

Abstract
DNA sequence evidence supports a superordinal clade of mammals that comprises elephants, sea cows, hyraxes, aardvarks, elephant shrews, golden moles, and tenrecs, which all have their origins in Africa, and therefore are dubbed Afrotheria. Morphologically, this appears an unlikely assemblage, which challenges—by including golden moles and tenrecs—the monophyly of the order Lipotyphla (Insectivora). We here identify in three proteins unique combinations of apomorphous amino acid replacements that support this clade. The statistical support for such “sequence signatures” as unambiguous synapomorphic evidence for the naturalness of the Afrotherian clade is reported. Using likelihood, combinatorial, and Bayesian methods we show that the posterior probability of the mammalian tree containing the Afrotherian clade is effectively 1.0, based on conservative assumptions. Presenting sequence data for another African insectivore, the otter shrew Micropotamogale lamottei, we demonstrate that such signatures are diagnostic for including newly investigated species in the Afrotheria. Sequence signatures provide “protein-morphological” synapomorphies that may aid in visualizing monophyletic groupings.
Molecular sequence data are increasingly used in mammalian phylogeny and recently have led to a number of unorthodox proposals (1–3). These proposals range from the claim that the guinea pig is not a rodent (4) to making whales and hippos sister groups (5). One of the most remarkable propositions is that of an “African clade” in which species as diverse as elephant shrews (Macroscelidea), golden moles (Chrysochloridae), and tenrecs (Tenrecidae) are grouped with aardvarks (Tubulidentata) and paenungulates (elephants, sea cows, and hyraxes; refs. 6 and 7). All of the African clade species find their fossil roots in Africa, and most are still confined to this continent, hence the name Afrotheria (7). The sequence evidence for Afrotheria is unanimous and strong, deriving from various nuclear and mitochondrial genes (6–10). Morphologically, however, there is no evidence whatsoever for a natural grouping of these taxa (11–14), prompting us to subject the molecular evidence to further scrutiny.
If Afrotheria is a real clade, it might be possible to find specific combinations of amino acid replacements in the proteins that support them. These replacements would represent synapomorphous character states, as remnants of mutational events during the last common ancestry of a clade. Several authors have used the concept of such “sequence signatures” qualitatively in molecular phylogeny (e.g., refs. 15–19), but thorough statistical interpretations are lacking.
We here search for the presence of unique Afrotherian sequence signatures in nine protein data sets—eight nuclear and one mitochondrial—that include at least four Afrotherian orders. Putative Afrotherian signatures were traced in αA-crystallin (CRYAA), aquaporin-2 (AQP2), and interphotoreceptor retinol-binding protein (IRBP). To demonstrate the diagnostic value of the signatures we seek their presence in CRYAA and AQP2 of other potential members of the African clade, including the otter shrew—representing the only tenrecid subfamily living outside of Madagascar. To assess the significance of the candidate signatures, we use likelihood methods (20) to reconstruct their most probable ancestral states at the basal node of the Afrotherian clade. These calculations use a phylogeny reconstructed independently of the protein under investigation. We further use likelihood and combinatorial methods to estimate the probability of the signatures on three alternative morphology-based trees that are incompatible with an African clade. We then combine the evidence from CRYAA, AQP2, and IRBP by using Bayesian techniques to yield a posterior probability for the Afrotherian clade. Demonstrating the statistical improbability of such events in the course of biological evolution (21) may help to escape from the current stalemating in the molecules-versus-morphology debate on vertebrate phylogeny (3).
Materials and Methods
Searching for Afrotherian Signatures.
Databases were searched for sets of protein sequences that included representatives of at least four Afrotherian orders, i.e., Proboscidea (elephants), Sirenia (sea cows), Hyracoidea (hyraxes), Tubulidentata, Macroscelidea, and Afrosoricida (golden moles and tenrecs; ref. 7). This yielded data sets of CRYAA, AQP2, IRBP, von Willebrand factor, α-2B adrenergic receptor, γ-fibrinogen, hemoglobin-α and -β, and cytochrome b. The AQP2 data set was complemented with newly determined sequences of pig, fin whale, and sperm whale (see below). From these data sets, one or, if available, two representatives of all included eutherian orders were taken. When more than two species were available for an order, only the two most divergent sequences were retained. This increases the homoplastic background, and thus the significance of retrieved signatures. Retaining all sequences would make the taxon representation unbalanced and hamper the signature searches. The selected sequences were aligned, using pileup, and manually edited. Where available, two divergent Marsupialia were included as outgroups. For full species names and accession numbers, and for protein alignments of CRYAA, AQP2, and IRBP, see Table 4 and Figs. 3–5, which are published as supplemental data on the PNAS web site, www.pnas.org.
