

Abstract
We have used the combination of single-molecule Förster resonance energy transfer and kinetic synchrotron radiation circular dichroism experiments to probe the conformational ensemble of the collapsed unfolded state of the small cold shock protein CspTm under near-native conditions. This regime is physiologically most relevant but difficult to access experimentally, because the equilibrium signal in ensemble experiments is dominated by folded molecules. Here, we avoid this problem in two ways. One is the use of single-molecule Förster resonance energy transfer, which allows the separation of folded and unfolded subpopulations at equilibrium and provides information on long-range intramolecular distance distributions. From experiments with donor and acceptor chromophores placed at different positions within the chain, we find that the distance distributions in unfolded CspTm agree surprisingly well with a Gaussian chain not only at high concentrations of denaturant, where the polypeptide chain is expanded, but also at low denaturant concentrations, where the chain is collapsed. The second, complementary approach is synchrotron radiation circular dichroism spectroscopy of collapsed unfolded molecules transiently populated with a microfluidic device that enables rapid mixing. The results indicate a β-structure content of the collapsed unfolded state of ≈20% compared with the folded protein. This suggests that collapse can induce secondary structure in an unfolded state without interfering with long-range distance distributions characteristic of a random coil, which were previously found only for highly expanded unfolded proteins.
Keywords: Gaussian chain, microfluidic mixing, protein folding, random coil, secondary structure
With the discovery of small proteins that fold in the absence of populated intermediates (1), our quantitative understanding of the elementary properties of protein folding reactions has made significant advances, including the structural characterization of transition states for folding (2) and the prediction of folding rates from native structure (3–5). One of the most severe limitations for the further development of these approaches is our ignorance about the energetic or structural properties of unfolded†† states of proteins. Because of the structural heterogeneity and complexity of the ensembles of conformations populated by unfolded proteins, their experimental characterization has proven extremely difficult. Traditional methods, such as small-angle scattering techniques (6), provide only global physical properties, e.g., the radius of gyration. In some cases, more detailed structural information can be obtained from NMR (7–10), but these studies usually provide information about the denatured state only under nonnative conditions, typically in the presence of large concentrations of denaturant, or through severe destabilization of the native state induced by covalent modification or mutations. The most interesting and physiologically relevant situation, however, is that of an unfolded state of a stable protein under native conditions. Unfortunately, the great majority of molecules will then be present in their native conformation, thus overwhelming the signal from unfolded molecules.
We avoid this problem by using two complementary optical techniques: single-molecule fluorescence and kinetic synchrotron radiation circular dichroism (SRCD) spectroscopy. Single-molecule spectroscopy has the inherent ability to separate the signals from subpopulations in heterogeneous mixtures and equilibria, which makes it ideally suited to analyze protein folding reactions (11, 12). Specifically, by using single-molecule Förster resonance energy transfer (FRET), intramolecular distances of the unfolded state can be measured even in the presence of a majority of folded molecules (13, 14). Recently, the collapse of unfolded molecules of the small cold shock protein CspTm at close to native conditions was discovered with this approach (14). This collapsed unfolded form also can be populated kinetically (15, 16) and has by now been found for a range of small proteins (16–20). It is unclear, however, whether this collapse is a nonspecific random heteropolymer collapse (21) or whether it is accompanied by the formation of specific structure. By placing FRET dye pairs in various positions of the protein, we obtain information on distance distributions in different segments of the unfolded polypeptide chain and their dependence on denaturant concentration.
A versatile method to complement such distance constraints with information about the secondary structure content is circular dichroism (CD) spectroscopy. In this case, however, we have to transiently populate the unfolded state under native conditions by using rapid mixing experiments. For this purpose, we use SRCD spectroscopy with a specifically designed microfluidic continuous-flow mixing system with millisecond dead time. In this way, the collapsed unfolded state, which is populated on a microsecond time scale or faster (14, 16), can be studied spectroscopically without interference from the signal of folded molecules, which form on a slower time scale. Importantly, SRCD gives us access to the far-UV wavelength range, where β-structure can be well distinguished from random coil (22, 23) but which is inaccessible with commercial stopped-flow instruments.
Results
Collapse from FRET Efficiency Histograms.
For the single-molecule FRET experiments, donor (Alexa Fluor 488) and acceptor (Alexa Fluor 594) dyes were placed specifically at different solvent-exposed positions of CspTm via pairs of Cys residues introduced by site-directed mutagenesis (Fig. 1). Upon unfolding of the protein, the average distance between the chromophores increases. As a consequence, the rate of energy transfer between them decreases, resulting in a reduced transfer efficiency E = nA/(nA + nD), where nA and nD are, respectively, the number of acceptor and donor photons emitted by the molecule [including corrections (24), see Materials and Methods]. In confocal single-molecule experiments, transfer efficiencies are determined from photon bursts originating from individual molecules freely diffusing through the focal spot of the laser beam (25). A histogram from a large number of such events shows distinct maxima corresponding to the subpopulations present in the sample (Fig. 2). The peak at high E corresponds to folded molecules, and the peak at intermediate E corresponds to unfolded molecules. The additional peak near E = 0 is thought to be caused by molecules lacking an active acceptor chromophore (24, 26) but does not interfere with our analysis.
Fig. 1.

Schematic of folded (Left) and unfolded (Right) CspTm with the sites for dye attachment for FRET indicated by colored spheres. For every variant investigated, one dye was reacted with Cys at position 67, and a second dye was reacted with a Cys at one of the other positions shown.
Fig. 2.

