

Abstract
We present a method for deriving energy functions for protein folding by maximizing the thermodynamic average of the overlap with the native state. The method has been tested by using the pairwise contact approximation of the energy function and generating alternative structures by threading sequences over a database of 1,169 structures. With the derived energy function, most native structures: (i) have minimal energy and (ii) are thermodynamically rather stable, and (iii) the corresponding energy landscapes are smooth. Precisely, 92% of the 1,013 x-ray structures are stabilized. Most failures can be attributed to the neglect of interactions between chains forming polychain proteins and of interactions with cofactors. When these are considered, only nine cases remain unexplained. In contrast, 38% of NMR structures are not assigned properly.
The starting point for folding proteins on a computer is to assume, according to Anfinsen's experiments (1), that the native state of the protein is in thermodynamic equilibrium and corresponds to the minimum free energy. The most straightforward approach considers a detailed atomistic model and follows its time evolution either by molecular dynamics (2, 3) or by Monte Carlo simulations (4, 5). To date, only for special regular structures (4, 5) and for small polypeptides (3), the experimentally known native structures have been reproduced in computer experiments. These simulations are still far from being routine methods of structure prediction. The reason is that a protein in solution is only marginally stable, and its behavior depends crucially on subtle details of the interaction. Thus, in most models even minor changes in the energy function can destabilize the native state (6, 7).
An alternative approach consists in adopting a coarse-grained (mesoscopic) description of the protein structure and using an energy function not derived from physical principles but obtained from the information contained in the Protein Data Bank (PDB) of native structures. To carry out this task, several authors assume that the structural motifs in the set of native protein structures follow a Boltzmann distribution, whose energy function is calculated from the observed frequencies (8–12). Here we follow a different approach and determine the energy parameters by an optimization scheme.
Two optimization schemes have been proposed so far. Their common goal is to obtain an energy function such that the ground state of the model corresponds to the observed native structure and is thermodynamically stable. A first optimization scheme, introduced by Maiorov and Crippen (13), requires that the native states of a target set of proteins have energies lower than for a set of alternative structures. This is obtained by solving a system of inequalities. Recently, this method has been improved by Domany and coworkers (14–16) and Maritan and coworkers (17, 18). In the new formulation, it is possible to answer rigorously the question whether the system of inequalities is solvable. If a solution exists, it is not unique, and it is possible to improve the method by choosing the solution of maximal stability, defined as the solution in which the stability gap of the least stable protein is maximized (16, 18).
A second class of methods (19–23) aims at providing the largest possible thermodynamic stability to the target proteins. These methods optimize quantities related to the Z score (24), measuring the difference between the energy of the native state and the average energy of the alternative states in units of the standard deviation of the energy. Goldstein et al. (19, 20), inspired by a spin-glass analysis, derived efficient parameters for fold recognition. More recently, the method was extended to protein structure prediction via simulated annealing (21). Hao and Scheraga applied a similar method to the more difficult problem of deriving an energy function for folding simulations of a single protein (22). Mirny and Shakhnovich proposed optimizing the Z score to obtain an energy function conferring large stability to most native states of a target set of proteins (23). These methods, however, do not guarantee that the native structure has the lowest energy among all alternative structures.
We present a third method, which combines the main advantages of the two previous ones. Here, the energy function is determined by maximizing the average native overlap Q. When Q is very close to 1, it is guaranteed that the native state and the ground state coincide. Moreover, a large value of Q also indicates that the native state is thermodynamically stable and suggests that the energy landscape is well correlated.
As a first application of the method, we determine optimal parameters for the pairwise contact approximation of the energy function. We consider a database of 1,169 protein chains (25, 26) and generate alternative structures by threading (9, 10, 13). Our energy function stabilizes 92% of the 1,013 x-ray structures even without considering interactions between different chains and with cofactors. These can explain the remaining cases with very few exceptions. On the other hand, only 62% of NMR structures are stable. This can be at least partially explained by the way in which these structures are represented in the PDB files.
In the next section, we define the theoretical framework of optimization methods. Then we describe our method and apply it to the determination of pairwise contact interactions. Finally, we discuss our results.
