

Abstract
We describe a green fluorescent protein (GFP)–based assay for investigating membrane traffic on the secretory pathway in plants. Expression of AtRab1b(N121I), predicted to be a dominant inhibitory mutant of the Arabidopsis Rab GTPase AtRab1b, resulted in accumulation of a secreted GFP marker in an intracellular reticulate compartment reminiscent of the endoplasmic reticulum. This accumulation was alleviated by coexpressing wild-type AtRab1b but not AtRab8c. When a Golgi-targeted and N-glycosylated variant of GFP was coexpressed with AtRab1b(N121I), the variant also accumulated in a reticulate network and an endoglycosidase H–sensitive population appeared. Unexpectedly, expression of AtRab1b(N121I), but not of the wild-type AtRab1b, resulted in a reduction or cessation of vectorial Golgi movement, an effect that was reversed by coexpression of the wild type. We conclude that AtRab1b function is required for transport from the endoplasmic reticulum to the Golgi apparatus and suggest that this process may be coupled to the control of Golgi movement.
INTRODUCTION
The structure and biosynthetic activities of the endoplasmic reticulum (ER) and Golgi apparatus in higher plants are well described, but the mechanisms that promote and regulate membrane traffic between these compartments are not well understood (reviewed in Robinson and Kristen, 1982; Staehelin and Moore, 1995; Dupree and Sherrier, 1998; Vitale and Denecke, 1999). The Golgi apparatus in higher plants exists as a large number of independent Golgi stacks distributed throughout the cytoplasm. Each Golgi stack typically comprises five to eight flattened cisternae, whose protein and polysaccharide composition may vary in a cis to trans direction across the stack (Zhang and Staehelin, 1992; Wee et al., 1998; Nebenführ et al., 1999). Analysis of tobacco cells in which the Golgi stacks were labeled with green fluorescent protein (GFP) revealed that each stack can travel as much as 2 to 4 μm sec−1 on an actin network that is coextensive with the dynamic ER network (Boevink et al., 1998; Nebenführ et al., 1999). Consequently, despite the dynamic organization of both the ER and Golgi, both organelles remain in close association. The movement of individual Golgi stacks appears to alternate between two modes: a directed vectorial mode, probably dependent on myosin motors, and a nonvectorial mode involving apparently random oscillations reminiscent of Brownian motion at particular points in the cytoplasm (Nebenführ et al., 1999). Video microscopy of GFP-labeled Golgi stacks in cultured tobacco cells suggests that the control of Golgi movement is devolved to the individual stacks because adjacent stacks can enter independently into phases of vectorial movement, and a stack undergoing vectorial movement can travel past another that is exhibiting the nondirected oscillatory type of motion (Nebenführ et al., 1999).
In contrast, the mammalian Golgi apparatus in general forms a single interconnected cluster of membranes at the microtubule organizing center. ER-derived COPII transport vesicles arising at peripheral sites fuse to form vesiculo-tubular clusters, which are transported along microtubules to the central Golgi cluster (Presley et al., 1997; Scales et al., 1997; Burkhardt, 1998; Infante et al., 1999). As the clusters are transported, they mature by the selective removal of noncargo molecules in COPI vesicles, which are targeted back to the ER (Letourneur et al., 1994; Martínez-Menárguez et al., 1999).
The GFP studies have led to formulation of two models to account for the membrane traffic between the ER and Golgi apparatus in higher plants. Both models invoke the existence of a vesicular intermediate, although candidate vesicles have yet to be unambiguously identified (Movafeghi et al., 1999) and direct tubular connections have been observed in some plant cell types (Satiat-Jeunemaitre and Hawes, 1996).
In the “vacuum cleaner” model (Boevink et al., 1998), derived from observations of leaf epidermal cells, Golgi stacks moving along the actin network are thought to encounter and fuse with relatively static transport vesicles released from the associated ER. In this model, transport vesicles can be produced at any point on the ER/actin network, and Golgi movement is considered essential for efficient targeting and consumption of transport vesicles. These vesicles may remain associated with the ER/actin network until they encounter and fuse with an itinerant Golgi stack, or the release of nascent vesicles from the ER may be coupled to an interaction with a passing Golgi stack.
The second model (Nebenführ et al., 1999) attaches particular significance to the alternation between vectorial and oscillatory modes of Golgi movement in cultured tobacco BY-2 cells and to the observation that particular regions of the cytoplasm can be identified at which successive Golgi stacks appear to cease vectorial movement and enter oscillatory motion (Nebenführ et al., 1999). Although the ER was not visible, this model proposes that vesicle exchange occurs during the periods of nonvectorial oscillatory movement, when Golgi stacks are probably positioned adjacent to particular sites on the ER or plasma membrane that are hypothesized to be specialized for vesicle exchange. Vectorial movement is thus proposed to allow the Golgi to move from sites of vesicle acquisition to sites of secretion to promote the distribution of Golgi-derived secretory vesicles around the cell. This model predicts that vesicle production is restricted to particular ER sites and that membrane exchange depends on the controlled arrest of vectorial Golgi movement at these sites.
Although these models are not mutually exclusive, the first model attaches no particular significance to the periods of nonvectorial oscillatory movement that may arise trivially when a Golgi stack encounters a discontinuity in the actin network and is temporarily arrested until the network is reengaged and vectorial movement resumes (Nebenführ et al., 1999).
Investigation of the validity of each of these models necessitates identifying the sites and mechanisms of vesicle production and exchange in membrane traffic. However, work to date has failed to identify a single molecule with a demonstrable function in directing vesicle traffic between the ER and Golgi. Work on yeasts and mammalian cells has identified many molecular components of a conserved mechanism that promotes efficiency and accuracy in vesicle transport between compartments of the endomembrane system. Some of the key components of this mechanism are encoded by multigene families in which individual members act to promote specific trafficking events in the cell. One important family is the Rab GTPase family, which is subdivided into several functionally distinct subclasses. Although Rab GTPases do not themselves encode specificity in vesicle targeting (reviewed in Pfeffer, 1999), they have been implicated in regulating the activity of important classes of molecules, including the tethering factors and the soluble N-ethylmaleimide-sensitive fusion attachment protein receptors (or SNAREs), which between them promote accurate vesicle targeting and vesicle fusion (although their detailed modes of action are still controversial [Mellman and Warren, 2000]). Some members of the Rab GTPase family have also been implicated in interactions between the vesicle transport machinery and the cytoskeleton (Schmitt et al., 1986; Bush et al., 1993; Valentijn et al., 2000).
All eukaryotes appear to have a Rab GTPase family, but mammals, brewer's yeast, and plants each possess a different complement of Rab GTPase subclasses, which may reflect the trafficking requirements of each organism. The 11 Rab GTPases in Saccharomyces are grouped into just seven functional subclasses, but >30 different subclasses are recognized in mammals (Lazar et al., 1997; Novick and Zerial, 1997). Sequencing expressed sequence tags and genomes suggests that species such as Arabidopsis encode >50 Rab GTPases. However, plant Rab GTPases are usually assigned to just eight subclasses (Bischoff et al., 1999), with each plant Rab subclass being more diverse and heterogeneous than any mammalian subclass. In plants, these subclasses are based almost entirely on sequence comparisons, and whether they represent functional groups is not clear. In the absence of effective in vivo trafficking assays, information on plant Rab GTPase function has been derived from complementation and expression studies in yeast (Palme et al., 1992; Bednarek et al., 1994; Ueda et al., 2000), immunolocalization data (Ueda et al., 1996), and expression studies (Terryn et al., 1993; Nagano et al., 1995; Moore et al., 1997). Transgenic approaches suggest that disrupting Rab function produces various developmental and cellular anomalies, but they have not identified the in vivo trafficking roles of any plant Rab GTPase (Cheon et al., 1993; Bischoff et al., 1999).
A GFP fusion translocated into the lumen of the ER reportedly fails to accumulate in a fluorescent form unless its transport to a post-Golgi compartment is prevented (Boevink et al., 1996, 1999). This has been achieved both by providing the GFP with targeting information that causes it to accumulate in either the ER or Golgi apparatus and by treating the cells with brefeldin A (BFA) (Boevink et al., 1996, 1999; Nebenführ et al., 1999). In these cases, a fluorescent form of the protein accumulates in either the ER or the Golgi apparatus, allowing these organelles to be readily visualized by fluorescence microscopy. We have used this observation to develop a visual and biochemical assay for in vivo transport in plant cells and have investigated the function of a member of the Arabidopsis Rab1 subclass in ER–Golgi traffic.
RESULTS
Rapid Membrane Trafficking Assay Is Based on Differential Fluorescence of Apoplastic and Intracellular GFP
To develop a versatile in vivo membrane trafficking assay based on the differential fluorescence of secreted and retained GFP, we constructed a series of vectors for Agrobacterium-mediated transient expression (Rossi et al., 1993; Kapila et al., 1997) of secreted and retained GFP fusions in tobacco leaves (Figure 1). The coding sequence of the ER-localized GFP variant mGFP5-ER, a thermotolerant derivative of mGFP4-ER (Haseloff et al., 1997), was modified such that its C-terminal ER retrieval signal was removed and replaced by a sequence encoding the c-Myc epitope tag. Because the N-terminal ER transit peptide of mGFP5-ER was left unaltered, this new GFP variant, secGFP, was expected to be translocated into the lumen of the ER and then transported by a default trafficking pathway (Wieland et al., 1987; Denecke et al., 1990; Martínez-Menárguez et al., 1999; Vitale and Denecke, 1999) to the apoplast. secGFP and its progenitor mGFP5-ER were cloned in a binary transformation vector pVKH18 (I. Moore, unpublished results) behind an enhanced cauliflower mosaic virus (CaMV) 35S promoter and tobacco mosaic virus–Ω translation enhancer (Mitsuhara et al., 1996) to generate pVKH-secGFP and pVKH-GFP-HDEL, respectively.
Figure 1.

Constructs for Transient Expression.
secGFP is derived from the ER-targeted GFP-HDEL by replacing the C-terminal tetrapeptide with a c-Myc epitope tag. N-secGFP, N-GFP- HDEL, and N-ST-GFP are derived from secGFP, GFP-HDEL, and ST-GFP (Boevink et al., 1998), respectively, by addition of an N-terminal peptide containing an N-glycosylation site. The steady state location and the fluorescence characteristics of each GFP in this location are indicated at right. All constructs were inserted under control of an enhanced CaMV promoter in an Agrobacterium binary vector (Hawes et al., 2000). NGT, single-letter amino acid code; ST, sialyltransferase.
