

Abstract
Mitotic cell death (MCD) is a prominent but poorly defined form of death that stems from aberrant mitosis. One of the early steps in MCD is premature mitosis and uneven chromatin condensation (UCC). The mechanism underlying this phenomenon is currently unknown. In this study, we show that DNA damage in cells with a compromised p53-mediated G2/M checkpoint triggers the unscheduled activation of cyclin-dependent kinase 1 (Cdk1), activation and chromatin loading of the condensin I complex, and UCC followed by the appearance of multimicronucleated cells, which is evidence of MCD. We demonstrate that these processes engage some of the players of normal mitotic chromatin packaging but not those that drive the apoptotic chromatin condensation. Our findings establish a link between the induction of DNA damage and mitotic abnormalities (UCC) through the unscheduled activation of Cdk1 and recruitment of condensin I. These results demonstrate a clear distinction between the mechanisms that drive MCD-associated and apoptosis-related chromatin condensation and provide mechanistic insights and new readouts for a major cell death process in treated tumors.
Introduction
Cell cycle progression after DNA damage is rapidly halted by checkpoint controls, which are relaxed after the damage has been assessed and processed. Cells with misrepaired or unrepaired DNA lesions are eliminated by different cell death mechanisms (Zhou and Elledge, 2000; Roninson et al., 2001; Bree et al., 2004). One such mechanism is mitotic cell death (MCD), which is also known as “mitotic catastrophe,” a prominent but poorly defined form of cell death that is described mainly in morphological terms. MCD is an outcome of aberrant mitosis that results in the formation of giant multimicronucleated cells (Erenpreisa and Cragg, 2001; Roninson et al., 2001). It is a major form of tumor cell death after treatments with ionizing radiation (IR) or certain chemotherapeutic agents (Torres and Horwitz, 1998; Roninson et al., 2001; Blank et al., 2003).
MCD has been shown to prevail in cells with impaired G1, G2, prophase, and mitotic spindle checkpoint functions (Chan et al., 2000; Roninson et al., 2001; Nitta et al., 2004). A prominent cell cycle checkpoint is activated by DNA double-strand breaks (DSBs) at the G2/M boundary. Activation of this checkpoint is driven by the nuclear protein kinase ataxia telangiectasia mutated (ATM), its downstream substrates p53 and the Chk1 and Chk2 kinases, Polo-like kinase 1 (Plk-1), and the p53-inducible proteins p21waf1 and 14-3-3-σ. The p53-mediated arm of the G2/M checkpoint was shown to be pivotal in preventing MCD in DNA-damaged cells (Chan et al., 2000; Fei and El-Deiry, 2003), although some studies challenge this observation (Andreassen et al., 2001; Castedo et al., 2004).
MCD has been assumed to result from the entry into mitosis of cells with unrepaired DNA damage, although a mechanism linking DNA lesions with mitotic abnormalities has yet to be uncovered. One of the early steps in the chain of events that culminates in MCD is cell entry into premature mitosis (Chan et al., 2000; Roninson et al., 2001; Nitta et al., 2004). To date, evidence of premature mitosis in damaged cells relies primarily on the appearance of uneven chromatin condensation (UCC), which is the formation of hypercondensed chromatin aggregates at nucleolar sites (Swanson et al., 1995; Ianzini and Mackey, 1997; Roninson et al., 2001). The mechanisms underlying this phenomenon are unknown.
During normal progression through mitosis, the structural reorganization of chromatin into condensed chromosomes entails the multiprotein complexes condensin I and II (Schmiesing et al., 1998; Swedlow and Hirano, 2003; Ono et al., 2004). In vitro studies showed that condensin I possesses a DNA-stimulated ATPase activity and is capable of introducing constrained, positive supercoils into DNA (Hirano, 2002). This activity is believed to be essential for initiating the assembly of mitotic chromosomes and for proper assembly and orientation of centromeres (Hagstrom et al., 2002; Ono et al., 2004). The two condensin complexes are each composed of five subunits conserved from yeast to mammals (Hirano et al., 1997; Schmiesing et al., 2000; Kimura et al., 2001). The core complex common to both condensins consists of the structural maintenance of chromosomes (SMC) proteins CAP-E/SMC2 and CAP-C/SMC4. Two other members of this family, SMC1 and SMC3, form the core of the cohesin complex that plays a central role in sister chromatid cohesion (Hirano, 2002). Each condensin complex then contains a regulatory subcomplex consisting of three non-SMC proteins. In condensin I, these are CAP-D2, -G, and -H. CAP-D2 and -G possess a highly degenerate repeating motif known as the HEAT repeat (Neuwald and Hirano, 2000), whereas CAP-H belongs to a recently identified superfamily of proteins termed kleisins (Schleiffer et al., 2003). In condensin II, the regulatory subcomplex contains the proteins CAP-D3, -G2, and -H2 (Fig. 1 A; Ono et al., 2003; Yeong et al., 2003).
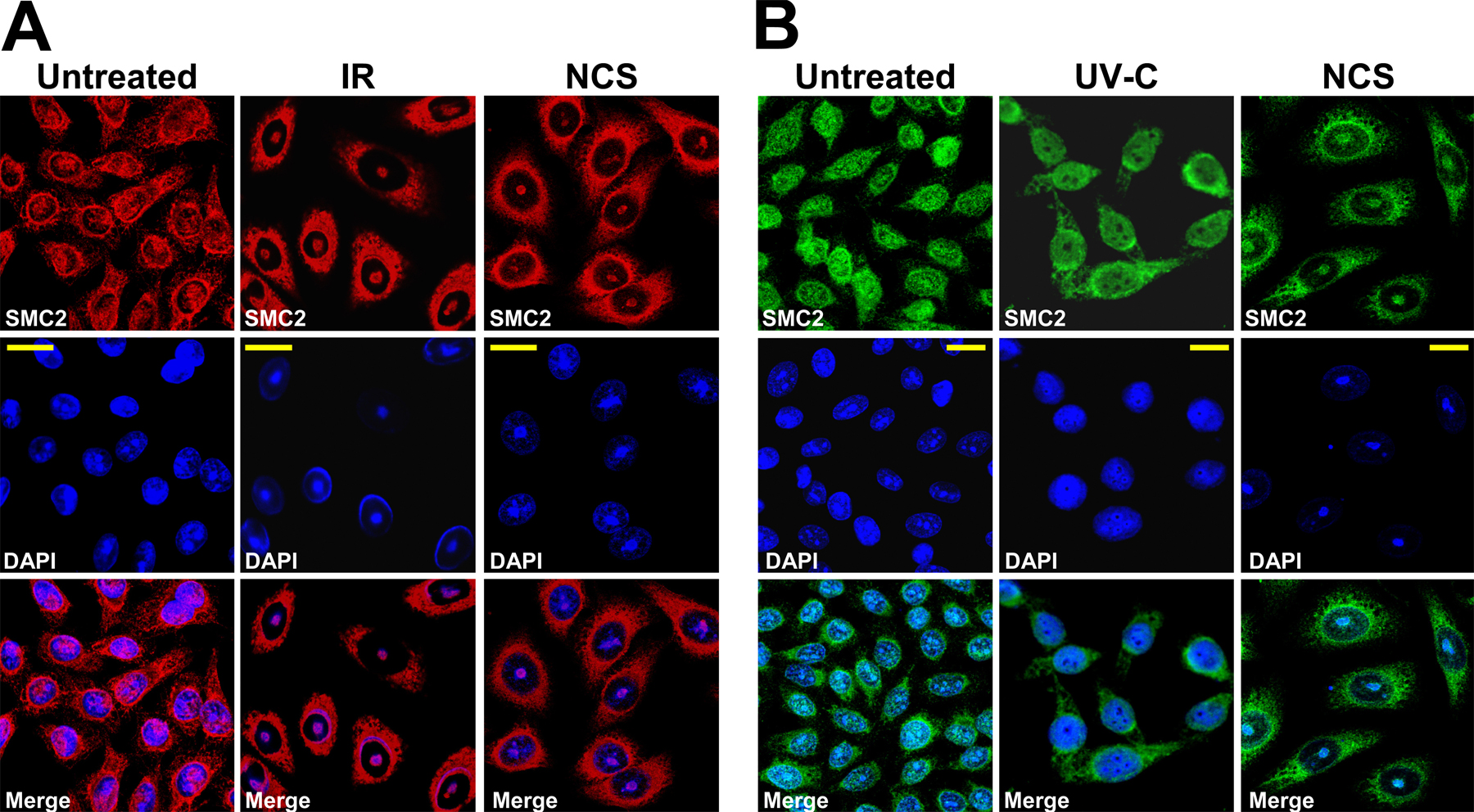
Figure 1.

DNA damage triggers the recruitment of condensin core subunits to damaged chromatin and the formation of UCC bodies. (A) Schematic of the condensin I and II complexes. H, G, and D2 denote condensin I non-SMC subunits hCAP-H, hCAP-G, and hCAP-D2, respectively. H2, G2, and D3 denote condensin II non-SMC subunits hCAP-H2, hCAP-G2, and hCAP-D3, respectively. (B) Recruitment of condensin proteins and UCC in HeLa cells after treatments with NCS. The cells were treated with 200 ng/ml NCS and immunostained 24 h later with anti-SMC2 antibody (red). DNA was counterstained with Yo-Pro-1 (green) or DAPI (blue). Bars, 8 μm. (C) Western blotting analysis of chromatin fractions obtained from untreated HeLa cells and 24 h after treatment with 200 ng/ml NCS, demonstrating recruitment of SMC2 and SMC4 to chromatin in response to the treatment. (D) Treatment of DNA-damaged cells with DNase-I leads to the dissociation of condensin proteins from DNA. Arrows indicate the nucleolar sites where UCC is formed. Arrows with asterisks in DNase-I–treated cells indicate nucleolar locations. Note the disappearance of condensin recruitment after DNase-I treatment. Bars, 20 μm.
