

Abstract
The tomato Cf-9 disease resistance gene encodes a type I membrane protein carrying a cytosolic dilysine motif. In mammals and yeast, this motif promotes the retrieval of type I membrane proteins from the Golgi apparatus to the endoplasmic reticulum (ER). To test whether the C-terminal KKXX signal of Cf-9 is functional as a retrieval motif and to investigate its role in plants, green fluorescent protein (GFP) was fused to the transmembrane domain of Cf-9 and expressed in yeast, Arabidopsis, and tobacco cells. The fusion protein was targeted to the ER in each of these expression systems, and mutation of the KKXX motif to NNXX led to secretion of the fusion protein. In yeast, the mutant protein reached the vacuole, but plants secreted it as a soluble protein after proteolytic removal of the transmembrane domain. Triple hemagglutinin (HA)–tagged full-length Cf-9 was also targeted to the ER in tobacco cells, and cleavage was also observed for the NNXX mutant protein, suggesting an endoprotease recognition site located within the Cf-9 lumenal sequence common to both the GFP- and the HA-tagged fusions. Our results indicate that the KKXX motif confers ER localization in plants as well as mammals and yeast and that Cf-9 is a resident protein of the ER.
INTRODUCTION
Proteins resident in the endoplasmic reticulum (ER) contain structural motifs responsible for their correct subcellular localization. Many soluble ER proteins, such as BiP or protein disulfide isomerase, carry a C-terminal tetrapeptide H/KDEL motif for localization in the ER (Munro and Pelham, 1987; Pelham et al., 1988; Mazzarella et al., 1990; Andres et al., 1991; Denecke et al., 1992, 1993; Napier et al., 1992). A membrane-bound receptor encoded by the yeast ERD2 gene retrieves escaping HDEL-marked proteins from the Golgi apparatus or salvage compartment back to the ER (Lewis et al., 1990; Semenza et al., 1990). Homologs of the Erd2 receptor have been cloned from mammals (Lewis and Pelham, 1990, 1992; Tang et al., 1993) and plants (Lee et al., 1993), highlighting conservation of the H/KDEL motif and retrieval mechanism.
In mammals and yeast, the cytosolic dilysine motif is critical for ER localization of type I membrane proteins (Nilsson et al., 1989; Jackson et al., 1990). The two lysine residues need to be in either the −3,−4 (KKXX) or −3,−5 (KXKXX) positions relative to the C terminus, and no other basic amino acid can be substituted (Jackson et al., 1990, 1993). Mutation of lysine residues leads to expression of the reporter protein on the cell surface in mammals (Nilsson et al., 1989; Jackson et al., 1990) and to its vacuolar delivery in yeast (Gaynor et al., 1994). Studies of the post-translational modification kinetics of dilysine-tagged reporter proteins have demonstrated a constant cycling of the reporter from the ER to the Golgi apparatus and back to the ER (Jackson et al., 1993; Gaynor et al., 1994). The retrieval mechanism is mediated by vesicular Golgi apparatus-to-ER retrograde transport, involving the coat protein I (COPI) complex (Cosson and Letourneur, 1994; Letourneur et al., 1994; Cosson et al., 1996). The dilysine motif interacts directly with COPI, which then drives the recycling of type I membrane proteins to the ER (Cosson and Letourneur, 1994; Letourneur et al., 1994; Cosson et al., 1996; Harter et al., 1996; Kappeler et al., 1997; Harter and Wieland, 1998). A recent study has also demonstrated that the KKAA motif of a chimeric reporter protein is localized in the ER by a retention mechanism, because it does not acquire any of the cis-Golgi apparatus modifications it would have if it were cycling from the ER to the Golgi apparatus and back to the ER (Andersson et al., 1999).
Although several studies have demonstrated that the C-terminal H/KDEL ER signal localizes soluble proteins to the ER in plants (Denecke et al., 1992, 1993; Napier et al., 1992; Gomord et al., 1997; Vitale and Denecke, 1999), there are no similar reports on the role of the dilysine motif for type I membrane proteins in plants. Interestingly, the tomato Cf-9 resistance gene, conferring resistance against the fungal pathogen Cladosporium fulvum, encodes a type I membrane protein that has a large extracytosolic leucine-rich repeat domain, a single transmembrane domain, and a cytosolic tail carrying a C-terminal dilysine (KKXX) motif (Jones et al., 1994). Consistent with Flor's gene-for-gene hypothesis (Flor, 1971), a specific interaction between Cf-9 and the fungal elicitor Avr9 is required to trigger plant defense mechanisms (Van Den Ackerveken et al., 1992). Knowing where Cf-9 functions is one of the first steps toward a better understanding of the activation of plant defenses and the resistance process.
To study the putative ER retrieval motif of Cf-9, fusions of green fluorescent protein (GFP) to the transmembrane domain and cytosolic tail of Cf-9 have been expressed in yeast, Arabidopsis, and tobacco cells. Fluorescence analysis revealed that the fusion protein was localized to the ER in each expression system. In yeast, mutation of the KKXX motif to NNXX resulted in vacuolar localization, but in plants, the GFP fusion was secreted. Surprisingly, proteolytic cleavage removed the transmembrane domain of the NNXX fusion, releasing a soluble form of GFP in plants. Triple hemagglutinin (HA) epitope–tagged Cf-9 was also targeted to the ER in tobacco cells, where a similar proteolytic cleavage occurred when the retrieval signal was mutated. These results suggest that an endoprotease recognition site is located in the lumenal portion of the Cf-9 sequence common to the GFP- and HA-tagged fusions. Altogether, our results demonstrate that plants make use of the dilysine motif to target type I membrane proteins to the ER and that Cf-9 is a resident protein of the ER.
RESULTS
Construction of GFP Fusions for Yeast Expression
GFP from Aequorea victoria is a useful reporter protein with which to study the subcellular localization of proteins in vivo. Several improved variants of GFP have been engineered (Heim et al., 1994, 1995; Haseloff et al., 1997; Kimata et al., 1997). For example, a point mutation in the chromophore, converting Ser 65 to Thr 65 (GFPS65T), increases the fluorescence signal >100-fold (Heim et al., 1995). To test whether the Cf-9 dilysine motif is functional in yeast, GFPS65T was fused to the transmembrane domain and cytosolic tail of Cf-9. One construct (Figure 1A) carried the original Cf-9 dilysine motif (KKRY), whereas a second construct (Figure 1B) contained a mutated motif (NNRY). These chimeric proteins are in-frame fusions of the preproleader sequence of the yeast α-factor precursor, the triple HA epitope tag, GFPS65T, and the transmembrane domain and cytosolic tail of Cf-9. The α-factor sequence carries a Kex2 cleavage site, the cleavage of which often provides a convenient means to assess whether a fusion protein has reached the late Golgi apparatus where the Kex2 protease is located (Graham and Emr, 1991). The GFP constructs were inserted into a high-copy-number URA3 yeast plasmid under the control of a copper-inducible promoter (Hottiger et al., 1994), resulting in plasmids pCBJ132 and pCBJ133.
Figure 1.

Fusion Protein Constructs Analyzed in This Study and Their Corresponding Plasmids.
(A) and (B) Constructs designed for expression in yeast, under the copper-inducible promoter. The first 85 amino acids of the yeast prepro-α factor (αF), containing a signal peptide and three N-glycosylation sites (Y), allow correct ER translocation. The triple HA epitope tag is indicated, and GFP is highlighted in black. The Cf-9 sequence is hatched, and the position of the transmembrane (TM) domain is also indicated. The arrow shows the position of the cleavage site for the late Golgi Kex2 endopeptidase.
(C) to (F) Constructs designed for expression in Arabidopsis and tobacco BY-2 cells, under the cauliflower mosaic virus (CaMV) 35S promoter. The Arabidopsis chitinase signal peptide (SP) confers efficient ER translocation.
Constructs in (A) to (D) carry only domains E (acidic), F (transmembrane), and G (basic, cytosolic) of Cf-9 (Jones et al., 1994), whereas constructs in (E) and (F) also carry domains B, C, and D of Cf-9 (Jones et al., 1994). The four amino acids corresponding to the Cf-9 C-terminal dilysine motif or its mutant derivative are shown at right.
The Cf-9 Dilysine Motif Is Functional as an ER Localization Signal in Yeast
The yeast strain BJ2168 was transformed with the plasmids pCBJ132 and pCBJ133. Expression of the fusion proteins was induced for 2 hr by addition of CuSO4 to a final concentration of 50 μM to midlogarithmic-phase growing cells. As shown in Figure 2, GFP fluorescence was visualized by confocal laser scanning microscopy, using identical image acquisition and processing variables in all analyses, so that relative intensities of fluorescence output could be accurately assessed to compare the GFP constructs.
Figure 2.
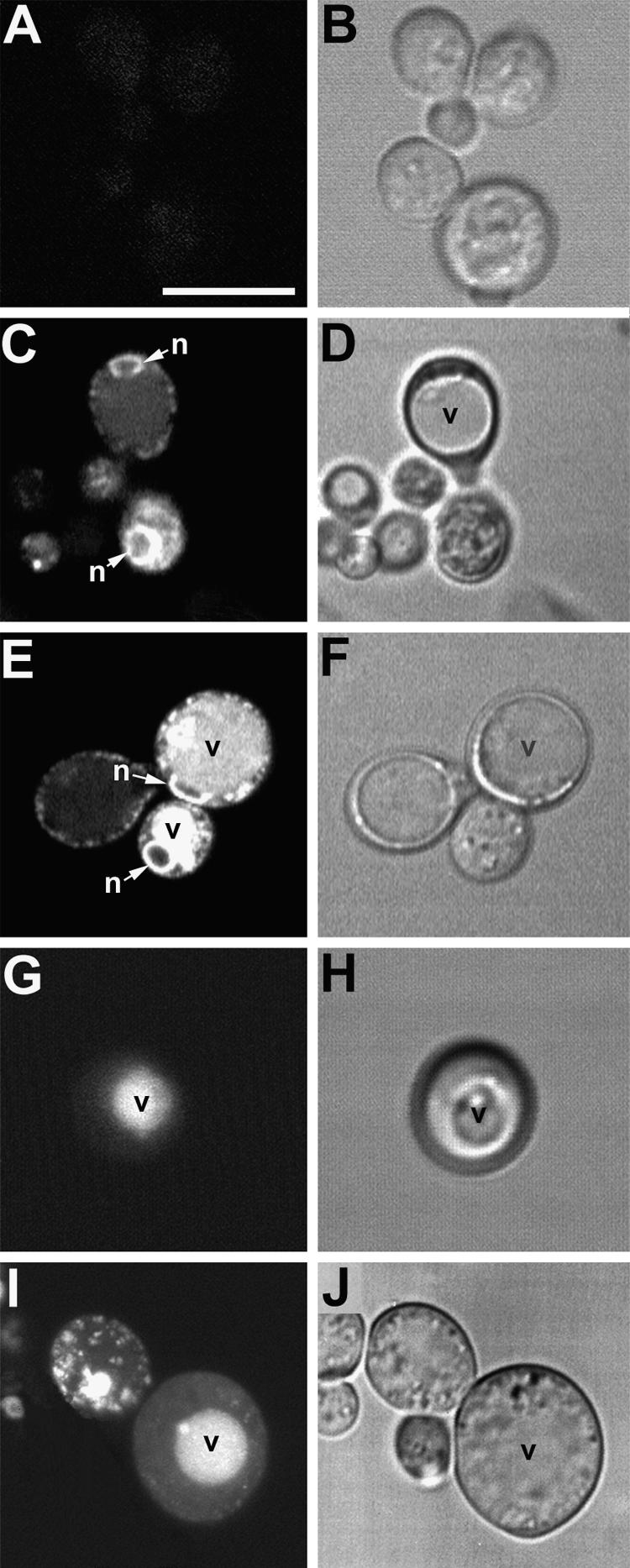
Distribution of GFP Fluorescence in Yeast Demonstrates That the Cf-9 Dilysine Motif Confers ER Localization to Membrane-Bound GFP.
(A) and (B) Nontransformed yeast cells are only weakly autofluorescent (∼1-μm section through midplanes of cells).
(C) and (D) Localization pattern of the KKRY fusion. A midplane section (∼1 μm) reveals a perinuclear (n) and punctate peripheral fluorescence pattern, consistent with ER localization. Note the absence of fluorescence in the vacuole (v), which is clearly visible in the bright- field image (D).
(E) and (F) Overexpression of the KKRY fusion results in secretion of the fusion protein into the vacuoles (v) of some cells. n, nucleus.
(G) and (H) Localization of the NNRY fusion. A projection of seven sequential scans through the entire cell (total depth, ∼6 μm) demonstrates a strictly vacuolar fluorescence pattern (v) with no ER distribution pattern.
(I) and (J) Overexpression of the NNRY fusion results in a punctate fluorescence in addition to the vacuolar localization pattern (v), consistent with Golgi localization. The confocal projection in (I) includes 10 consecutive sections (total depth, ∼8.5 μm).
Confocal fluorescence ([A], [C], [E], [G], and [I]) and bright-field (transmission mode) images ([B], [D], [F], [H], and [J]) were obtained using identical acquisition and image processing parameters. (G) and (I) are projections of several optical sections, so their background areas appear relatively bright.
 .
.
At the standard image acquisition settings used, autofluorescence from nontransformed yeast cells was negligible (Figure 2A), so almost all detectable fluorescence in the transformants was GFP specific. Transformants expressing the membrane-bound GFP fusion containing the KKRY motif (Figure 1A) displayed both a perinuclear and a reticulate cortical fluorescence pattern, which is indicative of ER (Figure 2C). When the same fusion protein was expressed in greater amounts because 500 μM CuSO4 was used, additional vacuolar fluorescence was observed in some cells (Figure 2E). In contrast, yeast cells expressing membrane-bound GFP carrying the NNRY motif (Figure 1B) exhibited no fluorescence in the ER, only vacuolar fluorescence (Figure 2G). Occasionally, overexpression of this fusion resulted in an additional punctate pattern of fluorescence corresponding to the Golgi apparatus (Figure 2I). These results suggest that the Cf-9 KKRY dilysine motif is necessary and sufficient to confer ER localization of membrane-bound GFP and also that the localization mechanism is saturable.
To further investigate the transit out of the ER of the NNRY fusion, we looked for evidence of Kex2 endoproteolytic cleavage. To reach the vacuole, a protein transits through the Golgi apparatus and thus through the Kex2 compartment (Graham and Emr, 1991). The Kex2 processing of both fusions was analyzed by protein gel blotting of proteins extracted from microsomal and soluble fractions of crude cell extracts. When GFP-specific antibodies were used, no signal was observed in the soluble fraction for either GFP fusion (data not shown). Analysis of the microsomal fraction for the KKRY fusion revealed a single band at 61 kD (Figure 3, lane 2). The predicted molecular mass of this chimeric protein, minus its signal peptide, is 46.7 kD. The additional 14 kD suggests that the three N-glycosylation sites of the pro-α-factor peptide are glycosylated. Mutation of the dilysine motif led to processing of the fusion, as shown in Figure 3, lane 3. Five bands were detected, at 61, 44, 37, 35, and 28 kD. The smear observed at ∼61 kD indicates that contrary to expectation, the pro-α-factor peptide has not been removed by Kex2 but the N-linked oligosaccharides have been elongated in the Golgi complex. This strongly supports the idea that the NNRY motif is unable to confer ER localization and thereby allows entry of the GFP fusion into the secretory pathway. Conceivably, the nature of the fusion precluded efficient processing by Kex2 during transit through the late Golgi apparatus. Kex2 processing should give a signal at 39 kD, so the band at 37 kD probably represents the Kex2-processed form of the fusion protein. The smear between 44 and 61 kD probably corresponds to various glycosylated forms of the fusion, ranging from zero to three N-glycans, that were further elongated in the Golgi apparatus. The bands at 35 and 28 kD probably correspond to vacuolar degradation products, although the yeast strain BJ1268 is deficient in the major vacuolar proteases. The signal at 28 kD corresponds well to the molecular mass of GFP itself, consistent with the suggestion that GFP is protease resistant because of its barrel-like structure (Ormö et al., 1996; Yang et al., 1996).
