

Abstract
Sequestration of misfolded proteins into pericentriolar inclusions called aggresomes is a means that cells use to minimize misfolded protein-induced cytotoxicity. However, the molecular mechanism by which misfolded proteins are recruited to aggresomes remains unclear. Mutations in the E3 ligase parkin cause autosomal recessive Parkinson's disease that is devoid of Lewy bodies, which are similar to aggresomes. Here, we report that parkin cooperates with heterodimeric E2 enzyme UbcH13/Uev1a to mediate K63-linked polyubiquitination of misfolded DJ-1. K63-linked polyubiquitination of misfolded DJ-1 serves as a signal for interaction with histone deacetylase 6, an adaptor protein that binds the dynein–dynactin complex. Through this interaction, misfolded DJ-1 is linked to the dynein motor and transported to aggresomes. Furthermore, fibroblasts lacking parkin display deficits in targeting misfolded DJ-1 to aggresomes. Our findings reveal a signaling role for K63-linked polyubiquitination in dynein-mediated transport, identify parkin as a key regulator in the recruitment of misfolded DJ-1 to aggresomes, and have important implications regarding the biogenesis of Lewy bodies.
Introduction
A prominent feature common to most neurodegenerative diseases is the accumulation of misfolded proteins in cytoplasmic inclusions, such as Lewy bodies in Parkinson's disease (PD) and neurofibrillary tangles in Alzheimer's disease (AD). The accumulation of misfolded proteins in these diseases most likely occurs due to a chronic imbalance in the generation and clearance of misfolded proteins. Misfolded proteins can be generated by genetic mutations (Goldberg, 2003) or oxidative modifications (Dalle-Donne et al., 2006). Misfolded proteins are prone to aggregation and have the potential to impair cellular functions (Gregersen et al., 2006). Cells combat the buildup of misfolded proteins either by chaperone-mediated refolding or by proteasomal degradation (Goldberg, 2003). Alternatively, when the proteasome becomes overwhelmed or impaired, misfolded proteins are transported by the retrograde dynein motor complex to pericentriolar inclusions called aggresomes (Johnston et al., 1998; Kopito, 2000; Garcia-Mata et al., 2002). It has been proposed that aggresome formation is a specific and active cellular response serving to sequester potentially toxic misfolded proteins (Kopito, 2000). Recent evidence suggests that there are similarities between Lewy bodies and aggresomes (Olanow et al., 2004). However, the molecular mechanisms involved in recognition and targeting of misfolded proteins to aggresomes remain unclear.
Ubiquitination is a dynamic post-translational modification that serves diverse cellular roles (Weissman, 2001; Pickart and Fushman, 2004). Ubiquitin is covalently attached to a substrate protein through the formation of an isopeptide bond between the C-terminal glycine residue of ubiquitin and the ɛ-amino group of a lysine residue on the substrate by a cascade of enzymatic reactions involving an E1 ubiquitin-activating enzyme, an E2 ubiquitin-conjugating enzyme, and an E3 ubiquitin protein ligase. Successive conjugation of ubiquitin molecules to one of the seven internal lysine residues (K6, K11, K27, K29, K33, K48, and K63) within the preceding ubiquitin molecule results in formation of a polyubiquitin chain (Pickart and Fushman, 2004). K48-linked polyubiquitination acts as the canonical signal for targeting the substrate to the proteasome for degradation. In contrast, K63-linked polyubiquitination has recently been shown to have a proteasome-independent role in the regulation of several cellular processes, including endocytosis, signal transduction, and DNA damage (Pickart and Fushman, 2004; Haglund and Dikic, 2005). Although K48-linked polyubiquitination has been extensively studied, relatively little is known about the molecular mechanisms underlying polyubiquitination via K63 or other lysine linkages.
Mutations in the E3 ligase parkin account for ∼50% of all recessively transmitted early-onset PD cases (Kitada et al., 1998; Lucking et al., 2000; Hattori and Mizuno, 2004). Interestingly, parkin-associated PD is devoid of Lewy bodies (Takahashi et al., 1994; Mori et al., 1998; Hayashi et al., 2000), suggesting that parkin function may be required for the formation of these inclusion bodies (Sulzer, 2007). Parkin is present in Lewy bodies in sporadic PD (Schlossmacher et al., 2002) and is recruited to aggresomes in cells treated with proteasome inhibitors (Junn et al., 2002; Ardley et al., 2003; Zhao et al., 2003; Muqit et al., 2004). However, the precise role of parkin in the biogenesis of Lewy bodies or aggresomes is unclear. Two studies suggest that parkin is capable of mediating K63-linked polyubiquitination (Doss-Pepe et al., 2005; Lim et al., 2005a). However, the cellular role of parkin-mediated K63-linked polyubiquitination and the molecular mechanism by which parkin mediates K63-linked polyubiquitination remain undefined.
DJ-1 is a ubiquitously expressed protein that is mutated in an autosomal recessive, early-onset form of PD (Abou-Sleiman et al., 2003; Bonifati et al., 2003). We and others have previously shown that the PD-linked L166P mutation disrupts DJ-1 protein folding, resulting in a misfolded protein that is prone to aggregation (Olzmann et al., 2004; Shendelman et al., 2004). The folding state of other parkin-interacting proteins has not been investigated. Therefore, in this study we investigated the role of parkin in ubiquitinating misfolded proteins, using the L166P mutant DJ-1 as a model. Our findings reveal a novel proteasome-independent role for parkin in the regulation of aggresome formation and have important implications for understanding the biogenesis of Lewy bodies in PD.
Results
Parkin selectively recognizes and ubiquitinates L166P mutant DJ-1, but not wild-type DJ-1
We and others have previously shown that the L166P mutation-induced misfolded DJ-1 is selectively polyubiquitinated and degraded by the proteasome (Baulac et al., 2004; Olzmann et al., 2004). The molecular machinery that recognizes and ubiquitinates L166P mutant DJ-1 is unknown. We have proposed that L166P mutant DJ-1 might be a substrate for the E3 ligase parkin (Olzmann et al., 2004). As shown in Fig. 1, immunoprecipitation of hemagglutinin (HA)-tagged L166P mutant DJ-1, but not wild-type DJ-1, was able to coprecipitate Myc-tagged parkin, providing evidence for an interaction of these two proteins in vivo (Fig. 1 A). Given the reported interaction of Parkin and L166P mutant DJ-1 with Hsp70 (Imai et al., 2002; Li et al., 2005; Moore et al., 2005), we considered the possibility that these three proteins might be present in a complex. However, coimmunoprecipitation experiments indicated that, even with high levels of Hsp70 overexpression, Hsp70 did not associate with L166P mutant DJ-1 under the experimental conditions used (Fig. S1, available at http://www.jcb.org/cgi/content/full/jcb.200611128/DC1). Furthermore, we found that GST-parkin bound Myc-tagged L166P mutant DJ-1 expressed in SH-SY5Y cells, but not Myc-tagged wild-type DJ-1 or endogenous DJ-1 (Fig. S2, available at http://www.jcb.org/cgi/content/full/jcb.200611128/DC1). In addition, GST-parkin was able to efficiently pull down in vitro translated [35S]-labeled L166P mutant DJ-1, but not wild-type DJ-1 (Fig. 1 B). Together, our data indicate that parkin directly binds L166P mutant DJ-1, but not wild-type DJ-1, and that this interaction does not require Hsp70 as a recruiting factor.
Figure 1.

Selective interaction and ubiquitination of L166P mutant DJ-1 by parkin. (A) Lysates from transfected SH-SY5Y cells were subjected to immunoprecipitation with anti-HA antibody followed by Western blotting. endo., endogenous; IB, immunoblot; IP, immunoprecipitation. (B) [35S]-labeled DJ-1 proteins were incubated with immobilized GST or GST-parkin fusion proteins. Bound DJ-1 proteins were detected by autoradiography. (C) Transfected SH-SY5Y cells were incubated in the absence or presence of 20 μM proteasome inhibitor MG132 for 8 h before harvest. Lysates were subjected to immunoprecipitation with anti-Myc antibody followed by Western blotting. The expression of parkin in the cell lysates is confirmed by immunoblotting with anti-parkin antibody. Ubn, polyubiquitin. (D) Lysates from MG132 (20 μM)- or vehicle (0.1% DMSO)-treated SH-SY5Y cells transfected with the indicated plasmids were analyzed by Western blotting. The relative level of wild-type or mutant DJ-1 was measured by quantification of the intensity of the HA-tagged DJ-1 or L166P band and normalized to the actin level in the corresponding cell lysate. The bar graph shows the results (mean ± SEM) from at least three independent experiments. (E) The half-life of Myc-tagged wild-type and L166P mutant DJ-1 expressed in SH-SY5Y cells was analyzed by pulse chase and the protein levels quantified and plotted relative to the corresponding DJ-1 levels at 0 h.
Next, we performed in vivo ubiquitination assays to examine whether the interaction with parkin targets misfolded DJ-1 for parkin-mediated ubiquitination. In agreement with previous reports (Baulac et al., 2004; Olzmann et al., 2004), we found that L166P mutant DJ-1, but not wild-type DJ-1, was selectively polyubiquitinated in cells (Fig. 1 C). The polyubiquitination of L166P mutant DJ-1 was significantly increased by coexpression of parkin in the presence of the proteasome inhibitor MG132 (Fig. 1 C). Under the same conditions, parkin did not promote the ubiquitination of wild-type DJ-1 (Fig. 1 C). Together, these results demonstrate that parkin selectively recognizes and ubiquitinates misfolded DJ-1 in vivo.