During interphase, both types of condensins appear to be localized in the cytosol and the nucleus (Cabello et al., 2001; Watrin and Legagneux, 2005), with condensin I being predominantly cytosolic and condensin II being primarily nuclear (Ono et al., 2003; Hirota et al., 2004). The two condensin complexes localize in different places along mitotic chromosomes assembled in vitro and in vivo, suggesting distinct functions in chromosome architecture (Hirota et al., 2004; Ono et al., 2003, 2004). Importantly, in vitro studies indicated that cyclin-dependent kinase 1 (Cdk1)–mediated phosphorylation of the non-SMC subunit set is required for chromosomal localization of condensin I and stimulation of its supercoiling activity (Kimura et al., 1998, 2001).
Another prominent type of chromatin condensation noted in mammalian cells is apoptosis-related condensation. It is believed to be mediated primarily by the nuclear protein acinus (apoptotic chromatin condensation inducer in the nucleus) after its cleavage by activated caspase 3 (Sahara et al., 1999). Acinus lacks the DNase activities exhibited by other cellular factors that can induce apoptotic chromatin condensation via DNA fragmentation. Therefore, acinus functions as a “pure” regulator of apoptosis-related chromatin condensation (Zamzami and Kroemer, 1999).
In this study, we show that induction of the highly cytotoxic DSBs in DNA of cells with compromised p53-mediated G2/M checkpoint functions triggers UCC and premature mitosis. We demonstrate that the unscheduled activation of Cdk1 and the recruitment of activated condensin I to damaged chromatin are specifically involved in UCC formation. Condensin II and cohesin proteins are not involved in this process. Importantly, the acinus-mediated pathway, which is responsible for apoptotic chromatin condensation, was also not found to operate in UCC. Using a panel of tumor cell lines, we show that the defective damage-induced, p53-mediated G2/M checkpoint is an important but not a sole requirement for the activation of this pathway.
Results
Condensin but not cohesin is recruited to chromatin during UCC induced by DSBs
DNA damage–induced MCD, with UCC as an early step, has been shown to occur at high rates in tumor cells in which cell cycle checkpoint functions are impaired (Roninson et al., 2001). We studied this phenomenon in human cervix carcinoma HeLa cells in which the G1 and G2 DNA damage checkpoint functions are compromised as a result of p53 inactivation by human papilloma virus E6 (Scheffner et al., 1990). UCC was induced in exponentially growing HeLa cells by treatment with 10 Gy IR or 200 ng/ml neocarzinostatin (NCS), a radiomimetic agent that intercalates into cellular DNA and induces DSBs (Goldberg, 1987). 24 h later, confocal microscopy revealed that cellular DNA was condensed into globular clumped structures typical of UCC (Fig. 1 B and Fig. S1 A, available at http://www.jcb.org/cgi/content/full/jcb.200604022/DC1). This phenomenon was observed with two different DNA dyes: DAPI and Yo-Pro-1, a monomeric green fluorescent cyanine with a high affinity for double-stranded DNA (Suzuki et al., 1997). Importantly, these globular structures contained the condensin core subunits SMC2 (Fig. 1 B) and SMC4 (Fig. S2), whose nuclear localization changed after treatment from a diffuse, pannuclear pattern to a compact one that overlapped the UCC bodies. Western blotting analysis of chromatin fractions confirmed the recruitment of SMC2 and SMC4 to chromatin in response to DNA damage (Fig. 1 C). Accordingly, DNase-I treatment in situ led to dissociation of the UCC bodies and release of the associated SMC2 (Fig. 1 D).
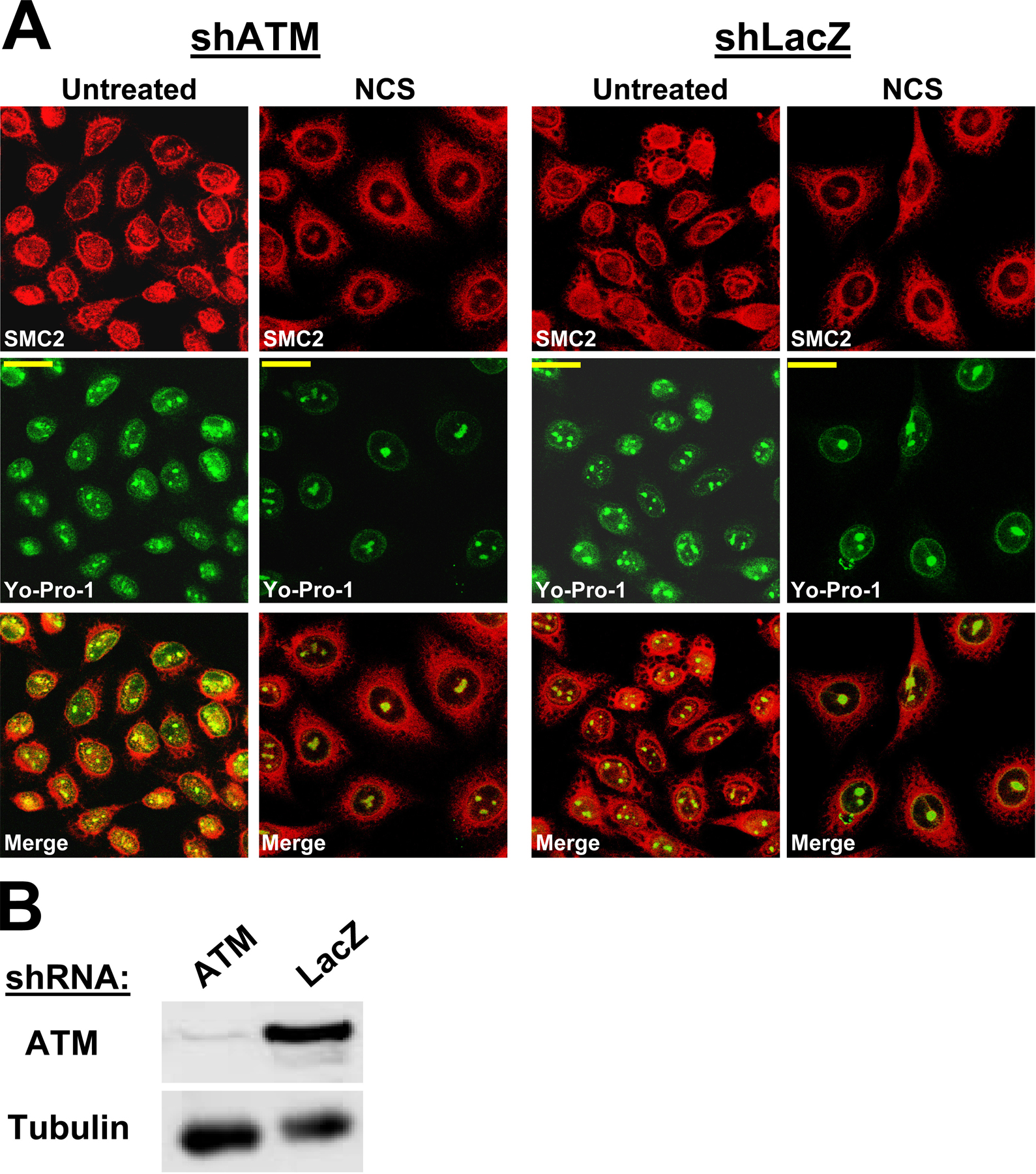
DSB-induced UCC was dose dependent and maximized at 24 h after treatment (Fig. S2). The percentage of cells exhibiting UCC increased from 5 ± 2% in untreated cells to 65.8 ± 25.1% 24 h after 200 ng/ml NCS (P < 0.01; n = 6). Importantly, the spatial distribution of the cohesin subunit SMC1 was not altered (unpublished data), suggesting that the cohesin complex was not involved in this phenomenon. UV irradiation did not induce this process (Fig. S1 B), suggesting that it was associated specifically with DSBs. Importantly, a similar rate of UCC induction was observed in both ATM-proficient and ATM-deficient HeLa cells in which ATM had been knocked down using stable expression of the appropriate short hairpin RNA (shRNA; Fig. S3, available at http://www.jcb.org/cgi/content/full/jcb.200604022/DC1).
Collectively, the results suggest that UCC is a delayed cellular response to DSBs that involves condensin but not cohesin complexes and apparently does not rely on the presence of ATM protein in the cells. It is noteworthy that UCC induction and recruitment of condensin proteins to chromatin after DNA damage were not strictly confined to HeLa but were also observed in other cell lines in which cell cycle checkpoints had been compromised, although at significantly lower rates compared with HeLa cells (see the last paragraph in Results).
Colocalization of UCC bodies with coalescent nucleoli
According to our Nomarski images (Fig. 1, B and D), UCC structures colocalized with nucleoli, a phenomenon that has been previously reported (Swanson et al., 1995; Ianzini and Mackey, 1997; Roninson et al., 2001). However, although the mean number of nucleoli in untreated cells was about three per nucleus, most of the nuclei with damage-induced UCC contained a single nucleolar body (Fig. 1, B and D). These nucleolar bodies were significantly larger than normal nucleoli: their major axis was measured as 5.82 ± 0.93 μm in damaged cells versus 3.06 ± 1.35 μm in untreated cells (P < 0.001). To examine this phenomenon further, nucleoli were visualized by immunostaining with antibodies against the nucleolar proteins GNL3 (guanine nucleotide-binding proteinlike 3; nucleostemin; Tsai and McKay, 2002; Tsai and McKay, 2005) and nucleolin (Tuteja and Tuteja, 1998). Although UV irradiation led to nucleolar disruption and release of nucleolar proteins into the nucleoplasm as previously described (Al-Baker et al., 2004; Tsai and McKay, 2005), in NCS-treated cells, the nucleolar proteins remained within a single nucleolar body that colocalized with the UCC bodies (Fig. 2).
Figure 2.