Optimizing Energy Parameters
We consider a chain of N amino acids and an effective energy function E(C, S) depending on the mesoscopic configuration Cε/ΩN and on the sequence S ={S1, … SN}εΣ. We choose as mesoscopic representation the contact map matrix C = f(Γ), where Γ is the microscopic state and
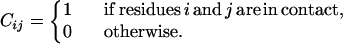 |
1 |
We consider two residues in contact if they are separated by more than two residues along the sequence and if any two heavy atoms belonging to them are closer than a threshold distance of 4.5 Å (16).
The ensemble of configurations ΩN is the ensemble of all contact maps realized by a given set of structures with N residues. We shall consider three possibilities:
(i) The threading set (9, 10, 13): backbone structures are obtained by cutting in all possible ways structures with length N′ > N contained in a subset of the PDB.
(ii) The lattice set: structures are self avoiding random walks (28) on a suitable lattice.
(iii) The off-lattice set: structures are obtained from Monte Carlo or molecular dynamics simulations that fulfill the physical constraints of excluded volume and of definite values for bond angles and lengths.
The present study will be limited to the simpler cases of lattice and threading sets. The more difficult case of an off-lattice set, relevant for folding simulations, will be studied in a forthcoming paper.
A central role in our method is played by the overlap q(C,C′), which measures the similarity between two contact maps.
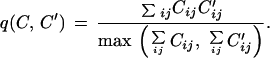 |
2 |
It holds q(C,C′)ɛ (0, 1) and q(C,C′) = 1 if and only if the two contact maps are equal.
We can compute formally the free energy FT(C, S)= ET(C, S) − TS(C) of the mesoscopic state C assuming that the protein-solvent system is in thermodynamic equilibrium at temperature T and that all microscopic configurations Γ and their energies ℰ(Γ) are known:
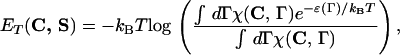 |
3 |
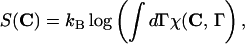 |
where we introduced the characteristic function of the contact map C, χ(C,Γ) = δ(1 − q(f(Γ),C)). The entropy S(C) increases as the compactness of C decreases (29). The above computation is only formal, however. We assume in the following that the effective energy function can be represented as the sum of contact interactions,
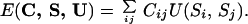 |
4 |
This expression depends on a set of 210 interaction energies U = {U(a,b)}, where a, b ε [1, 20] are 2 of the 20 types of amino acids, and the U(a,b) are temperature dependent. This is the functional form of the energy most studied in the literature. It has been proved by Domany and coworkers (14, 15) that this energy function is not accurate enough to allow off-lattice folding simulations. Following refs. 14 and 15, we consider Eq. 4 as a first step in a phenomenological approximation scheme and determine the best parameters at this level of description. The method that we develop can be applied to include additional energy terms. Another possibility would be to consider a more refined mesoscopic description where, for example, dihedral angles are also taken into account. However, for the simple case of the threading set, it turns out that the contact energy is a rather good approximation.
The Overlap Method.
Folding simulations are mainly aimed at finding low-energy structures of a protein model that are as similar as possible to the true native structure Cn(S). A measure of such similarity is the overlap q0 = q(C0(S),Cn(S)), where C0(S) is the lowest energy contact map for sequence S. If q0 = 1, we are guaranteed that the native state contact map has the lowest energy. In the spirit of our statistical mechanics approach, we optimize the similarity between the native structure and the whole Boltzmann ensemble obtained from a simulation or by threading. We define the average native overlap
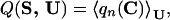 |
5 |
where qn(C) = q(C,Cn(S)), Cn(S) being the native state. The brackets denote a Boltzmann average in the ensemble Ω, 〈A(C)〉 = 1/z∑C A(C) exp(−F(C, S)) and z = ∑C exp(−F(C, S)), and the interaction parameters U are measured in units such that kBT = 1. For threading, the entropy of contact maps is zero, and we have F(C, S) = E(C, S, U). There are three advantages in optimizing Q(S, U) instead of q0: (i) Q(S, U) yields information about the thermodynamic stability of Cn(S). A value of Q close to 1 does not only mean that C0(S) and Cn(S) coincide, but also that Cn(S) has a large Boltzmann weight. (ii) If the temperature is not too low and the set of alternative conformations contains structures similar enough to the native one, this condition implies also that the low energy states are those similar to Cn(S). This is a way to obtain a smooth energy landscape, where the energy decreases on the average as Cn(S) is approached. For most reasonable dynamical rules, the correlation between E and q is expected to favor fast folding, in agreement with the funnel scenario proposed by Bryngelson and Wolynes (30) and with lattice models (31–36). (iii) Q(S, U) takes into account also the entropy of the native contact map.