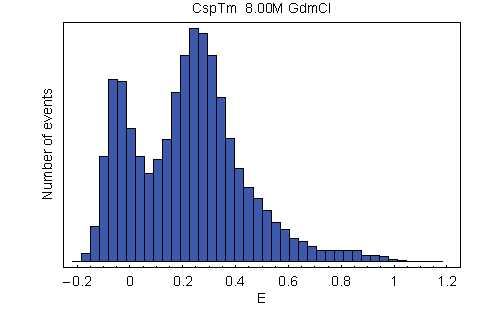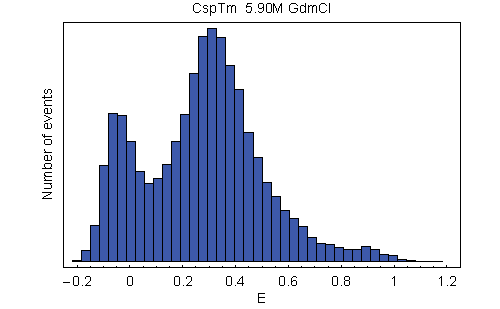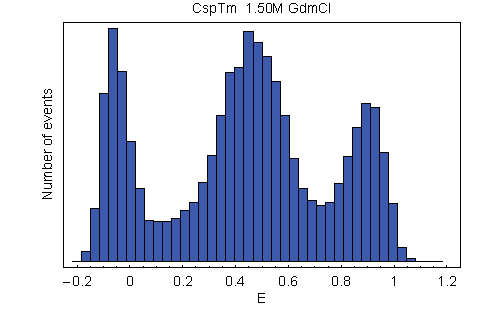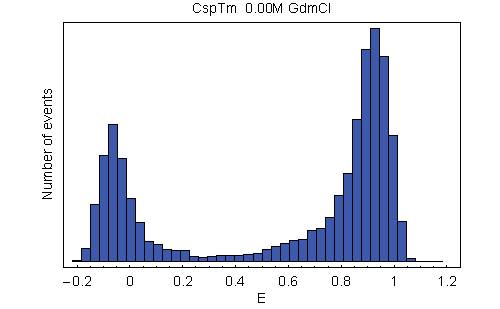
Energy transfer efficiency (E) histograms from single-molecule FRET measurements. (A) Examples from a GdmCl titration of variant C21C67, illustrating unfolded state collapse. See SI Movie 1 for a complete data set of C2C67. (B) Histograms of all variants at 1.5 M GdmCl. The peak at E ≈ 0.9 corresponds to folded molecules, and the peak at intermediate E corresponds to unfolded molecules. The peak at E ≈ 0 (shaded) originates from molecules with an inactive acceptor (26). To determine mean transfer efficiencies, the unfolded peak was fit to a normal distribution, and the other two peaks were fit to log normal functions (black lines) (14). The colors used correspond to those used in Fig. 1.
With increasing concentration of the denaturant guanidinium chloride (GdmCl), we observe a change in the relative amplitudes of the signal from native and unfolded molecules, corresponding to the expected redistribution of the two populations (Fig. 2A). However, whereas the mean transfer efficiency 〈E〉 of the folded state is invariant, the peak from unfolded molecules is continuously shifting to higher transfer efficiencies with decreasing GdmCl concentration. By comparison with stiff polyproline peptides, it has previously been shown that this increase in 〈E〉 corresponds to a collapse of the unfolded state in response to the altered solvent conditions (14). To address the question of whether this collapse is a global process that is evenly distributed across the polypeptide chain or whether it is due to compaction of a specific part of the molecule, we measured single-molecule transfer efficiency histograms of the labeled CspTm variants at a wide range of denaturant concentrations. At a given GdmCl concentration, 〈E〉 in the unfolded state shows an overall increase with decreasing sequence separation, as expected (Fig. 2B). Moreover, all variants exhibit the characteristic continuous collapse at low concentrations of denaturant, resulting in an increase in 〈E〉 [Fig. 3A and supporting information (SI) Movie 1].
Fig. 3.

Denaturant dependence of the mean transfer efficiencies 〈E〉 and persistence lengths lp. (A) 〈E〉 of the unfolded state of all variants as a function of GdmCl concentration. (B) lp calculated from 〈E〉 using Eqs. 1–4. (Inset) Shows the measured values of 〈E〉 for all variants at 8 M GdmCl, and as a black line 〈E〉 for a Gaussian chain calculated from Eqs. 1–4 for lp = 1.1 nm (the mean value of all variants) as a function of sequence separation n (number of peptide bonds excluding linkers).‡‡ The solid lines in the GdmCl titrations are fits to the empirical equation y = y0 [1 + ΔyKx/(1 + Kx)] used for interpolation; the solid black line is a fit to all data. Estimated error ranges for 〈E〉 and lp are indicated by dashed lines (see SI Materials and Methods). The colors used correspond to those used in Fig. 1.
Distance Distributions from Mean Transfer Efficiencies.
To analyze the mean transfer efficiencies in terms of distance distributions in the unfolded state, we use the Gaussian chain model, the simplest realistic model for describing large-scale properties of macromolecules (21), such as highly unfolded peptides and proteins (6, 27–31). 〈E〉 of the unfolded state at a given GdmCl concentration can be expressed in terms of the end-to-end distance probability distribution function of a Gaussian chain P(r) and the distance dependence of the transfer efficiency E(r) according to§§
 |
with
and
where r is the end-to-end distance, a is the distance of closest approach of the chain ends, lc is the contour length of the labeled polypeptide segment (with dyes and linkers, see Materials and Methods), and R0 is the Förster radius of the dye pair at the GdmCl concentration of the solution (R0 = 5.4 nm at 0 M GdmCl; see SI Materials and Methods for details). The mean squared end-to-end distance 〈r2〉 of a Gaussian chain can be expressed as
where n is the number of peptide bonds between the chromophores, l is the projection of the distance between two consecutive α-carbon atoms on the axis of the fully extended chain (0.38 nm), and lp (the only free parameter in the model) is a measure for the effective chain stiffness, which in our limit of lc ≫ lp is equivalent to the persistence length commonly used in the worm-like chain model (34). Fig. 3B Inset shows the measured 〈E〉 of our CspTm variants at 8 M GdmCl. The continuous line shows 〈E〉 as a function of n calculated according to Eqs. 1–4 with lp = 1.1 nm. All variants can be described with the same value of lp, indicative of isotropic distance distributions within unfolded CspTm, and in agreement with previous results on other proteins unfolded at high denaturant concentrations (27–30).
