

Abstract
The S-phase checkpoint kinases Mec1 and Rad53 in the budding yeast, Saccharomyces cerevisiae, are activated in response to replication stress that induces replication fork arrest. In the absence of a functional S-phase checkpoint, stalled replication forks collapse and give rise to chromosome breakage. In an attempt to better understand replication dynamics in S-phase checkpoint mutants, we developed a replication origin array for budding yeast that contains 424 of 432 previously identified potential origin regions. As expected, mec1-1 and rad53-1 mutants failed to inhibit late origin activation. Surprisingly however, 17 early-firing regions were not replicated efficiently in these mutants. This was not due to a lack of initiation, but rather to problems during elongation, as replication forks arrested in close proximity to these origins, resulting in the accumulation of small replication intermediates and eventual replication fork collapse. Importantly, these regions were not only prone to chromosome breakage in the presence of exogenous stress but also in its absence, similar to fragile sites in the human genome.
Keywords: DNA replication, fragile sites, Mec1, Rad53, replication stress
Introduction
Initiation of DNA replication in eukaryotic cells occurs at multiple origins that are distributed throughout the genome. Although all origins are licensed for a single round of replication during the G1 phase of the cell cycle, activation of replication origins in S phase underlies temporal regulation, such that some origins ‘fire' early and others late (Diffley and Labib, 2002; Shechter and Gautier, 2005). How origin timing is controlled in an unperturbed S phase is poorly understood, but recent evidence supports the view that different S-phase-specific cyclin-dependent kinase activities target early- and late-firing origins (Donaldson et al, 1998; Donaldson and Blow, 1999). When a cell is subjected to replication stress by exposure to hydroxyurea (HU), which causes a decrease in cellular deoxyribonucleotide triphosphates (dNTPs), S-phase checkpoint kinases are activated to inhibit S-phase progression (Weinert et al, 1994; Longhese et al, 2003). Kinase activation prevents late origins from firing (Santocanale and Diffley, 1998; Kihara et al, 2000; Duncker et al, 2002; Costanzo et al, 2003) and spindles from elongating (Krishnan et al, 2004). These actions lead to a mitotic delay, in an effort to ensure that replication is complete before cells divide. During this delay, one important function of the S-phase checkpoint is to stabilize replication forks that are stalled owing to the lack of dNTPs (Lopes et al, 2001; Sogo et al, 2002). Recent evidence suggests that the stabilization of stalled forks (and not the delay of mitosis) is the key step for retaining cell viability after removal of replication stress (Desany et al, 1998; Tercero et al, 2003). The loss of DNA polymerases, replication protein A and the putative helicase, the minichromosome maintenance (Mcm)2–7 complex, likely leads to replication fork collapse and chromosome breakage (Cobb et al, 2003, 2005; Lucca et al, 2004), resulting in elevated rates of gross chromosomal rearrangements (GCRs) in S-phase checkpoint mutants (Myung et al, 2001).
The current model for HU-induced replication arrest in wild-type budding yeast suggests that forks are generated at early-firing origins and move an average distance of 8–9 kb before they stall (Lengronne et al, 2001). Stalling is believed to occur stochastically and independently of Mec1 and Rad53 (Tercero et al, 2003). Fork progression rates appear to be similar in replication checkpoint mutants, as shown in a study of chromosome VI in Saccharomyces cerevisiae (although arrested forks eventually collapse in these mutants; Katou et al, 2003), suggesting that replication fork arrest in checkpoint mutants follows the same stochastic dynamics as in wild-type cells. Although it is unknown whether this is true for all chromosomes in S. cerevisiae, it is clearly not the case in mammalian cells. In human chromosomes, certain regions are particularly sensitive to replication inhibitors such as HU or aphidicolin (Yan et al, 1987). These fragile sites are highly unstable, are prone to breakage and can extend over large regions. Numerous studies support the view that fragile sites are frequently deleted or rearranged in cancer cells (Arlt et al, 2003). Importantly, the stability of fragile sites has been shown to depend on the human Mec1 homolog ATR (ataxia-telangiectasia and Rad3-related protein; Casper et al, 2002, 2004). These observations have culminated in a model according to which fragile sites encounter replication fork stalling with an unusually high frequency, which renders them especially sensitive to replication stress. However, this model has not been tested directly.
In S. cerevisiae, chromosome breakage has recently been observed in the so-called replication slow zones (RSZs) in the absence of functional Mec1 during an otherwise unperturbed S phase (Cha and Kleckner, 2002). Loss of Mec1 likely leads to the failure to upregulate dNTP levels in late S phase, which explains why breakage in these cells occurs in late-replicating regions and is alleviated by deletion of the ribonucleotide reductase inhibitor Sml1. Another region that displays fragility is located immediately adjacent to the early-firing origin ARS310 (Lemoine et al, 2005). These two studies in yeast (Cha and Kleckner, 2002; Lemoine et al, 2005), together with evidence from mammalian systems, suggest that chromosome fragility occurs at specific sites in the genome that are more sensitive to replication stress than other regions and thus require S-phase checkpoint proteins for timely fork progression (Casper et al, 2002).
To obtain a more comprehensive picture of replication dynamics in S-phase checkpoint mutants, we have devised a replication origin DNA microarray, representing 424 of 432 potential replication origins in the budding yeast genome (Wyrick et al, 2001). In the course of our origin activation studies, we have identified origin proximal regions that are highly sensitive to HU and cause premature replication fork arrest. Furthermore, these regions are prone to chromosome breakage even in the absence of HU, making them equivalent to fragile sites in humans (Casper et al, 2002). Thus, we believe that these fragile sites in yeast will provide an important model to better understand the mechanism underlying chromosome fragility.
Results
Genome-wide study of replication origin activation utilizing a customized DNA microarray
To facilitate the study of replication origin activation in budding yeast, we developed a replication origin DNA microarray. This microarray contains 424 of the previously mapped 432 potential replication origins (Wyrick et al, 2001). Moreover, 199 non-origin loci were added, including four gene sequences of Arabidopsis thaliana to serve as negative controls. A complete listing of all loci is available in Supplementary Table I. Origin activation was monitored by copy number change experiments (Pollack et al, 1999). Briefly, cells were arrested in G1 and released into S phase in the presence of HU. In these cells, activated origins are replicated and their copy number increases from one to a maximum of two depending on origin efficiency. As we utilized a relatively small array, commonly used global normalization procedures could not be applied. Instead, we normalized values to a defined set of negative controls. Normalized values were approximately the same, regardless of whether we used A. thaliana gene sequences or a set of S. cerevisiae non-origin sequences as negative controls (data not shown).
To validate the replication origin array, we analyzed the activation profile of two different wild-type strains (DCY1671 and SP1) in the presence of HU. Under these conditions, a replication checkpoint response is triggered and thus, early-firing origins are activated, but late-firing origins are inhibited (Santocanale and Diffley, 1998). We isolated DNA from cells that were either arrested in G1 in the presence of α-factor or arrested in S phase by α-factor release into HU. To control for differences in hybridization efficiency, replicating DNA from S-phase cells (labeled with Cy5) and non-replicating DNA from G1-arrested cells (labeled with Cy3) were co-hybridized. All resulting Cy5/Cy3 ratios were normalized using a set of 36 negative controls the average copy number of which was set to a value of 1.0 (indicated as squares in Supplementary Figure S3). Raw and normalized ratios resulted in the same general activation profile (Supplementary Figure S1A). Averages of three independent experiments and their respective P-value were calculated, as described in Materials and methods. Background values for non-replicating DNA were determined by co-hybridizing Cy5- and Cy3-labeled DNA from G1-arrested cells. We determined an average background value and defined ratios with P-values <0.05 and higher than one standard deviation above background as significant (Figure 1), similar to what was described previously (Pollack et al, 1999). Applying these criteria, in the yeast strain DCY1671, we identified 66 and 180 activated origins following α-factor release for either 80 or 120 min, respectively (Supplementary Table II). Similar results were obtained for the SP1 strain (data not shown). We then compared our data to an earlier study by Yabuki et al (2002), who had analyzed the strain W303 by high-density DNA microarrays 90 min after release from α-factor into HU. Figure 1 and Supplementary Figure S1B show a direct comparison of our data and that by Yabuki et al for chromosomes III and XIII. Although the profiles for chromosome III look similar, with the exception of ARS307, we detected differences for chromosome XIII in the initial activation pattern 80 min after G1 release (Figure 1). However, when we looked at a later time point, eight out of the 10 peaks that Yabuki et al (2002) had identified were observed (Supplementary Figure S1B). We found an additional 14 activated origins, likely because our cells were left longer in HU (120 versus 90 min), 12 of which were previously classified as early origins (Yabuki et al, 2002).
