

Abstract
A single-molecule analysis was applied to study the dynamics of synaptic and presynaptic DNA-protein complexes (binding of two DNA and one DNA duplex, respectively). In the approach used in this study, the protein was tethered to a surface, allowing a freely diffusing fluorescently labeled DNA to bind to the protein, thus forming a presynaptic complex. The duration of fluorescence burst is the measure of the characteristic lifetime of the complex. To study the formation of the synaptic complex, the two SfiI-bound duplexes with the labeled donor and acceptor were used. The synaptic complex formation by these duplexes was detected by the fluorescence resonance energy transfer approach. The duration of the fluorescence resonance energy transfer burst is the measure of the characteristic lifetime of the synaptic complex. We showed that both synaptic and presynaptic complexes have characteristic dissociation times in the range of milliseconds, with the synaptic SfiI-DNA complex having the shorter dissociation time. Comparison of the off-rate data for the synaptic complex with the rate of DNA cleavage led to the hypothesis that the complex is very dynamic, so the formation of an enzymatically active synaptic complex is a rather rare event in these series of conformational transitions.
INTRODUCTION
A synaptic DNA-protein complex is formed by two DNA regions brought together and stabilized by a specialized protein or protein complex. The formation of synaptic DNA-protein complexes is the key step of various genetic processes, such as site-specific recombination, genome integration, excision, and inversion of specific DNA regions (1). In fact, the formation of a synaptic complex is a more general phenomenon that is not limited to various site-specific recombination systems. There is a family of DNA restriction enzymes that requires the formation of synaptic complexes for further site-specific DNA cleavage (2–5). Despite a wide range of protein sizes and complexity of systems (dimers, homo and heterotetramers, or even higher stoichiometry), the formation of synaptic complexes is a dynamic process in which the creation of a complex with only one DNA duplex (presynaptic complex) is the first step to the formation of an active complete complex (1). However, this initial step varies for different systems. The presynaptic complex may be a part of the complete protein complex (e.g., a monomer) bound to the DNA template, so parts of association via the protein-protein interaction form the synaptic complex. This is typically observed in multiprotein complexes such as the synaptic system formed by two RAG1/RAG2 heterotetramers during the V(D)J recombination (6). An alternative pathway for the presynaptic complex formation involves the interaction of the complete protein complex with one of the DNA templates, so the synaptic complex is formed by the recruiting of the second DNA chain. This pathway is realized in a number of type II restriction enzymes, and SfiI is one of the best characterized enzymes of this type (7–9). There are a number of questions related to the mechanism of synaptic complex formation that have not been answered so far. For example, how dynamic are the entire complex and its intermediates? Given the requirements for conformational transitions within the synaptic complex (DNA cleavage and relegation with the formation of Holliday junction in the case of site-specific recombination), one anticipates a dynamic behavior of the synaptic complex. How fast are the structural transitions within the synaptic complex? How stable is the DNA-protein complex? Answering these questions is critical for understanding the molecular mechanisms of synaptic complex formation, but relies on the availability of methods that allow lifetime measuring of transiently formed states of molecular complexes.
It was recently demostrated that single-molecule imaging techniques are capable of detecting intermediate states of rather complex molecular systems (10–15) and can thus be applied for characterizing synaptic complexes. Single-molecule dynamic atomic force microscope (AFM) spectroscopy is another technique that provides measurements of pairwise interactions within the molecular complexes (16–19), and we have recently applied this technique to characterize the strength of the synaptic complex formed by SfiI restriction enzyme and its transient state, which is termed a presynaptic complex (20). These data also allowed us to estimate the characteristic dissociation rates for both synaptic and presynaptic complexes and led to the proposal that the complexes have a highly dynamic character. However, the extrapolation of the data to zero value of rupture force performed on the measurements over the range of relatively high loading rates is a serious limitation of this informative and useful single-molecule approach.
Here, we describe the results of the analysis of dynamics of synaptic SfiI-DNA complexes with the use of “tethered” single-molecule fluorescence approach including fluorescence resonance energy transfer (FRET) (10,14,21–28). With this approach we were able to detect the interaction of the system on a timescale exceeding the free diffusion time by orders of magnitude. We also employed single-molecule FRET to study dynamics of SfiI-DNA complex. The “tethered” single-molecule fluorescence approach can measure kinetic parameters on immobilized single molecules, and it was used to reveal the mechanisms of such biologically important processes as Holliday junction dynamics (29,30), branch migration (31), enzyme activity (26,32) and RNA translation (15,33). Conventional fluorescence correlation spectroscopy (FCS) on freely diffusing molecules is capable of analyzing the dynamics of isolated molecules on a millisecond timescale (34,35). However, this technique is limited to the lifetime of a ligand-receptor complex in the range of the diffusion time through the observation volume, which is typically 1 ms (36). FCS, in combination with total internal reflection, was applied to study reversible interaction of fluorescently labeled IgG with the mouse receptor FcγRII in substrate-supported planar membranes (37). In these studies, dissociation times in the range of hundreds of milliseconds were measured. We took advantage of this tethered technique and applied this approach to study synaptic complexes. To achieve this goal, one of the interacting components of the system (protein or DNA) was anchored to a surface, allowing the other freely diffusing partner (DNA duplex) to bind to the immobilized target. Using the “tethered” approach in combination with single-molecule FRET analysis, we were able to analyze the dynamics of the interaction within both presynaptic and synaptic complexes in real time.
METHODS
Preparation of oligonucleotides
DNA oligonucleotides containing a 13-base pair (bp) recognition site (5′-GGCCNNNNNGGCC-3′) and terminal amino modification at the 5′ end were purchased from Integrated DNA Technologies (Coralville, IA) as single-stranded complements. The sequence for a modified single-stranded oligonucleotide was 5′-/5AmMC6/CCGGCCTCGAGGGCCATT-3′. The complementary strand (5′-AATGGCCCGTCAGGCCGG-3′) did not contain a modification. Amino groups terminating single-stranded oligonucleotides were labeled with succinimide esters of Cy3 or Cy5 dye (Amersham Biosciences, Piscataway, NJ) according to the protocol provided. Dye-labeled oligonucleotides were separated from the unlabeled ones with reverse-phase high-performance liquid chromatography. Separation quality was controlled by measuring the absorption spectra; the molar ratios of attached dyes and DNAs were close to 1:1. Both Cy3- and Cy5-labeled single-stranded oligonucleotides were then annealed with their complements by heating to 98°C followed by slow cooling to room temperature.