Fig. 3B shows lp for every variant as a function of GdmCl concentration calculated from 〈E〉 (Fig. 3A) using Eqs. 1–4. The concomitant renormalization for chain length allows a direct comparison of the variants with different sequence separation of donor and acceptor. The values of lp averaged over all variants range from 1.1 ± 0.1 nm at 8 M GdmCl, to 0.39 ± 0.07 nm extrapolated to 0 M GdmCl (Fig. 3B), close the values found in atomic force microscopy experiments (0.33 to 0.42 nm; summarized in ref. 31) or unstructured peptides [0.64 nm (32)] at 0 M GdmCl. All variants show a similar denaturant dependence of lp, suggesting isotropic chain collapse. From these data alone we cannot exclude that the more pronounced differences at low denaturant concentrations, especially for variant C10C67, may indicate slight deviations from completely isotropic collapse, but several points suggest otherwise. First, it seems structurally implausible that C2C67 and C21C67, which flank the slightly deviating C10C67, exhibit distance distributions in accord with the remaining variants, whereas a large difference occurs between C2C67 and C10C67. Second, other effects, such as changes in the photophysical properties of the chromophores upon collapse, may play a role; e.g., the two lysine residues directly neighboring Cys-10 may act as fluorescence quenchers (35) whose electrostatic interaction with the negatively charged fluorophores may be shielded at high GdmCl concentrations. In view of the error ranges we estimate for this type of measurement (Fig. 3), we conclude that the data are in agreement with isotropic collapse and do not justify an interpretation beyond this simple model.
Subpopulation-Selective Fluorescence Lifetime Distribution Analysis.
Mean transfer efficiencies obtained from transfer efficiency histograms do not provide direct information about the shape of the distance distribution, because the dynamics of inter-dye distance fluctuations are fast compared with the millisecond observation time per burst (14, 36). Fluorescence intensity decays, on the other hand, do report directly on the shape of the distance distribution (37, 38), because distance fluctuations for long polypeptides are slow relative to the fluorescence lifetimes of the dyes (32, 33). Conventional approaches to the analysis of distance distributions from fluorescence lifetimes suffer from interference with signal from native molecules; however, in combination with the separation of subpopulations by virtue of single-molecule detection, we can selectively analyze distance distributions in the unfolded state while remaining unaffected by native signal (17). For every individual fluorescence burst, the mean donor fluorescence lifetime is estimated (39) in addition to E. From the resulting two-dimensional histograms (Fig. 4A), we select the bursts corresponding to the unfolded state and, by combining the photons from all unfolded molecules, obtain high-resolution fluorescence decays (Fig. 4B). Donor and acceptor decays are then analyzed in a global fit¶¶ that assumes a distribution of transfer rates resulting from the distribution of donor–acceptor distances (see SI Materials and Methods), analogous to the established procedures for ensemble data (17, 38, 40). Specifically, we use P(r) of a Gaussian chain (Eq. 3) with 〈r2〉 as the only free parameter.
Fig. 4.

Fluorescence lifetime distribution analysis. (A) All events corresponding to unfolded molecules were selected (dashed box) from a two-dimensional histogram (shown is C2C67 at 1.5 M GdmCl) of the number of bursts (color scale) with transfer efficiency E and donor fluorescence lifetime τD,burst. (B) The photons were combined to generate time-correlated single-photon counting histograms for donor (green) and acceptor (red) (see SI Movie 2 for a complete data set of C2C67). (C) The rms end-to-end distance was determined and converted to the apparent persistence length lp (Eq. 4) by using a global fit assuming the distance distribution of a Gaussian chain (black lines). Error bars indicate the uncertainty in the fits. The solid black line in is a fit to all data as described in Fig. 3. The colors used correspond to those used in Fig. 1.
The time-correlated single-photon counting histograms from unfolded molecules are fit well, if we assume the distance distribution of a Gaussian chain (Fig. 4B; see also SI Movie 2), and again we observe a continuous collapse of the chain for all variants, with the expected increase in 〈r2〉 with n (data not shown). To normalize the data for n and to facilitate direct comparison of lifetime and intensity results, we calculate lp from 〈r2〉 (Eq. 4) (Fig. 4C). The greater demands on the signal-to-noise ratio for this type of analysis reduces the number of suitable data sets, especially at low GdmCl concentrations, where the overlap between native and unfolded subpopulation increases, but nevertheless we again find a similar GdmCl-dependence of lp for all variants. The good overall agreement of fluorescence intensity (Fig. 3B) and lifetime (Fig. 4C) results and the lack of consistent deviations from the averages in Figs. 3B and 4C provide additional evidence that the remaining differences between variants seen in either analysis are within the errors of the methods. The combined results of single-molecule intensity and lifetime data therefore suggest that the chain dimensions of CspTm are isotropic and Gaussian chain-like on the length scale we are probing, even for strongly collapsed unfolded molecules. But does this random coil behavior exclude the presence of secondary structure in the collapsed unfolded state?
