

Abstract
Feline Leukemia Virus (FeLV) subgroup T uses both a multiple membrane-spanning receptor, FePit1, and a soluble cofactor, FeLIX, to enter feline cells. FeLIX is expressed from endogenous FeLV-related sequence and resembles the receptor binding domain (RBD) of the viral envelope protein. It remains unclear whether FeLV-T receptor activity requires specific residues within FePit1 and FeLIX and/or a threshold level of receptor/cofactor expression. To address this, we examined FeLV-T infection of cells expressing variable levels of FePit1 and other gammaretroviral receptors in the presence of variable amounts of soluble cofactor, either RBD or the envelope surface subunit (SU). Cofactor receptor-pairs fall into three groups with regards to mediating FeLV-T infection: those that are efficient at all concentrations tested, such as FePit1 and FeLIX; those requiring high expression of both cofactor and receptor, and those that are nonfunctional as receptors even at high expression. This suggests that both expression levels and specific interactions with receptor and cofactor are critical for mediating entry of FeLV-T.
Keywords: FeLV, gammaretrovirus, receptor, viral entry
INTRODUCTION
Most simple retroviruses, including the major subgroups of the gammaretrovirus feline leukemia virus (FeLV), appear to require a single, cell-surface, small molecule transporter protein for mediating the requisite receptor functions leading to entry into host cells (Overbaugh, Miller, and Eiden, 2001; Tailor et al., 2003). Receptor interaction is facilitated by the two subunits of the viral envelope protein: the surface subunit (SU), which binds to the receptor via the receptor binding domain (RBD); and the transmembrane domain (TM), which tethers the envelope to the plasma membrane and also contains the fusion machinery.
The four subgroups of FeLV use different receptors for entry into cells. FeLV-A, which is highly conserved across different decades and geographic locations (Donahue et al., 1988), is the transmissible form of FeLV, and uses the receptor FeTHTR1 (Mendoza, Anderson, and Overbaugh, 2006). Chronic infection by FeLV-A in cats gives rise to mutations in the envelope gene that lead to the development of new viral subgroups that recognize new cellular receptors (Overbaugh, Miller, and Eiden, 2001; Rohn, 1999a). For example, FeLV-C arises from FeLV-A by characteristic point mutations leading to usage of FLVCR, a heme transporter protein, for receptor function (Quigley et al., 2000; Quigley et al., 2004; Tailor, Willett, and Kabat, 1999). FeLV-A can also recombine with related endogenous sequences to form FeLV-B, which uses for a receptor either of two phosphate transporter proteins, FePit1 or FePit2, (Anderson et al., 2001; Takeuchi et al., 1992). The fourth subgroup, FeLV-T, is described below.
A combination of an insertion and single amino acid changes, including changes at a conserved PHQ motif at the N-terminus of SU (Overbaugh et al., 1988), are required to convert FeLV-A into FeLV-T (Cheng et al., 2006; Gwynn et al., 2000; Rohn et al., 1994; Rohn et al., 1998). FeLV-T was the first identified example of a naturally occurring simple retrovirus requiring two host proteins to enter and infect, thereby representing a non-classical entry pathway for gammaretroviruses (Anderson et al., 2000; Cheng et al., 2006). In addition to requiring the phosphate transporter FePit1, FeLV-T also requires the soluble cofactor, FeLIX, which is coded for by endogenous FeLV sequence resembling RBD of the envelope SU (Anderson et al., 2000).
Other soluble SUs from a diverse subset of gammaretroviruses paired with their cognate receptors (FeLV-A, GALV, amphotropic MLV) have been tested as cofactor/receptor combinations permissive for FeLV-T entry; among those, only cofactors derived from endogenous FeLV, such as FeLIX or FeLV-B SU, could function as efficient cofactors for FeLV-T infection (Lauring, Anderson, and Overbaugh, 2001). Moreover, these endogenous FeLV-derived cofactors could only mediate efficient FeLV-T infection in combination with the receptor FePit1 and not with the other FeLV-B receptor, FePit2 (Lauring, Anderson, and Overbaugh, 2001). This implied that FeLV-T has specific requirements for the cofactor/receptor combinations that will function to permit entry. However, a more recent report suggests that at least one other murine cofactor/receptor pair may function for entry of FeLV-T, suggesting some promiscuity in FeLV-T receptor requirements, at least in cells of other species (Barnett et al., 2003).
Other groups have shown that engineering a disruption or deletion of the histidine of the N-terminal PHQ motif conserved among gammaretroviruses resulted in murine leukemia viruses (MLVs) that were not infectious using only the cognate receptor (Bae, Kingsman, and Kingsman, 1997). However, these engineered MLVs could be rescued for infection by the addition of soluble RBDs containing a wildtype histidine, in the presence of the cognate cell-surface receptor (Barnett and Cunningham, 2001; Barnett, Davey, and Cunningham, 2001; Lavillette et al., 2000). Thus, the entry requirements appeared similar to those of naturally occurring FeLV-T. Interestingly, the engineered MLVs could be rescued by a variety of heterologous soluble RBD cofactors paired with cognate receptors, so long as the receptors for both the virus envelope disrupted in PHQ and the wildtype soluble RBD were present on the cell surface (Lavillette et al., 2000). This broader specificity for heterologous cofactor/receptor pairs was subsequently reported for other retroviruses, including two naturally occurring retroviruses: Mink cell focus-forming virus (MCF) and Porcine endogenous retroviruses (PERVs). MCF can utilize the RBD of its precursor virus form, Fr-MLV, bound to the Fr-MLV receptor MCAT in the absence of its own receptor, Syg1 for viral entry (Wensel, Li, and Cunningham, 2003). PERVs have also been shown to gain entry by this non-classical pathway using a combination of GALV sRBD and Pit1 (Lavillette and Kabat, 2004). These examples reflect less stringent cofactor/receptor requirements than we had observed for FeLV-T.
One possible explanation for the differences in specificity observed between our initial study and subsequent studies is the differences in length of the soluble envelope constructs used to generate soluble cofactors. We had previously used a soluble form of the envelope SU, as we were interested in what is occurring during natural infections in cats where this form of envelope could be present on the surface of infected cells. In contrast, MLV studies focused on an engineered form of the SU that was truncated to remove the C-terminus, thus included primarily the RBD. Additionally, differences in cell-surface expression levels of the receptors between cell lines may be critical for determining entry, and this has not been carefully addressed for FeLV-T.
To address these questions, we compared the effects of using soluble RBD as compared to soluble SU cofactors on FeLV-T infection. We constructed a number of different cell lines that express varying amounts of cell surface receptor, and found that there are distinct thresholds for both cell-surface receptor expression and cofactor levels that depend on the receptor examined. These studies suggest that cells in the cat that are infected by the other FeLV subgroups may all be targets of infection by FeLV-T.
