

Abstract
We examined gene regulation in murine lungs after hind-limb vessel occlusion and reperfusion. A rapid increase of transcript for the AMP deaminase 3 gene (AMPD3) and its enzymatic activity (EC 3.5.4.6) generating inosine monophosphate (IMP) were identified with transcripts located in bronchial and alveolar epithelium. AMP deaminase inhibitor decreased IMP levels and significantly enhanced neutrophil recruitment within lung tissue during reperfusion. In addition, IMP inhibited cytokine-initiated neutrophil infiltration in vivo and selectively attenuated neutrophil rolling by 90% in microvessels. We prepared labeled IMP and demonstrated that IMP specifically binds to neutrophils. IMP also stimulated binding of γ-[35S]thio-GTP, suggesting that IMP is a potent regulator of neutrophils. Taken together, these results elucidate a previously unrecognized mechanism that protects tissues from the potentially deleterious consequences of aberrant neutrophil accumulation. Moreover, they are relevant for new therapeutic approaches to regulate neutrophil responses in inflammation and vascular disease.
Reperfusion causes local and distant (remote) organ injury that is often a serious and widespread clinical event associated with various forms of vascular occlusion and surgical procedures such as cardiopulmonary bypass, transplantation, and aortic cross-clamping (1–3). In these settings, reperfusion can lead to severe neutrophil-mediated tissue damage in the lung, heart, brain, kidney, and intestine. Failure of transplanted organs is also attributed in part to this syndrome (1). Neutrophils play a key role in initiating remote organ injury by infiltrating and accumulating in high numbers within tissues upon resumption of flow to ischemic organs (1, 2, 4). Several aspects of neutrophil-dependent reperfusion injury resemble local acute inflammatory sequelae (5–7). When reperfusion begins, neutrophils that possess the ability to release a wide array of proinflammatory chemical mediators intended for host defense accumulate within remote organs by first interacting with vascular endothelium of the target organ. They release mediators (e.g., bioactive lipids, chemokines and cytokines, hydrolytic enzymes, and reactive oxygen species) that lead to tissue injury and dysfunction, the hallmark of reperfusion injury (1, 2). Blocking neutrophil entry in animal models by administering anti-adhesion molecule antibodies or depleting the peripheral blood neutrophils spares reperfusion-associated organ injury (8). The molecular events that regulate neutrophil accumulation in remote organs during reperfusion remain to be elucidated.
To understand the mechanisms of underlying neutrophil-mediated reperfusion injury, we set out to identify endogenous signaling systems. In the present report, we identified a gene and its products that are involved in regulating the extent of neutrophil accumulation in remote organ lung after occlusion of limb vessels and reperfusion. These results have implications in a wide range of disease mechanisms that involve aberrant neutrophil accumulation.
Materials and Methods
Murine Model of Reperfusion-Associated Remote Organ Injury: Hind-Limb Vascular Occlusion.
Male mice (BALB/c, 6–8 weeks of age, body weight 20 ± 2 g) were anesthetized with pentobarbital (50 mg/kg, i.p.). Both hind limbs of mice were banded and clipped with tourniquets (rubber bands) and surgical clamps in the hip region, occluding the vessels and reducing local blood circulation (9). After 3 h, the tourniquets were removed from the limbs and reperfusion was initiated for intervals of 0, 3, or 24 h. Control and test mice were then euthanized with an overdose of pentobarbital and immediately dissected to remove the organs of interest.
Neutrophil Accumulation: Myeloperoxidase (MPO).
Assessment of MPO activity was carried out as in Bradley et al. (10). MPO activity was converted into the number of neutrophils by using a standard calibration curve (11).
Differential Display of mRNA and Northern Analysis.
Differential display of reverse transcription (RT)-PCR (12) was performed by using the Hieroglyph mRNA Profile Kit (Genomyx, Foster City, CA). Total RNA isolated (13) from lungs of mice undergoing treatments (see Results) was treated with DNase I (Message Clean Kit, GenHunter, Nashville, TN) and then reverse transcribed with 1 of 12 anchored primers (5′ T7(dT12)AP 3′) (Genomyx) at 42°C for 60 min. PCR amplifications of the first strand cDNAs were continued with dNTPs including [α-33P]dATP (NEN), 1 of 20 arbitrary primers (5′ M13rARP 3′) (Genomyx), and the same anchored primer previously used for reverse transcription under the conditions recommended by the supplier. PCR products were loaded on a denaturing 4.5% polyacrylamide gel and run in a Genomyx LR DNA Sequencer. Gels were exposed to Kodak film, and differentially displayed bands (e.g., L5B1 and others) were identified, excised, and reamplified. The purified cDNA fragment of L5B1 was sequenced in both directions (by the Brigham and Women's Hospital DNA Sequencing Core) with the same primers used in RT-PCR. Northern analysis and RT-PCR (42°C for 60 min; then 30 cycles of 94°C for 30 sec, 56°C for 1 min, 72°C for 2 min) were performed mainly as reported (11). Sequences of cDNA fragments were analyzed by GenBank Database searches.
AMP Deaminase Activity.
AMP deaminase activity (EC 3.5.4.6) was assayed by using the method of ref. 14. Briefly, lung tissues from control and reperfusion mice were homogenized with 0.089 M potassium phosphate, pH 6.5/0.18 M KCl/0.1 mM DTT on ice. The mixtures were incubated with 50 mM cacodylic acid, pH 6.5/150 mM KCl/10 mM AMP at 25°C for 45 min. The levels of AMP deaminase activity were assessed by the amount of ammonia produced upon deamination of AMP, the ammonia being converted to indophenol, whose absorbance was measured at its λmax of 625 nm.