Candidate sequence signatures were retrieved from the alignments by using the spreadsheet signwin (available from the authors). No phylogenetic information is included in this search; signwin solely selects positions at which a designated number of in-group species have the same putatively apomorphous replacement, considering the out-group residue(s) as plesiomorphous condition. The selection window is set to be appropriate for the number of species for which the monophyly is investigated. Thus, when searching for positions that might support the monophyly of the five Afrotheria among the 26 selected eutherian CRYAA sequences (Figs. 1A and 3), the window is set at 5 ± 1. This allows for 20% back or otherwise superimposed replacements within a five-species clade, and the same absolute number of parallel replacements in the other 21 in-group sequences. Positions at which 4–6 species share the same apomorphy are then candidates for any Afrotherian sequence signature. Using a wider or narrower criterion would change our candidate signatures, but as seen in Results the candidate sites for a potential signature emerge clearly from the data.
Figure 1.

Afrotherian signatures in CRYAA, AQP2, and IRBP. (A) Positions from an alignment of 26 eutherian and two marsupial CRYAA sequences at which the same putatively apomorphous replacement occurs in 4–6 eutherian sequences. In black are replacements that occur in at least four of the five Afrotheria. Note that “.” indicates all residues that did not pass the 4–6 search window; they may be identical to the two top out-group sequences or be apomorphies occurring in <4 or >6 sequences; x is unassigned residue; * denotes species that are included in the trees in Fig. 2. (B) Apomorphous replacements occurring in 5–7 AQP2 sequences, considering armadillo as out-group for the other Eutheria; rodents as out-group yields the same signature (shown in Fig. 4). (C) Positions in IRBP that pass the search for four- to six-species eutherian clades. Note that at certain positions different putatively apomorphous replacements fulfil the search criterium and may set apart different clades (e.g., 13E for Afrotheria, 13G for Cetartiodactyla). (D and E) Afrotherian signature positions in newly determined CRYAA and AQP2 sequences, respectively.
Sequence Determination of CRYAA and AQP2.
CRYAA genomic sequences, coding for amino acid residues 64–94, were determined for Indian elephant (Elephas maximus), dugong (Dugong dugon), tail-less tenrec (Tenrec ecaudatus), small Madagascar hedgehog (Echinops telfairi), otter shrew (Micropotamogale lamottei), and golden mole (Amblysomus hottentotus). Otter shrew DNA was extracted from ethanol-preserved liver (voucher specimen IZEA-7083); sources of other DNA were as before (6, 7). Amplification was performed by using a forward primer hybridizing to exon 1 and a reverse primer complementary to the 3′ end of exon 2 (22). AQP2 was sequenced (23) for pig (Sus scrofa), sperm whale (Physeter macrocephalus), manatee (Trichechus manatus), fin whale (Balaenoptera physalus), tree hyrax (Dendrohyrax dorsalis), tail-less tenrec, small Madagascar hedgehog, and otter shrew.
Phylogenetic Tree Reconstruction.
To study the evolution of the candidate Afrotherian signatures found in CRYAA, AQP2, and IRBP (see Results), phylogenetic trees were constructed from a 5,708-bp data set of concatenated α-2B adrenergic receptor, von Willebrand factor, IRBP, and 12S rRNA-tRNA valine-16S rRNA sequences (10), taking those entries that corresponded most closely with the species in our CRYAA, AQP2, and IRBP data sets (see Table 5, which is published as supplemental data on the PNAS web site). In the case of the IRBP signature, phylogeny was constructed with exclusion of the IRBP sequences. Topologies and branch lengths of the obtained trees are thus independent of the protein sequences whose signatures we investigate. It also avoids the problem that covarion processes might influence our tree building (24).
We used a two-step procedure to derive the maximum likelihood (ML or maximum average likelihood sensu Steel and Penny, ref. 25) phylogeny from our sequence data. The size of our phylogenies precluded an exhaustive search of all possible topologies to find the global ML tree. We therefore first calculated the likelihood of the sequence data on starter topologies obtained from a simple neighbor-joining (minimum evolution) analysis. Likelihood calculations were done by using paup-ml (26) assuming the HKY85 model of evolution with gamma rate heterogeneity to allow for the possibility of unequal rates of evolution across sites. We estimated the shape parameter (α) of the gamma distribution, and the transition/transversion ratio, from the data. This yielded a candidate topology with branch lengths based on the ML distance calculation. Subsequently, we searched for better topologies in the region of the initial neighbor-joining topology by using the tree-bisection-reconnection branch swapping algorithm in paup-ml. We repeated this analysis procedure with random sequence input orders and always found the same ML tree. “Constrained trees” were constructed to conform with alternative morphology-based hypotheses for eutherian relationships (see Fig. 2). For these trees we supplied the topologies and reconstructed branch lengths by ML.
Figure 2.