Agrobacterium-mediated transient expression of T-DNA–encoded genes in tobacco leaves varies linearly over several orders of magnitude as the density of the bacterial inoculum is varied (Rossi et al., 1993). Tobacco leaves were inoculated with serial dilutions of Agrobacterium strains containing either pVKH-secGFP or pVKH-GFP-HDEL; 48 hr later, the abaxial epidermis was imaged by confocal laser scanning microscopy. As shown in Figure 2A, both strains directed readily detectable transient expression of GFP when infiltrated at OD600 ⩾ 0.1, although the secGFP fluorescence was usually weaker than that of GFP-HDEL. However, with inocula of OD600 0.01 to 0.033, little or no secGFP fluorescence was detectable at low magnification, but fluorescence of GFP-HDEL was still clearly detectable in most cells. Confocal imaging at greater resolution indicated that GFP-HDEL expression resulted in a dynamic reticulate pattern of GFP fluorescence typical of the ER of tobacco epidermal cells (Figure 2B) (Boevink et al., 1996, 1999). No GFP-HDEL fluorescence was visible in the apoplast, even at high inoculum densities. secGFP fluorescence, on the other hand, was observed principally in the apoplast (Figure 2B), and at the highest inoculum densities, faint fluorescence was also detectable in an intracellular reticulum typical of the ER (data not shown). At OD600 0.01, secGFP fluorescence was just barely imaged, even in the apoplast, but with high laser power, faint apoplastic GFP fluorescence was visible in many cells with no detectable intracellular accumulation (Figure 2B). The apoplastic location of this signal was confirmed by labeling the adjacent plasma membranes with FM4-64 dye (Figure 2B, inset). Fluorescence from both GFP fusions peaked at ∼48 hr, but secGFP fluorescence decreased to undetectable levels over a period of 5 days, whereas GFP-HDEL was still fluorescent in most cells as long as 7 days later (data not shown). This is consistent with decreased stability of fluorescent GFP secretion to the apoplast. In all subsequent experiments, strains containing GFP constructs were inoculated at the discriminating OD600 value of 0.01 unless otherwise stated.
Figure 2.

Agrobacterium-Mediated Transient Expression of secGFP and GFP-HDEL.
(A) Differential fluorescence signals from tobacco epidermal cells after transient expression of secGFP and GFP-HDEL using Agrobacterium strains infiltrated at various OD600 values, as indicated above each pair of images. Individual epidermal cells exhibiting GFP fluorescence in the apoplast (secGFP) and cortical cytoplasm (GFP-HDEL) can be seen. (Because the epidermal cells possess a large central vacuole with a thin layer of cortical cytoplasm, only the GFP-HDEL signal originating from the ER in the cortical cytoplasm adjacent to the anticlinal cell walls is clearly visible at this magnification.) The images are a projection of a 30 × 1-μm Z-series collected by confocal laser scanning microscopy.
(B) High-resolution image of epidermal cells inoculated with secGFP and GFP-HDEL at OD600 0.01, collected by confocal laser scanning microscopy. The inset shows secGFP fluorescence in the apoplast (green) imaged in the presence of 5 μg/mL plasma membrane–labeling dye FM4-64 (red). To show the typical reticulate organization of the ER, the GFP-HDEL image is taken from an optical section passing through the cortical cytoplasm that lies immediately adjacent to the outer periclinal cell walls; the secGFP image is taken from a section passing through the center of the cells and shows the anticlinal walls in cross-section with their associated secGFP fluorescence.
 ;
;  .
.
Export of secGFP from the ER Results in Proteolysis and Degradation in a Downstream Compartment
To test the interpretation that the reduced accumulation of fluorescent secGFP was associated with its transport to the apoplast, we incubated sections of infiltrated leaf either in water or, to inhibit export from the ER, in 10 μg/mL BFA (Boevink et al., 1999). The resulting marked intracellular accumulation of fluorescent secGFP was apparent after 6 hr and reached a maximum after 15 to 24 hr. As shown in Figure 3A, in the presence of BFA, secGFP fluorescence was observed in a network of tubules and planar structures typical of tobacco epidermal ER. If leaf pieces were incubated for 8 hr in BFA and then were washed and transferred to water without BFA for 7 hr, the accumulated secGFP fluorescence was lost (Figure 3A); in contrast, control samples that were returned to 10 μg/mL BFA after being washed showed no loss of secGFP accumulation during the 7-hr incubation. Meanwhile, the intensity of GFP-HDEL fluorescence was unaltered by incubation in BFA or by BFA washout (Figure 3A), although the use of BFA led to the appearance of small punctate structures closely associated with the ER network. Similar structures were observed to a lesser extent with secGFP in the presence of BFA. These structures, which were specific for BFA-treated cells, were clearly smaller and less mobile than GFP-labeled Golgi stacks and appeared to exclude Golgi markers (H.Q. Zheng, C. Saint-Jore, I. Moore, and C. Hawes, unpublished observations), but their identity remains to be established.
Figure 3.

secGFP Reports Reversible Inhibition of Membrane Traffic by BFA.
(A) Confocal microscopy of GFP fluorescence in epidermal cells of samples of leaves infiltrated with Agrobacterium strains carrying either secGFP (left) or GFP-HDEL (right) after either 15 hr of incubation in water (Water) or 8 hr of incubation in 10 μg/mL BFA followed by washing and a further 7-hr incubation in either 10 μg/mL BFA (BFA) or water (Washout).  .
.
(B) Protein extracts from samples incubated for either 15 hr in water (lanes 1), 15 hr in BFA (lanes 2), or 8 hr in BFA followed by washing and incubation for 7 hr in water (lanes 3) were subjected to SDS-PAGE and immunoblotted with antibodies raised against GFP. Three times as much extract was loaded in the samples from the uninfected control (lane C) and the secGFP-treated tissues because this protein accumulated consistently less than did GFP-HDEL. The arrow indicates the position of the 30-kD molecular marker, and the arrowheads denote the differences in mobility of the detected bands.
To investigate the molecular basis of the differential fluorescence of secGFP and GFP-HDEL, we analyzed protein extracts of inoculated tissues by SDS-PAGE, immunoblotting with anti-GFP antibodies. As indicated in Figure 3B (lane 1), GFP-HDEL accumulated predominantly as a single band of slightly less than 30 kD, as expected, although a faint band apparently 2 kD smaller was also detected in some experiments. The abundance of this latter band was not detectably altered by treatment with BFA or after BFA washout (lanes 2 and 3). In contrast, secGFP accumulated as two distinct bands, the upper of which had an apparent molecular mass consistent with our expectation (∼1 kD larger than GFP-HDEL because of the C-terminal c-Myc epitope tag; lane 1), whereas the lower band appeared to migrate at the same position as the minor band in the GFP-HDEL extracts. Both bands were considerably less intense than the major GFP-HDEL band, and in contrast to GFP-HDEL, the two bands in secGFP extracts were either similar in intensity or the lower band was more abundant. Strikingly, in the presence of BFA, the upper band accumulated at the expense of the lower band, an effect that was reversed when BFA was washed out (lanes 2 and 3). Thus, the decreased fluorescence observed from secGFP may reflect in part reduced protein accumulation within the cell, whereas transport out of the ER appears to result in proteolysis, which may also in part account for the fact that secGFP is less fluorescent than GFP-HDEL (but see Discussion).
These observations suggested that the accumulation of fluorescent secGFP might provide a simple, rapid assay for the ability of diverse inhibitors, mutants, and transgenes to perturb membrane traffic between the ER and the plasma membrane in plant cells. Furthermore, identifying the endomembrane organelles in which GFP accumulates would provide an initial indication of which membrane trafficking step is disrupted.
A Dominant Inhibitory Mutant of AtRAB1b Inhibits the Transport of secGFP to the Apoplast
To determine whether the assay described above could be used to investigate the membrane trafficking functions of the plant Rab GTPase family, we chose to study a member of the Arabidopsis Rab1 subclass AtRab1b (Bischoff et al., 1999). Members of this subclass in yeasts and mammals have been implicated in vesicular traffic in the early steps of the secretory pathway (Schmitt et al., 1988; Bacon et al., 1989; Plutner et al., 1991; Tisdale et al., 1992; Jedd et al., 1995). To investigate the trafficking function of AtRab1b, we generated a dominant inhibitory AtRAB1b mutant containing a single amino acid substitution, asparagine to isoleucine, in the conserved GTP binding motif GNKxD. On the basis of structure–function analyses of the Ras-like GTPase superfamily (Bourne et al., 1990, 1991), we predicted that the resulting allele, AtRAB1b(N121I), would be antimorphic, or dominant negative, when expressed in plant cells, inhibiting the activity of the wild-type GTPase. The equivalent asparagine-to-isoleucine substitution in many members of the Ras superfamily has provided much useful information about the cellular functions of the respective wild-type GTPases (Schmitt et al., 1988; Bucci et al., 1992, 2000; Tisdale et al., 1992). To investigate the effect of AtRab1b(N121I) on secGFP transport in tobacco, we cloned both the mutant and wild-type cDNAs, under control of an enhanced CaMV 35S promoter in a binary T-DNA vector. To achieve coexpression of secGFP and AtRab1b derivatives, we grew Agrobacterium strains containing the appropriate T-DNA vector and mixed them before leaf infiltration. Unless specified, the Agrobacterium mixtures contained the secGFP strain at a final OD600 of 0.01 and the test strain at OD600 0.03.
As shown in Figure 4A, when secGFP was coexpressed with wild-type AtRab1b, the fluorescence signal was unaltered in comparison with that of tissues inoculated with the secGFP strain alone or with the secGFP strain plus a control strain expressing β-glucuronidase (GUS) rather than AtRab1b (pVKH-En6-GUS) (data not shown). In contrast, coexpression of secGFP with AtRab1b(N121I) resulted in strong accumulation of intracellular secGFP in many cells of the infected region (Figure 4A). At higher resolution, GFP fluorescence was observed in a dynamic reticulate pattern that was very reminiscent of the ER visualized with GFP-HDEL (Figure 4B). This suggested that, like BFA, AtRab1b(N121I) may inhibit export of secGFP from the ER and thus lead to accumulation of a stable fluorescent form of the protein in this compartment. In contrast to some BFA experiments (Boevink et al., 1999), the morphology of the ER was not appreciably altered by expression of AtRab1b(N121I). We never observed secGFP accumulating in genuinely punctate structures such as those generated by BFA or by Golgi-targeted GFP fusions (Boevink et al., 1998, 1999; Nebenführ et al., 1999). Occasionally, both GFP-HDEL and the secGFP that accumulated in the presence of AtRab1b(N121I) appeared to reside in diffuse punctate structures of various dimensions, but whenever these were viewed at greater resolution, they always resolved into junctions of the ER network and were thus most probably imaging artifacts. The accumulation of secGFP fluorescence in the presence of AtRab1b(N121I) reached its maximum ∼2 days after infiltration, as with secGFP at high inoculum densities, but the fluorescence decreased much more slowly than for secGFP, being still visible 5 or 6 days later; this suggested that in the presence of the Rab mutant, the stability of fluorescent secGFP had been increased. Coexpression of GFP-HDEL with either wild-type or mutant AtRab1b resulted in no change in its apparent distribution or fluorescence intensity (data not shown), making it unlikely that the effect of AtRab1b(N121I) on secGFP fluorescence resulted from a general enhancement of GFP expression or detection.