Colocalization of UCC and coalesced nucleoli. (A) Simultaneous detection of the NCS-triggered recruitment of SMC2 (green) to UCC bodies and their colocalization with nucleoli marked by nucleostemin (NS) staining (red). DNA (DAPI) appears in blue. Note the nucleolar disruption after UV treatment versus nucleolar coalescence and colocalization of the resultant large nucleolar bodies with UCC bodies after NCS treatment. Bars, 20 μm. (B) Visualization of NCS-triggered UCC colocalized with coalesced nucleoli using immunostaining with an antinucleolin antibody. Nucleolin, green; SMC2, red; DNA (DAPI), blue. Bars, 8 μm.
Condensin proteins are not involved in apoptotic chromatin condensation
HeLa cells are capable of activating the apoptotic cell death pathway (Byun et al., 2001), making them suitable for determining whether the recruitment of condensin to chromatin is specific to UCC or also accompanies apoptotic chromatin condensation. HeLa cells were treated with NCS or staurosporine, a documented apoptosis inducer (Couldwell et al., 1994), and the induction of apoptosis was monitored by following poly (ADP-ribose)polymerase-1 (PARP-1) cleavage (Fig. 3 A; Soldani and Scovassi, 2002). The apoptotic chromatin condensation was morphologically distinct from damage-induced UCC and, unlike UCC and mitotic chromatin condensation, did not involve condensin recruitment (Fig. 3 C). In fact, in apoptotic cells, the chromatin-bound SMC2 fraction was reduced compared with untreated cells (Fig. 3 B), conceivably because of apoptosis-associated DNA fragmentation, which subsequently released SMC2 from chromatin.
Figure 3.

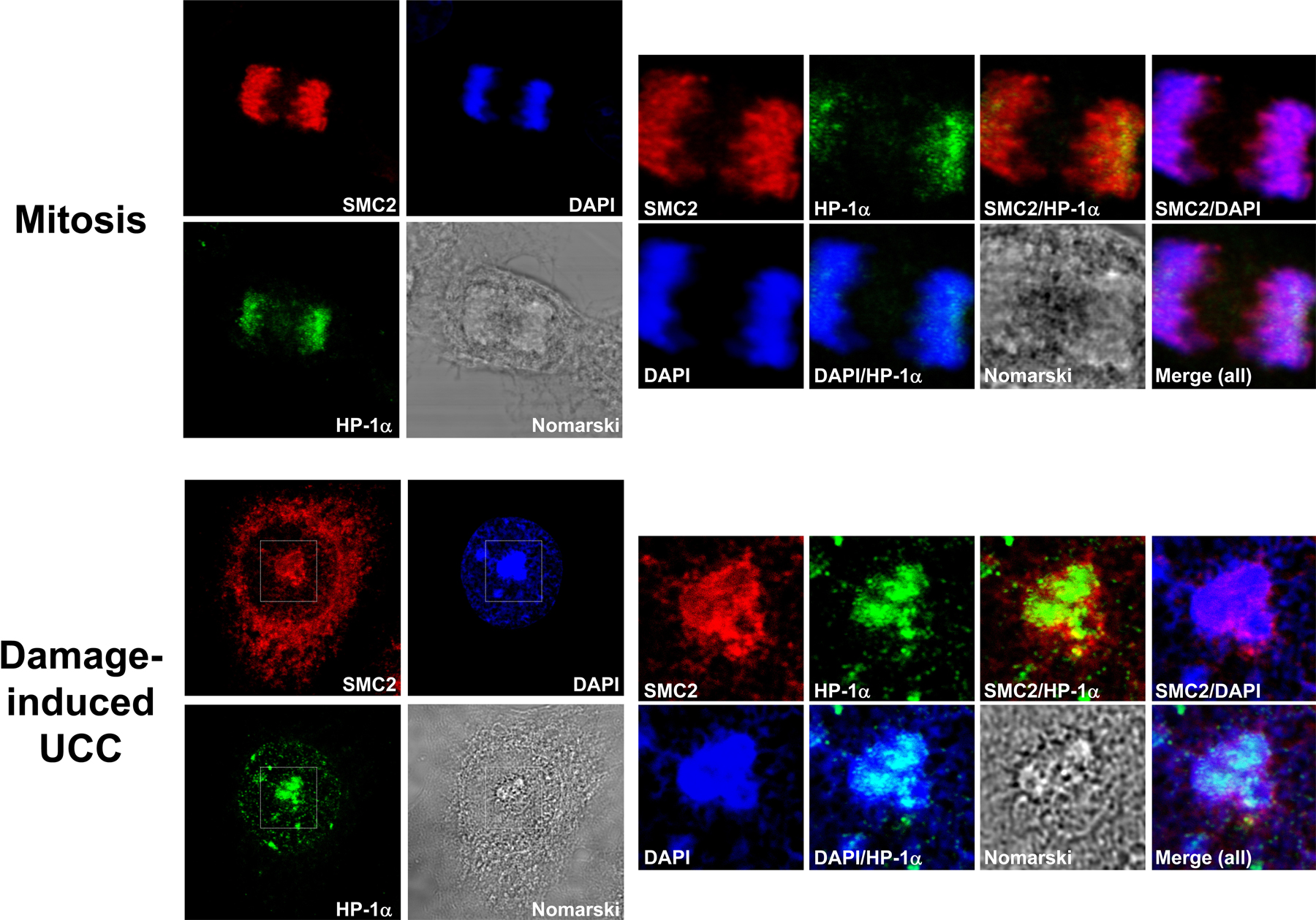
Condensin proteins participate in mitotic chromatin condensation and in UCC but not in apoptotic chromatin condensation. (A) Induction of apoptosis in HeLa cells by staurosporine treatment. PARP-1 cleavage is detected in staurosporine-treated cells (1 μM for 3.5 h). Note the absence of this response in NCS-treated cells (200 ng/ml). U, untreated; St, staurosporine treated. (B) Western blotting analysis of chromatin fractions from NCS- and staurosporine-treated HeLa cells demonstrates condensin recruitment to chromatin in damage-induced UCC but not in apoptotic chromatin condensation. (C) Three types of chromatin condensation in HeLa cells: mitotic, apoptotic, and damage-induced UCC. Immunofluorescence images taken from logarithmically growing HeLa cells treated with 1 μM staurosporine (for 3.5 h) or 200 ng/ml NCS (for 24 h). SMC2, red; DNA (Yo-Pro-1), green. Bars, 20 μm. (D) Visualization of three types of chromatin condensation in HeLa cells stained with both anti-SMC2 (red) and anti–HP-1α (green) antibodies. DNA (DAPI), blue. Bars, 8 μm.
To further demonstrate the differential recruitment of SMC2 to chromatin upon different types of chromatin condensation, we compared its distribution with that of HP-1α (heterochromatin protein 1α). HP-1α is known to constitutively associate with condensed chromatin in interphase and mitotic cells (Minc et al., 1999). We found that HP-1α was associated with condensed DNA in all three types of chromatin condensation that we studied (mitotic, apoptotic, and UCC), whereas SMC2 was loaded onto DNA in mitosis and upon UCC but not upon apoptotic chromatin condensation (Fig. 3 D). Interestingly, HP-1α and SMC2 were colocalized in normal mitotic chromosomes and in UCC bodies, but HP-1α was observed mainly in subcompartments of these structures, particularly in the inner core of the UCC bodies (Fig. S4, available at http://www.jcb.org/cgi/content/full/jcb.200604022/DC1).
Condensin I but not condensin II is involved in UCC
SMC2 and SMC4 constitute the common cores of the condensin I and II complexes (Ono et al., 2003; Yeong et al., 2003). The condensin I–related protein hCAP-H (human chromatin-associated protein H; kleisin-γ) and the condensin II–related protein hCAP-H2 (kleisin-β) are thought to be the first of the non-SMC subcomplex proteins to bind to SMC2/SMC4 heterodimers, subsequently recruiting other non-SMC subunits to form the complete type I or II condensin complexes, respectively (Nasmyth and Haering, 2005). We found that the condensin I–associated protein hCAP-H but not condensin II–related hCAP-H2 was associated with damage-induced UCC bodies (Fig. 4 A). Accordingly, biochemical analysis of cytosolic, nucleoplasmic, and chromatin fractions confirmed that hCAP-H, similar to condensin core subunits, was recruited to the damaged chromatin, but hCAP-H2 was not (Fig. 4 B). These results suggest that of the two condensin complexes, condensin I but not condensin II is involved in damage-induced UCC.
Figure 4.

Condensin I but not condensin II is recruited to chromatin after DNA damage. (A) NCS- (200 ng/ml for 24 h) and IR (10 Gy for 24 h)-induced DNA damage triggers the recruitment to DNA of condensin I subunit hCAP-H but not condensin II subunit hCAP-H2. Note that DNA staining (DAPI) reveals the formation of UCC bodies in both hCAP-H– and hCAP-H2–stained fields. Bars, 20 μm. (B) Western blotting analysis of chromatin fractions demonstrates recruitment of hCAP-H (condensin I) but not hCAP-H2 (condensin II) to damaged chromatin 24 h after NCS treatment. Protein loadings and degree of fractionations are demonstrated by using antibodies against the cytosolic α-tubulin, the nucleoplasmic ATM, and the chromatin component histone H2B. (C) λ-phosphatase (λ-PPase) treatment abolishes the shift of chromatin-bound hCAP-H. Western blotting analyses performed on chromatin fractions obtained from NCS-treated (200 ng/ml for 24 h) and untreated cells.