Alternative topologies used to calculate the likelihood of the CRYAA signature. Trees are constructed from a 5,708-bp concatenation of six genes representing the species as indicated by * in Fig. 1 A and D, using kangaroo and opossum as out-group. (A) Unconstrained ML tree. (B) Tree enforcing the association of Afrosoricida (Madagascar hedgehog and golden mole) with hedgehog. (C) Tree constrained to group elephant shrew with Glires. (D) Tree constrained to conform with morphological relationships of eutherian orders as proposed by Novacek (11). All trees present internal branch lengths proportional to likelihood; terminal branches are shortened, and related species combined (Paenungulata: African elephant, manatee and hyrax; primates: galago and human; Rodentia: mole rat and guinea pig; Chiroptera: fruit-eating bat and flying fox; Cetartiodactyla: minke whale, cow, and pig; Perissodactyla: rhinoceros and horse; Carnivora: mink and seal). Filled and open bars indicate where the QLLC signature is assumed to have evolved and disappeared, respectively. In B it is equally parsimonious to have the signature evolve twice, in the ancestor of Afrosoricida and the aardvark-elephant shrew-paenungulate clade, respectively. For complete CRYAA trees and corresponding AQP2 and IRBP trees see Fig. 6. The estimated posterior probabilities of observing the signature QLLC at the numbered nodes are (B) ≈0.0, 0.820, and 0.796 at nodes 1, 2, and 3; (C) ≈0.00, 0.982, and ≈0.00 at nodes 1, 2, and 3; (D) ≈0.0 at nodes 1–4, and 0.923 and 0.507 at nodes 5 and 6, respectively.
Computation of Ancestral States.
We conducted two sorts of likelihood computation to investigate which trees best described the CRYAA, AQP2, and IRBP sequence evolution. In one we calculated the overall likelihood of observing the protein sequence signature separately for CRYAA, AQP2, and IRBP, using as our model the empirical JTT substitution rate matrix (27). A separate likelihood was calculated on the unconstrained ML tree and on the three morphology-constrained topologies. The second set of computations involved the likelihood of the most probable ancestral character states of the candidate sequence signatures. These calculations used the same model of evolution, and followed established procedures of which the details have been described (20, 28, 29). These procedures calculate the likelihood of observing the protein sequence data given a topology and a specified amino acid at some node. A likelihood is calculated for each possible amino acid, with the largest corresponding to the ML estimate. The ratio of the largest likelihood to the sum over all amino acids (the total likelihood), each weighted by the prior probabilities of occurrence, is a measure of the posterior probability of that amino acid at that node. As is customary in such analyses, we assume equal prior probabilities for each amino acid, although basing our calculations on priors equal to the proportion of a given amino acid in the sequence does not alter our conclusions. The product of the probabilities over the separate amino acids that comprise a signature measures the probability of the entire signature at that node. By comparing probabilities at a pair of ancestral and descendant nodes it can be inferred whether the signature arose in the branch leading to the descendant node.
Results
Candidate Sequence Signatures in CRYAA, AQP2, and IRBP.
In the alignment of 28 mammalian CRYAA sequences, six positions were found to be relevant for distinguishing any possible five-species clade (Fig. 1A). The only group of five species set apart by a combination of two or more putative apomorphies, namely 70Q, 74L, and 142C, is formed by elephant, manatee, hyrax, aardvark, and elephant shrew. The combination QLC at positions 70, 74, and 142 thus is a unique feature for the Afrotheria in this CRYAA data set. All Afrotheria, apart from African elephant, share in addition the apomorphy 72L. We therefore investigated the phylogenetic value of 70Q, 72L, 74L, 142C as a putative Afrotherian signature in CRYAA. Among 20 aligned AQP2 sequences, we traced four positions at which putative apomorphies might be diagnostic for a six-species clade (Fig. 1B). The combination 10S, 55T and 104L perfectly set apart the Afrotheria, with exception of dugong, which only shares the 10S apomorphy. The signature STL was studied as an Afrotherian marker in AQP2. In the alignment of 28 IRBP sequences, 47 positions passed the search for a five-species grouping (Fig. 1C). There are 17 putative apomorphies in support of at least four of the five Afrotheria. The combination 18 M, 19S, 76S, 147G, 226I, 272D, and 328E is even perfectly unique for all five Afrotheria. At the 10 other positions the signature is affected by homoplasy, within the limits allowed by our search procedure (see Materials and Methods). The “degenerate” 17-residue signature is used in our further analyses. No signatures were detected in the other six proteins.
CRYAA and AQP2 Signatures in Other Afrotheria.