Figure 3.
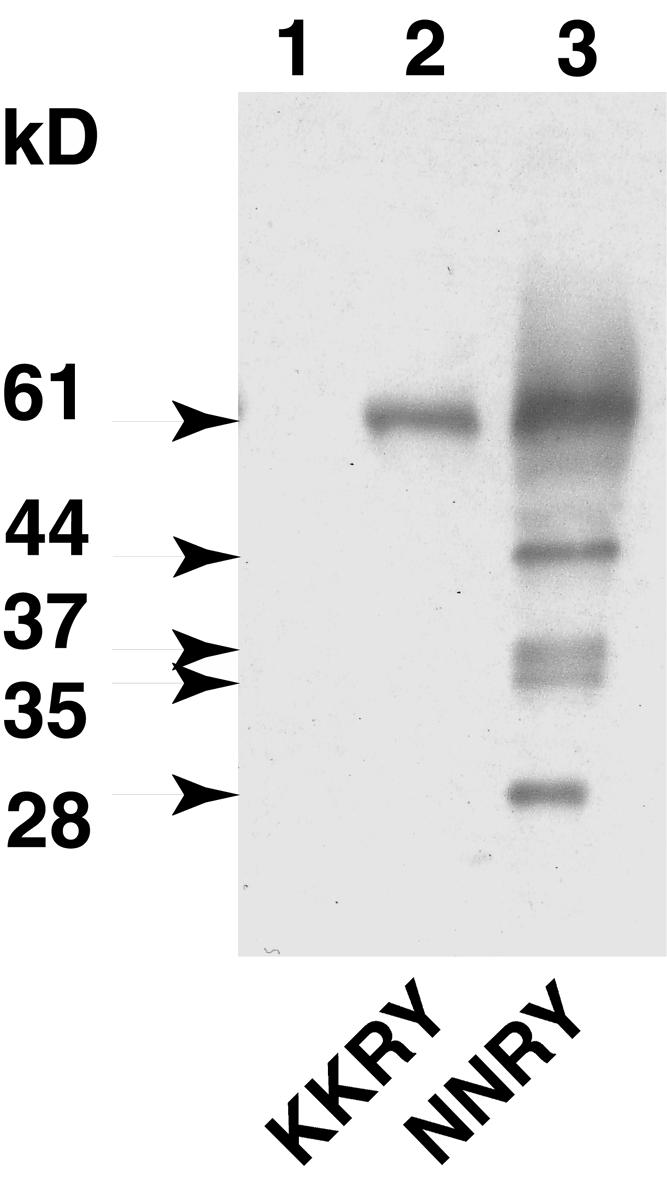
The Cf-9 Dilysine Motif Prevents Processing of Membrane-Bound GFP in Yeast.
Microsomal proteins of transformed BJ2168 yeast cells were analyzed by SDS-PAGE on a 12% gel, processed for protein gel blotting, and probed with anti-GFP-specific antibody. Lane 1 shows proteins from yeast transformed with YEplac195 (vector only); lane 2, proteins from yeast transformed with pCBJ132 (KKRY fusion); and lane 3, proteins from yeast transformed with pCBJ133 (NNRY fusion). The relative molecular masses of the protein bands are indicated at left in kilodaltons.
Construction of GFP Fusions for Expression in Plants
The strategy used to investigate the role of the KKXX motif in plants was also based on the use of membrane-bound GFP as a reporter molecule and mutation of the retrieval signal. An in-frame fusion was made between the Arabidopsis chitinase leader sequence for ER translocation, GFPS65T, and the transmembrane domain and cytosolic tail of Cf-9 (Figure 1C). As before, an NNRY version of this fusion was also produced (Figure 1D). The choice of the Arabidopsis chitinase leader sequence instead of the Cf-9 leader sequence was based on preliminary studies that revealed poor ER translocation of the fusion protein carrying the Cf-9 leader sequence in Arabidopsis (data not shown). In tomato, however, the Cf-9 leader sequence drives efficient ER translocation (M. Benghezal, unpublished observations).
The Cf-9 Dilysine Motif Confers ER Localization in Plants
Transformants of both Arabidopsis and cultured tobacco BY-2 cells expressing the GFP fusions were first analyzed by confocal laser scanning microscopy. Figure 4 compares KKRY and NNRY fusion protein fluorescence patterns in three cell types, including root cap epidermis (Figures 4A, 4D, and 4E [KKRY] and 4B, 4F, and 4G [NNRY]), cotyledon pavement cells (Figures 4H [KKRY] and 4I [NNRY]), and the ridged epidermal cells of petals (Figures 4J to 4L [KKRY] and 4M to 4O [NNRY]). Fluorescence was consistently stronger in the KKRY transformants (Figure 4A), but the NNRY signal intensity (Figure 4B) was well above the autofluorescence of nontransformed tissues under identical acquisition settings (Figure 4C). Scanning at higher magnifications revealed that GFP signals were localized in the ER in both KKRY and NNRY plants. Optical sections near the cell surface revealed a typically “reticulate” cortical ER (Figures 4D, 4F, and 4K), whereas optical sections closer to the midplane (Figures 4E, 4G, 4L, and 4O) revealed a perinuclear localization. The latter was remarkably clear in petal tissues (Figures 4J and 4M), in which the absence of chlorophyll autofluorescence meant that red fluorescence barrier filters, which reduce the signal intensity from leaf tissues, were not required.
Figure 4.

Differences in GFP Fluorescence in the ER of Arabidopsis Demonstrate That the Cf-9 Dilysine Motif Confers ER Localization to Membrane-Bound GFP.
Relative fluorescence of a KKRY transformant ([A], [D], [E], [H], and [J] to [L]) compared with a NNRY transformant ([B], [F], [G], [I], and [M] to [O]) in three different tissues.
(A) to (C) Root cap epidermal cells. At identical acquisition settings, the intensity of fluorescence is markedly greater in the KKRY transformant (A), despite greater expression levels of the NNRY fusion (B). Autofluorescence of a nontransformed plant (C) is distinct from that of the GFP fusion products, demonstrating that the fluorescence in (A) and (B) is GFP specific.
(D) and (E) Root cap epidermis of KKRY fusion. (D) shows an outer surface (∼6 μm thick) projection and (E) a midplane (∼3 μm) projection.
(F) and (G) Root cap epidermis of NNRY fusion. (F) shows an outer surface (∼6 μm thick) projection and (G) a midplane (∼3 μm) projection. Note cortical ER ([D] and [F]) and perinuclear ([E] and [G]) localization.
(H) and (I) Comparison of KKRY (H) and NNRY (I) fusion protein fluorescence in cotyledon pavement cells, abaxial surface. Note the weaker fluorescence in (I) and the strong delineation of the anticlinal cell surface in (H), despite identical thickness (19 μm) Z-series projections.
(J) to (O) Comparison of KKRY ([J] to [L]) and NNRY ([M] to [O]) fusion fluorescence in petal epidermal cells.
(J) KKRY fusion with projection of 27 1-μm-thick sections.
(K) and (L) KKRY fusion, with higher magnification image of petal epidermis, with 7.2-μm projection through cell periphery showing cortical ER network (K) and 10.4-μm projection through cell midplane showing strong perinuclear fluorescence (L).
(M) NNRY fusion, with as in (J), a projection of 27 1-μm-thick optical sections.
(N) and (O) NNRY fusion. (N) Higher magnification image of petal epidermis; 7.2-μm projection through cell periphery. Compared with (K), there is almost no fluorescence in the cortical ER. (O) Midplane projection (10.4 μm thick) immediately beneath the projection shown in (N). Some perinuclear fluorescence is detected.
 ;
; 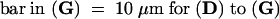 ;
; 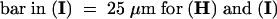 ;
; 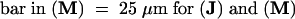 ;
; 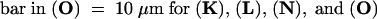 .
.
Importantly, despite the lower fluorescence output of the NNRY transformant, its protein expression levels were in fact greater than those in the KKRY transformant (see later results). Therefore, the most likely explanation for reduced fluorescence is that the NNRY fusion protein did not remain in the ER, which is consistent with the hypothesis that the dilysine motif confers ER localization. Just where the fusion protein was located could not be determined by fluorescence imaging, which suggests the quenching of GFP fluorescence or degradation of the fusion protein itself. No fluorescent material, for example, was detected in the vacuoles or apoplastic spaces in the NNRY transformants (see Figure 4G).
Figure 5 shows that tobacco BY-2 cells expressing the KKRY fusion also displayed an ER-specific pattern of fluorescence. Optical sections from the cell surface through the first 25 μm of two cells show cortical reticulae at low magnification in Figure 5B (upper projection) and at high magnification in Figure 5A as well as transvacuolar strands in Figure 5B. Sectioning deeper into the cells, from 26 to 42 μm, perinuclear strands become visible and transvacuolar strands are clearer (lower projection in Figure 5B). Whereas mutation of the dilysine motif in Arabidopsis transformants resulted in a greatly reduced signal but a similar localization to the ER, it completely eliminated any GFP-specific fluorescence in tobacco BY-2 cells (Figures 4 and 5C, respectively). Any signals observed were no different from those in nontransformed controls (data not shown), attributable to autofluorescent material in both cases.
Figure 5.
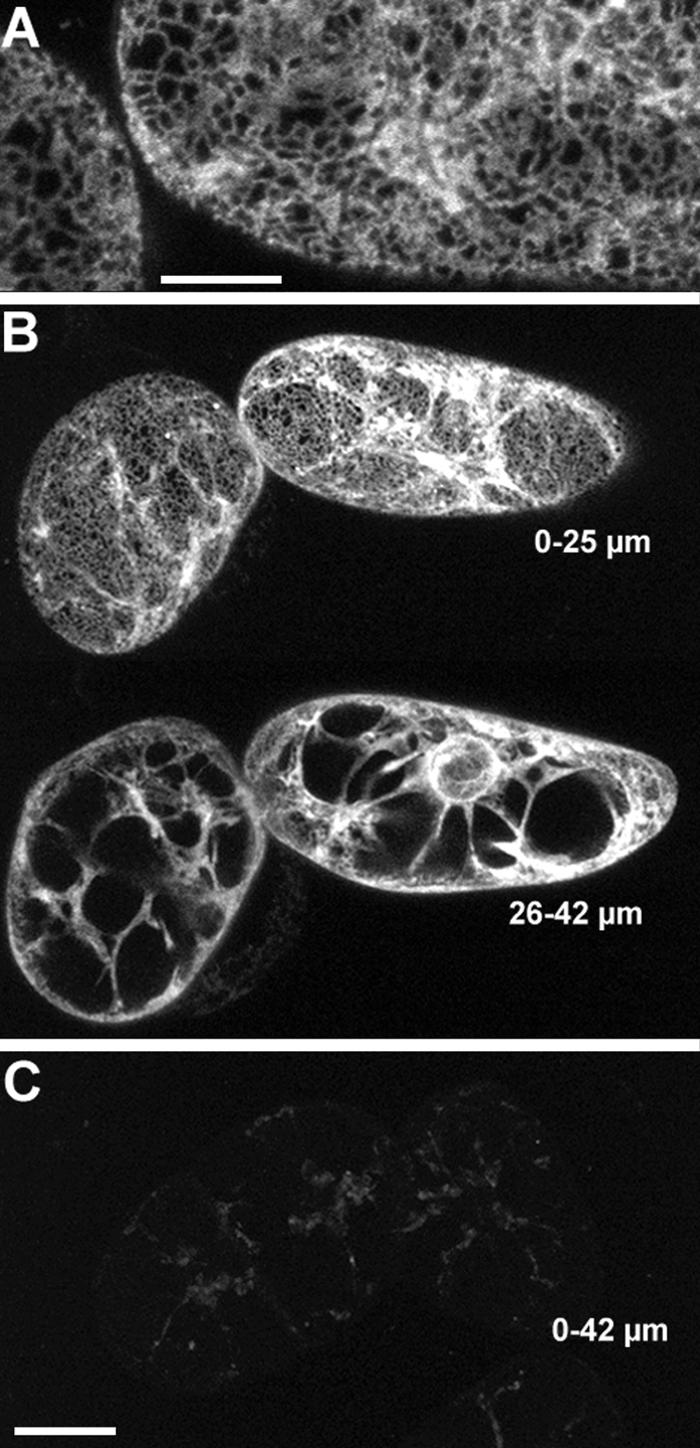
The Dilysine Motif Confers ER Localization to Membrane-Bound GFP in Tobacco BY-2 Cells.
(A) KKRY fusion. A projection of four 1-μm-thick sections through the cortex reveals that GFP fluorescence is localized in the ER.
(B) Lower magnification views of same cells depicted in (A). At the top, a projection through the first 25 μm shows cortical ER and some transvacuolar strands. At the bottom, a projection through the next 17 μm of optical sections clearly illustrates the fluorescence extending from the nuclear envelope through transvacuolar strands to the cortical ER.
(C) NNRY fusion. Using identical image acquisition and processing settings as in (B), virtually no fluorescence was attributable to the GFP. The same autofluorescent material was observed in nontransformed controls (data not shown). This is a 42-μm-thick projection, which is equal to the total depth shown in (B).
Bar in (A) = 10 μm; bar in (C) = 25 μm for (B) and (C).
Fluorescence analyses support the functionality of the dilysine motif in plants, because mutation of the two lysines markedly reduced fluorescent signals in Arabidopsis plants and suppressed them entirely in tobacco BY-2 cells. Further investigation, however, is necessary to elucidate the nature of this quenching.
Membrane-Bound GFP Colocalizes with BiP
To verify that the expressed GFP–KKRY fusion was confined to the ER, we performed double immunofluorescent labeling of GFP and BiP, an ER-resident chaperone. We first tested the reliability of the labeling procedure to distinguish mouse and rabbit antisera by performing actin–tubulin double labeling. Samples were first incubated with a mouse anti-actin together with a rabbit anti-tubulin and then with the same pair of secondary antibody conjugates used for BiP and GFP labeling: goat anti-rabbit–Alexa 568 and sheep anti-mouse–fluorescein isothiocyanate (FITC). In Figures 6A (Arabidopsis) and 6E (tobacco BY-2 cells), the species-specific secondary antibodies display virtually no cross-reactivity, and the FITC and Alexa 568 emissions are cleanly separated by the two confocal filter blocks. Thus, the almost identical distribution patterns of the BiP- and GFP-specific antibodies shown in Figures 6B to 6D and 6F to 6H can be interpreted with confidence.
Figure 6.

