


Abstract
Here, we describe novel puromycin derivatives conjugated with iminobiotin and a fluorescent dye that can be linked covalently to the C-terminus of full-length proteins during cell-free translation. The iminobiotin-labeled proteins can be highly purified by affinity purification with streptavidin beads. We confirmed that the purified fluorescence-labeled proteins are useful for quantitative protein–protein interaction analysis based on fluorescence cross-correlation spectroscopy (FCCS). The apparent dissociation constants of model protein pairs such as proto-oncogenes c-Fos/c-Jun and archetypes of the family of Ca2+-modulated calmodulin/related binding proteins were in accordance with the reported values. Further, detailed analysis of the interactions of the components of polycomb group complex, Bmi1, M33, Ring1A and RYBP, was successfully conducted by means of interaction assay for all combinatorial pairs. The results indicate that FCCS analysis with puromycin-based labeling and purification of proteins is effective and convenient for in vitro protein–protein interaction assay, and the method should contribute to a better understanding of protein functions by using the resource of available nucleotide sequences.
INTRODUCTION
An understanding of the rate and specificity of assembly of biomolecular complexes is essential for a full appreciation of the mechanisms of biological events. Further, currently available information on genome sequences of various organisms can be exploited as a resource for characterizing novel functions of proteins or hypothetical proteins. For this purpose, a high-throughput method is required for functional protein analysis. Fluorescence correlation spectroscopy (FCS) and fluorescence cross-correlation spectroscopy (FCCS) have recently been applied to such important biological problems (1–11). FCS allows monitoring of the individual movements of fluorescence-labeled molecules through a very tiny area (1,2). The time-dependent fluorescence autocorrelation function allows us to analyze the relative proportions of species involved in the diffusion. Changes of the proportions can be used to calculate the binding kinetics (3,4,8). FCCS utilizes separate channels to detect two distinct fluorophores, as well as the cross-correlated signals, in real time (5). With FCCS, bound molecules can be detected even if the differences of diffusion are not great. So far, FCCS has been applied to the studies of DNA hybridization (5), PCR (9), enzymatic cleavage of a DNA substrate by EcoRI endonuclease (6,10) and protein–DNA interactions (11).
Fluorescence labeling of proteins is a key step for the FCS and FCCS analysis of protein interactions. So far, chemical modifications (12,13) and recombinant fusion tagging with fluorescent proteins (14–17) have been used for fluorescence labeling of proteins. These methods are often useful, but the modifications of internal amino acid residues or the addition of relatively large fluorescent proteins may affect the functions of labeled proteins. As an alternative approach, we have previously developed a puromycin-based method for fluorescence labeling of proteins (18,19). By using this method, various fluorophores can be incorporated into full-length proteins in the presence of a low concentration of fluorophore-conjugated puromycin in a cell-free translation system (11). Small fluorescent probes are expected to be less likely to interfere with the structure or biological function of proteins and cell-free protein synthesis is suitable for a high-throughput format owing to its simplicity. We have previously reported the FCCS analysis of protein–DNA interactions between RhG (rhodamine green)-labeled proteins and Cy5-labeled DNA (11). Although high-throughput analysis of protein–protein interactions in solution using FCCS is of great interest, detection of cross-correlations between differently labeled proteins has been difficult, because the labeling efficiency of our method ranges from only 10 to 30% (11), and the remaining unlabeled proteins in solution inhibit the formation of the protein–protein complex carrying both RhG and Cy5. In this study, we have improved the purification process of fluorescence-labeled proteins by using novel iminobiotin-conjugated fluorescent puromycin derivatives to aid the removal of unlabeled proteins, thereby making protein–protein interaction assay using FCCS practically feasible. We used three model systems, proto-oncogenes c-Fos and c-Jun, archetypes of the family of Ca2+-modulated calmodulin (CaM) and CaM-related binding proteins, and the polycomb group (PcG) complex proteins to confirm the usefulness of our method.
MATERIALS AND METHODS
Synthesis of fluorescent puromycin derivatives
NHS-iminobiotin trifluoroacetamide was purchased from Pierce. Iminobiotin-T(Cy5)-dC-puromycin (Figure 1A) and iminobiotin-T(RhG)-dC-puromycin (data not shown) were synthesized and purified as described previously (11), with some modifications (see Supplementary Data). The structural identity of the synthesized fluorescent puromycin analogs was confirmed by MALDI-TOF mass spectrometry (Voyager; Perceptive Biosystems).
Figure 1.

Materials for fluorescence labeling. (A) The structure of a fluorescent puromycin derivative. A fluorophore (Cy5 or RhG) and iminobiotin were chemically conjugated to puromycin through a linker. (B) DNA construction for fluorescence labeling of proteins. Template DNA consists of SP6 promoter, Omega sequence and an open reading frame (ORF) with a T7·tag at the N-terminus and a polyhistidine tag at the C-terminus, followed by a XhoI restriction enzyme site.
Preparation of templates for translation
In a template DNA, two tags were added to the 5′- and 3′-termini of the open reading frame (11,19) (Figure 1B) by PCR and the fragment was subcloned into a pCR2.1Topo vector (Invitrogen). The DNA template was amplified from the clone by PCR and cleaved with XhoI. The purified DNA was transcribed in an SP6 large-scale RNA production system (Promega).