Glass surface modification
Square glass coverslips (Karl Hecht, Sondheim, Germany) were cleaned with 1:1 nitric acid/hydrogen peroxide mixture for 10 min and stored in deionized water until use. A 20-μL glass cell was assembled on the original sample holder (PicoQuant, Berlin, Germany) from the glass coverslip, 0.1-mm-thick teflon spacer (American Durafilm, Holliston, MA), and 25-mm-diameter quartz disk with two small holes to fill the observation cell. Glass slips were treated with 167 μM maleimide silatrane aqueous solution for 3 h and rinsed with dd-water resulting in a maleimide functionalized surface. For covalent attachment of SfiI, 8 nM solution of the protein (low bovine serum albumin content, New England Biolabs, Beverly, MA) was treated with TCEP-hydrochloride (Tris(2-carboxyethyl)phosphine) (Pierce, Rockford, IL) at 25°C for 7 min to reduce disulfide bonds of the protein. The maleimide-functionalized surface was then incubated with this solution for 1 h. The thiol groups of protein cysteine residues react with the maleimide units on the surface, leading to covalent immobilization of the protein. The modified glass cell was thoroughly rinsed with buffer solution (10 mM HEPES, 50 mM NaCl, 2 mM CaCl2, pH 7.0) to avoid bubbles and used immediately after preparation. Measurements were performed in SfiI binding buffer (10 mM HEPES, pH 7.0, and 50 mM NaCl) containing 2 mM CaCl2 at room temperature.
Peak durations and FRET measurements
Single-molecule FRET and FCS measurements were carried out on a confocal microscope built around an Olympus IX71 (Hitschfel Instruments, St. Louis, MO) inverted microscope body equipped with a piezo-driving scanning stage (Physik Instrumente, Karlsruhe, Germany). An oil immersion UPlanApo 100× objective with 1.35 numerical aperture (Olympus, Tokyo, Japan) was used for all measurements. Autocorrelation functions were typically obtained in 10 min using incident laser intensity of ∼100 μW provided by a 532-nm laser (Crystalaser, Reno, NV). This wavelength was optimal for the excitation of Cy3 dye (donor). Fluorescence was collected through a 30-μm-diameter pinhole placed in the focal plane of the right-side port. After the pinhole, a dichroic mirror (Chroma 630dcxr, Rockingham, VT) separated the emission into either a donor or acceptor channel, depending on the wavelength. The emission was then focused onto silicon avalanche photodiodes (SPCM-AQR-15, Perkin Elmer Optoelectronics, Freemont, CA) operating in single-photon counting mode. Data acquisition and preliminary analysis were performed using TimeHarp 200 PCI-board (PicoQuant, Berlin, Germany).
To measure peak duration, the fluorescence intensity time traces were averaged with 3-ms bin intervals. Data were corrected for the background level and for 20% bleed-through of the donor intensity into the acceptor channel, which was obtained from the donor-only labeled molecules. For each individual time trace, cut-off level was determined based on the average intensity of the signal, and duration of the peaks two times higher than cut-off level were measured. Donor and acceptor intensity time traces were analyzed in the exact same way.
To analyze peak duration, the normalized survival probability, Psurv, was evaluated as follows: the number of events to the right of a selected time at each time-bin interval was counted. These values, divided by the total number of events, were plotted against time according to the equation
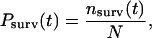 |
(1) |
where 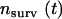 is the number of complexes that survive longer than time t, and N is the total number of complexes observed (38,39). The time dependence of Psurv cannot be approximated by a single exponential, and the curve was best fit with two decaying exponentials corresponding to species with different lifetimes,
is the number of complexes that survive longer than time t, and N is the total number of complexes observed (38,39). The time dependence of Psurv cannot be approximated by a single exponential, and the curve was best fit with two decaying exponentials corresponding to species with different lifetimes, 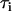 :
:
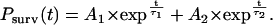 |
(2) |
Here, 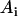 is the reflecting fraction of species with lifetime
is the reflecting fraction of species with lifetime 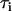 . The major feature of normalized survival probability (NSP) approach is that no assumption about the type of the distribution of the data is needed. Therefore, if the decay process is complex (e.g., multiexponential), the NSP approach is capable of retrieving characteristics for all components. All fitting algorithms were performed on Microcal Origin Version 6.0.
. The major feature of normalized survival probability (NSP) approach is that no assumption about the type of the distribution of the data is needed. Therefore, if the decay process is complex (e.g., multiexponential), the NSP approach is capable of retrieving characteristics for all components. All fitting algorithms were performed on Microcal Origin Version 6.0.
Apparent FRET efficiency (27)was estimated according to the equation
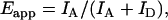 |
(3) |
where IA and ID are the corrected acceptor and donor intensities, respectively. The general equation for FRET efficiency is
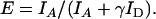 |
(4) |
The γ-factor in the Eq. 4 is equal to 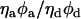 and accounts for quantum yields of donor and acceptor emission (φd and φa, respectively), as well as instrument detection efficiencies in both channels (ηd and ηa). This factor was estimated to be ∼1 for our experimental setup. However, the single-pair FRET technique on immobilized molecules should be used cautiously in absolute distance measurements, due to the effect of microenvironments, and it is more feasible for relative dynamic distance changes between the fluorescent dyes (25,26,40).
and accounts for quantum yields of donor and acceptor emission (φd and φa, respectively), as well as instrument detection efficiencies in both channels (ηd and ηa). This factor was estimated to be ∼1 for our experimental setup. However, the single-pair FRET technique on immobilized molecules should be used cautiously in absolute distance measurements, due to the effect of microenvironments, and it is more feasible for relative dynamic distance changes between the fluorescent dyes (25,26,40).