In the aforementioned experiments, we noticed slower gel migration of chromatin-bound hCAP-H compared with soluble hCAP-H (Fig. 4 B), suggesting that chromatin-bound hCAP-H underwent posttranslational modifications. During mitotic chromatin condensation, the non-SMC subunits of condensin I and presumably condensin II undergo Cdk1/cyclin B–mediated phosphorylation, which is required for condensin targeting to chromatin and its subsequent functionality (Kimura et al., 1998, 2001). We asked whether hCAP-H that had been recruited to chromatin upon UCC was phosphorylated. We treated the chromatin fractions with λ-phosphatase before Western blotting analysis and found that this treatment indeed abolished the shift in hCAP-H migration (Fig. 4 C). This result suggests that similar to its recruitment to mitotic chromatin, condensin I is recruited to chromatin during UCC as an activated complex.
Unscheduled Cdk1 activation, recruitment of condensin I to chromatin, and UCC are hallmarks of premature mitosis
UCC is a morphological hallmark of premature mitosis that is typical of cells with a defective G2/M checkpoint (Roninson et al., 2001). Our data suggest that recruitment of condensin I to chromatin is a biochemical marker of this process. To substantiate the link between a defective G2/M checkpoint and premature mitosis, we examined this process in HeLa cells, in which the G2/M checkpoint is compromised, and the human osteosarcoma cell line U2OS, in which p53 and the G2/M checkpoint are functional. Significantly, U2OS cells exhibited neither UCC nor condensin recruitment to chromatin after the same radiomimetic treatment that induced them in HeLa cells (Fig. 5, A and B), even after prolonged time periods (up to 48 h) following high NCS doses (≥200 ng/ml).
Figure 5.

Biochemical analysis of premature mitosis versus G2/M arrest and normal mitotic events. (A) Condensin recruitment and UCC develop in DNA-damaged HeLa but not in U2OS cells. Cells were treated with NCS (200 ng/ml for 24 h), immunostained with anti-SMC2 antibody (red), and counterstained with DAPI (blue). Bars, 20 μm. (B) Western blotting analyses of chromatin fractions from NCS-treated (200 ng/ml for 24 h) HeLa and U2OS cells. Note that in HeLa cells, recruitment of SMC2 to damaged chromatin coincides with the phosphorylation of histone 3 on Ser10 (p-H3). (C) Biparametric FACS analysis demonstrating that DNA damage drives HeLa cells into mitosis and arrests U2OS cells in G2/M. The figure represents one of three independent experiments. (D) Levels of expression of p53 and its inducible regulator p21waf1 in DNA-damaged U2OS cells. U24 and U72 represent two untreated controls obtained at 24 and 72 h, respectively. (E) Western blotting analysis of G2→M transition regulators in DNA-damaged cells. Total cell lysates were obtained from NCS-treated U2OS and HeLa cells 24–72 h after 200 ng/ml NCS administration. U24 and U72 represent two untreated controls obtained at 24 and 72 h, respectively. Hsc70 was used as a loading control. (F) Comparative analysis of Cdk1 phosphorylation pattern in DNA-damaged and mitotic-arrested HeLa cells. Cells were either treated with 200 ng/ml NCS (for 24 h) or arrested in metaphase using colcemid. Total cell lysates were analyzed by Western blotting. (G) DNA damage leads to the appearance of two Cdk1 pools marked with the activating phosphorylation on Thr161 or the inhibitory phosphorylation on Tyr15, each with a distinct subcellular localization. Cytosolic and nuclear fractions were prepared from untreated and NCS-treated HeLa cells 24–72 h after the treatment and subjected to Western blotting analysis. The degree of fractionation is demonstrated using antibodies against α-tubulin and PARP-1, reflecting cytosolic and nuclear protein fractions, respectively. Asterisks in the cytosol fraction of the PARP-1 immunoblot denote the cross-reactive bands.
One of the biochemical markers of the G2→M transition is elevated phosphorylation of histone 3 on Ser10, which has been associated with the loading of condensins onto chromatin and chromatin condensation at the early stages of mitosis (Sauve et al., 1999; Schmiesing et al., 2000). We noticed that UCC-inducing treatments led to histone 3 hyperphosphorylation in HeLa but not in U2OS cells (Fig. 5 B), suggesting that DNA damage in HeLa cells indeed leads to premature mitosis. To substantiate this conclusion, we used FACS analyses to demonstrate the G2/M and mitotic fractions in both cell types after DNA damage. The results (Fig. 5 C) showed that a vast majority of the HeLa cells were at G2/M 24 h after the treatment, with >70% of the cells being in mitosis. In contrast, the G2/M population in U2OS cells consisted mainly of cells at G2; here, the mitotic fraction decreased from 1.3 to 0.4% after NCS treatment. Accordingly, the G2/M arrest in U2OS cells was correlated with elevated levels of p53 and p21waf1 (Fig. 5 D), which are both required for sustaining the G2/M checkpoint (Bunz et al., 1998).
Because damage-induced premature mitosis in HeLa cells was characterized by histone 3 phosphorylation, a hallmark of normal mitosis, we examined whether premature mitosis was also associated with Cdk1 activation, which is an important requirement for the initiation of normal mitosis (Morgan, 1997). The activation status of Cdk1 can be monitored by comparing the extent of its phosphorylation on Tyr15 (inhibitory phosphorylation) and Thr161 (activating phosphorylation; Makela et al., 1994; Fletcher et al., 2002). Therefore, Cdk1 activation was compared in HeLa and U2OS cells after DNA damage. We found that in U2OS cells 24–48 h after treatment, Cdk1 exhibited the expected phosphorylation of Tyr15 that is typical for G2/M-arrested cells (Fig. 5 E). The level of cyclin B, Cdk1's regulatory subunit that is highly expressed at G2/M and decreases as the cells progress through mitosis, was also markedly elevated 24–48 h after NCS treatment and began to decrease thereafter (Fig. 5 E). These results indicate that the G2/M arrest in DNA-damaged U2OS cells lasted for at least 48 h after treatment. FACS analysis supported these conclusions (unpublished data). Interestingly, in HeLa cells, Cdk1 was hyperphosphorylated on both Tyr15 (inhibitory phosphorylation) and Thr161 (activating phosphorylation; Fig. 5 E). Cyclin B levels were markedly elevated 24 h after NCS treatment but declined significantly thereafter (Fig. 5 E).
We compared this unique Cdk1 phosphorylation pattern with Cdk1 phosphorylation in HeLa cells artificially arrested in mitosis using colcemid treatment. In metaphase-arrested cells, the expected dephosphorylation of Tyr15 occurred concomitantly with the hyperphosphorylation of Thr161 (Fig. 5 F). This experiment allowed us to demonstrate the sharp difference between this pattern of Cdk1 phosphorylation and the one observed in DNA-damaged cells, in which Cdk1 was hyperphosphorylated on both Thr161 and Tyr15 (Fig. 5 F).
Cell fractionation further revealed that after NCS treatment, cyclin B/pT161-Cdk1 (activated complex) was predominantly nuclear, whereas inactivated Cdk1, which was tagged by pTyr15, was largely sequestered in the cytosol (Fig. 5 G). These findings indicate that the induction of DSBs leads to unscheduled Cdk1 activation with the appearance of two Cdk1 pools exhibiting distinct subcellular localizations. Collectively, our data provide insights into the mechanism of damage-induced UCC and suggest that the unscheduled activation of Cdk1, recruitment of condensin I to chromatin, and UCC together lead to (and represent) a hallmark of premature mitosis and could serve as useful markers for distinguishing premature from regular mitotic events.
Premature mitosis precedes MCD rather than apoptosis
We asked whether cells that react to extensive damage with UCC and premature mitosis (HeLa) will activate a different death pathway compared with cells that activate the G2/M checkpoint in the face of such damage and do not exhibit premature mitosis (U2OS). The results are summarized in Fig. 6. 3 d after damage infliction, U2OS cells exhibited typical morphological and biochemical features of apoptosis: pyknosis with the condensation of chromatin, nuclear fragmentation, phosphorylation of Ser46 of p53 (Fridman and Lowe, 2003), activation of caspase 3, and subsequent cleavage of its downstream substrates such as acinus (a marker of apoptosis-related chromatin condensation) and PARP (Fig. 6, A and B). On the other hand, the same DNA damage led in HeLa cells to the appearance of giant multimicronucleated cells, which is a hallmark of MCD (Fig. 6, A and C). Caspase 3, acinus, and PARP remained intact in these cells (Fig. 6 B). FACS analysis supported the occurrence of apoptosis in U2OS cells, with the sub-G1 fraction increasing approximately ninefold, whereas the same treatment in HeLa cells resulted in only a 1.7-fold increase in the sub-G1 fraction (unpublished data).
Figure 6.

DNA damage–induced MCD engages mechanisms distinct from those operative in apoptosis. (A) HeLa and U2OS cells undergoing MCD and apoptosis after DNA damage, respectively. The cells were treated with 200 ng/ml NCS for 72 h, immunostained with SMC2 (red), and counterstained with DAPI. The figure represents merged images. Bottom panels show the appearance of apoptosis and MCD in NCS-treated cells stained with Hemacolor reagents and photographed under light microscopy. M-labeled arrows indicate MCD, and A-labeled arrows denote apoptotic features (pyknosis, apoptotic chromatin condensation, and nuclear fragmentation). Bars, 20 μm. (B) DNA-damaged U2OS but not HeLa cells exhibit biochemical markers of apoptosis. The cells were treated with 200 ng/ml NCS for periods ranging from 24 to 72 h. Total cell extracts were analyzed by Western blotting. U, untreated samples. Hsc70 was used as a loading control. (C) MCD develops in DNA-damaged HeLa cells (200 ng/ml NCS for 72 h) at high frequency. The percentages of mono-, bi-, and polynucleated (more than three nuclei) cells were calculated from a total of 300 counted cells (n = 4). Each bar represents the mean ± SD (error bars).
We concluded that the process that begins in HeLa cells with UCC and premature mitosis and eventually culminates in MCD involves mechanisms distinct from those operative in apoptosis, whereas G2/M arrest typical of U2OS cells leads to apoptotic cell death.