Fluorescence labeling and purification
Fluorescence labeling was carried out using the wheat germ extract translation system ‘Proteios’ (Toyobo, Japan) as described in the manufacturer's protocol, except that a fluorophore-conjugated puromycin was added. The translation was terminated by the addition of RNase A (1 μg/0.3 ml; Ambion). The purification of fluorescently labeled proteins was performed at 4°C. The mixture was dialyzed against nickel binding buffer (50 mM phosphate, 150 mM NaCl, 1 mM DTT and 0.05% NP-40, pH 8.0), followed by centrifugation at 16 000 g for 20 min. The supernatant was mixed with 10 μl of Ni-NTA agarose (20) (SuperFlow; Qiagen) for 1 h. The supernatant was removed, and the beads were washed three times, with agitation, in nickel binding buffer (1.0 ml) containing 2.5 mM imidazole and 300 mM NaCl. Proteins were eluted with 50 μl of buffer containing 0.5 M imidazole, pH 8.0. The fraction was mixed with 9 vol of 50 mM phosphate buffer (pH 8.0) containing 300 mM NaCl, 5 mM DTT and 0.05% NP-40, then 10 μl of streptavidin–Sepharose (21) (Amersham Pharmacia) was added and the mixture was rocked for 1 h. The beads were washed with the buffer three times. Protein was eluted with 50 μl of buffer (240 mM Tris–HCl, 150 mM NaCl, 0.1 M biotin, 5 mM DTT and 0.1% NP-40, pH 8.0). The protein fraction was mixed with 10 mM DTT and kept at 4°C before use.
Immunodetection and fluorescence determination
The proteins were detected by enhanced immunoblotting (22) with mouse anti-T7·tag antibody (Novagen) and horseradish peroxidase (HRP)-labeled goat anti-mouse IgG (Transduction Laboratories). The blot was determined semiquantitatively with the T7·tag positive control recombinant protein (Novagen), an ECL detection kit (Amersham Pharmacia) and a CCD camera (ChemiDoc; Bio-Rad). Proteins separated by SDS–PAGE were stained with SyproOrange (Molecular Probes) and detected using a fluorescence image analyzer (excitation at 488 nm and emission at 515–545 nm, Molecular Imager FX; Bio-Rad). The fluorescence yield was spectrophotometrically determined using the fluorescence image analyzer (Cy5 was detected with excitation at 635 nm and emission at 670–720 nm; RhG with excitation at 488 nm and emission at 515–545 nm) and a standard dye with molecular extinction coefficients of ɛ505 (RhG) = 68 000 cm−1 M−1 (measured at pH 8.0) and ɛ647 (Cy5) = 250 000 cm−1 M−1.
FCCS measurement
FCCS measurement was performed on a ConfoCor2 system (Carl Zeiss) as described previously (11). The two pinholes and the cross-correlated volume element were adjusted by measurement (5). All solutions were prepared in water (fluorescence analysis grade; Dojindo, Japan) and filtered through an Ultrafree-MC filter unit (Millipore). Fluorescently labeled proteins were dialyzed against 50 mM phosphate, 150 mM NaCl, 0.1% NP-40 and 1 mM DTT, pH 7.4. After centrifugation at 16 000 g for 20 min, differently labeled proteins were mixed in a Lab-Tek 8-well chamber (Nalge Nunc) and kept for 10 min. Interaction of c-Fos with c-Jun was also analyzed in the presence of DNA, poly(dI–dC)·poly(dI–dC) (2 μg/ml; Amersham Pharmacia) and the AP-1 synthetic oligonucleotides of 30 bp (Dateconcept, Sapporo, Japan) (23). CaM interactions were analyzed in the presence of 0.5 mM CaCl2. Two autocorrelation curves and the cross-correlation curve of FCCS data were analyzed by using fitting algorithm described below in the software package for ConfoCor2 (Carl Zeiss).
Theory and data calibration
The theoretical background of FCCS analysis has been des-cribed by Eigen et al. and Rigler et al. (5,6,9). The fluorescence autocorrelation function and the cross-correlation function were acquired from an online system-controlling computer software package. The normalized cross-correlation function G(τ) is given by
| 1 |
where the indices refer to one or two measured fluorescence signals, Ig and/or Ir. In the case of one fluorescent species, Equation 1 (r = g) defines normalized autocorrelation function in a single detection channel. ω1 is the radius and ω2 is half of the long axis of the confocal volume element. The structural parameter S is the ratio of ω2/ω1. Two-component model of the autocorrelation function for translational diffusion in a 3D Gaussian volume element is described as follows:
| 2 |
where and are the diffusion times of the faster component and slower component in the assay. Y represents the fraction of fluorescent protein with the diffusion time in the total number of fluorescent particles N. The values of ω1,i (i = g or r) were determined from the diffusion times of rhodamine 6G (Sigma Aldrich; diffusion coefficient D = 2.8 × 10−10 m2 s−1) and Cy5 (mono-reactive dye, Amersham Pharmacia; D = 3.16 × 10−10 m2 s−1).