Secondary Structure Content from SRCD.
An ideal method to quantify the secondary structure content of proteins is CD spectroscopy. Collapse of Csp has been shown to occur on a microsecond timescale or faster (12, 14, 16), whereas the folding rate at low GdmCl concentrations is in the tens of milliseconds range (41). In a kinetic mixing experiment with millisecond time resolution, the collapsed unfolded state is therefore formed during the dead time, and only the actual folding process is resolved. The dead time signal amplitude reports on the change in structure during collapse. However, although the formation of α-helical structure is readily observed with stopped-flow CD spectroscopy (typically at 222 nm), formation of β-sheet structure is often not accessible because of the small difference in CD signal between a random coil and β-structure as well as the strong influence of aromatic amino acids at wavelengths of >220 nm (42), the range available in conventional stopped-flow CD. Accordingly, previous attempts to measure secondary structure formation upon collapse of CspTm with CD spectroscopy have failed (B.S., unpublished results). To solve this problem, we have started to establish a methodology for which SRCD is used with microfabricated rapid mixing devices. SRCD has the advantage of high photon flux in the far UV below 220 nm, where the CD signal differences between random coil and β-structure become very pronounced (43), and the Gaussian beam shape allows focusing into microstructures. Microfluidic mixers (44) permit the implementation of sophisticated mixing strategies, making them the fastest method available for rapid-dilution experiments (45). They can be fabricated in fused silica with path lengths in the micrometer range, thus maximizing transmission in the far UV range. Because of the small feature size, sample consumption is drastically reduced compared with conventional continuous-flow mixing experiments (46), some of which have already led to substantial improvements both in time resolution and accessible wavelength range in kinetic CD experiments (47).
For refolding, CspTm in 4 M GdmCl was diluted to a final concentration of 0.8 M GdmCl (Fig. 5). The actual mixing region (Fig. 5B) consists of a narrow serpentine-shaped channel that performs mixing in the laminar flow regime by virtue of diffusion and chaotic advection (48). CD kinetics were measured by scanning the synchrotron radiation beam along the broader observation channel (Fig. 5C). Different positions in the channel thus correspond to different times after mixing, with a dead time of 1.3 ms. A kinetic trace taken at 205 nm (Fig. 5D), fit to a single exponential decay, resulted in a time constant of 19 ± 4 ms, in good agreement with the folding rates measured with stopped-flow fluorescence under identical conditions (41). However, the CD signal of −6 × 103 deg·cm2·dmol−1 at 1.3 ms does not coincide with the CD signal of CspTm unfolded in 4 M GdmCl (Fig. 5 D and E, dashed red line), indicating the formation of secondary structure before the actual folding reaction.
Fig. 5.

Secondary structure content of collapsed unfolded CspTm from SRCD. (A) Channel pattern of the microfluidic mixing device. To initiate refolding, unfolded protein injected into inlet 2 is diluted with buffer injected into inlet 1. (B) Rapid mixing occurs in the serpentine-shaped channel shown as a scanning electron micrograph. (C) The synchrotron radiation beam (white ellipse) is positioned in the observation channel. (D) Refolding kinetics of CspTm at 0.8 M GdmCl measured at 205 nm (blue line; error bars give one standard deviation calculated from eight measurements) and a single-exponential fit to the data (solid black line). (E) CD spectrum taken 1.3 ms after mixing (solid blue line), compared with equilibrium spectra under native (solid green line; 0.8 M GdmCl) and unfolding conditions (red line; 4 M GdmCl). The corresponding ellipticities at 205 nm are indicated in the spectra as dashed blue, green, and red lines, respectively. The shaded light blue, green, and red bands indicate one standard deviation from the equilibrium ellipticities of folded and unfolded CspTm, respectively, at 205 nm. The black dashed–dotted curve is a linear combination of the spectra at 0.8 M and 4 M GdmCl used to estimate the secondary structure content of collapsed unfolded CspTm.
For spectral information on collapsed unfolded CspTm, wavelength scans were taken 1.3 ms after mixing (Fig. 5E, solid blue line), when the fraction of native molecules calculated from the folding time is only 7%. Steady-state spectra of CspTm unfolded in 4 M GdmCl (Fig. 5E, solid red line), and native CspTm in 0.8 M GdmCl (Fig. 5E, solid green line) were recorded in the same mixing device and corrected for baselines taken without protein under otherwise identical conditions. These spectra illustrate the advantage gained by extending the wavelength range to <220 nm. Fitting the CD spectrum of collapsed unfolded CspTm to a linear combination of the spectra in 4 M and 0.8 M GdmCl (Fig. 5E, black dash–dotted curve) results in a signal change of ≈27% relative to the native structure (excluding the contribution from the 7% folded molecules). Relating this amplitude quantitatively to β-structure content is complicated by the contribution of aromatic amino acids to the native state signal, but from CD reference spectra for β-sheet (43) and the β-structure content from the NMR structure of CspTm (49), we estimate for the collapsed unfolded protein at 0.8 M GdmCl a β-structure content of ≈20% relative to the native protein (assuming no contribution from aromatic amino acids to the CD of collapsed unfolded CspTm). A very small population of additional α-helical structure cannot be excluded, but from the signal at 222 nm, the α-helix contribution is not more than approximately −1 × 103 deg·cm2·dmol−1, corresponding to less than ≈3% α-helix (43).