The structural similarity with the native state has also been used in the optimization procedure in the framework of the inequality method (13, 18). However, such method, at variance with the one presented here, does not use an optimization criterion based on statistical averages, as in Eq. 5, but imposes inequalities for each alternative structure. The difference might be relevant in the cases where some proteins whose native state is not very stable are included in the target set, as can happen in real applications.
In analogy to ref. 22, we obtain the gradient of Q(S, U) with respect to U and use it in the optimization.
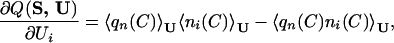 |
6 |
where Ui is the ith energy parameter and ni(C) counts how many contacts of type i are present in the configuration C. The optimization works iteratively. At each step, we compute the gradient and update the interaction parameters with a gradient descent algorithm. The new interaction matrix is then multiplied with a scalar 1/τ so that 𝒰2 = ∑a,bU2(a,b) is kept constant. This multiplication is equivalent to rescaling the temperature by τ without changing the interactions. In absence of it, 𝒰 would grow during the optimization, so that the effective temperature of the system decreases and the ground state becomes more stable. In other words, Q(S, U) possesses trivial local maxima for 𝒰 → ∞. In general, the system can reach local maxima lower than the desired one Q ≈ 1 when the structure of lowest energy is the most similar to Cn(S) among the thermodynamically relevant structures. In this situation, every change that makes this structure more stable produces an increase of Q. However, in analogy with ref. 22, we can drive the system toward the true maximum Q ≈ 1 by decreasing the energy of the native structure as well. To this end, we use the following optimization equation:
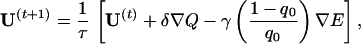 |
7 |
where q0 = q(C0,Cn(S)) is the overlap between the ground state and the target structure. When q0 ≈ 1, only the optimization of Q takes place.
In this study, we consider NS different sequences, and we optimize their average overlap 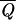 (U) = 1/NS∑SɛΣQ(S, U).
(U) = 1/NS∑SɛΣQ(S, U).
Results
Lattice Model.
We first tested the method on a lattice model with 36 residues (37). We considered a target structure C* and an appropriate sequence S*. After few iterations, we obtained an energy function such that C* was the ground state of S*, and it was thermodynamically very stable. Moreover, the energy landscape was very correlated (data not shown).
Protein Structures.
We considered three sets of proteins: 456 chains extracted from the whatif database (24) (database A), 713 chains contained in the latest release of the PDB select set (25) and which have less than 90% sequence homology with the chains in database A (database B), and the union of them, containing 1,169 chains (database C).
We show in Fig. 1 the number of intrachain contacts Nc vs. the number of residues N. For single-chain proteins, a good fit to the data is the line Nc/N ≈ a − bN−1/3, as expected from surface effects (38). The best fit values are a = 3.9 ± 0.1, and b = 5.8 ± 0.2 ≈ 3/2a. Thus, by using a threshold of 4.5 Å for contacts, residues in the interior of the globule have on the average nearly 8 contacts (to compute Nc, we divide the sum of these numbers by 2) plus 4 contacts with neighbors along the chain, not contributing to Nc, which makes roughly two contacts per Cartesian direction. However, this number depends strongly on the residue type. Proteins formed by several chains have on the average less intrachain contacts. Interchain interactions can be very important for the stability of the native structure, but they cannot be considered with threading. Also interactions with cofactors cannot be considered. Thus polychain proteins sometimes have a ground state different from the native one (gray symbols in Fig. 1). Structures determined by NMR have a distribution of contacts broader and with smaller average than that of x-ray structures.
Figure 1.

Fraction of intrachain contacts as a function of the number of residues to the power −1/3. Filled symbols are protein chains whose ground state does not coincide with the native structure.
Energy Parameters.