Membrane-Bound KKRY–GFP Colocalizes with the ER Marker Protein BiP in Arabidopsis and Tobacco BY-2 Transformants.
Arabidopsis and tobacco BY-2 transformants expressing the KKRY–GFP fusion were immunolabeled after aldehyde fixation with mouse monoclonal GFP–specific antibodies, rabbit polyclonal BiP–specific antibodies, mouse monoclonal actin–specific antibodies, and rabbit polyclonal tubulin–specific antibodies, as indicated. Primary antibodies were detected with either mouse species–specific FITC or rabbit species–specific Alexa 568 conjugates. Confocal laser images are therefore presented in green pseudocolor for both the KKRY–GFP fusion and actin and in red pseudocolor for BiP and tubulin. Colocalization is indicated by yellow where the green and red colors are superimposed.
(A) to (D) Arabidopsis cotyledon epidermis.
(A) Anti-actin and anti-tubulin double labeling. A pair of guard cells and surrounding pavement cells show distinct distribution patterns for microtubules and actin bundles. Note the clean separation of FITC (green) and Alexa 568 (red) signals. Some residual GFP fluorescence is visible along with the actin label in the 5-μm-thick projection.
(B) Anti-GFP and anti-BiP double labeling of a single pavement cell with a well-preserved cortical ER network in the upper 2.4 μm of the Z series. Autofluorescent cuticular material and chlorophyll are visible in the BiP and merged images.
(C) Anti-GFP and anti-BiP double labeling of a guard cell with a cortical focus of 0 to 2 μm. Note the relatively weak residual GFP fluorescence in the adjacent guard cell. The fact that BiP localization is restricted to the lower guard cell indicates that only this cell was permeabilized.
(D) Same region as shown in (C), with a midplane focus. Note the GFP- and BiP-specific fluorescence at the periphery of nuclei.
(E) to (H) Tobacco BY-2 cells.
(E) Anti-actin and anti-tubulin double labeling with clean separation of FITC and Alexa 568 signals in a 4-μm projection.
(F) Anti-GFP and anti-BiP double labeling in a cortical region, ∼3 μm from the upper surface of the cell.
(G) Anti-GFP and anti-BiP double labeling at the upper edge of the nucleus, ∼8 μm below the upper surface.
(H) Anti-GFP and anti-BiP double labeling with a midplane focus ∼18 μm below the upper surface. Note the fluorescence at the nuclear envelope, transvacuolar strands, and cell cortex.
 ;
;  ;
;  ;
; 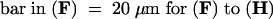 .
.
In Arabidopsis cotyledons (Figures 6B to 6D), fixation preserved the in vivo GFP patterns. Residual GFP activity was relatively weak after glutaraldehyde fixation but was sufficient to show essentially the same distribution as in living cells (cf. with Figures 4H to 4O). The sporadic permeabilization of single epidermal cells by the freeze–shatter method (Wasteneys et al., 1997) provided a convenient way to contrast antibody-specific fluorescence with GFP fluorescence. The upper guard cells shown in Figures 6C and 6D, for example, were not permeabilized and consequently display only residual GFP fluorescence, whereas the permeabilized lower guard cells (and pavement cell shown in Figure 6B) are brighter because the GFP has been labeled with FITC. The yellow color in the merged images (GFP + BiP) indicates sites of BiP (red) and GFP (green) colocalization. Cortical reticulae and perinuclear and transvacuolar strands are apparent in both pavement (Figure 6B) and guard cells (Figures 6C and 6D). Taking into account the presence of chlorophyll and cuticular wax, which fluoresce at similar wavelengths as the Alexa 568 fluorochrome (Figures 6B and 6C), the anti-BiP localized the same structures as the anti-GFP. Wild-type controls showed positive labeling of BiP epitopes but lacked any GFP-specific fluorescence (data not shown). GFP immunolabeling of transformants expressing the GFP–NNRY fusion revealed a weak GFP-specific fluorescent signal colocalized with BiP, but no other signal attributable to GFP immunolabeling could be observed (data not shown).
In tobacco BY-2 cells expressing KKRY–GFP, anti-BiP fluorescence also colocalized with anti-GFP fluorescence (Figures 6F to 6H). GFP and BiP epitopes were labeled at the cell cortex (Figure 6F) and through the transvacuolar strands to the perinuclear region (Figures 6G and 6H).
Mutation of the Dilysine Motif Affects Processing of Membrane-Bound GFP in Plants
To better understand the fate of the GFP fusion protein upon mutation of the dilysine motif, total microsomal and soluble protein extracts from Arabidopsis transformants and transformed tobacco BY-2 cells were analyzed by protein gel blotting with GFP-specific antibodies.
Protein gel blot analysis of total protein extract from Arabidopsis plants transformed with the KKRY construct (Figure 1C) revealed that the GFP fusion protein migrated at the predicted size of 36 kD (Figure 7A, lane 2) and appeared in the microsomal fraction (Figure 7A, lane 3) but not in the soluble protein fraction (Figure 7A, lane 4). The signal at 47 kD clearly represented cross-reactivity of the GFP-specific antibodies with an unrelated soluble protein, because it was also present in the total protein extract from a nontransformed Arabidopsis control plant (Figure 7A, lane 1). In the total protein extract from Arabidopsis plants expressing the NNRY fusion (Figure 1D), a weak signal at the predicted size of 36 kD and a strong signal at 28 kD could be detected (Figure 7A, lane 5). Surprisingly, the 28-kD protein appeared predominantly in the soluble protein fraction (Figure 7A, lane 7), in contrast to the 36-kD protein, which was absent from the soluble fraction (Figure 7A, lane 7). Taken together, the smaller size and fractionation with soluble proteins suggest that the NNRY fusion undergoes endoproteolytic cleavage to release a major portion of the protein from its membrane anchor. Importantly, the 8-kD reduction in size is consistent with the removal of the transmembrane domain and cytosolic tail of Cf-9, and 28 kD is approximately the same size as GFP itself. This strongly suggests that endoproteolytic cleavage of the NNRY fusion in a post-ER compartment releases a soluble form of GFP, because no such cleavage was detected for the KKRY fusion protein. The latter hypothesis is further supported by the detection of both an intact and a processed GFP fusion in the total protein extracts (Figure 7A, lane 5). Moreover, the ratio of processed to intact GFP fusion and their distribution into the microsomal and soluble protein fractions is consistent with proteolytic cleavage and passage of the processed GFP through the secretory pathway. Whether the NNRY fusion is cleaved by an endoprotease or undergoes autocatalytic cleavage is not known.
Figure 7.

Mutation of the Dilysine Motif Results in Endoproteolytic Processing of Membrane-Bound GFP in Plant Cells.
(A) Immunodetection of the GFP fusions in Arabidopsis. Lane 1, nontransformed Arabidopsis; lanes 2 to 4, Arabidopsis transformed with pCBJ98 (KKRY fusion); and lanes 5 to 7, Arabidopsis transformed with pCBJ99 (NNRY fusion).
(B) Immunodetection of the GFP fusions in tobacco BY-2 cells. Lane 1, nontransformed BY-2 cells; lanes 2 to 4, BY-2 cells transformed with pCBJ98 (KKRY fusion); and lanes 5 to 7, BY-2 cells transformed with pCBJ99 (NNRY fusion).
Crude total protein extracts (T) from Arabidopsis leaves and tobacco BY-2 cells were subjected to ultracentrifugation to obtain the microsomal pellet (M) and the soluble fraction (S). Aliquots of each fraction were analyzed by SDS-PAGE on a 6 to 15% gradient gel, processed for protein gel blotting, and probed with GFP-specific antibodies. The negative control (C) corresponds to the crude total protein extracts from nontransformed Arabidopsis or tobacco cells. The relative molecular masses of the protein bands in kilodaltons are indicated at left.
Clearly, given that the nonspecific signal at 47 kD provides an internal control for similar protein loading in each lane (Figure 7A, lanes 2 and 5), the amount of protein expression of the NNRY fusion is much greater than that of the KKRY fusion (Figure 7A, lane 2 versus lane 5). Consequently, we concluded that the 28-kD processed protein (Figure 7A, lane 5) has lost its fluorescent properties, given that the fluorescence output from KKRY plants is much greater than that from NNRY plants. Indeed, the relative intensities of the GFP fluorescence images shown in Figure 4 (strong ER signal in KKRY, weak in NNRY) correlate very well with the relative intensities of the 36-kD bands shown in Figure 7A (lane 3 versus lane 6). Furthermore, the relative intensities of the anti-GFP fluorescence images (strong ER signal in KKRY shown in Figure 6, weak ER signal in NNRY, not shown) also correlate very well with the relative intensities of the 36-kD bands shown in Figure 7A. This suggests not only that the 28-kD protein has lost its fluorescent properties but also that it is absent from the cytoplasm and more specifically from the ER, consistent with secretion into the apoplast, where it would be removed during the preparation of tissue for immunolocalization.
A similar experiment was performed with the transformed tobacco BY-2 cells. Signals at 36 and 28 kD were observed in the total protein extract of cells expressing the KKRY fusion (Figure 7B, lane 2). The 36-kD band appeared only in the microsomal fraction (Figure 7B, lane 3), whereas the 28-kD band was found only in the soluble fraction (Figure 7B, lane 4). In contrast, only the 28-kD band was detected in the total protein extract of cells expressing the NNRY fusion (Figure 7B, lane 5) but again the 28-kD band appeared only in the soluble fraction (Figure 7B, lane 7). Thus, the presence of an ER fluorescence signal in the KKRY fusion and its absence from the NNRY fusion (Figure 5) correlate with the presence of the 36-kD band in the KKRY fusion and its absence from the NNRY fusion (Figure 7B), reinforcing the conclusion that the 28-kD protein is not fluorescent. Moreover, the absence of the 28-kD protein from the microsomal fraction is consistent with passage through the secretory pathway. If the 28-kD protein had been retained in the ER, then a result similar to that observed in yeast would be expected, in which processed GFP located in the vacuole appeared in the microsomal fraction (Figure 3) and not in the soluble fraction (data not shown).
The weak processing of the KKRY fusion to the 28-kD form (Figure 7B, lane 2) may reflect saturation of the retrieval mechanism in tobacco cells because of overexpression of the fusion protein driven by the cauliflower mosaic virus (CaMV) 35S promoter. In Arabidopsis, the low amount of unprocessed 36-kD GFP fusion (Figure 7B, lane 5) observed for the NNRY fusion probably represents steady state protein synthesis in the ER, especially because protein expression is driven by the strong CaMV 35S promoter. Thus, the weak fluorescence of the cortical ER network observed in the NNRY plants (Figure 4) most likely reflects constant replenishment of the 36-kD protein, but a weak retention signal in the Cf-9 transmembrane domain cannot be ruled out.
To test whether soluble GFP is released into the tobacco cell culture medium, we collected and concentrated the liquid culture medium by precipitation with trichloroacetic acid. Protein gel blot analysis with GFP-specific antibodies showed that soluble GFP was recoverable from the culture medium of transformants expressing the NNRY fusion (Figure 8, lane 4) and, to a lesser extent, also from the culture medium of cells expressing the KKRY fusion (Figure 8, lane 3). The culture medium from nontransformed BY-2 control cells exhibited no signal (Figure 8, lane 2). The differential migration of the 36-kD KKRY fusion protein from the microsomes included on the same blot showed unambiguously that only the 28-kD product corresponding to soluble GFP could be recovered from the culture medium (Figure 8, lane 1). This indicates that the soluble GFP has been secreted into the apoplast and released into the culture medium. The absorbance and fluorescence properties of GFPS65T are very sensitive to pH, and its fluorescence is reported to be completely suppressed below pH 6.0 (Haupts et al., 1998; Kneen et al., 1998). Thus, pH sensitivity may be one explanation for the quenching observed when GFP was released into the apoplast of Arabidopsis plants or the tobacco cell culture medium, both of which are acidic. Indeed, the tobacco cell culture medium is buffered at pH 5.7, and the pH of the apoplast is thought to vary between 4.0 and 6.0 (McQueen-Mason et al., 1993; Bibikova et al., 1998; Fry, 1998).
Figure 8.

Mutation of the Dilysine Motif Leads to Secretion of Proteolytically Processed GFP Fusion Protein into the Culture Medium of Tobacco BY-2 Cells.
Aliquots of culture media in which tobacco BY-2 cells had been grown were analyzed by SDS-PAGE on a 6 to 15% gradient gel, processed for protein gel blotting, and probed with GFP-specific antibodies. Lane 1, microsomal pellet (M) from BY-2 cells transformed with pCBJ98 (KKRY fusion); lane 2, medium from nontransformed BY-2 cells; and lanes 3 and 4, medium from BY-2 cells transformed with pCBJ98 (KKRY fusion) or pCBJ99 (NNRY fusion), respectively. The relative molecular masses (in kilodaltons) of the protein bands are indicated at left.
Nevertheless, in conjunction with confocal laser microscopy analyses, these protein gel blot analyses demonstrate that the KKRY motif results in ER localization of an intact GFP fusion protein, whereas the NNRY motif allows exit of the GFP fusion protein from the ER, proteolytic release of GFP from its membrane anchor, and loss of its fluorescence properties.
Mutation of the Dilysine Motif Leads to Rapid Proteolytic Cleavage of Membrane-Bound GFP
To confirm that the dilysine motif is responsible for ER retrieval or retention of membrane-bound GFP, we looked at the kinetics of the proteolytic processing of the GFP fusion carrying the KKRY motif and its NNRY mutant derivative by using pulse–chase analysis. Tobacco BY-2 cells expressing each of the two GFP constructs were radiolabeled with 35S-labeled amino acids for 30 min and chased with excess unlabeled methionine and cysteine for various times. Fusion proteins were recovered by immunoprecipitation with GFP-specific antibodies, separated by SDS-PAGE, and processed for fluorography. Figure 9A shows that the signal for the 36-kD product corresponding to the unprocessed KKRY fusion was stable for 3 hr, and no proteolytic processing was observed in that period. In contrast, Figure 9B shows that the NNRY fusion underwent a rapid proteolytic processing in that a substantial proportion of the fusion protein was already observable as a signal for the 28-kD product during the 30 min of pulse labeling. After 30 min of chase, the signal for the 36-kD product, corresponding to the unprocessed fusion, was almost absent and was converted completely to the signal for the 28-kD product after 60 min. This result indicates that the NNRY mutation leads to rapid proteolytic processing of membrane-bound GFP. We interpret this as a rapid exit out of the ER to a distal compartment that contains the processing activity.
Figure 9.

Kinetics Analysis Shows That the Dilysine Motif Prevents Proteolytic Cleavage of Membrane-Bound GFP in Tobacco Cells.
Tobacco BY-2 cells expressing membrane-bound GFP were pulse-labeled for 30 min with 35S-labeled amino acids and chased with unlabeled methionine and cysteine for the indicated time. Immunoprecipitated proteins were separated by SDS-PAGE and processed for fluorography. The relative molecular masses (in kilodaltons) of the protein bands are indicated at left.
(A) KKRY fusion.
(B) NNRY fusion.