| 3 |
The volume elements V are calculated according to
| 4 |
| 5 |
The measured total number of autocorrelated particles NAC,i and complex cross-correlated particles Ncc is given by
| 6 |
where in the case of Ncc, Gi(0) indicates Ggr(0). The red emission excited by the green laser Qg (cross-talk fraction) was calculated from the mean count rates of the red channel when excited by both lasers (Cgr) and only the red laser (Cr), using a modification of the method in the application manual of ConfoCor2.
| 7 |
Calculated free molecules Ni and calculated complex molecules Ngr are as follows:
| 8 |
| 9 |
| 10 |
The concentrations of each fluorescent protein were calculated with the use of A (Avogadro's number) as follows:
| 11 |
The dissociation constants (Kds) are given by
| 12 |
RESULTS
Tandem affinity purification of fluorescently labeled proteins
c-Fos(118–211) and c-Jun(216–318) were translated in the presence of iminobiotin–fluorophore-conjugated puromycin, whose structure is presented in Figure 1A. The optimal concentrations of the puromycin derivatives were 12.5 μM as RhG and 25 μM as Cy5, respectively (data not shown). The reaction mixtures were purified by two steps of affinity purification with nickel-chelate beads (Figure 2A) and streptavidin-conjugated beads (Figure 2B). Excess unincorporated dyes and lower molecular weight proteins were not retained on nickel-chelate beads (Figure 2A, lane 2). The fractions eluted with 0.5 M imidazole (Figure 2A, lane 3) were further purified using streptavidin beads. The flow-through fraction contained <5% of the total fluorescence intensity, but ∼30% of the immunoblotting signal (Figure 2B, lane 2). The biotin-eluted fraction (Figure 2B, lane 3) showed a weaker signal than that of the applied fraction (Figure 2B, lane 1) in immunodetection. These results indicate that unlabeled proteins were successfully removed by the second step of affinity purification. The purified Cy5–protein fraction was identical to one band detected in SDS–PAGE by protein staining with SyproOrange (Figure 2C, lane 2). Similarly, CaM, CaM-binding proteins and PcG proteins were labeled and highly purified (data not shown).
Figure 2.

Purification of fluorescence-labeled proteins. Subscript Cy5 or RhG indicates a fluorophore linked to puromycin derivative. Proteins were separated on 15–25% continuous gradient SDS–PAGE and detected using a fluorescence imager (Cy5 or RhG) or αT7·tag antibody. (A) Affinity purification with nickel-chelate resin. Lane 1, in vitro translation products; lane 2, flow-through fractions; and lane 3, eluates with 0.5 M imidazole. (B) Affinity purification with streptavidin resin. Lane 1, nickel-chelate affinity-purified fractions; lane 2, flow-through; and lane 3, eluates with 0.1 M biotin. (C) Protein staining with SyproOrange. Lane 1, in vitro translation products; and lane 2, purified fractions.
Conditions of FCCS analysis with ConfoCor2
The pinhole diameters were adjusted to 70 μm in the green channel and 48 μm in the red channel to provide a sufficient observation volume for our system. The overlap of the excitation volumes between red and green laser lines was achieved by exciting the Cy5 dye with both wavelengths (5). The autocorrelation curve of the red channel with only the 633 nm laser (red line in Supplementary Figure 1) was coincident with the curve of the channel with only the 488 nm laser (blue line in Supplementary Figure 1). The particle numbers detected in the red channel and in the green channel were 24.1 and 23.7, respectively. The structural parameter S was calculated as 5 for the green channel and as 7 for the red channel. The diffusion time of ∼10 nM rhodamine 6G in the green channel was 30 μs and that of ∼10 nM Cy5 dye in the red channel was 44 μs in the laser power range used. The effective volume elements were Vg = ∼0.19 fl in the green channel, Vr = ∼0.41 fl in the red channel and Vgr = ∼0.26 fl in the cross-correlated channel (see Equations 4 and 5 in Materials and Methods for definitions). The differences between these detection volumes were accounted for in the data analysis according to Equation 11. The calculated diffusion coefficients of iminobiotin-T(RhG)-dC-puromycin and iminobiotin-T(Cy5)-dC-puromycin were 2.1 and 2.3 × 10−10 m2 s−1, respectively, using FCS analysis. The cross-talk from red to green was zero whereas the cross-talk from green to red was ∼10%: this was accounted for in the calculation of complex concentration according to Equation 10.