Parkin-mediated polyubiquitination has no apparent effect on the degradation of misfolded DJ-1
To determine whether parkin-mediated ubiquitination targets L166P mutant DJ-1 for degradation by the proteasome, we assessed the effect of parkin overexpression on the steady-state levels of wild-type and L166P mutant DJ-1 proteins (Fig. 1 D). As we and others reported previously (Miller et al., 2003; Moore et al., 2003; Gorner et al., 2004; Olzmann et al., 2004), the steady-state level of L166P mutant DJ-1 was substantially lower compared with wild-type DJ-1. Inhibition of proteasome function by MG132 selectively increased the steady-state level of the L166P mutant, but not wild-type DJ-1 (Fig. 1 D). Contrary to our expectation, we found that overexpression of parkin did not alter the steady-state levels of L166P mutant or wild-type DJ-1 (Fig. 1 D). Given the recent report that parkin promotes degradation of synphilin-1 only when the expression of parkin reaches very high levels (Lim et al., 2005a), we examined the effects of increased levels of parkin expression on the degradation of L166P mutant DJ-1 (Fig. S3, available at http://www.jcb.org/cgi/content/full/jcb.200611128/DC1). Even at extremely high levels of overexpression, parkin was unable to promote degradation of L166P mutant DJ-1 (Fig. S3). Consistent with our previous findings (Olzmann et al., 2004), pulse chase experiments indicated that L166P mutant DJ-1 was highly unstable compared with the wild-type DJ-1 (Fig. 1 E). Co-expression of parkin had no effect on the stability of L166P mutant DJ-1 (Fig. 1 E). Together these results suggest that parkin-mediated polyubiquitination of mutant DJ-1 may have a nondegradative role.
Parkin associates with the heterodimeric E2 enzyme UbcH13/Uev1a to mediate K63-linked polyubiquitination of misfolded DJ-1
One possibility that may account for the inability of parkin to promote the degradation of L166P mutant DJ-1 is that parkin may mediate an alternative form of polyubiquitination that is not associated with proteasomal degradation, such as K63-linked polyubiquitination. The best-characterized E2 enzyme in mediating K63-linked polyubiquitination is the UbcH13/Uev1a heterodimer, in which UbcH13 is the catalytic subunit (Deng et al., 2000; Wang et al., 2001). A recent in vitro study has shown that the GST-fused second RING domain of parkin can bind UbcH13 and this interaction recruits the UbcH13/Uev1a heterodimer to parkin (Doss-Pepe et al., 2005). However, it is unknown whether full-length parkin is able to bind UbcH13 in vivo. As shown (Fig. 2 A), UbcH13 readily coprecipitated with parkin, providing evidence for an interaction between these two proteins in vivo. In addition, we observed the interaction of parkin with UbcH7 and UbcH8, but not with UbcH5, consistent with previous studies (Shimura et al., 2000; Zhang et al., 2000). These data indicate that parkin binds multiple E2 enzymes in vivo and suggest that the ability of parkin to facilitate K48- or K63-linked polyubiquitination may be dependent on the selective recruitment of distinct E2 enzymes.
Figure 2.

Parkin mediates K63-linked polyubiquitination of L166P mutant DJ-1 in cooperation with the UbcH13/Uev1a E2 enzyme. (A) Lysates from transfected SH-SY5Y cells were subjected to immunoprecipitation with anti-Myc antibody followed by Western blotting. (B and C) Purified L166P mutant DJ-1 was subjected to in vitro ubiquitination in the presence of E1, E2 (UbcH13/Uev1a), GST-tagged parkin, and wild-type or mutant ubiquitin as indicated. Ubiquitinated mutant DJ-1 was detected by immunoblotting with anti-DJ-1 antibody. All experiments were replicated three times with similar results.
We next performed in vitro ubiquitination assays to determine if the UbcH13/Uev1a heterodimer is the cognate E2 enzyme for parkin-mediated ubiquitination of L166P mutant DJ-1. We found that parkin ubiquitinated L166P mutant DJ-1 in an UbcH13/Uev1a-dependent manner (Fig. 2 B). To investigate the linkage of parkin-mediated polyubiquitination of L166P mutant DJ-1, we used ubiquitin mutants Ub-K29, Ub-K48, and Ub-K63, which contain arginine substitutions of all of its lysine residues except the one at position 29, 48, and 63, respectively. The Ub-K29, Ub-K48, and Ub-K63 mutants are thus expected to only allow the formation of K29-, K48-, and K63-linked polyubiquitin chains, respectively. In vitro ubiquitination analyses using these ubiquitin mutants revealed that parkin was able to facilitate the polyubiquitination of L166P mutant DJ-1 in the presence of wild-type ubiquitin (Ub-WT) and Ub-K63 (Fig. 2 C). In contrast, in vitro ubiquitination reactions using Ub-K29 or Ub-K48 resulted in the accumulation of monoubiquitinated L166P mutant DJ-1 (Fig. 2 C). In addition, low levels of higher molecular weight bands were also observed at sizes corresponding to L166P mutant DJ-1 conjugated with three or four ubiquitin molecules (Fig. 2 C), and these bands could potentially represent monoubiquitination of L166P mutant DJ-1 at multiple lysine residues. Together these results demonstrate that parkin-mediated polyubiquitination of L166P mutant DJ-1 occurs in vitro via K63-linked ubiquitin chains.
We then used Ub-K48 and Ub-K63 mutants in in vivo ubiquitination assays to determine the linkage of parkin-mediated polyubiquitination of L166P mutant DJ-1 in cells (Fig. 3 A). In agreement with the results of the in vitro ubiquitination analysis (Fig. 2 C), parkin promoted robust polyubiquitination of L166P mutant DJ-1 in cells expressing Ub-WT or Ub-K63, but not in cells expressing Ub-K48 (Fig. 3 A). To provide further support for the identified K63 linkage of parkin-mediated polyubiquitination of L166P mutant DJ-1, we used another set of ubiquitin mutants, Ub-K29R, Ub-K48R, Ub-K63R, which contain a single lysine-to-arginine mutation at position 29, 48, and 63, respectively. The Ub-K29R, Ub-K48R, and Ub-K63R mutants are expected to solely disrupt the assembly of K29-, K48-, and K63-linked polyubiquitin chains, respectively. As a control, we also included a polymerization-defective mutant of ubiquitin (Ub-K0), in which all lysine residues of ubiquitin were changed to arginines, and therefore is unable to form polyubiquitin chains. We found that parkin-mediated polyubiquitination of L166P mutant DJ-1 was greatly reduced by replacement of Ub-WT with Ub-K63R, but not by the replacement with Ub-K29R (Fig. 3 B). As expected, parkin-mediated polyubiquitination of L166P mutant DJ-1 was virtually abolished by replacement of Ub-WT with Ub-K0 (Fig. 3, A and B). The low levels of L166P mutant DJ-1 ubiquitination observed with Ub-K63R could be due to non-K63-linked ubiquitination of mutant DJ-1 by another E3 ubiquitin-protein ligase(s) in SH-SY5Y cells. Consistent with this possibility, we found that the polyubiquitination of L166P mutant DJ-1 was reduced by replacement of Ub-WT with Ub-K48R mutant (Fig. 3 B). Furthermore, we and others have previously shown that L166P mutant DJ-1 associates with the E3 ligase CHIP (Moore et al., 2005; Olzmann et al., 2005). Collectively, our in vitro and in vivo data provide strong evidence that parkin facilitates K63-linked polyubiquitination of L166P mutant DJ-1 in cooperation with UbcH13/Uev1a.
Figure 3.

Parkin promotes K63-linked polyubiquitination of L166P mutant DJ-1 in vivo. (A and B) Transfected SH-SY5Y cells were treated with 20 μM MG132 for 8 h before harvest. Lysates (input) were subjected to immunoprecipitation with anti-Myc antibody followed by Western blotting. IgG HC, immunoglobulin heavy chain; IgG LC, immunoglobulin light chain.
L166P mutant DJ-1 and parkin colocalize in aggresomes induced by proteasome impairment
Our findings that parkin specifically binds and ubiquitinates L166P mutant DJ-1 prompted us to investigate the potential colocalization of these two proteins in SH-SY5Y cells by immunofluorescence confocal microscopy. HA-tagged L166P mutant DJ-1 exhibited a cytoplasmic staining pattern that displayed substantial overlap with that of Myc-tagged parkin (Fig. 4 A, top). In addition, treatment with the proteasome inhibitor MG132 caused a dramatic relocation of L166P mutant DJ-1 and parkin to a single prominent, perinuclear inclusion (Fig. 4 A, bottom).
Figure 4.