FCS measurements
FCS measurements were performed in the same way as described above. Fluorescence-intensity data were acquired with TimeHarp 200 PCI-board (PicoQuant). Data were stored in time-tagged time-resolved format, and fluctuation correlation functions were evaluated by the company-provided software according to the following equation:
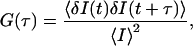 |
(5) |
where I is the fluorescence intensity and δI represents fluctuations of fluorescence intensity. The fluctuation correlation function shown differs from the autocorrelation function by a constant value of −1. To estimate averaged lifetimes of the molecules in the detection volume, a lateral diffusion approximation for intensity fluctuation correlation function was used:
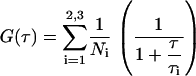 |
(6) |
Here, Ni is a fitting parameter reflecting fluorescent intensity of species i with lifetime τi, and τi is the average species lifetime in the detection volume. We used the fitting procedure described in Allen and Thompson (41) to analyze the form of G(τ) for freely diffusing fluorescent molecules excited with a focused laser beam when fluorescence is collected through a confocal pinhole. Fitting was performed on Microcal Origin Version 6.0.
RESULTS
Tethered single-molecule fluorescence approach
Fig. 1 shows schematics of the approach on tethered single molecules. The key point of this technique is anchoring the target molecule while the probe molecule diffuses freely in solution. In this particular case, SfiI is tethered to the surface via a flexible linker, which allows for observation of the interaction with fluorescent probes (DNA duplexes) over the selected area for an indefinite time (Fig. 1, A and B). The SfiI enzyme was covalently attached to a functionalized glass substrate via SH-group of the unique cysteine residue located near the C-terminus of the SfiI protein sequence (230th position out of 269 amino acids total) using the maleimide-silatrane approach described recently for mica surface functionalization (42). The fact that this residue is not involved in the recognition process (43) is important for assembly of the complexes. In addition, we have also shown that covalent immobilization of SfiI protein via this single cysteine residue does not affect the ability of the protein to form a complex with the DNA duplex containing a specific 13-bp recognition sequence (20).
FIGURE 1.

Scheme illustrating the procedures for studies of presynaptic (A) and synaptic (B) SfiI-DNA complexes. (A) The DNA duplex tagged with Cy3 (D in green burst) was used for detecting the formation of the presynaptic complex. (B) Two DNA duplexes tagged with Cy3 and Cy5 (A in red burst) dyes are bound to protein tetramer to form the Forster pair. The fluorescence of the acceptor molecule (Cy5) is observed only in the synaptic complex upon energy transfer from the excited donor molecule. The laser beam was focused directly on the surface (not shown). Diameter of the laser beam is ∼0.5 μm.
The presynaptic (Fig. 1 A) complex was formed upon adding the duplex labeled with Cy3. Analysis of the synaptic complex, which requires the binding of two DNA duplexes to the target, was performed with the use of duplexes labeled with Cy3 (donor) and Cy5 (acceptor) capable of producing FRET if the distance between the dyes is <10 nm (Fig. 1 B). According to recent crystallographic data (43) for SfiI-DNA synaptic complexes, the donor and acceptor can be separated by 6 nm, the distance easily detectable by our single-molecule fluorescence microscope (31).
Presynaptic SfiI-DNA complex
First, we analyzed the interaction of the DNA duplex with immobilized SfiI enzyme leading to the formation of the presynaptic complex, where only one DNA duplex binds to the enzyme (Fig. 1 A). A diluted solution of the DNA duplex fluorescently labeled with Cy3 dye was injected to allow binding to the target, and the microscope objective was focused on the surface so that the duplexes flowing in the surface proximity and bound to the enzyme were detected. A computer-controlled scan of the surface was performed to detect the positions of the enzymes capable of binding to the fluorescently labeled duplex. The developed surface modification and sample preparation procedure allowed us to prepare the surface with a low density of immobilized protein molecules. This is illustrated in Fig. S1 (Supplementary Material), which shows a 13 × 13 μm scan in which the distance between adjacent proteins (bright spots on the scans) is in the range of several microns. The laser beam was brought to one of the identified positions of the protein and the time-dependent fluorescence intensity was recorded. Typical diameter of the focused beam is in the range of 500 nm, ensuring the acquisition of data from an individual protein within the spot. The fluctuations of fluorescence intensity due to binding-unbinding events were collected with the avalanche photodiode detector of the single-molecule fluorescence setup (31). The nanomolar concentration of the duplex (typically used for free-diffusion, single-molecule FCS experiments) enabled us to detect individual complex-formation events. The sample stage was moved to another spot on the initial scan, and the data acquisition process was repeated with another enzyme molecule.
The fluorescence intensity fluctuation due to binding-unbinding events were converted into FCS correlation curves, as described in Materials and Methods, and one of these decay curves is shown in Fig. 2 A (1). This is a typical S-shaped FCS correlation curve, from which the characteristic time of 16.0 ± 0.2 ms was obtained; here ±0.2 ms is a fitting error for a single time trajectory. The data collected well above the surface produced a different autocorrelation curve. As is seen in Fig. 2 B, the characteristic time is considerably less, τ = 0.60 ± 0.01 ms. This value corresponds very well to the diffusion times obtained for freely diffusing DNA duplexes (21,44). Similar experiments with a longer duplex (39 bp) (Fig. 2 B (2)) yielded a larger time value, τ = 1.19 ± 0.01 ms, as anticipated for a longer DNA duplex. Another control experiment using the same 18-bp DNA duplex and the maleimide-functionalized glass surface with no immobilized enzyme is shown in Fig. 2 A (2). The characteristic time τ = 0.43 ± 0.02 ms is close to the value determined in the experiments described above (Fig. 2 B (1)). These observations suggest that DNA does not adsorb nonspecifically to maleimide-modified surface. It also ensures that only events of specific binding between DNA and SfiI are detected in the experiment with surface-bound SfiI. A slightly lower τ value for control experiments with no protein bound (0.43 ms) compared to that obtained for the off-the-surface experiments (0.60 ms) is explained by a smaller detection volume for the beam focused at the surface (37).
FIGURE 2.