Defective G2/M checkpoint is important but is not the only requirement for the UCC–MCD pathway
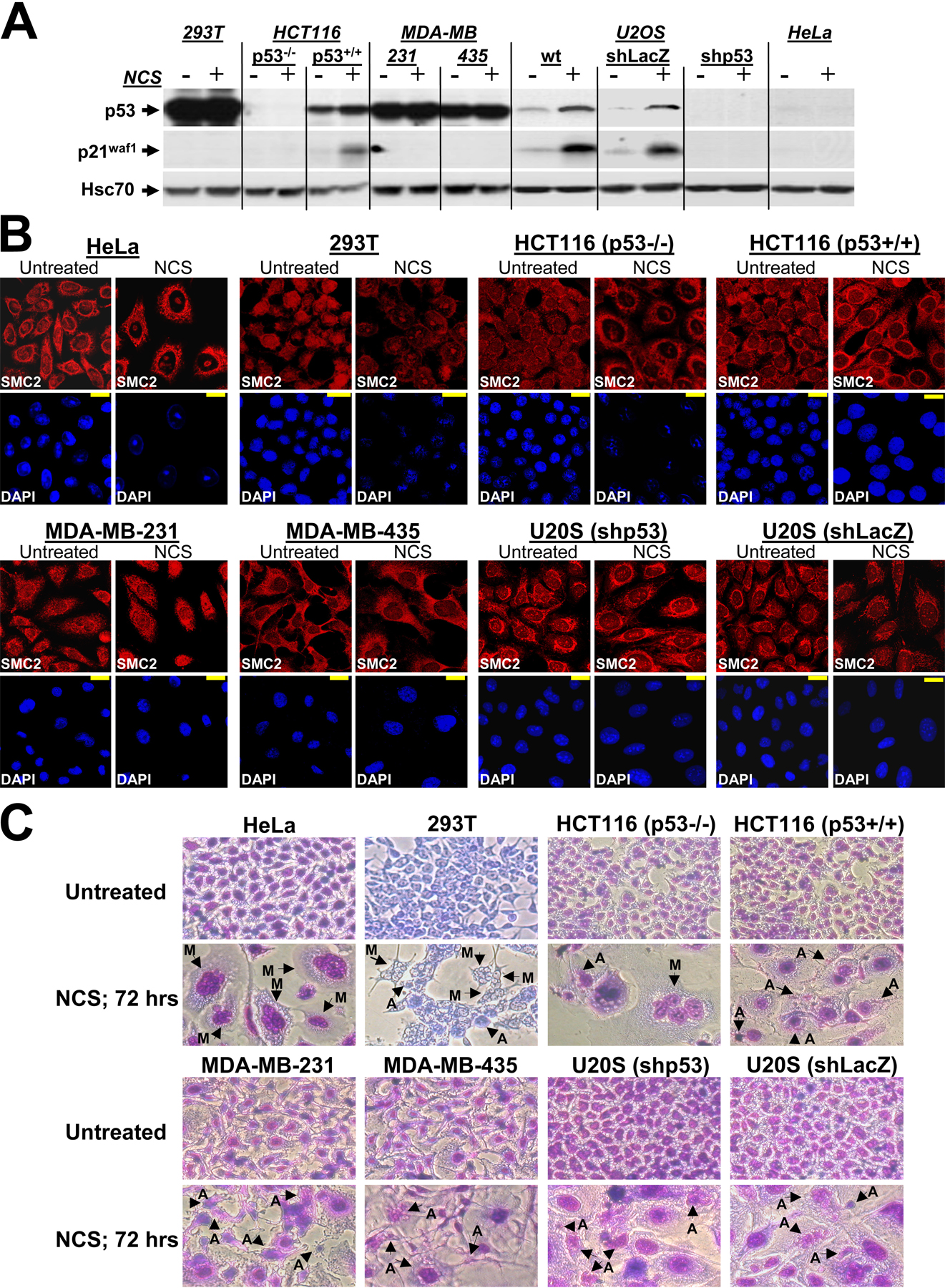
The role of the p53-mediated G2/M checkpoint in MCD is currently unresolved (Bunz et al., 1998; Chan et al., 1999; Andreassen et al., 2001; Castedo et al., 2004). Data reported here imply that a compromised p53-mediated G2/M checkpoint might be associated with MCD, but the role of p53 in preventing MCD requires further substantiation. We examined MCD in a panel of tumor cell lines with different p53 constitutions. We also stably knocked down p53 in U2OS cells by expressing in them an appropriate shRNA. The p53 status and functionality (measured by the ability to induce the gene encoding p21waf1) in these cell lines are demonstrated in Fig. S5 A (available at http://www.jcb.org/cgi/content/full/jcb.200604022/DC1). After NCS treatment, UCC was observed in three cell lines in which p53 was impaired—HeLa, 293T, and HCT166 (p53−/−) cells—but not in other p53-compromised cell lines (Table I and Fig. S5 B). Neither increasing the NCS dose (≥500 ng/ml) nor prolonging cell exposure to this drug (up to 48 h) changed these results (unpublished data). Accordingly, 72 h after NCS treatment, MCD was observed only in HeLa, 293T, and HCT166 (p53−/−) cells (Table I and Fig. S5 C) but not in the cell lines that did not exhibit UCC. Notably, the highest levels of MCD was observed in HeLa cells (60–70%), whereas MCD levels in 293T and HCT166 (p53−/−) cells reached only 15–20%. Apoptosis was the other prominent type of cell death noted to occur at high rates in all of these cell lines (with the exception of HeLa cells) after DNA damage (Fig. S5 C).
Table I.
Condensin recruitment and activation of the UCC–MCD pathway in a panel of human cell lines with different p53 constitutions treated with NCS
| Cell lines | p53 status | Condensin recruitment and UCC |
MCD |
|---|---|---|---|
| HeLa | Inactivated | + | + |
| U2OSwt | Wild type | – | – |
| U20SshLacZ | Wild type | – | – |
| U20Sshp53 | Knocked down | – | – |
| 293T | Inactivated | + | + |
| HCT116wt | Wild type | – | – |
| HCT116p53−/− | Knocked out | + | + |
| MDA-MB-231 | Mutated | – | – |
| MDA-MB-435 | Mutated | – | – |
Cells were treated with ≥200 ng/ml NCS and examined for UCC and MCD at 24 and 72 h, respectively. In HEK 293T cells (293T), p53 is stabilized and inactivated by SV40 large T antigen (Ahuja et al., 2005). In HCT116 (p53−/−) cells, the TP53 gene encoding p53 was inactivated using homologous recombination (Bunz et al., 1998). In MDA-MB-231 and -435 cells, both TP53 alleles are inactivated by mutations (Gartel et al., 2003). In HeLa cells, p53 is inactivated by the HPV-E6 protein (Scheffner et al., 1990).
Discussion
MCD results from aberrant mitosis, which fails to produce proper chromosome alignment and subsequent chromosome segregation, and culminates in the formation of large polynucleated cells (Erenpreisa and Cragg, 2001; Roninson et al., 2001). Abnormal mitosis in drug-treated or irradiated cells may proceed through several different pathways, but the final step is almost always the formation of nuclear envelopes around individual clusters of missegregated chromosomes (Roninson et al., 2001).
At least two mechanisms have been proposed for MCD. The first one is based on the aberrant duplication of centrosomes that leads to multipolar mitosis and subsequent formation of micronuclei (Sato et al., 2000). The second one is based on cell entry into premature mitosis, implying that cells proceed into mitosis before the completion of S or G2 phase. Such cells cannot properly compact their chromatin and segregate their chromosomes and must be eliminated. This mechanism offers an attractive explanation for DNA damage–induced MCD, but the evidence for premature mitosis in damaged cells has rested primarily on the morphological observations of UCC, whose mechanism is elusive (Roninson et al., 2001). Thus, elucidation of the molecular pathways underlying these processes is a prerequisite to understanding MCD. In this study, we provide some of the missing components of this process.
Although damage-induced UCC appears, at least morphologically, to differ from other types of chromatin condensation in eukaryotic cells such as mitotic or apoptotic chromatin condensation, the question arises whether the mechanisms involved in mitotic or apoptotic chromatin condensations are involved in MCD-related UCC. We demonstrated that UCC engages some of the proteins involved in mitotic chromatin packaging but not the acinus-mediated mechanism that operates in apoptotic chromatin condensation. Condensin recruitment was found to be a common denominator of mitotic chromatin condensation and UCC, but UCC involved activated condensin I and not condensin II, thereby differing from mitosis, which entails the recruitment of both condensins to chromatin (Hagstrom et al., 2002; Ono et al., 2004). Because condensin recruitment is important for proper chromosome alignment and subsequent segregation, the exclusive targeting of condensin I to damaged chromatin might lead to the formation of unaligned, hypercondensed chromatin aggregates typical of UCC.
Another difference between cell progression through normal mitosis and premature mitosis is unscheduled activation of Cdk1. In contrast to cells entering mitosis normally, Cdk1 in premature mitosis undergoes unscheduled activation, resulting in the appearance of two Cdk1 pools with different subcellular localizations. We showed that cyclin B/pThr161-activated Cdk1 is localized predominantly in the nucleus, whereas Cdk1, which is inactivated by Tyr15 phosphorylation, is sequestered primarily in the cytosol. This division of Cdk1 into two pools may reflect two populations of Cdk1 molecules, each characterized by different posttranslational modifications and each probably capable of responding to different stimuli or interacting with different regulators.