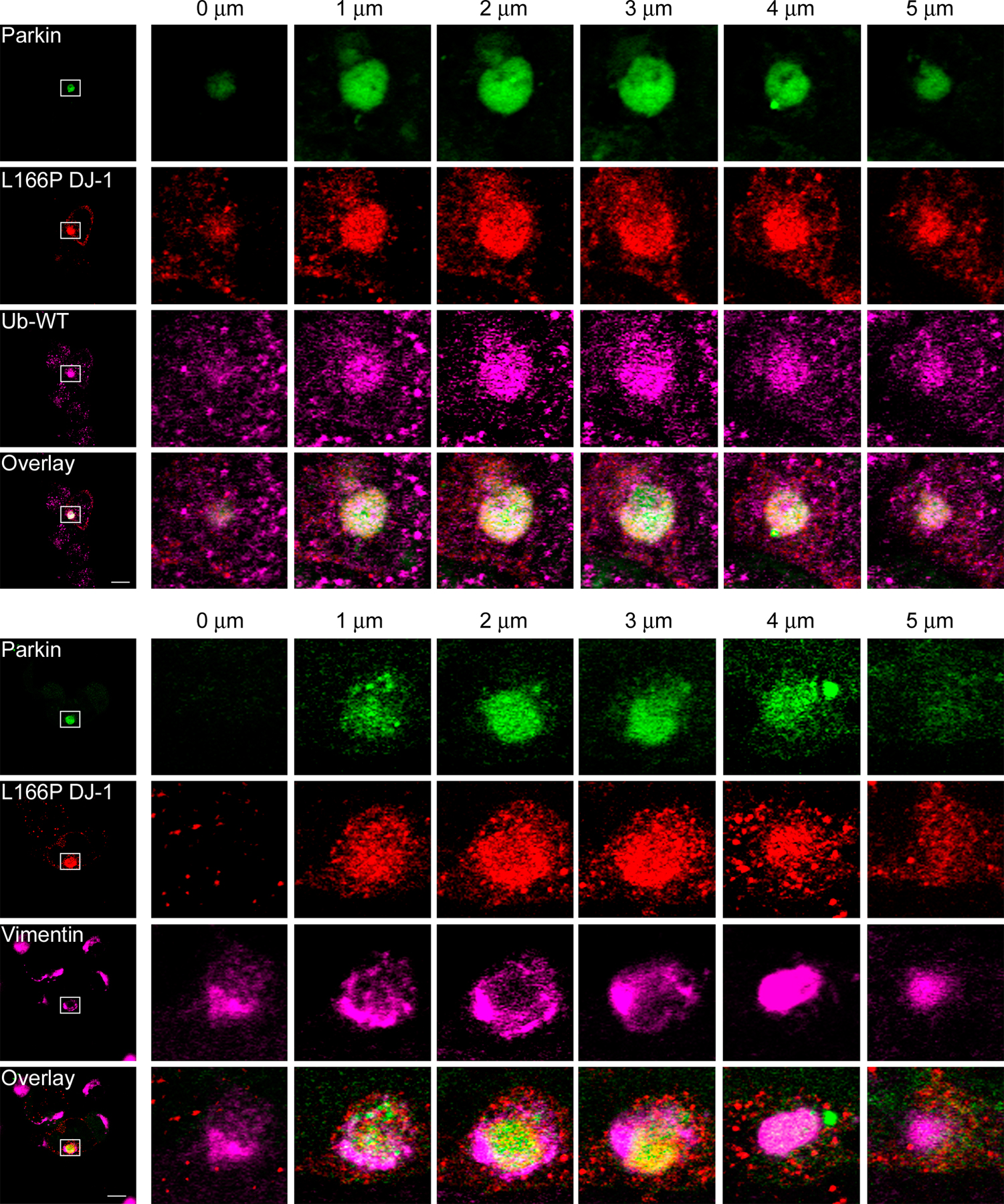
Colocalization of L166P mutant DJ-1 and parkin in perinuclear aggresomes. (A) SH-SY5Y cells coexpressing Myc-tagged parkin and HA-tagged L166P mutant DJ-1 were incubated in DME containing 0.1% DMSO, 20 μM MG132, 20 μM MG132 plus 5 μg/ml nocodazole for 16 h as indicated. In cells treated with MG132 a prominent L166P mutant DJ-1–containing aggresome is evident (arrowhead). However, cotreatment with nocodazole results in the accumulation of L166P mutant DJ-1–containing micro-aggregates (open arrowheads). (B) For further analysis of inclusions, SH-SY5Y cells coexpressing GFP-tagged parkin and Myc-tagged L166P were incubated in the presence of 20 μM MG132 for 16 h and subsequently processed for immunocytochemistry with the indicated antibodies. The colocalization between parkin and L166P mutant DJ-1 is indicated by the yellow color. The colocalization of parkin, L166P mutant DJ-1, and ubiquitin or hsp70 is indicated by the white color. Bar = 10 μm.
The morphology and localization of the L166P mutant DJ-1–containing inclusions appeared similar to that of aggresomes (Johnston et al., 1998; Kopito, 2000; Garcia-Mata et al., 2002). To test whether these inclusions were aggresomes, we performed additional immunofluorescence confocal microscopy experiments to further characterize these inclusions (Fig. 4 B). We found that the L166P mutant DJ-1-containing inclusions were highly enriched with ubiquitin and the chaperone protein Hsp70, and surrounded by a compacted cage of the intermediate filament protein vimentin. Lamp2-immunoreactive lysosomes did not colocalize with the inclusions, but were observed in close apposition and often tightly ringed the inclusions. This finding is consistent with recent studies indicating that aggresomes may represent substrates for autophagic degradation, a process involving fusion with lysosomes (Taylor et al., 2003). Moreover, serial 1-μm Z-sections revealed that the misfolded DJ-1 protein was present throughout the center of the aggresome, extensively colocalized with ubiquitin, and completely surrounded by vimentin (Fig. S4, available at http://www.jcb.org/cgi/content/full/jcb.200611128/DC1).
Given the reported role of microtubule-dependent retrograde transport in aggresome formation (Kopito, 2000), we examined the effects of the microtubule-depolymerizing drug nocodazole on parkin-induced recruitment of L166P mutant DJ-1 to aggresomes (Fig. 4 A). Depolymerization of microtubules by nocodazole in MG132-treated cells disrupted delivery of L166P mutant DJ-1 to the aggresome, leading to accumulation of misfolded DJ-1 in widely dispersed preaggresome particles (Fig. 4 A). Parkin colocalized with L166P mutant DJ-1 in these preaggresome particles (Fig. 4 A), demonstrating an association of these two proteins before their transport to the aggresome. Together, these data indicate that recruitment of L166P mutant DJ-1 to the aggresome is dependent on intact microtubules and that the inclusions containing L166P mutant DJ-1 are bona fide aggresomes.
Accumulating evidence indicates that aggresomes found in cultured cells are substrates for eventual autophagic degradation. To determine if L166P mutant DJ-1-containing aggresomes are sites of autophagy we used the widely used marker of autophagic vacuoles monodansyl cadaverine (MDC) (Biederbick et al., 1995; Jackson et al., 2005) (Fig. S5, available at http://www.jcb.org/cgi/content/full/jcb.200611128/DC1). The specificity of MDC staining is further confirmed by our results, which show that MDC staining does not overlap with lamp2 (Fig. S5), a protein present on late endosomes and lysosomes. Our findings indicate that under control conditions MDC displays a widely distributed, punctate distribution with relatively low fluorescence levels (Fig. S5). However, MG132-induced proteasomal impairment results in an increase in MDC fluorescence levels and a redistribution of MDC immunoreactivity to a perinuclear region that colocalizes with the L166P mutant DJ-1 aggresomes (Fig. S5). Together these findings indicate that L166P mutant DJ-1 aggresomes may be sites of autophagy.
Parkin selectively promotes the recruitment of L166P mutant DJ-1, but not wild-type DJ-1, to aggresomes
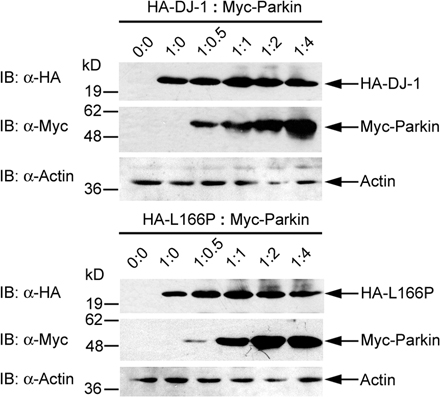
To determine whether parkin selectively targets L166P mutant DJ-1 to aggresomes, we assessed the effects of parkin overexpression on the accumulation of HA-tagged wild-type and L166P mutant DJ-1 in aggresomes formed in response to proteasome inhibition by MG132 (Fig. 5). In untreated cells the coexpression of parkin had no effect on the cytoplasmic distribution of wild-type or L166P mutant DJ-1 (Fig. 5, A and B). Treatment of the cells with MG132 did not alter the distribution of wild-type DJ-1, but caused the accumulation of L166P mutant DJ-1 in aggresomes in a small percentage of cells (Fig. 5, C and E). Interestingly, coexpression of parkin resulted in a dramatic increase in the percentage of cells containing L166P mutant DJ-1–positive aggresomes, but had no effect on the localization of wild-type DJ-1 (Fig. 5, D and E).
Figure 5.

Parkin promotes the accumulation of L166P mutant DJ-1 in aggresomes. (A–D) SH-SY5Y cells coexpressing HA-tagged wild-type DJ-1 or L166P mutant DJ-1 and Myc vector or Myc-tagged parkin were incubated in the presence and absence of 20 μM MG132 for 16 h and processed for immunofluorescence with anti-HA (red) and anti-Myc (green) antibodies. (A and B) In untreated cells HA-tagged wild-type DJ-1 and L166P mutant DJ-1 are distributed throughout the cytoplasm and colocalize with Myc-tagged parkin. (C and D) Inhibition of the proteasome coupled with overexpression of parkin resulted in the accumulation of L166P mutant DJ-1 in aggresomes (arrowheads). In cells that were not transfected with parkin L166P mutant DJ-1 mostly retained a diffuse distribution (open arrowheads). In contrast, the distribution of wild-type DJ-1 was unaffected, although these cells displayed robust parkin-containing aggresomes (arrowheads). Superimposed images revealed a colocalization (yellow) between L166P mutant DJ-1 and parkin in the perinuclear region in the presence of MG132. Bar = 10 μm. (E) Quantification shows that parkin selectively increases the targeting of L166P mutant DJ-1 to aggresomes. The asterisk indicates statistical significance (P < 0.05).
