

Abstract
Urea cycle disorders are a group of inborn errors of hepatic metabolism that result in often life-threatening hyperammonemia and hyperglutaminemia. Clinical and laboratory diagnosis of partial deficiencies during asymptomatic periods is difficult, and correlation of phenotypic severity with either genotype and/or in vitro enzyme activity is often imprecise. We hypothesized that stable isotopically determined in vivo rates of total body urea synthesis and urea cycle-specific nitrogen flux would correlate with both phenotypic severity and carrier status in patients with a variety of different enzymatic deficiencies of the urea cycle. We studied control subjects, patients, and their relatives with different enzymatic deficiencies affecting the urea cycle while consuming a low protein diet. On a separate occasion the subjects either received a higher protein intake or were treated with an alternative route medication sodium phenylacetate/benzoate (Ucephan), or oral arginine supplementation. Total urea synthesis from all nitrogen sources was determined from [18O]urea labeling, and the utilization of peripheral nitrogen was estimated from the relative isotopic enrichments of [15N]urea and [15N]glutamine during i.v. co-infusions of [5-(amide)15N]glutamine and [18O]urea. The ratio of the isotopic enrichments of 15N-urea/15N-glutamine distinguished normal control subjects (ratio = 0.42 ± 0.06) from urea cycle patients with late (0.17 ± 0.03) and neonatal (0.003 ± 0.007) presentations irrespective of enzymatic deficiency. This index of urea cycle activity also distinguished asymptomatic heterozygous carriers of argininosuccinate synthetase deficiency (0.22 ± 0.03), argininosuccinate lyase deficiency (0.35 ± 0.11), and partial ornithine transcarbamylase deficiency (0.26 ± 0.06) from normal controls. Administration of Ucephan lowered, and arginine increased, urea synthesis to the degree predicted from their respective rates of metabolism. The 15N-urea/15N-glutamine ratio is a sensitive index of in vivo urea cycle activity and correlates with clinical severity. Urea synthesis is altered by alternative route medications and arginine supplementation to the degree that is to be expected from theory. This stable isotope protocol provides a sensitive tool for evaluating the efficacy of therapeutic modalities and acts as an aid to the diagnosis and management of urea cycle patients.
Keywords: stable isotope, hyperammonemia, diagnosis, ornithine transcarbamylase, citrullinemia
The urea cycle consists of five sequential enzymatic conversions that incorporate two nitrogen atoms into urea (1). One nitrogen derives from ammonia and enters the cycle at the initial synthesis of carbamyl phosphate (catalyzed by carbamyl phosphate synthetase) whereas the other is derived from aspartate incorporated via argininosuccinate synthesis [catalyzed by argininosuccinate synthetase (ASS)]. In the long term, both of the urea nitrogen atoms derive from dietary protein, but the principal immediate sources are probably glutamine and alanine derived from peripheral amino acid catabolism, and ammonia derived from the intestinal first-pass metabolism of dietary protein (2, 3). Deficiencies of all of the respective enzymes have been identified, and, although each specific disorder results in the accumulation of different precursors, hyperammonemia is a common feature of most of these disorders. In affected individuals, morbidity and mortality are directly correlated with the duration and severity of hyperammonemic episodes (4, 5). Disorders in ASS, argininosuccinate lyase (ASL), and type-I arginase are inherited in autosomal recessive fashion whereas ornithine transcarbamylase (OTC) deficiency is an X-linked disorder. Hence, female OTC heterozygotes exhibit variable clinical severity depending on the pattern of X-inactivation in the liver (6–8).
Patients with urea cycle disorders present either in the neonatal period with massive hyperammonemia after initial protein intake or later in life with episodic hyperammonemic episodes that are often associated with intercurrent illness. Although the genetic loci for each of the conditions have been elucidated, individual genotypes may not be predictive of phenotypic severity. Moreover, DNA mutation analysis is currently only available for OTC deficiency (9, 10). In addition, other measures of metabolic status, such as the plasma amino acid profile and the presence of orotic aciduria or hyperammonemia, are relatively late indicators of metabolic derangement and therefore are inadequate for predicting phenotypic severity and prognosis. Indeed, in some cases, the basic metabolic derangement may not be initially recognized, and patients will present with recurrent episodes of altered mental status or behavioral abnormalities (11).