Membrane-Bound GFP Is Absent from the Plasma Membrane
Further biochemical characterization ruled out the presence of the membrane-bound GFP fusion protein carrying a dilysine motif at the plasma membrane (PM). Based on the properties of the PM, polyethylene glycol/dextran aqueous two-phase partitioning (Larsson et al., 1987) allows efficient enrichment of PM vesicles at the expense of intracellular vesicles. Therefore, microsomes from Arabidopsis transformants and transformed tobacco BY-2 cells expressing the KKRY fusion (Figure 1C) were isolated and submitted to aqueous two-phase partitioning. Protein extracts from total microsomes, from intracellular membrane vesicles, and from enriched PM vesicles were subjected to protein gel blot analysis. Anti-GFP was used to detect the GFP fusion protein, and the following antibodies were used to identify proteins that would mark different subcellular compartments: anti-BiP as a marker for the ER (Fontes et al., 1991), anti-vacuolar (V) H+-ATPase as a marker for the tonoplast (Ward et al., 1992), and anti-RD28 (Daniels et al., 1994) and anti-PM H+-ATPase (Palmgren et al., 1991) as markers for the PM of Arabidopsis and tobacco BY-2 cells, respectively.
GFP-specific antibodies detected a signal present in the total microsomal fraction and the intracellular membrane phase but not in the PM-enriched phase of transformed Arabidopsis (Figure 10), demonstrating that membrane-bound GFP is absent from the PM. The distribution of the marker proteins confirms this result. Only the PM aquaporin RD28 monomers (and dimers; data not shown) were detected in the PM-enriched phase (Figure 10). The presence of the RD28 protein in the intracellular membrane phase is the result of “outside in” PM vesicles that partition with the intracellular membrane vesicles (Larsson et al., 1987). Neither the BiP ER marker protein nor the V H+-ATPase tonoplast marker protein was present in the PM-enriched phase (Figure 10).
Figure 10.

Aqueous Two-Phase Partitioning of Arabidopsis and Tobacco Microsomes Demonstrates the Absence of Membrane-Bound KKRY–GFP from the PM.
The microsomal fractions from Arabidopsis leaves or tobacco BY-2 cells transformed with pCBJ98 (KKRY fusion) were subjected to polyethylene glycol (PEG)/dextran aqueous two-phase partitioning. Total protein extracts (T) and proteins from the intracellular membrane (I) and plasma membrane (P) fractions were analyzed by SDS-PAGE on a 6 to 15% gradient gel, processed for protein gel blotting, and probed with anti-GFP (α GFP) and three antibodies to various compartment marker proteins. The PM markers are RD28 for Arabidopsis and PM H+-ATPase for BY-2 cells. The ER marker is BiP, and the tonoplast marker is V H+-ATPase.
The behavior of the KKRY fusion protein in transformed tobacco BY-2 cells was identical to that in Arabidopsis. Membrane-bound GFP could be detected only in the total microsomal fraction and the intracellular membrane phase (Figure 10), consistent with the ER localization already demonstrated by confocal microscopy. As expected, BiP and the V H+-ATPase were also restricted to the total microsomal fraction and the intracellular membrane phase (Figure 10). The PM H+-ATPase was found in all three protein extracts (Figure 10), confirming the enrichment for PM vesicles.
Cf-9 Is Localized in the ER
To investigate the subcellular localization of Cf-9, we used the entire coding sequence of Cf-9 to produce an in-frame fusion between the Arabidopsis chitinase leader sequence, the triple HA epitope tag, and the Cf-9 coding sequence minus its own leader sequence (Figure 1E). The expression of the fusion was driven by the CaMV 35S promoter in transformed tobacco BY-2 cells. To assess the subcellular distribution of the HA-tagged Cf-9 (HA–Cf-9), tobacco membranes were separated on a 5% step sucrose gradient from 15 to 55% in the presence or absence of Mg2+. HA-specific antibodies were used to detect the HA–Cf-9 fusion protein, and antibodies to BiP, PM H+-ATPase, and vacuolar H+-pyrophosphatase (Maeshima and Yoshida, 1989) were used to identify the ER, the PM, and the vacuole, respectively. The Golgi apparatus was identified by Triton X-100–stimulated UDPase activity. Figure 11A shows that Cf-9 cofractionated with the ER marker BiP, and Figure 11B shows that BiP and HA–Cf-9 were shifted to the heavier fractions when Mg2+ was included in the homogenization buffer and the gradient (protein largely disappeared from fractions 5 to 9 inclusive, such that the medians of the protein distributions shifted from fraction 11/12 to fraction 13 and the modes from fraction 11 to fraction 15). The vacuolar marker showed no shift and very little overlap with BiP or HA–Cf-9, indicating clearly that HA–Cf-9 was not located in the vacuole. The Golgi marker also showed no shift but did show some overlap with HA–Cf-9, indicating that although some HA–Cf-9 was clearly located in the ER, some could also have been located in the Golgi apparatus.
Figure 11.

Cf-9 Is an ER Resident Protein.
(A) Subcellular fractionation of tobacco BY-2 cells expressing the HA–Cf-9 fusion in the presence of EDTA. Immunoblots of step sucrose gradient fractions, with fraction 1 corresponding to 15% sucrose and fraction 15 to 55% sucrose, show that Cf-9 cofractionates with the ER. Anti-HA identifies HA–Cf-9, anti-PM H+-ATPase identifies the PM, anti-BiP identifies the ER, and anti–H+-pyrophosphatase identifies the tonoplast. The Golgi apparatus is identified by Triton X-100–stimulated UDPase activity (micromoles per minute per fraction).
(B) Subcellular fractionation of tobacco BY-2 cells expressing the HA–Cf-9 fusion in the presence of Mg2+ results in a shift of the ER to the heavier fractions as well as a shift of HA–Cf-9.
(C) Aqueous two-phase partitioning of tobacco microsomes demonstrates the absence of HA–Cf-9 from the PM. The microsomal fractions from tobacco BY-2 cells expressing HA–Cf-9 were subjected to PEG/dextran aqueous two-phase partitioning. Total protein extracts (T) and proteins from the intracellular membrane (I) and PM (P) fractions were analyzed by SDS-PAGE on a 6 to 15% gradient gel, processed for protein gel blotting, and probed with HA-specific antibodies and three antibodies to various compartment marker proteins. The PM marker is PM H+-ATPase, the ER marker is BiP, and the tonoplast marker is H+-pyrophosphatase.
Because the sucrose gradient result does not rule out PM localization of HA–Cf-9, microsomes from transformed tobacco BY-2 cells were isolated and submitted to aqueous two-phase partitioning. Protein extracts from total microsomes, from intracellular membrane vesicles, and from enriched PM vesicles were subjected to protein gel blot analysis. HA-specific antibodies detected a signal in the total microsomal fraction and in the intracellular membrane phase but not in the PM-enriched phase of transformed tobacco cells (Figure 11C), thus demonstrating that HA–Cf-9 is absent from the PM. The distribution of the marker proteins confirms this result. Only the PM H+-ATPase was detected in the PM-enriched phase (Figure 11C). Neither BiP nor the vacuolar H+-pyrophosphatase was present in the PM-enriched phase (Figure 11C). Thus, the characteristic Mg2+ shift of the ER in the sucrose gradient analysis, together with the aqueous two-phase partitioning results, indicates that Cf-9 is most likely an ER resident protein.
Endoproteolytic Cleavage Takes Place within the Cf-9 Sequence
The endoproteolytic cleavage of the NNRY fusion observed in plants could occur in the Cf-9 sequence, in the GFP sequence, or at the GFP–Cf-9 junction. To determine whether the cleavage site is in the Cf-9 sequence, we also produced an additional HA–Cf-9 fusion carrying the NNRY mutation (Figure 1F). The expression of the fusion was also driven by the CaMV 35S promoter. Tobacco BY-2 cells were transformed with this construct, and proteins from microsomal and soluble fractions of both KKRY and NNRY transformants were analyzed by protein gel blotting with HA-specific antibodies. The KKRY fusion protein migrated as a sharp band at 140 kD in the microsomal fraction (Figure 12A, lane 2) but was absent from the soluble protein fraction (Figure 12A, lane 3), as was expected for an integral membrane protein. The predicted size of the fusion protein is 98 kD, indicating the presence of 42 kD of post-translational modifications. Considering that Cf-9 contains 22 potential glycosylation sites, apparently only a subset of them is being used. The NNRY fusion protein appeared in both the microsomal and the soluble protein fractions (Figure 12A, lanes 4 and 5). It migrated as a smear from 89 to 133 kD and exhibited two discrete bands of 89 and 98 kD. Its presence in the soluble protein fraction (Figure 12A, lane 5) suggests endoproteolytic removal of the transmembrane domain. However, most of the fusion is still associated with the microsomal fraction (Figure 12A, lane 4). Conceivably, the cleavage product might remain loosely associated with the membrane as a peripheral membrane protein.
Figure 12.

Mutation of the Dilysine Motif Leads to Endoproteolytic Processing of Triple HA–Tagged Cf-9 in Tobacco Cells.
(A) Immunodetection of the triple HA–tagged Cf-9. Tobacco BY-2 cells were homogenized, and crude protein extracts were subjected to ultracentrifugation to obtain the microsomal pellet (M) and the soluble fraction (S). Aliquots of proteins from each fraction were analyzed by SDS-PAGE on a 6% gel, processed for protein gel blotting, and probed with HA-specific antibodies. The negative control (C) corresponds to the crude total protein extracts from nontransformed tobacco cells. Lane 1, nontransformed BY-2 cells; lanes 2 and 3, BY-2 cells transformed with pCBJ100 (KKRY fusion); and lanes 4 and 5, BY-2 cells transformed with pCBJ118 (NNRY fusion).
(B) EndoH treatment. Tobacco BY-2 cells were homogenized and processed for immunoprecipitation with HA-specific antibodies. Immunoprecipitated proteins were split into two aliquots and incubated with (+) (lanes 2 and 4) and without (−) (lanes 1 and 3) endoH (EndoH). Lanes 1 and 2, BY-2 cells transformed with pCBJ100 (KKRY fusion); and lanes 3 and 4, BY-2 cells transformed with pCBJ118 (NNRY fusion).
The relative molecular masses of the protein bands (in kilodaltons) are indicated at left.
To further characterize the two fusion proteins, we treated them with endoglycosidase H (EndoH). While complex N-linked glycans are forming in the Golgi apparatus, they become resistant to EndoH (Faye et al., 1986; von Schaewen et al., 1993). The ER localization of the KKRY fusion should lead to the presence of high-mannose-type N-glycans only and therefore to EndoH sensitivity. However, transit of the NNRY fusion or its proteolytic derivative through the secretory pathway should lead to the modification of its N-glycans into complex glycans in the Golgi apparatus and therefore to EndoH resistance. After treatment with EndoH, the KKRY fusion protein shifted from 140 kD (Figure 12B, lane 1) to 98 kD (Figure 12B, lane 2), thus demonstrating that the protein is homogeneously glycosylated and carries ∼42 kD of N-linked oligosaccharides of the high-mannose type. Unexpectedly, the NNRY fusion protein also appeared to be sensitive to EndoH, because it migrated as a single band of 89 kD after treatment (Figure 12B, lane 4). This suggests proteolytic cleavage of the NNRY fusion because it is 9 kD smaller than the KKRY fusion after EndoH treatment. The completeness of the proteolytic cleavage indicates that the NNRY fusion has reached a compartment distal to the ER or the salvage compartment, because the ER-localized KKRY fusion is not proteolytically processed. The 9-kD difference in size between the deglycosylated KKRY and NNRY fusion proteins is consistent with the 8-kD reduction observed previously for GFP fused to the transmembrane and cytosolic domains of Cf-9, suggesting that the transmembrane domain has been removed by proteolytic cleavage at the same position.
These results also suggest that mutation of the KKXX retrieval motif leads to heterogeneous glycosylation of Cf-9, given that the NNRY fusion protein presented as a smear of variously glycosylated forms before EndoH treatment (Figures 12A, lane 4, and 12B, lane 3), whereas the KKRY fusion occurred as a uniformly glycosylated protein (Figures 12A, lane 2, and 12B, lane 1). The heterogeneous glycosylation of the NNRY fusion may reflect a rapid exit from the ER, preventing homogeneous N-glycosylation from taking place, as in the case of the KKRY fusion. The presence of only high-mannose N-glycans on the NNRY fusion protein suggests a protein conformation–related low accessibility of the N-glycans to modifying enzymes in the Golgi apparatus (Faye et al., 1986).
DISCUSSION
The C-terminal dilysine motif confers ER localization to type I membrane proteins in yeast and mammals (Nilsson et al., 1989; Jackson et al., 1990; Gaynor et al., 1994). Using GFP as a reporter fused to the transmembrane domain and cytosolic tail of Cf-9 carrying a C-terminal KKRY motif and a mutant derivative with a C-terminal NNRY motif, we present evidence that plants also recognize the C-terminal dilysine motif as an ER localization signal for type I membrane proteins. Expression of the GFP fusions in yeast demonstrates that the plant dilysine motif is functional, and mutation of the two lysines into asparagines allows exit out of the ER and vacuolar delivery of the chimeric protein, in agreement with a previous study in yeast (Gaynor et al., 1994). However, in our study, overexpression of the KKRY fusion protein resulted in both ER and vacuolar localization, suggesting that the retrieval/retention mechanism could become saturated. Alternatively, the sequence context of the Cf-9 dilysine motif could weaken the retrieval/retention mechanism, resulting in less efficient ER localization and leakage to the vacuole. The presence of two phenylalanines after the two lysines has been reported to reduce the ER retrieval capacity of the lectin ERGIC-53 and to act as an ER exit signal (Itin et al., 1995). Moreover, the two phenylalanines can be replaced by two tyrosines without grossly altering the ER exit signal (Itin et al., 1995). It may be significant that the Cf-9 KKRY motif contains tyrosine in the −1 position.
The Cf-9 dilysine motif also confers ER localization of type I membrane proteins in plants. Arabidopsis plants and tobacco BY-2 cells expressing membrane-bound GFP carrying the Cf-9 KKRY motif exhibited a typical ER fluorescence pattern, confirmed by the colocalization of BiP with the GFP fusion observed in the double-labeling immunofluorescence experiment. Mutation of the dilysine motif led to endoproteolytic removal of the transmembrane domain and release of soluble GFP, which was secreted into the apoplast, consistent with exit of the GFP fusion from the ER to a distal compartment containing the cleavage activity. The pulse–chase experiment with tobacco BY-2 cells demonstrated that the dilysine motif was responsible for the ER retrieval/retention of membrane-bound GFP harboring the KKRY motif because the GFP fusion was not proteolytically processed even after a 3-hr chase. In contrast, the NNRY mutation allowed rapid proteolytic processing, suggesting rapid exit out of the ER and progression through the secretory pathway. Therefore, we can rule out misfolding and association with BiP as a reason for retention of the GFP fusion. The site of the proteolytic processing and the fate of the remaining transmembrane domain are not known. Although unlikely, cleavage might take place in the ER, given that lack of interaction between Cf-9 and the KKXX retrieval mechanism could conceivably leave Cf-9 exposed to an endoproteolytic activity resident in the ER. This might account for the rapid processing of the NNRY fusion, which, if it occurred in a distal compartment, would suggest a more rapid exit from the ER than might be expected. However, Matsuoka et al. (1995) have shown similar kinetics for a protein targeted to and cleaved in the vacuole of tobacco BY-2 cells and argue that the rate of exit from the ER is dependent on protein-folding efficiency. Similarly, very rapid export from the ER has been observed in yeast and is also suggested to be dependent on protein-folding efficiency (Kjeldsen et al., 1999). Given that the GFP fusion protein has no glycosylation sites and no disulfide bonds, perhaps it could fold efficiently and exit the ER rapidly. The absence of a fluorescence signal and the presence of only the processed form of the GFP fusion in protein gel blots of tobacco cells expressing the NNRY fusion is consistent with a rapid exit of the NNRY fusion out of the ER into the compartment in which endoproteolytic cleavage takes place. However, the slow decrease in the soluble GFP signal observed in the pulse–chase experiment suggests slow secretion from the cell. Kjeldsen et al. (1999) have shown in yeast that although exit from the ER can be rapid, passage through the Golgi apparatus can be a rate-limiting step for secretion, which might explain the slow decline in the soluble GFP signal observed.