Aggresomes contain misfolded and aggregated proteins, and are often insoluble in detergents (Johnston et al., 2002). To determine if recruitment of L166P mutant DJ-1 to these aggresomes alters its solubility, cell lysates were separated into detergent soluble and insoluble fractions, and analyzed by Western blotting. Wild-type and L166P mutant DJ-1 were predominantly found in the soluble fraction in untreated cells (Fig. 6, A and B). However, MG132 treatment resulted in the selective accumulation of L166P mutant DJ-1 in the detergent insoluble fraction, and this accumulation was increased by coexpression of parkin (Fig. 6, A and B). These results demonstrate that parkin selectively promotes the redistribution of misfolded DJ-1 into a detergent-insoluble aggresome.
Figure 6.

Parkin increases the insolubility of L166P mutant, but not wild-type DJ-1. (A), Lysates from transfected SH-SY5Y cells were separated into detergent-soluble (S) and detergent-insoluble (I) fractions and analyzed by Western blotting. (B) The relative level of insoluble DJ-1 was measured by quantification of the intensity of the HA-tagged DJ-1 or L166P band in the detergent-insoluble fraction and normalized to the total level of HA-tagged wild-type or mutant DJ-1 in the corresponding cell lysate. The asterisk indicates a statistically significant (P < 0.05) difference compared with the corresponding control.
Parkin-mediated K63-linked polyubiquitination facilitates the formation of aggresomes
To investigate the role of parkin-mediated K63-linked polyubiquitination in aggresome formation, we analyzed and compared the formation of MG132-induced aggresomes in SH-SY5Y cells coexpressing L166P mutant DJ-1 and Myc-tagged parkin in the presence of either HA-tagged Ub-WT, Ub-K48, Ub-K63, Ub-K48R, or Ub-K63R. We observed the presence of a clearly visible aggresome containing immunoreactivity to HA-tagged ubiquitin and L166P mutant DJ-1 in most of the cells expressing Ub-WT (46%) after 16-h MG132 treatment (Fig. 7, A and B). Replacement of Ub-WT with Ub-K63R dramatically reduced aggresome formation and resulted in the accumulation of cytoplasmic preaggresomal particles, whereas replacement with Ub-K48R had no apparent effect (Fig. 7, A and B). Moreover, expression of Ub-K63 significantly enhanced aggresome formation compared with cells expressing Ub-K48, which were more likely to contain cytoplasmic preaggresomal particles (Fig. 7, A and B). Together, our results suggest that K63-linked polyubiquitination of misfolded DJ-1 by parkin promotes aggresome formation.
Figure 7.

Parkin-mediated K63 polyubiquitination promotes the formation of L166P mutant DJ-1 aggresomes. (A) SH-SY5Y cells coexpressing GFP-tagged parkin, Myc-tagged L166P mutant DJ-1, and the indicated HA-tagged ubiquitin mutants were incubated in the presence of 20 μM MG132 for 16 h, fixed, and immunostained with antibodies against Myc (green) and HA (purple). Bar = 10 μm. (B) The percentage of cells containing L166P mutant DJ-1-positive aggresomes was quantified from 50–100 transfected cells and indicates that K63-linked polyubiquitination is important for the targeting of the misfolded protein to aggresomes. The asterisk indicates a statistically significant decrease (P < 0.05) in the percentage of cells containing aggresomes in cells expressing HA-tagged Ub-K48 or Ub-K63R versus the cells expressing HA-tagged Ub-WT, Ub-K63, or Ub-K48R.
Parkin-mediated K63-linked polyubiquitination of misfolded DJ-1 serves as a signal for binding HDAC6
The observed microtubule-dependent recruitment of L166P mutant DJ-1 to the aggresome (Fig. 4) suggests that the retrograde dynein motor complex may be involved in the transport of misfolded DJ-1. Recent evidence indicates that histone deacetylase 6 (HDAC6) is critically involved in aggresome formation by acting as an adaptor between polyubiquitinated proteins and the dynein motor complex (Kawaguchi et al., 2003; Hideshima et al., 2005). We performed coimmunoprecipitation analyses to examine a potential interaction between Myc-tagged L166P mutant DJ-1 and endogenous HDAC6. We found that HDAC6 does not bind nonubiquitinated L166P mutant DJ-1 in untreated cells (Fig. 8 A). In contrast, HDAC6 strongly interacts with polyubiquitinated L166P mutant DJ-1 induced by parkin coexpression in MG132-treated cells (Fig. 8 A). Moreover, immunofluorescence confocal microscopic studies revealed that, in response to proteasome inhibition, both HDAC6 and dynein are recruited with L166P mutant DJ-1 to aggresomes (Fig. 8 B).
Figure 8.

Parkin-mediated K63-linked polyubiquitination of L166P mutant DJ-1 promotes an association with HDAC6. (A) Lysates from transfected SH-SY5Y cells incubated in the absence or presence of MG132 were subjected to immunoprecipitation with anti-Myc antibodies followed by Western blotting. (B) SH-SY5Y cells coexpressing GFP-tagged parkin and HA-tagged L166P mutant DJ-1 were incubated in the presence and absence of MG132, immunostained as indicated, and imaged by immunofluorescence confocal microscopy. Bar = 10 μm. (C) Lysates from transfected SH-SY5Y cells were subjected to immunoprecipitation with anti-HA antibodies and analyzed by Western blotting.
Although HDAC6 has been shown to bind polyubiquitinated proteins (Hook et al., 2002; Kawaguchi et al., 2003), it remains unknown whether HDAC6 specifically binds K48-linked and/or K63-linked polyubiquitin chains. Coimmunoprecipitation studies in SH-SY5Y cells expressing HA-tagged Ub-K48 or Ub-K63 indicate that HDAC6 interacts with both K48- and K63-linked polyubiquitin chains, but binds more strongly to K63-linked chains (Fig. 8 C), providing evidence that HDAC6 preferentially interacts with K63-linked polyubiquitinated proteins in vivo. Together, these data suggest that parkin-mediated K63-linked polyubiquitination of L166P mutant DJ-1 serves as a signal for binding HDAC6, and thus couples the misfolded DJ-1 to the dynein motor complex for transport to the aggresome.
Parkin is required for delivery of misfolded DJ-1 to aggresomes
Given our findings that parkin-mediated K63-linked polyubiquitination mediates the recognition of misfolded DJ-1 by HDAC6 and the dynein motor complex (Figs. 5, 7, and 8), we next assessed the role of parkin in the transport of misfolded proteins to the aggresome using mouse embryonic fibroblasts (MEFs) cultured from Parkin knockout (Parkin −/−) mice (Perez et al., 2005; Perez and Palmiter, 2005). Western blot analysis with anti-parkin antibodies confirmed the absence of parkin protein in Parkin −/− MEFs (Fig. 9 A). In addition, pulse chase analysis indicated that the absence of parkin had no effect on the stability of L166P mutant DJ-1 (Fig. 9 B). Immunofluorescence confocal microscopy showed that L166P mutant DJ-1 exhibited a diffuse cytoplasmic distribution in both untreated Parkin +/+ and Parkin −/− MEFs (unpublished data). In Parkin +/+ MEFs treated with MG132, L166P mutant DJ-1 accumulated in aggresomes (Fig. 9, C and D). In contrast, in Parkin −/− MEFs, Myc-tagged L166P mutant DJ-1 was mostly observed in small preaggresomal particles dispersed throughout the cytoplasm (Fig. 9, B and C). Furthermore, coexpression of untagged-parkin rescued the ability of Parkin −/− MEFs to properly target the misfolded DJ-1 to aggresomes (Fig. 9, C and D). Together these results indicate that parkin is critically involved in the transport of misfolded DJ-1 to aggresomes.
Figure 9.

Parkin is required for proper transport of misfolded proteins to aggresomes. (A) Lysates from MEFs cultured from Parkin −/− and Parkin +/+ were separated by SDS-PAGE and analyzed by Western blotting. (B) The half-life of Myc-tagged L166P mutant DJ-1 expressed in Parkin −/− and Parkin +/+ MEFs was analyzed by pulse chase and the protein levels quantified and plotted relative to the corresponding levels at 0 h. (C) Parkin −/− and Parkin +/+ MEFs were transfected with Myc-tagged L166P mutant DJ-1 and untagged-parkin as indicated, treated with 20 μM MG132 for 16 h, and processed for immunocytochemistry with anti-Myc (green) antibodies. Bar = 10 μm. (D) The percentage of cells containing L166P mutant DJ-1 aggresomes was quantified from 50–100 transfected cells. The asterisk indicates a statistically significant difference (P < 0.05).
Discussion
Although it is clear that cells sequester misfolded proteins into centrosomal aggresomes, how the cell recognizes these proteins for transport to the aggresome is poorly understood. Our findings support the model depicted in Fig. 10, and suggest that misfolded proteins are normally polyubiquitinated and efficiently degraded by the 26S proteasome. However, under pathogenic conditions in which proteasome function is impaired, parkin cooperates with UbcH13/Uev1a to mediate K63-linked polyubiquitination of misfolded proteins, promotes binding of HDAC6 and couples the misfolded protein to the dynein motor complex for transport to aggresomes.
Figure 10.

Model of parkin-dependent targeting of misfolded proteins to the aggresome. Under normal conditions misfolded proteins are recognized and conjugated with K48-linked polyubiquitin chains, resulting in efficient degradation by the 26S proteasome. However, under conditions of proteasomal impairment, parkin cooperates with Ubc13/Uev1a to mediate K63-linked polyubiquitination of misfolded proteins. This K63-linked polyubiquitin chain promotes binding of the dynein adaptor protein HDAC6, which effectively “loads” the misfolded protein onto the dynein motor complex for retrograde transport along microtubules to aggresomes.