Discussion
Over the past 40 years, many proteins unfolded at high concentrations of denaturant have been shown to obey Gaussian chain statistics on a global scale (6, 27–30, 50, 51). CspTm is no exception in this respect. Its radius of gyration in 6 M GdmCl calculated from all labeled variants [using Eq. 4 and Rg2 = 〈r2〉/6 (21)] is 2.9 (±0.1) nm excluding dye linkers, in agreement with the 2.4 (+0.4/−0.2) nm calculated from the scaling law given by Kohn et al. (30, 51). But how does the collapse of the unfolded state at low GdmCl concentrations affect the intramolecular distance distributions? Surprisingly, even under near-native conditions, the agreement with Gaussian chain behavior is good, and lp is similar for all intramolecular distance pairs (Figs. 3B and 4B). At 1 M GdmCl, for instance, the resulting radii of gyration equal 2.2 (±0.2) nm. The small variance suggests that the distance distributions within the unfolded protein are rather isotropic. We thus have no evidence for native state topology in collapsed unfolded CspTm, in contrast to NMR experiments on staphylococcal nuclease (8) and eglin (52) at high concentrations of urea and in contrast to suggestions from simulations (53). Similarly, there is no obvious relation to the transition state structure (54). Our observation is in contrast to recent measurements on chymotrypsin inhibitor 2 and acyl-CoA-binding protein, where indications were found for a substantial deviation of the collapsed denatured state from Gaussian chain behavior (17) possibly involving folding intermediates (55). In summary, we conclude from the single-molecule fluorescence data that unfolded CspTm is close to a random coil in terms of polymer physics, even under near-physiological conditions.
It may therefore come as a surprise that the collapsed unfolded state contains a significant amount of β-structure, as was observed in our kinetic SRCD experiments. How can we reconcile these two observations? Clearly, global random coil behavior does not exclude the presence of short structured segments (30, 56–58), even more so if these are only populated transiently. This argument has been used to resolve the seemingly conflicting views of residual structure observed in proteins under highly denaturing conditions on the one hand and the successful description of global properties of unfolded polypeptides with the random coil model on the other (59, 60). Our results suggest that there are cases for which we must extend this notion of a random coil with residual structure even to the collapsed unfolded state, populated under conditions that have so far evaded confrontation with the “reconciliation problem” (6). Compaction of the chain would be expected to contribute to the formation of local structure, because the increase in excluded volume effects will introduce more steric interference with non-nearest-neighbor residues (29, 60). As a result, the backbone will be forced even more into the core regions of the Ramachandran map, corresponding to extended structures that avoid such steric conflicts. The largest one of these regions, and thus entropically the most favorable one, is the extended structure of β-strands. The particular preference of CspTm for this conformation is reflected by its extremely low propensity for the formation of α-helices [<1% helical content predicted with AGADIR (61)].
Currently, we have no direct evidence for the detailed conformation and average length of β-strand segments populated in collapsed unfolded CspTm. Because Gaussian-distributed intramolecular distances can only be observed if the segment length is considerably less than the contour length (21), the stretches of β-conformation must be short relative to the sequence separation of our dye pairs. Details about more local distance distributions could be addressed by using FRET pairs with much smaller Förster radii, which are currently inaccessible to single-molecule spectroscopy. In a very recent study, the dead time amplitudes of stopped-flow ensemble FRET experiments were used to probe the first β-hairpin of a closely related cold shock protein for local conformational preferences in the collapsed unfolded state (62). Magg et al. (62) observed a stretching of the second β-strand upon collapse, suggesting that the CD signal observed in our experiments could be due to strands of lengths approaching those in the native state. The question of segment length could possibly also be addressed with new methods for the analysis of CD spectra in terms of the number and size of structured segments in proteins (63, 64), but the current quality of our CD spectra for collapsed unfolded CspTm does not yet warrant such detailed deconvolution. The further development of kinetic SRCD and its combination with single-molecule fluorescence will be an important complementation of NMR methods in clarifying these structural details and the question of whether the behavior of CspTm is an exception or possibly a more general characteristic of such small all-β proteins.
Materials and Methods
Synthesis and Labeling of CspTm Variants.
Cysteine residues were introduced by site-directed mutagenesis to provide functional groups for the specific attachment of the dyes essentially as described previously (14). Expression and purification of Cys variants and wild-type protein were performed as described by Kremer et al. (49) (for details, see SI Materials and Methods).
Confocal Fluorescence Spectroscopy.
Observations of single-molecule fluorescence were made with a MicroTime 200 confocal microscope (PicoQuant, Berlin, Germany) equipped with a 470-nm pulsed diode laser (LDH 470) operated at 40 MHz (average power, 130 μW) and an Olympus (Tokyo, Japan) UplanApo ×60, 1.20-W objective. Sample fluorescence was separated into donor and acceptor components with a dichroic mirror (Chroma 585DCXR) and two final filters (Chroma HQ525/50 and Omega 600ALP). Each component was focused onto an avalanche photodiode (SPCM-AQR-15; PerkinElmer Optoelectronics, Fremont, CA), and the arrival time of every detected photon was recorded relative to the exciting laser pulse with a time resolution of 38 ps. Samples of labeled protein were diluted to a concentration of ≈20 pM in 50 mM sodium phosphate buffer at the appropriate GdmCl (Pierce, Rockford, IL) concentration and individually adjusted to pH 7. Tween 20 (0.001%; Pierce) was added to prevent surface adhesion of the protein (14). Data were taken for 30–60 min.
Single-Molecule Data Reduction and Analysis.
Successive photons detected in either channel separated by <100 μs were combined into one burst. A burst was retained as a significant event if the total number of counts exceeded 50. Identified bursts were corrected for background, differences in quantum yields, the different collection efficiencies of the detection channels, cross-talk, and direct acceptor excitation with the matrix approach (24) (SI Materials and Methods). For determining lp, the length of dyes and linkers were assumed to be equivalent to an additional 9 aa total, comparable with previous estimates (14, 17, 51, 65). Fluorescence lifetime distribution analysis was performed as described by Laurence et al. (17), except that the time-correlated single-photon counting histograms obtained from the unfolded state subpopulations were analyzed in terms of donor–acceptor distance distributions of a Gaussian chain (38, 40) (for details, see SI Materials and Methods).