FCCS analysis of c-Fos and c-Jun
The fractions of fluorescently labeled proteins to fluorescent particles were c-FosCy5 71%, c-JunCy5 68%, c-FosRhG 69% and c-JunRhG 67% when the functions were fitted to two-component models with diffusion times corresponding to those of the fluorescent derivatives using FCS analysis (Figure 3). Diffusion coefficients of c-FosRhG, c-FosCy5, c-JunRhG and c-JunCy5 were calculated to be 7.6–8.1 × 10−11 m2 s−1. As shown in Supplementary Figure 2, the diffusion coefficients of fluorescent-puromycin-labeled proteins were consistent with the predictions of the Stokes–Einstein theory (24). Concentrations of fluorescently labeled proteins were calculated from the autocorrelation functions in the FCCS analysis (Figure 3A–C, upper panels). The apparent Kd values calculated with the equilibrium data are summarized in Table 1. The translational diffusion time of c-Fos homodimer was determined after the addition of AP-1 DNA by using the cross-correlation function (Figure 3A, lower panel). In the presence of the AP-1 DNA sequence, the Kd of the heterodimer was ∼80-fold lower than that of c-Fos homodimer (Table 1). In the case of the heterodimer and c-Jun homodimer, the Kd decreased to ∼70% after the addition of the AP-1 sequence. The cross-correlation function of c-FosRhG/c-JunCy5 gave the diffusion coefficient of the cross-correlated complex as 7.0 × 10−11 m2 s−1 in the absence of the AP-1 sequence and 4.4 × 10−11 m2 s−1 in its presence (Figure 3C, lower panel). The Kd of c-FosCy5/c-JunRhG was determined to be 7 × 10−8 M in the absence of the AP-1 sequence and 5 × 10−8 M in its presence (data not shown).
Figure 3.

FCCS analysis of AP-1-binding proteins. The autocorrelation function (upper panels) and cross-correlation function (lower panels) of c-FosCy5 and c-FosRhG (A), c-JunCy5 and c-JunRhG (B), and c-JunCy5 and c-FosRhG (C). In the autocorrelation plot, red and green lines represent Cy5 and RhG. The dashed curves (blue in cross-correlation function) represent data obtained after the addition of 50 nM AP-1 oligonucleotides.
Table 1.
Concentrations of fluorescent proteins and apparent Kd values determined using FCCS in this study
| Cy5-labeled protein (nM) | RhG-labeled protein (nM) | Addition | Kd (nM) |
|---|---|---|---|
| Fos (18.4) | Fos (18.0) | — | ND |
| Fos (19.2) | Fos (17.9) | AP-1 DNA | 3720 |
| Jun (8.1) | Jun (15.2) | — | 270 |
| Jun (7.8) | Jun (14.6) | AP-1 DNA | 190 |
| Jun (4.8) | Fos (12.3) | — | 69 |
| Jun (4.5) | Fos (11.5) | AP-1 DNA | 45 |
| Rab3A (4.8) | CaM (8.1) | — | ND |
| Rab3A (4.5) | CaM (7.8) | EGTA | ND |
| Caldesmon (6.1) | CaM (8.3) | — | 500 |
| Caldesmon (5.7) | CaM (7.5) | EGTA | 2300 |
| Calcineurin (11.2) | CaM (7.8) | — | 160 |
| Calcineurin (11.5) | CaM (7.9) | EGTA | 2200 |
| M33 (5.6) | Bmi1 (7.8) | — | 92 |
| M33 (4.6) | RYBP (5.3) | — | 70 |
| M33 (5.2) | Ring1A (7.7) | — | 51 |
| Ring1A (4.6) | RYBP (16.3) | — | 74 |
| Bmi1 (4.4) | RYBP (14.7) | — | 2300 |
| Bmi1 (4.1) | Ring1A (5.5) | — | 2000 |
ND; not determined.
FCCS analysis of CaM and CaM-binding proteins
The diffusion coefficients of CaM(1–149)RhG, calcineurin A(328–521)Cy5, Rab3A(1–219)Cy5 and caldesmon(302–564)Cy5 were calculated to be 7.6, 7.3, 6.8 and 7.0 × 10−11 m2 s−1, respectively. Variations of the cross-correlation function between CaM and CaM-binding proteins were observed in the presence of Ca2+ (see Figure 4A–C, solid curves). The diffusion times of the cross-correlated functions were determined, except for that of Rab3A, by using analyzing software. The amplitudes of the cross-correlation functions were reduced by the addition of EGTA (Figure 4B and C, dashed blue curves), indicating the involvement of Ca2+-mediated interactions. The calculated Kds after the addition of EGTA indicated a non-specific-binding interaction or the background of the detection procedure. The significant Kd values were determined to be 2–5 × 10−7 M in the assay (Table 1).
Figure 4.

Cross-correlation function between CaMRhG and CaM-binding proteins: Rab3ACy5 (A), caldesmonCy5 (B) and calcineurin AαCy5 (C). The dashed blue curves represent data obtained after the addition of 5 mM EGTA.
FCCS analysis of PcG complex proteins
The diffusion coefficients of fluorescently labeled proteins M33(1–519)Cy5, Bmi1(1–326)Cy5, Bmi1(1–326)RhG, Ring1A(201–377)Cy5, Ring1A(201–377)RhG, RYBP(92–228)Cy5 and RYBP(92–228)RhG were 4.1, 6.1, 6.6, 7.2, 7.4, 7.3 and 7.5 × 10−11 m2 s−1, respectively. Variations of cross-correlation function were observed for Bmi1RhG/M33Cy5, M33Cy5/Ring1ARhG, M33Cy5/RYBPRhG and RYBPRhG/Ring1ACy5 (solid curves shown in Figure 5A–D and Table 1). The significant interactions are shown schematically in Figure 6. M33 appeared to mediate the association. To confirm the role of M33, we examined the association with the mediator using FCCS. Interestingly, the amplitude of the cross-correlation function of Bmi1Cy5/Ring1ARhG was increased by the addition of non-labeled M33 (dashed blue curves shown in Figure 5F). The diffusion coefficient of the cross-correlated complex was 2.7 × 10−11 m2 s−1, corresponding to ∼120 kDa. The molecular brightness (C/M) was not altered by the addition of a non-labeled protein (19.5–20.0 kHz in the red channel and 12.7–11.3 kHz in the green channel). In contrast, the effect of the addition of M33 on the interactions of Ring1A/RYBP and Bmi1/RYBP was not significant (dashed blue curves in Figure 5D and E).