RESULTS
Other cofactors besides those derived from endogenous FeLV sequences can mediate FeLV-T infection
In order to determine whether the presence of the proline-rich region and C-terminal domain of SU could account for differences in the ability to serve as a cofactor for FeLV-T, we constructed full-length SU and RBD-length versions of various viral envelope proteins (Figure 1A). All of the cofactors were expressed as judged by Western blot, although there was a substantial increase in the expression of FeLV-C-FSC sRBD and GALV sRBD compared to the other proteins (data not shown). Because we used a quantitative method of Western blotting, the results of the initial Western blot could be used to adjust the volume of each preparation so that roughly comparable amounts of cofactor protein could be used for further studies (Figure 1B).
Figure 1. Panel of soluble gammaretroviral envelope RBDs and SUs tested as entry cofactors.
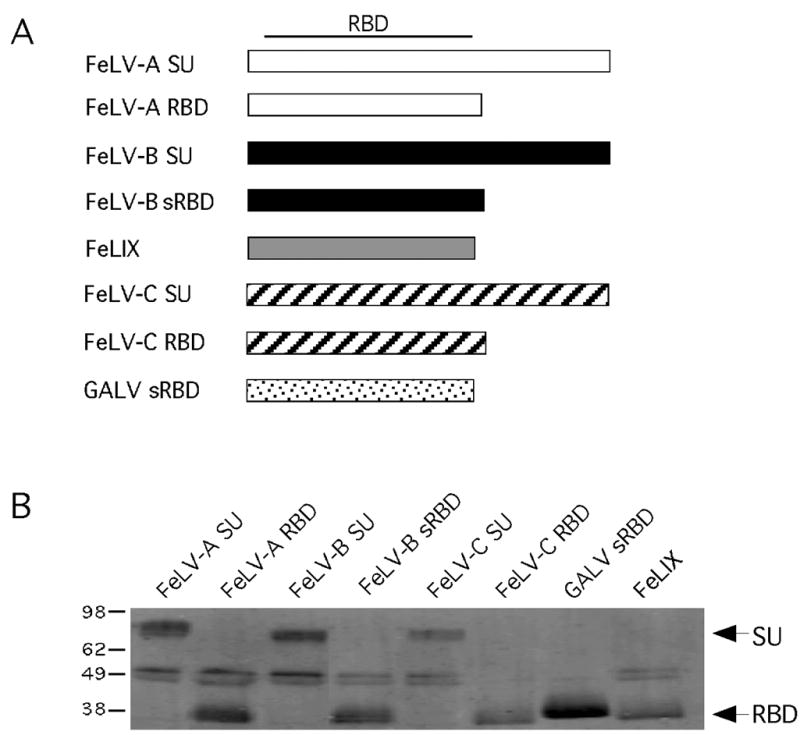
(A) Schematic representation of the structure of each envelope-derived cofactor. The name of each cofactor is indicated to the left of the boxes representing the linear DNA sequence. The approximate location of the receptor binding domain (RBD) within the SU is indicated at the top. (B) Western blot analysis of a volume of conditioned supernatant that corresponds to one equivalent unit of each envelope-derived cofactor. The amount loaded in each lane was determined from the results of quantitative Western blot analysis of equal amounts of each supernatant (not shown). Envelope-derived cofactors were detected with the monoclonal antibody HA.11 to the C-terminal HA-epitope tags as described in Materials and Methods. To the left of the blot are approximate kilodaltons; to the right are the expected sizes of the SU and sRBD constructs.
As seen previously, cofactors derived from endogenous FeLV sequences, FeLIX, FeLV-B-90Z SU and FeLV-B-90Z sRBD were able to mediate FeLV-T infection; in the presence of FePit1 yielding titers on the order of ~5x105 IU/ml (Figure 2). We also found that FeLV-T infected cells expressing FLVCR with FeLV-C-FSC SU or FeLV-C-FSC sRBD with titers nearing to FePit1/FeLIX (1–5x105 IU/ml; Figure 2). However, we did not observe FeLV-T infection on MDTF-FeTHTR1 cells with FeLV-A-61E SU or FeLV-A-61E sRBD cofactors (Figure 2). Also, in keeping with results from our previous study, there was no detectable infection with FePit1 using GALV sRBD as a cofactor or FePit2 in combination with FeLV-B cofactors (Figure 2) (Lauring, Anderson, and Overbaugh, 2001).
Figure 2.
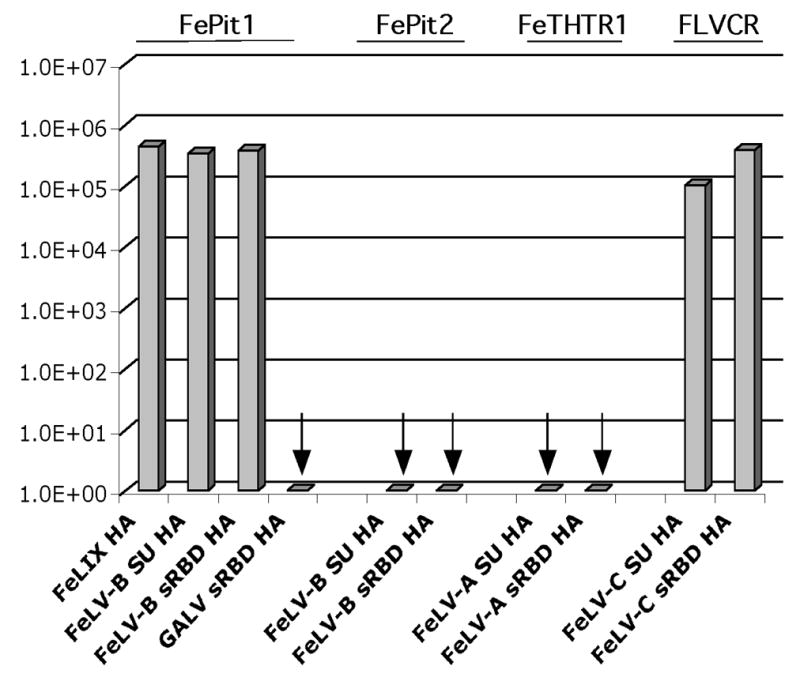
FeLV-T infection of MDTF cells in the present of different receptor/cofactor combinations. Results are shown as log10 titers of β-galactosidase-positive foci per ml in a single cycle infection assay. The receptor expressed in the cell line tested is shown above the graph, and the cofactor used with it is shown at the bottom. The amount of cofactor used was one equivalent, as shown in Figure 1B. Arrows denote combinations where less than 10 β-galactosidase-positive foci/ml were observed. The data shown are the results of triplicate experiments, and are typical of experiments performed on at least three occasions.