Microfluidic Mixing Devices and Synchrotron Radiation Circular Dichroism.
Mixers were fabricated by deep reactive ion etching of fused silica substrates (HPFS Standard Grade, Corning code 7980; Corning, Corning, NY) to a depth of 14.5 μm. Mixers were sealed by direct fusion wafer bonding to another fused silica substrate. A serpentine-shaped channel after the T region joining the inlet channels performs mixing in the laminar flow regime by diffusion and chaotic advection (48). Dean vortices in the transverse plane and corner vortices in the longitudinal plane accomplish mixing by stretching and folding the fluid streamlines. The widening of the channel after mixing slows down the flow, resulting in an accessible time window of ≈27 ms before the solution reaches the exit port (labeled “3” in Fig. 5A).
Microfluidic devices were mounted in the SRCD sample chamber (see SI Materials and Methods) via a custom-designed holder with connections to two syringe pumps (PHD22/2000; Harvard Apparatus, Holliston, MA). Motorized translation stages (M-111.1; PI, Karlsruhe, Germany) allowed reproducible positioning of the mixer relative to the beam. For refolding, a solution of 6.7 mM unfolded CspTm in 4 M GdmCl/50 mM sodium phosphate buffer (pH 7.0) was injected through inlet 2 (Fig. 5) at a flow rate of 30 μl/min and mixed with buffer solution without GdmCl injected through inlet 1 at a flow rate of 120 μl/min. Complete mixing was assessed via absorbance and CD scans orthogonal to the flow direction across the observation channel. Flow rates ≥150 μl/min [corresponding to flow velocities of 6.9 m/s in the mixing channel (25 μm wide) and 0.43 m/s in the observation channel (400 μm wide)] were found to result in uniform concentrations after the mixing region. The dead time calculated from these flow rates and the position of the synchrotron beam is 1.3 ms.
Supplementary Material
Acknowledgments
We thank Jana Kramer for excellent technical assistance. This work has been supported by the Human Frontier Science Program (A.H., D.E.H., O.B., and B.S.), the Schweizerische Nationalfonds (B.S.), and the Deutsche Forschungsgemeinschaft (J.L., B.S., and R.S.). O.B., A.K., and D.E.H. performed work under the auspices of the U.S. Department of Energy at University of California Lawrence Livermore National Laboratory under Contract W-7405-Eng-48, with funding from the Laboratory Research and Development Program and additional support by the National Science Foundation Center for Biophotonics Science and Technology through Cooperative Agreement PHY 0120999 (managed by the University of California, Davis). A.K. also was supported by the Student Employee Graduate Research Fellowship Program at Lawrence Livermore National Laboratory. A.K. and D.E.H.'s travel was supported by the International Institute for Complex Adaptive Matter. R.S., P.B., and the SRCD station at Berliner Elektronenspeicherring-Gesellschaft für Synchrotronstrahlung were supported by German Federal Ministry of Education and Research Contract 05 KS4IP1/2.
Abbreviations
- FRET
Förster resonance energy transfer
- CD
circular dichroism
- SRCD
synchrotron radiation CD.
Footnotes
The authors declare no conflict of interest.
This article is a PNAS direct submission.
This article contains supporting information online at www.pnas.org/cgi/content/full/0604353104/DC1.
We denote all nonnative conformations as “unfolded” to stress that for true two-state proteins, such as CspTm, there are no thermodynamically or kinetically distinguishable denatured states.
Note that our analysis does not take into account the effect of excluded volume on the length scaling of the end-to-end distance. Such differences become discernible only if unfolded proteins are investigated over a much wider range of chain lengths than here (30).
For this analysis, we assume intramolecular distance fluctuations to be slow relative to the fluorescence lifetime of the donor chromophore, as expected for long polypeptide chains (32, 33).
Note that a global analysis is substantially more robust than the individual fits and mitigates the common problems of quantitatively analyzing fluorescence decays deviating from single exponential behavior, especially for the short donor lifetime components, which result in a rise term in the acceptor intensity.