The management of these disorders is aimed at reducing hyperammonemic episodes by restriction of dietary protein intake, aggressive support during episodes of catabolic stress, and by treatment with sodium phenylacetate/benzoate (Ucephan) or sodium phenylbutyrate to stimulate the excretion of nitrogen as phenylacetylglutamine and hippuric acid (in the case of Ucephan) (12–14). Finally, citrulline or arginine are frequently given to fulfill the nutritional requirement for arginine and to provide precursors for enzymatic steps upstream of the putative block. The long term benefits of these treatments to IQ and survival have been demonstrated in a prospective study of OTC-deficient females (15). However, despite dietary and pharmacotherapy, patients continue to be at risk for recurrent hyperammonemia. Liver transplantation is an option but has its inherent long term morbidity (16, 17). Ultimately, gene replacement therapy may play a role in the long term treatment of these disorders.
The present work was undertaken to investigate whether a direct estimate of in vivo urea cycle activity might be of value in assessing these disorders. In so doing, we hypothesized that the measurement of total urea synthesis in combination with an independent estimate of the transfer of a specific nitrogen source to urea would be a direct physiologic correlate of residual urea cycle activity and, therefore, of phenotypic severity. Previous work by Yudkoff et al. has shown that the time course of 15N-urea enrichment after an oral dose of 15NH4Cl can distinguish symptomatic OTC-deficient females and one neonatal onset hemizygous male patient from normal controls (18). However, in this previous study, asymptomatic OTC-deficient females were not distinguishable from normal controls. We hypothesized that a more specific nitrogen source, in this case glutamine labeled with 15N in its amide group, which will preferentially donate labeled nitrogen via carbamyl phosphate synthesis, would provide a direct measure of the activity of the complete urea cycle and perhaps distinguish partial deficiencies, whether symptomatic or asymptomatic, from normal. To this end we measured (i) the rate of total body urea production by measuring blood 18O-urea isotopic enrichment during an infusion with the stable isotope 18O-urea and (ii) the proportional contribution of peripheral nitrogen to urea synthesis by measuring the incorporation of 15N from 5-15N-glutamine into 15N-urea.
Materials and Methods
The protocol received prior approval from the Institutional Review Boards for Human Subjects of Baylor College of Medicine and Texas Children's Hospital.
Study Subjects and Groups.
Twelve healthy adult control subjects (seven males, ages 19–39 years, and five females, ages 25–42 years) and six pediatric control subjects (five males, ages 6–15 years, and one female, age 14 years) were enrolled between 1995 and 1999. Twelve subjects with disorders affecting the urea cycle were also enrolled. This patient group comprised five OTC-deficient hemizygous males (one of neonatal presentation), five citrullinemic [argininosuccinate synthetase deficiency (ASSD)] individuals, and two patients with argininosuccinic aciduria (ASLD). We also investigated 10 OTC-deficient heterozygous females (2 symptomatic on pharmacotherapy, 8 asymptomatic and untreated), 5 obligate heterozygote ASSD, and 4 obligate heterozygote ASLD parents (Table 1).
Table 1.
Urea cycle patient subjects, clinical presentation, and molecular/enzymatic analyses
| Condition | Subject | Presentation* | Diagnostic studies† |
|---|---|---|---|
| OTC female | 1001 | Late onset, symptomatic | PAA, OA, Linkage |
| 1002 | Late onset, symptomatic | PAA, OA, DNA (IVS-1/codon 22 delG) | |
| 1003 | Late onset, asymptomatic | PAA, OA, Linkage | |
| 1004 | Late onset, asymptomatic | PAA, OA, Linkage | |
| 1005 | Late onset, asymptomatic | PAA, OA, Linkage | |
| 1006 | Late onset, asymptomatic | PAA, DNA (F354C) | |
| 1007 | Late onset, asymptomatic | PAA, DNA (F354C) | |
| 1008 | Late onset, asymptomatic | PAA, OA, Linkage | |
| 1009 | Late onset, asymptomatic | PAA, DNA (F354C) | |
| 1010 | Late onset, asymptomatic | PAA, DNA (F354C) | |
| OTC male | 1101 | Neonatal | PAA, OA |
| 1102 | Late onset | PAA, OA, Enz (220, control 3448)‡ | |
| 1103 | Late onset | PAA, OA, DNA (F354C) | |
| 1104 | Late onset | PAA, OA, DNA (F354C) | |
| 1105 | Late onset | PAA, OA, DNA (A336C) | |
| ASSD | 2001 | Neonatal | PAA, DNA (deletion exon 7), Enz (0, control 2.67)§ |
| 2002 | Neonatal | PAA, DNA (deletion exon 5), Enz (0, control 1.09)§ | |
| 2003 | Neonatal | PAA, DNA (deletion exon 5), | |
| 2004 | Neonatal | PAA | |
| 2005 | Late onset | PAA, Enz (0, control 3.36)§ | |
| ASLD | 3001 | Neonatal | PAA, Enz (0, control 11.9)§ |
| 3002 | Late onset | PAA |
Neonatal onset defined as symptomatic presentation in the first month of life, but invariably all neonatal patients presented in first week. Late onset is defined as second month and thereafter. Symptomatic refers to documented hyperammonemia requiring pharmacotherapy. Asymptomatic patients have not had documented hyperammonemia and do not require pharmacotherapy. However, these patients may exhibit signs of protein aversion.