Figure 1.
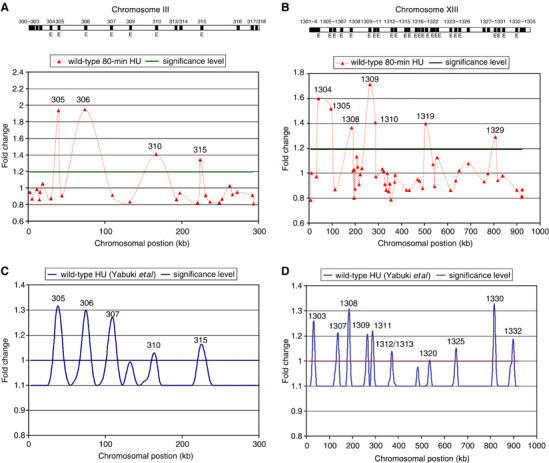
Origin activation profiles for wild-type chromosomes III and XIII. Origin activation profiles (80 min in 0.2 M HU) for chromosomes III (A) and XIII (B) are shown. The distribution of replication origins on each chromosome is indicated by black bars in the cartoon at the top. The letter E indicates early-firing origins (Raghuraman et al, 2001). Each point on the graph represents the average of three experiments. The dotted connecting line does not represent data points. The significance level used is one standard deviation above background (corresponding to approximately 20% of total signal). All scored origins had P-values <0.05. Replication profiles for chromosomes III (C) and XIII (D) based on an earlier study (Yabuki et al, 2002) are also shown. The significance level that was used by Yabuki and co-workers is 10% of total signal. Activated origins in panels A–D are indicated by their respective numbers.
To ascertain that most of the origins activated in our wild-type strain represented early-firing origins, we assigned replication times, as determined by two earlier studies (Raghuraman et al, 2001; Yabuki et al, 2002), to all origins that exhibited a copy number change on our array (Figure 2). Origins with a replication time of up to 28 min were defined as early, similar to a previous study (Raghuraman et al, 2001), because our cells required 55–60 min to complete S phase after release from G1 (Supplementary Figure S2D). Depending on which of the two studies we used for the replication time assignment, either 67 or 91% of the origins that we identified in HU-treated wild-type cells (80 min in HU) were activated early (Figure 2A). We obtained similar numbers for wild-type cells that were left in HU for 120 min: either 66 or 84% of all origins had activation times between 10 and 28 min (Figure 2B). Thus, the vast majority of activated origins we identified in our strain are located in early-replicating regions. Moreover, our peaks show a 74% (127/172) overlap with the early peaks determined by Raghuraman et al (2001). This overlap is greater than that between the peaks reported by Yabuki et al and Raghuraman et al, respectively (59%; 102/172)—again—probably because our cells remained in HU for 30 min longer.
Figure 2.
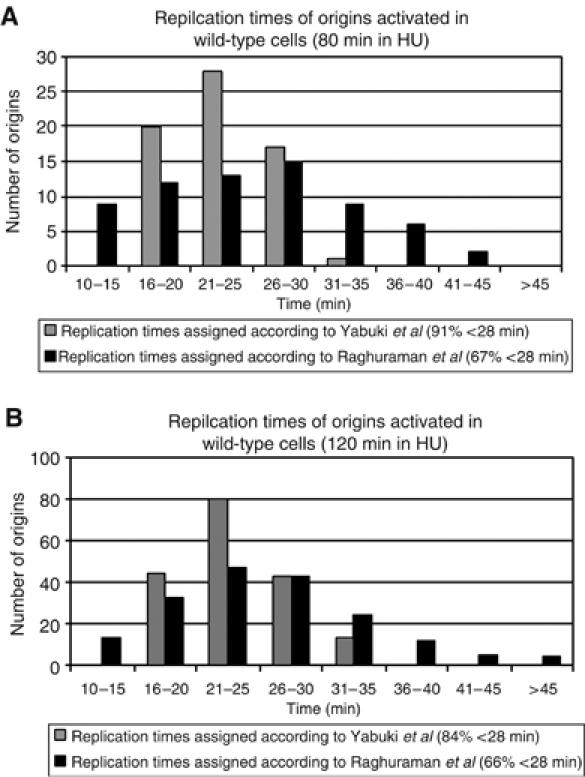
Replication times of origins activated in wild-type cells (DCY1671) assigned according to previously published timing data. Replication times were assigned to all activated origins identified in this study (Supplementary Table II) according to two earlier studies (Raghuraman et al, 2001; Yabuki et al, 2002). Replication times ranged from 10 to 60 min for Raghuraman et al and 16 to 34 min for Yabuki et al. The ‘cutoffs' for early-firing origins used by Raghuraman et al and Yabuki et al were 28 and 24 min, respectively. Assigned replication times are shown for origins activated in wild-type cells after 80 (A) or 120 min (B) in HU. (X% <28 min) indicates that X% of origins are activated within 28 min after G1 release.
Comparing S-phase dynamics of wild-type cells, mec1-1 and rad53-1 mutants
Accumulating evidence suggests that the checkpoint kinases Mec1 and Rad53 regulate late-firing origins in an unperturbed S phase and under conditions of replication stress or DNA damage (Santocanale and Diffley, 1998; Shirahige et al, 1998; Santocanale et al, 1999; Wang et al, 2001a; Katou et al, 2003; Aparicio et al, 2004; Gibson et al, 2004). A recent study demonstrates that rad53 mutants precociously activate late-firing origins genome-wide (Feng et al, 2006). However, this has not yet been shown for mec1 cells, and thus, it remained unclear whether the activation profiles of mec1 and rad53 mutants completely overlapped. To experimentally address these questions, we sought to compare the origin activation profiles of wild-type cells, mec1-1 and rad53-1 mutants in the presence of HU. In contrast to wild-type cells, these two mutants have lost the ability to stably arrest in S phase. Therefore, cell cycle dynamics in checkpoint mutants differ significantly from wild-type cells, as the mutants progress farther through S phase with continual origin firing and spindle elongation (Clarke et al, 1999, 2001; Shimada et al, 2002). To provide a meaningful comparison among wild-type and mutant strains with regard to origin activation, we wanted to ensure that all cells were analyzed at identical points in S phase. Thus, we released cells from G1 into HU and measured bud formation, spindle assembly and spindle elongation in 5-min intervals (Supplementary Figure S2A). Because similar experiments have been reported for different strains, a detailed description of the results can be found in Supplementary data. Importantly, we defined the time window of 0–80 min during which cell cycle dynamics appeared to be uniform for the three strains (Supplementary Figure S2A). Hence, for the following experiments, we chose the 80-min time point as our reference point to compare origin activation profiles of wild-type cells, mec1-1 and rad53-1 mutants.