References
- 1.Jackson SE. Fold Des. 1998;3:R81–R91. doi: 10.1016/S1359-0278(98)00033-9. [DOI] [PubMed] [Google Scholar]
- 2.Fersht AR. Structure and Mechanism in Protein Sci. New York: Freeman; 1998. [Google Scholar]
- 3.Galzitskaya OV, Finkelstein AV. Proc Natl Acad Sci USA. 1999;96:11299–11304. doi: 10.1073/pnas.96.20.11299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Baker D. Nature. 2000;405:39–42. doi: 10.1038/35011000. [DOI] [PubMed] [Google Scholar]
- 5.Muñoz V, Eaton WA. Proc Natl Acad Sci USA. 1999;96:11311–11316. doi: 10.1073/pnas.96.20.11311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Millett IS, Doniach S, Plaxco KW. Adv Protein Chem. 2002;62:241–262. doi: 10.1016/s0065-3233(02)62009-1. [DOI] [PubMed] [Google Scholar]
- 7.Shortle DR. Curr Opin Struct Biol. 1996;6:24–30. doi: 10.1016/s0959-440x(96)80091-1. [DOI] [PubMed] [Google Scholar]
- 8.Shortle D, Ackerman MS. Science. 2001;293:487–489. doi: 10.1126/science.1060438. [DOI] [PubMed] [Google Scholar]
- 9.Dyson HJ, Wright PE. Chem Rev. 2004;104:3607–3622. doi: 10.1021/cr030403s. [DOI] [PubMed] [Google Scholar]
- 10.Religa TL, Markson JS, Mayor U, Freund SM, Fersht AR. Nature. 2005;437:1053–1056. doi: 10.1038/nature04054. [DOI] [PubMed] [Google Scholar]
- 11.Jia YW, Talaga DS, Lau WL, Lu HSM, DeGrado WF, Hochstrasser RM. Chem Phys. 1999;247:69–83. [Google Scholar]
- 12.Schuler B. ChemPhysChem. 2005;6:1206–1220. doi: 10.1002/cphc.200400609. [DOI] [PubMed] [Google Scholar]
- 13.Deniz AA, Laurence TA, Beligere GS, Dahan M, Martin AB, Chemla DS, Dawson PE, Schultz PG, Weiss S. Proc Natl Acad Sci USA. 2000;97:5179–5184. doi: 10.1073/pnas.090104997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Schuler B, Lipman EA, Eaton WA. Nature. 2002;419:743–747. doi: 10.1038/nature01060. [DOI] [PubMed] [Google Scholar]
- 15.Lipman EA, Schuler B, Bakajin O, Eaton WA. Science. 2003;301:1233–1235. doi: 10.1126/science.1085399. [DOI] [PubMed] [Google Scholar]
- 16.Magg C, Schmid FX. J Mol Biol. 2004;335:1309–1323. doi: 10.1016/j.jmb.2003.11.050. [DOI] [PubMed] [Google Scholar]
- 17.Laurence TA, Kong XX, Jager M, Weiss S. Proc Natl Acad Sci USA. 2005;102:17348–17353. doi: 10.1073/pnas.0508584102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kuzmenkina EV, Heyes CD, Nienhaus GU. J Mol Biol. 2006;357:313–324. doi: 10.1016/j.jmb.2005.12.061. [DOI] [PubMed] [Google Scholar]
- 19.Sherman E, Haran G. Proc Natl Acad Sci USA. 2006;103:11539–11543. doi: 10.1073/pnas.0601395103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Qi PX, Sosnick TR, Englander SW. Nat Struct Biol. 1998;5:882–884. doi: 10.1038/2321. [DOI] [PubMed] [Google Scholar]
- 21.Grosberg AY, Khokhlov AR. Statistical Physics of Macromolecules. Woodbury, NY: Am Inst Phys; 1994. [Google Scholar]
- 22.Sutherland JC. In: Circular Dichroism and the Conformational Analysis of Biomolecules. Fasman GD, editor. New York: Plenum; 1996. pp. 599–633. [Google Scholar]
- 23.Wallace BA, Janes RW. Curr Opin Chem Biol. 2001;5:567–571. doi: 10.1016/s1367-5931(00)00243-x. [DOI] [PubMed] [Google Scholar]
- 24.Schuler B. In: Protein Folding Protocols. Bai Y, Nussinov R, editors. Vol 366. Totowa, NJ: Humana; 2006. [Google Scholar]
- 25.Deniz AA, Laurence TA, Dahan M, Chemla DS, Schultz PG, Weiss S. Annu Rev Phys Chem. 2001;52:233–253. doi: 10.1146/annurev.physchem.52.1.233. [DOI] [PubMed] [Google Scholar]
- 26.Schuler B, Lipman EA, Steinbach PJ, Kumke M, Eaton WA. Proc Natl Acad Sci USA. 2005;102:2754–2759. doi: 10.1073/pnas.0408164102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Tanford C. Adv Protein Chem. 1968;23:121–282. doi: 10.1016/s0065-3233(08)60401-5. [DOI] [PubMed] [Google Scholar]
- 28.Damaschun G, Damaschun H, Gast K, Zirwer D. Biochemistry (Moscow) 1998;63:259–275. [PubMed] [Google Scholar]
- 29.Zhou HX. J Phys Chem B. 2002;106:5769–5775. [Google Scholar]
- 30.Kohn JE, Millett IS, Jacob J, Zagrovic B, Dillon TM, Cingel N, Dothager RS, Seifert S, Thiyagarajan P, Sosnick TR, et al. Proc Natl Acad Sci USA. 2004;101:12491–12496. doi: 10.1073/pnas.0403643101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Zhou HX. Biochemistry. 2004;43:2141–2154. doi: 10.1021/bi036269n. [DOI] [PubMed] [Google Scholar]
- 32.Lapidus LJ, Steinbach PJ, Eaton WA, Szabo A, Hofrichter J. J Phys Chem B. 2002;106:11628–11640. [Google Scholar]
- 33.Möglich A, Joder K, Kiefhaber T. Proc Natl Acad Sci USA. 2006;103:12394–12399. doi: 10.1073/pnas.0604748103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Doi M, Edwards SF. The Theory of Polymer Dynamics. New York: Oxford Univ Press; 1988. [Google Scholar]
- 35.Ratner V, Amir D, Kahana E, Haas E. J Mol Biol. 2005;352:683–699. doi: 10.1016/j.jmb.2005.06.074. [DOI] [PubMed] [Google Scholar]