PAA, plasma amino acid profile; OA, urine orotic acid; Linkage, linkage analysis to independently diagnosed proband; DNA, mutation analysis of the respective genetic loci; Enz, enzymatic analysis for enzyme deficiency in the Baylor Biochemical Genetics Laboratory.
Enzyme assay on liver (activity in nmol/g tissue).
Enzyme assay on primary fibroblasts (activity in nmol/h/mg).
All adult controls were initially studied on a low protein intake [0.4 g/(kg⋅day)]. Eleven of these subjects were also randomly assigned to one of three groups for a second study. During this second isotope study, they either received a higher protein intake [0.8 g/(kg⋅day), n = 4], a low protein diet together with Ucephan treatment [250 mg/(kg⋅day), n = 4), or a low protein diet with arginine treatment [500 mg/(kg⋅day), n = 3]. Pediatric controls (n = 4), asymptomatic OTCD females (n = 4), and heterozygote carriers of citrullinemia (n = 4) and argininosuccinic aciduria (n = 4) were studied on low and high protein intakes. Two untreated asymptomatic OTC-deficient females were studied first on low protein intakes and then on low protein intake with Ucephan.
Infusion Protocol.
[5-15N]glutamine (98% isotopically pure, Cambridge Isotopes, Andover, MA) and [18O]urea (86% enriched with 18O, Cambridge Isotopes) were diluted in 1/2N saline and were prepared for infusion by the Texas Children's Hospital Investigational Pharmacy. The subjects were admitted into the Texas Children's Hospital General Clinical Research Center and were started on the study protocol after physical examination and informed consent. Each subject was then started on the assigned level of protein intake (0.4 or 0.8 g/kg/day) and medication if indicated for a 2-day period of stabilization. Symptomatic urea cycle patients continued on their respective medical regimens in all studies. Protein intake was monitored by weighing portions before and after each meal. On the third day of study, after an overnight fast and after a preinfusion blood sampling for the determination of baseline labeling of urea and glutamine, the subject consumed the first of four twice-hourly meals that each supplied 1/12 of their prior daily protein intake. A priming dose of 5-15N-glutamine (2 mg/kg) and 18O-urea (1 mg/kg) was given over a 10-minute period via a peripheral i.v. catheter. Immediately after completion of the priming infusion, a constant infusion of 5-15N-glutamine (2 mg/kg/h) and 18O-urea (0.1 mg/kg/h) was initiated and continued for 8 h. Blood for plasma amino acid, ammonia, and isotopic analyses was obtained at 0 h preinfusion, and then at 4, 6, and 7.5 h during the infusion.
Metabolite Analyses.
Mass spectrometric measurements were made on a Hewlett–Packard 9880 GC-quadrupole mass spectrometer. Urea was analyzed as the 2-pyrimidinol-tertburyl, dimethylsilane derivative by using electron impact ionization after separation on a DB 1701 GC column (19). Ions with mass-to-charge ratios of 153, 154 (15N1-urea), and 155 (18O-urea) were monitored. Glutamine was measured as the n-propyl ester of the heptafluorobutyramide derivative by using negative methane chemical ionization after separation on a DB 25 column (20). We monitored ions with mass-to-charge ratios of 346 and 347. The excess enrichment of each isotopomer was calculated by using the ion distribution in urea and glutamine isolated from each subject's baseline sample. Standard curves for 15N-glutamine, 15N-urea, and 18O-urea were analyzed at enrichments that we expected to observe in patient samples.
Calculations and Statistical Analyses.