Coformycin-Treated Mice.
Coformycin (15) (Calbiochem) was used for suppressing the activity of AMP deaminase in control and hind-limb reperfusion mice. Mice were anesthetized, then underwent 3 h with or without clamping of both hind limbs. Ten minutes before reperfusion, mice were injected via their tail veins with either coformycin (6 mg/kg in 120 μl of PBS) or 120 μl of vehicle. After 3 h of reperfusion, the mice were euthanized and the lung tissues were harvested and taken for MPO and AMP deaminase determinations as above.
Measurement of IMP in Tissue or Blood.
Tissue samples were prepared according to a modified method of Soussi et al. (16). Briefly, the control and reperfused tissues (70–180 mg) or blood (0.5 ml) from mice were isolated and homogenized with 1.5 M perchloric acid. After ultracentrifugation and neutralization, the extracts were injected into a high-performance liquid chromatography (HPLC) system. A Hewlett-Packard model 1050 system with an HP1100 diode array detector was used with a reverse-phase column (5x C18, 150 × 4.6 mm; Phenomenex). For the measurement of IMP, 0.1 M sodium phosphate buffer (pH 6.0) was used as mobile phase (1 ml/min). UV absorbance of IMP was measured at 260 nm. Its chromatogram was distinguished by the unique retention time and confirmed by co-eluting tissue samples with purified IMP. Tissue concentration of IMP was expressed as nmol/g of wet weight.
Mouse Air Pouch Inflammation Model.
The air pouch model was used in the method of refs. 17 and 18. Briefly, while the animals were under isoflurane anesthesia, first and second subcutaneous injections of air into the backs of mice (BALB/c, 20–25 g) were performed at 3-day intervals. IMP (10–1,000 μg per mouse) or vehicle (saline) was first injected into individual air pouch followed by the injection of tumor necrosis factor α (TNF-α; 20 ng per mouse) into each. Four hours after the injection of TNF-α, each mouse was euthanized and the air pouch cavity was washed three times with 3 ml of saline. Collected saline samples were centrifuged, and the total cell numbers were quantitated.
Microvascular Neutrophil Rolling.
Mice were anesthetized with pentobarbital (50 mg/kg, i.p.) containing heparin (1,000 units/kg, i.p.). Mesenteric microvessels (70–120 μm in diameter, 1 mm in length) were removed and incubated with 50 μM NG-nitro-l-arginine methyl ester (l-NAME; Sigma) at 37°C for 60 min to increase the expression of adhesion molecules on the endothelial cells (19). The microvessels were then placed within a vessel chamber (model CH/2/A; Living System Instrumentation, Burlington, VT) and perfused with Krebs buffer (perfusion speed: 13 μl/min). Freshly isolated human neutrophils (20) (0.3 ml of 3 × 106 cells per ml, purity ≈ 98%) were treated with 100 μM AMP, inosine, or IMP (0.1–100 μM) at 25°C for 5 min, then perfused into the lumen of the microvessels. Neutrophil rolling was recorded with a charge-coupled device camera (Sanyo) attached to a VCR system (Hitachi) and an inverted microscope. Rolling neutrophils were defined as those moving slower than the perfusion rate (21). The enumerated rolling cells are expressed as rolling cells per min per mm2.
[3H]IMP Synthesis and Binding.
[8-3H]IMP was prepared by the deamination of [8-3H]AMP. Briefly, [8-3H]AMP (15 Ci/mmol, American Radiolabeled Chemicals, St. Louis; 1 Ci = 37 GBq) was incubated with AMP deaminase (Sigma) in 10 mM Tris⋅HCl, pH 6.8/25 mM NaCl at 25°C for 10 min. [8-3H]IMP was isolated by TLC (PEI-cellulose) using 1 M acetic acid as the mobile phase. The specific binding of [8-3H]IMP to intact human neutrophils was evaluated as described for other neutrophil small molecule ligands (22). For competitive binding experiments, neutrophils were incubated with increasing concentrations of adenosine or unlabeled IMP in the presence of 200 nM [8-3H]IMP, and the data were analyzed by using nonlinear regression curve fitting for a one-site binding model (23).
Binding of γ-[35S]thio-GTP (GTP[γ35S]; 12.5 mCi/ml; NEN) binding was performed with isolated neutrophil membrane fractions as in Northup et al. (24).
In Situ Hybridization.
Preparation of tissue sections and hybridization conditions mainly followed the procedures of P. Komminoth (Application Manual, Roche). Briefly, lung tissues from control mice and mice that had undergone limb clamping–reperfusion were fixed in 4% paraformaldehyde and embedded in paraffin. Eight-micrometer-thick sections were prepared and deparaffinized. The rehydrated samples were treated with proteinase K at 37°C for 30 min. The specific antisense oligonucleotides for hybridizing in situ with mRNA transcribed from the mouse AMP deaminase 3 (AMPD3) gene in lung tissue sections were 5′-GAGACCCTCTTCCAAAGGCACACT-3′ (N-terminal coding sequence) and 5′-CTATTTGGTCAGGGCTGTGATCTC-3′ (C-terminal coding sequence). The sense oligonucleotides were used in control experiments. Signals were detected by using a digoxigenin (DIG) nucleic acid detection kit (Roche–Boehringer Mannheim).
Statistical Analysis.
All results are expressed as means ± SEM. Statistical comparisons were made with Student's t test or Dunn's method after the analysis of variance (ANOVA). The results were considered significantly different at P < 0.05.
Results
Neutrophils in Reperfusion-Induced Remote Organ Injury: A Murine Model.
