

Abstract
We have investigated the folding behavior of dimeric and covalently crosslinked versions of the 33-residue α-helical GCN4-p1 coiled coil derived from the leucine zipper region of the transcriptional activator GCN4. The effects of multisite substitutions indicate that folding occurs along multiple routes with nucleation sites located throughout the protein. The similarity in activation energies of the different routes together with an analysis of intrinsic helical propensities indicate that minimal helix is present before a productive collision of the two chains. However, approximately one-third to one-half of the total helical structure is formed in the postcollision transition state ensemble. For the crosslinked, monomeric version, folding occurs along a single robust pathway. Here, the region nearest the crosslink, with the least helical propensity, is structured in the transition state whereas the region farthest from the tether, with the most propensity, is completely unstructured. Hence, the existence of transition state heterogeneity and the selection of folding routes critically depend on chain topology.
Keywords: protein folding, nucleation, topology, secondary structure
The folding of many small globular proteins is kinetically two-state without the accumulation of intermediates. We and others have proposed that such folding reactions are nucleation processes (1–5), and the issue of uniqueness of the transition state (TS) nucleus has become a subject of much debate (6–9). The major method for characterizing TSs is protein engineering or mutational Φ analysis (10, 11). For some mutations, intermediate effects on refolding rates have been observed (7, 12–17). A crucial question is whether these fractional effects represent a single TS with partially formed structure or a heterogeneous population of TSs, some with the structure fully formed and others with it completely absent. Most (7, 18), but not all (19), folding experiments have been interpreted in the context of a homogeneous TS ensemble and a single dominant folding pathway. The small, but fractional, Φ values that we measured previously for the GCN4-p1 coiled coil (CC) allowed for the possibility that folding occurred along multiple pathways with nucleation sites located throughout the protein (14).
The importance of secondary structure relative to tertiary structure and chain topology in the determination of folding pathways and rates is another unsettled issue. The general insensitivity of refolding rates to helix-destabilizing substitutions in the CC indicated that a large fraction of the helical structure is not formed in the rate-limiting step (14). Based on these and other results with cytochrome c, we proposed that the critical element of the TS is the formation of the overall chain topology, established by pinning the chain by the interaction of a number of apolar side chains, rather than secondary structure formation (3, 5, 14). A strong correlation recently was noted for nearly a dozen proteins between the folding speed and the average sequence distance between residues, called the contact order (20). Although helical structure reduces the contact order of a protein and its stabilization can accelerate folding in some circumstances (15, 21), this correlation can be taken to suggest the importance of topology in the structure of the folding TS.
A third central question in protein folding is whether helical structure forms before hydrophobic collapse as envisioned by the diffusion-collision model (19, 22, 23). The likelihood of such a folding mechanism is increased if residual helical structure is present in the denatured state. In fact, isolated regions of GCN4-p1 sequence have been observed to be more than 50% helical (23). Also, the presence of this amount of residual structure in the denatured state could account for the insensitivity of folding rates to Ala-to-Gly substitution, because the free energy gap between the denatured state and the TS would be unchanged if helix is present in both states (24, 25).
To investigate the issues of TS heterogeneity, the role of topology, residual structure, and precollision helical structure, we have studied the folding of single-site and multisite mutants as well as a crosslinked derivative of the CC where the two helices have been covalently linked with an unstructured tether. We find that the dimeric version folds via multiple routes whereas the crosslinked version folds along a single robust pathway. The mutational data further indicate that minimal helix is formed before a productive collision but up to half of the molecule is helical in the folding TS. Although this helical region is predominantly near the carboxyl terminus in the dimeric version, the presence of the amino tether in the crosslinked version induces nucleation to occur exclusively at the amino terminus in spite of this region’s very low helical propensity. The switch in locality of the nucleus and loss of pathway heterogeneity emphasize the importance of topology in the determination of folding behavior even in the context of this elemental helical protein.
MATERIALS AND METHODS
Peptide Synthesis.
Peptides were prepared and characterized as described (26). GCN4-p1′ (Ac-RMKQLEDKVEELLSKNWHLENEVARLKKLVGER-NH2) included a tryptophan in place of the tyrosine at position 17. GCN4-p2′ contained an additional Cys-Gly-Gly tripeptide at the amino terminus but residue count begins at the Arg for direct comparison to GCN4-p1′. Crosslinked GCN4-p2′ was formed by bubbling oxygen under native conditions for 2 hr at neutral pH. Dimeric GCN4-p2′ was formed by reduction with 10-fold molar excess of Tris-(2-carboxyethyl)-phosphine hydrochloride. Peptide concentrations were determined by using an extinction coefficient of 5,700 M−1⋅cm−1 at 280 nm.