Total urea synthesis from all sources was calculated from the 18O-urea labeling with the standard equation
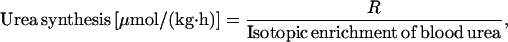 |
1 |
in which R is the rate of 18O-urea infusion (urea infusion × isotopic enrichment of infused urea). Urea synthesis from peripheral nitrogen sources was estimated as
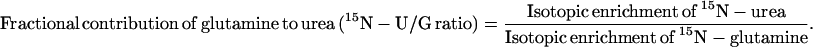 |
2 |
Total Incorporation of glutamine amide N into urea [μmol/(kg⋅h) = Eq. 1 × Eq. 2. Data are shown as mean ± 1 SD.
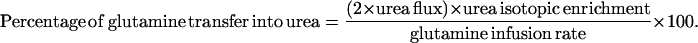 |
3 |
Different groups were initially compared with one way analysis of variance followed by post hoc two-tailed t tests. A P (two-tailed) of less 0.05 was taken as statistically significant. Within a group, treatment effects (e.g., effect of dietary protein) were assessed by paired t tests.
Results
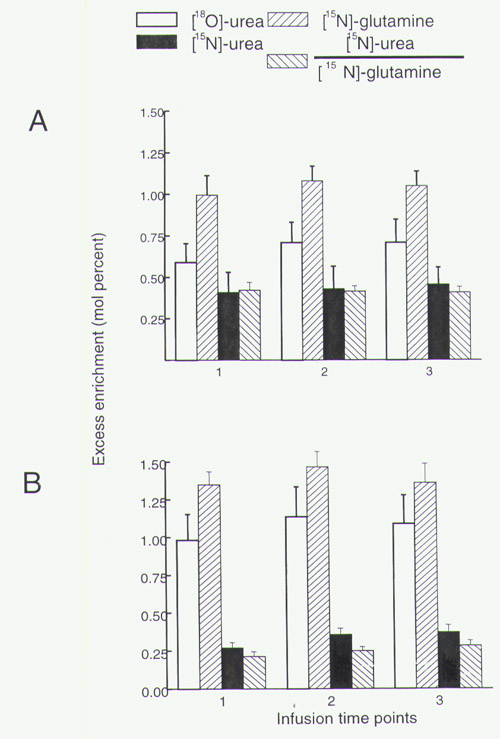
By 6 h of infusion the isotopic enrichment of both 18O- and 15N-urea had reached steady state (data not shown). The values for the isotopic enrichment of [15N]glutamine, urea, and their ratio [15N-urea/15N-glutamine (15N-U/G)] are shown in Fig. 1. Individual values are published as supplemental data on the PNAS web site at www.pnas.org.
Figure 1.

Isotopic enrichment and 15N-U/G ratio in subject groups on 0.4 g/kg/day protein intake. (Top) The respective steady state mole percent excess enrichment of blood [15N]glutamine is shown for each study group with the mean illustrated by bars. (Middle) The respective steady state mole percent excess enrichment of blood [15N]urea is shown for each study group. (Bottom) 15N-U/G ratio representing proportion of [15N]urea to [15N]glutamine labeling for the respective study groups.
Urea Synthesis in Control Subjects.
While consuming the low protein diet, adult and pediatric control subjects had similar total urea [132 ± 25 and 129 ± 12 μmol/(kg⋅h)] and glutamine fluxes [396 ± 74 and 349 ± 36 μmol/(kg⋅h)]. Because at this protein intake, the subjects would have been in negative nitrogen balance, the values for total urea synthesis exceeded the urea equivalent of the dietary protein intake [92 μmol/(kg⋅h)]. The proportion of total urea nitrogen directly derived from the amide-N of glutamine was slightly, but not significantly, lower in pediatric subjects as compared with adult controls (15N-U/G ratio 0.44 ± 0.07 vs. 0.37 ± 0.04). The mean value for both groups combined is listed in Table 2. Because only one of the two urea nitrogen atoms derives from carbamyl phosphate, this ratio would be expected to have a maximum value of 0.5. Table 2 also shows values for the percentage of the infused 15N-glutamine transferred to urea. This is calculated from the independent measures of total urea flux, urea 15N-enrichment, and the 15N-glutamine infusion rate (Eq. 3) and should be independent of the isotopic dilution of glutamine in the liver.
Table 2.