Figure 5.

Cross-correlation function of M33Cy5 and Bmi1RhG (A), M33Cy5 and Ring1ARhG (B), M33Cy5 and RYBPRhG (C), Ring1ACy5 and RYBPRhG (D), Bmi1Cy5 and RYBPRhG (E), and Bmi1RhG and Ring1ACy5 (F). Dashed blue curves represent data obtained after the addition of non-labeled M33 (2 nM).
Figure 6.
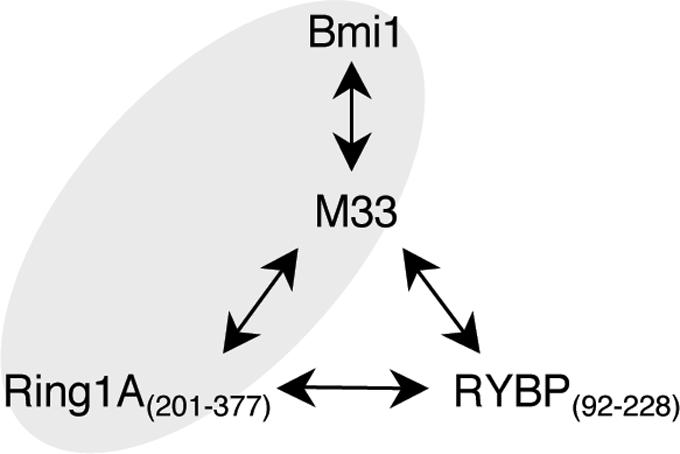
Schematic diagram of association of polycomb gene complex proteins. Arrows indicate interactions between the proteins as judged from the apparent Kd values in this study. Gray areas indicate triplet interaction detected using FCCS.
DISCUSSION
Purification of fluorescently labeled proteins by using a secondary affinity tag, iminobiotin, introduced on to fluorescent puromycin as described here, improved the sensitivity for FCCS analysis of interactions between two distinct fluorescence-labeled proteins. Indeed, the c-JunRhG/c-JunCy5 interactions both with and without non-labeled AP-1 oligonucleotide could be detected in this study, whereas the interaction among c-JunRhG/Cy5-labeled AP-1/non-labeled Jun was not detected in the previous study (11). The apparent Kd of c-Fos/c-Jun/AP-1 found in this study was in good agreement with reported values (25,26). The Kd of c-Jun homodimer and AP-1 sequence also coincided with the value of 140 nM determined previously (25). Further, the Kd of CaM and caldesmon was in agreement with the reported value of 550 nM (27). The apparent Kd was independent of the concentrations of fluorescence-labeled proteins (data not shown). These results indicate that FCCS analysis with puromycin-based labeling of proteins is effective and convenient for protein–protein interaction assay, and that the puromycin derivatives and affinity tags did not interfere substantially with the protein interactions. It should be noted that the Kd values obtained from FCCS are minimum estimates because small amounts of unlabeled proteins may remain.
The interaction of c-Fos homodimer and CaM/Rab3A mediated by Ca2+ could not be identified in this study. The Kd of c-Fos homodimer and AP-1 sequence was previously reported to be ∼6 μM (28). The Kd of CaM/Rab3A was also reported to be 20−50 μM (29,30). The interaction of c-Fos homodimer (and c-Jun homodimer) in this study might include interactions between single-colored proteins, but the molecular brightness was not greater than that of other probed proteins (data not shown). Such weak interactions might be detected if the concentrations of fluorescently labeled proteins were increased.
A surface plasmon resonance (SPR) biosensor allows real-time analysis of specific interactions on a solid phase, whereas FCS and FCCS detect interactions in solution. Schubert et al. (31) compared the entropic contribution to the free energy between SPR and FCS and concluded that the reaction entropy determined from an SPR experiment was lower than that from an FCS experiment. Indeed, the Kd between CaM and calcineurin was determined as 1.7 × 10−8 M by means of an SPR biosensor (32), and this is 10 times lower than our value using FCCS. Similarly, interaction assay of c-Fos/c-Jun heterodimer immobilized on a polystyrene tray gave a Kd of 1 nM (33), whereas our FCCS analysis gave 70 nM. Although the immobilizing method may be advantageous for the detection of protein interactions with low affinity, we believe that Kd values in living cells are likely to be more similar to those determined using FCCS in solution than to those determined on a solid phase.