We used as target set a subset of database A with 47 single-chain proteins, generating decoys from a set of 120 chains. For the resulting set of parameters U(1) only in 29 cases of 456, the ground state C0(S) differs from the native structure Cn(S), and all these are either chains belonging to polychain proteins or small proteins with cofactors, for which some of the native interactions are neglected. The protein chains for which ground state and native structure coincide have 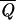 = 0.93, and thus most of them are rather stable. Using smaller target sets, we found almost the same parameters, and we did not obtain significantly better results even by using all database A as a target. Hence, the energy function remains stable by enlarging the target set. It is interesting that 27 proteins had q0 < 1 with all of the energy functions that we derived. We tested the energy parameters U(1) also on database B. It is convenient to divide these chains in three classes. For the 202 single chains, the ground state differs only in six cases from the native structure. All of these proteins but one contain cofactors. The fraction of proteins with Q > 0.6 is 95%. For 80% of the 377 chains belonging to polychain proteins, we found q0 = 1, and Q > 0.6 in 75% of the cases, whereas only 50% of the 136 protein structures determined by NMR have q0 = 1, and many of them are not very stable. These results show that predictions of NMR structures are more difficult than those of x-ray structures. The necessity of distinguishing between NMR and x-ray structures when one derives an energy function has already been pointed out by Godzik et al (39).
= 0.93, and thus most of them are rather stable. Using smaller target sets, we found almost the same parameters, and we did not obtain significantly better results even by using all database A as a target. Hence, the energy function remains stable by enlarging the target set. It is interesting that 27 proteins had q0 < 1 with all of the energy functions that we derived. We tested the energy parameters U(1) also on database B. It is convenient to divide these chains in three classes. For the 202 single chains, the ground state differs only in six cases from the native structure. All of these proteins but one contain cofactors. The fraction of proteins with Q > 0.6 is 95%. For 80% of the 377 chains belonging to polychain proteins, we found q0 = 1, and Q > 0.6 in 75% of the cases, whereas only 50% of the 136 protein structures determined by NMR have q0 = 1, and many of them are not very stable. These results show that predictions of NMR structures are more difficult than those of x-ray structures. The necessity of distinguishing between NMR and x-ray structures when one derives an energy function has already been pointed out by Godzik et al (39).
We performed another optimization run by using as a target all 1,079 chains with length N ≤ 455. The number of alternative structures, generated from the whole database C, varies from 38,000 for N = 455 to 209,000 for the shortest chain with N = 30. Because some chains are closely related in structure and in sequence, we should expect the recognition to become more difficult. We give below the results for the new energy function U(2).
(i) For 401 of the 406 single-chain proteins, q0 = 1 and Q(S) > 0.7. Exceptions are five proteins with cofactors.
(ii) 83% of the 515 chains in polychain proteins have q0 = 1 and Q(S) > 0.7.
(iii) 68% of the 153 NMR structures have the correct ground state, and only 60% have Q(S) > 0.7.
Energy Landscape and Stability.
Studies on lattice models suggest that energy correlations are a key ingredient for obtaining fast folding models (31–36). As discussed briefly above, for lattice models our method provides very well correlated energy landscapes. The same correlations are found with threading, although only few structures with large q are available. In Fig. 2, we plot in the plane (q, E) the lowest energy structures of six sequences selected so that alternative structures with q(C, Cn) > 0.6 exist, and the native structure has lowest energy. Under these conditions, structures with large overlap also have low energy.
Figure 2.

Normalized energy gap vs. the overlap q for six protein chains.
For each sequence, we define a dimensionless parameter α(S) such that
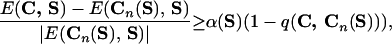 |
8 |
and α(S) = 0 in case the energy of the native structure is not lowest. This parameter can be used to characterize stability and fast folding. The smoother the energy landscape of chain S, the larger α(S). Related information is given by the Z score (27) and by the energy gap used by Shakhnovich and coworkers as a signature of fast folding (40), but the parameter α(S) is also sensitive to the presence of low energies structures unrelated to the native one. The value of α(S) depends on chain length N and it is seen to increase roughly as 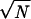 . It also decreases slightly when the number of alternative conformations used increases. Fig. 3 shows the distribution of α(S)/
. It also decreases slightly when the number of alternative conformations used increases. Fig. 3 shows the distribution of α(S)/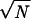 for chains for which native state and ground state coincide, distinguishing three classes of proteins. Single chains are most stable, followed by polychains and NMR structures.
for chains for which native state and ground state coincide, distinguishing three classes of proteins. Single chains are most stable, followed by polychains and NMR structures.
Figure 3.

Distribution of the normalized stability parameter α(S)/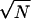 , where N is the number of residues and only chains with qo = 1 are included.
, where N is the number of residues and only chains with qo = 1 are included.
