

Abstract
Cofilin is the major mediator of actin filament turnover in vivo. However, the molecular mechanism of cofilin recruitment to actin networks during dynamic actin-mediated processes in living cells and cofilin's precise in vivo functions have not been determined. In this study, we analyzed the dynamics of fluorescently tagged cofilin and the role of cofilin-mediated actin turnover during endocytosis in Saccharomyces cerevisiae. In living cells, cofilin is not necessary for actin assembly on endocytic membranes but is recruited to molecularly aged adenosine diphosphate actin filaments and is necessary for their rapid disassembly. Defects in cofilin function alter the morphology of actin networks in vivo and reduce the rate of actin flux through actin networks. The consequences of decreasing actin flux are manifested by decreased but not blocked endocytic internalization at the plasma membrane and defects in late steps of membrane trafficking to the vacuole. These results suggest that cofilin-mediated actin filament flux is required for the multiple steps of endocytic trafficking.
Introduction
Actin filament assembly is essential for numerous cellular events, including the formation of endocytic vesicles at the plasma membrane (Perrais and Merrifield, 2005; Rodal et al., 2005; Yarar et al., 2005; Kaksonen et al., 2006). However, the role of actin filament disassembly (a key step in actin dynamics) during actin-mediated endocytosis is less clear. A major modulator of actin filament disassembly is the actin-depolymerization factor/cofilin family of proteins (hereafter referred to as cofilin). These small highly conserved actin-binding proteins are essential regulators of actin dynamics in living cells.
Cofilin and its role in actin dynamics have been studied extensively in vitro. Cofilin binds preferentially to ADP-actin subunits within actin filaments. This binding induces a twist in the filament, accelerates the release of Pi from ADP-Pi subunits, and severs actin filaments (Pollard and Borisy, 2003). Severing increases the number of actin filament pointed ends and, in conjunction with capping of barbed ends by capping proteins, stimulates filament disassembly (Andrianantoandro and Pollard, 2006).
The molecular determinants of cofilin recruitment to actin filaments in vivo have not been fully investigated. Svitkina and Borisy (1999) demonstrated the restriction of cofilin to zones slightly displaced from the leading edge in Xenopus laevis keratocytes. This localization was found to be unaffected by manipulations that were expected to change polymerization rates at the leading edge, which should alter the relative abundance of ATP-, ADP-Pi-, and ADP-actin subunits within the filaments. These observations led to the hypothesis that cofilin localization is largely dependent on accessory factors or other regulatory inputs, such as phosphorylation or pH.
In the budding yeast Saccharomyces cerevisiae, fixed cell imaging analysis demonstrated that cofilin localizes with actin at sites of endocytosis (Moon et al., 1993; Lappalainen and Drubin, 1997). Genetic and biochemical analysis of a conditional allele of cofilin showed that the actin filament disassembly activity of cofilin is essential for fluid-phase endocytosis (Lappalainen and Drubin, 1997). A later study indicated that at elevated temperatures, cofilin is important in the initial internalization step of endocytosis (Idrissi et al., 2002). However, this study did not address the molecular basis for a drastic block in the endocytic delivery of a fluid-phase marker to the vacuole observed in the cofilin mutant at permissive temperatures. Here, we sought to understand cofilin dynamics in live cells, to address the molecular determinants of its recruitment to actin structures in vivo, and to determine how cofilin influences actin dynamics, endocytic internalization, and delivery of cargo to the vacuole.
Results
Cofilin recruitment to actin structures is delayed relative to filament assembly in vivo
To investigate cofilin dynamics in live cells, we attempted to tag cofilin with GFP. However, both N- and C-terminal fusion proteins were nonfunctional. Therefore, we attempted to make a functional construct by creating internal in-frame fusions of GFP to cofilin. We inserted GFP flanked on both sides by either 12–, 7–, 4– or 0–amino acid linkers after amino acids D34, P48, P58, and N74. One construct, in which GFP was inserted between amino acids N74 and G75 and was flanked on both sides by 12–amino acid linkers (see Materials and methods section Media, plasmids, and strains), complemented a cof1Δ mutation when expressed from a high-copy plasmid (Fig. S1, available at http://www.jcb.org/cgi/content/full/jcb.200703092/DC1). The localization and dynamics of GFP-cofilin expressed from high-copy or low-copy plasmids were identical. However, the expression levels of GFP-cofilin from cell to cell were less variable using the low-copy plasmid. Therefore, we used the low-copy plasmid in COF1 wild-type cells to express GFP-cofilin for our microscopy studies. These cells exhibited normal cortical actin patch dynamics (unpublished data).