Fluorescence fluctuation correlation functions G(τ) of Cy3 labeled DNA duplexes. (A) FCS functions of labeled DNA duplexes near the surface: Curve 1 (blue diamonds) corresponds to the experiment performed in the presence of anchored SfiI tetramers. Curve 2 (red circles) represents data obtained in the absence of the attached protein. (B) FCS curves acquired from the freely diffusing in solution Cy3-labeled DNA duplexes of different lengths: Curve 1 (red circles), 18 bp; curve 2 (green triangles), 39 bp.
We also studied the interaction between SfiI and nonspecific DNA duplex of the same length (18 bp). The FCS analysis produced characteristic times of 0.38 ± 0.09 ms and 0.52 ± 0.09 ms for experiments performed by focusing the beam at the surface and in the bulk, respectively. The value obtained for a characteristic lifetime (0.38 ms) suggests that the interaction of the protein with the nonspecific duplex occurs in the submillisecond timescale. We did observe short spikes in the time traces, indicating the formation of complexes with such short lifetimes; however, these rare events cannot be reliably identified on the FCS curve as a step.
The experiments were performed as described over various positions on the same sample and different samples. The characteristic time averaged over a set of fluorescence measurements provides the mean value for the dissociation time <τ> = 13 ± 5 ms (5 ms is a standard deviation calculated from the set of 10 FCS experiments). A large variability in the characteristic time values, even those acquired for different positions on the same sample, primarily reflects the effect of the local microenvironmental conditions typically detected in single-molecule experiments (45).
An alternative way to analyze the surface-bound events is by direct analysis of the duration of the fluorescence bursts on the time trajectory data. A fragment of a typical fluorescence-intensity time trace obtained from a single experiment, corresponding to the FCS curve described in Fig. 2 A, is shown in Fig. 3 A. The temporal fluctuations of the fluorescence (IF) are characterized by a set of bursts, the duration of which corresponds to times when the fluorescently labeled DNA molecule enters and leaves the detection volume. The peaks shown in Fig. 3 A with intensity >40 total counts/3 ms were analyzed. The abruptness of the intensity spikes is the indication for the single-molecule events and is defined by the diffusion time over the focal area. The peak width corresponds to the time the fluorescently labeled molecule spends in the detection volume. For example, the burst widths are 9 ms for peak 1, 27 ms for peak 2, and 63 ms for peak 3. The diffusion time, ∼0.5 ms, is thus substantially smaller than the lifetime of the SfiI-DNA complex, which is why the peaks appear so sharp on the time trajectories. Characteristic lifetimes of the individual protein-DNA complexes deviate quite significantly due to the effect of microenvironment, as mentioned above. The fluorescent dyes under our experimental conditions bleach in the range of several seconds; therefore, the correction of the characteristic lifetime of the complex (10–20 ms) is ∼1%, which is also far beyond the accuracy of their determination.
FIGURE 3.

Direct analysis of the time traces obtained for the presynaptic DNA-SfiI complex. (A) Two examples of the fluorescence-intensity time traces selected from the full time trajectory acquired during 10 min of continuous observation. The parameters of all peaks were measured as described in the Methods section; lifetimes τ of three peaks are indicated, for example. Some peaks are marked with the numbers (see text). (B) Distribution of burst durations obtained from multiple time-trace trajectories. (C) Plot of normalized survival probability versus the lifetime of the complex. The characteristic lifetime of 14.5 ± 0.3 ms was determined from the two-exponentials fit as an average lifetime of the most abandoned fraction; ±0.3 is the error of the fit.
The lifetimes of DNA-protein complexes obtained from ∼5000 bursts are assembled as a histogram in Fig. 3 B. This data set was analyzed by the NSP approach utilized in other studies (38,39) for analysis of time-dependent events (see Methods for details). In the NSP approach, the number of events to the right of a selected time is counted. These values are plotted against time, producing a graph that can be fit with multiple exponential decays, enabling us to retrieve the characteristic times and ratios of each of these components. The NSP curve calculated for the dataset shown in Fig. 3 B is shown in Fig. 3 C. This curve provides the characteristic lifetime τ = 14.5 ± 0.3 ms. Lifetime value obtained using survival probability analysis is very close to τ = 13 ± 5 ms, determined by analysis using multiple autocorrelation functions. NSP analysis also reveals a second component in the decay, which comprises ∼4% of the total events, with a characteristic time of 142 ± 19 ms.
Synaptic SfiI-DNA complex
During analysis of the time trajectories, we found that there are rare but repeatable events when the heights of the peaks on the time trajectories are double the mean value, suggesting that binding of two DNA duplexes within the spot occurred. Some of these events might be due to the binding of two DNA duplexes to the same SfiI target, leading to the formation of the synaptic complex. To distinguish this event reliably from the simultaneous binding of DNA duplexes to different proteins within the observation area, and thus to ensure detection of the synaptic complex, the approach utilizing FRET was developed and applied. In these experiments, DNA duplexes labeled with Cy3 and Cy5 dyes were used. When a synaptic complex consisting of both DNA duplexes is formed, energy transfer from Cy3 (donor) to Cy5 (acceptor) occurs, leading to the FRET signal, as schematically shown in Fig. 1 B.
In these experiments, the complex was excited at the wavelength corresponding to the donor absorbance (Cy3), and the time-correlated fluorescence intensities from donor and acceptor were detected (31). Fig. 4 A shows two examples of typical single-molecule time dependences of fluorescence intensity for donor and acceptor (green and red lines, respectively). The fluorescence-intensity time trace of the donor (green) resembles that of the presynaptic complex (Fig. 3 A), with approximately the same frequency of fluorescence bursts. However, in addition to these peaks, the bursts of the acceptor appear (arrows). The intensity of the donor fluorescence is low when the acceptor peaks appear, and alternatively the donor fluorescence increases when the acceptor peaks drop and disappear, as can be seen in Fig. 4 A (peak 3). This is a clear indication of the FRET signal and thus of the formation of synaptic complexes. Typically, events with a FRET efficiency of 10–40% appear, but events with higher FRET efficiency were also observed. For example, peaks marked with 1 and 2 in Fig. 4 A have mean FRET efficiencies of ∼14% and ∼17%, respectively, but peak 3 has much larger FRET efficiency (∼70%). Such a difference in FRET values can be explained by variability of the local environment and is rather typically observed in single-molecule experiments (45). FRET events are rare, due to the low probability of binding simultaneously both donor- and acceptor-labeled DNA at the low DNA concentrations required for single-molecule experiments; therefore, multiple experiments were performed to accrue a reasonable number of FRET events. FRET values obtained for multiple experiments were assembled and plotted as a histogram (Fig. 4 B). This histogram has two distinct maxima corresponding to FRET values of 0.11 and 0.33. Based on the available crystallographic data of SfiI-DNA complex (43), it is reasonable to assign these two peaks in the FRET distribution to the formation of synaptic complexes with “antiparallel” or “parallel” orientations corresponding to donor-acceptor distances of ∼8 or ∼6.5 nm, respectively. We recently showed (46) that despite the asymmetry of the central part of the recognition site, both orientations of the duplexes within the synaptosomes can be formed.