Figure 4.
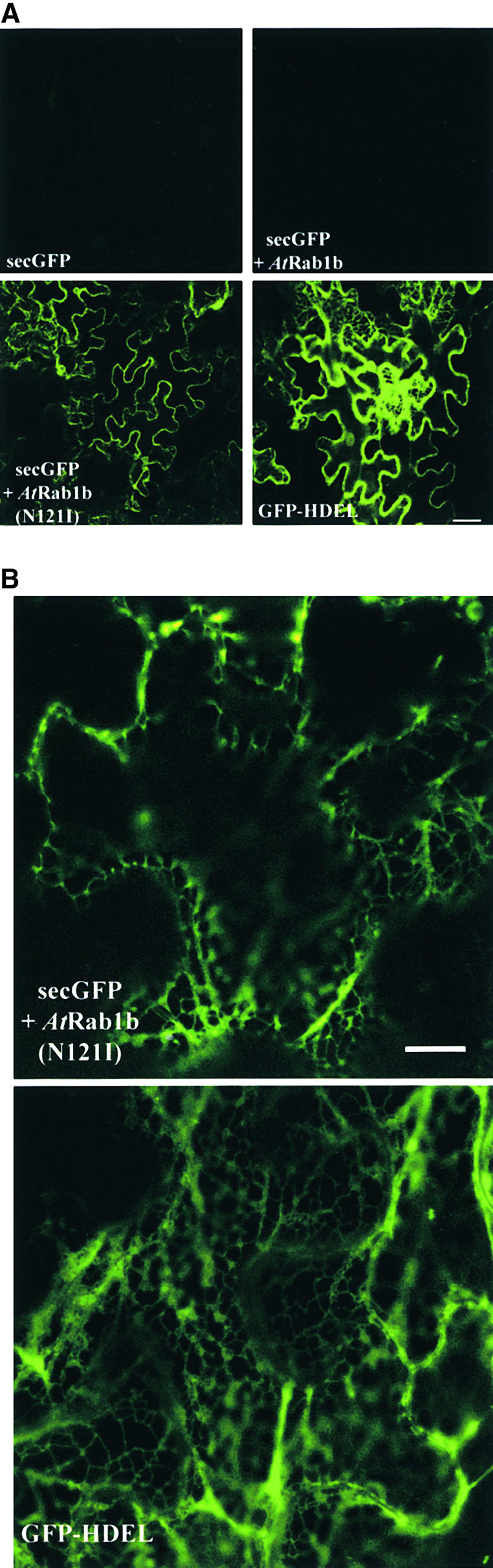
Inhibition of secGFP Transport by an AtRab1b Mutant.
(A) Low-magnification images of GFP fluorescence in epidermal cells of leaves infiltrated with strains carrying secGFP, GFP-HDEL, or secGFP plus either wild-type or mutant AtRab1b as indicated. AtRab1b(N121I) causes accumulation of secGFP in the ER. This is visible as a reticulate network when the optical section passes through the cytoplasm underlying the periclinal cell walls; in most cells, however, the section passes through the anticlinal walls, and the network in the underlying cortical cytoplasm is not resolved.
(B) High-resolution images of the intracellular accumulation of secGFP and GFP-HDEL. The distribution of secGFP accumulating in the presence of AtRab1b(N121I) resembles that of GFP-HDEL. In each case, the optical sections pass through a portion of the cortical cytoplasm beneath the periclinal cell walls.
 for all images;
for all images;  for both images.
for both images.
These data suggest that the dominant inhibitory mutant AtRab1b(N121I) inhibits membrane traffic to the plasma membrane and may act to prevent export of secGFP from the ER.
Inhibitory Effect of AtRab1b(N121I) Is Specifically Rescued by Coexpression of AtRab1b
Interpretation of the phenotypes of mutants such as AtRab1b(N121I) is complicated by the likelihood that they act by sequestering an essential cellular cofactor in a nonproductive complex and thus indirectly inhibit the capacity of the wild-type protein to perform its usual function. The Ras-like GTPases carrying the asparagine-to-isoleucine substitution that we introduced into AtRab1b(N121I) are predicted to act by inhibiting the activity of a guanine nucleotide exchange factor (GEF) that ordinarily promotes the conversion of the GTPase from its GDP-bound form to its activated GTP-bound form (Barbacid, 1987; Wilson et al., 1994; Jones et al., 1995). This poses a potential problem when the phenotype of such mutants is used to infer the normal cellular activity of the wild-type GTPase. If the GEF (or other cellular factor) sequestered by the mutant GTPase is required not only for the activity of the wild-type GTPase but also for the activity of one or more functionally distinct proteins in the cell, then the mutant phenotype that is observed could arise in part or in full from inhibition of this second activity. To infer the cellular function of the wild-type GTPase from this phenotype would thus be misleading.
In practice, that theoretical objection has not been a practical impediment to the use of dominant inhibitory Rab GTPases of this type, which have proved to be specific inhibitors of their respective Rab GTPase subclass when tested either in vivo or in vitro in other eukaryotic systems (Tisdale et al., 1992; Wilson et al., 1994; Jones et al., 1995). However, no information is available about the specificity of Rab GEFs in plants, nor do we know the biochemical basis of the antimorphic behavior of AtRab1b(N121I); therefore, we tested empirically for the specificity of AtRab1b(N121I) action in vivo.
Our starting assumption was that the interaction between AtRab1b(N121I) and a GEF or other essential cofactor is in equilibrium with the interaction between the same factor and the wild-type GTPase. If so, coexpressing the wild type in large amounts should alleviate the inhibitory effect of AtRab1b(N121I) on the activity of wild-type AtRab1b. If the cofactor sequestered by AtRab1b(N121I) is required only for the activity of either AtRab1b or proteins with the same function as AtRab1b, this coexpression of wild-type GTPase should completely restore the trafficking function that is disrupted by AtRab1b(N121I) in the secGFP transport assay. Alternately, perhaps AtRab1b(N121I) sequesters a GEF or other cofactor that is required for the activity of both AtRab1b and one or more proteins with a trafficking function distinct from that of AtRab1b; if so, the accumulation of secGFP could result in part or in full from the loss of this second activity. In this case, the disrupted trafficking function will not be restored by coexpression of AtRab1b and may even be further inhibited because of the increased competition for a common cofactor. Thus, by asking whether coexpression of wild-type AtRab1b could restore normal membrane traffic to an assay inhibited by AtRab1b(N121I), we were able to test empirically whether secGFP accumulates through loss of AtRab1b function.
Coexpression of AtRab1b(N121I) and the wild type at equal inoculum densities completely rescued the secGFP trafficking defect associated with AtRab1b(N121I), as shown in Figure 5A. When viewed at high resolution, secGFP fluorescence in these samples was comparable to that obtained with secGFP alone (Figure 2B; data not shown). Inoculation of AtRab1b(N121I) at twice its usual inoculum density, or coinoculation with control strains expressing GUS rather than AtRab1b, resulted in no decrease in secGFP accumulation (data not shown). Furthermore, when GFP-HDEL was coexpressed with AtRab1 and AtRab1(N121I), neither the intensity nor the distribution of the fluorescent signal was altered (Figure 5B), indicating that the absence of secGFP fluorescence in the rescue experiment is unlikely to have resulted from a nonspecific influence of the two AtRab1b constructs on GFP expression, detection, or localization. On the basis of these observations, we suggest that the trafficking defect associated with AtRab1b(N121I) is specific to a trafficking function in which AtRab1b can act. Consistent with this interpretation, the inhibition of secGFP transport by AtRab1b(N121I) was not rescued by coexpression with AtRab8c, a member of a different but closely related Rab subclass, Rab8 (Moore et al., 1995; Bischoff et al., 1999), which is predicted to perform a different trafficking function from that of AtRAB1b (Figure 5C).
Figure 5.

sec-GFP Accumulation Phenotype of AtRab1b(N121I) Is Rescued by Coexpression of Wild-Type AtRab1b but Not AtRab8c.
(A) Low-magnification images of epidermal cells expressing secGFP, either alone or in combination with the AtRab1b derivative, as indicated at bottom left. secGFP accumulates in the presence of AtRab1b(N121I) (bottom left), but when wild-type and mutant AtRab1b are coexpressed (bottom right), secGFP accumulation is suppressed to amounts similar to those observed in controls that express only secGFP (top left) or secGFP and wild-type AtRab1b alone (top right).
(B) Low-magnification (at top) and high-magnification (at bottom) confocal images of epidermal cells expressing GFP-HDEL alone or in combination with AtRab1b and AtRab1b(N121I), as specified at bottom left of each image. Coexpression with the AtRab1b constructs affected neither the intensity nor the pattern of GFP-HDEL accumulation, as indicated by the upper and lower images, respectively.
(C) Low-magnification images of epidermal cells expressing secGFP. The wild-type AtRab8c does not induce intracellular accumulation of secGFP (left) and cannot suppress the effects of AtRab1b(N121I) (right).
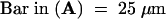 for all images;
for all images;  for the upper images and 10 μm for the lower images;
for the upper images and 10 μm for the lower images; 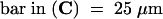 for both images.
for both images.
The rescue test thus demonstrates that AtRab1b is sufficient to restore the trafficking function inhibited by AtRab1b(N121I). On morphological grounds, we propose that AtRab1b(N121I) causes secGFP to accumulate in the ER, which suggests that AtRab1b promotes continued export of secGFP from the ER.