Actin assembly at cortical patches in budding yeast powers the invagination and vesicle scission steps of endocytosis (Kaksonen et al., 2003). To determine the timing of cofilin recruitment to actin patches, we used simultaneous two-color imaging of Abp1–monomeric RFP (mRFP) as a marker for actin at endocytic sites and GFP-cofilin. GFP-cofilin localized to actin patches in live cells (Fig. 1 A and Video 1, available at http://www.jcb.org/cgi/content/full/jcb.200703092/DC1), which is in agreement with previous studies in fixed cells (Moon et al., 1993; Lappalainen and Drubin, 1997). We then examined cofilin and Abp1 dynamics at the actin patch. Strikingly, GFP-cofilin associated with patches 3.6 s (±0.6 s) after the initial assembly of actin/Abp1p (Fig. 1 A, bottom). Analysis of cofilin spatiotemporal dynamics with kymographs (Fig. 1 A) indicated that cofilin associates with patches during the internalization/disassembly phase of the endocytic patch lifetime.
Figure 1.

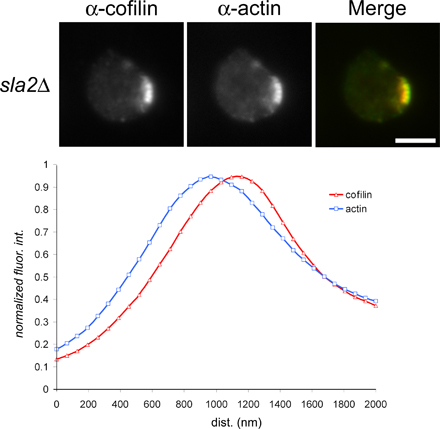
Visualization of cofilin dynamics in actin structures in vivo. (A) GFP-cofilin localization in cells expressing Abp1-mRFP was captured using simultaneous two-color imaging of a medial focal plane. The temporal maturation of a single actin patch is illustrated in a montage image in which each frame represents a 1-s exposure. The mean temporal delay in cofilin assembly at the actin patch is indicated (n = 25). A kymograph illustrates the spatiotemporal relationship between Abp1-mRFP and GFP-cofilin with the cell exterior on the top of the kymograph and the cell interior oriented downwards. dfc, distance from cortex. The bar on the x axis is 10 s, and the bar on the y axis is 200 nm. WT, wild type. (B) Simultaneous two-color imaging showing GFP-cofilin localization in sla2Δ cells expressing Abp1-mRFP (top) and GFP-actin localization in sla2Δ cells expressing Abp1-mRFP (bottom). Quantification of normalized fluorescence intensities along the length of the actin comet tails is presented in the graphs. The mean of three measurements per actin comet tail for 10 cells is reported. Bars, 4 μm.
To further document and analyze the temporal delay in cofilin recruitment to actin structures, we examined cofilin localization in sla2Δ mutant cells in which endocytic coat internalization is defective but actin still assembles at cortical patches. In this mutant background, actin comet tails stably associate with the endocytic machinery at the cell cortex (Kaksonen et al., 2003). Actin subunits actively treadmill through these elongated Arp2/3-dependent filamentous actin (F-actin) structures as assessed by fluorescence recovery after photobleaching, with new actin filament assembly taking place at the cell cortex (Kaksonen et al., 2003). Consistent with a temporal delay in cofilin association with actin patches, there is a significant (P < 0.01), spatially resolvable exclusion of cofilin from regions of actin comet tails adjacent to the cell cortex (Fig. 1 B). The spatial exclusion of untagged cofilin from regions of active actin assembly in sla2Δ cells was verified by immunolocalization of cofilin and actin (Fig. S2, available at http://www.jcb.org/cgi/content/full/jcb.200703092/DC1). The localization of these two proteins was quantified as a function of fluorescence intensity along the length of the actin tails, moving from the cell surface to the cell interior (Fig. 1 B). These observations further strengthened our conclusion that the low-copy GFP-cofilin construct is a faithful reporter of cofilin dynamics in vivo.
