genet. 1989;6:87–103. doi: 10.1002/prot.340060202. [DOI] [PubMed] [Google Scholar]
- 44.Feng Y, Sligar S G, Wand A J. Nat Struct Biol. 1994;1:30–35. doi: 10.1038/nsb0194-30. [DOI] [PubMed] [Google Scholar]
- 45.Redfield C, Smith R A, Dobson C M. Nat Struct Biol. 1994;1:23–29. doi: 10.1038/nsb0194-23. [DOI] [PubMed] [Google Scholar]
- 46.Baum J, Dobson C M, Evans P A, Hanley C. Biochemistry. 1989;28:7–13. doi: 10.1021/bi00427a002. [DOI] [PubMed] [Google Scholar]
- 47.Jeng M F, Englander S W, Elove G A, Wand A J, Roder H. Biochemistry. 1990;29:10433–10437. doi: 10.1021/bi00498a001. [DOI] [PubMed] [Google Scholar]
- 48.Bai Y, Sosnick T R, Mayne L, Englander S W. Science. 1995;269:192–197. doi: 10.1126/science.7618079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Bai Y, Englander S W. Proteins Struct Funct Genet. 1996;24:145–151. doi: 10.1002/(SICI)1097-0134(199602)24:2<145::AID-PROT1>3.0.CO;2-I. [DOI] [PubMed] [Google Scholar]
- 50.Milne J S, Mayne L, Roder H, Wand A J, Englander S W. Protein Sci. 1998;7:739–745. doi: 10.1002/pro.5560070323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Chamberlain A K, Handel T M, Marqusee S. Nat Struct Biol. 1996;3:782–787. doi: 10.1038/nsb0996-782. [DOI] [PubMed] [Google Scholar]
- 52.Fuentes E J, Wand A J. Biochemistry. 1998;37:3687–3698. doi: 10.1021/bi972579s. [DOI] [PubMed] [Google Scholar]
- 53.Llinas M, Gillespie B, Dahlquist F W, Marqusee S. Nat Struct Biol. 1999;6:1072–1078. doi: 10.1038/14956. [DOI] [PubMed] [Google Scholar]
- 54.Hiller R, Zhou Z H, Adams M W, Englander S W. Proc Natl Acad Sci USA. 1997;94:11329–11332. doi: 10.1073/pnas.94.21.11329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Bhuyan A K, Udgaonkar J B. Proteins Struct Funct Genet. 1998;30:295–308. [PubMed] [Google Scholar]
- 56.Hollien J, Marqusee S. Biochemistry. 1999;38:3831–3836. doi: 10.1021/bi982684h. [DOI] [PubMed] [Google Scholar]
- 57.Milne J S, Xu Y, Mayne L C, Englander S W. J Mol Biol. 1999;290:811–822. doi: 10.1006/jmbi.1999.2924. [DOI] [PubMed] [Google Scholar]
- 58.Fuentes E J, Wand A J. Biochemistry. 1998;37:9877–9883. doi: 10.1021/bi980894o. [DOI] [PubMed] [Google Scholar]
- 59.Clarke J, Fersht A R. Folding Des. 1996;1:243–254. doi: 10.1016/S1359-0278(96)00038-7. [DOI] [PubMed] [Google Scholar]
- 60.Chamberlain A K, Fischer K F, Reardon D, Handel T M, Marqusee A S. Protein Sci. 1999;8:2251–2257. doi: 10.1110/ps.8.11.2251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Elöve G A, Roder H. In: Protein Refolding. Georgiou G, De Bernardez-Clark E, editors. Washington, DC: Am. Chem. Soc. Symposium Series; 1991. pp. 50–63. [Google Scholar]
- 62.Sosnick T R, Mayne L, Englander S W. Proteins Struct Funct Genet. 1996;24:413–426. doi: 10.1002/(SICI)1097-0134(199604)24:4<413::AID-PROT1>3.0.CO;2-F. [DOI] [PubMed] [Google Scholar]