Equilibrium Measurements.
Equilibrium free energies were determined from guanidinium chloride (GdmCl) denaturation profiles monitoring tryptophan fluorescence and CD at 222 nm (27). CD measurements were conducted at 1- to 2-nm resolution with a pathlength of 0.1 cm. Peptide concentrations ranged from 2 to 42 μM, and experiments were carried out in 20 mM sodium acetate, 150–200 mM sodium chloride at pH 5.5, 10°C.
Stopped-Flow Spectroscopy.
Rapid mixing fluorescence experiments used a Biologic (Grenoble, France) SFM-4 stopped-flow apparatus interfaced with a Jasco (Easton, MD) model 715 CD spectropolarimeter as described (27). Protein concentrations ranged from 5 μM to 35 μM.
RESULTS AND DISCUSSION
Two-State Folding Behavior. To investigate the role of topology and the generality of our previous work with the dimeric GCN4-p1′ CC, we studied a modified version that folds either as a crosslinked monomer or as a dimer. This modified species contains an amino-terminal Cys-Gly-Gly linker that forms a disulfide bond between the Cys residues under oxidizing conditions. Under reducing conditions, the disulfide crosslink is not formed and the folding behavior is the same as for the tetherless version (27).
Stopped-flow fluorescence spectroscopy was used to measure the folding kinetics of the tryptophan containing GCN4-p2′ molecule (Fig. 1). Crosslinked GCN4-p2′ exhibits first-order behavior consistent with the unfolded ↔ native unimolecular nature of this folding reaction. Dimeric GCN4-p2′ exhibited second-order folding and first-order unfolding behavior consistent with the 2(monomer) ↔ dimer bimolecular nature of this reaction. The folding rates of the dimeric and crosslinked versions are 2 × 106 M−1⋅s−1 and 7.5 × 103 s−1, respectively (extrapolated to zero denaturant). These rates equate to an effective chain concentration of 4 mM for the crosslinked version.
Figure 1.

Chevron plot of folding kinetics of dimeric and crosslinked GCN4-p2′ in 20–100 mM sodium acetate, 150 mM sodium chloride, pH 5.5, 10°C. Symbols are the same in the left and right panels. The measured bimolecular folding rates for the dimeric CC have been scaled to 5.5 μM protein concentration. The solid line in lower left represents the predicted folding rates for the GGG mutant determined from the difference in equilibrium stability and activation energy for unfolding according to ΔG‡f = ΔG‡u + ΔG0. To measure the unfolding rates of the marginally stable dimeric GGG version, 5% (vol/vol) of 2,2,2-trifluoroethanol was added. Previous work demonstrated that 2,2,2-trifluoroethanol does not affect unfolding rates (34).
Current and previous kinetic studies demonstrate that the folding of both versions obeys a thermodynamically and kinetically two-state transition between an unfolded state and a fully helical native state (14, 27, 28). This demonstration is accomplished by using the chevron analysis with a linear dependence of the equilibrium and activation free energies for folding (f) and unfolding (u) on GdmCl concentration (10)
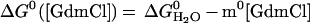 |
1a |
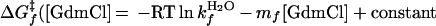 |
1b |
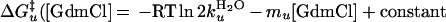 |
1c |
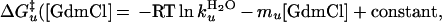 |
1d |
where Eqs. 1c and 1d apply to the unfolding of the dimeric and crosslinked versions, respectively. When folding is effectively two-state, the equilibrium values for the change in free energy and surface burial can be calculated from kinetic measurements according to: −ΔG0H2O = ΔG‡u − ΔG‡f and m0 = mu − mf. The equivalence of thermodynamically and kinetically determined values for ΔG0H2O and m0 demonstrates the applicability of a two-state model for GCN4-p2′ folding (see supplemental Tables 2 and 3, which are available on the PNAS web site, www.pnas.org).