Actin nucleotide state is important for cofilin recruitment in vivo
We next focused on the molecular mechanisms responsible for the delay in cofilin recruitment to actin filaments. This delay could reflect regulation by phosphorylation, pH, accessory proteins, or the nucleotide-bound state of actin subunits within the filaments. In S. cerevisiae, cofilin is not phosphorylated, and its severing activity is not regulated by pH (Moseley and Goode, 2006; Pavlov et al., 2006). Therefore, we examined the contributions of two proteins that are important for cofilin function in vivo, Srv2p and Aip1p, to cofilin localization within actin tails in sla2Δ cells (Rodal et al., 1999; Balcer et al., 2003). Previously, we showed that in the absence of Aip1p, cofilin, which is normally restricted to cortical actin patches, becomes distributed on both patches and cytoplasmic cables (Rodal et al., 1999). When either Aip1p or Srv2p was eliminated by gene deletion in sla2Δ cells, no detectable effect on cofilin localization within actin comet tails was observed (unpublished data). In vitro experiments have shown that cofilin has preferential affinity for ADP-actin, but the in vivo relevance of this observation has never been established (Blanchoin and Pollard, 1999). To test this relevance, we used an allele of actin, act1-V159N, which forms filaments that structurally mimic ADP-Pi-actin and that have reduced F-actin disassembly rates in vitro and in vivo (Belmont and Drubin, 1998; Belmont et al., 1999). Strikingly, cofilin localization to F-actin tails is quantifiably reduced in sla2Δ cells with act1-V159N as the sole source of actin (Fig. 2 A). Consistent with the observation in sla2Δ act1-V159N double mutants, GFP-cofilin in SLA2 act1-V159N cells also showed an increase in cytoplasmic localization and, in addition, in a subset of cells, was assembled into aberrant cablelike structures (unpublished data).
Figure 2.

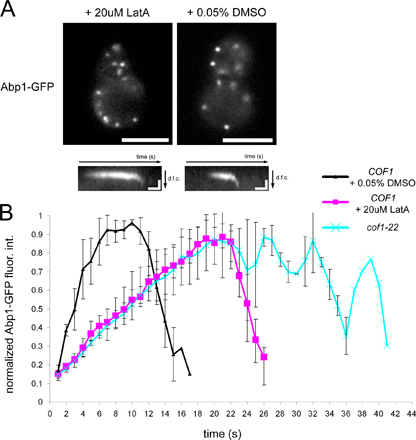
Actin nucleotide state controls cofilin association with F-actin structures in vivo. (A) GFP-cofilin and Abp1-mRFP localization in sla2Δ act1-V159N cells was captured by simultaneous two-color imaging. The ratio of the fluorescence intensity of GFP-cofilin in actin tails and in the cytoplasm was quantified in 28 cells for sla2Δ mutants and act1-V159N sla2Δ mutant cells. The data are presented as range bar graphs in red, where the range of the entire dataset from minimum to maximum is given. Means of the data (black squares) with standard deviations (error bars) are overlaid on the range bar graphs. (B) The redistribution of GFP-cofilin localization within actin comet tails labeled by Abp1-mRFP in sla2Δ pdr5Δ snq2Δ cells upon treatment with jasplakinolide at t = 0 s. Cells were immobilized in a flow chamber (see Materials and methods), and 50 μM jasplakinolide was flowed through the chamber while simultaneously imaging both fluorophores. Single frames from the time-lapse analysis at the given time points are shown. Quantification of the normalized fluorescence intensities as a function of length along the actin tail for five cells (three measurements per tail) 5 min after treatment with jasplakinolide is presented in the graph. Bars, 4 μm.