Total urea synthesis and the transfer of 15N from [5-15N]glutamine to urea in control subjects and patients with disorders of the urea cycle of varying severity
| Subject category | Urea flux, μmol/(kg⋅h) | Glutamine flux, μmol/(kg⋅h) | 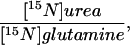 |
Transfer of glutamine to urea, percent dose |
|---|---|---|---|---|
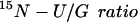 |
||||
| Healthy controls | 131 ± 21 | 380 ± 63 | 0.42 ± 0.06 | 27.9 ± 7.2 |
| Null patients | 82 ± 17‡ | 456 ± 103§ | 0.003 ± 0.007*** | −0.2 ± 1.2*** |
| Partial OTC males | 97 ± 45* | 454 ± 51 | 0.16 ± 0.04*** | 6.8 ± 2.4*** |
| Symptomatic OTC females | 108 ± 9 | 442 ± 68 | 0.18 ± 0.01*** | 9.3 ± 1.8*** |
| Asymptomatic OTC females | 99 ± 27* | 380 ± 89 | 0.26 ± 0.04***,† | 12.6 ± 2.8***,† |
| ASLD heterozygous carriers | 100 ± 15* | 373 ± 28 | 0.35 ± 0.11* | 13.7 ± 4.3*** |
| ASSD heterozygous carriers | 108 ± 36 | 282 ± 46 | 0.22 ± 0.03*** | 14.3 ± 8.4** |
Differences from control values are indicated; *, P < 0.05; **, P < 0.01; ***, P < 0.001. Differences from symptomatic OTC females: †, P < 0.01.
Reflects urea synthesis from therapeutic arginine or citrulline. When this is taken into account endogenous urea synthesis was −0.7 ± 16 μmol/(kg⋅h).
Reflects the effect of Ucephan on glutamine flux.
Urea Cycle Patients.
On the basis of age of diagnosis and symptoms, patients were initially classified either as having either null (neonatal onset, requiring medication) or partial (later onset, minimally symptomatic) urea cycle activity. Some of the null patients had previously been shown to have absent enzyme activity in liver biopsy or fibroblast assays (Table 1). Based on these criteria, one OTC male, four ASSD patients, and one ASLD patient were classified as null. In these subjects, the 15N-U/G ratio (0.003 ± 0.007) was not significantly different from zero, consistent with negligible conversion of glutamine to urea (Table 2). These subjects were, however, receiving arginine or citrulline therapy and hence had measurable urea synthesis [mean 82 ± 17 μmol/(kg⋅h)]. However, when the dose of amino acid was taken into account, their urea synthesis from endogenous nitrogen [−0.7 ± 16 μmol/(kg⋅h)] was not significantly different from zero.
The patients who were classified as partially affected included 10 OTC females (2 symptomatic and 8 asymptomatic), 4 late-onset OTC males, 1 citrullinemic female, and 1 ASLD male. Total urea synthesis in the OTC females [98 ± 20 μmol/(kg⋅h)] and late onset OTC males [96 ± 28 μmol/(kg⋅h)] as well as in the ASLD heterozygous subjects [100 ± 15 μmol/(kg⋅h)] was slightly but significantly lower than control values [131 ± 21 μmol/(kg⋅h)]. However, the transfer of glutamine amide nitrogen, as measured by the 15N-U/G ratio, revealed differences among these groups of subjects. Thus, in late onset OTC males and symptomatic OTC females, the ratio (0.16 ± 0.04 and 0.18 ± 0.01, respectively) was significantly lower than in asymptomatic OTC females (0.26 ± 0.04). It was of particular interest that heterozygous carriers of both ASSD (0.22 ± 0.03) and ASLD (0.35 ± 0.11) both exhibited significantly lower ratios than control subjects. Importantly, the percentage of the glutamine-15N converted to urea showed similar differences among the groups (Table 2). Collectively, these data suggest that the flux from 15N glutamine to 15N urea is a sensitive measure of urea cycle activity and is capable of differentiating severely affected, partially affected, and unaffected individuals (Table 2).
Effects of Protein Restriction and Medical Therapy.
Subgroups of eight controls (four adult and four pediatric subjects), four asymptomatic OTC females, and four heterozygous ASSD and ASLD subjects were studied on a higher protein intake [0.8 g/(kg⋅day)] (Table 3). In comparison to the study on low protein, total urea synthesis was highly significantly increased in all groups whereas the glutamine flux was unaltered. The increase in urea synthesis in the ASSD and ASLD heterozygotes [+57 ± 8 μmol/(kg⋅h)] was similar to that in control subjects [+64 ± 7 μmol/(kg⋅h)] and, in these three groups, the 15N-U/G ratio remained constant (mean difference −0.012 ± 0.036). However, in the OTC females, the increase in urea synthesis [+44 ± 5 μmol/(kg⋅h)] was significantly less, and there was a small but statistically significant fall in the 15N-U/G ratio (−0.057 ± 0.006) (Table 3).
Table 3.