An N-Glycosylated GFP Variant Is Processed to an Endoglycosidase-H–Resistant Form in the Golgi
To investigate the hypothesis that the secGFP accumulating in the presence of AtRab1b(N121I) has not been transported through the Golgi stack, we developed an N-glycosylated GFP derivative. Before the action of mannosidase II in the Golgi, N-glycans are sensitive to removal by endoglycosidase H (endoH) (Staehelin and Moore, 1995; Dupree and Sherrier, 1998). Consequently, if membrane traffic is inhibited upstream of the Golgi compartment in which mannosidase II acts, then a secreted N-glycosylated GFP is expected to accumulate an endoH-sensitive form. Where in the Golgi stack the mannosidase II acts is not known, but a medial compartment is thought likely (Staehelin and Moore, 1995; Dupree and Sherrier, 1998).
We initially modified the coding sequence of GFP-HDEL to introduce a potential N-glycosylation site in a 10-residue synthetic peptide incorporated between the cleavage site of the ER targeting signal and the GFP coding region. The resulting protein, N-GFP-HDEL, retained bright fluorescence in the ER (Figure 6) and acquired an N-glycan that remained in an endoH-sensitive form, as shown in Figure 7A. When the equivalent synthetic peptide was included in secGFP, the resulting protein, N-secGFP, was glycosylated and showed fluorescence and stability properties similar to those described for secGFP. In the presence of AtRab1b(N121I), N-secGFP accumulated in a typical reticulate pattern, an accumulation inhibited by coexpression with AtRab1b but not AtRab8c, as previously observed with secGFP (data not shown). In the presence of AtRab1b(N121I), N-secGFP accumulated in a form that was sensitive to digestion by endoH, consistent with an ER location (data not shown). Unfortunately, however, trafficking of N-secGFP to the apoplast in control treatments appeared to result in loss of the synthetic glycosylated peptide from the N terminus of the fusion protein (data not shown), so we were unable to determine whether the glycan on N-secGFP ordinarily is processed to an endoH-resistant form during transport through the Golgi stack. In an attempt to solve this problem, we introduced several other synthetic and natural N-glycosylation sequences into the N terminus of secGFP; however, we were unable to identify a variant that exhibited fluorescence and retained the N-terminal glycosylated extension when transported to the apoplast.
Figure 6.
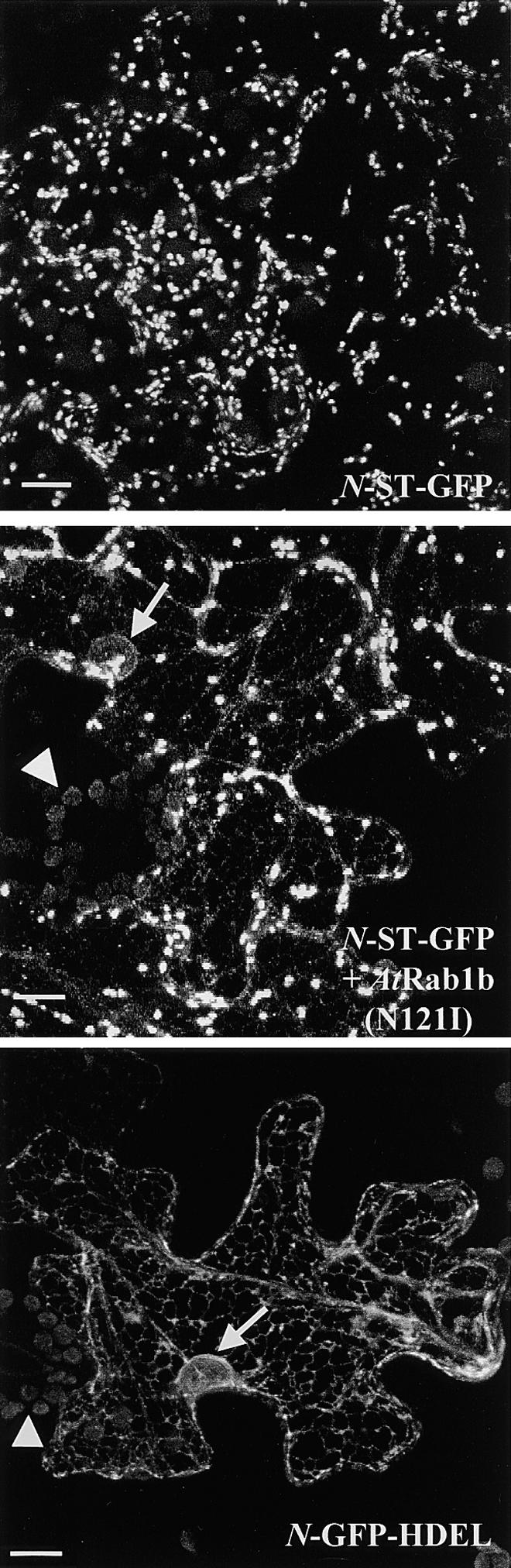
Effects of AtRab1b(N121I) on Intracellular Distribution of N-ST-GFP.
Projections of 24 × 1-μm confocal optical sections through epidermal cells expressing N-ST-GFP, N-GFP-HDEL, or N-ST-GFP plus AtRab1b(N121I), as indicated. In the cells expressing N-ST-GFP alone, fluorescence is observed in a series of discrete mobile punctate structures typical of the plant Golgi apparatus. In the presence of AtRab1b(N121I), the Golgi are still labeled, but fluorescence is also seen in a reticulate network similar to that observed with N-GFP-HDEL. The arrow points to a nuclear membrane present in the section of one of these cells. Autofluorescence is visible in the chloroplasts of guard cells (arrowheads). 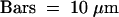 .
.
Figure 7.

Effect of AtRab1b(N121I) on Processing of N-Glycosylated GFP Derivatives.
(A) Extracts of leaf tissues expressing N-GFP-HDEL or N-ST-GFP were incubated either with (+) or without (−) endoH and analyzed by SDS-PAGE and immunoblotting with a GFP C-terminal antibody (N15; ABCAM). The N-glycans on GFP-HDEL, but not those on N-ST-GFP, are sensitive to removal by endoH, as illustrated by a shift of ∼2 kD in the apparent molecular mass of the major band.
(B) Cells expressing N-ST-GFP alone or N-ST-GFP plus AtRab1b(N121I) were analyzed as described for (A). Coexpression of N-ST-GFP with AtRab1b(N121I) resulted in the accumulation of an endoH-sensitive population of N-ST-GFP. This example shows that slightly less than half of the N-ST-GFP had accumulated in an endoH-sensitive form. Although in other experiments the endoH-sensitive population was sometimes slightly more abundant than the resistant form, it was never observed to be the predominant species.
Because GFP is more stable when targeted to the plant Golgi (Boevink et al. 1998, 1999), we suspect that proteolysis of secGFP and N-secGFP may occur in a post-Golgi compartment. To generate a stable glycosylated GFP for which the glycan was demonstrably modified in the Golgi, we replaced the N-terminal signal peptide of N-secGFP with the N-terminal 53 residues of the rat trans-Golgi enzyme sialyltransferase (ST) to generate N-ST-GFP (Figure 1). This enzyme is targeted to the trans-Golgi cisternae when expressed in Arabidopsis (Wee et al., 1998). These N-terminal 53 residues, which include the cytoplasmic and transmembrane domains, are sufficient to target GFP to the Golgi in tobacco (Boevink et al., 1998) and were expected to target N-ST-GFP to this organelle. As shown in Figure 6, confocal imaging of epidermal cells transiently expressing N-ST-GFP revealed that fluorescence was confined to numerous mobile punctate structures typical of Golgi stacks labeled with GFP (Boevink et al., 1998; Nebenführ et al., 1999). Furthermore, immunoblot analysis showed that N-ST-GFP accumulated in an endoH-resistant form, which migrated as expected with an apparent molecular mass of 35 kD (Figure 7A). These observations suggest that N-ST-GFP resides in or passes through the mannosidase II–containing compartment of the Golgi.
Cells consistently accumulated less N-ST-GFP than GFP-HDEL (Figure 7A), even when the expression of N-ST-GFP was increased until its accumulation in the Golgi was maximized and the Golgi were brighter than the ER of cells expressing N-GFP-HDEL, as in Figure 6. This suggests either that GFP is more fluorescent in the Golgi than in the ER or, more probably, that the total volume of the Golgi apparatus is less than that of the ER, such that GFP is at a relatively high concentration in each Golgi stack, accounting for the relative brightness of the stacks. Our inability to increase the total quantity of N-ST-GFP in the Golgi to amounts similar to those in the ER suggests a limit on the capacity of the Golgi to accumulate this protein. We next asked whether coexpression with AtRab1b(N121I) alters the transport and processing of N-ST-GFP.
N-ST-GFP Accumulates an EndoH-Sensitive Form in the Presence of AtRab1b(N121I)
Coexpression of N-ST-GFP with AtRab1b(N121I) changed its intracellular distribution, causing it to accumulate additionally in a typically ER-like reticulate network associated with the Golgi stacks (Figure 6). This reticulate network was not observed when N-ST-GFP was coexpressed with wild-type AtRab1b or when the wild-type and mutant Rab proteins were coexpressed together with N-ST-GFP; therefore, this network most probably represents the ER. This interpretation was supported by immunoblot analysis of N-ST-GFP, which revealed that coexpression with AtRab1b(N121I) induced the accumulation of an additional endoH-sensitive form of N-ST-GFP that apparently had not encountered Golgi mannosidase II (Figure 7B). The residual endoH-resistant form of N-ST-GFP presumably represents the Golgi-localized population identified in confocal images (Figure 6). We observed that the labeling of the reticulate compartment in the presence of AtRab1b(N121I) was far more transient with N-ST-GFP than with secGFP or N-secGFP, and that the loss of N-ST-GFP fluorescence in this compartment was accompanied by the loss of the endoH-sensitive form. This is consistent with the view that the sensitive form resides in the network, the resistant form in the Golgi.
These observations suggest that AtRab1b(N121I) inhibits the transport of GFP fusions before they reach the mannosidase II compartment of the Golgi. They strongly suggest that the reticulate compartment in which the GFP fusions accumulate is pre-Golgi and therefore is most probably the ER. They also show that AtRab1b(N121I) can inhibit the normal trafficking of both a membrane protein such as N-ST-GFP and soluble proteins such as secGFP and N-secGFP.