Increasing cofactor concentration can allow FeLV-A sRBD and FeTHTR1 to act as FeLV-T receptors
To determine if increasing amounts of cofactor would impact receptor specificity or efficiency, we tested a range of concentrations of cofactors with their cognate receptors. For each cofactor, we considered the volume per sample used on the normalized Western blot in Figure 1B, and for studies in Figure 2, to be equal to one equivalent. We tested two, four and eight times that equivalent. Increasing the amount of cofactor in this way typically increased the level of infection, although generally in increments of 2- to, at most, 10-fold (Figure 3). For example, titers of FeLV-T on MDTF-FePit1 with one to eight equivalents of FeLIX ranged from 6x105 to 2x106 IU/ml. Titers of FeLV-T on MDTF-FLVCR with one to eight equivalents of FeLV-C-SU ranged from 5x105 to 2x106 IU/ml and FeLV-C-SU increased from 1 to 5x105 IU/ml. The exception was FeLV-A-61E-sRBD, which increased from no detectable infection with one equivalent of cofactor to ~103 IU/ml with two equivalents, ~104 IU/ml with four equivalents, and almost 105 IU/ml with eight equivalents. Thus, the amount of cofactor had varying impacts on infectivity, depending on the receptor/cofactor combination.
Figure 3.
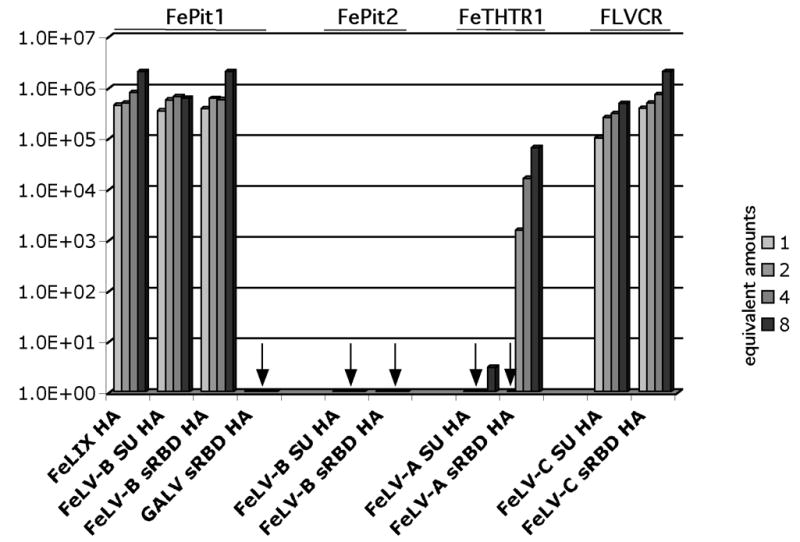
FeLV-T infection of MDTF cells expressing receptor and incubated with increasing amounts of envelope-derived cofactor. Figure is the same as described in the legend to Figure 2, except that results using variable amounts of cofactor (1, 2, 4 and 8 equivalents) are shown. The amount of cofactor added is shown in a particular grayscale, as shown to the right. Results are representative of triplicate experiments, and are typical of experiments performed on at least three occasions.
Soluble envelope RBD and facilitates FeLV-T infection more efficiently than soluble SU
We found that, in most cases, the full-length soluble SU cofactor and the corresponding soluble sRBD cofactor were comparable in their ability to mediate FeLV-T infection. For example, FeLV-T infection was similar with one equivalent of either the SU or sRBD form of FeLV-B-90Z in the presence of FePit1 (~5x105 IU/ml; Figure 3). However, there were subtle differences (~five-fold) in the infectivity of the SU as compared to the sRBD form of FeLV-C-FSC in the presence of FLVCR. One equivalent of FeLV-C-FSC sRBD facilitated titers of 1x105 IU/ml whereas the same amount of FeLV-C-FSC SU FeLV-T facilitated titers of 5x105 IU/ml (Figure 3). Most notably, the sRBD form of FeLV-A-61E facilitated a 1000-fold increase over the SU form in FeLV-T infectious titer on MDTF-FeTHTR1 cells, 5x104 IU/ml as compared to less than 10 IU/ml at eight equivalents of cofactor.
A threshold amount of FeTHTR1 receptor and FeLV-A cofactor is required for FeLV-T infection
Given that cofactor concentration impacted FeLV-T receptor use for FeTHTR1/FeLV-A-61E sRBD, we wanted to test whether levels of cell surface receptor might have a similar impact. For this purpose, we sorted for individual cells expressing high, medium and low amounts of FeTHTR1. To confirm the differences in receptor expression, we bound saturating amounts of cofactor to cells, detected bound cofactor with an antibody to an HA tag incorporated into each cofactor, and compared the amount of mean fluorescence (Figure 4). The results of this analysis show differing amounts of mean fluorescence, indicative of variable amounts of receptor expression (Figure 4A).
Figure 4.