The effect of a higher protein intake on urea synthesis and the transfer of 15N from [5-15N]glutamine to urea
| Subject category | Change in
|
||
|---|---|---|---|
| Urea flux, μmol/(kg⋅h) | Glutamine flux, μmol/(kg⋅h) | 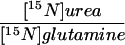 |
|
| Healthy controls | +64 ± 7 | +11 ± 26 | −0.027 ± 0.02 |
| ASSD heterozygous carriers | +58 ± 12 | +7 ± 80 | −0.017 ± 0.05 |
| ASLD heterozygous carriers | +56 ± 3 | +26 ± 16 | −0.029 ± 0.013 |
| Asymptomatic OTCD carriers | +44 ± 5* | −1 ± 5 | −0.057 ± 0.006* |
Significant difference from control values. *, P < 0.05.
Four control and two asymptomatic OTC females were treated with sodium phenylacetate/benzoate while ingesting the low protein diet. This treatment is expected to conjugate precursor glutamine and glycine in the nitrogen pool and stimulate alternative renal excretion of the conjugated products. The expectation is that nitrogen will be diverted away from urea synthesis. In all of these individuals, the measured decrease in urea flux caused by treatment with 250 mg/kg/day each of sodium phenylacetate/benzoate was 41 ± 9 μmol/kg/h. This was virtually identical to the expected decline in urea production (40 μmol/kg/h). Both groups exhibited a significant increase in glutamine flux [+136 ± 52 μmol/(kg⋅h)], but the 15N-U/G ratios was unchanged by this manipulation of nitrogen metabolism (0.45 ± 0.05 vs. 0.41 ± 0.08 for controls and 0.26 ± 0.04 vs. 0.25 ± 0.05 for OTC females with and without sodium phenylacetate/benzoate, respectively). Finally, we gave arginine at an hourly rate of 60 μmol/(kg⋅h) to three control patients. This treatment should increase total urea synthesis without affecting the use of glutamine. It is expected, therefore, that the 15N-U/G ratio should fall during arginine administration. Arginine treatment increased urea synthesis by 50 ± 3 μmol/(kg⋅h), and the 15N-U/G ratio fell highly significantly to 0.29 ± 0.08. These data show that the 15N-U/G ratio is relatively insensitive to treatment with sodium phenylacetate/benzoate and protein intake at the levels studied, but, as expected, treatment with arginine decreases the ratio. Importantly, the measured differences in urea production correlate well with the expected stoichiometric conversion of the drugs to their respective end products.
Discussion
We have made use of stable isotope precursor/product relationships to correlate in vivo urea cycle activity with clinical severity in null and partial activity urea cycle patients and control subjects. Total urea production correlated well with protein intake and, importantly, with the expected effects of pharmacotherapy (sodium phenylacetate/benzoate and arginine). Moreover, the contribution of glutamine amide-N to urea synthesis (as measured by the 15N-U/G ratio), and the proportion of the dose of glutamine transferred to urea, differentiated between control subjects and individuals affected to varying degrees by disorders of the urea cycle. Critically, this measure of urea synthesis proved to be relatively insensitive to small changes in protein intake or treatment with medications that stimulate alternative routes of nitrogen disposal (sodium phenylacetate/benzoate). It appears that the application of this specific measure of urea cycle activity will be a useful adjunct to the diagnosis and prospective management of these patients.
In some respects it is to be expected that some female carriers of OTCD might have abnormal urea nitrogen metabolism, but an unexpected finding of the present study was the ability of the 15N-U/G ratio to distinguish heterozygous carriers of two autosomal recessive conditions (ASS and ASL deficiencies) both from their affected offspring and from controls. Critically, these subjects had relatively normal absolute rates of urea synthesis so that they were deriving more urea from non-glutamine sources. At this stage, we hypothesize that OTCD-female carriers as well as ASS and ASL heterozygotes derive a higher than normal proportion of their urea from nitrogen metabolites generated by the first-pass metabolism of dietary protein. Such a proposition would be compatible with the clinical observation that these individuals are often clinically less sensitive to minimal variations in dietary protein vs. peripheral mobilization caused by catabolic stress.