Recently, a study in tobacco showed that when carrying a mutated dilysine retrieval motif, yeast invertase fused to the transmembrane domain and cytoplasmic tail of yeast Wbp1 (an integral membrane protein in the oligosaccharyltransferase complex) was delivered to the vacuole and converted to a soluble protein (Barrieu and Chrispeels, 1999). The most likely explanation for the vacuolar delivery of invertase, which otherwise behaves like a secreted protein in plants, was that the Wbp1 membrane anchor promoted delivery of the fusion to the tonoplast, where a vacuolar protease released the soluble invertase into the vacuole. This suggests that in plants, as in yeast (Gaynor et al., 1994), the tonoplast is the default destination for membrane proteins with short transmembrane domains such as that of Wbp1, which is predicted to consist of ∼21 amino acids. Cf-9 is also predicted to have a short transmembrane domain of approximately the same number of amino acids, suggesting that the vacuole could be the default destination for Cf-9 if it escaped the ER. Because soluble GFP was delivered to the apoplast and not the vacuole in the NNRY mutant, this suggests cleavage preceding delivery to the tonoplast, most probably in the Golgi apparatus.
Endoproteolytic cleavage clearly occurred within the Cf-9 sequence, because it was also observed for triple HA–tagged Cf-9 carrying the NNRY motif. The reduction in size by ∼8 kD observed for both membrane-bound GFP and triple HA–tagged Cf-9 and the fact that both NNRY fusions were converted to soluble proteins (Figures 7A, lane 7, 7B, lane 7, and 12A, lane 5) strongly suggest that the same cleavage site on the lumenal side of the transmembrane domain was targeted by an endoprotease or subjected to autocatalytic cleavage. The abolition of ER retrieval/retention was required for efficient endoproteolytic cleavage, indicating that processing most likely takes place in a compartment distal to the ER and, in the case of a retrieval mechanism, distal to the salvage compartment. As shown in Figure 13, the 23 amino acids of lumenal Cf-9 sequence left in the membrane-bound GFP fusion protein are proposed to contain the cleavage site. A characteristic feature of this domain is its high content of acidic amino acids, which are thought to contribute to the tight anchoring of the transmembrane domain of Cf-9 in conjunction with the basic residues of the cytosolic tail (Jones et al., 1994).
Figure 13.

Model for Processing Membrane-Bound NNRY–GFP.
The GFP fusion to the transmembrane and cytosolic domain of Cf-9 carrying the C-terminal NNRY motif is subject to endoproteolytic cleavage between GFP and the transmembrane domain in plants. The 23–amino acid sequence of this region is shown relative to the C terminus of the construct. SP denotes the Arabidopsis chitinase signal peptide. The GFP is highlighted in black, and the Cf-9 sequence is hatched. The position of the transmembrane (TM) domain is also indicated.
The dilysine motif also confers ER localization in tobacco BY-2 cells expressing intact Cf-9 in that two-phase partitioning shows Cf-9 is absent from the PM and sucrose gradient centrifugation shows Cf-9 is absent from the tonoplast and present in the ER. Although the Golgi apparatus could not be fully excluded by this analysis, it is unlikely, given that cleavage of Cf-9 would be expected to occur if Cf-9 escaped into the Golgi apparatus and none was observed except when the KKRY motif was mutated to NNRY. In addition to Cf-9 (Jones et al., 1994) and Cf-4 (Thomas et al., 1997), two tomato resistance genes against the fungal pathogen C. fulvum, at least six other plant genes encoding membrane proteins exhibiting a C-terminal KKXX or KXKXX motif have been cloned. The Arabidopsis ECA1 gene (Liang et al., 1997; Liang and Sze, 1998) and the tomato LCA1 gene (Wimmers et al., 1992) encode ER Ca2+ pump ATPases, somewhat homologous to the rabbit SERCA1 gene, which also encodes an ER P-type Ca2+ pump ATPase (Liang et al., 1997). A set of membrane proteins from Arabidopsis involved in lipid biosynthesis—C-8,7 sterol isomerase (Grebenok et al., 1998), Δ7-sterol-C-5-desaturase (Gachotte et al., 1995, 1996), ω-3 fatty acid desaturase (Arondel et al., 1992; Yadav et al., 1993; Nishiuchi et al., 1994), and cytidine-5′-diphosphate-diacylglycerol synthase (Kopka et al., 1997)—also carry a dilysine motif. These proteins are hypothesized to function in the ER, consistent with the C-terminal dilysine motif playing a role in the subcellular localization of type I membrane-anchored proteins in plants. Furthermore, the mouse C-8,7 sterol isomerase homolog mSI (Silve et al., 1996) and the yeast Δ7-sterol-C-5-desaturase homolog Erg3p/Syr1p (Arthington et al., 1991; Taguchi et al., 1994) also contain a dilysine motif at their C termini. Of the plant proteins listed above, only the ECA1 gene product has so far been shown to be localized in the ER by biochemical methods (Liang et al., 1997).
Conservation of the retrieval machinery between plants, mammals, and yeast further supports the functionality of the dilysine motif in plants. The COPI complex (Duden et al., 1991; Waters et al., 1991; Rothman, 1994) is known to bind the dilysine motif through γ-COP (Harter et al., 1996; Harter and Wieland, 1998). A γ-COP homolog from Arabidopsis, AtSec21p, has been cloned recently and partially characterized (Movafeghi et al., 1999). Also, a homolog of the small GTP binding protein Arf, required for assembly of the seven COPI complex subunits, has been identified in Arabidopsis (Regad et al., 1993). Finally, several expressed sequence tags homologous with other COPI subunits have been identified (Andreeva et al., 1998).
The endoproteolytic removal of the transmembrane domain of Cf-9, although surprising, could potentially have a functional role. Although the function of Cf-9 is beyond the scope of this study, it is involved in the activation of plant defense mechanisms that lead eventually to cell death. Therefore, its expression and localization are likely to be tightly controlled by the cell, and endoproteolytic cleavage may regulate Cf-9 expression or function. The specific interaction between Cf-9 and the fungal elicitor Avr9 would seem to require the presence of Cf-9 at the PM for a direct physical interaction, because the Avr9 peptide is secreted by the fungus into the apoplast. In that case, a mechanism to escape ER retrieval must be postulated. One such mechanism has been described for the human oligomeric high-affinity receptor for immunoglobulin E (Letourneur et al., 1995). The cytoplasmic tail of the α chain contains a dilysine motif responsible for its ER localization. After assembly with the γ chain, the dilysine motif of the α chain becomes sterically masked and nonfunctional, allowing transport out of the ER for cell surface expression of the assembled receptor. This process is thought to be a quality control mechanism to distinguish between assembled and nonassembled receptors, and it it possible that a similar mechanism could be used by the plant cell for strict control of the cell surface expression of a Cf-9 receptor complex. Endoproteolytic cleavage of Cf-9 could be irrelevant for an assembled receptor complex, because the endoprotease recognition site may be buried in the complex, but it would not be irrelevant for partially assembled or unassembled Cf-9 molecules. However, our study shows Cf-9 localized to the ER in tobacco cells, and we were unable to detect its presence at the PM. Moreover, although Cf-9 is known to function in tobacco (Hammond-Kosack et al., 1998), suggesting that any other components required for formation of a complex receptor are available in tobacco, we were unable to detect any epitope-tagged Cf-9 that had not been endoproteolytically processed in tobacco cells transformed with the NNRY fusion. This argues in favor of a more simplistic model in which Cf-9 is a resident protein of the ER and proteolytic cleavage is only an artifact resulting from mutation of the dilysine motif. In that case, a mechanism for uptake and delivery of Avr9 to the ER or additional components for an indirect interaction with Cf-9 must be postulated. Interestingly, several toxins have been found to be transported all the way from the cell surface, through endosomes and the Golgi apparatus, to the ER: the Shiga toxin (Sandvig et al., 1992), the cholera toxin (Sandvig et al., 1996), and ricin (Rapak et al., 1997). Avr9 binding studies, using purified PM, showed that tomato plants exhibited a high-affinity binding site for Avr9 whether or not they carried Cf-9 (Kooman-Gersmann et al., 1996). This observation, combined with the ER subcellular localization of Cf-9, suggests that the capacity of Cf-9 to bind Avr9 remains largely untested. It also suggests the presence of an Avr9 receptor, different from Cf-9, at the PM. We find it tempting to postulate that the Avr9 binding site at the cell surface of tomato might represent a target for endocytosis of Avr9.
In conclusion, our data demonstrate that the dilysine motif is functional in plants as an ER localization signal for type I membrane proteins and that as a consequence, Cf-9 is an ER resident protein. We hope to gain greater insight into Cf-9–Avr9 interaction and subsequent plant defense activation by looking at the possible delivery of Avr9 into the plant ER and possible direct physical interaction with Cf-9 in the ER.
METHODS
Construction and Modification of a Cf-9 Cassette
The Cf-9 gene was cloned as a 7.8-kb PstI fragment from cosmid 2/9-34 (Thomas et al., 1995) into dark Bluescript KS+ (Jones et al., 1992) to generate pSLJ7584. A ClaI site was introduced at the start codon by site-directed mutagenesis (Kunkel et al., 1987), and the 3.6-kb HindIII fragment, containing the promoter and 5′ end of the coding region, was cloned in inverted orientation into a derivative of dark Bluescript KS+ in which the EcoRI site had been removed by cutting with SmaI and EcoRV, treating with the Klenow fragment, and self-ligating to generate pSLJ9542. In parallel, the BglII site in the 3′ end of the coding region of pSLJ7584 was removed by site-directed mutagenesis, after which the 1.6-kb EcoRI-BamHI fragment, containing the 3′ end of the coding region and the terminator, was ligated along with the 3-kb XbaI-EcoRI fragment of pSLJ9542, containing the promoter and 5′ coding region, into dark Bluescript KS+ to generate pSLJ9595. This generated a minimal Cf-9 cassette that could be excised with BamHI or XbaI. pSLJ9597, which carries this cassette in the opposite orientation, was generated by cutting pSLJ9595 with BamHI and religating.
To enable in-frame fusions between the green fluorescent protein (GFP) and Cf-9, XbaI sites were introduced into pSLJ9595 by site-directed mutagenesis at the junction between Cf-9 domains A and B (Jones et al., 1994) with the oligonucleotide 5′-CTTGCTTTATCCTCA-TCCGAATTTTCTAGATTGTGCCCCGAA-3′ to generate pSLJ10142 and between domains D and E with the oligonucleotide 5′-AAACTTTGTGGTGGTTCTAGACAAGTGACAACT-3′ to generate pSLJ10151. These mutant Cf-9 cassettes were then ligated as BamHI fragments into a derivative of dark Bluescript KS+ in which the XbaI site had been removed by cutting with XbaI and SmaI, treating with the Klenow fragment, and self-ligating to generate pSLJ10191 and pSLJ10201, derived from pSLJ10142 and pSLJ10151, respectively.
To alter the C-terminal KKRY motif to NNRY, the 3′ end of the coding sequence in pSLJ9595 was altered by site-directed mutagenesis using the oligonucleotide 5′-CATAATTACTACGAAAAGGAAAAAGCGCAATAATAGATATTAGTG-3′, which encodes KRKKRNNRY rather than KMKKHKKRY, to generate pSLJ9701. This mutation, which mimics the C terminus of Cf-2 (Dixon et al., 1996), destroys the KKXX retrieval motif but maintains the number of positively charged residues in the cytoplasmic domain of Cf-9.
Construction and Modification of a GFP Cassette
mGFP5 (Siemering et al., 1996) was cloned as a BamHI-SacI fragment into Bluescript SK− to generate pSLJ10232, which was then modified by site-directed mutagenesis to produce an XbaI GFP cassette lacking the N-terminal leader sequence and the C-terminal KDEL motif of mGFP5 and to destroy an internal ClaI site in the GFP sequence. The mutagenic oligonucleotides used were 5′-GAAAAGTTCTTCTCCTCTAGAGAATTCATCTAA-3′, 5′-GAGCTCTTAGAGTTCTCTAGATTTGTATAGTTC-3′, and 5′-TCCGTCCTCCTTGAAGTCGAT-TCCCT-3′. The resulting plasmid, pSLJ10245, was further mutagenized to convert S65 of GFP to T65 by using the oligonucleotide 5′-AGCATTGAACACCATATGTGAAAGTAGTG-3′ to generate pCBJ4.
Construction of Cf-9–GFP Fusion Cassettes
pCBJ5 (with GFP inserted between domains A and B of Cf-9) and pCBJ7 (with GFP inserted between domains D and E of Cf-9) were constructed by inserting the 711-bp XbaI GFP cassette from pCBJ4 into XbaI-cut pSLJ10191 and pSLJ10201, respectively. pCBJ84 (with GFP inserted between domains D and E of Cf-9 and carrying the NNRY mutation) was constructed by ligation of the 2922-bp ClaI-BamHI fragment from Bluescript SK−, the 3249-bp ClaI-DraI fragment from pCBJ7, and the 473-bp BamHI-DraI fragment from pSLJ9701.
Construction of a Yeast Expression Vector for Hemagglutinin-Tagged Proteins
A BamHI fragment containing the copper-inducible promoter (Hottiger et al., 1994) was treated with the Klenow fragment and inserted into the YEplac195 (Gietz and Sugino, 1988), which had been cleaved with EcoRI and HindIII and also treated with the Klenow fragment. Clones were screened for the “+” orientation corresponding to a parallel orientation of the lacZ gene and the copper-inducible promoter to produce the yeast expression vector designated CUP195+. pCBJ11 (triple hemagglutinin [HA]–tagged Cf-9) was constructed by ligating the following: a 6166-bp BglII-SalI fragment of CUP195+; a 111-bp BglII-AscI triple HA fragment obtained by polymerase chain reaction (PCR) with pMPY-3xHA (Schneider et al., 1995) as template and the oligonucleotides 5′-AGAGATCTTGCTGGATAAAAGATACCCATACGATGTTGC-3′ and 5′-ATAGGCGCGCCCAGCGTAATCTGG-AACG-3′ as primers; a 341-bp AscI-PflMI Cf-9 fragment obtained by PCR with pSLJ9597 as template and the oligonucleotides 5′-GCT-GGGCGCGCCTTGCCTCATTTGTGCC-3′ and M13 forward as primers; and the 2780-bp PflMI-SalI Cf-9 fragment from pSLJ9595.