Many of the native structures whose energies are not minimal are only moderately unstable. We tried to see whether the interchain contacts and the cofactors, not taken into account with threading, are able to stabilize the native states. We computed the interaction energies between different chains and estimated the interactions with cofactors, assuming that one contact with a cofactor contributes an energy equal to the average energy of a native contact. This, however, may underestimate the interactions if the cofactors are charged or covalently bound. For 72 of 87 problematic x-ray structures, the coordinates of all chains and cofactors are available, and we saw that the corrected native energy is lower than the ground state energy of the single chain in all cases but nine. The PDB codes of the nine exceptions are: 3cyr (cytochrome c3 has a heme covalently bound), 1ail (small fragment), 1cbn (crambin), 1ajj (fragment of a receptor protein with charged cofactors), 1aws A (isomerase bound to HIV capside protein, q0 = 0.94), 1rfb A (interferon γ), 1isu A (iron–sulfur cluster), 1tgs I and 1fle I (inhibitors bound to an enzyme).
Structure Prediction.
The usefulness of the parameter set U(2) for structure prediction is illustrated in Fig. 4. We imagine considering sequences S, whose native structure Cn(S) is unknown, and we search for the lowest energy contact map different from the native one, C1(S, U). A histogram of q1 = q(Cn(S),C1(S, U)) is shown in Fig. 4. We note that the goodness of the prediction, q1, depends on the maximal similarity qmax between the native structure and the other structures in the threading set, whose histogram is also shown in Fig. 4. For only a few chains are there alternative structures with q > 0.6. In Fig. 4 Inset, we show the average ratio between the best prediction q1 and the best possible one, qmax, as a function of qmax. The correlation between q1 and qmax is nearly absent for qmax < 0.6 and rather strong for qmax > 0.6, in particular for single chains. For qmax > 0.8, the best prediction coincides in most cases with the best possible one. Thus, provided that structures with q > 0.8 respect to the structure of an unknown protein exist in the database, our energy function singles them out with large probability. This result suggests that the limitation to the prediction ability is posed by the limitations of threading more than by the accuracy of the energy function. Better strategies to generate alternative structures would be very useful. We must however remark that structures with q > 0.8 are derived from proteins related also in sequence to the target protein, so that the easier strategy of homology modeling would exhibit a comparable performance.
Figure 4.

Histograms of the overlap q1 between the best prediction and the native structure and of the maximal overlap qmax for single chains. (Inset) Average value of q1/qmax as a function of qmax.
Conclusions
We have presented a method for deriving energy parameters based on the maximization of the average overlap with the native structure for a target set of proteins. We tested the method on a database of 1,169 structures by using a contact approximation for the energy. For 92% of the 1,013 x-ray structures, the native state and the ground state coincide. Exceptions are mainly because we neglected interchain contacts and interactions with cofactors. When these are taken into account, only nine cases remain unexplained. Thus, surprisingly, it seems possible that the contact interaction energy, although unsuitable for off-lattice simulations (17, 18), is sufficient to account for the stability of x-ray structures if competing structures are generated by threading, and all relevant interactions are considered properly. The last condition was absent in ref. 16, hence the difference in the present results.
For NMR structures, the situation is different. In about one-third of the cases, we found alternative structures with an energy lower than the native structure. Interchain and cofactor interactions explain only nine cases. We also used the 136 NMR structures alone in the target set, but the number of misfolded chains decreased only from 49 to 43, still considerably worse than for x-ray structures. A simple difference between the two is the way in which they are represented in the PDB. For NMR structures, instead of an average structure, a list of conformers, typically 20, is provided. The contact maps used in this work were obtained from the first conformer of the list. The contact map can vary considerably from one conformer to the other, and the same happens with the contact energy. Moreover, NMR structures are generally more disordered than x-ray structures, and they often belong to small proteins, which are difficult to crystallize. Thus, the best strategy is probably to select only x-ray structures for the target set. However, this is not necessary. Our algorithm is able to identify the difficult cases in the target set even without external information. If incorrect matches of structure and sequence are present in the target set, we do not get an energy function performing worse for all of the chains, but we get a large value of Q for some chains and a low value for others. The results are robust with respect to changes of the target set.
The optimization procedure is rather efficient. One iteration for the target set with 1,000 proteins takes 3 h on a SGI Unix workstation with a R4000 central processing unit and less than 10 min for the target set with 47 chains. Less than 10 iterations are sufficient for convergence.