Our results demonstrate that parkin directly binds the misfolded L166P mutant, but not wild-type DJ-1. We find that parkin selectively promotes K63-linked polyubiquitination of L166P mutant DJ-1 in an UbcH13/Uev1a-dependent manner. K63-linked polyubiquitination has been proposed to play a role in the formation of inclusion bodies (Lim et al., 2005b), but the underlying molecular mechanisms remain unknown. Our data show that parkin promotes the accumulation of misfolded DJ-1 into aggresomes. Furthermore, the ability of parkin to promote the sequestration of misfolded DJ-1 into aggresomes is dependent on K63-linked polyubiquitination, and expression of Ub-K48 or Ub-K63R results in the accumulation of small preaggresomal particles dispersed throughout the cytoplasm. Previous studies suggest that misfolded proteins form small aggregates within the cytoplasm, which are then transported to the forming aggresome (Kopito, 2000; Garcia-Mata et al., 2002). Disruption of transport by incubation with nocodazole (Johnston et al., 1998), overexpression of dynamitin (Garcia-Mata et al., 1999; Johnston et al., 2002), siRNA-mediated depletion (Kawaguchi et al., 2003), or chemical inhibition (Wang et al., 2005) of HDAC6 results in the accumulation of small cytoplasmic aggregates. Thus the preaggresomal particles that accumulate upon expression of Ub-K48 or Ub-K63R may represent L166P mutant DJ-1-containing small aggregates. In addition to L166P mutant DJ-1, these preaggresomal particles also contained parkin, which suggests that K63-linked polyubiquitination may be important for the transport of parkin to the aggresome. Hampe et al. reported that parkin catalyzes multi-monoubiquitination of itself (auto-ubiquitination) in vitro and a fraction of parkin is multi-monoubiquitinated in vivo under basal conditions (Hampe et al., 2006). However, under conditions of proteasomal impairment, they found that parkin is mostly polyubiquitinated in vivo (Hampe et al., 2006), which might be mediated by another E3 ligase or by parkin in cooperation with additional factor(s) such as E4 (Imai et al., 2002; Richly et al., 2005). Furthermore, Lim et al. reported that parkin is modified by K63-linked polyubiquitination in vivo, which was suggested to be mediated by parkin itself (Lim et al., 2005a). K63-linked polyubiquitination of parkin under conditions of proteasomal impairment would promote the transport of parkin to aggresomes, and this could explain the reduced formation of parkin-positive aggresomes when K63-linked polyubiquitination was inhibited.
One possibility is that K63-linked polyubiquitination may block K48-linked polyubiquitination, resulting in decreased proteasomal degradation and increased substrate accumulation. However, this does not address how these proteins are recognized and targeted to the aggresome. Our results indicate that K63-linked polyubiquitination plays an active signaling role, and that under conditions of proteasomal impairment facilitates an interaction between L166P mutant DJ-1 and the dynein adaptor protein HDAC6. HDAC6 links polyubiquitinated proteins, to the molecular motor dynein by simultaneously binding ubiquitin via a zinc-finger ubiquitin binding domain and dynein via a distinct dynein motor binding domain (Kawaguchi et al., 2003). In addition, HDAC6 has been found to localize to Lewy bodies in sporadic PD patients, suggesting that HDAC6 may also play a similar role in the biogenesis of these hallmark PD inclusions. Our results further demonstrate that HDAC6 preferentially binds K63-linked polyubiquitinated proteins in vivo, suggesting that K63-linked polyubiquitination may act as a novel signal for the dynein-mediated transport and sequestration of misfolded proteins in the aggresome.
Based on the localization of parkin in aggresomes/inclusions, it has been proposed that parkin is first recruited to aggresomes in response to proteasome inhibition and then ubiquitinates misfolded proteins in these inclusion bodies (Lim et al., 2005a). In this case, one would expect that targeting of misfolded proteins to aggresomes in Parkin −/− MEFs would be unaffected, but that there would be a decrease in the ubiquitination of the inclusions. However, we observed that the percentage of cells containing L166P mutant DJ-1 aggresomes was dramatically reduced in Parkin −/− MEFs. These findings support a role for parkin in the transport of misfolded DJ-1 to aggresomes rather than in the ubiquitination of the misfolded proteins in aggresomes. In addition, when transport of misfolded proteins to the aggresome was disrupted with nocodazole we observed that parkin colocalized with misfolded DJ-1 in ubiquitin-positive, preaggresomal particles, suggesting that parkin associates with and polyubiquitinates misfolded proteins before their transport to aggresomes.
Whether protein inclusions are cytoprotective or cytotoxic is highly controversial. Studies indicate that several intermediates are generated during the process of protein aggregation, and suggest that the small-intermediate oligomers may be the principle toxic species (Caughey and Lansbury, 2003). Accumulation of aggregated proteins can cause global impairment of the ubiquitin-proteasome system (Bence et al., 2001; Bennett et al., 2005) and aggregated proteins also directly interfere with proteasomal function, possibly due to an inability to properly enter or exit the proteasome (Holmberg et al., 2004; Venkatraman et al., 2004; Kristiansen et al., 2007). Sequestration into inclusions like aggresomes may render these oligomers inert and protect an impaired proteasomal system from further damage. Interestingly, recent evidence suggests that aggresomes might also act as a staging area for disposal by autophagy (Taylor et al., 2003). In support of this possibility, we find that L166P mutant DJ-1 aggresomes stained with MDC, a marker of autophagic vacuoles. Consistent with the findings of Iwata et al. (Iwata et al., 2005), we observed that MG132-mediated inhibition of the proteasome resulted in a redistribution of lysosomes, and often lysosomes tightly encircled the aggresome. A protective role for aggresomes has been further supported by the finding that disruption of aggresome formation using small molecule inhibitors of HDAC6 (Hideshima et al., 2005) or by knockdown of HDAC6 protein levels by RNA interference (Kawaguchi et al., 2003) increases the susceptibility to apoptosis induced by proteasome inhibition or misfolded protein stress.
It is widely believed that loss of parkin function would lead to decreased proteasomal degradation of a cytotoxic substrate protein, resulting in selective dopaminergic neurodegeneration (Kahle and Haass, 2004). Studies have found that Parkin promotes the proteasomal degradation of several putative substrate proteins (Kahle and Haass, 2004). However, the accumulation of these substrates in parkin knockout mouse brain and human PD brains has been conflicting (Goldberg et al., 2003; Ko et al., 2005, 2006; Periquet et al., 2005). Thus the role these substrates play in the pathogenesis of PD remains to be determined. Recent reports indicate that parkin is able to mediate monoubiquitination (Hampe et al., 2006; Matsuda et al., 2006) and K63-linked polyubiquitination (Doss-Pepe et al., 2005; Lim et al., 2005a; Fig. 2 and 3), two forms of ubiquitination that are not associated with proteasomal degradation. In addition, parkin has been implicated in nonproteasomal processes, including endocytosis and signal transduction (Fallon et al., 2006). Our findings further indicate that parkin plays a regulatory role in trafficking of misfolded DJ-1 to aggresomes. Our data reveal a critical role for parkin-mediated K63 polyubiquitination in earmarking misfolded proteins for dynein-mediated transport to the aggresome and suggest that loss of parkin function may impair sequestration of toxic misfolded proteins, thereby resulting in an increased susceptibility to aggregated protein-induced cellular dysfunction.
Materials and methods
Plasmids and antibodies
The expression vectors encoding HA- and Myc-tagged wild-type and L166P mutant DJ-1 were described previously (Olzmann et al., 2004). Myc-tagged parkin and HA-tagged UbcH13 were provided by T. Suzuki (Tokyo Metropolitan Institute of Medical Science, Tokyo, Japan) and Z. Chen (University of Texas Southwestern, Dallas, TX), respectively. HA-tagged Ub-WT, Ub-K48, Ub-K63, and Ub-K0 were provided by T. Dawson (Johns Hopkins University, Baltimore, MD); and Ub-K29R, Ub-K48R, and Ub-K63R were provided by M. Wooten (Auburn University, Auburn, AL). Anti–DJ-1 antibodies P7F and P7C were described previously (Olzmann et al., 2004, 2007). Other antibodies used in this study include the following: anti-HA (12CA5), anti-Myc (9E10.3, Neomarkers), anti-parkin (Cell Signaling), anti-Hsp70 (Stressgen), anti-ubiquitin (P4G7, Covance), anti-vimentin (Sigma-Aldrich), anti–β-tubulin (Boehringer Mannheim), anti-HDAC6 (Santa Cruz Biotechnology, Inc.), anti-lamp2 (Iowa Developmental Studies Hybridoma Bank), and anti-dynein (Sigma-Aldrich); all secondary antibodies were purchased from Jackson ImmunoResearch Laboratories, Inc.
Cell transfections and immunoprecipitation
SH-SY5Y cells and mouse embryonic fibroblasts (MEFs) were transfected with the indicated plasmids using Lipofectamine 2000 (Invitrogen) or TransIt-Neural (Mirus), respectively, according to the manufacturer's instructions. Cell lysates were prepared from transfected cells and immunoprecipitations performed as described previously (Olzmann et al., 2004).
Recombinant protein purification and in vitro binding assays
GST-tagged parkin fusion proteins were expressed in Escherichia coli BL21 cells and affinity purified as described previously (Chin et al., 2002; Kim et al., 2007). Immobilized GST or GST-Parkin (∼5 μg) were incubated with [35S]Methionine-labeled Myc-tagged DJ-1 or L166P mutant DJ-1, generated using the TNT Quick Coupled Transcription/Translation System (Promega), in phosphate buffered saline for 2 h at 4°C. After extensive washes, bound proteins were separated by SDS-PAGE and visualized by autoradiography.