A murine model of remote organ reperfusion injury was set up (see Materials and Methods). Neutrophil accumulation was found within the lungs after 3 (or 24) h of reperfusion following 3 h of the hind-limb occlusion. In contrast, mice that underwent 3 h of limb tourniquet clamping alone without reperfusion or mice without limb clamping did not show significant accumulation of neutrophils within their pulmonary tissues (Fig. 1). Maximum accumulation of neutrophils in lungs was observed at 3 h of reperfusion, representing a 2.5-fold increase compared with control mice or mice with limb clamping alone without reperfusion. Histological evaluation of lung biopsies from reperfused mice confirmed the presence of significant numbers of neutrophils, inflammation, and hemorrhage within lung tissues (Fig. 1 Inset). The correlation between neutrophil accumulation and the lung tissue damage observed at the level of light microscopy confirmed in this model that lung tissue injury was associated with neutrophil infiltration. These pathophysiologic findings from murine tissues are consistent with findings in patients suffering ischemia–reperfusion, particular those undergoing orthopedic surgery (3), as well as in other animal models (25), validating its use for molecular analysis.
Figure 1.

Neutrophil accumulation in murine lungs after reperfusion. Mouse lungs were collected and homogenized for MPO determination after limb clamping with increased intervals of reperfusion (0, 3, and 24 h). Neutrophil numbers were obtained by using a calibration curve for MPO values. Neutrophils in lung biopsies from control mice (that received only pentobarbital, 50 mg/kg, i.p., n = 13 mice) and mice that underwent 3 h of limb clamping then continued with 0 (n = 13 mice), 3 (n = 16 mice), or 24 (n = 13 mice) h of reperfusion are indicated. Results are mean ± SEM of 13 or 16 mice. Asterisks indicate that the differences between 0 and 3 h of reperfusion as well as between 0 and 24 h of reperfusion are significant (P < 0.01). N.S., not significant. (Inset) Hematoxylin/eosin-stained lung biopsy from control mouse lung (Left) and lungs at 3 h clamping and 3 h reperfusion (Right). Arrowheads point to neutrophils within lung tissue. (×400.)
AMPD3 Gene and Its Product Are Up-Regulated in Reperfusion-Injured Lungs.
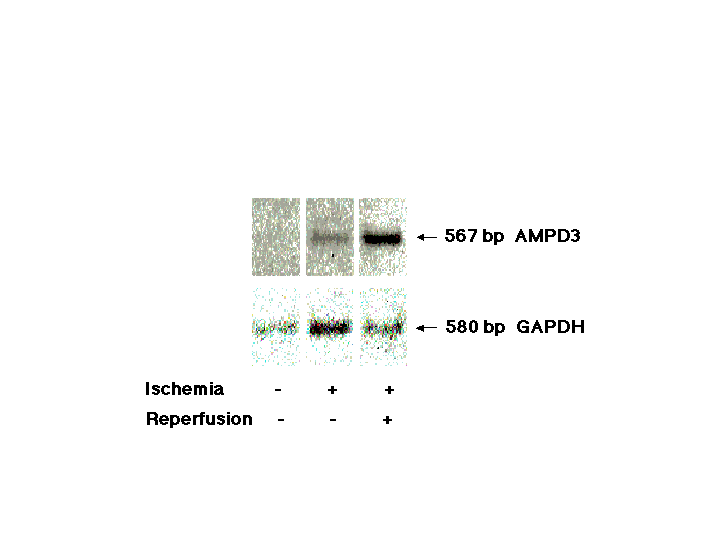
Using this model and mRNA differential display (12), we investigated whether specific genes were differentially expressed in lung tissues of mice undergoing limb vascular occlusion and reperfusion. Several dozen differentially displayed cDNA fragments were identified (see our web site at serhan.bwh.harvard.edu). In particular, one of them, denoted L5B1, was expressed at higher levels in lungs of mice that underwent reperfusion compared with control mice or mice that underwent only limb vessel occlusion without reperfusion (Fig. 2A). Northern analysis performed with the probe L5B1 showed that a 4-kb transcript was abundantly expressed in the lung after hind-limb reperfusion compared with controls (Fig. 2B). DNA sequencing analysis revealed that L5B1 (476 nt) was identical to the 3′ untranslated region of exon 15 of mouse AMPD3 (heart-type) gene (26, 27) (Fig. 2C). Knowing that the AMPD3 gene is expressed in only low levels in healthy lung and its coding sequences are homologous with AMPD1 (muscle-type) and AMPD2 (liver-type) at C-terminal regions and divergent at N-terminal domains (27), we performed quantitative RT-PCR with a pair of unique primers representing 5′-end coding sequences of the AMPD3 gene. Differentially expressed cDNA fragment (567 nt) was isolated and sequenced (sequencing data not shown), and the results confirmed that the AMPD3 gene was up-regulated in lungs after reperfusion of the hind limbs (see Fig. 6 in the supplementary materials at www.pnas.org). Further, an in situ hybridization of lung tissue section with antisense oligonucleotides of AMPD3 mRNA revealed that the up-regulated AMPD3 gene transcripts were predominantly located in bronchial and alveolar epithelium but not associated with the infiltrated neutrophils or vascular endothelium (Fig. 2D). Also, AMP deaminase activity was increased in reperfusion-injured lung tissues (Fig. 3A), a finding in line with the up-regulation of this gene product.
Figure 2.