Construction of Cf-9 Membrane-Bound GFP Fusions for Expression in Yeast
pCBJ132 (GFP fused to the transmembrane and cytosolic domains of Cf-9) was constructed by ligating the 418-bp AscI-PflMI fragment (obtained by PCR using pCBJ5 as template and the two oligonucleotides 5′-ATTTGGCGCGCCTCTAGAGGAGAAGAACTTTTC-3′ and 5′-GAAGAGATGTTTACAGATTCAAGG-3′ as primers), the 652-bp BseRI-SalI fragment from pSLJ9595, the 2339-bp AscI-BseRI fragment from pCBJ11, the 340-bp PflMI-BseRI fragment from pCBJ7, and the 2533-bp BseRI fragment from pCBJ11. pCBJ133 (GFP fused to the transmembrane and cytosolic domains of Cf-9 and carrying the NNRY mutation) was constructed by ligation of the 6695-bp SalI-PflMI fragment from pCBJ132, the 340-bp PflMI-BseRI fragment from pCBJ132, and the 652-bp BseRI-SalI fragment from pSLJ9701.
Construction of a Plant Expression Vector
pCBJ30, a binary vector for plant transformation carrying the cauliflower mosaic virus (CaMV) 35S promoter, was derived from pSLJ755I5 (described in http://www.jic.bbsrc.ac.uk/sainsbury-lab/jonathan-jones/plasmid-list/plasmid.htm), a derivative of pJJ1881 (Jones et al., 1992) that carries a resistance gene (White et al., 1990) to phosphinotricin (Basta; Hoechst Schering AgrEvo, Glen Iris, Australia) driven by the nopaline synthase promoter and also the dark Bluescript KS+ lacZ gene and multiple cloning sites. To generate pCBJ28, pSLJ755I5 was digested with ClaI and HindIII, treated with the Klenow fragment, and religated to remove the ClaI site. The CaMV 35S promoter was derived from pSLJ4J8 (Jones et al., 1992) as a 1.4-kb EcoRI-ClaI fragment cloned into dark Bluescript KS+ and from there as a PstI-SalI fragment cloned into pUC119 to generate pSLJ10122. The 1.4-kb EcoRI-BamHI fragment from pSLJ10122 containing the CaMV 35S promoter was ligated into pCBJ28 digested with EcoRI and BamHI to generate pCBJ30.
Construction of Cf-9 Membrane-Bound GFP Fusions for Expression in Plants
pCBJ94 (with the Arabidopsis chitinase signal peptide and GFP fused to the transmembrane domain and cytosolic tail of Cf-9) was constructed by ligation of the 2943-bp BamHI-SalI fragment of pCBJ5, the 1028-bp NcoI-SalI fragment from pCBJ7, and the 248-bp BamHI-NcoI fragment of mGFP5 carrying the chitinase signal peptide. pCBJ98, the binary plant expression vector carrying this fusion, was constructed by inserting the 1443-bp BamHI DNA fragment of pCBJ94 into pCBJ30 cleaved with BamHI.
pCBJ91 (with the Arabidopsis chitinase signal peptide and GFP fused to the transmembrane domain and cytosolic tail of Cf-9 and carrying the NNRY mutation) was constructed by ligation of the 2958-bp Bluescript SK− vector cleaved with BamHI, the 1195-bp BamHI-NcoI fragment from pCBJ84, and the 248-bp BamHI-NcoI fragment from mGFP5. pCBJ99, the binary plant expression vector carrying this fusion, was constructed by inserting the 1443-bp BamHI DNA fragment from pCBJ91 into pCBJ30 cleaved with BamHI.
Construction of Triple HA–Tagged Cf-9 Fusions for Expression in Plants
pCBJ83 (triple HA–tagged Cf-9) was constructed in two steps. First, the 88-bp BamHI-BglII fragment obtained by PCR with mGFP5 as template and the oligonucleotides 5′-AGCGGATAACAATTTCACACAGG-3′ and 5′-GACAGATCTGAGAATTCGGCCGAGGATAA-3′ as primers was ligated to the 625-bp BglII-SacI fragment obtained by PCR with pCBJ11 as template and the oligonucleotides 5′-ACAAGATCTGTACCCATACGATGTTGCT-3′ and 5′-GAAGAGATGTTTACAGATTCAAGG-3′ as primers and to Bluescript SK− cleaved with BamHI. The resulting plasmid was cleaved with KpnI and SacI to give a 779-bp fragment that was ligated with the 2470-bp SacI-XhoI fragment from pSLJ9595 and the 2943-bp KpnI-XhoI fragment from Bluescript SK−. pCBJ100, the binary plant expression vector carrying this fusion, was constructed by inserting the 3204-bp ClaI fragment from pCBJ83 into pCBJ30 cut with ClaI.
pCBJ113 (triple HA–tagged Cf-9 carrying the NNRY mutation) was constructed by ligation of the 2913-bp BamHI-SalI fragment from Bluescript SK−, the 572-bp SalI-PflMI fragment from pCBJ83, and the 2605-bp PflMI-BamHI fragment from pSLJ9701. pCBJ118, the binary plant expression vector carrying this fusion, was constructed by inserting the 2980-bp ClaI-XbaI DNA fragment from pCBJ113 into pCBJ30 cleaved with ClaI and XbaI.
Sequences of the DNA constructs are available upon request. All mutations and constructions were verified by sequencing with ABI Prism BigDye Terminator Cycle Sequencing Kits (Perkin-Elmer, Norwalk, CT) according to the manufacturer's instructions.
Yeast Strains, Growth Conditions, and Transformation
The Saccharomyces cerevisiae strain used was BJ2168 (Mat a, prc1-407, prb1-1122, pep4-3, leu2, trp1, ura3-52, gal2). Cultures were maintained on YPD plates (Wickerham, 1946) and were grown at 24°C in SD medium (Wickerham, 1946) containing salts, vitamins, trace elements with or without copper, and 2% glucose as carbon source. The optical density of diluted cell suspensions was measured in a 1-cm-pathlength cuvette at 600 nm; 1 OD600 unit of cells corresponds to 107 to 2.5 × 107 cells, depending on the strain. Yeast cells were transformed by electroporation (Becker and Guarente, 1991) and plated onto SDCA-selective plates (SDCA is SD medium supplemented with 1% casein hydrolysate and adenine at 40 μg/mL). For expression of fusion proteins, copper in the trace elements was omitted, and yeast cells were induced for 2 hr at 24°C with CuSO4 to a final concentration of 50 or 500 μM.
Growth and Transformation of Arabidopsis Plants
Arabidopsis plants (Arabidopsis thaliana ecotype Columbia) were grown at 21°C with 16 hr of light (100 μmol of photons m−2 sec−1) per day and 60% humidity.
Plant transformation was performed by the vacuum infiltration technique (Bechtold et al., 1993) using Agrobacterium tumefaciens strain AGL-1 (Lazo et al., 1991). Transformants were recovered by sowing seeds into soil previously moistened with an aqueous solution of phosphinotricin (White et al., 1990) at a concentration of 20 mg/L. Recovery of several dozen transformants from a population of ∼10,000 seedlings was routine. For each transformation, ∼24 transformants per transformation were screened for expression of GFP fluorescence, and three to six were further tested by protein gel blotting for the presence of the GFP fusion protein. One transformant per transformation was chosen for detailed characterization. Plants were grown for 4 to 7 days on plates containing sterile solidified Murashige and Skoog (MS) medium (Murashige and Skoog, 1962) supplemented with 3% sucrose and 20 mg/L phosphinotricin in preparation for microscopic analysis.
Growth and Transformation of Tobacco BY-2 Cells
Tobacco BY-2 cells were grown at 27°C and subcultured weekly in liquid MS medium containing 3% sucrose, 200 mg/L KH2PO4, 100 mg/L myo-inositol, 1 mg/L thiamine, and 0.2 mg/L 2,4-dichlorophenoxyacetic acid (Sigma) and adjusted to pH 5.7 with 1 M KOH. Transformation was performed using A. tumefaciens strain LB4404. Briefly, 6 mL of 3-day-old BY-2 cells were distributed into a Petri dish, and 0.1 mL of midlogarithmic-phase LB4404 cells, containing the binary of interest, was added. After 3 days of coincubation at 27°C, the BY-2 cells were washed five times with 25 mL of culture medium, and aliquots were plated onto solid MS medium containing 3% sucrose, 5 μg/mL phosphoinotricin , and 500 μg/mL carbenicillin (Sigma). Plates were incubated for 3 to 4 weeks at 27°C, and transformants were transferred twice onto new selective plates before reinitiating a liquid culture. Twelve transformants were screened for GFP fluorescence and expression of the fusion protein, and one transformant was chosen for further characterization.
Microscopy
Micrographs were obtained by using a confocal laser scanning system (MRC-Bio-Rad 600; Bio-Rad Microscience Division, Hemel Hempstead, UK) coupled to an Axiovert IM-10 (Zeiss, Oberhochen, Germany). GFP images were collected after 488-nm excitation from the ArKr laser, with use of the BHS filter block and additional neutral density filters to minimize damage to living tissues. In the case of photosynthetic material, an additional red barrier filter was used to eliminate chlorophyll autofluorescence. For double-label immunofluorescence, the K1 and K2 filter blocks were used to separate the emission spectra from the fluorescein isothiocyanate (FITC) and Alexa 568 fluorochromes. Bright-field images were collected in the transmission mode of the confocal system. Digital images were recorded by using the confocal microscope operating system software. In all cases, acquisition settings, including filter combinations, gain, black levels, laser dwell time, sample averaging, and pinhole and Z-step sizes, were first optimized for KKRY transformants; the identical settings were then used for NNRY transformants and nontransformed controls so that relative signal intensities could be accurately compared. For Arabidopsis tissues and BY-2 cells, Z-series parameters were also standardized. Yeast images were all collected by using the ×100 oil objective lens. Arabidopsis images used the ×40, ×63, and ×100 lenses (all oil immersion). Tobacco BY-2 images used the ×63 and ×100 lenses. Projections were generated with Confocal Assistant Software 4.02 (Bio-Rad). For final processing, we used Adobe Photoshop 4.0 (Adobe Systems, Mountain View, CA).
Immunofluorescence Techniques
Transformed Arabidopsis seedlings were fixed, permeabilized by freeze-shattering in liquid nitrogen, and immunolabeled as described previously (Wasteneys et al., 1997). Double immunolabeling of membrane-bound GFP and endoplasmic reticulum (ER)–specific BiP epitopes was achieved by combining two mouse GFP-specific monoclonal antibodies (each at a working dilution of 1:200; Boehringer Mannheim, Mannheim, Germany; Clontech, Palo Alto, CA) with the rabbit anti-BiP antibodies used in immunoblotting experiments (1:1000 dilution). After an overnight incubation in primary antibodies and washing in incubation buffer (PBS containing 50 mM glycine), samples were incubated for 3 hr at 37°C in a combination of species-specific secondary antibody conjugates: a sheep anti–mouse FITC (product code DDF; Amrad Biotech, Boronia, Australia) diluted 1:30 and a goat anti–rabbit Alexa 568 (product code A-11011; Molecular Probes, Eugene OR) diluted 1:100. To test the specificity of the secondary antibodies, we also performed double labeling of actin filaments and microtubules. Mouse anti-actin (clone C4; ICN Biomedicals, Costa Mesa, CA) was combined with a rabbit anti–soy tubulin (generously provided by Dr. Richard Cyr, Pennsylvania State University, University Park). Both antisera were diluted to a final working solution of 1:200.
Before the transformed tobacco BY-2 cells were immunolabeled, m-maleimidobenzoyl N-hydroxysuccinimide ester was added at a final concentration of 100 μM to a 3-day-old suspension of cells, which were then incubated for 10 min at room temperature. Cells were collected by centrifugation at 500g for 1 min and resuspended in PME buffer (100 mM PIPES, pH 6.9, 5 mM EGTA, and 2 mM MgCl2) containing 1.6% formaldehyde and 0.5% glutaraldehyde (electromicroscopy grade). Cells were then incubated for 40 min and washed once with PME buffer and once with cell wall buffer (10 mM MES, pH 5.7, 30 mM CaCl2, 0.1% BSA, 5 mM β-mercaptoethanol, and 0.4 M mannitol). Cells were then digested in the cell wall buffer plus 1% cellulase and 0.1% pectolyase Y-23 for 30 min at 30°C, washed twice with PBS, extracted for 1 hr with PBS containing 1% Triton X-100, washed once with PBS, reduced for 30 min by addition of NaBH4 at a final concentration of 1 mg/mL, and washed twice with PBS containing 50 mM glycine. Immunolabeling was performed as described above for Arabidopsis, using the same antibodies at identical working dilutions.
Preparation of Protein Extracts
Protein extraction from yeast cells was performed as described previously (Benghezal et al., 1996), and for SDS-PAGE (Laemmli, 1970), each lane was loaded with the protein equivalent of cells that would give a value of 1 OD600 . Plant tissue (3 g/mL buffer) from 3-week-old Arabidopsis plants or 3-day-old BY-2 cells was homogenized on ice in TEPI buffer (50 mM Tris-HCl, pH 8.5, 5 mM EDTA, 2 mM phenyl- methylsulfonyl fluoride, and 20 μg/mL each leupeptin, pepstatin, and antipain) by using a Sorvall Omni Mixer (Sorvall Products, Newtown, CT). The lysate was centrifuged for 10 min at 10,000g at 4°C, and the resulting supernatant constituted the crude total protein extract. If microsomes were to be isolated, then the supernatant was centrifuged for 1 hr at 100,000g at 4°C. The pellet, which corresponded to the microsomes, was resuspended in reducing sample buffer (SB) (Laemmli, 1970), and SB was added to the remaining supernatant to prepare the soluble protein fraction. For SDS-PAGE (Laemmli, 1970), 30 μg of protein from the crude total protein extract, the corresponding soluble protein fraction, or the microsomal fraction was loaded per lane.
Two-Phase Partitioning
Aqueous two-phase partitioning using microsomes prepared as described above was performed as described previously (Larsson et al., 1987), using a polyethylene glycol (PEG)/dextran concentration of 6.2% (w/v). After partitioning, the upper and lower phases were diluted 1:10 with TEPI and centrifuged for 1 hr at 100,000g at 4°C. The pellet was resuspended in SB (Laemmli, 1970), and equivalent protein loadings (30 μg) were analyzed by SDS-PAGE (Laemmli, 1970).
Subcellular Fractionation of Tobacco BY-2 Cells
Separation of membranes on a step sucrose gradient was performed as described previously (Ahmed et al., 1997). The step gradient was modified by using nine increments of 5% sucrose, from 55 to 15%, of 1.35 mL each. Four millimeters depth of total microsomes from 3-day-old BY-2 cells prepared in the presence of 1 mM EDTA or 3 mM MgCl2 was layered on top of sucrose gradients containing 1 mM EDTA or 3 mM MgCl2. After centrifugation, 15 fractions of 0.85 mL each were collected, starting from the top of the gradient. Aliquots of 20 μL were separated by SDS-PAGE, and the distribution of HA-tagged Cf-9 and other marker proteins was analyzed by immunoblotting. The assay of Triton X-100–stimulated UDPase activity, characteristic of the Golgi apparatus, was performed as described previously (Briskin et al., 1987). To prepare total microsomes, 3-day-old BY-2 cells expressing HA-tagged Cf-9 were first washed in the lysis buffer containing 1 mM EDTA or 3 mM MgCl2 and then homogenized with a Sorvall Omni Mixer. The lysate was centrifuged for 10 min at 1000g at 4°C, the resulting supernatant was centrifuged for 1 hr at 100,000g at 4°C, and the microsomal pellet was resuspended in the lysis buffer.