Origin activation profiles of S-phase checkpoint mutants
To obtain a comprehensive picture of how the S-phase checkpoint controls origin activation in budding yeast, we analyzed replication profiles for wild-type, mec1-1 and rad53-1 cells as described for Figure 1. For some chromosomes (e.g. chromosome III), the origin activation profiles of mec1-1 and rad53-1 mutants looked almost identical (Figure 3A). Other chromosomes, such as chromosome XIII, showed higher variation between the respective profiles (Figure 3B). Both mutants activated early- as well as late-firing origins (the latter are indicated by the letter ‘L' in Figure 3). A complete set of origin activation profiles for all 16 chromosomes is compiled in Supplementary Figure S3, and assigned replication times of all activated origins are documented in Supplementary Figure S4. For the following analysis, we defined ‘early' and ‘late' according to a previous report by Raghuraman et al (2001). In mec1-1 cells, 107 early- and 88 late-firing origins were activated. In rad53-1 cells, we identified 142 early- and 133 late-firing origins. A total of 88% (58/66) of all early-firing origins activated in wild-type cells were also activated in the mutants (Figure 4A). Unexpectedly, however, the two mutants showed an overlap of only 46% (84/182) for early- and also 46% (70/151) for late-firing origins (Figure 4A and C), indicating that Mec1 and Rad53 control origin activation through common and independent pathways. Strikingly, rad53-1 mutants activated many more late-firing origins than mec1-1 cells (Figure 4C). A detailed list of early- and late-firing origins activated in the respective mutants is provided in Supplementary Tables III and IV.
Figure 3.
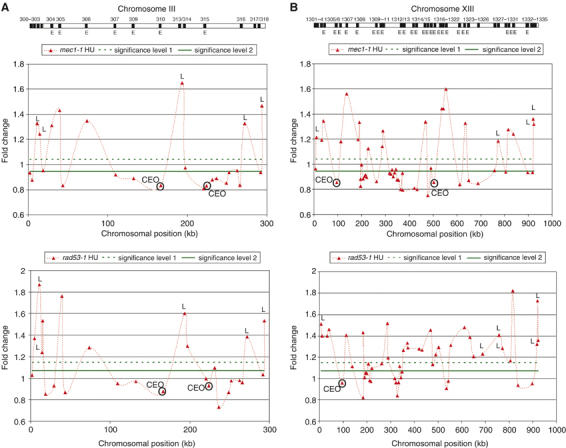
Origin activation profiles for chromosomes III and XIII in mec1-1 and rad53-1 strains. Chromosomes III (A) and XIII (B) are shown as cartoons at the top of the figure (see Figure 1 for details). Each point on the graph represents the average of three experiments. The dotted connecting line does not represent data points. Significance levels 1 and 2 correspond to one and one-half of a standard deviation above background, respectively. Late origins are activated in rad53-1 and mec1-1 mutants (P-values <0.05) and are marked by the letter L. Compromised early-firing origins (CEOs), which are activated in wild-type cells (Figure 1) but not efficiently replicated in the checkpoint mutants (copy number change <significance level 2 and P-value <0.05), are indicated by CEO and highlighted by a circle over the corresponding point on the graph.
Figure 4.
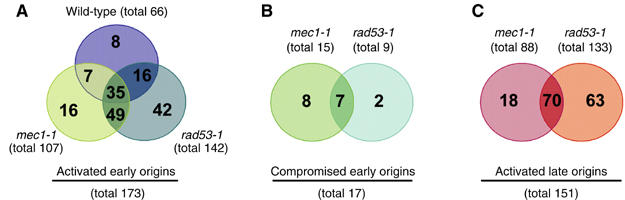
Overlap of activated and compromised replication origins between wild-type cells and S-phase checkpoint mutants. Venn diagrams showing the overlap for activated early origins between wild-type, mec1-1 and rad53-1 (A), or CEOs (B) and activated late origins (C) between mec1-1 and rad53-1. The total number of origins activated/compromised in each strain is indicated. The definition of early or late is based on our array results and on a previous study (Raghuraman et al, 2001). All origins activated in our wild-type strain (DCY1671) at the 80 min time point (66 in total) were deemed early. However, 22 of these origins are considered late in the KK14-3a background (Raghuraman et al, 2001) and six of these origins are defined as late in the W303 background (Yabuki et al, 2002).
A second unexpected finding was the disappearance of certain early-replicating peaks in the origin activation profiles of the two S-phase checkpoint mutants. On chromosome III, two regions, one over ARS310 and a second peak over ARS315, did not display any significant copy number change (Figure 3A, regions marked as ‘CEO' for compromised early origin). Importantly, we observed these changes in both mec1-1 and rad53-1 mutants (Table I). Even after a 120-min incubation of rad53-1 mutants in HU, we were unable to detect peaks over these CEOs, although additional late-firing origins were activated (data not shown). We classified a total of 17 loci as CEOs, because they showed a significant reduction in their ratios when compared to wild type (P<0.05 and a ratio below half of one standard deviation above background; significance level 2 in Figure 3 and Supplementary Figure S3). mec1-1 and rad53-1 mutants displayed 15 versus nine CEOs, respectively, with a 41% (7/17) overlap (Figure 4B).
Table 1.
CEOs in the yeast genome
| Chromosome | mec1-1a | rad53-1a,b |
|---|---|---|
| I | — | — |
| II | 208 | 208 |
| III | 310, 315 | 310, 315 |
| IV | — | — |
| V | — | — |
| VI | 606 | 606 |
| VII | 718, 728 | — |
| VIII | 806 | 806 |
| IX | 909 | 909 |
| X | — | — |
| XI | — | — |
| XII | — | — |
| XIII | 1305, 1319 | 1305 |
| XIV | 1407, 1416, 1426 | — |
| XV | 1510, 1512 | — |
| XVI | — | 1622, 1629 |
| All CEOs had a copy number change below one-half of a standard deviation above background (significance level 2 in Figure 3 and Suppplementary Figure S3) and P-values in the range of 0.039–0.0002. | ||
| These CEOs were verified in the rad53-1 sml1Δ strain. |
Replication forks stall in close proximity to CEOs
Theoretically, the lack of an activation peak could be explained by two different phenomena. Either an origin fails to fire or an origin initiates DNA replication properly, but elongation is inhibited. To distinguish between these possibilities, we examined replication intermediates by alkaline agarose electrophoresis and subsequent Southern blot analysis (Santocanale and Diffley, 1998). This assay provides a simple albeit qualitative assessment of DNA synthesis over specific regions of the yeast genome. Wild-type cells and rad53-1 mutants were arrested in G1 and released into 0.2 M HU. To slow down S-phase progression, cells were cultured at 25°C. Samples were taken in 30-min intervals and total DNA was isolated. Note that because cells were not grown under the same conditions as for the microarray experiments, the time points in this experiment are not directly comparable to the time points chosen for the microarray analyses (replication is delayed by about 30 min at 25°C; see Supplementary Figure S2C). Parental high molecular weight DNA was separated from single-stranded nascent DNA on alkaline agarose gels. Over time, these nascent DNA smears increase in size, indicative of replication elongation. For example, over ARS305, a well-characterized early-firing origin, wild-type cells initiate DNA synthesis between 30 and 60 min following G1 release (Figure 5A). By 60 min, nascent DNA strands had already reached a size of 2–3 kb and they increased up to 12 kb during the following 60 min. In rad53-1 mutants, we observed a very similar pattern (Figure 5A). However, this was not the case for the CEO ARS310 (Figure 5B). In the S-phase checkpoint mutant, replication initiated slightly earlier, because we observed a higher proportion of nascent DNA than in wild-type cells 90 min after release from G1 (Figure 5B, rad53-1, 90 min). Importantly, we detected predominantly small nascent DNA strands, approximately 0.5–2.5 kb in size (Figure 5B, rad53-1, 90 and 120 min). These small DNA strands were not efficiently elongated but instead accumulated, indicative of replication fork stalling (Figure 5B). We observed a similar pattern at a second CEO, ARS1305 (Figure 5C). The comparison between ARS305, ARS310 and ARS1305 suggested that replication initiated at all three origins in rad53-1 cells, but fork progression rates were dramatically slowed, likely owing to fork stalling, in the vicinity of the two CEOs.