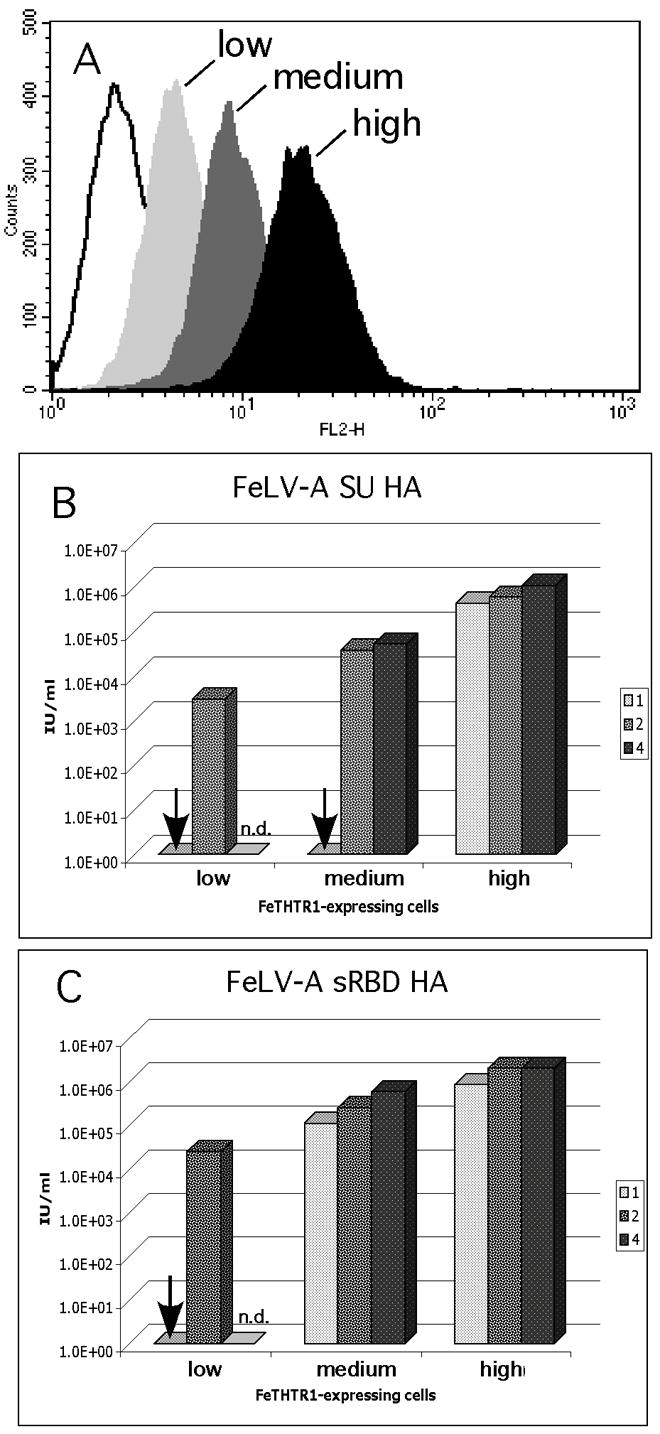
Analysis of FeLV-T infection of MDTF cells expressing increasing FeTHTR1 in the presence of variable amounts of FeLV-A envelope-derived cofactors. (A) Fluorescence histograms representing saturation binding of the FeLV-A SU HA to single clone populations of MDTF cells; cell lines were derived from single clones isolated by flow cytometry, as described in Materials and Methods. The black histogram represents high expressing cells, dark gray histogram represents medium expressing cells, light gray histograms represent low expressing cells, and open histograms represent the high expressing cells with no cofactor added. (B) Infection of MDTF-FeTHTR1 cells using cells from panel A and different equivalent amounts of the FeLV-A SU HA cofactor. (C) Infection of MDTF-FeTHTR1 cells using cells from panel A and different equivalent amounts of the FeLV-A sRBD HA cofactor. For panel B and C, the results for each amount of cofactor added are shown in the grayscale indicated to the right of the figure. Results are shown as log10 β-galactosidase-positive foci per ml (IU/ml). Results are representative of triplicate experiments, and are typical of experiments performed on at least three occasions. An arrow indicates infection was less than 10 IU/ml. N.d. indicates experiment not done.
We then tested these cell lines for the ability to mediate FeLV-T infection with variable amounts of FeLV-A-61E SU and FeLV-A-61E sRBD. Infection of cells was observable with FeLV-A-61E SU only when the cells expressed the highest levels of FeTHTR1 (H2) or when at least two equivalents of FeLV-A-61E SU were introduced (Figure 4B). Hence, the longer SU forms of envelope can function in FeLV-T infection, but either the receptor or the cofactor must be at a medium level, and it is most efficient if they are both present at high levels. As noted above, FeLV-T infection is higher in the presence of the FeLV-A sRBD versus the SU, and this was particularly true at lower levels of receptor or cofactor (Figure 4C).
High expression of FePit1/GALV sRBD cannot rescue FeLV-T infection
We constructed cell lines with variable expression of FePit1 in order to test whether increased expression might rescue infection with GALV sRBD, as they had for the high-expressing FeTHTR1-H cells with FeLV-A SU and sRBD. The expression levels of FePit1 on the selected cell clones was verified by FACS analysis using saturating levels of the FeLV-B SU (Figure 5A).
Figure 5.
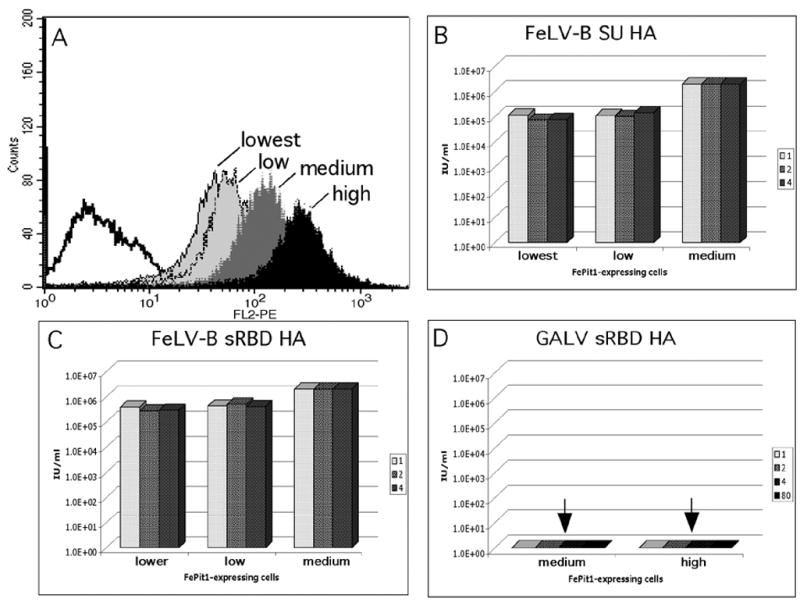
FeLV-T infection of cells expressing variable levels of FePit1 and with variable levels of cofactors. (A) Fluorescence histograms collected through flow cytometry representing saturation binding of the FeLV-B SU HA to single clone populations of MDTF cells expressing different amounts of the FePit1 receptor, as described in Materials and Methods. The black histogram represents high expressing cells, dark gray histogram represents medium expressing cells, light gray histograms represent low and lowest expressing cells. (B) Infection of MDTF-FePit1 cells using cells from panel A and different equivalent amounts of the FeLV-B SU HA cofactor. (C) Infection of MDTF-FePit1 cells using cells from panel A and different equivalent amounts of the FeLV-B sRBD HA cofactor. (D) Infection of MDTF-FePit1 cells using cells from panel A and different equivalent amounts of the GALV sRBD HA cofactor. For panels B-D, the results for each amount of cofactor added are shown in the grayscale indicated to the right of the figure. Results are shown as log10 β-galactosidase-positive foci per ml (IU/ml). Results are also representative of triplicate experiments, and are typical of experiments performed on at least three occasions. An arrow indicates infection was less than 10 IU/ml.
There was some impact of FePit1 expression level on FeLV-T infection in the presence of the FeLV-B cofactors, either FeLV-B SU (Figure 5B or FeLV-B sRBD (Figure 5C): FeLV-T infection was ~five-fold higher in the presence of these cofactors with cells expressing medium levels of FePit1 versus low levels. There was no difference between the medium- and high-receptor expressing cells (data not shown). We found that even with high levels of FePit1 expression, GALV sRBD was not able to mediate FeLV-T infection, even at greater than eight equivalents (Figure 5D). This suggests that there is an absolute block to infection that cannot be overcome by increasing the receptor or cofactor.