Analysis of steady-state protein levels
Transfected SH-SY5Y cells were incubated for 8 h at 37°C with the proteasome inhibitor MG132 (20 μM, Calbiochem) or vehicle dimethyl sulfoxide (DMSO, final concentration 0.1%) and protein levels analyzed as described previously (Olzmann et al., 2004).
[35S]Methionine pulse-chase experiments
As described previously (Olzmann et al., 2004), transfected SH-SY5Y cells or MEFs were labeled by incubation with Met/Cys-free DME (Invitrogen) containing 100 μCi of Met/Cys Tran35S-label (MP Biomedicals) for 1 h. Cells were washed and then incubated with DME containing 5× the normal concentrations of methionine and cysteine. At the indicated chase times, equal amount of proteins were immunoprecipitated with anti-Myc antibody, separated by SDS-PAGE, and analyzed by autoradiography.
In vitro and in vivo ubiquitination assays
In vitro ubiquitination assays were performed using a well-established reconstitution system (Shimura et al., 2000, 2001; Pridgeon et al., 2003). In brief, L166P mutant DJ-1 was purified as described previously (Olzmann et al., 2004). 1.5 μg of purified L166P mutant DJ-1 was incubated at 37°C in 60 μl of reaction buffer (50 mM Tris-HCl, pH 7.5, 2.5 mM MgCl2, 2 mM DTT, and 2 mM ATP) containing 200 ng of E1 (Boston Biochem), 400 ng of E2 ubiquitin-conjugating enzyme (UbcH13/Uev1a) (Boston Biochem), 1.5 μg of purified GST-tagged parkin, and 10 μg ubiquitin or ubiquitin mutants (Boston Biochem). After incubation for 2 h the reaction was stopped by addition of loading buffer. Reaction products were analyzed by immunoblotting with anti-DJ-1 antibody to detect ubiquitin-conjugated DJ-1 proteins. In vivo ubiquitination assays were performed as described previously (Wheeler et al., 2002; Olzmann et al., 2004).
Cell fractionation
Transfected SH-SY5Y cells were lysed in a buffer containing 50 mM Tris-HCl pH 7.6, 150 mM NaCl, 1.0% Triton X-100, and a cocktail of protease inhibitors. Lysates were centrifuged at 100,000 g for 30 min at 4°C and separated into detergent-soluble and insoluble fractions as described (Junn et al., 2002). Fractions were analyzed by Western blotting, and protein levels quantified using Scion Image. An ANOVA with a Tukey's posthoc test was used for statistical comparison.
Immunofluorescence confocal microscopy
SH-SY5Y cells or MEFs were processed and stained as previously described (Chin et al., 2000). Hoechst 33258 (Molecular Probes) was used to stain nuclei. MDC (Sigma-Aldrich) staining and costaining was performed as described (Jackson et al., 2005). Analysis and acquisition was performed using a confocal laser-scanning microscope (Axiovert 100 M; Carl Zeiss MicroImaging, Inc.) with a 63×, 1.4 NA, oil-immersion objective (Carl Zeiss MicroImaging, Inc.) at room temperature. Images were exported in TIFF format using LSM-510 software (Carl Zeiss MicroImaging, Inc.), and Adobe Photoshop Version 7.0 software was used to adjust the contrast and brightness.
Parkin knockout mice and generation of mouse embryonic fibroblasts
Parkin knockout mice, containing a targeted deletion of exon 2, were generated on a coisogenic background (129S4/SvJaeSor) and characterized as described previously (Pawlyk et al., 2003; Perez et al., 2005; Perez and Palmiter, 2005). MEF cultures were prepared using well-established methods (Bi et al., 2004; Ma et al., 2004). Early passage MEFs were used for all experiments.
Analysis of aggresome formation
Transfected SH-SY5Y cells or MEFs were incubated in the presence and absence of 20 μM MG132 for 16 h, and then processed for immunofluorescence microscopic analysis of aggresome formation. An aggresome was defined as a single, perinuclear inclusion containing L166P mutant DJ-1. For each experiment, 50–100 transfected cells were randomly selected and scored for the presence of an aggresome in a blinded manner. Experiments were repeated three times, and the data were subjected to statistical analysis by ANOVA with a Tukey's posthoc test.
Online supplemental material
Figure S1 shows how L166P mutant DJ-1 and parkin are not in a complex with Hsp70. Figure S2 shows that Parkin selectively pulls down L166P mutant DJ-1, but not wild-type DJ-1. Figure S3 shows how high levels of parkin overexpression have no effect on the degradation of L166P mutant DJ-1. Figure S4 shoes that the misfolded L166P mutant DJ-1 colocalizes with ubiquitin and is concentrated in the center of the aggresome. Figure S5 shows how L166P mutant DJ-1-containing aggresomes stain with the autophagosomal-marker monodansyl cadaverine. Online supplemental material is available at http://www.jcb.org/cgi/content/full/jcb.200611128/DC1.
Supplementary Material
Acknowledgments
We thank Drs. Zhijian Chen, Toshiaki Suzuki, Ted Dawson, and Marie Wooten for providing plasmids.
This work was supported by National Institutes of Health grants NS054597 (to J.A. Olzmann), NS050650 (to L.-S. Chin), AG021489 (to L. Li), and NS047199 (to L. Li).
Abbreviations used in this paper: HA, hemagglutinin; HDAC6, histone deacetylase 6; MDC, monodansyl cadaverine; MEF, mouse embryonic fibroblast; PD, Parkinson's disease.
References
- Abou-Sleiman, P.M., D.G. Healy, N. Quinn, A.J. Lees, and N.W. Wood. 2003. The role of pathogenic DJ-1 mutations in Parkinson's disease. Ann. Neurol. 54:283–286. [DOI] [PubMed] [Google Scholar]
- Ardley, H.C., G.B. Scott, S.A. Rose, N.G. Tan, A.F. Markham, and P.A. Robinson. 2003. Inhibition of proteasomal activity causes inclusion formation in neuronal and non-neuronal cells overexpressing Parkin. Mol. Biol. Cell. 14:4541–4556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baulac, S., M.J. LaVoie, J. Strahle, M.G. Schlossmacher, and W. Xia. 2004. Dimerization of Parkinson's disease-causing DJ-1 and formation of high molecular weight complexes in human brain. Mol. Cell. Neurosci. 27:236–246. [DOI] [PubMed] [Google Scholar]
- Bence, N.F., R.M. Sampat, and R.R. Kopito. 2001. Impairment of the ubiquitin-proteasome system by protein aggregation. Science. 292:1552–1555. [DOI] [PubMed] [Google Scholar]
- Bennett, E.J., N.F. Bence, R. Jayakumar, and R.R. Kopito. 2005. Global impairment of the ubiquitin-proteasome system by nuclear or cytoplasmic protein aggregates precedes inclusion body formation. Mol. Cell. 17:351–365. [DOI] [PubMed] [Google Scholar]
- Bi, Y., R.D. Palmiter, K.M. Wood, and Q. Ma. 2004. Induction of metallothionein I by phenolic antioxidants requires metal-activated transcription factor 1 (MTF-1) and zinc. Biochem. J. 380:695–703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Biederbick, A., H.F. Kern, and H.P. Elsasser. 1995. Monodansylcadaverine (MDC) is a specific in vivo marker for autophagic vacuoles. Eur. J. Cell Biol. 66:3–14. [PubMed] [Google Scholar]
- Bonifati, V., P. Rizzu, M.J. van Baren, O. Schaap, G.J. Breedveld, E. Krieger, M.C. Dekker, F. Squitieri, P. Ibanez, M. Joosse, et al. 2003. Mutations in the DJ-1 gene associated with autosomal recessive early-onset parkinsonism. Science. 299:256–259. [DOI] [PubMed] [Google Scholar]
- Caughey, B., and P.T. Lansbury. 2003. Protofibrils, pores, fibrils, and neurodegeneration: separating the responsible protein aggregates from the innocent bystanders. Annu. Rev. Neurosci. 26:267–298. [DOI] [PubMed] [Google Scholar]
- Chin, L.S., R.D. Nugent, M.C. Raynor, J.P. Vavalle, and L. Li. 2000. SNIP, a novel SNAP-25-interacting protein implicated in regulated exocytosis. J. Biol. Chem. 275:1191–1200. [DOI] [PubMed] [Google Scholar]
- Chin, L.S., J.P. Vavalle, and L. Li. 2002. Staring, a novel E3 ubiquitin-protein ligase that targets syntaxin 1 for degradation. J. Biol. Chem. 277:35071–35079. [DOI] [PubMed] [Google Scholar]
- Dalle-Donne, I., G. Aldini, M. Carini, R. Colombo, R. Rossi, and A. Milzani. 2006. Protein carbonylation, cellular dysfunction, and disease progression. J. Cell. Mol. Med. 10:389–406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deng, L., C. Wang, E. Spencer, L. Yang, A. Braun, J. You, C. Slaughter, C. Pickart, and Z.J. Chen. 2000. Activation of the IkappaB kinase complex by TRAF6 requires a dimeric ubiquitin-conjugating enzyme complex and a unique polyubiquitin chain. Cell. 103:351–361. [DOI] [PubMed] [Google Scholar]