- 63.Colon W, Elove G A, Wakem L P, Sherman F, Roder H. Biochemistry. 1996;35:5538–5549. doi: 10.1021/bi960052u. [DOI] [PubMed] [Google Scholar]
- 64.Pielak G J, Auld D S, Beasley J R, Betz S F, Cohen D S, Doyle D F, Finger S A, Fredericks Z L, Hilgenwillis S, Saunders A J, et al. Biochemistry. 1995;34:3268–3276. doi: 10.1021/bi00010a017. [DOI] [PubMed] [Google Scholar]
- 65.Jennings P A, Wright P E. Science. 1993;262:892–896. doi: 10.1126/science.8235610. [DOI] [PubMed] [Google Scholar]
- 66.Hughson F M, Wright P E, Baldwin R L. Science. 1990;249:1544–1548. doi: 10.1126/science.2218495. [DOI] [PubMed] [Google Scholar]
- 67.Eliezer D, Chung J, Dyson H J, Wright P E. Biochemistry. 2000;39:2894–2901. doi: 10.1021/bi992545f. [DOI] [PubMed] [Google Scholar]
- 68.Raschke T M, Marqusee S. Nat Struct Biol. 1997;4:298–304. doi: 10.1038/nsb0497-298. [DOI] [PubMed] [Google Scholar]
- 69.Dabora J M, Pelton J G, Marqusee S. Biochemistry. 1996;35:11951–11958. doi: 10.1021/bi9611671. [DOI] [PubMed] [Google Scholar]
- 70.Schulman B A, Kim P S, Dobson C M, Redfield C. Nat Struct Biol. 1997;4:630–634. doi: 10.1038/nsb0897-630. [DOI] [PubMed] [Google Scholar]
- 71. Hodsdon, M. E. & Frieden, C. (2000) Biochemistry, in press. [DOI] [PubMed]
- 72.Oesterhelt F, Oesterhelt D, Pfeiffer M, Engel A, Gaub H E, Muller D J. Science. 2000;288:143–146. doi: 10.1126/science.288.5463.143. [DOI] [PubMed] [Google Scholar]
- 73.Zimm G H, Bragg J K. J Chem Phys. 1959;31:526–535. [Google Scholar]
- 74.Lifson S, Roig A. J Chem Phys. 1961;34:1963–1974. [Google Scholar]
- 75.Eaton W A, Munoz V, Thompson P A, Henry E R, Hofrichter J. Acc Chem Res. 1998;31:745–754. [Google Scholar]
- 76.Jackson S E. Folding Des. 1998;3:R81–R91. doi: 10.1016/S1359-0278(98)00033-9. [DOI] [PubMed] [Google Scholar]
- 77.Laurents D V, Bruix M, Jamin M, Baldwin R L. J Mol Biol. 1998;283:669–678. doi: 10.1006/jmbi.1998.2118. [DOI] [PubMed] [Google Scholar]
- 78.Bai Y. Proc Natl Acad Sci USA. 1999;96:477–480. doi: 10.1073/pnas.96.2.477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Bai Y. Protein Sci. 2000;9:194–196. doi: 10.1110/ps.9.1.194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Jamin M, Baldwin R L. Nat Struct Biol. 1996;3:613–618. doi: 10.1038/nsb0796-613. [DOI] [PubMed] [Google Scholar]
- 81.Tsui V, Garcia C, Cavagnero S, Siuzdak G, Dyson H J, Wright P E. Protein Sci. 1999;8:45–49. doi: 10.1110/ps.8.1.45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Heidary D K, Gross L A, Roy M, Jennings P A. Nat Struct Biol. 1997;4:725–731. doi: 10.1038/nsb0997-725. [DOI] [PubMed] [Google Scholar]
- 83.Weissman J S, Kim P S. Nat Struct Biol. 1995;2:1123–1130. doi: 10.1038/nsb1295-1123. [DOI] [PubMed] [Google Scholar]