Feline fibroblast cells express an intermediate level of FeTHTR1 and FePit1 relative to engineered cell lines
We have shown that receptor and cofactor expression are critical to the efficiency of FeLV-T infection. In order to determine how these receptor levels compare to the endogenous levels in cat cells, we compared the AH927 feline fibroblast cell line to the cell lines expressing variable amounts of receptor (low, medium and high levels of FeTHTR1 or FePit1) in a single FACS experiment using the same amounts of cofactor (Figure 6). The expression of FeTHTR1 on AH927 is in the range between the medium- and high-expressing MDTF-FeTHTR1 cells (Figure 6). The expression of FePit1 on AH927 is in the range between the low- and medium-expressing MDTF-FePit1 cells (Figure 6). Thus, the MDTF cells engineered to express the various receptors did not express these receptors at levels that were aberrantly high compared to those found in a susceptible feline cell line.
Figure 6.
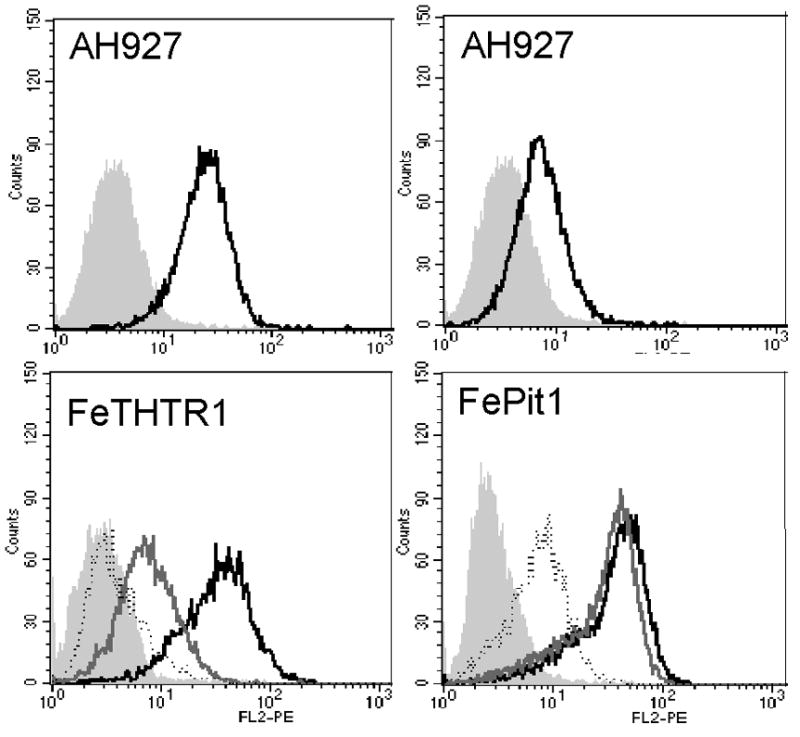
Relative expression of FeTHR1 and FePit1 on feline fibroblasts, AH927, compared to the MDTF cell lines. Top left: Fluorescence histograms representing saturation binding of 61E sRBD HA on AH927 and MDTF alone (gray filled profile), MDTF-FeTHTR1-L (dotted outline), MDTF-FeTHTR1-M (gray solid outline), and MDTF-FeTHTR1-H (black solid outline). Bottom left: Fluorescence histograms representing AH927 alone (gray filled profile) and AH927 with saturation binding of FeLV-A-61E sRBD HA (black solid profile). Top right: Fluorescence histograms representing saturation binding of FeLV-B-90Z sRBD HA on MDTF alone (gray filled profile), MDTF-FePit1-L (dotted outline), MDTF-FePit1-M (gray solid outline), and MDTF-Fepit1-H (black solid outline). Bottom right: Fluorescence histograms representing AH927 alone (gray filled profile) and AH927 with saturation binding of FeLV-B-90Z sRBD HA (black solid profile).
DISCUSSION
The naturally arising FeLV-T requires two host cell proteins for entry, a multiple membrane spanning transport protein and a soluble cofactor. This is in contrast to its progenitor virus, FeLV-A, which only requires the membrane spanning receptor. To better understand the basis for FeLV-T entry and specificity, we examine the specific cofactor/receptor requirements, as well as how cofactor and receptor levels influence FeLV-T specificity. We found that FeLV-T can utilize other combinations of cofactor and cell-surface receptor that were previously untested. For example, FeLV-T can enter cells using the FLVCR and either FeLV-C-FSC SU or sRBD with comparable efficiency to FePit1/FeLIX. We also found that increasing the receptor expression and cofactor concentration can render some previously inefficient cofactor-receptor pairs, such as FeTHTR1 with FeLV-A-61E SU or sRBD, more efficient for FeLV-T entry. Finally, we found that even using very high amounts of receptor and cofactor, some combinations of cofactor-receptor pairs remain unable to mediate FeLV-T infection. A caveat to interpreting our study is that, even thought the FeLV-T particles and the cofactor were both generated by similar transfection methods, we did not define precise ratios of the FeLV-T envelope, the cofactor and the receptor. Future studies of this type would allow a more direct comparison of the stoichiometry of the critical proteins.
We noted, in some cases, that FeLV-T infection was higher in the presence of equivalent levels of sRBD compared to SU. This was most pronounced for FeTHTR1/FeLV-A sRBD. This suggests that there is some reduction in efficiency to cofactor rescue in the presence of the C-terminus that is not simply due to differences in sRBD versus the full-length SU. Rather, this difference may be a reflection of the regulatory interactions between N- and C-termini in the gammaretroviral entry, as has been proposed previously for variants of MLV that require a soluble cofactor (Barnett and Cunningham, 2001; Lu and Roth, 2003).
The levels of receptor expression in the cell lines engineered to express the receptors examined here were similar to the endogenous levels found in a feline cell line, AH927. AH927 cells expressed levels of FePit1 that would be predicted to permit efficient entry of FeLV-T, even in the presence of lower levels of soluble cofactor. FeTHTR1 levels were also moderate to high in feline cells, but they could still be inadequate to permit a high level of FeLV-T infection, particularly when the full-length FeLV-A SU is the entry cofactor. Indeed, this may explain why in previous studies using AH927 cells to test the potential of FeLV-A to function as a cofactor, which were done prior to the identification of the FeLV-A receptor (Mendoza, Anderson, and Overbaugh, 2006), infection with FeLV-T was negligible (Lauring, Anderson, and Overbaugh, 2001). These studies underscore the importance of both receptor expression and cofactor concentration in defining which cells are infectable by FeLV-T in vivo. The importance of cell-surface receptor concentrations to the efficiency of infection has been previously shown for a number of other retroviruses (Chung et al., 1999; Kuhmann, 2000; Kurre, 1999; Platt, 1998; Tailor, Nouri, and Kabat, 2000). With the requirement of a second, soluble cofactor, it seems reasonable to suppose that the overall efficiency of entry should depend on the concentrations of both cell-surface receptor and soluble cofactor, as we have shown here.