Figure 5.

Analysis of replication intermediates from wild-type and rad53-1 cells in the presence of HU. G1-arrested cells were released into 0.2 M HU for 0–120 min at 25°C (A–C) or at 30°C (D, E). Replication intermediates (RIs) isolated from these cells at the indicated time points were studied using alkaline agarose gel electrophoresis (Santocanale and Diffley, 1998). RIs are shown for the early-firing origin ARS305 (A, D), and CEOs ARS310 (B, E) and ARS1305 (C).
To obtain a more accurate picture of replication fork progression rates under the same experimental conditions used for the microarray analysis, we compared ARS305 and ARS310 in wild-type and rad53-1 cells in the presence of 0.2 M HU at 30°C (Figure 5D and E). This analysis revealed that fork progression in rad53-1 mutants outside ARS310 is even slower at 30°C than at 25°C, likely owing to a temperature-dependent decrease in fork stability. Moreover, at 30°C, ARS305 and ARS310 initiate replication around the same time. Taken together, these results indicated that our failure to detect CEO activation in S-phase checkpoint mutants by microarray analysis did not stem from a lack of replication initiation but rather from inefficient elongation. To confirm this notion, we employed two additional assays: (1) two-dimensional (2D) gel analysis of replication intermediates (Friedman and Brewer, 1995) and (2) chromatin immunoprecipitation (ChIP) of the replication factor Mcm10 to monitor fork progression (Ricke and Bielinsky, 2004). For both procedures, cells were arrested in G1 and released in the presence of 0.2 M HU for 80 min at 30°C. For 2D gels, DNA was isolated and cleaved with suitable restriction enzymes, as indicated in Figure 6. Analysis of the CEO ARS310 showed that the locus was duplicated in our strain, resulting in a composite pattern that was difficult to resolve (data not shown). However, we observed a severe accumulation of replication forks close to the CEOs ARS315 and ARS1305, whereas the non-CEOs ARS305 and ARS1413 (a late-firing origin that was activated only in mec1-1 cells) showed no unusual replication intermediates (Figure 6 and Supplementary Figure S5). Moreover, at the Rad53-dependent CEO ARS1622 (Table I), we detected a strong signal for accumulating forks, but no or minor signals in wild-type and mec1-1 cells (Supplementary Figure S5), confirming our predictions from our microarray data. Consistent with the aberrant replication fork progression at CEOs, we further found that Mcm10, a fork component essential for S-phase progression, could be recovered by ChIP 1.5 kb away from ARS315, but not beyond, before it was lost from chromatin in both checkpoint mutants, between 40 and 80 min after release from G1 (Figure 7). In contrast, Mcm10 in wild-type cells associated with regions as far as 10 kb away from ARS315 (Figure 7). Similarly, at ARS305, Mcm10 displayed normal progression rates in all three strains, but was lost from chromatin in mec1-1 and rad53-1 cells between 40 and 80 min after release from G1, consistent with previous reports (Cobb et al, 2003, 2005; Katou et al, 2003; Lucca et al, 2004). Interestingly, when we placed ARS315 into the URA3 locus, we restored replication fork kinetics in the checkpoint mutants, suggesting that the ARS315 flanking regions have an innate propensity to interfere with replication fork progression.
Figure 6.
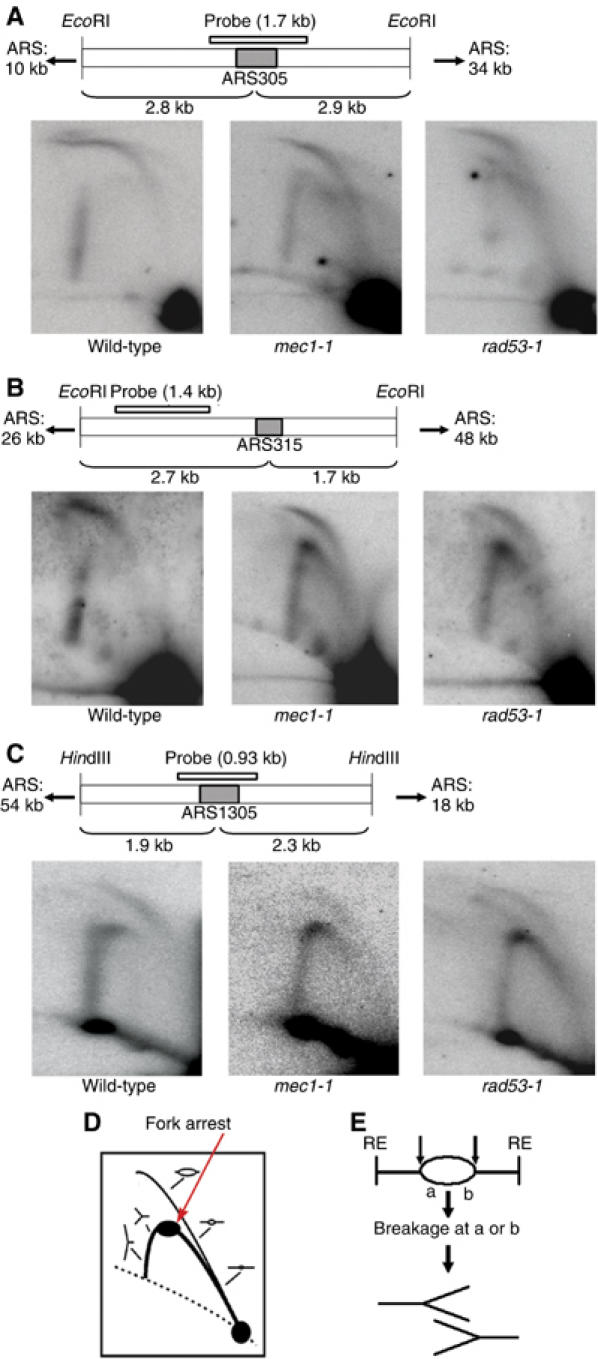
2D gel electrophoresis of replication intermediates reveals the accumulation of replication forks at CEOs in S-phase checkpoint mutants. 2D gel electrophoresis was carried out on replication intermediates isolated from wild-type, mec1-1 and rad53-1 cells arrested in 0.2 M HU for 80 min. The maps above the 2D gels indicate the locations of the restriction sites and probes used for each origin. The distance to neighboring origins is indicated as well. Replication patterns are shown for an early-firing origin, ARS305 (A), and CEOs, ARS315 (B) and ARS1305 (C). (D) A cartoon depicting the replication pattern of a restriction fragment containing a CEO. Accumulation of arrested forks is indicated by the spot on the fork arc. (E) A model illustrating fork arrest in CEO-flanking regions at points a and b. We hypothesize that forks collapse, inducing breakage at a or b, which results in the accumulation of fork structures as shown in panel D. RE indicates restriction enzyme cleavage sites. Please note that the width of the fork arcs at the CEOs is narrower in the mutants than wild-type cells.
Figure 7.

ChIP analysis shows accumulation of replication forks adjacent to a CEO in S-phase checkpoint mutants. Wild-type, mec1-1 and rad53-1 cells expressing Myc-tagged Mcm10 were synchronized in G1 phase and released into 0.2 M HU at 30°C. Samples were taken at the indicated time points. ChIP was performed using an anti-Myc antibody. PCR products were visualized on agarose gels. Results are shown for origin and origin-flanking regions at an early-firing origin, ARS305 (A), a CEO, ARS315, at its native locus (B), and ARS315 integrated into the URA3 locus (C). The distance to the origin is indicated and R11 was used as a non-origin control.