Single-Site Mutations. The two-state folding behavior provides a simple framework in which to interpret the kinetic effects of mutations and characterize the TS of the folding reactions. In our previous studies of the dimeric version, destabilizing Gly substitutions were made at external surface positions (14). The relatively small change in folding rates compared with the unfolding rates was taken as evidence that no large fraction of helix is present in the rate-limiting TS. The effect of each mutation was quantified by the parameter Φf, given by the change in folding activation free energy, ΔΔG‡f, divided by the change in global stability, ΔΔG0. A Φf value is the degree to which the total energetic effect of the substitution is realized in the TS. Generally, a value of zero is considered to mean that the mutated residue is unstructured in the TS whereas a value of one is consistent with this residue sensing a native-like environment.
Mutations were designed to compare Ala-to-Gly substitutions because these residues have a large difference in helix propensity. In the dimeric CC, Ala-to-Gly substitutions at the seventh, 14th, and 24th positions had only a small effect on folding rates, with measured Φf values of 0.07 ± 0.03, 0.16 ± 0.04, and 0.25 ± 0.01, respectively (14). The corresponding single-site substitutions in the crosslinked version are quite different (Fig. 2A, Table 1) with Φfvalues of 0.74 ± 0.02, 0.54 ± 0.05, and 0.00 ± 0.02, respectively. The trend in the Φf values has shifted from a slight bias toward the carboxyl terminus in the dimeric CC to a strong bias and high Φf values at the amino terminus, the location of the tether, in the crosslinked version.
Figure 2.

(A) Single-site Φf values for the dimeric (hatched bars) and the crosslinked CC (open bars). The slight bias in Φf values toward the carboxyl terminus in the dimeric version is shifted to a strong bias toward the amino, tethered end of the crosslinked version. (B) Simplified folding models. The dimeric CC folds with a heterogeneous TS ensemble with multiple nucleation sites whereas the crosslinked version folds along a single route. The width of arrows represents the approximate flux down each route and reflects the magnitude of the single-site Φf values.
Table 1.
Φf-values for GCN4-p2′ folding
| Substitution | Dimeric Φf value | Cross-linked Φf value |
|---|---|---|
| A7G | 0.07 ± 0.03 | 0.74 ± 0.02 (2.8 M, 5.6 M) |
| A14G | 0.16 ± 0.04 | |
| S14A | 0.54 ± 0.05 (4.5 M, 6.5 M) | |
| A24G | 0.25 ± 0.01 | 0.003 ± 0.05 (2.8 M, 5.3 M) |
| AAA:GGG | 0.46 ± 0.02 | 0.40 ± 0.02 (2.5 M, 4.5 M) |
| AAA:TTT | 0.51 ± 0.03 | 0.57 ± 0.02 (3.5 M, 6 M) |
| AAG:GGG | 0.72 ± 0.02 | 0.73 ± 0.02 (2.5 M, 4.5 M) |
Calculated according to Φf = −ΔΔG‡f/(ΔΔG‡u − ΔΔG‡f). The ΔΔG‡f and ΔΔG‡u values are calculated at 0.9 and 3.5 M GdmCl, respectively, for the dimeric version and at the concentration noted in parentheses for the crosslinked version, to reduce extrapolation errors and sensitivity to slight differences in m values.
TS Heterogeneity. The high Φfvalues and the spatially localized nucleus in the crosslinked species led us to re-examine the results for the dimeric molecule. Although the Φf values are small in the dimeric system, they are not zero. These marginal effects indicate that the mutated residues are either partially constrained in the TS, or fully constrained in a fraction of a heterogeneous population of TSs. A subcategory of the latter possibility pictures helix nucleation at multiple alternative positions along the chain (Fig. 2B) (14). If a destabilizing mutation is present at a given position, another region then might serve to nucleate helix formation and folding rates would be only marginally retarded.
We examined the possibility of TS heterogeneity by simultaneously probing the helicity at multiple sites by using variants with triple-site substitutions (Fig. 1). The first variant, AAA, contains alanines in the seventh, 14th, and 24th positions (six positions total). The second variant, GGG, contains glycines at these positions. The third variant, TTT, contains threonines.
The AAA-to-GGG substitution results in a change in equilibrium stability of 5–6 kcal/mol and nearly identical ΦfAAA/GGG values of 0.46 ± 0.02 and 0.40 ± 0.02 for the dimeric and crosslinked versions, respectively. The AAA-to-TTT substitution results in a change in stability of about 2–3 kcal/mol and ΦfAAA/TTT values of 0.51 ± 0.03 and 0.57 ± 0.02, respectively.