These findings have implications for the tropism of FeLV-T in the infected cat. FeLV-T was originally identified as a T-cell tropic variant (Donahue et al., 1991; Overbaugh et al., 1988), which was subsequently explained by the fact that the endogenous cofactor FeLIX is expressed at the highest levels in T cells (Anderson et al., 2000; Lauring, Anderson, and Overbaugh, 2001). The studies described here suggest that other cells may be targets of infection in FeLV-T infected cats that are co-infected with FeLV-A and FeLV-C. FeLV-C is relatively uncommon, but, when present, largely targets hematopoetic cells; thus in cats that harbor both FeLV-T and FeLV-C, these cells could be targets of co-infection. In fact, because FeLV-T does not cause superinfection interference, presumably due to the unique nature of its unusual receptor requirements, it is possible that these co-infected cells would then accumulate a high copy number of FeLV-T, leading to greater cytopathic effect. A recent study provides biochemical evidence that reduced binding affinity of the TR1.3 MLV leads to decreased superinfection interference and greater cytopathicity (Murphy et al., 2006). To date, we have not been able to reliably show binding of FeLV-T to the FeLIX/FePit1 complex by flow cytometry (Cheng, H.H. and Overbaugh J., unpublished data), suggesting that affinity of the virus for the cofactor/receptor complex is likely to be low. By analogy to the TR1.3 MLV, reduced binding affinity may also contribute to the cytopathic properties of FeLV-T.
While cases of FeLV-C infection in cats are rare, all FeLV-infected cats harbor FeLV-A (Rohn, 1999b). Because FeLV-A has a broad cell tropism for feline cells, a range of cells expressing FeTHTR1 and infected with FeLV-A are potential targets of FeLV-T. However, infection in the presence of the full-length FeLV-A SU cofactor is inefficient, and it is the truncated version of FeLV-A envelope that is most effective as an entry cofactor. Such truncated versions of the FeLV-A envelope have been detected in tumors (Rohn et al., 1994), but their potential impact on FeLV-T infection in the infected cat is unknown.
Here we show that specificity of FeLV-T for its receptor is of three varieties: the first are receptor/cofactor pairs where infection is less sensitive to expression levels, suggesting high-affinity, optimal interactions between the virus and the receptor/cofactor. A second variety includes receptor/cofactor combinations where FeLV-T infection is more dependent on expression levels to meet the threshold for infection, suggesting that viral envelope may have lower affinity for these receptor complexes. A third variety are receptor/cofactor pairs that are not functional at any concentration, suggesting FeLV-T cannot productively interact with these proteins to gain entry into the cell. The presumed endogenous receptor complex, FePit1/FeLIX, is likely to play a critical role in FeLV-T infection in the cat, but other receptor/cofactor combinations may also contribute to replication of FeLV-T in cats that are co-infected with multiple FeLV variants. Thus, the targets for FeLV-T infection in the host may be defined in part by how the infecting FeLV-A virus itself has diversified.
MATERIALS and METHODS
Cell culture
The MDTF murine fibroblast cell line has been described previously (Lander and Chattopadhyay, 1984), as has the MDTF-FePit1 line (Anderson et al., 2001), MDTF-FePit2 (Anderson et al., 2001), MDTF-FLVCR (Cheng et al., 2006), and MDTF-FeTHTR1 (Mendoza, Anderson, and Overbaugh, 2006). All receptor-expressing MDTF cell lines, including those described below, were maintained in complete DMEM (Dulbecco’s modified Eagle’s medium supplemented with 10% fetal bovine serum, 100U penicillin/ml, 100μg streptomycin/ml, 0.25mg amphotericin fungicide/ml, and 2mM L-glutamine), containing 1.0mg G418/ml (Geneticin, Gibco).
Construction of cell lines with variable amounts of receptor expression
Cells expressing variable receptor levels, either FePit1 or FeTHTR1, were made by introducing retroviral vectors L(FePit1)SN (Anderson et al., 2001) or L(FeTHTR1)SN (Mendoza, Anderson, and Overbaugh, 2006) into MDTF cells. Particles containing the relevant vector genome were generated in 293T cells using transient transfection with the following plasmids: amphotropic MuLV envelope (SV-A-MLV-env (Landau, Page, and Littman, 1991)), a construct encoding FeLV gag and pol (61E-LTR-ΔΨ gag-pol (Sugai et al., 2001), and either L(FeTHTR1)SN or L(FePit1)SN as appropriate. Two x105 MDTF cells were transduced with these particles in a 4 cm tissue culture dish and grown in selection media until approximately 75–80% confluence—about 4–6 days (Anderson et al., 2001), at which time they were sorted by flow cytometry. For that purpose, 2 mls of conditioned media containing FeLV-A-61E SU HA or FeLV-B-90Z SU HA were incubated with a pool of 2x105 cells transduced with L(FeTHTR1)SN or L(FePit1)SN respectively, in a 5ml Falcon 2054 tube for 1 hour on a rotator at 4°C. Cells were pelleted in a swing-bucket tabletop centrifuge at 1200 RPM for 5 minutes at 4°C, and media was aspirated. Washes were done by gently adding 1ml of cold Hanks buffered saline solution with magnesium and calcium and 2% fetal bovine serum (WB) to resuspend the cells. Cells were pellet again at 1200 RPM for 5 minutes at 4°C and WB was aspirated. Cells were then resuspended in 200μl of a 1:1000 dilution of an ascites concentrate of monoclonal antibody HA.11 (Covance, Berkeley, CA) in WB and incubated on ice for a minimum of 90 minutes. Primary antibody incubation was followed by two washes with 1ml cold WB. Cells were resuspended in 150μl of a 1:100 dilution of R-phycoerythrin-conjugated goat anti-mouse antibody (DAKO, Carpinteria, CA) in WB and incubated on ice and protected from light for a minimum of 30 minutes. Secondary antibody incubation was followed by a final wash in cold WB, and then resuspended in 300μl WB and stored on ice and protected from light until flow cytometric analysis. Cells were analyzed on a Vantage Flow Cytometer, and individually sorted based on high, medium and low fluorescence into a wells of a 96-well tissue culture dish with each well containing 200μl of conditioned selection media that was filtered through a 0.22μM filter. After 5 days, surviving clonal populations were expanded in selection media to a 12-well dish.
Clonal populations underwent a second round of screening for receptor expression by repeating the binding assay from above, except that 1x105 cells derived from each single clone were used. A subset of cells were frozen for storage at the time of the second screening. After the second screen, three or four representative clonal populations were selected, expanded and frozen back. A third binding assay was performed using 1ml of cofactor (at least 100x the minimal volume to detect maximal binding in previous experiments using polyclonal cells) on each of these final cell lines as confirmation of their receptor expression.