DNA damage–induced UCC was noted previously to develop in close proximity to the nucleolar site (Roninson et al., 2001). Our findings, although confirming these observations, also indicate that the number of nucleoli in such cells was markedly reduced, mainly to a single enlarged nucleolus. We propose that the UCC might entail coalescence of the nucleoli. A similar change in nucleolar morphology resulting in a single, enlarged nucleolar body was shown in HeLa cells after DNA damage induced by the alkylating agent MNNG (Alvarez-Gonzalez et al., 1999), but the underlying mechanisms were unknown. Unscheduled activation of Cdk1 in cells with UCC might explain this phenomenon. Because Cdk1 was shown to play a role in the maintenance of functional nucleoli and perturbation of its activation was associated with dramatic changes in nucleolar organization (Sirri et al., 2002), it is reasonable to assume that the unscheduled activation of Cdk1 could lead to the changes in nucleolar morphology. Furthermore, alteration of the nucleolar morphology after DNA damage could result from the marked changes in the spatial distribution of condensin. It has been recently shown that condensin could take part in nucleolar organization by the arrangement of rDNA gene repeats into heterochromatic-like structures thorough its interactions with Sir2p (Machin et al., 2004), a histone deacetylase that deacetylates histone tails to generate a hypoacetylated histone environment characteristic of heterochromatin.
The UCC–MCD pathway was primarily observed in cells with compromised p53-mediated G2/M checkpoint functions (Chan et al., 2000; Fei and El-Deiry, 2003), although its pivotal role in preventing MCD has recently been questioned (Andreassen et al., 2001; Castedo et al., 2004). In our panel of several human cell lines with compromised p53, an impaired p53-mediated G2/M checkpoint was not the sole requirement for activation of the UCC–MCD pathway after extensive DNA damage. Presumably, the combination of compromised p53 and impaired function of other mediators of G2/M progression are responsible for this phenomenon. Among the possible candidates for such mediators are Chk1, Plk-1, and the Aurora A kinase, which were recently shown to play a key role in the reactivation of Cdk1 and onset of mitosis after DNA damage (Krystyniak et al., 2006). In addition, it has recently been shown that in cells with compromised p53, p73, another member of the p53-like family of proteins, can at least partly substitute for p53 in cell cycle regulation (Ozaki and Nakagawara, 2005), making p73 still another possible candidate for involvement in IR-induced MCD.
It is noteworthy that ATM, the chief regulator of cellular responses to DSBs, was uninvolved in this pathway. In the face of DSBs, ATM mediates the pathways leading to cellular rescue and survival on the one hand and apoptosis on the other (Bree et al., 2004; Rashi-Elkeles et al., 2006). The ATM-dependent apoptotic response relies on functional p53 (Morgan and Kastan, 1997; Fridman and Lowe, 2003). Our findings suggest that the UCC–MCD pathway is turned on in cells that are destined to die and cannot activate the ATM–p53-mediated apoptotic pathway. This is probably one of the very few DSB-triggered pathways that are ATM independent. This observation draws a line between DSB-induced processes that are under ATM jurisdiction and those that are not.
In summary, our findings disclose a cell death pathway that is triggered by the extensive induction of DSBs, is ATM independent, and is distinct from the apoptotic pathway. It leads to the unscheduled activation of Cdk1 followed by premature mitosis characterized by UCC that is associated with recruitment of condensin I to chromatin and ultimately to MCD (Fig. 7). Thus, the unscheduled activation of Cdk1 and phosphorylation of the non-SMC subunits of condensin I could be sequentially linked. Considering the importance of this pathway in the response of tumor cells to radiotherapy and chemotherapy, further elucidation of its molecular steps is crucial for understanding the action of anticancer drugs and the rational design of therapeutic regimens.
Figure 7.

Proposed mechanisms of premature mitosis and MCD in DNA-damaged cells.
Materials and methods
Antibodies and reagents
The following antibodies were used in this study: α-SMC2 (BL548) and α-SMC4 (BL551) were obtained from Bethyl Laboratories, Inc.; anti–hCAP-H and hCAP-H2 were provided by J.-M. Peters (Research Institute of Molecular Pathology, Vienna, Austria); α-SMC1 was a gift from R. Jessberger (Mount Sinai School of Medicine, New York, NY); α-nucleolin (7G2) was a gift from Y. Shav-Tal (Bar-Ilan University, Ramat-Gan, Israel); α–HP-1α was purchased from Chemicon; α-nucleostemin was obtained from R&D Systems; α-phospho-Cdk1 (pTyr15), α-phospho-Cdk1 (pThr161), α-phospho- p53 (Ser46), α-cleaved caspase 3 (5A1), and α–PARP-1 were obtained from Cell Signaling Technology, Inc.; anti-H2B, α-p53 (DO-1), α-Hsc70, and α-Cdc2 p34 were obtained from Santa Cruz Biotechnology, Inc.; α-p21waf1 (C19) was obtained from Delta Biolabs; α-acinus (C terminus) and α-phospho-histone H3 were purchased from Upstate Biotechnology; α-tubulin was obtained from Sigma-Aldrich; and α–cyclin B was obtained from BD Transduction Laboratories. Secondary HRP-conjugated as well as rhodamine red– and FITC-conjugated antibodies were purchased from Jackson ImmunoResearch Laboratories.
DNase-free RNase, DNase-I RNase free, and DAPI were purchased from Roche Diagnostics; Yo-Pro-1 was obtained from Invitrogen. Staurosporine was obtained from Cell Signaling Technology, colcemid was purchased from Calbiochem, and NCS was purchased from Kayaka Chemicals.
Cell cultures
HeLa and U2OS cells were obtained from American Type Culture Collection, MDA-MD-231 and MDA-MB-435 cells were a gift from I. Tsarfaty (Tel Aviv University, Tel Aviv, Israel), and HCT116 cells and their p53-null variant (HCT116 p53+/+ and HCT116 p53−/−, respectively) were gifts from B. Vogelstein (Johns Hopkins University, Baltimore, MD). Cells were cultured in the recommended growth media under standard conditions.
DNA damage
DNA damage was induced either by adding NCS to the cultures, by X irradiation using an irradiator (MGC40; Philips), or by UV-C irradiation using an irradiator (FLX-35M; Vilber Lourmat) at dose ranges of 30–50 J/m2.
Confocal and light microscopy
Cells were fixed in methanol-acetone (1:1) at −20°C for 5 min. After RNase or DNase-I treatments, the cells were blocked in 3% BSA and immunostained. DNA was counterstained with 1 μM Yo-Pro-1 or 0.1 μg/ml DAPI. The samples were mounted using an aqueous mounting medium (Biomeda) and subjected to confocal microscopy using the AOBS system (TCS SP2; Leica) with a HCXPLAPOL NA 1.2 objective lens. All confocal images were taken with a 63× water correction objective and operated by confocal software (LCS Lite, version 2.5 Build 1347; Leica).
Apoptosis and MCD were visualized by light microscopy. The cells were fixed in methanol, stained with Hemacolor reagents (Merck) as previously described (Blank et al., 2003), and photographed under a light microscope (Eclipse TE 2000-5; Nikon) with a 20× NA 0.45 plain Fluor objective. All comparative images (treated vs. untreated samples) were obtained under identical microscope and camera settings.
DNase treatment
Cells grown on coverslips were treated with DNase-I diluted in Tris-buffered saline (25 mM Tris-HCl, pH 7.5) containing 1% BSA and supplemented with 5 mM MgSO4 for 1 h at 23°C) as described previously (Barrett et al., 2001). The cells were either directly processed for immunostaining or were processed after extraction of their cleaved DNA (with buffer containing 20 mM Tris-HCl, pH 7.5, 0.25 M ammonium sulfate, and 0.2 mM MgCl2).
Preparation of protein extracts and immunoblotting
Total cell lysates were prepared using radioimmunoprecipitation buffer (50 mM Tris-HCl, pH 7.5, 150 mM NaCl, 1% IGEPAL, 0.1% SDS, and 0.5% sodium deoxycholate) supplemented with a mixture of protease and phosphatase inhibitors. Nuclear and cytosolic extracts were obtained as previously described (Blank et al., 2003). In brief, cytosolic extracts were prepared using buffer A (10 mM Hepes, pH 7.9, 10 mM KCl, 1 mM EDTA, 1 mM EGTA, and 1 mM DTT supplemented with a mixture of protease and phosphatase inhibitors and 0.6% NP-40). Nuclear extracts were prepared by the dissolution of nuclei in buffer C (20 mM Hepes, pH 7.9, 400 mM KCl, 1 mM EDTA, 1 mM EGTA, 1 mM DTT, and a mixture of protease and phosphatase inhibitors) using vortex at 4°C for 45 min. Soluble chromatin fractions were subsequently prepared from the previous step by sonication (40% amplitude; three pulses at 20 s each on ice) in radioimmunoprecipitation buffer. Protein content was calibrated using the bicinchoninic acid protein assay reagent kit (Pierce Chemical Co.). The samples were subjected to standard Western blotting analysis. Immunoblots (polyvinylidene difluoride) were visualized using enhanced chemiluminescence (SuperSignal, Pierce Chemical Co.).
Phosphatase treatment
Reactions were performed on solubilized chromatin fractions in 1× PTase buffer (50 mM Tris-HCl, pH 7.5, 100 mM NaCl, 0.1 mM EGTA, 2 mM DTT, and 0.01% Brij 35) supplemented with MnCl2 using 400 U λ-protein phosphatase (λ-PPase; New England Biolabs, Inc.) per 100 μg of extracted proteins at 30°C for 1 h and were stopped by supplementation to 50 mM EDTA. The extracts were analyzed directly by immunoblotting.
Biparametric flow cytometric analysis
Cells were analyzed by two-dimensional flow cytometry after costaining with an antibody against phosphorylated histone 3 (H3-p, a mitotic marker) and propidium iodide (PI) for DNA quantitation. In brief, cells were fixed, permeabilized with 0.25% Triton X-100, incubated with the H3-p antibody, labeled with FITC-conjugated anti–rabbit IgG secondary antibody, treated with 5 μg/ml DNase-free RNase (37°C for 30 min), and stained with PI. Data were collected using FACSort flow cytometry (Becton Dickinson) with H3-p in the first and PI in the second dimension (10,000 events/sample). Cell cycle gating and analysis were performed using WinMDI software.