To perform meaningful likelihood calculations for the retrieved signatures it was desirable to broaden the Afrotherian representation by sequencing CRYAA and IRBP in golden mole and tenrec, and AQP2 in tenrec. This would also be a test for the diagnostic value of these signatures; if they are genuine synapomorphies for Afrotheria, one expects to find them, completely or partially, in CRYAA, AQP2, and IRBP from other members of this clade. We sequenced exon 2 of the CRYAA gene, which encodes the signature positions 70, 72, and 74, in Indian elephant, dugong, golden mole, and three Tenrecidae, including the otter shrew. All new CRYAA sequences were found to code for 70Q, 72L, and 74L, including that of Indian elephant, suggesting that 72V in African elephant is a back mutation (Fig. 1D). For AQP2, additional sequences were obtained for manatee, tree hyrax, and three tenrecs, again including otter shrew. All of these species have the STL signature, apart from manatee, which like dugong AQP2 misses 55T and 104L (Fig. 1E). Unfortunately, sequences for golden mole and tenrec IRBP could not be obtained.
These new sequences illustrate that signatures, even in short proteins like CRYAA and AQP2, have the potential to identify newly investigated species as belonging to a specific clade. These data confirm that golden moles and tenrecs associate with Afrotheria, and indicate that the otter shrew joins this clade.
Likelihoods of the CRYAA, AQP2 and IRBP Signatures.
To calculate the likelihood of the signatures in the Afrotherian species we needed topologies representing alternative hypotheses about their relationships. To construct these alternative topologies we used a 5,708-bp concatenation of six genes (10) that is the only extensive sequence data set available for most taxa that are relevant for our calculations (indicated by asterisks in Fig. 1). It allowed us to make trees with topologies and branch lengths independent of the particular signature under investigation. Fig. 2A shows the topology of the ML tree used for calculating the likelihoods of the CRYAA signature. In this tree the African clade receives bootstrap support of 100%. The principle morphologically favored alternatives are to group Afrosoricida with hedgehog in a monophyletic Lipotyphla, and elephant shrew with Glires (rabbits and rodents; refs. 11–14). The trees in Fig. 2 B–D are constrained to comply with these morphology-based hypotheses. Similar sets of alternative trees were constructed for the AQP2 and IRBP data sets (see Fig. 6, which is published as supplemental data on the PNAS web site).
The log-likelihoods of the CRYAA, AQP2, and IRBP signatures were separately calculated on the corresponding ML and constrained trees (Table 1). The signatures fit in every case the unconstrained ML tree substantially better than any of the constrained trees, providing independent support in three proteins for the Afrotherian clade.
Table 1.
Likelihood of the signatures in CRYAA, AQP2, and IRBP when reconstructed on alternative tree topologies
| Protein | Topology* | Log-likelihood† | Log-difference from best tree† |
|---|---|---|---|
| CRYAA (n = 27) | A | −90.07 (−1540.09) | 0.00 (0.00) |
| B | −100.12 (−1548.75) | 10.05 (8.66) | |
| C | −103.65 (−1563.69) | 13.58 (23.60) | |
| D | −119.17 (−1667.25)‡ | 29.1 (127.16)‡ | |
| AQP2 (n = 20) | A | −51.95 (−876.97) | 0.00 (0.00) |
| B | −61.83 (−891.97) | 9.88 (15.00) | |
| C | −61.52 (−896.81) | 9.57 (19.84) | |
| D | −98.78 (−1001.13)‡ | 46.83 (124.16)‡ | |
| IRBP (n = 28) | A | −541.55 (−6118.73) | 0.00 (0.00) |
| C | −590.41 (−6217.33) | 48.86 (98.27) | |
| D | −735.74 (−6419.23)‡ | 194.19 (300.46)‡ |
n, Number of sequences used for tree constructions.
Topologies as explained in legends of Fig. 2; tree B is lacking for IRBP, Afrosoricida not being available.
Likelihoods in parentheses are those calculated for the entire protein sequence and agree in every case with those calculated for the signature sequence alone.
Likelihood of tree D adjusted to have same number of branches as other trees.
Likelihoods of Ancestral State Reconstructions.
If the signatures in CRYAA, AQP2, and IRBP are synapomorphies of Afrotheria they should have evolved in the branch leading to the basal node of the Afrotherian clade. The estimated posterior probabilities of observing the signature QLLC at nodes 1 and 2 in Fig. 2A are 3.0 × 10-9 and 0.984, respectively. For the AQP2 and IRBP ML trees the corresponding probabilities are 2.0 × 10-6 and 0.987, and 7.8 × 10-32 and 0.391, respectively. The sequence signatures of all three proteins thus have a high probability of evolving in the branch leading to the basal node of the Afrotheria. Probabilities this high for the CRYAA and AQP2 signatures imply that each amino acid replacement in the signatures has a near 1.0 probability of having evolved in that branch. Even for the IRBP signature, which requires 17 separate events in a specified branch, the combined probability is 0.391. Removing just two of the more variable sites (e.g., 59S and 326E, each of which has an approximately 0.65 probability of having evolved in the branch), the combined probability rises to 0.94.