Based on our evidence that DNA elongation was severely compromised in CEO-flanking regions, we predicted that these regions accumulate double-strand breaks (DSBs) in S-phase checkpoint mutants. To test whether this occurred even in the absence of HU, we analyzed these regions in asynchronously growing cells. As DSBs generated as a consequence of replication fork collapse have been shown to require RAD52 for repair (Lambert et al, 2005), we constructed strains that carry a deletion in the RAD52 gene to increase the half-life of DSBs. These strains have similar S-phase dynamics as their RAD52 counterparts (Supplementary Figure S2B). We analyzed four CEOs that were compromised in both mec1-1 and rad53-1 mutants (Table I and Figure 8A). DNA of asynchronous RAD52 and rad52Δ strains that were either S-phase checkpoint proficient or carried mutations in MEC1 and RAD53, respectively, was isolated and linkers were ligated to all free DNA ends. A two-step linker-mediated (LM)PCR protocol was employed to detect DSBs (Figure 8A). We detected LMPCR products ranging from 0.2 kb upwards in repair-deficient mec1-1 and rad53-1 mutants at regions flanking the CEOs ARS310, ARS315, ARS1305 and ARS606 (Figure 8A). For ARS310, we even detected DSBs in repair-proficient S-phase checkpoint mutants, albeit at a lower frequency than in the corresponding rad52Δ strains. No breaks were detected at the uncompromised early origins ARS1, ARS121 and ARS305 (Figure 8A). To ensure that this did not stem from the use of inefficient primer combinations, we cut the DNA of all six strains with suitable restriction enzymes ∼1 kb downstream of the respective origins and performed the same LMPCR experiment. Similar amounts of PCR products were obtained in all samples regardless of the locus that was investigated (Figure 8A). In addition, DSB formation was clearly dependent on replication, as deletion of ARS315 alleviated breakage at this locus (Figure 8B, upper panel), whereas ARS310 remained unaffected (Figure 8B, lower panel). Placing ARS315 into the URA3 locus did not trigger DSBs (Figure 8C, upper panel), consistent with the normal fork progression rates we measured by ChIP. Again, the pattern at ARS310 did not change (Figure 8C, lower panel).
Figure 8.
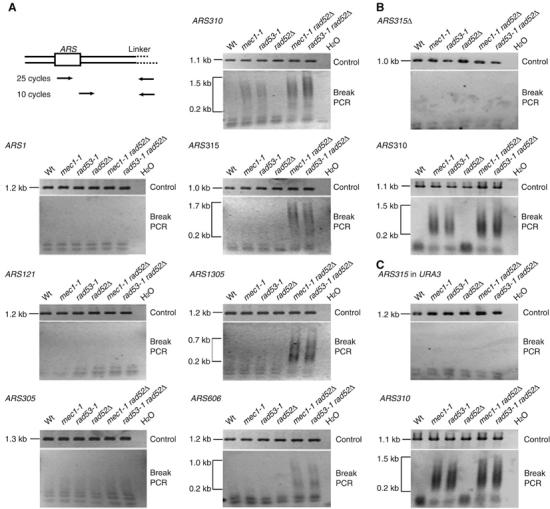
Chromosome fragility adjacent to CEOs in the absence of HU. (A) A two-step LMPCR method was used to detect DSBs near replication origins in asynchronous wild-type, mec1-1 and rad53-1 cells in the presence and absence of Rad52. DNA was isolated and a linker was ligated to all double-stranded DNA ends. DNA was amplified first by a 25-cycle PCR reaction with one origin- and one linker-specific primer, followed by a nested 10-cycle PCR reaction using a second origin-specific and the same linker-specific primer. PCR fragments were fractionated on agarose gels. As a control, breaks were introduced at a specific restriction site ∼1 kb downstream of the origin, and the same LMPCR was performed. Results are shown for early origins ARS305, ARS121 (proARS1021) and ARS1 (proARS416), and for CEOs ARS310, ARS1305, ARS315 and ARS606. (B) LMPCR analysis of DSBs adjacent to the CEO ARS315 in asynchronous wild-type, mec1-1 and rad53-1 cells (in the presence and absence of Rad52), containing a deletion of ARS315. As a control, DSBs were detected at the CEO ARS310 in these strains. (C) LMPCR analysis of DSBs in asynchronous wild-type, mec1-1 and rad53-1 cells (in the presence and absence of Rad52), containing an extra copy of the CEO ARS315 integrated into the URA3 locus. LMPCR analysis of ARS310 was conducted in the same strains as a positive control.
We then asked whether DSBs were introduced in S phase. To this end, we arrested cells in G1 and released them into 0.2 M HU for 80 min. Whereas we observed DSBs in S-phase cells, we did not observe any significant breakage in G1 (Supplementary Figure S6), arguing that breakage occurs during DNA replication. Moreover, we detected breaks regardless of whether cells carried a deletion of the SML1 gene or not (Supplementary Figure S6C). Taken together, all of these results argued that CEO-flanking regions displayed intrinsic chromosome fragility in S-phase checkpoint mutants, and that this fragility was dependent on DNA replication. Importantly, however, it was not triggered by nucleotide shortage.
Discussion
In this study, we have utilized a customized DNA microarray to analyze origin activation in S-phase checkpoint mutants of budding yeast. In the course of our experiments, we made two important observations: first, the origin activation profiles of mec1-1 and rad53-1 mutants partially overlapped, but also showed some distinct differences. Second, our analysis revealed fragile sites that are characterized by their high propensity of replication fork stalling and subsequent chromosome breakage in the absence of Mec1 and/or Rad53. Seven out of 17 loci that we have shown or predicted to be fragile sites are common between mec1-1 and rad53-1 mutants, whereas eight out of the remaining 10 were only detected in mec1-1 and two in rad53-1 mutants. These results might reflect the different roles proposed for Mec1 and Rad53 in stabilizing replication forks (Cobb et al, 2003; Lucca et al, 2004) and may also account for the differences in GCR frequencies between these mutants (Myung et al, 2001).
Usefulness of a customized DNA microarray for the study of DNA replication
Compared to high-density DNA microarrays, which represent the entire genome of budding yeast, our replication origin microarray covers only a small fraction (∼600 sites) of the yeast genome. Although this considerably simplifies data analysis, it was important to demonstrate the validity of this array. To this end, we performed a copy number change experiment with DNA isolated from HU-treated wild-type cells. Origins that are activated in the presence of HU are generally defined as early firing. We observed 180 early-firing origins in DCY1671 cells. This result is in good agreement with previous studies that estimated the number of early-firing origins to be 143/260 (Yabuki et al, 2002), 172/332 (Raghuraman et al, 2001) and 190 (Lengronne et al, 2001) either by microarray or DNA combing experiments. Furthermore, 127 out of our 180 peaks overlap with the 172 early peaks found in an independent genome-wide study (Raghuraman et al, 2001). These comparisons and our ability to monitor changes in the activation pattern when cells are deficient in responding to replication stress (Figure 2 and Supplementary Figure S4) demonstrate the usefulness of our array for the global analysis of replication origin activation in S. cerevisiae.