Generation of mitosis-sequestered cells
Logarithmically growing cultures of HeLa cells were mitotically arrested by treatment with 0.1 μg/ml colcemid for 17 h as described previously (Duesbery et al., 1997), collected by shake-off, and subjected to Western blotting analysis. FACS analysis confirmed that >90% of colcemid-treated cells were in M phase.
RNA interference
p53 was stably knocked down in U20S cells by expressing the appropriate shRNA in them using the pRETRO-SUPER retroviral vector (Brummelkamp et al., 2002). The sequence 5′-GATCCCCGACTCCAGTGGTAATCTACTTCAAGAGAGTAGATTACCACTG GAGTCTTTTTGGAAA-′3 was cloned in the vector using BglII and HindIII restriction sites.
ATM was knocked down in HeLa cells using a combination of two different shRNAs: ATM-I (7218), 5′-GATCCCCCTGGTTAGCAGAAACGTGCTTCAAGAGAGCA CGTTTCTGCTAACCAGTTTTTGGAAA-′3; and ATM-II (p480), 5′-GATCCCCGATACCAGATCCTTGGAGATTCAAGAG ATCTCCAAGGATCTGGTATCTTTTTGGAAA-3′ (a gift from R. Agami, Netherlands Cancer Institute, Amsterdam, Netherlands).
Irrelevant shRNA against the lacZ operon was used as a control. Cells were infected with retroviral particles according to standard protocols and subjected to selection with puromycin (for shp53 cells) or a combination of puromycin and hygromycin (for shATM cells).
Statistics
Data were analyzed using the two-tailed t test. P values of <0.05 were considered statistically significant.
Online supplemental material
Fig. S1 shows that IR and NCS but not UV trigger the recruitment of condensin core subunits to damaged chromatin and the formation of UCC bodies. Fig. S2 shows that condensin recruitment and UCC develop in HeLa cells as a delayed cellular response to severe DNA damage. Fig. S3 shows that ATM knockdown does not alter the rate of DSB-induced UCC. Fig. S4 shows that condensin and HP-1α strictly colocalize in normal mitosis but occupy different compartments in UCC bodies associated with premature mitosis. Fig. S5 shows that the defective p53-mediated G2/M checkpoint is important for condensin recruitment, UCC, and MCD but is not the sole requirement. Online supplemental material is available at http://www.jcb.org/cgi/content/full/jcb.200604022/DC1.
Supplementary Material
Acknowledgments
We are thankful to G. Lavie, R. Elkon, and I. Tsarfaty for comments on the manuscript. We are also grateful to J.-M. Peters, R. Jessberger, B. Vogelstein, I. Tsarfaty, and Y. Shav-Tal for providing antibodies and cell lines.
This work was supported by research grants from the A-T Medical Research Foundation, the AT Children's Project, and the National Institutes of Health (grant NS31763).
Abbreviations used in this paper: ATM, ataxia telangiectasia mutated; Cdk1, cyclin-dependent kinase 1; DSB, double-strand break; hCAP, human chromatin-associated protein; IR, ionizing radiation; MCD, mitotic cell death; NCS, neocarzinostatin; PARP, poly(ADP-ribose)polymerase; PI, propidium iodide; shRNA, short hairpin RNA; SMC, structural maintenance of chromosomes; UCC, uneven chromatin condensation.
References
- Ahuja, D., M.T. Saenz-Robles, and J.M. Pipas. 2005. SV40 large T antigen targets multiple cellular pathways to elicit cellular transformation. Oncogene. 24:7729–7745. [DOI] [PubMed] [Google Scholar]
- Al-Baker, E.A., J. Boyle, R. Harry, and I.R. Kill. 2004. A p53-independent pathway regulates nucleolar segregation and antigen translocation in response to DNA damage induced by UV irradiation. Exp. Cell Res. 292:179–186. [DOI] [PubMed] [Google Scholar]
- Alvarez-Gonzalez, R., H. Spring, M. Muller, and A. Burkle. 1999. Selective loss of poly(ADP-ribose) and the 85-kDa fragment of poly(ADP-ribose) polymerase in nucleoli during alkylation-induced apoptosis of HeLa cells. J. Biol. Chem. 274:32122–32126. [DOI] [PubMed] [Google Scholar]
- Andreassen, P.R., F.B. Lacroix, O.D. Lohez, and R.L. Margolis. 2001. Neither p21WAF1 nor 14-3-3sigma prevents G2 progression to mitotic catastrophe in human colon carcinoma cells after DNA damage, but p21WAF1 induces stable G1 arrest in resulting tetraploid cells. Cancer Res. 61:7660–7668. [PubMed] [Google Scholar]
- Barrett, K.L., J.M. Willingham, A.J. Garvin, and M.C. Willingham. 2001. Advances in cytochemical methods for detection of apoptosis. J. Histochem. Cytochem. 49:821–832. [DOI] [PubMed] [Google Scholar]
- Blank, M., M. Mandel, Y. Keisari, D. Meruelo, and G. Lavie. 2003. Enhanced ubiquitinylation of heat shock protein 90 as a potential mechanism for mitotic cell death in cancer cells induced with hypericin. Cancer Res. 63:8241–8247. [PubMed] [Google Scholar]
- Bree, R.T., C. Neary, A. Samali, and N.F. Lowndes. 2004. The switch from survival responses to apoptosis after chromosomal breaks. DNA Repair (Amst.). 3:989–995. [DOI] [PubMed] [Google Scholar]
- Brummelkamp, T.R., R. Bernards, and R. Agami. 2002. A system for stable expression of short interfering RNAs in mammalian cells. Science. 296:550–553. [DOI] [PubMed] [Google Scholar]
- Bunz, F., A. Dutriaux, C. Lengauer, T. Waldman, S. Zhou, J.P. Brown, J.M. Sedivy, K.W. Kinzler, and B. Vogelstein. 1998. Requirement for p53 and p21 to sustain G2 arrest after DNA damage. Science. 282:1497–1501. [DOI] [PubMed] [Google Scholar]
- Byun, Y., F. Chen, R. Chang, M. Trivedi, K.J. Green, and V.L. Cryns. 2001. Caspase cleavage of vimentin disrupts intermediate filaments and promotes apoptosis. Cell Death Differ. 8:443–450. [DOI] [PubMed] [Google Scholar]
- Cabello, O.A., E. Eliseeva, W.G. He, H. Youssoufian, S.E. Plon, B.R. Brinkley, and J.W. Belmont. 2001. Cell cycle-dependent expression and nucleolar localization of hCAP-H. Mol. Biol. Cell. 12:3527–3537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Castedo, M., J.L. Perfettini, T. Roumier, A. Valent, H. Raslova, K. Yakushijin, D. Horne, J. Feunteun, G. Lenoir, R. Medema, et al. 2004. Mitotic catastrophe constitutes a special case of apoptosis whose suppression entails aneuploidy. Oncogene. 23:4362–4370. [DOI] [PubMed] [Google Scholar]
- Chan, T.A., H. Hermeking, C. Lengauer, K.W. Kinzler, and B. Vogelstein. 1999. 14-3-3Sigma is required to prevent mitotic catastrophe after DNA damage. Nature. 401:616–620. [DOI] [PubMed] [Google Scholar]
- Chan, T.A., P.M. Hwang, H. Hermeking, K.W. Kinzler, and B. Vogelstein. 2000. Cooperative effects of genes controlling the G(2)/M checkpoint. Genes Dev. 14:1584–1588. [PMC free article] [PubMed] [Google Scholar]
- Couldwell, W.T., D.R. Hinton, S. He, T.C. Chen, I. Sebat, M.H. Weiss, and R.E. Law. 1994. Protein kinase C inhibitors induce apoptosis in human malignant glioma cell lines. FEBS Lett. 345:43–46. [DOI] [PubMed] [Google Scholar]
- Duesbery, N.S., T. Choi, K.D. Brown, K.W. Wood, J. Resau, K. Fukasawa, D.W. Cleveland, and G.F. Vande Woude. 1997. CENP-E is an essential kinetochore motor in maturing oocytes and is masked during mos- dependent, cell cycle arrest at metaphase II. Proc. Natl. Acad. Sci. USA. 94:9165–9170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Erenpreisa, J., and M.S. Cragg. 2001. Mitotic death: a mechanism of survival? A review. Cancer Cell Int. 1:1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fei, P., and W.S. El-Deiry. 2003. P53 and radiation responses. Oncogene. 22:5774–5783. [DOI] [PubMed] [Google Scholar]
- Fletcher, L., Y. Cheng, and R.J. Muschel. 2002. Abolishment of the Tyr-15 inhibitory phosphorylation site on cdc2 reduces the radiation-induced G(2) delay, revealing a potential checkpoint in early mitosis. Cancer Res. 62:241–250. [PubMed] [Google Scholar]
- Fridman, J.S., and S.W. Lowe. 2003. Control of apoptosis by p53. Oncogene. 22:9030–9040. [DOI] [PubMed] [Google Scholar]
- Gartel, A.L., C. Feliciano, and A.L. Tyner. 2003. A new method for determining the status of p53 in tumor cell lines of different origin. Oncol. Res. 13:405–408. [DOI] [PubMed] [Google Scholar]
- Goldberg, I.H. 1987. Free radical mechanisms in neocarzinostatin-induced DNA damage. Free Radic. Biol. Med. 3:41–54. [DOI] [PubMed] [Google Scholar]
- Hagstrom, K.A., V.F. Holmes, N.R. Cozzarelli, and B.J. Meyer. 2002. C. elegans condensin promotes mitotic chromosome architecture, centromere organization, and sister chromatid segregation during mitosis and meiosis. Genes Dev. 16:729–742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirano, T. 2002. The ABCs of SMC proteins: two-armed ATPases for chromosome condensation, cohesion, and repair. Genes Dev. 16:399–414. [DOI] [PubMed] [Google Scholar]