- Doss-Pepe, E.W., L. Chen, and K. Madura. 2005. Alpha-synuclein and parkin contribute to the assembly of ubiquitin lysine 63-linked multiubiquitin chains. J. Biol. Chem. 280:16619–16624. [DOI] [PubMed] [Google Scholar]
- Fallon, L., C.M. Belanger, A.T. Corera, M. Kontogiannea, E. Regan-Klapisz, F. Moreau, J. Voortman, M. Haber, G. Rouleau, T. Thorarinsdottir, et al. 2006. A regulated interaction with the UIM protein Eps15 implicates parkin in EGF receptor trafficking and PI(3)K-Akt signalling. Nat. Cell Biol. 8:834–842. [DOI] [PubMed] [Google Scholar]
- Garcia-Mata, R., Z. Bebok, E.J. Sorscher, and E.S. Sztul. 1999. Characterization and dynamics of aggresome formation by a cytosolic GFP-chimera. J. Cell Biol. 146:1239–1254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcia-Mata, R., Y.S. Gao, and E. Sztul. 2002. Hassles with taking out the garbage: aggravating aggresomes. Traffic. 3:388–396. [DOI] [PubMed] [Google Scholar]
- Goldberg, A.L. 2003. Protein degradation and protection against misfolded or damaged proteins. Nature. 426:895–899. [DOI] [PubMed] [Google Scholar]
- Goldberg, M.S., S.M. Fleming, J.J. Palacino, C. Cepeda, H.A. Lam, A. Bhatnagar, E.G. Meloni, N. Wu, L.C. Ackerson, G.J. Klapstein, et al. 2003. Parkin-deficient mice exhibit nigrostriatal deficits but not loss of dopaminergic neurons. J. Biol. Chem. 278:43628–43635. [DOI] [PubMed] [Google Scholar]
- Gorner, K., E. Holtorf, S. Odoy, B. Nuscher, A. Yamamoto, J.T. Regula, K. Beyer, C. Haass, and P.J. Kahle. 2004. Differential effects of Parkinson's disease-associated mutations on stability and folding of DJ-1. J. Biol. Chem. 279:6943–6951. [DOI] [PubMed] [Google Scholar]
- Gregersen, N., P. Bross, S. Vang, and J.H. Christensen. 2006. Protein misfolding and human disease. Annu. Rev. Genomics Hum. Genet. 7:103–124. [DOI] [PubMed] [Google Scholar]
- Haglund, K., and I. Dikic. 2005. Ubiquitylation and cell signaling. EMBO J. 24:3353–3359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hampe, C., H. Ardila-Osorio, M. Fournier, A. Brice, and O. Corti. 2006. Biochemical analysis of Parkinson's disease-causing variants of Parkin, an E3 ubiquitin-protein ligase with monoubiquitylation capacity. Hum. Mol. Genet. 15:2059–2075. [DOI] [PubMed] [Google Scholar]
- Hattori, N., and Y. Mizuno. 2004. Pathogenetic mechanisms of parkin in Parkinson's disease. Lancet. 364:722–724. [DOI] [PubMed] [Google Scholar]
- Hayashi, S., K. Wakabayashi, A. Ishikawa, H. Nagai, M. Saito, M. Maruyama, T. Takahashi, T. Ozawa, S. Tsuji, and H. Takahashi. 2000. An autopsy case of autosomal-recessive juvenile parkinsonism with a homozygous exon 4 deletion in the parkin gene. Mov. Disord. 15:884–888. [DOI] [PubMed] [Google Scholar]
- Hideshima, T., J.E. Bradner, J. Wong, D. Chauhan, P. Richardson, S.L. Schreiber, and K.C. Anderson. 2005. Small-molecule inhibition of proteasome and aggresome function induces synergistic antitumor activity in multiple myeloma. Proc. Natl. Acad. Sci. USA. 102:8567–8572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holmberg, C.I., K.E. Staniszewski, K.N. Mensah, A. Matouschek, and R.I. Morimoto. 2004. Inefficient degradation of truncated polyglutamine proteins by the proteasome. EMBO J. 23:4307–4318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hook, S.S., A. Orian, S.M. Cowley, and R.N. Eisenman. 2002. Histone deacetylase 6 binds polyubiquitin through its zinc finger (PAZ domain) and copurifies with deubiquitinating enzymes. Proc. Natl. Acad. Sci. USA. 99:13425–13430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Imai, Y., M. Soda, S. Hatakeyama, T. Akagi, T. Hashikawa, K.I. Nakayama, and R. Takahashi. 2002. CHIP is associated with Parkin, a gene responsible for familial Parkinson's disease, and enhances its ubiquitin ligase activity. Mol. Cell. 10:55–67. [DOI] [PubMed] [Google Scholar]
- Iwata, A., B.E. Riley, J.A. Johnston, and R.R. Kopito. 2005. HDAC6 and microtubules are required for autophagic degradation of aggregated huntingtin. J. Biol. Chem. 280:40282–40292. [DOI] [PubMed] [Google Scholar]
- Jackson, W.T., T.H. Giddings Jr., M.P. Taylor, S. Mulinyawe, M. Rabinovitch, R.R. Kopito, and K. Kirkegaard. 2005. Subversion of cellular autophagosomal machinery by RNA viruses. PLoS Biol. 3:e156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnston, J.A., C.L. Ward, and R.R. Kopito. 1998. Aggresomes: a cellular response to misfolded proteins. J. Cell Biol. 143:1883–1898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnston, J.A., M.E. Illing, and R.R. Kopito. 2002. Cytoplasmic dynein/dynactin mediates the assembly of aggresomes. Cell Motil. Cytoskeleton. 53:26–38. [DOI] [PubMed] [Google Scholar]
- Junn, E., S.S. Lee, U.T. Suhr, and M.M. Mouradian. 2002. Parkin accumulation in aggresomes due to proteasome impairment. J. Biol. Chem. 277:47870–47877. [DOI] [PubMed] [Google Scholar]
- Kahle, P.J., and C. Haass. 2004. How does parkin ligate ubiquitin to Parkinson's disease? EMBO Rep. 5:681–685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawaguchi, Y., J.J. Kovacs, A. McLaurin, J.M. Vance, A. Ito, and T.P. Yao. 2003. The deacetylase HDAC6 regulates aggresome formation and cell viability in response to misfolded protein stress. Cell. 115:727–738. [DOI] [PubMed] [Google Scholar]
- Kim, B.Y., J.A. Olzmann, G.S. Barsh, L.S. Chin, and L. Li. 2007. Spongiform neurodegeneration-associated E3 ligase Mahogunin ubiquitylates TSG101 and regulates endosomal trafficking. Mol. Biol. Cell. 18:1129–1142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kitada, T., S. Asakawa, N. Hattori, H. Matsumine, Y. Yamamura, S. Minoshima, M. Yokochi, Y. Mizuno, and N. Shimizu. 1998. Mutations in the parkin gene cause autosomal recessive juvenile parkinsonism. Nature. 392:605–608. [DOI] [PubMed] [Google Scholar]
- Ko, H.S., R. von Coelln, S.R. Sriram, S.W. Kim, K.K. Chung, O. Pletnikova, J. Troncoso, B. Johnson, R. Saffary, E.L. Goh, et al. 2005. Accumulation of the authentic parkin substrate aminoacyl-tRNA synthetase cofactor, p38/JTV-1, leads to catecholaminergic cell death. J. Neurosci. 25:7968–7978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ko, H.S., S.W. Kim, S.R. Sriram, V.L. Dawson, and T.M. Dawson. 2006. Identification of far upstream element-binding protein-1 as an authentic Parkin substrate. J. Biol. Chem. 281:16193–16196. [DOI] [PubMed] [Google Scholar]
- Kopito, R.R. 2000. Aggresomes, inclusion bodies and protein aggregation. Trends Cell Biol. 10:524–530. [DOI] [PubMed] [Google Scholar]
- Kristiansen, M., P. Deriziotis, D.E. Dimcheff, G.S. Jackson, H. Ovaa, H. Naumann, A.R. Clarke, F.W.B. van Leeuwen, V. Menendez-Benito, N.P. Dantuma, et al. 2007. Disease-associated prion protein oligomers inhibit the 26S proteasome. Mol. Cell. 26:175–188. [DOI] [PubMed] [Google Scholar]
- Li, H.M., T. Niki, T. Taira, S.M. Iguchi-Ariga, and H. Ariga. 2005. Association of DJ-1 with chaperones and enhanced association and colocalization with mitochondrial Hsp70 by oxidative stress. Free Radic. Res. 39:1091–1099. [DOI] [PubMed] [Google Scholar]
- Lim, K.L., K.C. Chew, J.M. Tan, C. Wang, K.K. Chung, Y. Zhang, Y. Tanaka, W. Smith, S. Engelender, C.A. Ross, et al. 2005. a. Parkin mediates nonclassical, proteasomal-independent ubiquitination of synphilin-1: implications for Lewy body formation. J. Neurosci. 25:2002–2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lim, K.L., V.L. Dawson, and T.M. Dawson. 2005. b. Parkin-mediated lysine 63-linked polyubiquitination: a link to protein inclusions formation in Parkinson's and other conformational diseases? Neurobiol. Aging. 27:524–529. [DOI] [PubMed] [Google Scholar]
- Lucking, C.B., A. Durr, V. Bonifati, J. Vaughan, G. De Michele, T. Gasser, B.S. Harhangi, G. Meco, P. Denefle, N.W. Wood, et al. 2000. Association between early-onset Parkinson's disease and mutations in the parkin gene. French Parkinson's Disease Genetics Study Group. N. Engl. J. Med. 342:1560–1567. [DOI] [PubMed] [Google Scholar]