For the Ala-to-Gly substitutions, where the decrease in helical propensity results from a increase in backbone entropy in the unfolded state, Φf values are sensitive to the decrease in chain entropy in the TS rather than to helix formation per se. For the Ala-to-Thr substitutions, where the decrease in propensity results from the loss of side-chain entropy in the helical conformation (29), Φf values more specifically reflect helix formation in the TS. The sizable triple-site ΦfAAA/TTT values indicate that the TS has helical structure rather than merely a partially constrained backbone.
Homogeneous and Heterogeneous Nuclei. For the crosslinked CC, the change in the kinetic behavior caused by the triple Ala-to-Gly substitution reflects the independent and additive effects caused by each of the single-site substitutions. A composite triple-site ΦfAAA/GGG value can be predicted from the effects of the single-site mutations according to 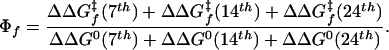 The predicted composite ΦfAAA/GGG value of 0.34 is very close to the observed value of 0.40 (using the values for measured S14A substitution in place of the unmeasured A14G substitution). This additivity, and also the lack of significant change in the kinetic mf value, indicate that a single robust folding pathway exists for the crosslinked CC.
The predicted composite ΦfAAA/GGG value of 0.34 is very close to the observed value of 0.40 (using the values for measured S14A substitution in place of the unmeasured A14G substitution). This additivity, and also the lack of significant change in the kinetic mf value, indicate that a single robust folding pathway exists for the crosslinked CC.
A different result is found for the dimeric system. The observed ΦfAAA/GGG value of 0.46 is much larger than 0.18, the composite value predicted from the single-site Φf values assuming their effects are independent and additive. In fact, the Φf values for both the triple-site substitutions are larger than any single-site value. When a single-site mutation is made that disrupts folding through one region, the bulk of the folding events proceeds through other regions and only a minimal decrease in folding rates is observed. In the case of the triple-site substitutions, however, a large decrease in the folding rates is observed because folding must proceed through at least one of the destabilized regions. These results point to heterogeneous nucleation in the dimeric CC, with multiple, alternative nucleation sites.
The multisite hypothesis was explicitly tested with an additional triple-site variant, D7A/S14A/A24G. This AAG variant has a destabilizing glycine mutation near the polypeptide’s carboxyl terminus in the region most likely to be nucleated (highest single-site Φf value). We hypothesized that the destabilizing substitution would block nucleation at this site in the dimeric CC and shift nucleation toward the amino regions, which then should exhibit heightened sensitivity to destabilization, just as in the crosslinked CC. The triple-site variant contained alanines at both the seventh and 14th positions to ensure that these two positions are probed.
In the background of the A24G substitution in the dimeric CC, the double-site Ala-to-Gly comparison at the seventh and 14th positions yields a high ΦfAAG/GGG value, 0.72 ± 0.02, indicating that these two regions become structured in a large fraction of the TSs. The same comparison in the crosslinked system, where nucleation occurs only at the amino tethered end, yields the same double-site ΦfAAG/GGG value, 0.73 ± 0.02. Hence, the A24G substitution in the dimeric CC shifts the nucleation site from one end of the molecule to the other, just as the tether does in the crosslinked CC.
These results demonstrate that multiple nucleation sites do exist and that the relative importance of each one is subject to manipulation. The existence of multiple nucleation sites in the dimeric CC is presumably a consequence of the system’s translational symmetry and the length of the helices. Nucleation can occur at essentially any position within the 10 turns of helix. Upon introduction of an unstructured crosslink, the translational symmetry of the CC is broken. A large difference in effective local concentration then results in a relatively homogeneous TS ensemble with a strong bias toward nucleation near the tethered end.
Our results are quantitatively consistent with this view (Fig. 2B). In the simple scenario where a particular region is structured in the TS for 50% of the folding routes, a 10-fold destabilization of this region will increase the activation energy of these routes by 1.3 kcal/mol. This will largely block folding along these pathways and decrease the net folding rate by nearly a factor of two (ΔΔG‡f = 0.33 kcal/mol), leading to a Φf value of 0.26. This reasoning suggests that folding of the dimeric CC can be approximated by three independent routes with nucleation occurring at the amino terminus, the center, and the carboxyl terminus with relative probabilities of 1/6, 1/3, and 1/2, respectively. These values for the relative fluxes equate to Φf values of 0.08, 0.12, and 0.16, for the seventh, 14th, and 24th positions, respectively, similar to the experimental values. Recently, Matthews and coworkers (30) also concluded that the TS is approximately 30% native-like with the two carboxyl-terminal heptads being the likely nucleation site.