Expression of AtRab1b(N121I) Disrupts Normal Movement of Golgi Stacks on the ER Network
Plant Golgi stacks are characterized by periods of rapid vectorial movement punctuated by periods of relative stasis in which individual stacks cease directed movement and oscillate in a fashion resembling Brownian motion within a small area little larger than the Golgi stack itself (Boevink et al., 1998; Nebenführ et al., 1999). However, the significance of this Golgi behavior for membrane traffic between the ER and Golgi in higher plants is unclear. Unexpectedly, confocal imaging of cells coexpressing N-ST-GFP and AtRab1b(N121I) revealed that vectorial Golgi movement was markedly reduced and in most cells was essentially abolished. In these cells, the Golgi stacks simply underwent minor oscillatory movements reminiscent of the movements that characterize periods of stasis in control cells. Apart from the altered Golgi behavior, the cells appeared to be otherwise healthy; they continued to exclude propidium iodide and appeared to maintain an unaltered ER network that exhibited its characteristic movement. Moving images of these cells are viewable at http://www.plants.ox.ac.uk/moore/index.html. To convey the extent of Golgi movement in a static image, we took 60 successive images of a single optical section every 1.5 sec and projected them on top of each other (shown in Figure 8). In this projected image, Golgi stacks that exhibited rapid vectorial movement over the period of observation appeared at many different positions, tracing out their path across the composite image as depicted in Figure 8A. In control cells expressing only N-ST-GFP, the movement of Golgi stacks was so extensive that entire regions of cytoplasm were covered by these tracks (Figure 8A). In contrast, stacks that did not exhibit vectorial movement throughout the 90-sec period of observation appear in the projection as single dots, the diameter of which reflects the amplitude of oscillation of the Golgi stack. We estimate that in control cells, ⩽2% of the total population of Golgi are static. Strikingly, in the projections of cells coexpressing N-ST-GFP and AtRab1b(N121I), between 50 and 90% of the Golgi are static over the 90-sec observation period (Figures 8B and 8C). In cells that exhibit vectorial Golgi movement, the extent of movement is much less than for the controls (cf. Figures 8A and 8B).
Figure 8.

AtRab1b(N116I) Inhibits Normal Golgi Movement.
Confocal laser scanning microscopy of cells coexpressing N-ST-GFP with wild-type AtRab1b (A), AtRab1b(N121I) (B) and (C), or wild-type AtRab1b plus AtRab1b(N121I) (D). Cells were imaged every 1.5 sec over a 90-sec period. At left in (A) to (D) is the first image from each series, and at right is the final image. At center is the projection of the entire series of 60 images in which moving Golgi trace out their tracks. Cells such as those in (B) and at the bottom of (C) represent ∼20% of the epidermal cell population and exhibited some Golgi movement, although this was less extensive than in controls (cf. rows [A] or [D] with [B] and [C]), whereas the other two cells shown in (C) exhibited essentially no Golgi movement and such cells represented ∼60% of the epidermal cell population.
As with the secGFP transport assay, wild-type AtRab1b did not alter Golgi movement (data not shown), but it could restore normal Golgi movement when coexpressed with AtRab1b(N121I) (Figure 8D). This suggests that Golgi movement can be inhibited by loss of AtRab1b activity alone, which implies that AtRab1b activity is required for the normal regulation of plant Golgi movement. Thus, in addition to the acto-myosin system, AtRab1b is now also implicated in the control of plant Golgi movements, providing experimental evidence of an intriguing link between Golgi mobility and the control of membrane traffic from the ER.
DISCUSSION
Investigation of membrane traffic on the biosynthetic secretory pathway in plants has been hindered by the absence of simple in vitro or in vivo transport assays. Here, we show that secreted and N-glycosylated variants of GFP can be used effectively to report on membrane traffic between the ER and the Golgi apparatus in tobacco epidermal cells. The basis of the assay is the observation that secreted GFP exhibits less fluorescence than do the forms retained within the ER or Golgi apparatus. This differential fluorescence provides a convenient assay of the ability of drugs or transgenes to interfere with normal trafficking of GFP to the apoplast. The use of transient expression allows transgenes to be tested rapidly and is applicable for use with transgenes whose expression would preclude the regeneration of transgenic plants. The weaker fluorescence observed with apoplastic GFP may arise at least in part from the lesser stability of the protein in this compartment compared with that in the ER. secGFP consistently accumulated in lesser amounts than did the ER-localized GFP-HDEL, and its predominant form was a proteolytic product estimated to be ∼2 kD smaller than expected. This product was not detected by an antiserum raised against a C-terminal peptide of GFP (H. Batoko and I. Moore, unpublished results), so this unexpected form is probably truncated at the C terminus. The truncation may contribute to the low stability or weak fluorescence of apoplastic GFP, although some GFP variants that have lost ∼10 residues from their C terminus are known to retain fluorescence in the ER and cytoplasm (Gadella et al., 1999). Thus, perhaps the apoplast provides a poor environment for the formation or maintenance of fluorescent GFP molecules.
Fusions to ST show that GFP can be stable in the lumen of the plant Golgi apparatus (Boevink et al., 1998), probably until reaching the trans-Golgi compartments (Wee et al., 1998); therefore, the secGFP assay potentially can detect perturbation in membrane trafficking before this compartment is reached. Still to be determined is whether the assay is able to report on subsequent steps in the secretory pathway, given uncertainty as to whether secGFP would emit a greater fluorescent signal in post-Golgi structures than in the apoplast.
We have used the GFP assay to identify a function for an Arabidopsis Rab GTPase in membrane traffic to the Golgi apparatus. In the presence of the antimorphic allele AtRAB1b(N121I), secGFP accumulated in a reticulate network that was morphologically indistinguishable from the ER. Consistent with this interpretation, in the presence of AtRab1b(N121I), an N-glycosylated derivative of GFP accumulated in an endoH-sensitive form that had not been modified by Golgi mannosidase II. Where mannosidase II acts within the plant Golgi stack is not known, although a medial location is considered likely (Staehelin and Moore, 1995; Dupree and Sherrier, 1998). The accumulation of an endoH-resistant form would also be observed if AtRab1b(N121I) were to inhibit transport between cis- and medial Golgi compartments rather than inhibit the transport from ER to Golgi, as we propose. We think inhibition of the former transport is unlikely, given recent demonstrations that a GFP fusion to mannosidase I accumulates in cis-Golgi compartments and results in a typically punctate distribution, as has been described for the ST-GFP fusion (Boevink et al., 1998; Nebenführ et al., 1999). However, in the presence of AtRab1b(N121I), we observed no such punctate distribution of secGFP, only a reticulate network typical of ER-targeted GFP fusions.
Notably, the AtRab1(N121I) did not appear to result in the vesiculation of the Golgi apparatus, which is seen with the equivalent Hs-Rab1a mutant in mammalian cells (Wilson et al., 1994) or in the accumulation in the ER, as happens with the equivalent YPT1 mutant in Saccharomyces cerevisiae (Schmitt et al., 1988; Jedd et al., 1995). Confirming these observations by electron microscopy of tobacco epidermal cells will be of interest. However, the apparently different effects of dominant inhibitory Rab1 mutants in the various organisms may reflect specific differences in the organization of ER–Golgi membrane traffic in each, and the molecular basis of these differences may become clearer as more factors involved in ER–Golgi traffic in plants are characterized.
The mechanism of action of AtRab1b(N121I) remains to be determined, but antimorphic GTPase mutants such as this are believed to act by inhibiting the activity of a GEF (Jones et al., 1995; Rybin et al., 1996; Cool et al., 1999). GEFs promote the conversion of the GTPase to its active GTP-bound form by catalyzing the release of GDP from the inactive GDP-bound form, facilitating the binding of GTP. The asparagine-to-isoleucine substitution in the GNKxD motif of the GTPase appears to reduce the affinity of the protein for guanine nucleotides (Cherfils and Chardin, 1999) and stabilizes the nucleotide-free form of the protein that arises, usually only transiently, during the nucleotide exchange reaction. This nucleotide-free state forms a high-affinity complex with the GEF; accordingly, the mutant is believed to competitively sequester the GEF in a nonproductive complex that precludes normal rates of nucleotide exchange on the wild-type GTPase.
Although no functional data are yet available on regulatory proteins of plant Rab GTPases, the inhibitory activity of AtRab1b(N121I) probably results from the titration of some essential cofactor (such as a GEF) with which AtRab1b interacts, although the identity of this factor is not known. When interpreting the phenotypes of such antimorphic alleles, one must consider the possibility that the mutant allele elicits a pleiotropic phenotype through titration of a cofactor that is required by more than one Rab GTPase or titration of some other cellular factor. Whatever the mechanism of action of AtRab1b(N121I), the ability of AtRab1b, but not AtRab8c, to rescue the secGFP accumulation phenotype indicates that the AtRab1b(N121I) phenotype probably arises specifically from the loss of an activity that can be supplied by the wild-type AtRab1b protein; this would reveal a role for AtRab1b in transport between the ER and Golgi. The failure of AtRab8c to rescue the phenotype is important because it is a member of the plant Rab subclass that is most closely related to the Rab1 subclass, which includes AtRab1b (Bischoff et al., 1999). This supports the view that the AtRab1b(N121I) phenotype is specific for the Rab1 subclass. Inhibition of a GEF specific for the Rab1 subclass would be consistent with the current understanding of Rab GTPase action in other eukaryotes. Once considered promiscuous, Rab GEFs in yeasts and mammals are now thought probably to be specific for individual Rab subclasses. This specificity is inferred from the specificity of action of antimorphic alleles, such as AtRab1b(N121I), that inhibit Rab GEF activity (Jones et al., 1995; Walch-Solimena et al., 1997; Day et al., 1998) and from the direct analysis of the few Rab GEFs that have been identified (Walch-Solimena et al., 1997; Wang et al., 2000).
Why transport of N-ST-GFP to the Golgi was not completely inhibited by AtRab1b(N121I) or why it accumulated in the ER more transiently than secGFP did is not clear. Perhaps before the membrane traffic was inhibited by mutant AtRab1b, some N-ST-GFP was transported to the Golgi, where it accumulated in a stable and fluorescent form; in contrast, secGFP transported to the apoplast was unstable and therefore was not detected. Although we cannot exclude this trivial explanation, we were not able to prevent some residual accumulation in the Golgi either by varying the inoculum of the AtRab1b(N121I) strain or by infiltrating leaves with the AtRab1b(N121I) strain at 1 or 2 days before exposure to the N-ST-GFP strain. Consequently, perhaps AtRab1b(N121I) never completely inhibits export from the ER. Possibly, tobacco epidermal cells possess a second ER export mechanism that is not dependent on AtRAB1b, and N-ST-GFP and maybe secGFP can be transported by this mechanism. Interestingly, the Rab1 subclass is unusually large and diverse in plants (e.g., Arabidopsis has at least six genes sharing as little as 74% identity compared with two loci that are 91% identical in mammals and a single locus in Saccharomyces and Schizosaccharomyces); perhaps these additional Rab1 proteins are involved in mechanisms of ER–Golgi traffic that are insensitive to AtRab1b(N121I).