Although cases of urea cycle disorders that present in the immediate neonatal period have characteristic amino acid abnormalities, late-onset cases with partial activity often have few laboratory alterations (6–8). In the case of OTC females, provocative testing, including protein loads and allopurinol challenge, has been used successfully to diagnose partial activity OTC patients (14, 21, 22). However, the specificity to OTC and utility of this approach in other urea cycle disorders is unclear (7). In principle, a direct measure of in vivo urea cycle activity should allow for delineation of different degrees of urea cycle deficiency. However, absolute measurements themselves may be subject to fluctuations in the metabolic status of subjects. For example, even on stable protein intakes, a slightly catabolic state would increase the flux through the nitrogen precursor pool. In developing the isotopic method, we argued that it was likely that physiological variations in urea synthesis would not alter the relative contributions of different nitrogen precursors, so that the use of a specific nitrogen tracer in combination with measurement of total urea synthesis from all nitrogen sources would be of use in assessing urea synthetic capacity. Indeed, in the present study, we found that the increase in urea synthesis associated with a higher protein diet was lower in asymptomatic OTCD carriers than in the other groups, including ASSD and ASLD carriers. Furthermore, in one severely affected OTCD female subject, we observed large differences in urea and glutamine flux during two serial studies, during one of which she was clearly in a catabolic state, as evidenced by significant weight loss. However, the 15N-U/G ratio was unchanged (0.174 vs. 0173), indicating that this measures the underlying metabolic phenotype, irrespective of the current clinical status of the patient.
Although glutamine flux as determined by excess 15N glutamine enrichment were not significantly different in each of the patient groups, the urea flux as determined by 15N urea enrichments were significantly different between controls and patient subjects. Moreover, the proportionate transfer of nitrogen from glutamine to urea (15N-U/G ratio) specifically differentiated null activity, partial activity patients, and normal controls, and does so even when the patients are asymptomatic. Thus, the method could be used directly for diagnosis of individuals who may be currently asymptomatic but whose history may be suggestive of a disorder of urea cycle metabolism. Furthermore, this indicator of urea cycle activity may allow for presymptomatic diagnosis in related family members, not only in the case of partial OTC deficiency, but also for heterozygous carriers of ASS or ASL deficiency.
Importantly, the in vivo measures of urea flux correlated directly with the expected decrease and increase, respectively, caused by the molar conversion of sodium phenylacetate/benzoate and arginine. In addition to supporting the basis for the use of these medications in these conditions, the method is a direct measure of putative therapeutic interventions on total urea production and the proportionate contribution of urea cycle specific production. This advantage will be especially relevant for evaluating outcomes of new therapeutic interventions such as gene replacement therapy. Finally, the metabolic status as it relates to nitrogen balance may be directly quantified by measures of glutamine and total body urea flux in the same patient at different times. As data such as these are collected for larger cohorts, management strategies for modifying dietary and pharmacotherapy based on these indicators of flux through the precursor nitrogen pool, the urea cycle, and the total body urea pool may be developed.
Supplementary Material
Acknowledgments
We thank J. Stuff, H. Jennings, and K. Lamance for their clinical support and R. Dawn for administrative support. The work was supported in part by federal funds from the U.S. Department of Agriculture Agricultural Research Service, Cooperative Agreement No. 58-6258-6001, by the National Institutes of Health (Grants DK54450 and DK02407), and by the Baylor College of Medicine General Clinical Research Center (RR00188), Mental Retardation Research Center, and Child Health Research Center. The contents of this publication do not necessarily reflect the views or policies of the U.S. Department of Agriculture, nor does mention of trade names, commercial products, or organizations imply endorsement by the U.S. Government.
Abbreviations
- ASS
argininosuccinate synthetase
- ASL
argininosuccinate lyase
- OTC
ornithine transcarbamylase
- ASLD
argininosuccinic aciduria
- ASSD
argininosuccinate synthetase deficiency
- 15N-U/G
15N-urea/15N-glutamine
Footnotes
This paper was submitted directly (Track II) to the PNAS office.
Article published online before print: Proc. Natl. Acad. Sci. USA, 10.1073/pnas.140082197.