(A) Representative differential gene expression in murine lungs after reperfusion: mRNA differential display. Total RNAs were isolated from the lungs of control, clamping alone, or clamping–reperfusion mice. RNA was reverse transcribed, amplified, and displayed in a sequencing gel. The oligonucleotides 5′-ACGACTCACTATAGGGCTTTTTTTTTTTTGG-3′ and 5′-ACAATTTCACACAGGAGCTAGCATGG-3′ were used as anchored and arbitrary primers, respectively. Autoradiogram illustrates the prominent differential display of RT-PCR products designated L5B1, which is expressed in lungs of mice that underwent 3 h of hind-limb clamping and 3 h of reperfusion. (B) Northern analysis of L5B1 mRNA expression in lung tissues of control mice and mice that underwent lower-limb ischemia–reperfusion. Total RNAs were isolated and samples (15 μg per lane) were loaded and run on a formaldehyde/agarose gel, then transferred onto a Hybond-N+ membrane (Amersham Life Science) and hybridized with [α-32P]dCTP-labeled L5B1 cDNA fragment (specific activity ≈ 1.2 × 109 cpm/μg; 30 × 106 cpm/10 ml of hybridization buffer). A unique ≈4-kb mRNA of L5B1 was detected (arrow). RNA loading was monitored by ethidium bromide-stained 28S and 18S rRNA on the same gel, displayed beside the Northern hybridization blot. (C) DNA sequence comparison between L5B1 cDNA fragment and mouse AMP deaminase 3 (heart-type) gene exon 15. The 476-nt sequence of the L5B1 cDNA fragment is aligned with the 3′ untranslated sequence of exon 15 (GenBank accession no. D88984S11) of the mouse AMPD3 (heart-type) gene. Identical nucleotides found for L5B1 cDNA are indicated by vertical lines. AATAAA, the putative polyadenylation signal sequence, is double underlined, and the sequence of the poly(A) tail is underlined with dashes. (D) Distribution of the up-regulated AMPD3 gene transcripts in remote lung tissue after ischemia–reperfusion. Left and Center represent lung biopsy sections from the same reperfused mouse hybridized with sense and antisense oligonucleotides for AMPD3 mRNA, respectively. In Right, lung tissue section from control mouse (without ischemia–reperfusion) was hybridized with the same antisense oligonucleotides used in Center. The dark blue-stained spots are selectively distributed in bronchial and alveolar epithelium. (×200.)
Figure 3.

(A) Increase in AMP deaminase activity in murine lung with limb clamping and reperfusion. Lung tissue extracts from control mice (0 h of clamping and 0 h of reperfusion) and hind-limb reperfused mice (3 h of clamping and 3 h of reperfusion) were prepared. The AMP deaminase activities were assessed by the absorbance at 625 nm of the blue chromophore produced by the conversion of the ammonia to indophenol. Percent increases in AMP deaminase activity are expressed as mean ± SEM, n = 6. (B) Enhanced neutrophil accumulation in reperfused lungs of mice treated with an AMP deaminase inhibitor. Neutrophils in lung biopsies were obtained from mice that underwent 3 h of limb clamping and reperfusion and treated with vehicle (n = 8 mice) or coformycin (n = 9 mice), as well as control mice treated with vehicle (n = 8 mice) or coformycin (6 mg/kg; n = 4 mice). The mean values (n = 4, 8, or 9 mice) are indicated by the horizontal black lines. Asterisks denote significant differences (P < 0.01) between values for vehicle versus those from coformycin-treated mice that underwent hind-limb clamping and reperfusion. In the scheme at Right, the action site of coformycin in AMP metabolism is shown (see text for details). (C) IMP increases in reperfused lung and decreases with administration of AMP deaminase inhibitor. Lung tissues were obtained from control (n = 4 mice), ischemia alone (n = 4 mice), ischemia and reperfusion mice (n = 5 mice), as well as ischemia-reperfusion mice with coformycin treatment (6 mg/kg; n = 4 mice). The chromatographic values of IMP by HPLC analysis are presented as nmol/g wet weight of lung. Asterisk denotes the significant differences (P < 0.01) of the values for lung IMP from mice that underwent ischemia–reperfusion versus those from control mice or mice that underwent ischemia alone or ischemia–reperfusion with coformycin.
Administration of AMP Deaminase Inhibitor Enhances Neutrophil Accumulation in Lungs.
It was deemed essential to determine whether the prominently induced AMPD3 gene product might play a functional role within lung tissues during remote organ reperfusion. To address this question, we injected these mice intravenously with an AMP deaminase inhibitor (coformycin) (15) immediately before ischemic hind-limb reperfusion. Of interest, we found statistically significant increases in neutrophil accumulation evident in remote lung tissues compared with mice injected with vehicle alone before reperfusion (P < 0.01) (Fig. 3B). Also, treatment with coformycin did not lead to significant increases in neutrophil values within lungs from the control groups, suggesting that the additional accumulation of neutrophils in reperfused lungs was not the result of toxicity or other potentially nonspecific actions of coformycin (Fig. 3B). These results revealed a clear correlation between administration of the AMP deaminase inhibitor (see below) and enhanced neutrophil accumulation within reperfused lung tissues of mice that underwent hind-limb vascular occlusion, suggesting that this enzyme or its product may play a protective role.
IMP Increases in Lung During Reperfusion.