- 36.Gopich IV, Szabo A. J Phys Chem B. 2003;107:5058–5063. [Google Scholar]
- 37.Haas E, Wilchek M, Katchalskikatzir E, Steinberg IZ. Proc Natl Acad Sci USA. 1975;72:1807–1811. doi: 10.1073/pnas.72.5.1807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Van Der Meer BW, Coker G, III, Chen SYS. Resonance Energy Transfer: Theory and Data. New York: VCH; 1994. [Google Scholar]
- 39.Eggeling C, Berger S, Brand L, Fries JR, Schaffer J, Volkmer A, Seidel CA. J Biotechnol. 2001;86:163–180. doi: 10.1016/s0168-1656(00)00412-0. [DOI] [PubMed] [Google Scholar]
- 40.Beechem JM, Haas E. Biophys J. 1989;55:1225–1236. doi: 10.1016/S0006-3495(89)82918-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Perl D, Welker C, Schindler T, Schröder K, Marahiel MA, Jaenicke R, Schmid FX. Nat Struct Biol. 1998;5:229–235. doi: 10.1038/nsb0398-229. [DOI] [PubMed] [Google Scholar]
- 42.Woody RW, Dunker AK. In: Circular Dichroism and the Conformational Analysis of Biomolecules. Fasman GD, editor. New York: Plenum; 1996. pp. 109–158. [Google Scholar]
- 43.Greenfield NJ. Methods Enzymol. 2004;383:282–317. doi: 10.1016/S0076-6879(04)83012-X. [DOI] [PubMed] [Google Scholar]
- 44.Knight JB, Vishwanath A, Brody JP, Austin RH. Phys Rev Lett. 1998;80:3863–3866. [Google Scholar]
- 45.Hertzog DE, Michalet X, Jager M, Kong XX, Santiago JG, Weiss S, Bakajin O. Anal Chem. 2004;76:7169–7178. doi: 10.1021/ac048661s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Shastry MG, Luck SD, Roder H. Biophys J. 1998;74:2714–2721. doi: 10.1016/S0006-3495(98)77977-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Kimura T, Uzawa T, Ishimori K, Morishima I, Takahashi S, Konno T, Akiyama S, Fujisawa T. Proc Natl Acad Sci USA. 2005;102:2748–2753. doi: 10.1073/pnas.0407982102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Chamarthy P, Wereley ST. 2004 Proceedings of the ASME International Mechanical Engineering Congress and Exposition. 3 Vols. New York: ASME Press; 2004. CD-ROM, art no 61902. [Google Scholar]
- 49.Kremer W, Schuler B, Harrieder S, Geyer M, Gronwald W, Welker C, Jaenicke R, Kalbitzer HR. Eur J Biochem. 2001;268:2527–2539. doi: 10.1046/j.1432-1327.2001.02127.x. [DOI] [PubMed] [Google Scholar]
- 50.Wilkins DK, Grimshaw SB, Receveur V, Dobson CM, Jones JA, Smith LJ. Biochemistry. 1999;38:16424–16431. doi: 10.1021/bi991765q. [DOI] [PubMed] [Google Scholar]
- 51.McCarney ER, Werner JH, Bernstein SL, Ruczinski I, Makarov DE, Goodwin PM, Plaxco KW. J Mol Biol. 2005;352:672–682. doi: 10.1016/j.jmb.2005.07.015. [DOI] [PubMed] [Google Scholar]
- 52.Ohnishi S, Lee AL, Edgell MH, Shortle D. Biochemistry. 2004;43:4064–4070. doi: 10.1021/bi049879b. [DOI] [PubMed] [Google Scholar]
- 53.Zagrovic B, Snow CD, Khaliq S, Shirts MR, Pande VS. J Mol Biol. 2002;323:153–164. doi: 10.1016/s0022-2836(02)00888-4. [DOI] [PubMed] [Google Scholar]
- 54.Perl D, Holtermann G, Schmid FX. Biochemistry. 2001;40:15501–15511. doi: 10.1021/bi011378s. [DOI] [PubMed] [Google Scholar]
- 55.Teilum K, Poulsen FM, Akke M. Proc Natl Acad Sci USA. 2006;103:6877–6882. doi: 10.1073/pnas.0509100103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Fitzkee NC, Rose GD. Proc Natl Acad Sci USA. 2004;101:12497–12502. doi: 10.1073/pnas.0404236101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Jha AK, Colubri A, Freed KF, Sosnick TR. Proc Natl Acad Sci USA. 2005;102:13099–13104. doi: 10.1073/pnas.0506078102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Ding F, Jha RK, Dokholyan NV. Structure (London) 2005;13:1047–1054. doi: 10.1016/j.str.2005.04.009. [DOI] [PubMed] [Google Scholar]
- 59.McCarney ER, Kohn JE, Plaxco KW. Crit Rev Biochem Mol Biol. 2005;40:181–189. doi: 10.1080/10409230591008143. [DOI] [PubMed] [Google Scholar]
- 60.Fitzkee NC, Fleming PJ, Gong HP, Panasik N, Street TO, Rose GD. Trends Biochem Sci. 2005;30:73–80. doi: 10.1016/j.tibs.2004.12.005. [DOI] [PubMed] [Google Scholar]
- 61.Muñoz V, Serrano L. Biopolymers. 1997;41:495–509. doi: 10.1002/(SICI)1097-0282(19970415)41:5<495::AID-BIP2>3.0.CO;2-H. [DOI] [PubMed] [Google Scholar]
- 62.Magg C, Kubelka J, Holtermann G, Haas E, Schmid FX. J Mol Biol. 2006;360:1067–1080. doi: 10.1016/j.jmb.2006.05.073. [DOI] [PubMed] [Google Scholar]
- 63.Pancoska P, Janota V, Keiderling TA. Anal Biochem. 1999;267:72–83. doi: 10.1006/abio.1998.2960. [DOI] [PubMed] [Google Scholar]
- 64.Sreerama N, Venyaminov SY, Woody RW. Protein Sci. 1999;8:370–380. doi: 10.1110/ps.8.2.370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Schröder GF, Alexiev U, Grubmüller H. Biophys J. 2005;89:3757–3770. doi: 10.1529/biophysj.105.069500. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}
{kind=link}