- 84.Heidary D K, O'Niell J C J, Roy M, Jennings P A. Proc Natl Acad Sci USA. 2000;97:5866–5870. doi: 10.1073/pnas.100547697. . (First Published May 16, 2000; 10.1073/pnas.100547697) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Xu Y, Mayne L, Englander S W. Nat Struct Biol. 1998;5:774–778. doi: 10.1038/1810. [DOI] [PubMed] [Google Scholar]
- 86.Bai Y, Milne J S, Mayne L, Englander S W. Proteins Struct Funct Genet. 1994;20:4–14. doi: 10.1002/prot.340200103. [DOI] [PubMed] [Google Scholar]
- 87.Mayne L, Englander S W. Protein Sci. 2000;9:1873–1877. doi: 10.1110/ps.9.10.1873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Cohen D S, Pielak G J. J Am Chem Soc. 1995;117:1675–1677. [Google Scholar]
- 89.Pappu R V, Srinivasan R, Rose G D. Proc Natl Acad Sci USA. 2000;97:12565–12570. doi: 10.1073/pnas.97.23.12565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Crippen G M. J Mol Biol. 1978;126:315–332. doi: 10.1016/0022-2836(78)90043-8. [DOI] [PubMed] [Google Scholar]
- 91.Rose G D. J Mol Biol. 1979;134:447–470. doi: 10.1016/0022-2836(79)90363-2. [DOI] [PubMed] [Google Scholar]
- 92.Lesk A M, Rose G D. Proc Natl Acad Sci USA. 1981;78:4304–4308. doi: 10.1073/pnas.78.7.4304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Baldwin R L, Rose G D. Trends Biochem Sci. 1999;24:26–33. doi: 10.1016/s0968-0004(98)01346-2. [DOI] [PubMed] [Google Scholar]
- 94.Baldwin R L, Rose G D. Trends Biochem Sci. 1999;24:77–83. doi: 10.1016/s0968-0004(98)01345-0. [DOI] [PubMed] [Google Scholar]
- 95.Chamberlain A K, Marqusee S. Structure (London) 1997;5:859–863. doi: 10.1016/s0969-2126(97)00240-2. [DOI] [PubMed] [Google Scholar]
- 96.Burton R E, Huang G S, Daugherty M A, Fullbright P W, Oas T G. J Mol Biol. 1996;263:311–322. doi: 10.1006/jmbi.1996.0577. [DOI] [PubMed] [Google Scholar]
- 97.Cavagnero S, Dyson H J, Wright P E. J Mol Biol. 1999;285:269–282. doi: 10.1006/jmbi.1998.2273. [DOI] [PubMed] [Google Scholar]
- 98.Garcia C, Nishimura C, Cavagnero S, Dyson H J, Wright P E. Biochemistry. 2000;39:11227–11237. doi: 10.1021/bi0010266. [DOI] [PubMed] [Google Scholar]
- 99.Nishimura C, Prytulla S, Dyson J H, Wright P E. Nat Struct Biol. 2000;7:679–686. doi: 10.1038/77985. [DOI] [PubMed] [Google Scholar]
- 100.Xiong H, Buckwalter B L, Shieh H M, Hecht M H. Proc Natl Acad Sci USA. 1995;92:6349–6353. doi: 10.1073/pnas.92.14.6349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Sosnick T R, Mayne L, Hiller R, Englander S W. Nat Struct Biol. 1994;1:149–156. doi: 10.1038/nsb0394-149. [DOI] [PubMed] [Google Scholar]
- 102.Elove G A, Bhuyan A K, Roder H. Biochemistry. 1994;33:6925–6935. doi: 10.1021/bi00188a023. [DOI] [PubMed] [Google Scholar]
- 103.Nall B T. Commun Mol Cell Biophys. 1985;3:123–143. [Google Scholar]
- 104.Balbach J, Steegborn C, Schindler T, Schmid F X. J Mol Biol. 1999;285:829–842. doi: 10.1006/jmbi.1998.2364. [DOI] [PubMed] [Google Scholar]