FIGURE 4.

Results of the experiments for the synaptic SfiI-DNA complexes. (A) Fluorescence-intensity time traces of both donor (green) and acceptor (red) selected from 600 s of total acquisition time. Peak durations were measured from acceptor time trajectories as described in Methods: measured peaks are marked with arrows; lifetimes (τ) of three peaks are indicated and peaks are numbered (see text). (B) A histogram of the apparent FRET efficiencies obtained from the donor and acceptor intensities of measured peaks. The maxima of the histogram were obtained from the Gaussian distributions; ±0.01 is the fitting error of the peak positions. (C) A histogram of the acceptor fluorescence-burst duration. (D) The survival probability plot versus time. The characteristic lifetime of the major fraction, τ = 6.8 ± 0.4 ms, was determined from the double-exponential fit; ±0.4 is the fitting error.
The assembled widths of each FRET peak over the same series of time trajectories are shown in Fig. 4 C. The data was analyzed using the NSP approach and the survival probability plot is shown in Fig. 4 D. The data were fitted with two exponential decay curves, with a characteristic time of 6.8 ± 0.3 ms, corresponding to ∼99% of the events. Note that the mean lifetime value is less than the lifetime for the presynaptic complex.
DISCUSSION
The results obtained in this work show that the disassembly of synaptic and presynaptic complexes occurs on the millisecond timescale, suggesting that the SfiI-DNA complexes are very dynamic. We recently applied single-molecule AFM dynamic force spectroscopy to analyze the stability of SfiI-DNA complexes (20). The characteristic dissociation time value for the synaptic complex was 26 ± 4 ms, that is, in the same time range as the value obtained in this study, 6.8 ± 0.3 ms, and the 3.5-times difference in the data can be attributed to the difference in design of the DNA duplexes used in the two works (47). Duplexes (40 bp) with the asymmetric position of the 13-bp recognition sequence having 4-bp and 23-bp flanking sequences were used in the force spectroscopy study. In this work, a design with a symmetric position of the recognition region within the 18-bp duplex was used. However, we propose an alternative explanation, which seems reasonable to consider. The force spectroscopy data for the complex lifetimes were obtained by extrapolation to the zero rupture force of the experimental AFM pull-off force values obtained at relatively high pulling rates. The data were approximated by one line, suggesting that the energy profile for the dissociation of SfiI-DNA complexes has only one barrier, although we admitted the existence of another barrier, which might not have been detected by AFM force spectroscopy. The difference between the AFM force spectroscopy results and single-molecule fluorescence data suggests that the SfiI-DNA dissociation energy profile may have two different transient states separated by a relatively low barrier.
An interesting finding in this work is that the synaptic complex has a shorter lifetime than the presynaptic complex. The difference is only twofold, but given that the presynaptic complex is considered a transient state for the synaptic complex, one could anticipate higher stability of a productive synaptic complex compared to the presynaptic one. Our data do not support this hypothesis. The structures of both complexes are apparently different, and our data show that this difference is in favor of higher stability of the synaptic complex. Clearly, an elevated stability of the transient state (presynaptic complex) increases the probability of association of this complex with another DNA duplex leading to the formation of the synaptic complex, but to determine which structural differences provide this effect will require additional study.
Our single-molecule dynamics data were obtained for the DNA duplexes with lengths corresponding to the maximum of the enzyme cleavage efficiency (48). It is instructive to compare our results on the lifetime of synaptic complexes with the SfiI cleavage rates. The kinetics data obtained by Williams et al. (49) showed that the rate constants of DNA cleavage vary between ∼1 min−1 and ∼1 s−1, depending on the sequence within the nonspecific region. The off-rate constant for the complex dissociation determined in our experiments is 100 s−1. If all synaptic complexes were equivalent in their structure, DNA should be cleaved during the complex lifetime, and the cleavage rate would be at least the same as the complex dissociation rate, i.e., in the 10-ms timescale. A plausible explanation for such low efficiency of the productive complex formation is that the SfiI-DNA complex is quite dynamic, allowing various conformational states to form. There are two major pieces of evidence supporting this view. First, each elementary enzymatic chemical process of the catalytic bond scission step occurs on the subnanosecond scale, so the millisecond lifetime would be sufficient for DNA cleavage to occur. Second, the cleavage reaction rate depends on the sequence of the central nonessential part of the DNA's cognate site, and we showed recently (50) that this effect correlates with the synaptic complex lifetime. The cleavage of the substrate is the last step in a long chain of events starting with the formation of the productive synaptic complex and ending with DNA cleavage. Note that the catalytic step, the cleavage reaction per se, is a complex process in which the residues of the enzyme involved in direct and indirect readout communicate with the catalytic center and trigger conformational changes that are required for the initiation of phosphodiester bond cleavage (51). Proper orientation of interacting groups within the catalytic center of the enzyme is the key step allowing fast cleavage of the substrate. Coupling between the recognition (the formation of the synaptic complex) and catalytic steps is the key feature of the enzymology of type II restriction enzymes, and is yet to be clarified. The characterization of the complex dynamics is the first step in understanding the enzyme cleavage mechanism and we believe that further experiments with this system will allow us to further characterize this dynamic process.