Following ref. 41, we compared our interaction matrix to hydrophobicity and to other known potentials. The correlation is good with respect to the hydrophobicity measured by Fauchere and Pliska (42), if we exclude the cysteine–cysteine interaction, which is much stronger than expected on the basis of hydrophobicity. The correlation coefficient between the hydrophobicity h(a) and the self-interaction term U(a,a) is r = 0.72, and between h(a) and the average interaction 1/20∑bU(a,b), it is r = 0.77. Also the correlation with respect to other knowledge-based potentials is good if we exclude the cysteine–cysteine interaction, which is much stronger in our potential. The correlation coefficient is r = 0.74 with the Miyazawa–Jernigan potential (8), r = 0.83 with the Skolnick et al. potential (11), and r = 0.70 with the Thomas and Dill potential (12). This comparison also reveals two interesting points. First, the ranking of single-chain proteins, polychain proteins, and NMR structures is confirmed with all of the above potentials. Second, all of the proteins that have q0 < 1 with our potential also have q0 < 1 with at least two of the three other potentials.
The real challenge is to apply this method to real space simulations of protein folding. This will be the subject of forthcoming work.
Acknowledgments
U.B. is supported by the Deutsche Forschungsgemeinschaft SFB 312, Project D7. M.V. is supported by the Biotechnology and Biological Sciences Research Council and by the European Moleuclar Biology Organization.
Abbreviation
- PDB
Protein Data Bank
References
- 1.Anfinsen C-B. Science. 1973;181:223–230. doi: 10.1126/science.181.4096.223. [DOI] [PubMed] [Google Scholar]
- 2.Lazaridis T, Karplus M. Science. 1997;278:1928–1931. doi: 10.1126/science.278.5345.1928. [DOI] [PubMed] [Google Scholar]
- 3.Duan Y, Kollman P-A. Science. 1998;282:740–744. doi: 10.1126/science.282.5389.740. [DOI] [PubMed] [Google Scholar]
- 4.Skolnick J, Kolinski A. Science. 1990;250:1121–1125. doi: 10.1126/science.250.4984.1121. [DOI] [PubMed] [Google Scholar]
- 5.Hoffman D, Knapp E-W. Phys Rev E. 1996;53:4221–4224. doi: 10.1103/physreve.53.4221. [DOI] [PubMed] [Google Scholar]
- 6.Pande V S, Grosberg A Yu, Tanaka T. Folding Des. 1997;2:109–114. doi: 10.1016/s1359-0278(97)00015-1. [DOI] [PubMed] [Google Scholar]
- 7.Vendruscolo M, Maritan A, Banavar J R. Phys Rev Lett. 1997;78:3967–3970. [Google Scholar]
- 8.Miyazawa S, Jernigan R-L. Macromolecules. 1985;18:534–552. [Google Scholar]
- 9.Sippl M-J. J Mol Biol. 1990;213:859–883. doi: 10.1016/s0022-2836(05)80269-4. [DOI] [PubMed] [Google Scholar]
- 10.Heindlich M, Lackner P, Weitckus S, Floeckner H, Froschauer R, Gottsbacher K, Casari G, Sippl M-J. J Mol Biol. 1990;216:167–180. doi: 10.1016/S0022-2836(05)80068-3. [DOI] [PubMed] [Google Scholar]
- 11.Skolnick J, Jaroszweski L, Kolinski A, Godzik A. Protein Sci. 1997;6:676–688. doi: 10.1002/pro.5560060317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Thomas P-D, Dill K-A. Proc Natl Acad Sci USA. 1996;93:11628–11633. doi: 10.1073/pnas.93.21.11628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Maiorov V-N, Crippen G-M. J Mol Biol. 1992;227:876–888. doi: 10.1016/0022-2836(92)90228-c. [DOI] [PubMed] [Google Scholar]
- 14.Vendruscolo M, Domany E. J Chem Phys. 1998;109:11101–11108. [Google Scholar]
- 15.Vendruscolo M, Najmanovich R, Domany E. Phys Rev Lett. 1999;82:656–659. [Google Scholar]