The binding assay used for confirmation of the newly constructed cell lines is the same assay used to analyze receptor expression of other cell lines, and has been previously described (Lauring, Anderson, and Overbaugh, 2001).
Expression of HA-tagged FeLIX and viral RBDs and SUs
The expression constructs CS2-FeLV-B-90Z-SU-HA, CS2-FeLV-B-90Z-RBD-HA, CS2-FeLV-A-61E-SU-HA, CS2-FeLIX-HA, CS2-GALV-SU1–262-HA have been previously described (Lauring, Anderson, and Overbaugh, 2001). CS2-FeLV-A-61E-sRBD-HA, CS2-FeLV-C-FSC-SU HA and CS2-FeLV-C-FSC-sRBD HA were made using the same method (primers available on request). All constructs were verified by nucleotide sequence analyses. Conditioned media containing soluble retrovirus sRBDs, SUs and FeLIX were generated with the plasmids mentioned above by transient transfection of 293T cells using a calcium phosphate protocol as described previously (Lauring, Anderson, and Overbaugh, 2001).
Virus production and infection assays
Pseudotyped virus was made by calcium phosphate transfection of 293T cells (Mammalian Transfection Kit, Stratagene) with FeLV-61E-gag-pol (61E-LTR-ΔΨ-gal-pol), a MLV-derived reporter gene that expresses β-galactosidase (pRT43.2Tnlsβgal-1) and the desired FeLV-T envelope construct, as described previously (Sugai et al., 2001). Viral infectivity was assayed using a single-cycle infection assay described previously (Anderson et al., 2001).
Immunoprecipitation and Western blot analysis
Initial steps of immunoprecipitation and Western blot analysis was performed as described previously (Lauring, Anderson, and Overbaugh, 2001). The blot membrane was probed with 10μl rabbit polyclonal HA.11 antibody (Covance, Berkeley, CA), followed by thorough washing and probing with 1.8μl of Alexa Fluor 680 goat anti-rabbit secondary antibody (Molecular Probes, Eugene, OR) in 10ml of antibody dilution buffer for one hour at room temperature, protected from light. The blot was washed again with phosphate buffered saline (PBS) containing 0.1% Tween 20, and rinsed in PBS. Proteins bound with antibodies were detected and quantified using the Odyssey Infrared Imaging System according to manufacturer’s protocol (LI-COR Biosciences, Lincoln, NB)
Acknowledgments
We thank Dusty Miller and Ramon Mendoza for helpful discussions. We are grateful to Weldon DeBusk and Cyntia de Barros for their assistance with cell sorting, and to Pei-Feng Cheng for her technical advice as well as the protocol and reagents for the quantitative Western blot. We thank Nick Rabena and Brannon Orton for technical assistance. We are indebted to Jan Abkowitz for providing the LX(FLVCR)SN clone, and to Jim Mullins for providing the FeLV-C-Sarma plasmid. This work was supported by NIH grant CA 51080. HHC was supported by a fellowship from the Paul Allen Foundation.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Anderson MM, Lauring AS, Burns CC, Overbaugh J. Identification of a cellular cofactor required for infection by feline leukemia virus. Science. 2000;287(5459):1828–30. doi: 10.1126/science.287.5459.1828. [DOI] [PubMed] [Google Scholar]
- Anderson MM, Lauring AS, Robertson S, Dirks C, Overbaugh J. Feline Pit2 functions as a receptor for subgroup B feline leukemia viruses. J Virol. 2001;75(22):10563–72. doi: 10.1128/JVI.75.22.10563-10572.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bae Y, Kingsman SM, Kingsman AJ. Functional dissection of the Moloney murine leukemia virus envelope protein gp70. J Virol. 1997;71(3):2092–9. doi: 10.1128/jvi.71.3.2092-2099.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barnett AL, Cunningham JM. Receptor binding transforms the surface subunit of the mammalian C-type retrovirus envelope protein from an inhibitor to an activator of fusion. J Virol. 2001;75(19):9096–105. doi: 10.1128/JVI.75.19.9096-9105.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barnett AL, Davey RA, Cunningham JM. Modular organization of the Friend murine leukemia virus envelope protein underlies the mechanism of infection. Proc Natl Acad Sci U S A. 2001;98(7):4113–8. doi: 10.1073/pnas.071432398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barnett AL, Wensel DL, Li W, Fass D, Cunningham JM. Structure and mechanism of a coreceptor for infection by a pathogenic feline retrovirus. J Virol. 2003;77(4):2717–29. doi: 10.1128/JVI.77.4.2717-2729.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng HH, Anderson MM, Hankenson FC, Johnston L, Kotwaliwale CV, Overbaugh J. Envelope determinants for dual-receptor specificity in feline leukemia virus subgroup A and T variants. J Virol. 2006;80(4):1619–28. doi: 10.1128/JVI.80.4.1619-1628.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chung M, Kizhatil K, Albritton LM, Gaulton GN. Induction of syncytia by neuropathogenic murine leukemia viruses depends on receptor density, host cell determinants, and the intrinsic fusion potential of envelope protein. J Virol. 1999;73(11):9377–85. doi: 10.1128/jvi.73.11.9377-9385.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Donahue PR, Hoover EA, Beltz GA, Riedel N, Hirsch VM, Overbaugh J, Mullins JI. Strong sequence conservation among horizontally transmissible, minimally pathogenic feline leukemia viruses. J Virol. 1988;62(3):722–31. doi: 10.1128/jvi.62.3.722-731.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Donahue PR, Quackenbush SL, Gallo MV, deNoronha CM, Overbaugh J, Hoover EA, Mullins JI. Viral genetic determinants of T-cell killing and immunodeficiency disease induction by the feline leukemia virus FeLV-FAIDS. J Virol. 1991;65(8):4461–9. doi: 10.1128/jvi.65.8.4461-4469.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gwynn SR, Hankenson FC, Lauring AS, Rohn JL, Overbaugh J. Feline leukemia virus envelope sequences that affect T-cell tropism and syncytium formation are not part of known receptor-binding domains. J Virol. 2000;74(13):5754–61. doi: 10.1128/jvi.74.13.5754-5761.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuhmann SE, Platt EJ, Kozak SL, Kabat D. Cooperation of Multiple CCR5 Coreceptors is Required for Infections by Human Immunodeficiency Virus Type 1. Journal of Virology. 2000;74(15):7005–7015. doi: 10.1128/jvi.74.15.7005-7015.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kurre P, Kiem H-P, Morris J, Heyward S, Battini J-L, Miller AD. Efficient Transduction by an Amphotropic Retrovirus Vector is Dependent on High-Level Expression of the Cell Surface Virus Receptor. Journal of Virology. 