Residual Structure and Marginal Φf Values. If residual helical structure is present in both the denatured state and the TS, then a different explanation is possible for the marginal Φf values observed in the dimeric CC (25). In this case, the activation energy for folding would be unchanged for any given surface substitution and only a negligible change in folding rates would be observed. Another consequence of residual structure would be a negligible change in equilibrium stability because helix is present in both the unfolded and native states. For helix altering mutations in the protein CheY, both of these unusual behaviors have been observed (24). Also, there is an accompanying “rollover” region in the chevron plot at low denaturant concentrations, consistent with decreased surface burial in the folding TS.
For a peptide containing residues 16–32 of GCN4-p1, a considerable amount of ellipticity, Θ222 ≈ −15,000 deg⋅cm2/dmol, was observed under extremely low ionic conditions at pH 7, 3°C (23). Further, the agadir program (31) predicts that in isolated monomers, the region encompassing residues 22–30 should have helical structure 60–70% of the time (Fig. 3A). Hence, residual structure may be present in the denatured state and could be responsible for the marginal Φf values that we observe.
Figure 3.

Residual structure in the denatured state. (A) Predicted helicity for monomers of GCN4-p1′ and the A24G variant at pH 5.5, 10°C, 0.2 M ionic strength, and for the related leucine zipper with a strengthened hydrophobic core (32) at pH 4.8, 25°C, 0.1 M ionic strength, calculated by using agadir (31). (B) CD spectra of an 18-residue peptide encompassing the most helical region of GCN4-p1′ (residues 16–33 with N16D substitution) in 20 mM sodium acetate, pH 5.5, 10°C in 0.2 M NaCl (83 μM); 180 mM NaCl, 0.6 M GdmCl (73 μM), and 6 M GdmCl (43 μM). (C) Denaturation profile of peptide 16–33.
To investigate this possibility, we synthesized a peptide containing residues 16–33. This peptide does have a partially helical spectrum under our conditions (pH 5.5, 10°C) with Θ222 = −8,000 deg⋅cm2/dmol (Fig. 3B). However, the signal is very denaturant sensitive; ellipticity is reduced to −3,000 deg⋅cm2/dmol in 1 M GdmCl (Fig. 3C), well within the folding arm of the chevron plot.
A variety of other results also indicate that under the conditions used in our study, only minor amounts of helical structure exist in the denatured state. The GCN4-p2′ chevron plots do not exhibit rollover behavior. All of the single-site Ala-to-Gly substitutions (two total per CC) result in destabilization of at least 1.2 kcal/mol, the appropriate ΔΔGo for such mutations. Millisecond (burst phase) stopped-flow CD folding studies of the entire CC indicate that the ellipticity of the denatured state is smaller than −3,000 deg⋅cm2/dmol at 0.5 M GdmCl (28). These results demonstrate that little residual helical structure exists in the denatured state under our refolding conditions and cannot provide an explanation for the fractional single-site Φf values.
Precollision Helix Formation. Although minimal residual structure exists in the denatured state, transient helix formation still may be required for a productive collision in the folding of the dimeric CC (25). In diffusion-collision models (22), the folding rate is the probability of two monomers being helical multiplied by the success frequency and the diffusion-limited encounter rate, ≈109 M−1⋅s−1. The agadir program (31) predicts 60–70% helix for residues 22–30 in the absence of denaturant. However, all other residues are predicted to be much less helical (Fig. 3A), and the probability of a helical stretch with 11 or more residues is only 4%. This analysis sets an upper limit of 11 residues as the maximum length of precollision helical structure consistent with the dimeric folding rate, 2 × 106 M−1⋅s−1 (extrapolated to 0 M denaturant and assuming all collisions are productive).
However, the mutational results are inconsistent with this amount of precollision helical structure. According to diffusion-collision models, the majority of productive folding events in the CC should involve the most helical region, residues 22–30, and the effect of a mutation in this region on folding rates should be determined by the change in the region’s helicity. The A24G substitution reduces helicity in this region from 69% to 29% (Fig. 3A). The folding rate then should decrease by a factor of 5.7 (ΔΔG‡f = 0.97 kcal/mol). Likewise, the glycine substitution reduces the stability of the dimeric CC by 1.9 kcal/mol (2 RT ln [KeqA24/KeqA24G]). This analysis predicts a ΦfA24G value of 0.51, double the observed value of 0.25. Hence, diffusion-collision models using the high agadir-calculated levels of helix incorrectly predict the observed Φf A24G value.