These results confirm that the absence of 72L in African elephant CRYAA must be a loss of L at that site. Similarly, the absence of 55T and 104L in dugong AQP2 is reconstructed as a loss in the branch leading to that species. We also infer that elephant shrew IRBP has lost 59S and 326E, and other instances of homoplasy arise (compare Fig. 1C). However, none constitutes an alternative to the signatures we investigate here.
The morphology-constrained trees each require that the signatures evolved more than once or have evolved and been lost again. Reconstructions of ancestral states similar to those for the ML tree support this interpretation, as shown for CRYAA by the probabilities at the nodes numbered in Fig. 2 B–D, and given in the legends. Comparable values were found for the constrained AQP2 and IRBP trees (Fig. 6). However, to reject the constrained trees solely on the basis that they require more than one gain or loss of the signatures requires a framework within which to consider the probability of a signature event occurring more than once on a tree. If this probability is high, then the alternative topologies are not ruled out by our data.
Phylogenetic Value of the Afrotherian Signatures.
Is it unlikely that the signatures we observed have evolved more than once? To answer this question we developed a methodology that takes account of all possible ways a signature could have arisen given the number of elements (i.e., amino acid replacements in the signature) and the length of the protein. This removes the possibility that we have capitalized on chance. First, we calculate the probability of a given class of signature events arising once. Let r be the number of apomorphic elements in a signature. The class of r-events (i.e., all of the possible signatures of size r) need not be unlikely itself, but for the signature to be an unambiguous marker of a clade the probability must be low that the same (identical) member of the class arises twice.
Given V variable sites in a sequence, and a signature of size r there are (rV) possible signatures of size r. Each signature has probability prqV−r of occurring in any given branch, where p is the probability of an amino acid replacement at a given site in a branch, and q = 1 − p. We assume that p is constant across sites. The product
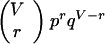 |
1 |
gives the probability of an r-event. Summing this product over r, allowing r to range from r to V, gives the probability of a signature of length r or greater. Call this probability ps, where s denotes signature.
The probability that a signature of length r or greater will arise at least once in a given tree is calculated as follows. Let there be Nb branches in the tree. Then
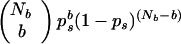 |
2 |
gives the probability of observing a signature of length r or greater in b branches of the tree. Summing this product over b ranging from 1 to N gives the probability of observing on the tree at least one signature of length r or greater. Call this quantity pt, where the t denotes the tree.
We estimated p for each protein, from the number of sites in the sequence, the total number of changes reconstructed on the ML tree, and the number of branches in the tree. We then applied this estimate of p to all sites to calculate ps and pt (Table 2). The results show that none of our signature classes alone is improbable. Thus, given as many variable sites as we observe in each protein, signatures of the sort we have detected or longer, are expected somewhere on each tree.
Table 2.
Probabilities of signatures occurring in the ML trees
| Protein (length) | Variable sites | Total changes on tree* | p† | ps‡ | pt§ | Probability
p for same signature of r sites to occur
|
||
|---|---|---|---|---|---|---|---|---|
| Twice | Three times | Four times | ||||||
| CRYAA (173) | 57 | 123 | 0.014 | 0.008 | 0.328 | 2.80 × 10−7 | 1.22 × 10−13 | 3.43 × 10−20 |
| AQP2 (111) | 31 | 78 | 0.018 | 0.019 | 0.523 | 6.76 × 10−5 | 4.47 × 10−9 | 1.92 × 10−13 |
| IRBP (334) | 227 | 912 | 0.051 | 0.069 | 0.979 | 9.48 × 10−26 | 4.51 × 10−51 | 1.40 × 10−76 |
Calculated by reconstructing the most parsimonious set of amino acid replacements on the ML tree.
Probability of a substitution per site per branch. Calculated as total changes/length of protein/no. of branches in tree. Our conclusions are unaltered if we calculate p using the number of variable sites rather than the length of the protein.
Probability of a signature of length r; for CRYAA, AQP2, and IRBP, r is 4, 3, and 17, respectively.
Probability of any ps event at least once on a tree (further defined in the text).