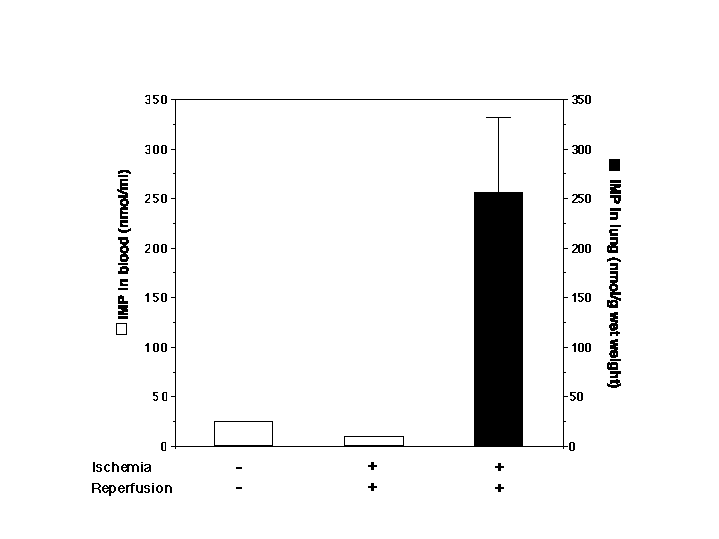
IMP is the unique product of AMP deaminase (see scheme in Fig. 3B) (28). Therefore, we examined IMP levels in the lung after reperfusion of mouse hind limbs. Fig. 3C indicates that the levels of IMP in lung tissues of mice after hind-limb ischemia–reperfusion were 5-fold higher than those of control or nonreperfused mice. IMP levels in reperfused lung tissues were lower after injection of the AMP deaminase inhibitor (i.e., coformycin) (Fig. 3C). These results suggested that IMP levels in the lung are associated with increased AMP deaminase activity after its gene up-regulation. In contrast, the level of IMP in blood (see Fig. 7 in the supplementary materials at www.pnas.org) and hind-limb muscles (data not shown) did not alter after 3 h of ischemia and 3 h of reperfusion in comparison with mice that did not undergo ischemia–reperfusion. These results indicate that IMP is predominantly increased in the microenvironment of the lung after hind-limb reperfusion.
IMP Inhibits Neutrophil Trafficking.
To directly investigate whether IMP has a role in reperfusion-associated inflammation, we first tested the stability of IMP in vivo. We found that IMP levels in blood quickly declined to approximately basal levels within 15 min after injection of IMP (200 μg per mouse, i.v.; see Fig. 8 in the supplementary materials at www.pnas.org), confirming that nucleotides are short-lived in whole blood (29). Given that IMP is not stable in vivo, we hypothesized that if IMP plays a role in reperfusion it will likely be within the local milieu of neutrophil recruitment. It is also known that the cytokine TNF-α evokes neutrophil infiltration in lung (30) and can stimulate neutrophils to release proinflammatory mediators that can give rise to tissue damage during reperfusion injury. Therefore, we examined the impact of IMP on TNF-α-induced leukocyte infiltration in the murine air pouch model of acute inflammation. Injection of TNF-α into the dorsal cavity induced a marked increase in neutrophil accumulation within the dorsal air pouch (17, 18). IMP significantly inhibited TNF-α-induced neutrophil infiltration and accumulation in a dose-dependent fashion (IC50 ≈ 140 μg) (Fig. 4A). These results demonstrate that local administration of IMP prevents neutrophil traffic in vivo.
Figure 4.

(A) Dose–response inhibition of IMP on neutrophil accumulation in mouse air pouch. The numbers of TNF-α (20 ng per mouse) -induced neutrophil infiltration in the air pouch with IMP (n = 3 for each dose) or without IMP (n = 5) treatment are expressed as mean ± SEM from each set of mice. Asterisk denotes significant differences (P < 0.01) of the values from IMP-treated (300–1000 μg per mouse) mouse versus vehicle-treated mouse. (Inset) percent inhibition of IMP on neutrophil infiltration in the air pouch model. (B) IMP selectively inhibits neutrophil rolling in microvessels. Isolated human neutrophils were incubated (25°C, 5 min) with each purine (100 μM), then perfused into mouse microvessels treated with 50 μM NG-nitro-l-arginine methyl ester (l-NAME). Rolling neutrophils were enumerated by using video microscopy and are expressed as rolling cells per min per mm2. Asterisk denotes P < 0.01 versus control. Each column represents the mean ± SEM (n = 7–11). In the absence of l-NAME and purine treatment, only 7 to 8 neutrophils roll per min per mm2 within microvessels. (Inset) Concentration-dependent inhibition for IMP (0.1–100 μM) on neutrophil rolling (n = 4–10).
IMP Regulates Neutrophil Rolling.