Under our experimental conditions where the amount of residual structure is minimal, an even stronger argument can be made against the requirement of precollision helical structure. When Keq ≪1, the occurrence of precollision helical structure for a destabilizing mutation decreases as the ratio Keqmutant/KeqWT. The folding rate and the stability both decrease as the square of this ratio. Therefore, the Φf value for the A24G substitution should be near unity according to diffusion-collision models. Because observed value is much less, either the precollision helix formation occurs in regions having much less intrinsic helicity, or more probably, the helical structure present in the TS forms after the initial collision.
The present analysis differs from the recent analysis by Myers and Oas (25), who proposed that precollision helix formation is consistent with the observed rate and single-site mutational data for the dimeric CC (25). Although they also used agadir to calculate the average helicity for the GCN4-p1 sequence, their subsequent analysis assumed a uniform helical propensity. This analysis spreads the helicity more uniformly than that predicted by agadir (Fig. 3A), which results in an overestimation of the probability of encountering long helical stretches in isolated monomers. More importantly, it also underestimates the effects of mutation in the region having the most intrinsic helicity (e.g., A24G), particularly under the present conditions where there is minimal residual structure.
Finally, a modified version of the CC with a strengthened hydrophobic core (32) has a maximum helicity 10-fold lower than GCN4-p1′ (Fig. 3A). This variant folds about 20-fold faster than GCN4-p1′, rather than 102-fold slower as would be predicted by a diffusion-collision model. Further, the near diffusion-limited folding rate found for this CC (3 × 108 M−1⋅s−1) and for a hydrophobic variant of Arc repressor (2 × 108 M−1⋅s−1) (33) indicate that the major fraction of chain encounters are productive so that folding is too fast to be contingent on transient preformed structure.
Secondary Structure, Folding Rates, and Topology. Intrinsic helical propensity does not dominate the choice of folding routes for either CC. For the crosslinked molecule, the region with the highest intrinsic helicity is completely unstructured in the TS. Effective chain concentration, determined by chain connectivity, governs which pathway is selected and folding begins from the least helical region. For the dimeric CC, the region between residues 22 and 30 has the highest intrinsic helicity even with the A24G substitution (Fig. 3A), but less helical regions can comprise the majority of the nucleation sites (ΦfAAG/GGG = 0.72).
Whether the stabilization of local interactions increases folding rates depends on whether a particular element is structured in the TS. For example, the 24th position is unstructured in the TS of the crosslinked CC, and the ≈3 kcal/mol destabilization for the glycine substitution has no effect on folding rates. Furthermore, the version of the dimeric CC with a strengthened hydrophobic core but having only 20% of the intrinsic helicity of GCN4-p1 (Fig. 3A) folds about 20 times faster (32). Evidently, hydrophobicity plays a more important role than intrinsic helicity in the determination of folding rates for this version of the CC.
Even with its simple topology, the folding rate of the crosslinked CC and its contact order of 10% agree well with the correlation between folding speed and the average sequence distance between contacts noted for other proteins (20). Although the topology of the CC should be reasonably well defined once the tether bends and residues in the amino region contact each, the TS has additional requirements. More than a single turn of helix must be formed and about 50% of the denaturant-sensitive surface must be buried (mf/m0) before additional folding steps can proceed in a thermodynamically downhill manner. The helix-stabilizing cosolvent 2,2,2-trifluoroethanol interacts to nearly the same degree with the TS as with folded state in both species (Φfsolvent ≈ 1; ref. 34 and unpublished data), indicating that a high degree of backbone desolvation occurs in the TS. A quantitative connection between folding rates, topology, secondary structure formation, and surface burial remains to be determined.
Recently, β-turns have been postulated to be folding initiation sites because they have some intrinsic stability and are the only structures completely formed in the TS of three proteins (7, 35, 36). Although the tether is unstructured in the crosslinked CC, it increases the local chain concentration and serves the same purpose as a β-turn. Hence, initiation sites need not have any intrinsic stability, and folding can begin from the least helical region of the molecule. These considerations bear on the considerable effort being directed at identifying relatively stable regions of proteins as possible folding initiation sites.