When AtRab1b(N121I) was coexpressed with the glycosylated Golgi marker N-ST-GFP, we consistently observed a residual endoH-resistant form in addition to the new endoH-sensitive form. Perhaps some of the endoH-resistant molecules were generated in the ER, through either redistribution of Golgi processing enzymes to this compartment or the accumulation in this compartment of newly synthesized processing enzymes for which transport to the Golgi had been inhibited. However, coexpression of AtRab1b(N121I) with N-GFP-HDEL did not result in the appearance of endoH-resistant forms of this marker, although whether N-GFP-HDEL is a substrate for the glycan-processing activities normally found in the Golgi remains to be established. However, given the clear presence of residual Golgi-localized N-ST-GFP in the presence of AtRab1b(N121I), the simplest interpretation is that the endoH-resistant molecules represent this Golgi-localized population.
Our analysis of N-ST-GFP transport unexpectedly revealed that expression of AtRab1b(N121I) resulted in either partial or complete inhibition of Golgi movement on the ER network. The movement of plant Golgi stacks on an actin network coextensive with the ER has been demonstrated recently by the expression of ST-GFP in tobacco epidermal cells. Individual Golgi stacks alternate between periods of rapid vectorial movement and periods of nondirected oscillation reminiscent of Brownian motion (Boevink et al., 1998). A recent analysis of Golgi movement in cultured tobacco cells also identified these two modes of Golgi movement and demonstrated that the probability of individual stacks entering periods of vectorial movement or stasis varied markedly in different regions of the cytoplasm of a single cell (Nebenführ et al., 1999). Certain cytoplasmic positions could be identified in which a succession of Golgi stacks individually entered periods of stasis and then resumed vectorial movement. Furthermore, an individual Golgi stack was able to remain in a period of stasis while an adjacent stack engaged in rapid vectorial movement, suggesting that the control of Golgi movement is devolved to the individual stack. These unexpected observations have led to the formulation of at least two distinct hypotheses to account for membrane exchange between the ER and the highly dynamic Golgi stacks of plant cells. The vacuum cleaner model postulates that rapidly moving Golgi stacks encounter and fuse with ER-derived vesicles as they travel on the actin/ER network (Boevink et al., 1998). The alternate “filling station” model postulates that membrane traffic between these two organelles occurs during the periods of stasis, when the Golgi may be positioned near putative export sites on the ER (Nebenführ et al., 1999). However, another explanation of these periods of stasis is that they simply represent discontinuities in the actin network, in which Golgi stacks are immobilized before reengaging the network (Nebenführ et al., 1999). So although the distribution of the ER and the movement of the Golgi stacks clearly are closely associated with the same actin network, no evidence links any of these properties to membrane traffic between the two organelles. Our observation that expression of AtRab1b(N121I) inhibits the trafficking of secGFP from ER to Golgi and also results in the inhibition or cessation of Golgi movement provides evidence of a possible mechanistic link between Golgi traffic and movement. To investigate these possibilities more fully, it is important to quantify the rate of ER–Golgi traffic in cells in which their Golgi are immobilized by treating the cells with inhibitors of the acto-myosin system or by expression of AtRab1b(N121I). Identification of sites of vesicle production on the ER will also be informative, as will establishing whether these coincide with the positions at which Golgi stacks cease their bouts of rapid vectorial movement.
METHODS
Molecular Cloning and Generation of Mutant AtRab1b(N121I)
Standard molecular techniques as described by Ausubel et al. (1999) were followed. The wild-type cDNA of AtRAB1b (Bischoff et al., 1999) was amplified by the polymerase chain reaction (PCR) from the expressed sequence tag clone z26415 by using the upstream primer PAR1 (5′-ACCGCTCGAGAACCATGAATCCTGAGTACG-3′) and the downstream primer PAR2 (5′-TAGATGTCGACGGATCCTCAAGTTGAGCAGCAGCCG-3′). The PCR product was digested and subcloned as a XhoI-SalI fragment into pBluescript SK+ (Stratagene). A double-stranded oligonucleotide constructed with PARNI1 (5′-AACAAACTCCTTGTTGGAATCAAGTCAGATCTTACTGAAAACAGAGCCATTCCTTATGAA-3′) and PARNI2 (5′-CTTGGCAGTTTCATA-AGGAATGGCTCTGTTTTCAGTAAGATCTGACTTGATTGATTCCAACAAG-3′) was used to replace a HincII-StyI fragment in the wild-type coding region. The double-stranded oligonucleotide introduced both a single-point mutation, the asparagine-to-isoleucine substitution (letters in boldface), and an additional HinfI restriction site (underlined letters) to generate AtRab1b(N121I). The wild-type and the mutant cDNAs were checked by sequencing with primers T3 and T7 and then were cloned as an XhoI-BamHI fragment into SalI and BamHI sites of the expression cassette of the binary vector pVKH18-EN6-GUS, under control of an enhanced cauliflower mosaic virus (CaMV) 35S promoter, a tobacco mosaic virus–Ω sequence as translation enhancer, and a nopaline synthase polyadenylation signal, all derived from pE6113GUS (Mitsuhara et al., 1996). Plasmid pVKH18-EN6-GUS had been generated by inserting the expression cassette of pE6113GUS (Mitsuhara et al., 1996) as a PvuII-HindIII fragment into the Ecl136II-HindIII sites in the polylinker of pVKH18, a derivative of pVK18 (Moore et al., 1998) that carries a hygromycin-selectable marker in place of the methotrexate resistance marker of pVK18 (I. Moore, unpublished results).
To create pVKH-GFP-HDEL, the coding region of mGFP5 fused to an Arabidopsis chitinase signal peptide from plasmid pBIN-mGFP5-ER (gift of Dr. J. Haseloff, University of Cambridge, UK) was subcloned into the expression vector pVKH18-EN6 as an XbaI and SacI fragment, replacing the β-glucuronidase (GUS) coding region to yield green fluorescent protein (GFP)–HDEL (Figure 1). The C-terminal tetrapeptide HDEL of GFP-HDEL was replaced by a c-Myc epitope by PCR with primers 5′-CCGGATCCAACAATGAAGACTAATCT-3′ and 5′-TTGGATCCTTACAAATCCTCCTCAGAGATAAGCTTCTGCTC-3′. The PCR product was cloned into pVKH18-EN6-gus, replacing the GUS coding sequence to generate pVKH-secGFP.
To engineer the glycosylated form of GFP, we amplified the signal peptide in mGFP5-ER by using Taq/Pwo (Hybaid Ltd., Ashford, UK) and primer SGFP1 (5′-GCTCTAGAACCACCATGAAGACTAATCTTT-TTC-3′) combined with SGFP2′ (5′-AACCTCGAGCCGGTAACGGTTCCATTTGAAACAAGTTCTCCAGTCGCAGTGAATTCGGCCGAGGAT-AAT-3′). The resulting PCR product, which encoded the signal peptide with a 10-residue linker downstream, was digested with XbaI and XhoI and then was used to replace the signal peptide of secGFP to yield pVKH-N-secGFP. The new GFP coding region is thus extended at the N terminus by a 10-residue region containing an N-glycosylation site (see Figure 1). The synthetic peptide harboring the glycosylation site was designed according to Gavel and von Heijne (1990). Primers SGFP1 and HGFP3′ (5′-AAGGATCCTTAAAGCTCAT-CATGAAGATCTTCGGAAATAAGCTTCTGCTCTTTGTATAGTTCATC-3′) were used to amplify the N-secGFP coding region, introducing a C-terminal HDEL tetrapeptide to generate N-GFP-HDEL. The PCR product was digested by XbaI and BamHI and then was cloned into pVKH18-En6-gus, replacing the GUS coding region. N-ST-GFP was constructed by replacing the signal peptide of N-secGFP with the sialyltransferase (ST) transmembrane domain (Boevink et al., 1998) and leaving the 10-residue N-glycosylated linker between the transmembrane domain and the GFP. Details on all of the constructs are available on request.
Plant Material and Agrobacterium tumefaciens–Mediated Transient Expression
Nicotiana tabacum SR1 (cv Petit Havana) seeds were disinfected with 70% ethanol and plated on Murashige and Skoog basal salt medium (Sigma) containing 0.8% agar (Difco). After incubation for 2 weeks at 20 to 22°C, the seedlings were transferred to soil and the resulting plants were maintained at 21°C. Each expression vector was introduced into A. tumefaciens strain GV3101 (pMP90) (Koncz and Schell, 1986) by electroporation. A single colony from the transformants was used to inoculate 2 mL of YEB medium (per liter: 5 g of beef extract, 1 g of yeast extract, 5 g of sucrose, and 0.5 g of MgSO4·7H2O), supplemented with 100 μg/mL kanamycin, 10 μg/mL gentamycin, and 1 mM isopropyl-β-d-thiogalactopyranoside. The bacterial culture was incubated at 28 to 30°C with agitation until reaching the stationary growth phase. One milliliter of culture was transferred into an Eppendorf tube, and the bacteria were pelleted by centrifugation at 2200g for 5 min in a microcentrifuge at room temperature. The pellet was washed twice with 1 mL of the infiltration buffer (50 mM Mes, pH 5.6, 2 mM Na3PO4, 0.5% glucose [w/v], and 100 μM acetosyringone [Aldrich]) and then resuspended in 1 mL of the same buffer. The bacterial suspension was diluted with infiltration buffer to adjust the inoculum concentration to the stated final OD600 value. The inoculum was delivered to the lamina tissues of tobacco leaves by gentle pressure infiltration through the stomata of the lower epidermis, by using a 1-mL syringe without a needle. For experiments requiring coinfection of more than one construct, bacterial strains containing the constructs were mixed before performing the leaf infection, with the inoculum of each construct adjusted to the required final OD600. The infected area of the leaf was delimited and labeled with an indelible pen, and the plant was incubated under normal growing conditions as above.