For the identical signature to arise twice in a tree of Nb branches, any of the r-length events can happen first and anywhere on the tree, but the second r-length event can only be one of the (rV) possible signatures of size r. Each of these occurs in any give branch with probability (prqV−r); call this probability pb, where b denotes branch. Then, the probability of the identical signature arising twice is given by the product of pt and all possible ways of the second signature arising in the Nb−1 remaining branches. (In fact the number of branches in which the second signature can arise typically will be less than Nb−1 because the first signature will usually be present in more than one branch of the tree, owing to identity by descent. This makes our calculations conservative.) This product is written as
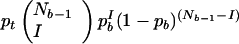 |
3 |
and the symbol I takes the value 1 to account for one additional signature arising. Using the same logic, Eq. 3 can be used to calculate the probability of the same signature arising three or four times by allowing I = 2 or I = 3. Table 2 reports the resulting probability for two, three, and four identical r-events. In these calculations we have replaced the pb of Eq. 3 with pb summed over all signatures of length r or greater. The calculations reported in Table 2 reveal that, although the class of r-events (pt) is not improbable, the probability of the identical r- or greater-length event occurring twice or more by chance is always small and often negligible.
Combining Results from the Three Proteins.
How do these results alter our view about the likelihood of Afrotherian monophyly? Using Bayes' rule (30) we can combine the signature probabilities from Table 2 to arrive at a posterior probability for the Afrotherian hypothesis. From Bayes' rule
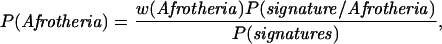 |
4 |
where P(Afrotheria) is the posterior probability of the Afrotheria signature, w(Afrotheria) is our prior belief in the Afrotherian hypothesis, P(signature/Afrotheria) is the probability of the Afrotherian signature given the unconstrained ML tree, and P(signatures) is the combined probabilities of the signatures summed over all four trees, weighted by their prior probabilities. P(signature/Afrotheria) is obtained from the pt column in Table 2, and P(signatures) by combining the Afrotherian results with those from the appropriate column of Table 2, corresponding to the number of times a signature has appeared in the three alternative trees.
Let our prior belief be skeptical to adopt a conservative view against the Afrotherian hypothesis. Let w(Afrotheria) be 0.001. Let our prior belief in the morphology-based hypotheses represented by the other trees be higher, at 0.333 each (0.4995 for IRBP). These weights then sum to 1.0 as they must. Table 3 reports that for all three proteins the posterior belief in Afrotheria is strong and substantially altered from the prior. Calculating the combined posterior support of the three proteins for the Afrotherian hypothesis yields P ∼1.0, even when a prior weight of only 0.0001 is used. Thus, the combined data effectively rule out support for polyphyly of the Afrotherian species.
Table 3.
Bayesian analysis of the evidence for the monophyly of Afrotheria (see text for explanation)
| Protein | Prior weight for Afrotherian clade | Posterior probability of Afrotheria |
|---|---|---|
| CRYAA | 0.001 | 0.999 |
| AQP2 | 0.001 | 0.921 |
| IRBP | 0.001 | ∼1.00 |
| Combined | 0.0001 | 1.000 |
Discussion
The sequence signatures that we identified in CRYAA, AQP2, and IRBP (Fig. 1), without resorting to prior phylogenetic analyses, provide independent evidence for the Afrotherian clade. The signatures are specific to Afrotheria, they arose with high probability at the basal node of the Afrotherian clade, and it is highly improbable that they would have arisen more than once as is required by the morphologically favored tree hypotheses. We demonstrated their predictive value by finding them in several species for which sequence data on the CRYAA and AQP2 proteins did not previously exist. Notably, the finding of the Afrotherian signatures in the otter shrew—for which no other sequence data have yet been published—supports the inclusion of this African insectivore in the Afrotheria.
Can the Afrotherian signatures be dismissed as homoplasy? The parallel appearance of signatures in a data set could be caused by the admixture of paralogous sequences, convergence, covarion processes, lineage sorting, or even to bias in base composition or differences in mutational mechanisms or repair systems (31). However, it seems highly implausible that such evolutionary mechanisms would cause similarly misleading signatures in three functionally independent proteins in precisely the same set of species.
At a methodological level our assumption that sites evolve independently may be questioned. An extensive literature deals with the correlated evolution of amino acid residues in a protein (e.g., refs. 32 and 33). Such mutual dependence makes it understandable that two or more replacements can originate or disappear in concert. To the extent that the amino acid replacements we have identified do change in a correlated manner, our calculations may underestimate the true probabilities of the signatures arising twice. Similarly, we have used a single estimate of the probability of a substitution to characterize every site and every branch. To the extent that the true probability varies our estimates may be affected. However, we reiterate that we have found similar highly improbable signature patterns in three independent proteins and always in the same set of species. Even using our simplifying assumptions, the results are congruent across trees and proteins. Further, our approach uses a statistical methodology that controls for the problem of capitalizing on chance that arises when searching for signatures of unknown length and composition.