- 16.Vendruscolo M, Najmanovich R, Domany E. Proteins. 2000;38:134–148. doi: 10.1002/(sici)1097-0134(20000201)38:2<134::aid-prot3>3.0.co;2-a. [DOI] [PubMed] [Google Scholar]
- 17.van Mourik J, Clementi C, Maritan A, Seno F, Banavar J R. J Chem Phys. 1999;110:10123–10133. [Google Scholar]
- 18.Settanni G, Micheletti C, Banavar J R, Maritan A. preprint available at http://xxx.lanl.gov/list/cond-mat/9902?300. 1999. [Google Scholar]
- 19.Goldstein R, Luthey-Shulten Z-A, Wolynes P-G. Proc Natl Acad Sci USA. 1992;89:4918–4922. doi: 10.1073/pnas.89.11.4918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Goldstein R, Luthey-Shulten Z-A, Wolynes P-G. Proc Natl Acad Sci USA. 1992;89:9029–9033. doi: 10.1073/pnas.89.19.9029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Koretke K K, Luthey-Shulten Z-A, Wolynes P-G. Proc Natl Acad Sci USA. 1998;89:2932–2937. doi: 10.1073/pnas.95.6.2932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hao M-H, Scheraga H-A. Proc Natl Acad Sci USA. 1996;93:4984–4989. doi: 10.1073/pnas.93.10.4984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Mirny L, Shakhnovich E-I. J Mol Biol. 1996;264:1164–1179. doi: 10.1006/jmbi.1996.0704. [DOI] [PubMed] [Google Scholar]
- 24.Bowie J-U, Lüthy R, Eisenberg D. Science. 1991;253:164–170. doi: 10.1126/science.1853201. [DOI] [PubMed] [Google Scholar]
- 25.Hooft R W, Vriend G, Sander C, Abola E E. Nature (London) 1996;381:272. doi: 10.1038/381272a0. [DOI] [PubMed] [Google Scholar]
- 26.Hobohm U, Sander C. Protein Sci. 1994;3:522–524. doi: 10.1002/pro.5560030317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hao M-H, Scheraga H-A. Curr Opin Struct Biol. 1999;9:184–188. doi: 10.1016/s0959-440x(99)80026-8. [DOI] [PubMed] [Google Scholar]
- 28.De Gennes P-G. Scaling Concepts in Polymer Physics. Ithaca, NY : Cornell Univ. Press; 1979. [Google Scholar]
- 29.Vendruscolo M, Subramanian B, Kanter I, Domany E, Lebowitz J L. Phys Rev E. 1999;59:977–984. [Google Scholar]
- 30.Bryngelson J-D, Wolynes P-G. Proc Natl Acad Sci USA. 1987;84:7524–7528. doi: 10.1073/pnas.84.21.7524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Socci N D, Onuchic J N, Wolynes P G. Proteins. 1998;32:136–158. [PubMed] [Google Scholar]
- 32.Mirny L-A, Abkevich V, Shakhnovich E-I. Folding Des. 1996;1:103–116. doi: 10.1016/S1359-0278(96)00019-3. [DOI] [PubMed] [Google Scholar]
- 33.Dill K-A, Chan H-S. Nat Struct Biol. 1997;4:10–19. doi: 10.1038/nsb0197-10. [DOI] [PubMed] [Google Scholar]
- 34.Govindarajan S, Goldstein R-A. Proc Natl Acad Sci USA. 1998;95:5545–5549. doi: 10.1073/pnas.95.10.5545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Cieplak M, Henkel M, Karbowski J, Banavar J R. Phys Rev Lett. 1998;80:3654–3657. [Google Scholar]
- 36.Bastolla U, Frauenkron H, Gerstner E, Grassberger P, Nadler W. Proteins. 1998;32:52–66. [PubMed] [Google Scholar]
- 37.Abkevich V-I, Gutin A-M, Shakhnovich E-I. J Chem Phys. 1994;101:6052–6062. [Google Scholar]
- 38.Vendruscolo M, Kussell E, Domany E. Folding Des. 1997;2:295–306. doi: 10.1016/S1359-0278(97)00041-2. [DOI] [PubMed] [Google Scholar]
- 39.Godzik A, Kolimski A, Skolmick J. Protein Sci. 1995;4:270–277. [Google Scholar]
- 40.Dinner A-R, Abkevich V, Shakhnovich E-I, Karplus M. Proteins. 1999;35:34–40. doi: 10.1002/(sici)1097-0134(19990401)35:1<34::aid-prot4>3.0.co;2-q. [DOI] [PubMed] [Google Scholar]
- 41.Betancourt M-R, Thirumalai D. Protein Sci. 1998;8:361–369. doi: 10.1110/ps.8.2.361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Fauchere J-L, Pliska V. Eur J Med Chem. 1983;18:369–375. [Google Scholar]