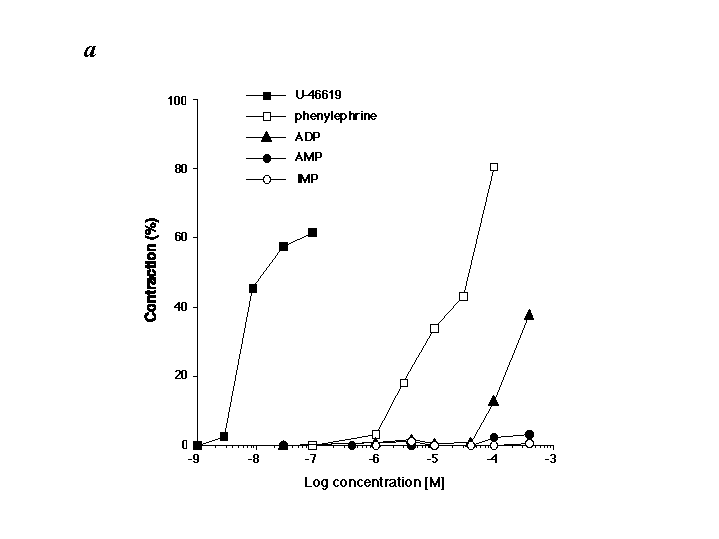
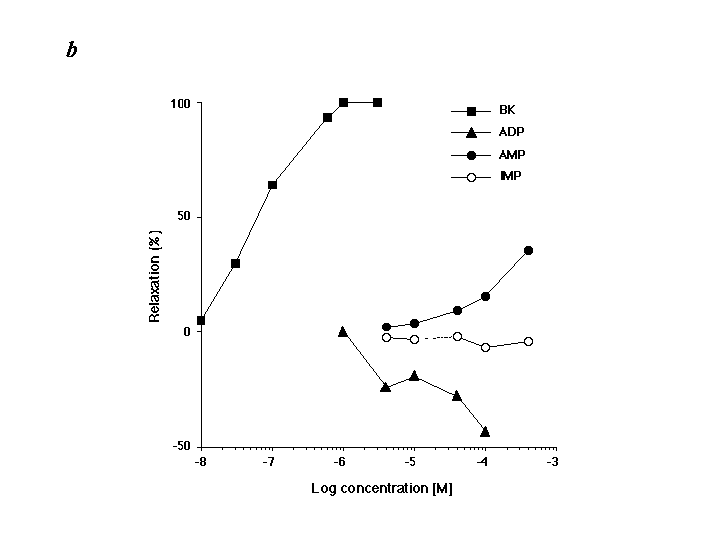
Neutrophils can damage organ tissues after recruitment to the target tissue, where they initially enter by means of rolling and adherence to vascular endothelial cells (1). Thus, we examined IMP–neutrophil interactions within isolated microvessels. IMP significantly reduced neutrophil rolling on microvessels in a concentration-dependent fashion (Fig. 4B) compared with other structurally related purines (such as AMP or inosine). Neutrophil rolling was inhibited by 70% and 90% when the cells were exposed to 1 μM and 10 μM IMP, respectively. In sharp contrast, significant inhibition of neutrophil rolling was not observed with either inosine or AMP at concentrations as high as 100 μM. We also assessed the potential cytotoxic actions of IMP with microvessels and neutrophils. After exposure and washout of IMP, isolated microvessels were still able to support statistically equivalent numbers of neutrophils rolling on their endothelial cells compared with microvessels that were not exposed to IMP. Neutrophils incubated with IMP (100 μM; at 37°C for 30 min) did not alter their viability as determined by their ability to exclude trypan blue. These results indicate that the reduction of neutrophil rolling by IMP was not a result of cytotoxic events. We also examined the action(s) of IMP on microvascular function and found that IMP (1–500 μM) had no impact on contraction or relaxation of mouse mesenteric microvessels (see Fig. 9 in the supplementary materials at www.pnas.org). In addition, human neutrophils exposed to IMP (1 or 100 μM) did not generate superoxide anion (see Fig. 10 in the supplementary materials), indicating that IMP is not a proinflammatory agonist.
IMP Specifically Binds to Neutrophils.
We investigated whether IMP directly binds to specific sites on neutrophils, and to this end prepared [8-3H]IMP for direct analysis. [8-3H]IMP specifically bound to a high-affinity recognition site on neutrophils (Fig. 5) with a Kd of 251.3 ± 27.7 nM and a Bmax of 4.5 ± 0.2 pmol/mg of protein. In addition, adenosine did not effectively compete with the specific binding of [3H]IMP (IC50 = 122 nM for IMP vs. 19.2 μM for adenosine), indicating that the binding site of IMP is specific (Fig. 5 Upper Right Inset). To obtain evidence that this specific binding reflects receptor activation by IMP, GTP[γ35S] binding experiments were performed with neutrophils. IMP increased GTP[γ35S] binding to neutrophil membranes, as did other neutrophil ligands such as leukotriene B4 (n = 6–9; see Fig. 11 in the supplementary materials at www.pnas.org), suggesting that IMP does act by means of a membrane receptor that is associated with a GTP[γS]-bound G protein, as is the case for many G protein-coupled receptors (31).
Figure 5.

Specific binding of [8-3H]IMP to neutrophils. [8-3H]IMP was prepared by biogenic synthesis. Structure of IMP with the 3H label position 8 is indicated in the Upper Right Inset. Isolated human neutrophils (8 × 106 cells in 0.4 ml) were incubated with [8-3H]IMP (0–500 nM) at 4°C for 120 min in the presence or absence of a 1000-fold excess of unlabeled IMP. (Lower Right Inset) Scatchard analysis for [8-3H]IMP binding. The Kd is 251.3 ± 27.7 nM and the Bmax is 4.5 ± 0.2 pmol/mg of protein (n = 4). (Upper Left Inset) Competition of unlabeled IMP (●) or adenosine (○) for specific [3H]IMP (200 nM) binding to intact neutrophils (n = 4). The IC50 value for unlabeled IMP was ≈122 nM and that for adenosine was ≈19.2 μM (n = 4).
Discussion
In this report, we found that up-regulation of AMPD3 gene transcript and the enzymatic activity of the gene product accompanies remote organ lung injury after murine hind-limb ischemia–reperfusion. Administration of AMP deaminase inhibitor (coformycin) before reperfusion significantly enhanced neutrophil accumulation in the lung. This result suggests that the up-regulated AMPD3 gene product may be required for controlling neutrophil infiltration and accumulation, to protect the lung from activated neutrophils. The AMPD3 gene product functions in the purine metabolism pathway. AMP deaminase converts AMP to IMP by deamination of the substrate (14). We obtained evidence that IMP levels in lung tissues were significantly increased as AMP deaminase was activated during reperfusion and leukocyte infiltration of the tissues (Figs. 2 and 3). Taken together, these results suggest that IMP plays a direct role in regulating neutrophil diapedesis in lung tissues after hind-limb ischemia–reperfusion. In support of this concept, we also found that low levels of IMP were present in whole blood during reperfusion, and the retention of IMP in blood was short-lived after i.v. injection of IMP. These two points indicate that the levels of IMP in blood circulation could not account for the increases in IMP monitored by HPLC analyses of the pulmonary tissues (see Fig. 7 in the supplementary materials).