Protein Immunoprecipitation and Endoglycosidase H Treatment
Three-day-old BY-2 cells were washed once with TEPI, and 0.6 mL of wet cells was homogenized in 0.5 mL of ice-cold TEPI. SDS was added to a final concentration of 1% (w/v) before heating the sample for 10 min at 95°C. The protein extract was then cleared by centrifugation at 10,000g for 15 min at 4°C, and the supernatant was diluted to 5 mL with TNET buffer (50 mM Tris-HCl, pH 8.5, 150 mM NaCl, 5 mM EDTA, and 1% Triton X-100) and incubated overnight with 2.5 μg of HA-specific antibodies (Boehringer Mannheim). The anti-HA–protein conjugate was immunoprecipitated by incubation with 10 mg of protein A–Sepharose resin (Pharmacia Biotech AB, Uppsala, Sweden) for 2 hr at 4°C. The resin was washed five times with TNET and resuspended in 50 μL of 50 mM sodium citrate buffer, pH 5.5, containing 0.5 mM phenylmethylsulfonyl fluoride. The sample was split in half and incubated with or without 5 mU of endoglycosidase H (EndoH) (Boehringer Mannheim) overnight at 37°C with shaking. The reaction was stopped by adding SB and boiling for 2 min.
Pulse–Chase Experiment
Processing membrane-bound GFP in transformed tobacco BY-2 cells by pulse–chase labeling with 35S-labeled amino acids was performed as described previously (Matsuoka et al., 1995), using Tran35S-label (ICN Biomedicals). Immunoprecipitation of GFP fusions was performed as described above, with 4 μL of monoclonal GFP-specific antibodies (Clontech) per sample. The immunoprecipitated proteins were separated by SDS-PAGE and processed for fluorography by using Amplify fluorographic reagent (Amersham Life Sciences, Little Chalfont, UK).
Recovery of Soluble GFP from Tobacco Cell Culture Medium
The culture medium from a 3-day-old culture of BY-2 cells was collected after an initial centrifugation of the cell suspension at 800g for 2 min at 27°C to remove the cells and a subsequent centrifugation of the supernatant at 10,000g for 15 min at 4°C. The supernatant was then concentrated by precipitation with trichloroacetic acid, 10% (w/v) final concentration, and prepared for SDS-PAGE (Laemmli, 1970) by adding SB and boiling for 5 min.
Immunoblotting
As required, proteins were separated by SDS-PAGE on 6, 12, or 6 to 15% gradient gels and transferred onto polyvinylidene fluoride membrane by electroblotting (Towbin et al., 1979). The blots were blocked overnight with PBS buffer containing 3% (w/v) nonfat dry milk powder and 0.1% PEG 35,000. The blots were further washed in PBS containing 0.05% (w/v) Tween 20 (PBST) and incubated for 2 hr at room temperature with the primary antibody diluted in PBST. After washing twice with PBST, the blots were incubated for 1 hr at room temperature with anti–mouse or anti–rabbit IgG–horseradish peroxidase conjugate, as appropriate, and diluted 1:3000 in PBST. Blots were washed with PBST, and bands were visualized using an electrochemiluminescence kit (Pierce Chemical Co., Rockford, IL). The GFP-specific antibodies were monoclonal antibodies from Clontech and Boehringer Mannheim. Kaleidoscope prestained protein standards (Bio-Rad) were used as size markers.
Acknowledgments
We thank Drs. Ramon Serrano (Universidad de Valencia-CSIC, Spain) for the gift of the PM H+-ATPase antiserum, Rebecca Boston (North Carolina State University, Raleigh) for the BiP antiserum, Heven Sze (University of Maryland, College Park) for the V H+-ATPase antiserum, Maarteen Chrispeels (University of California, San Diego) for the RD28 antiserum, Masayaoshi Maeshima (Nagoya University, Japan) for the H+ pyrophosphatase antiserum, and Richard Cyr (Pennsylvania State University, University Park) for providing rabbit anti–soy tubulin polyclonal serum. We thank Hoechst Schering AgrEvo (Glen Iris, Australia) for the generous gift of Basta. pSLJ constructs, apart from pSLJ7291, pSLJ7292, and pSLJ755I5, were made by D.A.J. while working with Jonathan Jones at the Sainsbury Laboratory, John Innes Centre (Norwich, UK). The introduction of the ClaI site into Cf-9 was performed by Miguel Angel-Torres, and the removal of the BglII site was done by Saijun Tang, also working with Jonathan Jones. We are grateful to Jonathan Jones for the use of pSLJ constructs. We thank Helen Kilborn for her technical assistance and Andreas Betzner and Brian Gunning for critical reading of the manuscript. This work is supported by a grant from the Scientific Swiss National Foundation and a Human Frontier Science Program Long-Term Fellowship (Grant No. LT0463/98-M).
References
- Ahmed, S.U., Bar-Peled, M., and Raikhel, N.V. (1997). Cloning and subcellular location of an Arabidopsis receptor-like protein that shares common features with protein-sorting receptors of eukaryotic cells. Plant Physiol. 114, 325–336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andersson, H., Kappeler, F., and Hauri, H.-P. (1999). Protein targeting to endoplasmic reticulum by dilysine signals involves direct retention in addition to retrieval. J. Biol. Chem. 274, 15080–15084. [DOI] [PubMed] [Google Scholar]
- Andreeva, A.V., Kutuzov, M.A., Evans, D.E., and Hawes, C.R. (1998). Proteins involved in membrane transport between the ER and the Golgi apparatus: 21 putative plant homologues revealed by dbEST searching. Cell. Biol. Int. 22, 145–160. [DOI] [PubMed] [Google Scholar]
- Andres, D.A., Rhodes, J.D., Meisel, R.L., and Dixon, J.E. (1991). Characterization of the carboxyl-terminal sequences responsible for protein retention in the endoplasmic reticulum. J. Biol. Chem. 266, 14277–14282. [PubMed] [Google Scholar]
- Arondel, V., Lemieux, B., Hwang, I., Gibson, S., Goodman, H.M., and Somerville, C.R. (1992). Map-based cloning of a gene controlling omega-3 fatty acid desaturation in Arabidopsis. Science 258, 1353–1355. [DOI] [PubMed] [Google Scholar]
- Arthington, B.A., Bennett, L.G., Skatrud, P.L., Guynn, C.J., Barbuch, R.J., Ulbright, C.E., and Bard, M. (1991). Cloning, disruption and sequence of the gene encoding yeast C-5 sterol desaturase. Gene 102, 39–44. [DOI] [PubMed] [Google Scholar]
- Barrieu, F., and Chrispeels, M.J. (1999). Delivery of a secreted soluble protein to the vacuole via a membrane anchor. Plant Physiol. 120, 961–968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bechtold, N., Ellis, J., and Pelletier, G. (1993). In planta Agrobacterium mediated gene transfer by infiltration of adult Arabidopsis thaliana plants. C. R. Acad. Sci. Paris Life Sci. 316, 1194–1199. [Google Scholar]
- Becker, D.M., and Guarente, L. (1991). High-efficiency transformation of yeast by electroporation. Methods Enzymol. 194, 182–187. [DOI] [PubMed] [Google Scholar]
- Benghezal, M., Benachour, A., Rusconi, S., Aebi, M., and Conzelmann, A. (1996). Yeast Gpi8p is essential for GPI anchor attachment onto proteins. EMBO J. 15, 6575–6583. [PMC free article] [PubMed] [Google Scholar]
- Bibikova, T.N., Jacob, T., Dahse, I., and Gilroy, S. (1998). Localized changes in apoplastic and cytoplasmic pH are associated with root hair development in Arabidopsis thaliana. Development 125, 2925–2934. [DOI] [PubMed] [Google Scholar]
- Briskin, D.P., Leonard, R.T., and Hodges, T.K. (1987). Isolation of the plasma membrane: Membrane markers and general principles. Methods Enzymol. 148, 542–548. [Google Scholar]
- Cosson, P., and Letourneur, F. (1994). Coatomer interaction with di-lysine endoplasmic reticulum retention motifs. Science 263, 1629–1631. [DOI] [PubMed] [Google Scholar]
- Cosson, P., Démollière, C., Hennecke, S., Duden, R., and Letourneur, F. (1996). δ- and ζ-COP, two coatomer subunits homologous to clathrin-associated proteins, are involved in ER retrieval. EMBO J. 15, 1792–1798. [PMC free article] [PubMed] [Google Scholar]
- Daniels, M.J., Mirkov, T.E., and Chrispeels, M.J. (1994). The plasma membrane of Arabidopsis thaliana contains a mercury-insensitive aquaporin that is a homolog of the tonoplast water channel protein TIP. Plant Physiol. 106, 1325–1333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Denecke, J., De Rycke, R., and Botterman, J. (1992). Plant and mammalian sorting signals for protein retention in the endoplasmic reticulum contain a conserved epitope. EMBO J. 11, 2345–2355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Denecke, J., Ek, B., Caspers, M., Sinjorgo, K.M.C., and Palva, E.T. (1993). Analysis of sorting signals responsible for the accumulation of soluble reticuloplasmins in the plant endoplasmic reticulum. J. Exp. Bot. 44, 213–221. [Google Scholar]
- Dixon, M.S., Jones, D.A., Keddie, J.S., Thomas, C.M., Harrison, K., and Jones, J.D.G. (1996). The tomato Cf-2 disease resistance locus comprises two functional genes encoding leucine-rich repeat proteins. Cell 84, 451–459. [DOI] [PubMed] [Google Scholar]
- Duden, R., Griffiths, G., Frank, R., Argos, P., and Kreis, T.E. (1991). β-COP, a 110 kd protein associated with non-clathrin-coated vesicles and the Golgi complex, shows homology to β-adaptin. Cell 64, 649–665. [DOI] [PubMed] [Google Scholar]
- Faye, L., Sturm, A., Bollini, R., Vitale, A., and Chrispeels, M.J. (1986). The position of the oligosaccharide side-chains of phytohemagglutinin and their accessibility to glycosidases determines their subsequent processing in the Golgi. Eur. J. Biochem. 158, 655–661. [DOI] [PubMed] [Google Scholar]
- Flor, H.H. (1971). Current status of the gene-for-gene concept. Annu. Rev. Phytopathol. 9, 275–296. [Google Scholar]
- Fontes, E.B., Shank, B.B., Wrobel, R.L., Moose, S.P., OBrian, G.R., Wurtzel, E.T., and Boston, R.S. (1991). Characterization of an immunoglobulin binding protein homolog in the maize floury-2 endosperm mutant. Plant Cell 3, 483–496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fry, S.C. (1998). Oxidative scission of plant cell wall polysaccharides by ascorbate-induced hydroxyl radicals. Biochem. J. 332, 507–515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gachotte, D., Meens, R., and Benveniste, P. (1995). An Arabidopsis mutant deficient in sterol biosynthesis: Heterologous complementation by ERG3 encoding a Δ7-sterol-C-5-desaturase from yeast. Plant J. 8, 407–416. [DOI] [PubMed] [Google Scholar]
- Gachotte, D., Husselstein, T., Bard, M., Lacroute, F., and Benveniste, P. (1996). Isolation and characterization of an Arabidopsis thaliana cDNA encoding a Δ7-sterol-C-5-desaturase by functional complementation of a defective yeast mutant. Plant J. 9, 391–398. [DOI] [PubMed] [Google Scholar]
- Gaynor, E.C., te Heesen, S., Graham, T.R., Aebi, M., and Emr, S.D. (1994). Signal-mediated retrieval of a membrane protein from the Golgi to the ER in yeast. J. Cell. Biol. 127, 653–665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gietz, R.D., and Sugino, A. (1988). New yeast–Escherichia coli shuttle vectors constructed with in vitro mutagenized yeast genes lacking six–base pair restriction sites. Gene 74, 527–534. [DOI] [PubMed] [Google Scholar]
- Gomord, V., Denmat, L.-A., Fitchette-Lainé, A.-C., Satiat-Jeune-maitre, B., Hawes, C., and Faye, L. (1997). The C-terminal HDEL sequence is sufficient for retention of secretory proteins in the endoplasmic reticulum (ER) but promotes vacuolar targeting of proteins that escape the ER. Plant J. 11, 313–325. [DOI] [PubMed] [Google Scholar]
- Graham, T.R., and Emr, S.D. (1991). Compartmental organization of Golgi-specific protein modification and vacuolar protein sorting events defined in a yeast sec18 (NSF) mutant. J. Cell Biol. 114, 207–218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grebenok, R.J., Ohnmeiss, T.E., Yamamoto, A., Huntley, E.D., Galbraith, D.W., and Della Penna, D. (1998). Isolation and characterization of an Arabidopsis thaliana C-8,7 sterol isomerase: Functional and structural similarities to mammalian C-8,7 sterol isomerase/emopamil-binding protein. Plant Mol. Biol. 38, 807–815. [DOI] [PubMed] [Google Scholar]
- Hammond-Kosack, K.E., Tang, S., Harrison, K., and Jones, J.D.G. (1998). The tomato Cf-9 disease resistance gene functions in tobacco and potato to confer responsiveness to the fungal avirulence gene product Avr9. Plant Cell 10, 1251–1266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harter, C., and Wieland, F.T. (1998). A single binding site for dilysine retrieval motifs and p23 within the γ subunit of coatomer. Proc. Natl. Acad. Sci. USA 95, 11649–11654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harter, C., Pavel, J., Coccia, F., Draken, E., Wegehingel, S., Tschochner, H., and Wieland, F. (1996). Nonclathrin coat protein γ, a subunit of coatomer, binds to the cytoplasmic dilysine motif of membrane proteins of the early secretory pathway. Proc. Natl. Acad. Sci. USA 93, 1902–1906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haseloff, J., Siemering, K.R., Prasher, D.C., and Hodge, S. (1997). Removal of a cryptic intron and subcellular localization of green fluorescent protein are required to mark transgenic Arabidopsis plants brightly. Proc. Natl. Acad. Sci. USA 94, 2122–2127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haupts, U., Maiti, S., Schwille, P., and Webb, W.W. (1998). Dynamics of fluorescence fluctuations in green fluorescent protein observed by fluorescence correlation spectroscopy. Proc. Natl. Acad. Sci. USA 95, 13573–13578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heim, R., Prasher, D.C., and Tsien, R.Y. (1994). Wavelength mutations and posttranslational autoxidation of green fluorescent protein. Proc. Natl. Acad. Sci. USA 91, 12501–12504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heim, R., Cubitt, A.B., and Tsien, R.Y. (1995). Improved green fluorescence. Nature 373, 663–664. [DOI] [PubMed] [Google Scholar]
- Hottiger, T., Fürst, P., Pohlig, G., and Heim, J. (1994). Physiological characterization of the yeast metallothionein (CUP1) promoter, and consequences of overexpressing its transcriptional activator, ACE1. Yeast 10, 283–296. [DOI] [PubMed] [Google Scholar]