The phylogenetic signal contained in sequence signatures, if present in a protein, contributes in any conventional phylogenetic analysis to the topology that is eventually reconstructed. What then is added by identifying and analyzing signatures on their own? It appears that the quantitative approach of analyzing ever longer sequences is not in all instances the panacea of molecular phylogeny, as in the case of deeper level analyses of mitochondrial protein sequences (e.g., refs. 34–38). If one accepts that synapomorphies are the cornerstones of phylogeny reconstruction, it is logical then to additionally search for mutational events that act as qualitative sequence characteristics for a specific clade. Such can be retropositions (39), specific insertions or deletions (e.g., refs. 10, 19, and 22), and the sequence signatures as discussed here. These molecular character-state data may allow a better discrimination between homoplasy and homology, a prerequisite for finding “true” trees (31). Where conventional analyses combine all of the site-by-site information into a single result, the signature approach highlights a concrete set of events whose most plausible evolutionary explanation can help to choose among competing phylogenetic hypotheses.
The “protein morphological” evidence provided by the signatures in CRYAA, AQP2, and IRBP may give an impetus to reevaluate the apparent absence of any morphological synapomorphies for the African clade against the backdrop of the various scenarios for lipotyphlan phylogeny (14, 40).
Supplementary Material
Acknowledgments
We thank Collin van Asten for writing signwin, Marcel Sweers for technical help, and Peter Vogel for otter shrew tissue. This work was supported by grants from the Netherlands Foundation for Life Sciences (to W.W.d.J.), the European Commission (to W.W.d.J., F.C. and M.J.S.), and the Leverhulme Trust and the Natural Environment Research Council (to M.P.).
Abbreviations
- CRYAA
αA-crystallin
- AQP2
aquaporin-2
- IRBP
interphotoreceptor retinol-binding protein
- ML
maximum likelihood
Footnotes
This paper was submitted directly (Track II) to the PNAS office.
See commentary on page 1.
Data deposition: The sequences reported in this paper have been deposited in the GenBank database (accession nos. AJ251100–AJ251106, AJ277647, and AJ270463–AJ270468).
Article published online before print: Proc. Natl. Acad. Sci. USA, 10.1073/pnas.250216797.
Article and publication date are at www.pnas.org/cgi/doi/10.1073/pnas.250216797
References
- 1.de Jong W W. Trends Ecol Evol. 1998;13:270–275. doi: 10.1016/s0169-5347(98)01335-4. [DOI] [PubMed] [Google Scholar]
- 2.Waddel P J, Okada N, Hasegawa M. Syst Biol. 1999;48:1–5. [PubMed] [Google Scholar]
- 3.Allard M W, Honeycutt R L, Novacek M J. Cladistics. 1999;15:213–219. doi: 10.1111/j.1096-0031.1999.tb00264.x. [DOI] [PubMed] [Google Scholar]
- 4.D'Erchia A M, Gissi C, Pesole G, Saccone C, Arnason U. Nature (London) 1996;381:597–600. doi: 10.1038/381597a0. [DOI] [PubMed] [Google Scholar]
- 5.Gatesy J. Mol Biol Evol. 1997;14:537–543. doi: 10.1093/oxfordjournals.molbev.a025790. [DOI] [PubMed] [Google Scholar]
- 6.Springer M S, Cleven G C, Madsen O, de Jong W W, Waddell V G, Amrine H M, Stanhope M J. Nature (London) 1997;388:61–64. doi: 10.1038/40386. [DOI] [PubMed] [Google Scholar]
- 7.Stanhope M J, Wadell V G, Madsen O, de Jong W W, Hedges S B, Cleven G C, Kao D, Springer M S. Proc Natl Acad Sci USA. 1998;95:9967–9972. doi: 10.1073/pnas.95.17.9967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Gatesy J, Milinkovitch M, Waddell V, Stanhope M J. Syst Biol. 1999;48:6–20. doi: 10.1080/106351599260409. [DOI] [PubMed] [Google Scholar]
- 9.Springer M S, Amrine H M, Burk A, Stanhope M J. Syst Biol. 1999;48:65–75. doi: 10.1080/106351599260445. [DOI] [PubMed] [Google Scholar]
- 10.Madsen, O., Scally, M., Kao, D. J., DeBry, R. W., Douady, C. J., Adkins, R., Amrine, H., Stanhope, M. J., de Jong, W. W. & Springer, M. S. (2001) Nature (London), in press. [DOI] [PubMed]