Overlapping and distinct roles for Mec1 and Rad53 in origin activation
Our analysis revealed that Mec1 and Rad53 share a subset of early- and late-replicating origins that they control in the presence of HU. However, a considerable number of origins appear to be exclusively regulated by Mec1 or Rad53. Given that Rad53 is not the only downstream target of Mec1, and that Rad53 appears to have Mec1-independent functions (Dohrmann et al, 1999; Gunjan and Verreault, 2003), this result is perhaps not so surprising. Whereas Mec1 phosphorylates Chk1 in response to DNA damage (Sanchez et al, 1997), Rad53 is the best-characterized target of Mec1 in response to replication stress (Sanchez et al, 1996). Besides Rad53, a second protein, Pds1/securin, is activated in a Mec1-dependent manner in late S phase to prevent spindle elongation and loss of sister chromatid cohesion (Clarke, 2003). Whether Pds1 has some capacity to regulate origin firing in early S phase in the absence of Rad53 remains to be seen. Of course, it is also possible that a second, as of yet, unidentified pathway exists that blocks origin firing in a Mec1-dependent, but Rad53-independent manner.
Interestingly, we found that significantly more late-firing origins were activated in rad53-1 than in mec1-1 mutants (Figure 4C). We do not believe that this is caused by different dNTP levels in these mutants, as mec1-1 cells that carry an SML1 deletion activate fewer origins than rad53-1 mutants. We favor the possibility that another checkpoint kinase, Tel1, the yeast homolog of human ATM (ataxia-telangiectasia-mutated), partially substitutes for the loss of Mec1 and causes activation of Rad53 in mec1-1 mutants. Recent precedence exists in support of this hypothesis (Clerici et al, 2004). We speculate that Tel1 might partially activate Rad53 in HU-treated mec1-1. In contrast, rad53-1 mutants largely lack the capacity to block origin firing in late S phase and would therefore display a larger number of precociously activated late origins, which is what we have observed in our experiments (Figure 4C).
CEO-flanking regions differ from RSZs
The most important finding of this study is the identification of fragile sites in S-phase checkpoint mutants that promote replication fork arrest. This is particularly well illustrated with the example of ARS310 (Figure 5B and E). Our results point to the fact that replication is initiated at ARS310 but nascent strands fail to be elongated, thus accumulating as small fragments. The accumulation of small fragments accounts for why we did not observe a significant hybridization signal on our microarrays. Short fragments hybridize with little efficiency to our array (Supplementary Figure S7). We believe that the larger replication intermediates that are present at CEOs are not sufficiently abundant to create significant signals.
As the fragile sites that we identified are not randomly distributed, they may share common characteristics that render them intrinsically difficult to replicate. This is different from the breakage that occurs at genetically programmed RSZs. RSZs coincide with replication termination regions in which elongating forks converge and are replicated late during S phase (Cha and Kleckner, 2002). In the absence of Mec1, RSZs are prone to chromosome breakage; however, the deletion of the ribonuclease reductase inhibitor Sml1 reverses this fragility (Cha and Kleckner, 2002). These results suggest that RSZs are especially sensitive to low dNTP levels. In contrast, we have demonstrated that CEO-flanking regions elicit DSBs in the absence of HU in mec1-1 sml1Δ and rad53-1 sml1Δ cells (Figure 8A and Supplementary Figure S6). Our conclusions, therefore, are that Mec1 and Rad53 regulate fork progression not only in late-replicating RSZs (Cha and Kleckner, 2002), but also in early-replicating regions of the genome.
DNA determinants of chromosome fragility
One of the CEOs that we have analyzed in detail in this study is ARS310. Importantly, ARS310 has recently been associated with chromosome fragility in yeast. Specifically, Lemoine et al (2005) have mapped a fragile site directly adjacent to ARS310 in a yeast strain that carried Ty retrotransposon insertions nearby and expressed DNA polymerase (pol)-α at very low levels. The authors demonstrated that chromosome breakage in this strain was dependent on the presence of two Ty elements in head to head orientation. They further hypothesized that these Ty elements could give rise to either hairpins or cruciform structures when replication forks uncouple leading from lagging strand synthesis because of insufficient amounts of pol-α. It remains to be seen whether Ty elements are necessary to induce fragility at ARS310 or other CEOs in S-phase checkpoint mutants; however, our strain, which carries a duplication of ARS310 has multiple Ty elements in the origin flanking regions (M Raveendranathan and A-K Bielinsky, unpublished observation). Ty elements have been shown to integrate preferably into tRNA genes (Kim et al, 1998). Under the assumption that the position of Ty elements and tRNA genes in the sequenced strain S288C may be similar to our strain, we compared CEOs to all other origins that were activated in S-phase checkpoint mutants and found that the frequency of tRNA genes and/or Ty elements within a distance of 10 kb was significantly higher in CEOs (71 versus 34%; Supplementary Tables V and VI). We speculate that these elements may be a determinant of some fragile sites, but are likely not the only factor contributing to chromosome fragility as other CEOs (e.g. ARS315) do not contain Ty elements nor tRNA genes in their immediate vicinity. However, ARS315 is flanked by repetitive elements that may affect chromosome fragility (data not shown).
As tRNA genes are known to impose replication fork pausing when the orientation of transcription opposes the direction of replication (Deshpande and Newlon, 1996; Wang et al, 2001b; Ivessa et al, 2003), it is reasonable to ask whether fragile sites correspond to natural replication pause sites. If this were the case, then we would expect that stable fork pausing is dependent on the S-phase checkpoint. This does not seem to be true, at least in budding yeast (Calzada et al, 2005; Szyjka et al, 2005). Furthermore, because there are an estimated 1400 natural pause sites in the yeast genome (Deshpande and Newlon, 1996; Wang et al, 2001b; Ivessa et al, 2003), one would expect to see a significant delay in replication elongation in rad53 mutants when DNA synthesis is measured across the entire genome. However, this is not the case (Versini et al, 2003). Although these considerations argue that natural pause sites are likely not the cause of replication fork stalling in S-phase checkpoint mutants, it is important to mention that tRNA clusters have been implicated in chromosome fragility (Admire et al, 2006).
At this point, we favor a model in which sequence characteristics of fragile sites, possibly caused by Ty insertions or other repetitive elements, trigger secondary structure formation during DNA unwinding that can interfere with replication fork assembly (very close to the origin) and/or replication fork stability.
Materials and methods
Replication origin arrays
The replication origin array used in this study was designed in our laboratory. A detailed description is available in Supplementary data.
Yeast strains and culture
All strains are derivatives of BF264-15DU MATa ade1 his2 leu2-3, 112 trp1-1aura3 Δns (Richardson et al, 1989). Details are described in Supplementary data.
FACS analysis and fluorescence microscopy
Fluorescence microscopy and flow cytometry were carried out as described (Clarke et al, 2001). For details, see Supplementary data.
DNA isolation and labeling of DNA with fluorescent nucleotides
DNA isolation and labeling procedures are described in Supplementary data.
Array hybridization and data analysis
We followed the procedures as previously described (Pollack et al, 1999). A detailed protocol is provided in Supplementary data.
Alkaline gel electrophoresis of replicating DNA
Replication intermediates from wild-type and rad53-1 cells were analyzed using alkaline agarose gel electrophoresis as described (Santocanale and Diffley, 1998). For further details, see Supplementary data.
Neutral/neutral 2D agarose gel electrophoresis
Replication intermediates were analyzed using neutral/neutral 2D agarose gel electrophoresis as described (Friedman and Brewer, 1995; Wu and Gilbert, 1995; Tourriere et al, 2005).
Chromatin immunoprecipitations
ChIP was performed as described (Ricke and Bielinsky, 2004). DNA fragments were on average 500 bp in size. Primer sequences are available upon request.
LMPCR analysis of DNA breaks
The presence of DNA breaks adjacent to replication origins was tested using a two-step LMPCR method. Details are described in Supplementary data.