The PcG proteins form multimeric complexes that bind to specific genomic sites of polycomb repressive elements (34). We applied FCCS to analyze in detail the individual associations of some PcG proteins by interaction assay of the pairs under homogeneous conditions. As shown in Table 1, significant interactions were found among M33/Bmi1, M33/Ring1A, M33/RYBP and Ring1A/RYBP, respectively, as previously confirmed by the yeast two-hybrid method and protein pulldown assay (35,36). It appears that M33 is a mediator in the association of these proteins (Figure 6), but only the association of Bmi1/M33/Ring1A was confirmed (Figure 5F). The association of Bmi1/M33/Ring1A was also supported by applying a three-component model to fit the autocorrelation function of Bmi1 after the addition of non-labeled M33 (data not shown). Bmi1, M33 and Ring1A are components of a stable core PcG repressive complex, according to a biochemical study (37). Interestingly, our results suggest that RYBP may interact with M33 or Ring1A in the free form without the formation of a core complex. This is consistent with the idea that RYBP plays a role in recruiting PcG components (38). The FCCS analysis of the components of PcG complex proteins presented here should be a good model for detailed analysis of other protein complexes. For example, use of puromycin-based fluorescently labeled proteins would allow FCCS analysis, as well as FCS analysis, of the dynamics of complex formation of retinoblastoma tumor suppressor complex (39). The range of detectable interactions should be improved by using FCCS.
The tandem affinity purification method using a polyhistidine tag and an iminobiotin tag was further applied to over 30 proteins and all but three were sufficiently purified for FCCS analysis. We also observed the interactions between IgG and its binding domain ZZ region (40), and between Smac (second mitochondria-derived activator of caspase or DIABLO) and XIAP (X-linked inhibitor of apoptosis protein, data not shown) (41). Combinations of two affinity tags are expected to help high-throughput purification of the fluorescently labeled proteins, because nickel-chelate beads and streptavidin beads for high-throughput robotic systems are already available from several vendors. Thus, the method presented in this paper should be applicable to a large-scale analysis of protein–protein interactions and should also contribute to the elucidation of protein functions in the post-genomic era.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
Acknowledgments
We thank Megumi Nakamura for the preparation of puromycin derivatives and Yuko Oishi for the preparation of CaM-binding protein plasmid DNAs. This work was supported by Special Coordination Funds of the Science and Technology Agency (Ministry of Education, Culture, Sports, Science and Technology) of the Japanese Government. Funding to pay the Open Access publication charges for this article was provided by Keio University.
Conflict of interest statement. None declared.
REFERENCES
- 1.Magde D., Elson E.L., Webb W.W. Fluorescence correlation spectroscopy. II. An experimental realization. Biopolymers. 1974;13:29–61. doi: 10.1002/bip.1974.360130103. [DOI] [PubMed] [Google Scholar]
- 2.Eigen M., Rigler R. Sorting single molecules: application to diagnostics and evolutionary biotechnology. Proc. Natl Acad. Sci. USA. 1994;91:5740–5747. doi: 10.1073/pnas.91.13.5740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Pack C.G., Nishimura G., Tamura M., Aoki K., Taguchi H., Yoshida M., Kinjo M. Analysis of interaction between chaperonin GroEL and its substrate using fluorescence correlation spectroscopy. Cytometry. 1999;36:247–253. doi: 10.1002/(sici)1097-0320(19990701)36:3<247::aid-cyto15>3.3.co;2-r. [DOI] [PubMed] [Google Scholar]
- 4.Wolcke J., Reimann M., Klumpp M., Gohler T., Kim E., Deppert W. Analysis of p53 ‘latency’ and ‘activation’ by fluorescence correlation spectroscopy. Evidence for different modes of high affinity DNA binding. J. Biol. Chem. 2003;278:32587–32595. doi: 10.1074/jbc.M303615200. [DOI] [PubMed] [Google Scholar]
- 5.Schwille P., Meyer-Almes F.J., Rigler R. Dual-color fluorescence cross-correlation spectroscopy for multicomponent diffusional analysis in solution. Biophys. J. 1997;72:1878–1886. doi: 10.1016/S0006-3495(97)78833-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kettling U., Koltermann A., Schwille P., Eigen M. Real-time enzyme kinetics monitored by dual-color fluorescence cross-correlation spectroscopy. Proc. Natl Acad. Sci. USA. 1998;95:1416–1420. doi: 10.1073/pnas.95.4.1416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Koltermann A., Kettling U., Bieschke J., Winkler T., Eigen M. Rapid assay processing by integration of dual-color fluorescence cross-correlation spectroscopy: high throughput screening for enzyme activity. Proc. Natl Acad. Sci. USA. 1998;95:1421–1426. doi: 10.1073/pnas.95.4.1421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kinjo M., Nishimura G., Koyama T., Mets Ü, Rigler R. Single-molecule analysis of restriction DNA fragments using fluorescence correlation spectroscopy. Anal. Biochem. 1998;260:166–172. doi: 10.1006/abio.1998.2652. [DOI] [PubMed] [Google Scholar]