- 11.Novacek M J. Nature (London) 1992;356:121–125. doi: 10.1038/356121a0. [DOI] [PubMed] [Google Scholar]
- 12.McKenna M C, Bell S K. Classification of Mammals Above the Species Level. New York: Columbia Univ. Press; 1997. [Google Scholar]
- 13.Liu F R, Miyamoto M M. Syst Biol. 1999;48:54–64. doi: 10.1080/106351599260436. [DOI] [PubMed] [Google Scholar]
- 14.Asher R J. Cladistics. 1999;15:231–252. doi: 10.1111/j.1096-0031.1999.tb00266.x. [DOI] [PubMed] [Google Scholar]
- 15.Woese C R, Kandler O, Wheelis M L. Proc Natl Acad Sci USA. 1990;87:4576–4579. doi: 10.1073/pnas.87.12.4576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Germot A, Philippe H, Le Guyader H. Proc Natl Acad Sci USA. 1996;93:14614–14617. doi: 10.1073/pnas.93.25.14614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Gupta R S, Johari V. J Mol Evol. 1998;46:716–720. doi: 10.1007/pl00006352. [DOI] [PubMed] [Google Scholar]
- 18.Luckett W P, Hong J. J Mamm Evol. 1998;5:127–182. [Google Scholar]
- 19.Huchon D, Catzeflis F M, Douzery E J. Proc R Soc London Ser B. 2000;267:393–402. doi: 10.1098/rspb.2000.1014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Pagel M. Syst Biol. 1999;48:612–622. [Google Scholar]
- 21.Pagel M. Nature (London) 1999;401:877–884. doi: 10.1038/44766. [DOI] [PubMed] [Google Scholar]
- 22.van Dijk M A M, Paradis E, Catzeflis F, de Jong W W. Syst Biol. 1999;48:94–106. doi: 10.1080/106351599260463. [DOI] [PubMed] [Google Scholar]
- 23.Madsen O, Deen P M T, Pesole G, Saccone C, de Jong W W. Mol Biol Evol. 1997;14:363–371. doi: 10.1093/oxfordjournals.molbev.a025772. [DOI] [PubMed] [Google Scholar]
- 24.Lockhart P J, Steel M A, Barbrook A C, Huson D H, Charleston M A, Howe C J. Mol Biol Evol. 1998;15:1183–1188. doi: 10.1093/oxfordjournals.molbev.a026025. [DOI] [PubMed] [Google Scholar]
- 25.Steel M, Penny D. Mol Biol Evol. 2000;17:839–850. doi: 10.1093/oxfordjournals.molbev.a026364. [DOI] [PubMed] [Google Scholar]
- 26.Swofford D L. paup*: Phylogenetic Analysis using Parsimony (* and other methods) Sunderland, MA: Sinauer; 1998. , Version 4. [Google Scholar]
- 27.Jones D T, Taylor W R, Thornton J M. Comput Appl Biosci. 1992;8:275–282. doi: 10.1093/bioinformatics/8.3.275. [DOI] [PubMed] [Google Scholar]
- 28.Yang Z, Kumar S, Nei M. Genetics. 1995;141:1641–1650. doi: 10.1093/genetics/141.4.1641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Koshi J M, Goldstein R A. J Mol Evol. 1996;42:313–320. doi: 10.1007/BF02198858. [DOI] [PubMed] [Google Scholar]
- 30.Bain L J, Engelhardt M. Introduction to Probability and Mathematical Statistics. Boston: Duxbury; 1987. [Google Scholar]
- 31.Doyle J J. In: Homoplasy: The Recurrence of Similarity in Evolution. Sanderson M J, Hufford L, editors. San Diego: Academic; 1996. pp. 37–66. [Google Scholar]
- 32.Mateu M G, Fersht A R. Proc Natl Acad Sci USA. 1999;96:3595–3599. doi: 10.1073/pnas.96.7.3595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Pollock D D, Taylor W R, Goldman N. J Mol Biol. 1999;287:187–198. doi: 10.1006/jmbi.1998.2601. [DOI] [PubMed] [Google Scholar]
- 34.Philippe H. J Mol Evol. 1997;45:712–715. [PubMed] [Google Scholar]
- 35.Naylor G J P, Brown W M. Syst Biol. 1998;47:61–76. doi: 10.1080/106351598261030. [DOI] [PubMed] [Google Scholar]
- 36.Curole A P, Kocher T D. Trends Ecol Evol. 1999;14:394–398. doi: 10.1016/s0169-5347(99)01660-2. [DOI] [PubMed] [Google Scholar]
- 37.Takezaki N, Gojobori T. Mol Biol Evol. 1999;16:590–601. doi: 10.1093/oxfordjournals.molbev.a026141. [DOI] [PubMed] [Google Scholar]
- 38.Waddell P J, Cao Y, Hauf J, Hasegawa M. Syst Biol. 1999;48:31–53. doi: 10.1080/106351599260427. [DOI] [PubMed] [Google Scholar]
- 39.Hillis D M. Proc Natl Acad Sci USA. 1999;96:9979–9981. doi: 10.1073/pnas.96.18.9979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Seiffert E R, Simons E L. Proc Natl Acad Sci USA. 2000;97:2646–2651. doi: 10.1073/pnas.040549797. . (First Published February 29, 2000; 10.1073/pnas.040549797) [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.