Also, the results of “add back” experiments indicate that IMP significantly inhibits TNF-α-induced local neutrophil infiltration in acute inflammation (Fig. 4). This in vivo evidence provides support for IMP as a regulator of neutrophil traffic, and it follows that diminished accumulation of neutrophils may spare tissue damage. We also found that IMP, but not AMP or inosine, attenuates neutrophil rolling in microvessels. These findings indicate that IMP serves as a local or short-range mediator regulating neutrophil rolling (i.e., transient adherence) (Fig. 4), the initial step in neutrophil infiltration and tissue accumulation (1–3). It is noteworthy that IMP, at concentrations as high as 100 μM, did not stimulate neutrophil responses in vitro (i.e., generation of superoxide anion) (see Fig. 10 in the supplemental materials), indicating that IMP serves as an antagonist for neutrophils in inflammation. Hence the findings (i) that IMP acts on neutrophils to inhibit their trafficking in vivo, (ii) that IMP acts on neutrophils to reduce rolling in microvessels, and (iii) that up-regulation of AMP deaminase and its inhibition correlate with decreased IMP levels as well as increased neutrophil accumulation in vivo, when taken together, provide strong evidence that IMP generation by up-regulated AMPD3 gene expression during reperfusion constitutes a newly identified protective system in lung reperfusion injury.
Determination of the localization of up-regulated AMPD3 gene transcripts in bronchial and alveolar epithelium provided evidence for a potential cellular mechanism, namely for the topography of IMP and AMP deaminase action. In view of these results, it is likely that increased AMP deaminase and IMP are generated in epithelium of lung in response to overt neutrophil infiltration. Thus, IMP may also regulate epithelial functions relevant to neutrophil penetration into pulmonary alveoli (1). Alternatively, IMP, like other nucleotides, can be released by injured cells into the microenvironment, where it regulates the functions of neutrophils and vascular endothelium as well as other cell types that are involved in wound healing (32). Along these lines, we demonstrated that IMP specifically binds to neutrophils. The results of [8-3H]IMP binding and adenosine competition strongly indicate that IMP has a specific recognition site on neutrophils distinct from adenosine receptors. Also, IMP stimulates GTP[γ35S] binding to neutrophil membrane, suggesting involvement of a G protein-coupled receptor. The present results with IMP and neutrophils are of interest in view of earlier findings with cyclic IMP-sensitive kinases reported in brain (33), erythrocytes (34), and neutrophils (35) that, when taken together with our present findings, suggest that inosine and its phosphorylated forms (i.e., IMP) may carry information in signaling systems.
In conclusion, we have uncovered a previously unappreciated regulatory system governing neutrophil traffic into ischemia–reperfused lung that involves transcriptional regulation for the AMPD3 gene enhancing IMP generation. These results provide the evidence that, in lung tissues during reperfusion, the AMPD3 gene is up-regulated and its enzymatic product IMP specifically binds to neutrophils, down-modulating rolling within microvessels and blocking trafficking in vivo, thereby limiting entry of neutrophils into the tissue. These findings open opportunities for using the endogenous IMP-AMP deaminase system to control neutrophil tissue entry, a critical step in minimizing reperfusion-associated injury and inflammation. Knowledge of these endogenous protective systems can provide new approaches to prevent and control neutrophil-mediated tissue damage associated with inflammation as well as certain cardiovascular and neurodegenerative diseases.
Supplementary Material
Acknowledgments
We thank Stephanie J. Andrews, Kristina Ware, Rachel Cochran, and Alexis Teplick for their skillful technical assistance and Mary H. Small for expert assistance in the preparation of this manuscript. We also gratefully acknowledge Dr. Birgitta Schmidt, Department of Pathology, Brigham and Women's Hospital, for expert histologic evaluation of tissue sections and Dr. Sean Colgan for helpful discussions regarding quantitation of purine nucleotide analysis. This study was supported in part by National Institutes of Health Grants GM38765 (C.N.S.), DK50305 (C.N.S.), and HL52886 (G.L.S.).
Abbreviations
- MPO
myeloperoxidase
- RT-PCR
reverse transcription–PCR
- TNF-α
tumor necrosis factor α
- GTP[γ35S]
γ-[35S]thio-GTP
Footnotes
This paper was submitted directly (Track II) to the PNAS office.