- Ma, Q., K. Kinneer, Y. Bi, J.Y. Chan, and Y.W. Kan. 2004. Induction of murine NAD(P)H:quinone oxidoreductase by 2,3,7,8-tetrachlorodibenzo-p-dioxin requires the CNC (cap ‘n’ collar) basic leucine zipper transcription factor Nrf2 (nuclear factor erythroid 2-related factor 2): cross-interaction between AhR (aryl hydrocarbon receptor) and Nrf2 signal transduction. Biochem. J. 377:205–213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsuda, N., T. Kitami, T. Suzuki, Y. Mizuno, N. Hattori, and K. Tanaka. 2006. Diverse effects of pathogenic mutations of Parkin that catalyze multiple monoubiquitylation in vitro. J. Biol. Chem. 281:3204–3209. [DOI] [PubMed] [Google Scholar]
- Miller, D.W., R. Ahmad, S. Hague, M.J. Baptista, R. Canet-Aviles, C. McLendon, D.M. Carter, P.P. Zhu, J. Stadler, J. Chandran, et al. 2003. L166P mutant DJ-1, causative for recessive Parkinson's disease, is degraded through the ubiquitin-proteasome system. J. Biol. Chem. 278:36588–36595. [DOI] [PubMed] [Google Scholar]
- Moore, D.J., L. Zhang, T.M. Dawson, and V.L. Dawson. 2003. A missense mutation (L166P) in DJ-1, linked to familial Parkinson's disease, confers reduced protein stability and impairs homo-oligomerization. J. Neurochem. 87:1558–1567. [DOI] [PubMed] [Google Scholar]
- Moore, D.J., L. Zhang, J. Troncoso, M.K. Lee, N. Hattori, Y. Mizuno, T.M. Dawson, and V.L. Dawson. 2005. Association of DJ-1 and parkin mediated by pathogenic DJ-1 mutations and oxidative stress. Hum. Mol. Genet. 14:71–84. [DOI] [PubMed] [Google Scholar]
- Mori, H., T. Kondo, M. Yokochi, H. Matsumine, Y. Nakagawa-Hattori, T. Miyake, K. Suda, and Y. Mizuno. 1998. Pathologic and biochemical studies of juvenile parkinsonism linked to chromosome 6q. Neurology. 51:890–892. [DOI] [PubMed] [Google Scholar]
- Muqit, M.M., S.M. Davidson, M.D. Payne Smith, L.P. MacCormac, S. Kahns, P.H. Jensen, N.W. Wood, and D.S. Latchman. 2004. Parkin is recruited into aggresomes in a stress-specific manner: over-expression of parkin reduces aggresome formation but can be dissociated from parkin's effect on neuronal survival. Hum. Mol. Genet. 13:117–135. [DOI] [PubMed] [Google Scholar]
- Olanow, C.W., D.P. Perl, G.N. DeMartino, and K.S. McNaught. 2004. Lewy-body formation is an aggresome-related process: a hypothesis. Lancet Neurol. 3:496–503. [DOI] [PubMed] [Google Scholar]
- Olzmann, J.A., K. Brown, K.D. Wilkinson, H.D. Rees, Q. Huai, H. Ke, A.I. Levey, L. Li, and L.S. Chin. 2004. Familial Parkinson's disease-associated L166P mutation disrupts DJ-1 protein folding and function. J. Biol. Chem. 279:8506–8515. [DOI] [PubMed] [Google Scholar]
- Olzmann, J.A., J. Pridgeon, L. Li, and L.S. Chin. 2005. CHIP ubiquitinates Parkinson's disease-associated L166P mutant DJ-1 in a chaperone- independent manner. In American Society for Cell Biology, San Francisco. Abstr. 2143.
- Olzmann, J.A., J.R. Bordelon, E.C. Muly, H.D. Rees, A.I. Levey, L. Li, and L.S. Chin. 2007. Selective enrichment of DJ-1 protein in primate striatal neuronal processes: implications for Parkinson's disease. J. Comp. Neurol. 500:585–599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pawlyk, A.C., B.I. Giasson, D.M. Sampathu, F.A. Perez, K.L. Lim, V.L. Dawson, T.M. Dawson, R.D. Palmiter, J.Q. Trojanowski, and V.M. Lee. 2003. Novel monoclonal antibodies demonstrate biochemical variation of brain parkin with age. J. Biol. Chem. 278:48120–48128. [DOI] [PubMed] [Google Scholar]
- Perez, F.A., and R.D. Palmiter. 2005. Parkin-deficient mice are not a robust model of parkinsonism. Proc. Natl. Acad. Sci. USA. 102:2174–2179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perez, F.A., W.R. Curtis, and R.D. Palmiter. 2005. Parkin-deficient mice are not more sensitive to 6-hydroxydopamine or methamphetamine neurotoxicity. BMC Neurosci. 6:71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Periquet, M., O. Corti, S. Jacquier, and A. Brice. 2005. Proteomic analysis of parkin knockout mice: alterations in energy metabolism, protein handling and synaptic function. J. Neurochem. 95:1259–1276. [DOI] [PubMed] [Google Scholar]
- Pickart, C.M., and D. Fushman. 2004. Polyubiquitin chains: polymeric protein signals. Curr. Opin. Chem. Biol. 8:610–616. [DOI] [PubMed] [Google Scholar]
- Pridgeon, J.W., T. Geetha, and M.W. Wooten. 2003. A method to identify p62's UBA domain interacting proteins. Biol. Proced. Online. 5:228–237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richly, H., M. Rape, S. Braun, S. Rumpf, C. Hoege, and S. Jentsch. 2005. A series of ubiquitin binding factors connects CDC48/p97 to substrate multiubiquitylation and proteasomal targeting. Cell. 120:73–84. [DOI] [PubMed] [Google Scholar]
- Schlossmacher, M.G., M.P. Frosch, W.P. Gai, M. Medina, N. Sharma, L. Forno, T. Ochiishi, H. Shimura, R. Sharon, N. Hattori, et al. 2002. Parkin localizes to the Lewy bodies of Parkinson disease and dementia with Lewy bodies. Am. J. Pathol. 160:1655–1667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shendelman, S., A. Jonason, C. Martinat, T. Leete, and A. Abeliovich. 2004. DJ-1 is a redox-dependent molecular chaperone that inhibits alpha-synuclein aggregate formation. PLoS Biol. 2:e362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shimura, H., N. Hattori, S. Kubo, Y. Mizuno, S. Asakawa, S. Minoshima, N. Shimizu, K. Iwai, T. Chiba, K. Tanaka, and T. Suzuki. 2000. Familial Parkinson disease gene product, parkin, is a ubiquitin-protein ligase. Nat. Genet. 25:302–305. [DOI] [PubMed] [Google Scholar]
- Shimura, H., M.G. Schlossmacher, N. Hattori, M.P. Frosch, A. Trockenbacher, R. Schneider, Y. Mizuno, K.S. Kosik, and D.J. Selkoe. 2001. Ubiquitination of a new form of alpha-synuclein by parkin from human brain: implications for Parkinson's disease. Science. 293:263–269. [DOI] [PubMed] [Google Scholar]
- Sulzer, D. 2007. Multiple hit hypotheses for dopamine neuron loss in Parkinson's disease. Trends Neurosci. 30:244–250. [DOI] [PubMed] [Google Scholar]
- Takahashi, H., E. Ohama, S. Suzuki, Y. Horikawa, A. Ishikawa, T. Morita, S. Tsuji, and F. Ikuta. 1994. Familial juvenile parkinsonism: clinical and pathologic study in a family. Neurology. 44:437–441. [DOI] [PubMed] [Google Scholar]
- Taylor, J.P., F. Tanaka, J. Robitschek, C.M. Sandoval, A. Taye, S. Markovic-Plese, and K.H. Fischbeck. 2003. Aggresomes protect cells by enhancing the degradation of toxic polyglutamine-containing protein. Hum. Mol. Genet. 12:749–757. [DOI] [PubMed] [Google Scholar]
- Venkatraman, P., R. Wetzel, M. Tanaka, N. Nukina, and A.L. Goldberg. 2004. Eukaryotic proteasomes cannot digest polyglutamine sequences and release them during degradation of polyglutamine-containing proteins. Mol. Cell. 14:95–104. [DOI] [PubMed] [Google Scholar]
- Wang, C., L. Deng, M. Hong, G.R. Akkaraju, J. Inoue, and Z.J. Chen. 2001. TAK1 is a ubiquitin-dependent kinase of MKK and IKK. Nature. 412:346–351. [DOI] [PubMed] [Google Scholar]
- Wang, L., T.V. Nguyen, R.W. McLaughlin, L.A. Sikkink, M. Ramirez-Alvarado, and R.M. Weinshilboum. 2005. Human thiopurine S-methyltransferase pharmacogenetics: variant allozyme misfolding and aggresome formation. Proc. Natl. Acad. Sci. USA. 102:9394–9399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weissman, A.M. 2001. Themes and variations on ubiquitylation. Nat. Rev. Mol. Cell Biol. 2:169–178. [DOI] [PubMed] [Google Scholar]
- Wheeler, T.C., L.S. Chin, Y. Li, F.L. Roudabush, and L. Li. 2002. Regulation of synaptophysin degradation by mammalian homologues of seven in absentia. J. Biol. Chem. 277:10273–10282. [DOI] [PubMed] [Google Scholar]
- Zhang, Y., J. Gao, K.K. Chung, H. Huang, V.L. Dawson, and T.M. Dawson. 2000. Parkin functions as an E2-dependent ubiquitin-protein ligase and promotes the degradation of the synaptic vesicle-associated protein, CDCrel-1. Proc. Natl. Acad. Sci. USA. 97:13354–13359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao, J., Y. Ren, Q. Jiang, and J. Feng. 2003. Parkin is recruited to the centrosome in response to inhibition of proteasomes. J. Cell Sci. 116:4011–4019. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}