Available structural data on the SfiI-DNA complex (43) provides a couple of potential pathways for structural rearrangements within the complex that may be involved in its dynamics. First, x-ray crystallography revealed two critical conformational states of the protein tetramer. They involve a conformational change of one of the loops that bring the catalytic residues closer to the scissile phosphodiester bond located within the spacer sequence. Therefore, we hypothesize that the flip between these two states is one potential dynamic path contributing to the formation of the fully productive complex. Second, in the crystal, the DNA molecule is bent at an angle of 25° to provide specific DNA-protein bonds; this is a substantial bend angle over short DNA segments, creating the bend tension to the duplex. Thus, our second hypothesis is that DNA bending and straightening is another pathway contributing to synaptic complex dynamics. Two DNA duplexes undergo this conformational transition, so their independent movement is a factor decreasing the probability of the formation of productive complex. Multiplying this probability by the probability of the formation of the appropriate protein conformation further decreases the chances of formation of the productive synaptic complex.
The SfiI cleavage reaction requires cutting of four DNA strands, which, according to the article by Nobbs et al. (52), occurs sequentially with the rate constants for hydrolysis of each separate phosphodiester bond in the range 0.1–0.2 s−1. To reconcile these data with a much higher dissociation rate of the complex, we propose that the complex lifetime increases after hydrolysis of the very first phosphodiester bond. An alternative hypothesis implying the complex dissociation after each cleavage event seems unrealistic because it would lead to the accumulation of intermediates with nicked duplexes; this was not observed experimentally (52). The hypothesis of an increase of the lifetime of the partially digested complex is supported by the finding that SfiI remains bound to the DNA duplexes even after completion of the cleavage (52). Moreover, the lifetime for such complexes is in an hour range that is >100 times larger than the characteristic time for the cleavage reaction. Additional support for the hypothesis on the elevated lifetime of the complex with partially cleaved DNA comes from the fact that a single-stranded break increases DNA flexibility, facilitating bending of the DNA duplex with the complex. Direct experiments with the synaptic complex containing the nick at the cleavage position may provide the answer, and experiments to this effect are in progress.
Our experiments with nonspecific DNA duplexes showed that if there is a complex formation between SfiI and nonspecific DNA, the dissociation time for this complex is less than the characteristic diffusion time, i.e., <0.5 ms. Currently, three models for the search by a site-specific protein for its cognate site on the DNA template are proposed (53,54): one-dimensional diffusion (sliding), hopping (dissociation), and intersegmental transfer. The fast dissociation of the complex observed in this article (10 ms) is seemingly in favor of the hopping model. However, we cannot rule out that the dissociation of the complex occurs via sliding of the protein from the specific site toward the duplex ends. Our data provide some estimates to this pathway as well. The experiments with nonspecific duplex showed that such a complex dissociates in a time frame comparable to that for free diffusion. We assume that if both mechanisms are involved, their characteristic time is not larger than 1 ms, the characteristic time determined for the nonspecific SfiI-DNA complex. Experiments with different lengths of DNA duplexes may provide evidence for a sliding mechanism that will allow us to estimate its characteristic times. These studies are currently in progress.
We showed in this work that the “tethered” single-molecule fluorescence method was a very useful tool for analyzing the dynamics of interactions within the SfiI-DNA synaptic complex. This system has many features in common with other synaptic complexes, those involved in site-specific DNA recombination in the first place. Therefore, we believe that this successfully tested “tethered” approach is applicable to any of these systems. It has a number of useful features. First of all, it is a single-molecule technique, with the advantages provided by this type of method. Second, it can be coupled with FRET to provide additional structural information for synaptic complexes. For example, we were able to distinguish between the presynaptic and the synaptic complex. Our time range was limited to a free-diffusion timescale from the short-time side; however, faster dynamics can be analyzed using the scanning FCS approach (55). Although we tested the “tethered” approach only on synaptic complexes, we do not anticipate problems applying this approach to any type of specific protein-DNA complexes. The simplicity of surface-immobilization procedures for DNA dramatically facilitates the application of the “tethered” method to almost any type of DNA-protein system.
SUPPLEMENTARY MATERIAL
An online supplement to this article can be found by visiting BJ Online at http://www.biophysj.org.
Supplementary Material
Acknowledgments
We thank E. Evans for valuable comments on the use of the NSP approach for the data analysis, A. Bogdanov and L. Shlyakhtenko for stimulating discussions of the manuscript, and A. Portillo for editing the manuscript.
This work was supported by grants from the National Institutes of Health (GM 062235) and the National Science Foundation (No. 0615590) to Y.L.L.