- 9.Rigler R., Foldes-Papp Z., Meyer-Almes F.J., Sammet C., Volcker M., Schnetz A. Fluorescence cross-correlation: a new concept for polymerase chain reaction. J. Biotechnol. 1998;63:97–109. doi: 10.1016/s0168-1656(98)00079-0. [DOI] [PubMed] [Google Scholar]
- 10.Winkler T., Kettling U., Koltermann A., Eigen M. Confocal fluorescence coincidence analysis: an approach to ultra high-throughput screening. Proc. Natl Acad. Sci. USA. 1999;96:1375–1378. doi: 10.1073/pnas.96.4.1375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Doi N., Takashima H., Kinjo M., Sakata K., Kawahashi Y., Oishi Y., Oyama R., Miyamoto-Sato E., Sawasaki T., Endo Y., et al. Novel fluorescence labeling and high-throughput assay technologies for in vitro analysis of protein interactions. Genome Res. 2002;12:487–492. doi: 10.1101/gr.218802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Patel L.R., Curran T., Kerppola T.K. Energy transfer analysis of Fos-Jun dimerization and DNA binding. Proc. Natl Acad. Sci. USA. 1994;91:7360–7364. doi: 10.1073/pnas.91.15.7360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Diebold R.J., Rajaram N., Leonard D.A., Kerppola T.K. Molecular basis of cooperative DNA bending and oriented heterodimer binding in the NFAT1–Fos–Jun–ARRE2 complex. Proc. Natl Acad. Sci. USA. 1998;95:7915–7920. doi: 10.1073/pnas.95.14.7915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Haupts U., Maiti S., Schwille P., Webb W.W. Dynamics of fluorescence fluctuations in green fluorescent protein observed by fluorescence correlation spectroscopy. Proc. Natl Acad. Sci. USA. 1998;95:13573–13578. doi: 10.1073/pnas.95.23.13573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kohl T., Heinze K.G., Kuhlemann R., Koltermann A., Schwille P. A protease assay for two-photon crosscorrelation and FRET analysis based solely on fluorescent proteins. Proc. Natl Acad. Sci. USA. 2002;99:12161–12166. doi: 10.1073/pnas.192433499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kim S.A., Heinze K.G., Waxham M.N., Schwille P. Intracellular calmodulin availability accessed with two-photon cross-correlation. Proc. Natl Acad. Sci. USA. 2004;101:105–110. doi: 10.1073/pnas.2436461100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kogure T., Karasawa S., Araki T., Saito K., Kinjo M., Miyawaki A. A fluorescent variant of a protein from the stony coral Montipora facilitates dual-color single-laser fluorescence cross-correlation spectroscopy. Nat. Biotechnol. 2006;24:577–581. doi: 10.1038/nbt1207. [DOI] [PubMed] [Google Scholar]
- 18.Nemoto N., Miyamoto-Sato E., Yanagawa H. Fluorescence labeling of the C-terminus of proteins with a puromycin analogue in cell-free translation systems. FEBS Lett. 1999;462:43–46. doi: 10.1016/s0014-5793(99)01474-x. [DOI] [PubMed] [Google Scholar]
- 19.Miyamoto-Sato E., Takashima H., Fuse S., Sue K., Ishizaka M., Tateyama S., Horisawa K., Sawasaki T., Endo Y., Yanagawa H. Highly stable and efficient mRNA templates for mRNA–protein fusions and C-terminally labeled proteins. Nucleic Acids Res. 2003;31:e78. doi: 10.1093/nar/gng078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hochuli E., Dobeli H., Schacher A. New metal chelate adsorbent selective for proteins and peptides containing neighbouring histidine residues. J. Chromatogr. 1987;411:177–184. doi: 10.1016/s0021-9673(00)93969-4. [DOI] [PubMed] [Google Scholar]
- 21.Hofmann K., Wood S.W., Brinton C.C., Montibeller J.A., Finn F.M. Iminobiotin affinity columns and their application to retrieval of streptavidin. Proc. Natl Acad. Sci. USA. 1980;77:4666–4668. doi: 10.1073/pnas.77.8.4666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Oyama R., Yamamoto H., Titani K. Glutamine synthetase, hemoglobin alpha-chain, and macrophage migration inhibitory factor binding to amyloid beta-protein: their identification in rat brain by a novel affinity chromatography and in Alzheimer's disease brain by immunoprecipitation. Biochim. Biophys. Acta. 2000;1479:91–102. doi: 10.1016/s0167-4838(00)00057-1. [DOI] [PubMed] [Google Scholar]