Article and publication date are at www.pnas.org/cgi/doi/10.1073/pnas.140082197
References
- 1.Brusilow S W, Horwich A L. In: The Molecular and Metabolic Basis of Inherited Disease. Scriver C R, Beaudet A L, Sly W S, Valle D, editors. New York: McGraw–Hill; 1995. pp. 1187–1232. [Google Scholar]
- 2.Jahoor F, Jackson A A, Golden M H N. Ann Nutr Metab. 1988;32:240–244. doi: 10.1159/000177447. [DOI] [PubMed] [Google Scholar]
- 3.Cooper A J, Nieves E, Coleman A E, Filc-DeRicco S, Gelbard A S. J Biol Chem. 1987;262:1073–1080. [PubMed] [Google Scholar]
- 4.Batshaw M L, Roan Y, Jung A L, Rosenberg L A, Brusilow S W. N Engl J Med. 1980;302:482–485. doi: 10.1056/NEJM198002283020902. [DOI] [PubMed] [Google Scholar]
- 5.Msall M, Batshaw M L, Suss R, Brusilow S W, Mellits E D. N Engl J Med. 1984;310:1500–1505. doi: 10.1056/NEJM198406073102304. [DOI] [PubMed] [Google Scholar]
- 6.Maestri N E, Lord C, Glynn M, Bale A, Brusilow S W. Medicine (Baltimore) 1998;77:389–397. [PubMed] [Google Scholar]
- 7.Ahrens M J, Berry S A, Whitley C B, Markowitz D J, Plante R J, Tuchman M. Am J Med Genet. 1996;66:311–315. doi: 10.1002/(SICI)1096-8628(19961218)66:3<311::AID-AJMG14>3.0.CO;2-P. [DOI] [PubMed] [Google Scholar]
- 8.Rowe P C, Newman S L, Brusilow S W. N Engl J Med. 1986;314:541–547. doi: 10.1056/NEJM198602273140903. [DOI] [PubMed] [Google Scholar]
- 9.Tuchman M, Morizono H, Rajagopal B S, Plante R J, Allewell N M. J Inherited Metab Dis. 1997;20:525–527. doi: 10.1023/a:1005301513465. [DOI] [PubMed] [Google Scholar]
- 10.Tuchman M, Plante R J, Garcia-Perez M A, Rubio V. Hum Genet. 1996;97:274–276. doi: 10.1007/BF02185751. [DOI] [PubMed] [Google Scholar]
- 11.DiMagno E P, Lowe J E, Snodgrass P J, Jones J D. N Engl J Med. 1986;315:744–747. doi: 10.1056/NEJM198609183151207. [DOI] [PubMed] [Google Scholar]
- 12.Batshaw M L, Brusilow S, Waber L, Blom W, Brubakk A M, Burton B K, Cann H M, Kerr D, Mamunes P, Matalon R, et al. N Engl J Med. 1982;306:1387–1392. doi: 10.1056/NEJM198206103062303. [DOI] [PubMed] [Google Scholar]
- 13.Brusilow S W, Danney M, Waber L J, Batshaw M, Burton B, Levitsky L, Roth K, McKeethren C, Ward J. N Engl J Med. 1984;310:1630–1634. doi: 10.1056/NEJM198406213102503. [DOI] [PubMed] [Google Scholar]
- 14.Brusilow S W, Maestri N E. Adv Pediatr. 1996;43:127–170. [PubMed] [Google Scholar]
- 15.Maestri N E, Brusilow S W, Clissold D B, Bassett S S. N Engl J Med. 1996;335:855–859. doi: 10.1056/NEJM199609193351204. [DOI] [PubMed] [Google Scholar]
- 16.Todo S, Starzl T E, Tzakis A, Benkov K J, Kalousek F, Saheki T, Tanikawa K, Fenton W A. Hepatology. 1992;15:419–422. doi: 10.1002/hep.1840150311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Jan D, Poggi F, Jouvet P, Rabier D, Laurent J, Beringer A, Hubert P, Saudubray J M, Revillon Y. Transplantation Proc. 1994;26:189–190. [PubMed] [Google Scholar]
- 18.Yudkoff M, Daikhin Y, Nissim I, Jawad A, Wilson J, Batshaw M. J Clin Invest. 1996;98:2167–2173. doi: 10.1172/JCI119023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lee B, Dennis J A, Healy P J, Mull B, Pastore L, Yu H, Aguilar-Cordova E, O'Brien W, Reeds P, Beaudet A L. Proc Natl Acad Sci USA. 1999;96:3981–3986. doi: 10.1073/pnas.96.7.3981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Reeds P J, Burrin D G, Stoll B, Jahoor F, Wykes L, Henry J, Frazer M E. Am J Physiol. 1997;273:E408–E415. doi: 10.1152/ajpendo.1997.273.2.E408. [DOI] [PubMed] [Google Scholar]
- 21.Hauser E R, Finkelstein J E, Valle D, Brusilow S W. N Engl J Med. 1990;322:1641–1645. doi: 10.1056/NEJM199006073222305. [DOI] [PubMed] [Google Scholar]
- 22.Burlina A B, Ferrari V, Dionisi-Vici C, Bordugo A, Zacchello F, Tuchman M. J Inherited Metab Dis. 1992;15:707–712. doi: 10.1007/BF01800010. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}