References
- 1.Grindley, N. D., K. L. Whiteson, and P. A. Rice. 2006. Mechanisms of site-specific recombination. Annu. Rev. Biochem. 75:567–605. [DOI] [PubMed] [Google Scholar]
- 2.Bilcock, D. T., and S. E. Halford. 1999. DNA restriction dependent on two recognition sites: activities of the SfiI restriction-modification system in Escherichia coli. Mol. Microbiol. 31:1243–1254. [DOI] [PubMed] [Google Scholar]
- 3.Embleton, M. L., V. Siksnys, and S. E. Halford. 2001. DNA cleavage reactions by type II restriction enzymes that require two copies of their recognition sites. J. Mol. Biol. 311:503–514. [DOI] [PubMed] [Google Scholar]
- 4.Bath, A. J., S. E. Milsom, N. A. Gormley, and S. E. Halford. 2002. Many type IIs restriction endonucleases interact with two recognition sites before cleaving DNA. J. Biol. Chem. 277:4024–4033. [DOI] [PubMed] [Google Scholar]
- 5.Gormley, N. A., A. L. Hillberg, and S. E. Halford. 2002. The type IIs restriction endonuclease BspMI is a tetramer that acts concertedly at two copies of an asymmetric DNA sequence. J. Biol. Chem. 277:4034–4041. [DOI] [PubMed] [Google Scholar]
- 6.Swanson, P. C. 2004. The bounty of RAGs: recombination signal complexes and reaction outcomes. Immunol. Rev. 200:90–114. [DOI] [PubMed] [Google Scholar]
- 7.Wentzell, L. M., T. J. Nobbs, and S. E. Halford. 1995. The SfiI restriction endonuclease makes a four-strand DNA break at two copies of its recognition sequence. J. Mol. Biol. 248:581–595. [DOI] [PubMed] [Google Scholar]
- 8.Wentzell, L. M., and S. E. Halford. 1998. DNA looping by the Sfi I restriction endonuclease. J. Mol. Biol. 281:433–444. [DOI] [PubMed] [Google Scholar]
- 9.Milsom, S. E., S. E. Halford, M. L. Embleton, and M. D. Szczelkun. 2001. Analysis of DNA looping interactions by type II restriction enzymes that require two copies of their recognition sites. J. Mol. Biol. 311:515–527. [DOI] [PubMed] [Google Scholar]
- 10.Ha, T., X. Zhuang, H. D. Kim, J. W. Orr, J. R. Williamson, and S. Chu. 1999. Ligand-induced conformational changes observed in single RNA molecules. Proc. Natl. Acad. Sci. USA. 96:9077–9082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Myong, S., I. Rasnik, C. Joo, T. M. Lohman, and T. Ha. 2005. Repetitive shuttling of a motor protein on DNA. Nature. 437:1321–1325. [DOI] [PubMed] [Google Scholar]
- 12.Tomschik, M., H. Zheng, K. van Holde, J. Zlatanova, and S. H. Leuba. 2005. Fast, long-range, reversible conformational fluctuations in nucleosomes revealed by single-pair fluorescence resonance energy transfer. Proc. Natl. Acad. Sci. USA. 102:3278–3283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Yasuda, R., T. Masaike, K. Adachi, H. Noji, H. Itoh, and K. Kinosita, Jr. 2003. The ATP-waiting conformation of rotating F1-ATPase revealed by single-pair fluorescence resonance energy transfer. Proc. Natl. Acad. Sci. USA. 100:9314–9318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Zhuang, X., L. E. Bartley, H. P. Babcock, R. Russell, T. Ha, D. Herschlag, and S. Chu. 2000. A single-molecule study of RNA catalysis and folding. Science. 288:2048–2051. [DOI] [PubMed] [Google Scholar]
- 15.Blanchard, S. C., R. L. Gonzalez, H. D. Kim, S. Chu, and J. D. Puglisi. 2004. tRNA selection and kinetic proofreading in translation. Nat. Struct. Mol. Biol. 11:1008–1014. [DOI] [PubMed] [Google Scholar]
- 16.Ratto, T. V., K. C. Langry, R. E. Rudd, R. L. Balhorn, M. J. Allen, and M. W. McElfresh. 2004. Force spectroscopy of the double-tethered concanavalin-A mannose bond. Biophys. J. 86:2430–2437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Schwesinger, F., R. Ros, T. Strunz, D. Anselmetti, H. J. Guntherodt, A. Honegger, L. Jermutus, L. Tiefenauer, and A. Pluckthun. 2000. Unbinding forces of single antibody-antigen complexes correlate with their thermal dissociation rates. Proc. Natl. Acad. Sci. USA. 97:9972–9977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kuhner, F., L. T. Costa, P. M. Bisch, S. Thalhammer, W. M. Heckl, and H. E. Gaub. 2004. LexA-DNA bond strength by single molecule force spectroscopy. Biophys. J. 87:2683–2690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hinterdorfer, P., W. Baumgartner, H. J. Gruber, K. Schilcher, and H. Schindler. 1996. Detection and localization of individual antibody-antigen recognition events by atomic force microscopy. Proc. Natl. Acad. Sci. USA. 93:3477–3481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Krasnoslobodtsev, A. V., L. S. Shlyakhtenko, and Y. L. Lyubchenko. 2007. Probing interactions within the synaptic DNA-SfiI complex by AFM force spectroscopy. J. Mol. Biol. 365:1407–1416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Eigen, M., and R. Rigler. 1994. Sorting single molecules: application to diagnostics and evolutionary biotechnology. Proc. Natl. Acad. Sci. USA. 91:5740–5747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Rigler, R., U. Mets, J. Widengren, and P. Kask. 1993. Fluorescence correlation spectroscopy with high count rate and low-background: analysis of translational diffusion. Eur. Biophys. J. Biophys. Lett. 22:169–175. [Google Scholar]
- 23.Krichevsky, O., and G. Bonnet. 2002. Fluorescence correlation spectroscopy: the technique and its applications. Rep. Prog. Phys. 65:251–297. [Google Scholar]
- 24.Haustein, E., and P. Schwille. 2004. Single-molecule spectroscopic methods. Curr. Opin. Struct. Biol. 14:531–540. [DOI] [PubMed] [Google Scholar]
- 25.Ha, T., T. Enderle, D. F. Ogletree, D. S. Chemla, P. R. Selvin, and S. Weiss. 1996. Probing the interaction between two single molecules: fluorescence resonance energy transfer between a single donor and a single acceptor. Proc. Natl. Acad. Sci. USA. 93:6264–6268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ha, T., A. Y. Ting, J. Liang, W. B. Caldwell, A. A. Deniz, D. S. Chemla, P. G. Schultz, and S. Weiss. 1999. Single-molecule fluorescence spectroscopy of enzyme conformational dynamics and cleavage mechanism. Proc. Natl. Acad. Sci. USA. 96:893–898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ha, T. 2001. Single-molecule fluorescence resonance energy transfer. Methods. 25:78–86. [DOI] [PubMed] [Google Scholar]