Supplementary Material
Supplementary Figure S1
Supplementary Figure S2
Supplementary Figure S3
Supplementary Figure S4
Supplementary Figure S5
Supplementary Figure S6
Supplementary Figure S7
Supplementary Data
Acknowledgments
We thank Dr J Berman for Arabidopsis clones, A Krieger and M Abdihalim for help with PCR, Dr E Bensen for help with microarray protocols, Dr B Calvi for advice on LMPCR, A Deshpande for printing arrays, Dr A Khodursky for advice on statistics, Dr K Kitada for data files, Dr P Pasero for protocols and R Ahmed for computational analysis. We are also indebted to the staff at the Advanced Genetic Analysis Center, the Biomedical Image Processing Lab and the Flow Cytometry Facility of the University of Minnesota. We especially thank Dr Y Haoyu at the Supercomputing Institute for advice on normalization procedures, Drs R Cha and C Pearson for discussions on fragile sites and Drs EA Hendrickson and D Livingston as well as Bielinsky lab members for critical reading of the manuscript. This work was supported in part by a Grant-in-Aid of the University of Minnesota (AKB), by an ACS grant RSG0216601 (AKB) and by NIH grant CA099033 (DJC).
References
- Admire A, Shanks L, Danzl N, Wang M, Weier U, Stevens W, Hunt E, Weinert T (2006) Cycles of chromosome instability are associated with a fragile site and are increased by defects in DNA replication and checkpoint controls in yeast. Genes Dev 20: 159–173 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aparicio JG, Viggiani CJ, Gibson DG, Aparicio OM (2004) The Rpd3-Sin3 histone deacetylase regulates replication timing and enables intra-S origin control in Saccharomyces cerevisiae. Mol Cell Biol 24: 4769–4780 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arlt MF, Casper AM, Glover TW (2003) Common fragile sites. Cytogenet Genome Res 100: 92–100 [DOI] [PubMed] [Google Scholar]
- Calzada A, Hodgson B, Kanemaki M, Bueno A, Labib K (2005) Molecular anatomy and regulation of a stable replisome at a paused eukaryotic DNA replication fork. Genes Dev 19: 1905–1919 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Casper AM, Durkin SG, Arlt MF, Glover TW (2004) Chromosomal instability at common fragile sites in Seckel syndrome. Am J Hum Genet 75: 654–660 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Casper AM, Nghiem P, Arlt MF, Glover TW (2002) ATR regulates fragile site stability. Cell 111: 779–789 [DOI] [PubMed] [Google Scholar]
- Cha RS, Kleckner N (2002) ATR homolog Mec1 promotes fork progression, thus averting breaks in replication slow zones. Science 297: 602–606 [DOI] [PubMed] [Google Scholar]
- Clarke DJ (2003) Establishment of dependence relationships between genome replication and mitosis. J Cell Biochem 88: 95–103 [DOI] [PubMed] [Google Scholar]
- Clarke DJ, Segal M, Jensen S, Reed SI (2001) Mec1p regulates Pds1p levels in S phase: complex coordination of DNA replication and mitosis. Nat Cell Biol 3: 619–627 [DOI] [PubMed] [Google Scholar]
- Clarke DJ, Segal M, Mondesert G, Reed SI (1999) The Pds1 anaphase inhibitor and Mec1 kinase define distinct checkpoints coupling S phase with mitosis in budding yeast. Curr Biol 9: 365–368 [DOI] [PubMed] [Google Scholar]
- Clerici M, Baldo V, Mantiero D, Lottersberger F, Lucchini G, Longhese MP (2004) A Tel1/MRX-dependent checkpoint inhibits the metaphase-to-anaphase transition after UV irradiation in the absence of Mec1. Mol Cell Biol 24: 10126–10144 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cobb JA, Bjergbaek L, Shimada K, Frei C, Gasser SM (2003) DNA polymerase stabilization at stalled replication forks requires Mec1 and the RecQ helicase Sgs1. EMBO J 22: 4325–4336 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cobb JA, Schleker T, Rojas V, Bjergbaek L, Tercero JA, Gasser SM (2005) Replisome instability, fork collapse, and gross chromosomal rearrangements arise synergistically from Mec1 kinase and RecQ helicase mutations. Genes Dev 19: 3055–3069 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Costanzo V, Shechter D, Lupardus PJ, Cimprich KA, Gottesman M, Gautier J (2003) An ATR- and Cdc7-dependent DNA damage checkpoint that inhibits initiation of DNA replication. Mol Cell 11: 203–213 [DOI] [PubMed] [Google Scholar]
- Desany BA, Alcasabas AA, Bachant JB, Elledge SJ (1998) Recovery from DNA replicational stress is the essential function of the S-phase checkpoint pathway. Genes Dev 12: 2956–2970 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deshpande AM, Newlon CS (1996) DNA replication fork pause sites dependent on transcription. Science 272: 1030–1033 [DOI] [PubMed] [Google Scholar]
- Diffley JF, Labib K (2002) The chromosome replication cycle. J Cell Sci 115: 869–872 [DOI] [PubMed] [Google Scholar]
- Dohrmann PR, Oshiro G, Tecklenburg M, Sclafani RA (1999) RAD53 regulates DBF4 independently of checkpoint function in Saccharomyces cerevisiae. Genetics 151: 965–977 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Donaldson AD, Blow JJ (1999) The regulation of replication origin activation. Curr Opin Genet Dev 9: 62–68 [DOI] [PubMed] [Google Scholar]
- Donaldson AD, Raghuraman MK, Friedman KL, Cross FR, Brewer BJ, Fangman WL (1998) CLB5-dependent activation of late replication origins in S. cerevisiae. Mol Cell 2: 173–182 [DOI] [PubMed] [Google Scholar]
- Duncker BP, Shimada K, Tsai-Pflugfelder M, Pasero P, Gasser SM (2002) An N-terminal domain of Dbf4p mediates interaction with both origin recognition complex (ORC) and Rad53p and can deregulate late origin firing. Proc Natl Acad Sci USA 99: 16087–16092 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feng W, Collingwood D, Boeck ME, Fox LA, Alvino GM, Fangman WL, Raghuraman MK, Brewer BJ (2006) Genomic mapping of single-stranded DNA in hydroxyurea-challenged yeasts identifies origins of replication. Nat Cell Biol 8: 148–155 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Friedman KL, Brewer BJ (1995) Analysis of replication intermediates by two-dimensional agarose gel electrophoresis. Methods Enzymol 262: 613–627 [DOI] [PubMed] [Google Scholar]
- Gibson DG, Aparicio JG, Hu F, Aparicio OM (2004) Diminished S-phase cyclin-dependent kinase function elicits vital Rad53-dependent checkpoint responses in Saccharomyces cerevisiae. Mol Cell Biol 24: 10208–10222 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gunjan A, Verreault A (2003) A Rad53 kinase-dependent surveillance mechanism that regulates histone protein levels in S. cerevisiae. Cell 115: 537–549 [DOI] [PubMed] [Google Scholar]
- Ivessa AS, Lenzmeier BA, Bessler JB, Goudsouzian LK, Schnakenberg SL, Zakian VA (2003) The Saccharomyces cerevisiae helicase Rrm3p facilitates replication past nonhistone protein–DNA complexes. Mol Cell 12: 1525–1536 [DOI] [PubMed] [Google Scholar]
- Katou Y, Kanoh Y, Bando M, Noguchi H, Tanaka H, Ashikari T, Sugimoto K, Shirahige K (2003) S-phase checkpoint proteins Tof1 and Mrc1 form a stable replication-pausing complex. Nature 424: 1078–1083 [DOI] [PubMed] [Google Scholar]