Sampling, Brefeldin A Treatment, and Imaging
If not otherwise stated, GFP-dependent fluorescence was analyzed 48 hr after infection in cells of the lower epidermis. Routinely, the expression pattern and intracellular distribution of GFP fluorescence were initially assessed by using an epifluorescence microscope before the confocal microscope. Pieces of leaf were randomly cut from the infected area and mounted in water for microscopic observations. For treatment with brefeldin A (BFA), leaf pieces were excised 35 hr after infection and floated on 10 μg/mL BFA solution or water as control. The BFA effect, as assessed by GFP fluorescence in the cell under an epifluorescence microscope, was evident after 6 hr. To monitor the reversibility of BFA effects, we thoroughly washed the leaf piece in water after 8 hr of incubation with the drug; half of the piece was kept in the BFA solution and the other half was incubated in water. The samples were examined 7 hr later. Samples that received 15 hr of BFA treatment gave the same result as those incubated with the drug for only 8 hr.
Confocal imaging (with a Zeiss [Wellwyn, UK] CLSM 410 or Bio-Rad MRC 1024) was performed on samples mounted in water. Optical sectioning and time-lapse scanning were acquired by using the Bio-Rad MRC 1024 imaging system. The software Confocal Assistant (Bio-Rad), LSMdummy (Zeiss), and Photoshop 5.0 were used for postacquisition image processing.
Protein Extraction, Endoglycosidase Treatment, and Protein Gel Blot Analysis
Proteins were extracted from BFA-treated and control samples (∼200 mg fresh tissues) by grinding them in liquid N2 followed by homogenization in two volumes of the extraction buffer: 75 mM Tris, pH 6.8, 1 mM EDTA, 2% SDS, 3% β-mercaptoethanol, and 1 mM phenylmethylsulfonyl fluoride. The homogenate was cleared by centrifugation for 5 min at 1100g in a microcentrifuge, and soluble proteins in the supernatant were precipitated in 6% trichloroacetic acid (v/v) for 20 min on ice. The mixture was centrifuged for 15 min at 12,000g, and the resulting pellet was washed twice with acetone, dissolved in loading buffer for SDS-PAGE, and boiled for 5 min before loading.
Protein extracts for endoglycosidase H (endoH) digestion were prepared from samples of infected leaf areas (300 to 400 mg fresh weight) frozen in liquid N2 . The tissue was ground in a microcentrifuge tube by using a plastic pestle in the presence of two volumes of the following extraction buffer: 50 mM sodium citrate, pH 5.5, 5% SDS (w/v), 0.01% BSA (w/v), 2% β-mercapthoethanol, 150 mM NaCl, and 10 μL (per 300 mg of plant material) of a protease inhibitor cocktail (Sigma). The mixture was boiled for 10 min and cleared by centrifugation at 4°C in a cooled microcentrifuge. The supernatant was transferred to a microcentrifuge tube and stored on ice. Aliquots were frozen in liquid N2 and stored at −80°C until used. For endoglycosidase digestion, buffer G5 (New England BioLabs, Hitchin, UK) was added to the protein extract thawed on ice to achieve a working concentration of buffer, which resulted in a twofold dilution of the protein extract. Ten to 40 μL of the extract was incubated for 1 hr at 37°C in the presence of 1 μL (1000 units) of EndoHf (New England BioLabs). Control samples were treated identically, but no endoglycosidase was added. The reaction was stopped by adding an equal volume of 2 × SDS-PAGE loading buffer.
For SDS-PAGE, we used a 12% acrylamide gel and Protean III apparatus (Bio-Rad). After electrophoresis, proteins were transferred to polyvinylidene difluoride or nitrocellulose membrane for 2 hr in transfer buffer according to Burnette (1981). The filter was blocked for 1 hr in Tris-buffered saline containing 0.1% (v/v) Tween 20 (Sigma), 1% BSA ([w/v]; Fraction V; Roche Biochemicals, Lewes, UK), and 0.1% goat serum (v/v) (Sigma). Antibody to full-length GFP (Santa Cruz Biotechnology, Santa Cruz, CA, or Molecular Probes, Leiden, The Netherlands) or an antibody raised against the GFP C-terminal peptide (N15; ABCAM, Cambridge, UK) was used at 1/2000 dilution to probe the membrane for 1 hr. Alkaline phosphatase–conjugated secondary antibody (Sigma) was used at 1/10,000 dilution (1-hr incubation), and the results were determined with the Sigma Fast 5-bromo-4-chloro-3-indolyl phosphate/nitroblue tetrazolium method, used according to the manufacturer's recommendations.
Acknowledgments
We thank Dr. Masashi Ugaki for the enhanced 35S cassette, Dr. Jim Haseloff for pBIN-mGFP5ER, Claude Saint-Jore for the ST-GFP construct, Dr. Edmund Wee for cloning N-ST-GFP, and Dr. Andreas Meyer for assistance with confocal imaging. This work was supported by a grant to I.M. from the Biotechnology and Biological Sciences Research Council (UK).
References
- Ausubel, F., Brent, R., Kingston, R.E., Moore, J.G., Seidman, J.G., Smith, J.A., and Struhl, J.G. (1999). Current Protocols in Molecular Biology. (New York: John Wiley).
- Bacon, R.A., Salminen, A., Ruohola, H., Novick, P., and Ferro-Novick, S. (1989). The GTP-binding protein Ypt1 is required for transport in vitro: The Golgi apparatus is defective in ypt1 mutants. J. Cell Biol. 109, 1015–1022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barbacid, M. (1987). ras genes. Annu. Rev. Biochem. 56, 779–827. [DOI] [PubMed] [Google Scholar]
- Bednarek, S.Y., Reynolds, T.L., Schroeder, M., Grabowski, R., Hengst, L., Gallwitz, D., and Raikhel, N.V. (1994). A small GTP-binding protein from Arabidopsis thaliana functionally complements the yeast YPT6 null mutant. Plant Physiol. 104, 591–596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bischoff, F., Molendijk, A., Rajendrakumar, C.S.V., and Palme, K. (1999). GTP binding proteins in plants. Cell. Mol. Life Sci. 55, 233–256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boevink, P., Cruz, S.S., Hawes, C., Harris, N., and Oparka, K.J. (1996). Virus-mediated delivery of the green fluorescent protein to the endoplasmic reticulum of plant cells. Plant J. 10, 935–941. [Google Scholar]
- Boevink, P., Oparka, K., Cruz, S.S., Martin, B., Betteridge, A., and Hawes, C. (1998). Stacks on tracks: The plant Golgi apparatus traffics on an actin/ER network. Plant J. 15, 441–447. [DOI] [PubMed] [Google Scholar]
- Boevink, P., Martin, B., Oparka, K., Cruz, S.S., and Hawes, C. (1999). Transport of virally expressed green fluorescent protein through the secretory pathway in tobacco leaves is inhibited by cold shock and brefeldin A. Planta 208, 392–400. [Google Scholar]
- Bourne, H.R., Sanders, D.A., and McCormick, F. (1990). The GTPase superfamily: A conserved switch for diverse cell functions. Nature 348, 125–131. [DOI] [PubMed] [Google Scholar]
- Bourne, H.R., Sanders, D.A., and McCormick, F. (1991). The GTPase superfamily: Conserved structure and molecular mechanism. Nature 349, 117–127. [DOI] [PubMed] [Google Scholar]
- Bucci, C., Parton, R.G., Mather, I.H., Stunnenberg, H., Simons, K., Hoflack, B., and Zerial, M. (1992). The small GTPase rab5 functions as a regulator in the early endocytic pathway. Cell 70, 715–728. [DOI] [PubMed] [Google Scholar]
- Bucci, C., Thomsen, P., Nicoziani, P., McCarthy, J., and van Deurs, B. (2000). Rab7: A key to lysosome biogenesis. Mol. Biol. Cell 11, 467–480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burkhardt, J.K. (1998). The role of microtubule-based motor proteins in maintaining the structure and function of the Golgi complex. Biochim. Biophys. Acta 1404, 113–126. [DOI] [PubMed] [Google Scholar]
- Burnette, W.N. (1981). Western blotting: Electrophoretic transfer of proteins from sodium dodecyl sulfate polyacrylamide gels to unmodified nitrocellulose and radiographic detection with antibody and radioiodinated protein A. Anal. Biochem. 112, 195–203. [DOI] [PubMed] [Google Scholar]
- Bush, J., Franek, K., Daniel, J., Spiegelman, G.B., Weeks, G., and Cardelli, J. (1993). Cloning and characterization of 5 novel Dictyostelium discoideum rab-related genes. Gene 136, 55–60. [DOI] [PubMed] [Google Scholar]
- Cheon, C., Lee, N.-G., Siddique, A.-B.M., Bal, A.K., and Verma, D.P.S. (1993). Roles of plant homologs of Rab1p and Rab7p in the biogenesis of the peribacterioid membrane, a subcellular compartment formed de novo during root nodule symbiosis. EMBO J. 12, 4125–4135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cherfils, J., and Chardin, P. (1999). GEFs: Structural basis for their activation of small GTP-binding proteins. Trends Biochem. Sci. 24, 306–311. [DOI] [PubMed] [Google Scholar]
- Cool, R.H., Schmidt, G., Lenzen, C.U., Prinz, H., Vogt, D., and Wittinghofer, A. (1999). The Ras mutant D119N is both dominant negative and activated. Mol. Cell. Biol. 19, 6297–6305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Day, G.-J., Mosteller, R.D., and Broek, D. (1998). Distinct subclasses of small GTPases interact with their guanine nucleotide exchange factors in a similar manner. Mol. Biol. Cell 18, 7444–7454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Denecke, J., Botterman, J., and Deblaere, R. (1990). Protein secretion in plant cells can occur via a default pathway. Plant Cell 2, 51–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dupree, P., and Sherrier, D.J. (1998). The plant Golgi apparatus. Biochim. Biophys. Acta Mol. Cell. Res. 1404, 259–270. [DOI] [PubMed] [Google Scholar]
- Gadella, T.W.J., van der Krogt, G.N.M., and Bisseling, T. (1999). GFP-based FRET microscopy in living plant cells. Trends Plant Sci. 4, 287–291. [DOI] [PubMed] [Google Scholar]
- Gavel, Y., and von Heijne, G. (1990). Sequence differences between glycosylated and non-glycosylated Asn-X-Thr/Ser acceptor sites: Implications for protein engineering. Protein Eng. 3, 433–442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haseloff, J., Siemering, K.R., Prasher, D.C., and Hodge, S. (1997). Removal of a cryptic intron and subcellular localization of green fluorescent protein are required to mark transgenic Arabidopsis plants brightly. Proc. Natl. Acad. Sci. USA 94, 2122–2127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hawes, C., Boevink, P., and Moore, I. (2000). Green fluorescent protein in plants. In Protein Localisation by Fluorescence Microscopy—A Practical Approach, V.J. Allan, ed (Oxford, UK: Oxford University Press), pp. 163–177.