Conclusions. A heterogeneous TS ensemble with multiple nucleation sites located throughout the molecule can explain the minimal effect of helix-destabilizing substitutions on folding rates of the dimeric CC. However, this pathway heterogeneity critically depends on connectivity and is lost on the introduction of an unstructured tether between the two helices.
We have seen that helix formation is unlikely to occur before a productive collision. Yet, one-third to one-half of the molecule becomes helical in the postcollision TS. Although secondary structure stability can influence the selection of pathways in the dimeric CC, the major fraction of nucleation sites need not occur in the most helical region of the molecule. For the crosslinked CC, in fact, the region having the highest helicity is the last to fold. We find it remarkable that such unexpected and complex behavior can be generated from such a simple system.
Supplementary Material
Acknowledgments
We thank S. W. Englander, W. F. DeGrado, N. Kallenbach, M. Weiss, D. Baker, L. Mayne, T. Oas, J. Myers, R. Bhattacharyya, R. L. Baldwin, V. Munoz, and X. Fang for numerous enlightening discussions; J. Myers and T. Oas for communicating their unpublished manuscript; B. Krantz, T. Pan, and S. W. Englander for useful comments on the manuscript; and S. Jackson and R. Wilk for generously providing synthesized peptides. We especially thank V. Munoz for assistance with the agadir calculation. This work was supported in part by National Institutes of Health Research Grants GM55694 (T.R.S.) and GM54616 (W. DeGrado), and Grant CA14599 (T.R.S.) from the National Cancer Institute to the University of Chicago Cancer Research Center.
ABBREVIATIONS
- CC
coiled coil
- GdmCl
guanidinium chloride
- TS
transition state
References
- 1.Abkevich V I, Gutin A M, Shakhnovich E I. Biochemistry. 1994;33:10026–10036. doi: 10.1021/bi00199a029. [DOI] [PubMed] [Google Scholar]
- 2.Guo Z Y, Thirumalai D. Biopolymers. 1995;36:83–102. [Google Scholar]
- 3.Sosnick T R, Mayne L, Hiller R, Englander S W. In: Peptide and Protein Folding Workshop. DeGrado W F, editor. Philadelphia: International Business Communications; 1995. pp. 52–80. [Google Scholar]
- 4.Fersht A R. Proc Natl Acad Sci USA. 1995;92:10869–10873. doi: 10.1073/pnas.92.24.10869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Sosnick T R, Mayne L, Englander S W. Proteins. 1996;24:413–426. doi: 10.1002/(SICI)1097-0134(199604)24:4<413::AID-PROT1>3.0.CO;2-F. [DOI] [PubMed] [Google Scholar]
- 6.Onuchic J N, Socci N D, Luthey-Schulten Z, Wolynes P G. Fold Des. 1996;1:441–450. doi: 10.1016/S1359-0278(96)00060-0. [DOI] [PubMed] [Google Scholar]
- 7.Martinez J C, Pisabarro M T, Serrano L. Nat Struct Biol. 1998;5:721–729. doi: 10.1038/1418. [DOI] [PubMed] [Google Scholar]
- 8.Shakhnovich E I. Fold Des. 1998;3:R108–R111. doi: 10.1016/s1359-0278(98)00056-x. [DOI] [PubMed] [Google Scholar]
- 9.Thirumalai D, Klimov D K. Fold Des. 1998;3:R112–R118. doi: 10.1016/S1359-0278(98)00057-1. [DOI] [PubMed] [Google Scholar]
- 10.Matthews C R. Methods Enzymol. 1987;154:498–511. doi: 10.1016/0076-6879(87)54092-7. [DOI] [PubMed] [Google Scholar]
- 11.Fersht A R, Matouschek A, Serrano L. J Mol Biol. 1992;224:771–782. doi: 10.1016/0022-2836(92)90561-w. [DOI] [PubMed] [Google Scholar]
- 12.Otzen D E, Itzhaki L S, elMasry N F, Jackson S E, Fersht A R. Proc Natl Acad Sci USA. 1994;91:10422–10425. doi: 10.1073/pnas.91.22.10422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Milla M E, Brown B M, Waldburger C D, Sauer R T. Biochemistry. 1995;34:13914–13919. doi: 10.1021/bi00042a024. [DOI] [PubMed] [Google Scholar]