- 105.Shtilerman M, Lorimer G H, Englander S W. Science. 1999;284:822–825. doi: 10.1126/science.284.5415.822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Sosnick T R, Mayne L C, Hiller R, Englander S W. In: Peptide and Protein Folding Workshop. Degrado W F, editor. Philadelphia: IBC; 1995. [Google Scholar]
- 107.Englander S W, Sosnick T R, Mayne L C, Shtilerman M, Qi P X, Bai Y W. Acc Chem Res. 1998;31:737–744. [Google Scholar]
- 108.Abkevich V I, Gutin A M, Shakhnovich E I. Biochemistry. 1994;33:10026–10036. doi: 10.1021/bi00199a029. [DOI] [PubMed] [Google Scholar]
- 109.Thirumalai D, Guo Z. Biopolymers. 1995;35:137–140. [Google Scholar]
- 110.Itzhaki L S, Otzen D E, Fersht A R. J Mol Biol. 1995;254:260–288. doi: 10.1006/jmbi.1995.0616. [DOI] [PubMed] [Google Scholar]
- 111.Hagen S J, Hofrichter J, Eaton W E. J Phys Chem B. 1997;101:2352–2365. [Google Scholar]
- 112.Lapidus L J, Eaton W A, Hofrichter J. Proc Natl Acad Sci USA. 2000;97:7220–7225. doi: 10.1073/pnas.97.13.7220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Riddle D S, Grantcharova V P, Santiago J V, Alm E, Ruczinski I, Baker D. Nat Struct Biol. 1999;6:1016–1024. doi: 10.1038/14901. [DOI] [PubMed] [Google Scholar]
- 114.Martínez J C, Serrano L. Nat Struct Biol. 1999;6:1010–1016. doi: 10.1038/14896. [DOI] [PubMed] [Google Scholar]
- 115.Chiti F, Taddei N, White P M, Bucciantini M, Magherini F, Stefani M, Dobson C M. Nat Struct Biol. 1999;6:1005–1009. doi: 10.1038/14890. [DOI] [PubMed] [Google Scholar]
- 116.Goldenberg D P. Nat Struct Biol. 1999;6:987–990. doi: 10.1038/14866. [DOI] [PubMed] [Google Scholar]
- 117.Plaxco K W, Simons K T, Baker D. J Mol Biol. 1998;277:985–994. doi: 10.1006/jmbi.1998.1645. [DOI] [PubMed] [Google Scholar]
- 118.Baker D. Nature (London) 2000;405:39–42. doi: 10.1038/35011000. [DOI] [PubMed] [Google Scholar]
- 119.Sosnick T R, Shtilerman M D, Mayne L, Englander S W. Proc Natl Acad Sci USA. 1997;94:8545–8550. doi: 10.1073/pnas.94.16.8545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Qi P X, Sosnick T R, Englander S W. Nat Struct Biol. 1998;5:882–884. doi: 10.1038/2321. [DOI] [PubMed] [Google Scholar]
- 121.Takei J, Chu R-A, Bai Y. Proc Natl Acad Sci USA. 2000;97:10796–10801. doi: 10.1073/pnas.190265797. . (First Published September 12, 2000; 10.1073/pnas.190265797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Jeng M-F, Englander S W. J Mol Biol. 1991;221:1045–1061. doi: 10.1016/0022-2836(91)80191-v. [DOI] [PubMed] [Google Scholar]
- 123.Bai Y, Milne J S, Mayne L, Englander S W. Proteins Struct Funct Genet. 1993;17:75–86. doi: 10.1002/prot.340170110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Connelly G P, Bai Y, Jeng M F, Englander S W. Proteins Struct Funct Genet. 1993;17:87–92. doi: 10.1002/prot.340170111. [DOI] [PubMed] [Google Scholar]
- 125.Bushnell G W, Louie G V, Brayer G D. J Mol Biol. 1990;213:585–595. doi: 10.1016/0022-2836(90)90200-6. [DOI] [PubMed] [Google Scholar]