References
- 1.Cotran R S, Kumar V, Collins T, editors. Robbins Pathologic Basis of Disease. Philadelphia: Saunders; 1999. pp. 1–30. [Google Scholar]
- 2.Gallin J I, Snyderman R. In: Inflammation: Basic Principles and Clinical Correlates. Gallin J I, Snyderman R, Fearon D T, Haynes B F, Nathan C, editors. Philadelphia: Lippincott Williams & Wilkins; 1999. pp. 1047–1061. [Google Scholar]
- 3.Gelman S. Anesthesiology. 1995;82:1026–1060. doi: 10.1097/00000542-199504000-00027. [DOI] [PubMed] [Google Scholar]
- 4.Seekamp A, Ward P A. Agents Actions. 1993;41:137–152. [PubMed] [Google Scholar]
- 5.Weiss S J. N Engl J Med. 1989;320:365–376. doi: 10.1056/NEJM198902093200606. [DOI] [PubMed] [Google Scholar]
- 6.Patarroyo M, Lindbom L, Lundberg C. Adv Exp Med Biol. 1991;314:1–17. doi: 10.1007/978-1-4684-6024-7_1. [DOI] [PubMed] [Google Scholar]
- 7.Weissmann G, Smolen J E, Korchak H M. N Engl J Med. 1980;303:27–34. doi: 10.1056/NEJM198007033030109. [DOI] [PubMed] [Google Scholar]
- 8.Lefer A M. Agents Actions. 1993;41,Suppl.:127–135. [PubMed] [Google Scholar]
- 9.Townsend H S, Goodman S B, Schurman D J, Hackel A, Rock-Utne J G. Acta Anaesthesiol Scand. 1996;40:1234–1237. doi: 10.1111/j.1399-6576.1996.tb05556.x. [DOI] [PubMed] [Google Scholar]
- 10.Bradley P P, Priebat D A, Christensen R D, Rothstein G. J Invest Dermatol. 1982;78:206–209. doi: 10.1111/1523-1747.ep12506462. [DOI] [PubMed] [Google Scholar]
- 11.Takano T, Fiore S, Maddox J F, Brady H R, Petasis N A, Serhan C N. J Exp Med. 1997;185:1693–1704. doi: 10.1084/jem.185.9.1693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Liang P, Pardee A B. Science. 1992;257:967–971. doi: 10.1126/science.1354393. [DOI] [PubMed] [Google Scholar]
- 13.Chomczynski P, Sacchi N. Anal Biochem. 1987;161:156–159. doi: 10.1006/abio.1987.9999. [DOI] [PubMed] [Google Scholar]
- 14.Raffin J P, Thebault M T. Comp Biochem Physiol B. 1991;99:125–127. doi: 10.1016/0305-0491(91)90016-7. [DOI] [PubMed] [Google Scholar]
- 15.Overgaard-Hansen K, Klenow H. J Cell Physiol. 1993;154:71–79. doi: 10.1002/jcp.1041540110. [DOI] [PubMed] [Google Scholar]
- 16.Soussi B, Lagerwall K, Idström J-P, Scherstén T. Am J Physiol. 1993;265:H1074–H1081. doi: 10.1152/ajpheart.1993.265.4.H1074. [DOI] [PubMed] [Google Scholar]
- 17.Cronstein B N, Naime D, Ostad E. J Clin Invest. 1993;92:2675–2682. doi: 10.1172/JCI116884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Tessier P A, Naccache P H, Clark-Lewis I, Gladue R P, Neote K S, McColl S R. J Immunol. 1997;159:3595–3602. [PubMed] [Google Scholar]
- 19.Scalia R, Gefen J, Petasis N A, Serhan C N, Lefer A M. Proc Natl Acad Sci USA. 1997;94:9967–9972. doi: 10.1073/pnas.94.18.9967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Böyum A. Scand J Clin Lab Invest Suppl. 1968;21:77–89. [PubMed] [Google Scholar]
- 21.Hickey M J. J Immunol. 1997;158:3391–3400. [PubMed] [Google Scholar]
- 22.Fiore S, Maddox J F, Perez H D, Serhan C N. J Exp Med. 1994;180:253–260. doi: 10.1084/jem.180.1.253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Munson P J. Methods Enzymol. 1983;92:543–576. doi: 10.1016/0076-6879(83)92044-x. [DOI] [PubMed] [Google Scholar]
- 24.Northup J K, Smigel M D, Gilman A G. J Biol Chem. 1982;257:11416–11423. [PubMed] [Google Scholar]
- 25.Klausner J M, Anner H, Paterson I S, Kobzik L, Valeri C R, Shepro D, Hechtman H B. Ann Surg. 1988;208:761–767. doi: 10.1097/00000658-198812000-00015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Sermsuvitayawong K, Wang X, Nagabukuro A, Matsuda Y, Morisaki H, Toyama K, Mukai T, Morisaki T. Mamm Genome. 1997;8:767–769. doi: 10.1007/s003359900562. [DOI] [PubMed] [Google Scholar]
- 27.Wang X, Morisaki H, Sermsuvitayawong K, Mineo I, Toyama K, Ogasawara N, Mukai T, Morisaki T. Gene. 1997;188:285–290. doi: 10.1016/s0378-1119(96)00818-9. [DOI] [PubMed] [Google Scholar]
- 28.Kyriazi H T, Basford R E. Anal Biochem. 1985;144:477–482. doi: 10.1016/0003-2697(85)90144-7. [DOI] [PubMed] [Google Scholar]
- 29.Soderback U, Sollevi A, Fredholm B B. Acta Physiol Scand. 1987;129:189–194. doi: 10.1111/j.1748-1716.1987.tb08058.x. [DOI] [PubMed] [Google Scholar]
- 30.Seekamp A, Warren J S, Remick D G, Till G O, Ward P A. Am J Pathol. 1993;143:453–463. [PMC free article] [PubMed] [Google Scholar]
- 31.Cerione R A. Biochim Biophys Acta. 1991;1071:473–501. doi: 10.1016/0304-4157(91)90007-j. [DOI] [PubMed] [Google Scholar]
- 32.Enjyoji K, Sevigny J, Lin Y, Frenette P S, Christie P D, Esch J S, 2nd, Imai M, Edelberg J M, Rayburn H, Lech M, et al. Nat Med. 1999;5:1010–1017. doi: 10.1038/12447. [DOI] [PubMed] [Google Scholar]
- 33.Miyamoto E, Kuo J F, Greengard P. J Biol Chem. 1969;244:6395–6402. [PubMed] [Google Scholar]
- 34.Rubin C S, Erlichman J, Rosen O M. J Biol Chem. 1972;247:6135–6139. [PubMed] [Google Scholar]
- 35.Tsung P-K, Hermina N, Weissmann G. Biochem Biophys Res Commun. 1972;49:1657–1662. doi: 10.1016/0006-291x(72)90533-5. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}