- Kihara M, Nakai W, Asano S, Suzuki A, Kitada K, Kawasaki Y, Johnston LH, Sugino A (2000) Characterization of the yeast Cdc7p/Dbf4p complex purified from insect cells. Its protein kinase activity is regulated by Rad53p. J Biol Chem 275: 35051–35062 [DOI] [PubMed] [Google Scholar]
- Kim JM, Vanguri S, Boeke JD, Gabriel A, Voytas DF (1998) Transposable elements and genome organization: a comprehensive survey of retrotransposons revealed by the complete Saccharomyces cerevisiae genome sequence. Genome Res 8: 464–478 [DOI] [PubMed] [Google Scholar]
- Krishnan V, Nirantar S, Crasta K, Cheng AY, Surana U (2004) DNA replication checkpoint prevents precocious chromosome segregation by regulating spindle behavior. Mol Cell 16: 687–700 [DOI] [PubMed] [Google Scholar]
- Lambert S, Watson A, Sheedy DM, Martin B, Carr AM (2005) Gross chromosomal rearrangements and elevated recombination at an inducible site-specific replication fork barrier. Cell 121: 689–702 [DOI] [PubMed] [Google Scholar]
- Lemoine FJ, Degtyareva NP, Lobachev K, Petes TD (2005) Chromosomal translocations in yeast induced by low levels of DNA polymerase a model for chromosome fragile sites. Cell 120: 587–598 [DOI] [PubMed] [Google Scholar]
- Lengronne A, Pasero P, Bensimon A, Schwob E (2001) Monitoring S phase progression globally and locally using BrdU incorporation in TK(+) yeast strains. Nucleic Acids Res 29: 1433–1442 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Longhese MP, Clerici M, Lucchini G (2003) The S-phase checkpoint and its regulation in Saccharomyces cerevisiae. Mutat Res 532: 41–58 [DOI] [PubMed] [Google Scholar]
- Lopes M, Cotta-Ramusino C, Pellicioli A, Liberi G, Plevani P, Muzi-Falconi M, Newlon CS, Foiani M (2001) The DNA replication checkpoint response stabilizes stalled replication forks. Nature 412: 557–561 [DOI] [PubMed] [Google Scholar]
- Lucca C, Vanoli F, Cotta-Ramusino C, Pellicioli A, Liberi G, Haber J, Foiani M (2004) Checkpoint-mediated control of replisome–fork association and signalling in response to replication pausing. Oncogene 23: 1206–1213 [DOI] [PubMed] [Google Scholar]
- Myung K, Datta A, Kolodner RD (2001) Suppression of spontaneous chromosomal rearrangements by S phase checkpoint functions in Saccharomyces cerevisiae. Cell 104: 397–408 [DOI] [PubMed] [Google Scholar]
- Pollack JR, Perou CM, Alizadeh AA, Eisen MB, Pergamenschikov A, Williams CF, Jeffrey SS, Botstein D, Brown PO (1999) Genome-wide analysis of DNA copy-number changes using cDNA microarrays. Nat Genet 23: 41–46 [DOI] [PubMed] [Google Scholar]
- Raghuraman MK, Winzeler EA, Collingwood D, Hunt S, Wodicka L, Conway A, Lockhart DJ, Davis RW, Brewer BJ, Fangman WL (2001) Replication dynamics of the yeast genome. Science 294: 115–121 [DOI] [PubMed] [Google Scholar]
- Richardson HE, Wittenberg C, Cross F, Reed SI (1989) An essential G1 function for cyclin-like proteins in yeast. Cell 59: 1127–1133 [DOI] [PubMed] [Google Scholar]
- Ricke RM, Bielinsky AK (2004) Mcm10 regulates the stability and chromatin association of DNA polymerase-alpha. Mol Cell 16: 173–185 [DOI] [PubMed] [Google Scholar]
- Sanchez Y, Desany BA, Jones WJ, Liu Q, Wang B, Elledge SJ (1996) Regulation of RAD53 by the ATM-like kinases MEC1 and TEL1 in yeast cell cycle checkpoint pathways. Science 271: 357–360 [DOI] [PubMed] [Google Scholar]
- Sanchez Y, Wong C, Thoma RS, Richman R, Wu Z, Piwnica-Worms H, Elledge SJ (1997) Conservation of the Chk1 checkpoint pathway in mammals: linkage of DNA damage to Cdk regulation through Cdc25. Science 277: 1497–1501 [DOI] [PubMed] [Google Scholar]
- Santocanale C, Diffley JF (1998) A Mec1- and Rad53-dependent checkpoint controls late-firing origins of DNA replication. Nature 395: 615–618 [DOI] [PubMed] [Google Scholar]
- Santocanale C, Sharma K, Diffley JF (1999) Activation of dormant origins of DNA replication in budding yeast. Genes Dev 13: 2360–2364 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shechter D, Gautier J (2005) ATM and ATR check in on origins: a dynamic model for origin selection and activation. Cell Cycle 4: 235–238 [PubMed] [Google Scholar]
- Shimada K, Pasero P, Gasser SM (2002) ORC and the intra-S-phase checkpoint: a threshold regulates Rad53p activation in S phase. Genes Dev 16: 3236–3252 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shirahige K, Hori Y, Shiraishi K, Yamashita M, Takahashi K, Obuse C, Tsurimoto T, Yoshikawa H (1998) Regulation of DNA-replication origins during cell-cycle progression. Nature 395: 618–621 [DOI] [PubMed] [Google Scholar]
- Sogo JM, Lopes M, Foiani M (2002) Fork reversal and ssDNA accumulation at stalled replication forks owing to checkpoint defects. Science 297: 599–602 [DOI] [PubMed] [Google Scholar]
- Szyjka SJ, Viggiani CJ, Aparicio OM (2005) Mrc1 is required for normal progression of replication forks throughout chromatin in S. cerevisiae. Mol Cell 19: 691–697 [DOI] [PubMed] [Google Scholar]
- Tercero JA, Longhese MP, Diffley JF (2003) A central role for DNA replication forks in checkpoint activation and response. Mol Cell 11: 1323–1336 [DOI] [PubMed] [Google Scholar]
- Tourriere H, Versini G, Cordon-Preciado V, Alabert C, Pasero P (2005) Mrc1 and Tof1 promote replication fork progression and recovery independently of Rad53. Mol Cell 19: 699–706 [DOI] [PubMed] [Google Scholar]
- Versini G, Comet I, Wu M, Hoopes L, Schwob E, Pasero P (2003) The yeast Sgs1 helicase is differentially required for genomic and ribosomal DNA replication. EMBO J 22: 1939–1949 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Beerman TA, Kowalski D (2001a) Antitumor drug adozelesin differentially affects active and silent origins of DNA replication in yeast checkpoint kinase mutants. Cancer Res 61: 3787–3794 [PubMed] [Google Scholar]
- Wang Y, Vujcic M, Kowalski D (2001b) DNA replication forks pause at silent origins near the HML locus in budding yeast. Mol Cell Biol 21: 4938–4948 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weinert TA, Kiser GL, Hartwell LH (1994) Mitotic checkpoint genes in budding yeast and the dependence of mitosis on DNA replication and repair. Genes Dev 8: 652–665 [DOI] [PubMed] [Google Scholar]
- Wu JR, Gilbert DM (1995) Rapid DNA preparation for 2D gel analysis of replication intermediates. Nucleic Acids Res 23: 3997–3998 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wyrick JJ, Aparicio JG, Chen T, Barnett JD, Jennings EG, Young RA, Bell SP, Aparicio OM (2001) Genome-wide distribution of ORC and MCM proteins in S. cerevisiae: high-resolution mapping of replication origins. Science 294: 2357–2360 [DOI] [PubMed] [Google Scholar]
- Yabuki N, Terashima H, Kitada K (2002) Mapping of early firing origins on a replication profile of budding yeast. Genes Cells 7: 781–789 [DOI] [PubMed] [Google Scholar]
- Yan ZA, Li XZ, Zhou XT (1987) The effect of hydroxyurea on the expression of the common fragile site at 3p14. J Med Genet 24: 593–596 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Figure S1
Supplementary Figure S2
Supplementary Figure S3
Supplementary Figure S4
Supplementary Figure S5
Supplementary Figure S6
Supplementary Figure S7
Supplementary Data