- Itin, C., Schindler, R., and Hauri, H.-P. (1995). Targeting of protein ERGIC-53 to the ER/ERGIC/cis-Golgi recycling pathway. J. Cell Biol. 131, 57–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jackson, M.R., Nilsson, T., and Peterson, P.A. (1990). Identification of a consensus motif for retention of transmembrane proteins in the endoplasmic reticulum. EMBO J. 9, 3153–3162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jackson, M.R., Nilsson, T., and Peterson, P.A. (1993). Retrieval of transmembrane proteins to the endoplasmic reticulum. J. Cell Biol. 121, 317–333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones, D.A., Thomas, C.M., Hammond-Kosack, K.E., Balint-Kurti, P.J., and Jones, J.D.G. (1994). Isolation of the tomato Cf-9 gene for resistance to Cladosporium fulvum by transposon tagging. Science 266, 789–793. [DOI] [PubMed] [Google Scholar]
- Jones, J.D.G., Shlumukov, L., Carland, F., English, J., Scofield, S.R., Bishop, G.J., and Harrison, K. (1992). Effective vectors for transformation, expression of heterologous genes, and assaying transposon excision in transgenic plants. Transgenic Res. 1, 285–297. [DOI] [PubMed] [Google Scholar]
- Kappeler, F., Klopfenstein, D.R.C., Foguet, M., Paccaud, J.-P., and Hauri, H.-P. (1997). The recycling of ERGIC-53 in the early secretory pathway. ERGIC-53 carries a cytosolic endoplasmic reticulum–exit determinant interacting with COPII. J. Biol. Chem. 272, 31801–31808. [DOI] [PubMed] [Google Scholar]
- Kimata, Y., Iwaki, M., Lim, C.R., and Kohno, K. (1997). A novel mutation which enhances the fluorescence of green fluorescent protein at high temperatures. Biochem. Biophys. Res. Commun. 232, 69–73. [DOI] [PubMed] [Google Scholar]
- Kjeldsen, T., Pettersson, A.F., and Hach, M. (1999). The role of leaders in intracellular transport and secretion of the insulin precursor in the yeast Saccharomyces cerevisiae. J. Biotechnol. 75, 195–208. [DOI] [PubMed] [Google Scholar]
- Kneen, M., Farinas, J., Li, Y., and Verkman, A.S. (1998). Green fluorescent protein as a noninvasive intracellular pH indicator. Biophys. J. 74, 1591–1599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kooman-Gersmann, M., Honée, G., Bonnema, G., and De Wit, P.J.G.M. (1996). A high-affinity binding site for the AVR9 peptide elicitor of Cladosporium fulvum is present on the plasma membranes of tomato and other solanaceous plants. Plant Cell 8, 929–938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kopka, J., Ludewig, M., and Müller-Röber, B. (1997). Complementary DNAs encoding eukaryotic-type cytidine-5′-diphosphate-diacylglycerol synthases of two plant species. Plant Physiol. 113, 997–1002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kunkel, T.A., Roberts, J.D., and Zakour, R.A. (1987). Rapid and efficient site-specific mutagenesis without phenotypic selection. Methods Enzymol. 154, 367–382. [DOI] [PubMed] [Google Scholar]
- Laemmli, U.K. (1970). Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature 227, 680–685. [DOI] [PubMed] [Google Scholar]
- Larsson, C., Widell, S., and Kjellbom, P. (1987). Preparation of high-purity plasma membranes. Methods Enzymol. 148, 558–568. [Google Scholar]
- Lazo, G.R., Stein, P.A., and Ludwig, R.A. (1991). A DNA transformation-competent Arabidopsis genomic library in Agrobacterium. Bio/Technology 9, 963–967. [DOI] [PubMed] [Google Scholar]
- Lee, H.-I., Gal, S., Newman, T.C., and Raikhel, N.V. (1993). The Arabidopsis endoplasmic reticulum retention receptor functions in yeast. Proc. Natl. Acad. Sci. USA 90, 11433–11437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Letourneur, F., Gaynor, E.C., Hennecke, S., Démollière, C., Duden, R., Emr, S.D., Riezman, H., and Cosson, P. (1994). Coatomer is essential for retrieval of dilysine-tagged proteins to the endoplasmic reticulum. Cell 79, 1199–1207. [DOI] [PubMed] [Google Scholar]
- Letourneur, F., Hennecke, S., Démollière, C., and Cosson, P. (1995). Steric masking of a dilysine endoplasmic reticulum retention motif during assembly of the human high affinity receptor for immunoglobulin E. J. Cell Biol. 129, 971–978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lewis, M.J., and Pelham, H.R.B. (1990). A human homologue of the yeast HDEL receptor. Nature 348, 162–163. [DOI] [PubMed] [Google Scholar]
- Lewis, M.J., and Pelham, H.R.B. (1992). Sequence of a second human KDEL receptor. J. Mol. Biol. 226, 913–916. [DOI] [PubMed] [Google Scholar]
- Lewis, M.J., Sweet, D.J., and Pelham, H.R.B. (1990). The ERD2 gene determines the specificity of the luminal ER protein retention system. Cell 61, 1359–1363. [DOI] [PubMed] [Google Scholar]
- Liang, F., and Sze, H. (1998). A high-affinity Ca2+ pump, ECA1, from the endoplasmic reticulum is inhibited by cyclopiazonic acid but not by thapsigargin. Plant Physiol. 118, 817–825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liang, F., Cunningham, K.W., Harper, J.F., and Sze, H. (1997). ECA1 complements yeast mutants defective in Ca2+ pumps and encodes an endoplasmic reticulum-type Ca2+-ATPase in Arabidopsis thaliana. Proc. Natl. Acad. Sci. USA 94, 8579–8584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maeshima, M., and Yoshida, S. (1989). Purification and properties of vacuolar membrane proton-translocating inorganic pyrophosphatase from mung bean. J. Biol. Chem. 264, 20068–20073. [PubMed] [Google Scholar]
- Matsuoka, K., Bassham, D.C., Raikhel, N.V., and Nakamura, K. (1995). Different sensitivity to wortmannin of two vacuolar sorting signals indicates the presence of distinct sorting machineries in tobacco cells. J. Cell Biol. 130, 1307–1318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mazzarella, R.A., Srinivasan, M., Haugejorden, S.M., and Green, M. (1990). ERp72, an abundant luminal endoplasmic reticulum protein, contains three copies of the active site sequences of protein disulfide isomerase. J. Biol. Chem. 265, 1094–1101. [PubMed] [Google Scholar]
- McQueen-Mason, S.J., Fry, S.C., Durachko, D.M., and Cosgrove, D.J. (1993). The relationship between xyloglucan endotransglycosylase and in-vitro cell wall extension in cucumber hypocotyls. Planta 190, 327–331. [DOI] [PubMed] [Google Scholar]
- Movafeghi, A., Happel, N., Pimpl, P., Tai, G.H., and Robinson, D.G. (1999). Arabidopsis sec21p and sec23p homologs. Probable coat proteins of plant COP-coated vesicles. Plant Physiol. 119, 1437–1446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Munro, S., and Pelham, H.R.B. (1987). A C-terminal signal prevents secretion of luminal ER proteins. Cell 48, 899–907. [DOI] [PubMed] [Google Scholar]
- Murashige, T., and Skoog, F. (1962). A revised medium for rapid growth and bioassays with tobacco tissue cultures. Physiol. Plant. 15, 473–497. [Google Scholar]
- Napier, R.M., Fowke, L.C., Hawes, C., Lewis, M., and Pelham, H.R.B. (1992). Immunological evidence that plants use both HDEL and KDEL for targeting proteins to the endoplasmic reticulum. J. Cell. Sci. 102, 261–271. [DOI] [PubMed] [Google Scholar]
- Nilsson, T., Jackson, M., and Peterson, P.A. (1989). Short cytoplasmic sequences serve as retention signals for transmembrane proteins in the endoplasmic reticulum. Cell 58, 707–718. [DOI] [PubMed] [Google Scholar]
- Nishiuchi, T., Nishimura, M., Arondel, V., and Iba, K. (1994). Genomic nucleotide sequence of a gene encoding a microsomal ω-3 fatty acid desaturase from Arabidopsis thaliana. Plant Physiol. 105, 767–768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ormö, M., Cubitt, A.B., Kallio, K., Gross, L.A., Tsien, R.Y., and Remington, S.J. (1996). Crystal structure of the Aequorea victoria green fluorescent protein. Science 273, 1392–1395. [DOI] [PubMed] [Google Scholar]
- Palmgren, M.G., Sommarin, M., Serrano, R., and Larsson, C. (1991). Identification of an autoinhibitory domain in the C-terminal region of the plant plasma membrane H+-ATPase. J. Biol. Chem. 266, 20470–20475. [PubMed] [Google Scholar]
- Pelham, H.R.B., Hardwick, K.G., and Lewis, M.J. (1988). Sorting of soluble ER proteins in yeast. EMBO J. 7, 1757–1762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rapak, A., Falnes, P.O., and Olsnes, S. (1997). Retrograde transport of mutant ricin to the endoplasmic reticulum with subsequent translocation to cytosol. Proc. Natl. Acad. Sci. USA 94, 3783–3788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Regad, F., Bardet, C., Tremousaygue, D., Moisan, A., Lescure, B., and Axelos, M. (1993). cDNA cloning and expression of an Arabidopsis GTP-binding protein of the ARF family. FEBS Lett. 316, 133–136. [DOI] [PubMed] [Google Scholar]
- Rothman, J.E. (1994). Mechanisms of intracellular protein transport. Nature 372, 55–63. [DOI] [PubMed] [Google Scholar]
- Sandvig, K., Garred, O., Prydz, K., Kozlov, J.V., Hansen, S.H., and Van Deurs, B. (1992). Retrograde transport of endocytosed Shiga toxin to the endoplasmic reticulum. Nature 358, 510–512. [DOI] [PubMed] [Google Scholar]
- Sandvig, K., Garred, O., and Van Deurs, B. (1996). Thapsigargin-induced transport of cholera toxin to the endoplasmic reticulum. Proc. Natl. Acad. Sci. USA 93, 12339–12343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schneider, B.L., Seufert, W., Steiner, B., Yang, Q.H., and Futcher, A.B. (1995). Use of polymerase chain reaction epitope tagging for protein tagging in Saccharomyces cerevisiae. Yeast 11, 1265–1274. [DOI] [PubMed] [Google Scholar]
- Semenza, J.C., Hardwick, K.G., Dean, N., and Pelham, H.R.B. (1990). ERD2, a yeast gene required for the receptor-mediated retrieval of luminal ER proteins from the secretory pathway. Cell 61, 1349–1357. [DOI] [PubMed] [Google Scholar]
- Siemering, K.R., Golbik, R., Sever, R., and Haseloff, J. (1996). Mutations that suppress the thermosensitivity of green fluorescent protein. Curr. Biol. 6, 1653–1663. [DOI] [PubMed] [Google Scholar]
- Silve, S., Dupuy, P.H., Labit-Lebouteiller, C., Kaghad, M., Chalon, P., Rahier, A., Taton, M., Lupker, J., Shire, D., and Loison, G. (1996). Emopamil-binding protein, a mammalian protein that binds a series of structurally diverse neuroprotective agents, exhibits Δ8-Δ7 sterol isomerase activity in yeast. J. Biol. Chem. 271, 22434–22440. [DOI] [PubMed] [Google Scholar]
- Taguchi, N., Takano, Y., Julmanop, C., Wang, Y., Stock, S., Takemoto, J., and Miyakawa, T. (1994). Identification and analysis of the Saccharomyces cerevisiae SYR1 gene reveals that ergosterol is involved in the action of syringomycin. Microbiology 140, 353–359. [DOI] [PubMed] [Google Scholar]
- Tang, B.L., Wong, S.H., Qi, X.L., Low, S.H., and Hong, W. (1993). Molecular cloning, characterization, subcellular localization and dynamics of p23, the mammalian KDEL receptor. J. Cell Biol. 120, 325–328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas, C.M., Vos, P., Zabeau, M., Jones, D.A., Norcott, K.A., Chadwick, B.P., and Jones, J.D.G. (1995). Identification of amplified restriction fragment polymorphism (AFLP) markers tightly linked to the tomato Cf-9 gene for resistance to Cladosporium fulvum. Plant J. 8, 785–794. [DOI] [PubMed] [Google Scholar]
- Thomas, C.M., Jones, D.A., Parniske, M., Harrison, K., Balint-Kurti, P.J., Hatzixanthis, K., and Jones, J.D.G. (1997). Characterization of the tomato Cf-4 gene for resistance to Cladosporium fulvum identifies sequences that determine recognitional specificity in Cf-4 and Cf-9. Plant Cell 9, 2209–2224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Towbin, H., Staehelin, T., and Gordon, J. (1979). Electrophoretic transfer of proteins from polyacrylamide gels to nitrocellulose sheets: Procedure and some applications. Proc. Natl. Acad. Sci. USA 76, 4350–4354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Den Ackerveken, G.F.J.M., Van Kan, J.A.L., and De Wit, P.J.G.M. (1992). Molecular analysis of the avirulence gene Avr9 of the fungal tomato pathogen Cladosporium fulvum fully supports the gene-for-gene hypothesis. Plant J. 2, 359–366. [DOI] [PubMed] [Google Scholar]
- Vitale, A., and Denecke, J. (1999). The endoplasmic reticulum—Gateway of the secretory pathway. Plant Cell 11, 615–628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- von Schaewen, A., Sturm, A., O'Neill, J., and Chrispeels, M.J. (1993). Isolation of a mutant Arabidopsis plant that lacks N-acetyl glucosaminyl transferase I and is unable to synthesize Golgi-modified complex N-linked glycans. Plant Physiol. 102, 1109–1118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ward, J., Reinders, A., Hsu, H., and Sze, H. (1992). Dissociation and reassembly of the vacuolar H+-ATPase complex from oat roots. Plant Physiol. 99, 161–169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wasteneys, G.O., Willingale-Theune, J., and Menzel, D. (1997). Freeze shattering: A simple and effective method for permeabilizing higher plant cell walls. J. Microsc. 188, 51–61. [DOI] [PubMed] [Google Scholar]
- Waters, M.G., Serafini, T., and Rothman, J.E. (1991). “Coatomer”: A cytosolic protein complex containing subunits of non-clathrin-coated Golgi transport vesicles. Nature 349, 248–251. [DOI] [PubMed] [Google Scholar]
- White, J., Chang, S.Y., and Bibb, M.J. (1990). A cassette containing the bar gene of Streptomyces hygroscopicus: A selectable marker for plant transformation. Nucleic Acids Res. 18, 1062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wickerham, L.J. (1946). A critical evaluation of the nitrogen assimilation tests commonly used in the classification of yeasts. J. Bacteriol. 52, 293–301. [DOI] [PubMed] [Google Scholar]
- Wimmers, L.E., Ewing, N.N., and Bennett, A.B. (1992). Higher plant Ca2+-ATPase: Primary structure and regulation of mRNA abundance by salt. Proc. Natl. Acad. Sci. USA 89, 9205–9209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yadav, N.S., et al. (1993). Cloning of higher plant ω-3 fatty acid desaturases. Plant Physiol. 103, 467–476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang, F., Moss, L.G., and Phillips, G.N., Jr. (1996). The molecular structure of green fluorescent protein. Nat. Biotechnol. 14, 1246–1251. [DOI] [PubMed] [Google Scholar]