- 28.Zhuang, X., T. Ha, H. D. Kim, T. Centner, S. Labeit, and S. Chu. 2000. Fluorescence quenching: a tool for single-molecule protein-folding study. Proc. Natl. Acad. Sci. USA. 97:14241–14244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Joo, C., S. A. McKinney, D. M. Lilley, and T. Ha. 2004. Exploring rare conformational species and ionic effects in DNA Holliday junctions using single-molecule spectroscopy. J. Mol. Biol. 341:739–751. [DOI] [PubMed] [Google Scholar]
- 30.McKinney, S. A., A. C. Declais, D. M. Lilley, and T. Ha. 2003. Structural dynamics of individual Holliday junctions. Nat. Struct. Biol. 10:93–97. [DOI] [PubMed] [Google Scholar]
- 31.Karymov, M., D. Daniel, O. F. Sankey, and Y. L. Lyubchenko. 2005. Holliday junction dynamics and branch migration: single-molecule analysis. Proc. Natl. Acad. Sci. USA. 102:8186–8191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Edman, L., Z. Foldes-Papp, S. Wennmalm, and R. Rigler. 1999. The fluctuating enzyme: a single molecule approach. Chem. Phys. 247:11–22. [Google Scholar]
- 33.Blanchard, S. C., H. D. Kim, R. L. Gonzalez, Jr., J. D. Puglisi, and S. Chu. 2004. tRNA dynamics on the ribosome during translation. Proc. Natl. Acad. Sci. USA. 101:12893–12898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Hertzog, D. E., X. Michalet, M. Jager, X. Kong, J. G. Santiago, S. Weiss, and O. Bakajin. 2004. Femtomole mixer for microsecond kinetic studies of protein folding. Anal. Chem. 76:7169–7178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Li, H., X. Ren, L. Ying, S. Balasubramanian, and D. Klenerman. 2004. Measuring single-molecule nucleic acid dynamics in solution by two-color filtered ratiometric fluorescence correlation spectroscopy. Proc. Natl. Acad. Sci. USA. 101:14425–14430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kim, H. D., G. U. Nienhaus, T. Ha, J. W. Orr, J. R. Williamson, and S. Chu. 2002. Mg2+-dependent conformational change of RNA studied by fluorescence correlation and FRET on immobilized single molecules. Proc. Natl. Acad. Sci. USA. 99:4284–4289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Lieto, A. M., R. C. Cush, and N. L. Thompson. 2003. Ligand-receptor kinetics measured by total internal reflection with fluorescence correlation spectroscopy. Biophys. J. 85:3294–3302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Guo, B., and W. H. Guilford. 2006. Mechanics of actomyosin bonds in different nucleotide states are tuned to muscle contraction. Proc. Natl. Acad. Sci. USA. 103:9844–9849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Bayas, M. V., A. Leung, E. Evans, and D. Leckband. 2006. Lifetime measurements reveal kinetic differences between homophilic cadherin bonds. Biophys. J. 90:1385–1395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Deniz, A. A., M. Dahan, J. R. Grunwell, T. Ha, A. E. Faulhaber, D. S. Chemla, S. Weiss, and P. G. Schultz. 1999. Single-pair fluorescence resonance energy transfer on freely diffusing molecules: observation of Forster distance dependence and subpopulations. Proc. Natl. Acad. Sci. USA. 96:3670–3675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Allen, N. W., and N. L. Thompson. 2006. Ligand binding by estrogen receptor β attached to nanospheres measured by fluorescence correlation spectroscopy. Cytometry A. 69:524–532. [DOI] [PubMed] [Google Scholar]
- 42.Kransnoslobodtsev, A. V., L. S. Shlyakhtenko, E. Ukraintsev, T. O. Zaikova, J. F. Keana, and Y. L. Lyubchenko. 2005. Nanomedicine and protein misfolding diseases. Nanomedicine. 1:300–305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Vanamee, E. S., H. Viadiu, R. Kucera, L. Dorner, S. Picone, I. Schildkraut, and A. K. Aggarwal. 2005. A view of consecutive binding events from structures of tetrameric endonuclease SfiI bound to DNA. EMBO J. 24:4198–4208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Sevenich, F. W., J. Langowski, V. Weiss, and K. Rippe. 1998. DNA binding and oligomerization of NtrC studied by fluorescence anisotropy and fluorescence correlation spectroscopy. Nucleic Acids Res. 26:1373–1381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Deniz, A. A., T. A. Laurence, G. S. Beligere, M. Dahan, A. B. Martin, D. S. Chemla, P. E. Dawson, P. G. Schultz, and S. Weiss. 2000. Single-molecule protein folding: diffusion fluorescence resonance energy transfer studies of the denaturation of chymotrypsin inhibitor 2. Proc. Natl. Acad. Sci. USA. 97:5179–5184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Lushnikov, A. Y., V. N. Potaman, E. A. Oussatcheva, R. R. Sinden, and Y. L. Lyubchenko. 2006. DNA strand arrangement within the SfiI-DNA complex: atomic force microscopy analysis. Biochemistry. 45:152–158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Embleton, M. L., S. A. Williams, M. A. Watson, and S. E. Halford. 1999. Specificity from the synapsis of DNA elements by the Sfi I endonuclease. J. Mol. Biol. 289:785–797. [DOI] [PubMed] [Google Scholar]
- 48.Moreira, R. F., and C. J. Noren. 1995. Minimum duplex requirements for restriction enzyme cleavage near the termini of linear DNA fragments. Biotechniques. 19:56–59. [PubMed] [Google Scholar]
- 49.Williams, S. A., and S. E. Halford. 2001. SfiI endonuclease activity is strongly influenced by the non-specific sequence in the middle of its recognition site. Nucleic Acids Res. 29:1476–1483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Krasnoslobodtsev, A. V., L. S. Shlyakhtenko, and Y. L. Lyubchenko. Probing interactions within the synaptic DNA-SfiI complex by AFM force spectroscopy. J. Mol. Biol. In press. [DOI] [PMC free article] [PubMed]
- 51.Pingoud, A., M. Fuxreiter, V. Pingoud, and W. Wende. 2005. Type II restriction endonucleases: structure and mechanism. Cell. Mol. Life Sci. 62:685–707. [DOI] [PubMed] [Google Scholar]
- 52.Nobbs, T. J., M. D. Szczelkun, L. M. Wentzell, and S. E. Halford. 1998. DNA excision by the Sfi I restriction endonuclease. J. Mol. Biol. 281:419–432. [DOI] [PubMed] [Google Scholar]
- 53.Halford, S. E. 2001. Hopping, jumping and looping by restriction enzymes. Biochem. Soc. Trans. 29:363–374. [DOI] [PubMed] [Google Scholar]
- 54.Halford, S. E., and J. F. Marko. 2004. How do site-specific DNA-binding proteins find their targets? Nucleic Acids Res. 32:3040–3052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Xiao, Y., V. Buschmann, and K. D. Weston. 2005. Scanning fluorescence correlation spectroscopy: a tool for probing microsecond dynamics of surface-bound fluorescent species. Anal. Chem. 77:36–46. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.