- 23.Yen J., Wisdom R.M., Tratner I., Verma I.M. An alternative spliced form of FosB is a negative regulator of transcriptional activation and transformation by Fos proteins. Proc. Natl Acad. Sci. USA. 1991;88:5077–5081. doi: 10.1073/pnas.88.12.5077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Krouglova T., Vercammen J., Engelborghs Y. Correct diffusion coefficients of proteins in fluorescence correlation spectroscopy. Application to tubulin oligomers induced by Mg2+ and Paclitaxel. Biophys. J. 2004;87:2635–2646. doi: 10.1529/biophysj.104.040717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.John M., Leppik R., Busch S.J., Granger-Schnarr M., Schnarr M. DNA binding of Jun and Fos bZip domains: homodimers and heterodimers induce a DNA conformational change in solution. Nucleic Acids Res. 1996;24:4487–4494. doi: 10.1093/nar/24.22.4487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kwon H., Park S., Lee S., Lee D.K., Yang C.H. Determination of binding constant of transcription factor AP-1 and DNA. Application of inhibitors. Eur. J. Biochem. 2001;268:565–572. doi: 10.1046/j.1432-1327.2001.01897.x. [DOI] [PubMed] [Google Scholar]
- 27.Shirinsky V.P., Bushueva T.L., Frolova S.I. Caldesmon–calmodulin interaction. Study by the method of protein intrinsic tryptophan fluorescence. Biochem. J. 1988;255:203–208. [PMC free article] [PubMed] [Google Scholar]
- 28.O'Shea E.K., Rutkowski R., Stafford W.F., III, Kim P.S. Preferential heterodimer formation by isolated leucine zippers from fos and jun. Science. 1989;245:646–648. doi: 10.1126/science.2503872. [DOI] [PubMed] [Google Scholar]
- 29.Park J.B., Farnsworth C.C., Glomset J.A. Ca2+/calmodulin causes Rab3A to dissociate from synaptic membranes. J. Biol. Chem. 1997;272:20857–20865. doi: 10.1074/jbc.272.33.20857. [DOI] [PubMed] [Google Scholar]
- 30.Coppola T., Perret-Menoud V., Luthi S., Farnsworth C.C., Glomset J.A., Regazzi R. Disruption of Rab3–calmodulin interaction, but not other effector interactions, prevents Rab3 inhibition of exocytosis. EMBO J. 1999;18:5885–5891. doi: 10.1093/emboj/18.21.5885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Schubert F., Zettl H., Hafner W., Krauss G., Krausch G. Comparative thermodynamic analysis of DNA–protein interactions using surface plasmon resonance and fluorescence correlation spectroscopy. Biochemistry. 2003;42:10288–10294. doi: 10.1021/bi034033d. [DOI] [PubMed] [Google Scholar]
- 32.Takano E., Hatanaka M., Maki M. Real-time-analysis of the calcium-dependent interaction between calmodulin and a synthetic oligopeptide of calcineurin by a surface plasmon resonance biosensor. FEBS Lett. 1994;352:247–250. doi: 10.1016/0014-5793(94)00965-1. [DOI] [PubMed] [Google Scholar]
- 33.Heuer K.H., Mackay J.P., Podzebenko P., Bains N.P., Weiss A.S., King G.F., Easterbrook-Smith S.B. Development of a sensitive peptide-based immunoassay: application to detection of the Jun and Fos oncoproteins. Biochemistry. 1996;35:9069–9075. doi: 10.1021/bi952817o. [DOI] [PubMed] [Google Scholar]
- 34.Levine S.S., King I.F., Kingston R.E. Division of labor in polycomb group repression. Trends Biochem. Sci. 2004;29:478–485. doi: 10.1016/j.tibs.2004.07.007. [DOI] [PubMed] [Google Scholar]
- 35.Hashimoto N., Brock H.W., Nomura M., Kyba M., Hodgson J., Fujita Y., Takihara Y., Shimada K., Higashinakagawa T. RAE28, BMI1, and M33 are members of heterogeneous multimeric mammalian polycomb group complexes. Biochem. Biophys. Res. Commun. 1998;245:356–365. doi: 10.1006/bbrc.1998.8438. [DOI] [PubMed] [Google Scholar]
- 36.Alkema M.J., Bronk M., Verhoeven E., Otte A., van't Veer L.J., Berns A., van Lohuizen M. Identification of Bmi1-interacting proteins as constituents of a multimeric mammalian polycomb complex. Genes Dev. 1997;11:226–240. doi: 10.1101/gad.11.2.226. [DOI] [PubMed] [Google Scholar]
- 37.Levine S.S., Weiss A., Erdjument-Bromage H., Shao Z., Tempst P., Kingston R.E. The core of the polycomb repressive complex is compositionally and functionally conserved in flies and humans. Mol. Cell. Biol. 2002;22:6070–6078. doi: 10.1128/MCB.22.17.6070-6078.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.García E., Marcos-Gutiérrez C., del Mar Lorente M., Moreno J.C., Vidal M. RYBP, a new repressor protein that interacts with components of the mammalian polycomb complex, and with the transcription factor YY1. EMBO J. 1999;18:3404–3418. doi: 10.1093/emboj/18.12.3404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Angus S.P., Solomon D.A., Kuschel L., Hennigan R.F., Knudsen E.S. Retinoblastoma tumor suppressor: analyses of dynamic behavior in living cells reveal multiple modes of regulation. Mol. Cell. Biol. 2003;23:8172–8188. doi: 10.1128/MCB.23.22.8172-8188.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Nilsson B., Moks T., Jansson B., Abrahmsen L., Elmblad A., Holmgren E., Henrichson C., Jones T.A., Uhlen M. A synthetic IgG-binding domain based on staphylococcal protein A. Protein Eng. 1987;1:107–113. doi: 10.1093/protein/1.2.107. [DOI] [PubMed] [Google Scholar]
- 41.Du C., Fang M., Li Y., Li L., Wang X. Smac, a mitochondrial protein that promotes cytochrome c-dependent caspase activation by eliminating IAP inhibition. Cell. 2000;102:33–42. doi: 10.1016/s0092-8674(00)00008-8. [DOI] [PubMed] [Google Scholar]