1999;73(1):495–500. doi: 10.1128/jvi.73.1.495-500.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Landau NR, Page KA, Littman DR. Pseudotyping with human T-cell leukemia virus type I broadens the human immunodeficiency virus host range. J Virol. 1991;65(1):162–9. doi: 10.1128/jvi.65.1.162-169.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lander MR, Chattopadhyay SK. A Mus dunni cell line that lacks sequences closely related to endogenous murine leukemia viruses and can be infected by ectropic, amphotropic, xenotropic, and mink cell focus-forming viruses. J Virol. 1984;52(2):695–8. doi: 10.1128/jvi.52.2.695-698.1984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lauring AS, Anderson MM, Overbaugh J. Specificity in receptor usage by T-cell-tropic feline leukemia viruses: implications for the in vivo tropism of immunodeficiency-inducing variants. J Virol. 2001;75(19):8888–98. doi: 10.1128/JVI.75.19.8888-8898.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lavillette D, Kabat D. Porcine endogenous retroviruses infect cells lacking cognate receptors by an alternative pathway: implications for retrovirus evolution and xenotransplantation. J Virol. 2004;78(16):8868–77. doi: 10.1128/JVI.78.16.8868-8877.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lavillette D, Ruggieri A, Russell SJ, Cosset FL. Activation of a cell entry pathway common to type C mammalian retroviruses by soluble envelope fragments. J Virol. 2000;74(1):295–304. doi: 10.1128/jvi.74.1.295-304.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu CW, Roth MJ. Functional interaction between the N- and C-terminal domains of murine leukemia virus surface envelope protein. Virology. 2003;310(1):130–40. doi: 10.1016/s0042-6822(03)00111-9. [DOI] [PubMed] [Google Scholar]
- Mendoza R, Anderson MM, Overbaugh J. A putative thiamine transport protein is a receptor for feline leukemia virus subgroup A. J Virol. 2006;80(7):3378–85. doi: 10.1128/JVI.80.7.3378-3385.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murphy SL, Chung-Landers M, Honczarenko M, Gaulton GN. Linkage of reduced receptor affinity and superinfection to pathogenesis of TR1.3 murine leukemia virus. J Virol. 2006;80(9):4601–9. doi: 10.1128/JVI.80.9.4601-4609.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Overbaugh J, Donahue PR, Quackenbush SL, Hoover EA, Mullins JI. Molecular cloning of a feline leukemia virus that induces fatal immunodeficiency disease in cats. Science. 1988;239(4842):906–10. doi: 10.1126/science.2893454. [DOI] [PubMed] [Google Scholar]
- Overbaugh J, Miller AD, Eiden MV. Receptors and entry cofactors for retroviruses include single and multiple transmembrane-spanning proteins as well as newly described glycophosphatidylinositol-anchored and secreted proteins. Microbiol Mol Biol Rev. 2001;65(3):371–89. doi: 10.1128/MMBR.65.3.371-389.2001. table of contents. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Platt EJ, Wehrly K, Kuhmann SE, Chesebro B, Kabat D. Effects of CCR5 and CD4 Cell Surface Concentrations on Infections by Macrophagetropic Isolates of Human Immunodeficiency Virus Type 1. Journal of Virology. 1998;72(4):2855–2864. doi: 10.1128/jvi.72.4.2855-2864.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quigley JG, Burns CC, Anderson MM, Lynch ED, Sabo KM, Overbaugh J, Abkowitz JL. Cloning of the cellular receptor for feline leukemia virus subgroup C (FeLV-C), a retrovirus that induces red cell aplasia. Blood. 2000;95(3):1093–9. [PubMed] [Google Scholar]
- Quigley JG, Yang Z, Worthington MT, Phillips JD, Sabo KM, Sabath DE, Berg CL, Sassa S, Wood BL, Abkowitz JL. Identification of a human heme exporter that is essential for erythropoiesis. Cell. 2004;118(6):757–66. doi: 10.1016/j.cell.2004.08.014. [DOI] [PubMed] [Google Scholar]
- Rohn JL, Overbaugh JM. Feline Leukemia Virus and Feline Immunodeficiency Virus. In: Chen RAaI., editor. Persistent Viral Infections. John Wiley & Sons Ltd; 1999a. pp. 379–408. [Google Scholar]
- Rohn JL, Linenberger ML, Hoover EA, Overbaugh J. Evolution of feline leukemia virus variant genomes with insertions, deletions, and defective envelope genes in infected cats with tumors. J Virol. 1994;68(4):2458–67. doi: 10.1128/jvi.68.4.2458-2467.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rohn JL, Moser MS, Gwynn SR, Baldwin DN, Overbaugh J. In vivo evolution of a novel, syncytium-inducing and cytopathic feline leukemia virus variant. J Virol. 1998;72(4):2686–96. doi: 10.1128/jvi.72.4.2686-2696.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rohn JL, A JMO. Pathogenic feline retroviruses: feline leukemia virus and feline immunodeficiency virus. In: A ISY, Chen RA, editors. Persistent Viral Infections. John Wiley and Sons; 1999b. pp. 379–408. [Google Scholar]
- Sugai J, Eiden M, Anderson MM, Van Hoeven N, Meiering CD, Overbaugh J. Identification of envelope determinants of feline leukemia virus subgroup B that permit infection and gene transfer to cells expressing human Pit1 or Pit2. J Virol. 2001;75(15):6841–9. doi: 10.1128/JVI.75.15.6841-6849.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tailor CS, Lavillette D, Marin M, Kabat D. Cell surface receptors for gammaretroviruses. Curr Top Microbiol Immunol. 2003;281:29–106. doi: 10.1007/978-3-642-19012-4_2. [DOI] [PubMed] [Google Scholar]
- Tailor CS, Nouri A, Kabat D. Cellular and species resistance to murine amphotropic, gibbon ape, and feline subgroup C leukemia viruses is strongly influenced by receptor expression levels and by receptor masking mechanisms. J Virol. 2000;74(20):9797–801. doi: 10.1128/jvi.74.20.9797-9801.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tailor CS, Willett BJ, Kabat D. A putative cell surface receptor for anemia-inducing feline leukemia virus subgroup C is a member of a transporter superfamily. J Virol. 1999;73(8):6500–5. doi: 10.1128/jvi.73.8.6500-6505.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takeuchi Y, Vile RG, Simpson G, O'Hara B, Collins MK, Weiss RA. Feline leukemia virus subgroup B uses the same cell surface receptor as gibbon ape leukemia virus. J Virol. 1992;66(2):1219–22. doi: 10.1128/jvi.66.2.1219-1222.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wensel DL, Li W, Cunningham JM. A virus-virus interaction circumvents the virus receptor requirement for infection by pathogenic retroviruses. J Virol. 2003;77(6):3460–9. doi: 10.1128/JVI.77.6.3460-3469.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]