- Infante, C., Ramos-Morales, F., Fedriani, C., Bornens, M., and Rios, M.M. (1999). GMAP-210, a cis-Golgi network associated protein, is a minus end microtubule-binding protein. J. Cell Biol. 145, 83–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jedd, G., Richardson, C., Litt, R., and Segev, N. (1995). The Ypt1 GTPase is essential for the first two steps of the yeast secretory pathway. J. Cell Biol. 131, 583–590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones, S., Litt, R.J., Richardson, C.J., and Segev, N. (1995). Requirement of nucleotide exchange factor for Ypt1 GTPase mediated protein transport. J. Cell Biol. 130, 1051–1061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kapila, J., De Rycke, R., Van Montagu, M., and Angenon, G. (1997). An Agrobacterium-mediated transient gene expression system for intact leaves. Plant Sci. 122, 101–108. [Google Scholar]
- Koncz, C., and Schell, J. (1986). The promoter of TL-DNA gene 5 controls the tissue-specific expression of chimaeric genes carried by a novel Agrobacterium binary vector. Mol. Gen. Genet. 204, 383–396. [Google Scholar]
- Lazar, T., Götte, M., and Gallwitz, D. (1997). Vesicular transport: How many Ypt/Rab-GTPases make a eukaryotic cell? Trends Biochem. Sci. 22, 468–472. [DOI] [PubMed] [Google Scholar]
- Letourneur, F., Gaynor, E.C., Hennecke, S., Demolliere, C., Duden, R., Emr, S.D., Riezman, H., and Cosson, P. (1994). Coatomer is essential for retrieval of dilysine-tagged proteins to the endoplasmic reticulum. Cell 79, 1199–1207. [DOI] [PubMed] [Google Scholar]
- Martínez-Menárguez, J.A., Geuze, H.J., Slot, J.W., and Klumperman, J. (1999). Vesicular tubular clusters between the ER and Golgi mediate concentration of soluble secretory proteins by exclusion from COPI-coated vesicles. Cell 98, 81–90. [DOI] [PubMed] [Google Scholar]
- Mellman, I., and Warren, G. (2000). The road taken: Past and future foundations of membrane traffic. Cell 100, 99–112. [DOI] [PubMed] [Google Scholar]
- Mitsuhara, I., et al. (1996). Efficient promoter cassettes for enhanced expression of foreign genes in dicotyledonous and monocotyledonous plants. Plant Cell Physiol. 37, 49–59. [DOI] [PubMed] [Google Scholar]
- Moore, I., Schell, J., and Palme, K. (1995). Subclass-specific sequence motifs identified in Rab GTPases. Trends Biochem. Sci. 20, 10–12. [DOI] [PubMed] [Google Scholar]
- Moore, I., Diefenthal, T., Zarsky, V., Schell, J., and Palme, K. (1997). A homolog of the mammalian GTPase Rab2 is present in Arabidopsis and is expressed predominantly in pollen grains and seedlings. Proc. Natl. Acad. Sci. USA 94, 762–767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moore, I., Gälweiler, L., Grosskopf, D., Schell, J., and Palme, K. (1998). A transcription activation system for regulated gene expression in transgenic plants. Proc. Natl. Acad. Sci. USA 95, 376–381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Movafeghi, A., Happel, N., Pimpl, P., Tai, G.-H., and Robinson, D.G. (1999). Arabidopsis Sec21p and Sec23p homologs. Probable coat proteins of plant COP-coated vesicles. Plant Physiol. 119, 1437–1445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagano, Y., Okada, Y., Narita, H., Asaka, Y., and Sasaki, Y. (1995). Location of light-repressible, small GTP-binding protein of the YPT/rab family in the growing zone of etiolated pea stems. Proc. Natl. Acad. Sci. USA 92, 6314–6318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nebenführ, A., Gallagher, L.A., Dunahay, T.G., Frohlick, J.A., Mazurkiewicz, A.M., Meehl, J.B., and Staehelin, L.A. (1999). Stop-and-go movements of the plant Golgi stacks are mediated by the acto-myosin system. Plant Physiol. 121, 1127–1141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Novick, P., and Zerial, M. (1997). The diversity of Rab proteins in vesicle transport. Curr. Opin. Cell Biol. 9, 496–504. [DOI] [PubMed] [Google Scholar]
- Palme, K., Diefenthal, T., Vingron, M., Sander, C., and Schell, J. (1992). Molecular cloning and structural analysis of genes from Zea mays (L.) coding for members of the ras-related YPT gene family. Proc. Natl. Acad. Sci. USA 89, 787–791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pfeffer, S.R. (1999). Transport-vesicle targeting: Tethers before SNAREs. Nat. Cell Biol. 1, E17–E22. [DOI] [PubMed] [Google Scholar]
- Plutner, H., Cox, A.D., Pind, S., Khosravi-Far, R., Bourne, J.R., Schwaninger, R., Der, C.J., and Balch, W.E. (1991). Rab1B regulates vesicular transport between the endoplasmic reticulum and successive Golgi compartments. J. Cell Biol. 115, 31–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Presley, J.F., Cole, N.B., Schroer, T.A., Hirchberg, K., Zaal, K.J.M., and Lippincott-Schartz, J. (1997). ER-to-Golgi transport visualized in living cells. Nature 389, 81–85. [DOI] [PubMed] [Google Scholar]
- Robinson, D.G., and Kristen, U. (1982). Membrane flow via the Golgi apparatus of plant cells. Int. Rev. Cytol. 77, 89–127. [Google Scholar]
- Rossi, L., Escudero, J., Hohn, B., and Tinland, B. (1993). Efficient and sensitive assay for T-DNA–dependent transient gene expression. Plant Mol. Biol. Rep. 11, 220–229. [Google Scholar]
- Rybin, V., Ullrich, O., Rubino, M., Alexandrov, K., Simon, I., Seabra, M.C., Goody, R., and Zerial, M. (1996). GTPase activity of Rab5 acts as a timer for endocytic membrane fusion. Nature 383, 266–269. [DOI] [PubMed] [Google Scholar]
- Satiat-Jeunemaitre, B., and Hawes, C. (1996). Brefeldin A effects in plant and fungal cells: Something new about vesicle trafficking? J. Microsc. 181, 162–177. [DOI] [PubMed] [Google Scholar]
- Scales, S.J., Perpperkok, R., and Kreis, T.E. (1997). Visualization of ER-to-Golgi transport in living cells reveals a sequential mode of action for COPII and COPI. Cell 90, 1137–1148. [DOI] [PubMed] [Google Scholar]
- Schmitt, H.D., Wagner, P., Pfaff, E., and Gallwitz, D. (1986). The ras-related YPT1 gene product in yeast: A GTP-binding protein that might be involved in microtubule organization. Cell 47, 401–412. [DOI] [PubMed] [Google Scholar]
- Schmitt, H.D., Puzicha, M., and Gallwitz, D. (1988). Study of a temperature-sensitive mutant of the ras-related YPT1 gene product in yeast suggests a role in the regulation of intracellular calcium. Cell 53, 635–647. [DOI] [PubMed] [Google Scholar]
- Staehelin, L.A., and Moore, I. (1995). The plant Golgi apparatus: Structure, functional organization and trafficking mechanisms. Annu. Rev. Plant Physiol. Plant Mol. Biol. 46, 261–288. [Google Scholar]
- Terryn, N., Arias, M.B., Engler, G., Tire, C., Villarroel, R., Van Montagu, M., and Inzé, D. (1993). RHA1, a gene encoding a small GTP-binding protein from Arabidopsis, is expressed primarily in developing guard cells. Plant Cell 5, 1761–1769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tisdale, E.J., Bourne, J.R., Khosravi-Far, R., Der, C.J., and Balch, W.E. (1992). GTP-binding mutants of Rab1 and Rab2 are potent inhibitors of vesicular transport from the endoplasmic reticulum to the Golgi complex. J. Cell Biol. 119, 749–761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ueda, T., Anai, T., Tsukaya, H., Hirata, A., and Uchimya, H. (1996). Characterisation and subcellular localisation of a small GTP-binding protein (Ara4) from Arabidopsis: Conditional expression under the control of the promoter of the gene for heat-shock protein HSP81-1. Mol. Gen. Genet. 250, 533–539. [DOI] [PubMed] [Google Scholar]
- Ueda, T., Matsuda, N., Uchimya, H., and Nakano, A. (2000). Modes of interaction between the Arabidopsis Rab protein, Ara4, and its putative regulator molecules revealed by a yeast expression system. Plant J. 21, 341–349. [DOI] [PubMed] [Google Scholar]
- Valentijn, J.A., Valentijn, K., Pastore, L.M., and Jamieson, J.D. (2000). Actin coating of secretory granules during regulated exocytosis correlates with the release of rab3D. Proc. Natl. Acad. Sci. USA 97, 1091–1095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vitale, A., and Denecke, J. (1999). The endoplasmic reticulum—Gateway of the secretory pathway. Plant Cell 11, 615–628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walch-Solimena, C., Collins, R., and Novick, P. (1997). Sec2 mediates nucleotide exchange on Sec4p and is involved in polarized delivery of post-Golgi vesicles. J. Cell Biol. 137, 1495–1509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang, W., Sacher, M., and Ferro-Novick, S. (2000). TRAPP stimulates guanine nucleotide exchange on Ypt1p. J. Cell Biol. 151, 289–295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wee, E.G.T., Sherrier, D.J., Prime, T.A., and Dupree, P. (1998). Targeting of active sialyltransferase to the plant Golgi apparatus. Plant Cell 10, 1759–1768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wieland, F.T., Gleason, M.L., Serafini, T.A., and Rothman, J.E. (1987). The rate of bulk flow from the endoplasmic reticulum to the cell surface. Cell 50, 289–300. [DOI] [PubMed] [Google Scholar]
- Wilson, B., Nuoffer, C., McCaffery, M., Feramisco, J., Balch, W., and Farquhar, M. (1994). A Rab1 mutant affecting guanine nucleotide exchange promotes disassembly of the Golgi apparatus. J. Cell Biol. 125, 557–571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang, G.F., and Staehelin, L.A. (1992). Functional compartmentation of the Golgi apparatus of plant cells. Plant Physiol. 99, 1070–1083. [DOI] [PMC free article] [PubMed] [Google Scholar]