- Hirano, T., R. Kobayashi, and M. Hirano. 1997. Condensins, chromosome condensation protein complexes containing XCAP-C, XCAP-E and a Xenopus homolog of the Drosophila Barren protein. Cell. 89:511–521. [DOI] [PubMed] [Google Scholar]
- Hirota, T., D. Gerlich, B. Koch, J. Ellenberg, and J.M. Peters. 2004. Distinct functions of condensin I and II in mitotic chromosome assembly. J. Cell Sci. 117:6435–6445. [DOI] [PubMed] [Google Scholar]
- Ianzini, F., and M.A. Mackey. 1997. Spontaneous premature chromosome condensation and mitotic catastrophe following irradiation of HeLa S3 cells. Int. J. Radiat. Biol. 72:409–421. [DOI] [PubMed] [Google Scholar]
- Kimura, K., M. Hirano, R. Kobayashi, and T. Hirano. 1998. Phosphorylation and activation of 13S condensin by Cdc2 in vitro. Science. 282:487–490. [DOI] [PubMed] [Google Scholar]
- Kimura, K., O. Cuvier, and T. Hirano. 2001. Chromosome condensation by a human condensin complex in Xenopus egg extracts. J. Biol. Chem. 276:5417–5420. [DOI] [PubMed] [Google Scholar]
- Krystyniak, A., C. Garcia-Echeverria, C. Prigent, and S. Ferrari. 2006. Inhibition of Aurora A in response to DNA damage. Oncogene. 25:338–348. [DOI] [PubMed] [Google Scholar]
- Machin, F., K. Paschos, A. Jarmuz, J. Torres-Rosell, C. Pade, and L. Aragon. 2004. Condensin regulates rDNA silencing by modulating nucleolar Sir2p. Curr. Biol. 14:125–130. [PubMed] [Google Scholar]
- Makela, T.P., J.P. Tassan, E.A. Nigg, S. Frutiger, G.J. Hughes, and R.A. Weinberg. 1994. A cyclin associated with the CDK-activating kinase MO15. Nature. 371:254–257. [DOI] [PubMed] [Google Scholar]
- Minc, E., Y. Allory, H.J. Worman, J.C. Courvalin, and B. Buendia. 1999. Localization and phosphorylation of HP1 proteins during the cell cycle in mammalian cells. Chromosoma. 108:220–234. [DOI] [PubMed] [Google Scholar]
- Morgan, D.O. 1997. Cyclin-dependent kinases: engines, clocks, and microprocessors. Annu. Rev. Cell Dev. Biol. 13:261–291. [DOI] [PubMed] [Google Scholar]
- Morgan, S.E., and M.B. Kastan. 1997. p53 and ATM: cell cycle, cell death, and cancer. Adv. Cancer Res. 71:1–25. [DOI] [PubMed] [Google Scholar]
- Nasmyth, K., and C.H. Haering. 2005. The structure and function of smc and kleisin complexes. Annu. Rev. Biochem. 74:595–648. [DOI] [PubMed] [Google Scholar]
- Neuwald, A.F., and T. Hirano. 2000. HEAT repeats associated with condensins, cohesins, and other complexes involved in chromosome-related functions. Genome Res. 10:1445–1452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nitta, M., O. Kobayashi, S. Honda, T. Hirota, S. Kuninaka, T. Marumoto, Y. Ushio, and H. Saya. 2004. Spindle checkpoint function is required for mitotic catastrophe induced by DNA-damaging agents. Oncogene. 23:6548–6558. [DOI] [PubMed] [Google Scholar]
- Ono, T., A. Losada, M. Hirano, M.P. Myers, A.F. Neuwald, and T. Hirano. 2003. Differential contributions of condensin I and condensin II to mitotic chromosome architecture in vertebrate cells. Cell. 115:109–121. [DOI] [PubMed] [Google Scholar]
- Ono, T., Y. Fang, D.L. Spector, and T. Hirano. 2004. Spatial and temporal regulation of Condensins I and II in mitotic chromosome assembly in human cells. Mol. Biol. Cell. 15:3296–3308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ozaki, T., and A. Nakagawara. 2005. p73, a sophisticated p53 family member in the cancer world. Cancer Sci. 96:729–737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rashi-Elkeles, S., R. Elkon, N. Weizman, C. Linhart, N. Amariglio, G. Sternberg, G. Rechavi, A. Barzilai, R. Shamir, and Y. Shiloh. 2006. Parallel induction of ATM-dependent pro- and antiapoptotic signals in response to ionizing radiation in murine lymphoid tissue. Oncogene. 25:1584–1592. [DOI] [PubMed] [Google Scholar]
- Roninson, I.B., E.V. Broude, and B.D. Chang. 2001. If not apoptosis, then what? Treatment-induced senescence and mitotic catastrophe in tumor cells. Drug Resist. Updat. 4:303–313. [DOI] [PubMed] [Google Scholar]
- Sahara, S., M. Aoto, Y. Eguchi, N. Imamoto, Y. Yoneda, and Y. Tsujimoto. 1999. Acinus is a caspase-3-activated protein required for apoptotic chromatin condensation. Nature. 401:168–173. [DOI] [PubMed] [Google Scholar]
- Sato, N., K. Mizumoto, M. Nakamura, H. Ueno, Y.A. Minamishima, J.L. Farber, and M. Tanaka. 2000. A possible role for centrosome overduplication in radiation-induced cell death. Oncogene. 19:5281–5290. [DOI] [PubMed] [Google Scholar]
- Sauve, D.M., H.J. Anderson, J.M. Ray, W.M. James, and M. Roberge. 1999. Phosphorylation-induced rearrangement of the histone H3 NH2- terminal domain during mitotic chromosome condensation. J. Cell Biol. 145:225–235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scheffner, M., B.A. Werness, J.M. Huibregtse, A.J. Levine, and P.M. Howley. 1990. The E6 oncoprotein encoded by human papillomavirus types 16 and 18 promotes the degradation of p53. Cell. 63:1129–1136. [DOI] [PubMed] [Google Scholar]
- Schleiffer, A., S. Kaitna, S. Maurer-Stroh, M. Glotzer, K. Nasmyth, and F. Eisenhaber. 2003. Kleisins: a superfamily of bacterial and eukaryotic SMC protein partners. Mol. Cell. 11:571–575. [DOI] [PubMed] [Google Scholar]
- Schmiesing, J.A., A.R. Ball Jr., H.C. Gregson, J.M. Alderton, S. Zhou, and K. Yokomori. 1998. Identification of two distinct human SMC protein complexes involved in mitotic chromosome dynamics. Proc. Natl. Acad. Sci. USA. 95:12906–12911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmiesing, J.A., H.C. Gregson, S. Zhou, and K. Yokomori. 2000. A human condensin complex containing hCAP-C-hCAP-E and CNAP1, a homolog of Xenopus XCAP-D2, colocalizes with phosphorylated histone H3 during the early stage of mitotic chromosome condensation. Mol. Cell. Biol. 20:6996–7006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sirri, V., D. Hernandez-Verdun, and P. Roussel. 2002. Cyclin-dependent kinases govern formation and maintenance of the nucleolus. J. Cell Biol. 156:969–981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soldani, C., and A.I. Scovassi. 2002. Poly(ADP-ribose) polymerase-1 cleavage during apoptosis: an update. Apoptosis. 7:321–328. [DOI] [PubMed] [Google Scholar]
- Suzuki, T., K. Fujikura, T. Higashiyama, and K. Takata. 1997. DNA staining for fluorescence and laser confocal microscopy. J. Histochem. Cytochem. 45:49–53. [DOI] [PubMed] [Google Scholar]
- Swanson, P.E., S.B. Carroll, X.F. Zhang, and M.A. Mackey. 1995. Spontaneous premature chromosome condensation, micronucleus formation, and non-apoptotic cell death in heated HeLa S3 cells. Ultrastructural observations. Am. J. Pathol. 146:963–971. [PMC free article] [PubMed] [Google Scholar]
- Swedlow, J.R., and T. Hirano. 2003. The making of the mitotic chromosome: modern insights into classical questions. Mol. Cell. 11:557–569. [DOI] [PubMed] [Google Scholar]
- Torres, K., and S.B. Horwitz. 1998. Mechanisms of Taxol-induced cell death are concentration dependent. Cancer Res. 58:3620–3626. [PubMed] [Google Scholar]
- Tsai, R.Y., and R.D. McKay. 2002. A nucleolar mechanism controlling cell proliferation in stem cells and cancer cells. Genes Dev. 16:2991–3003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsai, R.Y., and R.D. McKay. 2005. A multistep, GTP-driven mechanism controlling the dynamic cycling of nucleostemin. J. Cell Biol. 168:179–184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tuteja, R., and N. Tuteja. 1998. Nucleolin: a multifunctional major nucleolar phosphoprotein. Crit. Rev. Biochem. Mol. Biol. 33:407–436. [DOI] [PubMed] [Google Scholar]
- Watrin, E., and V. Legagneux. 2005. Contribution of hCAP-D2, a non-SMC subunit of condensin I, to chromosome and chromosomal protein dynamics during mitosis. Mol. Cell. Biol. 25:740–750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yeong, F.M., H. Hombauer, K.S. Wendt, T. Hirota, I. Mudrak, K. Mechtler, T. Loregger, A. Marchler-Bauer, K. Tanaka, J.M. Peters, and E. Ogris. 2003. Identification of a subunit of a novel Kleisin-beta/SMC complex as a potential substrate of protein phosphatase 2A. Curr. Biol. 13:2058–2064. [DOI] [PubMed] [Google Scholar]
- Zamzami, N., and G. Kroemer. 1999. Condensed matter in cell death. Nature. 401:127–128. [DOI] [PubMed] [Google Scholar]
- Zhou, B.B., and S.J. Elledge. 2000. The DNA damage response: putting checkpoints in perspective. Nature. 408:433–439. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}