- 14.Sosnick T R, Jackson S, Wilk R M, Englander S W, DeGrado W F. Proteins. 1996;24:427–432. doi: 10.1002/(SICI)1097-0134(199604)24:4<427::AID-PROT2>3.0.CO;2-B. [DOI] [PubMed] [Google Scholar]
- 15.Burton R E, Huang G S, Daugherty M A, Calderone T L, Oas T G. Nat Struct Biol. 1997;4:305–310. doi: 10.1038/nsb0497-305. [DOI] [PubMed] [Google Scholar]
- 16.Kim D E, Yi Q, Gladwin S T, Goldberg J M, Baker D. J Mol Biol. 1998;284:807–815. doi: 10.1006/jmbi.1998.2200. [DOI] [PubMed] [Google Scholar]
- 17.Goldberg J M, Baldwin R L. Proc Natl Acad Sci USA. 1999;96:2019–2024. doi: 10.1073/pnas.96.5.2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Fersht A R, Itzhaki L S, elMasry N F, Matthews J M, Otzen D E. Proc Natl Acad Sci USA. 1994;91:10426–10429. doi: 10.1073/pnas.91.22.10426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Burton R E, Myers J K, Oas T G. Biochemistry. 1998;37:5337–5343. doi: 10.1021/bi980245c. [DOI] [PubMed] [Google Scholar]
- 20.Plaxco K W, Simons K T, Baker D. J Mol Biol. 1998;277:985–994. doi: 10.1006/jmbi.1998.1645. [DOI] [PubMed] [Google Scholar]
- 21.Viguera A R, Villegas V, Aviles F X, Serrano L. Fold Des. 1997;2:23–33. doi: 10.1016/S1359-0278(97)00003-5. [DOI] [PubMed] [Google Scholar]
- 22.Karplus M, Weaver D L. Protein Sci. 1994;3:650–668. doi: 10.1002/pro.5560030413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kammerer R A, Schulthess T, Landwehr R, Lustig A, Engel J, Aebi U, Steinmetz M O. Proc Natl Acad Sci USA. 1998;95:13419–13424. doi: 10.1073/pnas.95.23.13419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lopez-Hernandez E, Cronet P, Serrano L, Munoz V. J Mol Biol. 1997;266:610–620. doi: 10.1006/jmbi.1996.0793. [DOI] [PubMed] [Google Scholar]
- 25.Myers J K, Oas T G. J Mol Biol. 1999;289:205–209. doi: 10.1006/jmbi.1999.2747. [DOI] [PubMed] [Google Scholar]
- 26.Choma C T, Lear J D, Nelson M J, Dutton L P, Robertson D E, DeGrado W F. J Am Chem Soc. 1994;116:856–865. [Google Scholar]
- 27.Bhattacharyya R P, Sosnick T R. Biochemistry. 1999;38:2601–2609. doi: 10.1021/bi982209j. [DOI] [PubMed] [Google Scholar]
- 28.Zitzewitz J A, Bilsel O, Luo J, Jones B E, Matthews C R. Biochemistry. 1995;34:12812–12819. doi: 10.1021/bi00039a042. [DOI] [PubMed] [Google Scholar]
- 29.Creamer T P, Rose G D. Proteins. 1994;19:85–97. doi: 10.1002/prot.340190202. [DOI] [PubMed] [Google Scholar]
- 30.Zitzewitz J A, Ibarra-Molero B, Fishel D R, Terry K L, Matthews C R. Protein Sci. 1999;8:265. -S (abstr.). [Google Scholar]
- 31.Munoz V, Serrano L. Biopolymers. 1997;41:495–509. doi: 10.1002/(SICI)1097-0282(19970415)41:5<495::AID-BIP2>3.0.CO;2-H. [DOI] [PubMed] [Google Scholar]
- 32.Durr E, Jelesarov I, Bosshard H R. Biochemistry. 1999;38:870–880. doi: 10.1021/bi981891e. [DOI] [PubMed] [Google Scholar]
- 33.Waldburger C D, Jonsson T, Sauer R T. Proc Natl Acad Sci USA. 1996;93:2629–2634. doi: 10.1073/pnas.93.7.2629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kentsis A, Sosnick T R. Biochemistry. 1998;37:14613–14622. doi: 10.1021/bi981641y. [DOI] [PubMed] [Google Scholar]
- 35.Gu H, Kim D, Baker D. J Mol Biol. 1997;274:588–596. doi: 10.1006/jmbi.1997.1374. [DOI] [PubMed] [Google Scholar]
- 36.Grantcharova V P, Riddle D S, Santiago J V, Baker D. Nat Struct Biol. 1998;5:714–720. doi: 10.1038/1412. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.