To further test whether cofilin binds preferentially to ADP-actin filaments in vivo, we used the actin filament–stabilizing molecule jasplakinolide. Jasplakinolide blocks F-actin disassembly, which is expected to deplete the assembly-competent actin monomer pool and stop actin subunit flux through filament networks. Actin nucleotide hydrolysis would be expected to convert ATP-actin into ADP-actin subunits within jasplakinolide-stabilized filaments, which should eliminate the zone of the actin tails that lacks associated cofilin. We generated an sla2Δ strain, which expressed Abp1-mRFP and GFP-cofilin and was sensitized to jasplakinolide by elimination of two multidrug transporters, Snq2 and Pdr5 (Ayscough, 2000). When these cells were treated with jasplakinolide, actin tails began to lengthen, indicating that F-actin disassembly was blocked. After 5 min, no actin subunit flux was detectable by following fiduciary marks in the elongated actin tails using kymographs (unpublished data). Strikingly, cofilin was found to be uniformly distributed along the elongated actin tails (Fig. 2 B and Video 2, available at http://www.jcb.org/cgi/content/full/jcb.200703092/DC1). This localization effect is quantitatively documented in Fig. 2 B. As a complimentary analysis, treatment of sla2Δ cells with the actin filament assembly inhibitor latrunculin A (lat A) is also predicted to eliminate ATP and ADP-Pi from actin filaments because new assembly is blocked. Treatment of sla2Δ cells with lat A caused the tails to begin to shorten, and, over time, cofilin was no longer restricted to regions distal to the cell cortex (Fig. S3 and Video 3A). These independent pharmacological approaches to modify actin dynamics through the monomer pool with lat A and through the filamentous population with jasplakinolide implicate the actin filament nucleotide state in controlling cofilin localization in vivo.
Cofilin is the major mediator of actin subunit flux in vivo
Having established the molecular determinants of cofilin recruitment to F-actin in vivo, we wanted to better understand how cofilin contributes to actin subunit flux through filament networks. To do this, we examined the effects of a mutant of cofilin, Cof1-22p, on actin flux in actin comet tails and on tail morphology in sla2Δ mutant cells (Lappalainen and Drubin, 1997). In vitro studies suggested that Cof1-22p has weakened interactions with actin filaments and is defective in actin filament disassembly (Lappalainen and Drubin, 1997; Lappalainen et al., 1997). sla2Δ cof1-22 cells expressing actin-GFP had drastic alterations to actin tail morphology (Fig. 3 A), and the tails were significantly elongated (∼3 μm; P < 0.01) compared with tails in cells with wild-type cofilin function (∼700 nm; Fig. 3 C). Actin subunit flux rates, as assessed using fluorescence recovery after photobleaching analysis, were also significantly perturbed in sla2Δ cof1-22 cells (P < 0.01; Fig. 3 B and Video 4, available at http://www.jcb.org/cgi/content/full/jcb.200703092/DC1) and averaged ∼55 nm/s in cells with wild-type cofilin function compared with ∼10 nm/s in cells expressing Cof1-22p (Fig. 3 C). Interestingly, despite the previously reported contribution of Srv2p and Aip1p to cofilin-dependent actin filament turnover (Okada et al., 2002; Balcer et al., 2003), these proteins did not contribute to actin comet tail morphology or subunit flux in sla2Δ cells (unpublished data). These studies provide evidence that cofilin-dependent actin filament disassembly contributes to actin subunit flux in vivo and suggest that cofilin is the major mediator of this process in S. cerevisiae.
Figure 3.

Defects in actin flux in vivo in a cofilin mutant. (A) Clustered actin comet tails labeled with GFP-actin in sla2Δ COF1 cells and sla2Δ cof1-22 cells. Representative budded and unbudded cells are shown. (B) Fluorescence recovery after photobleaching of a cluster of actin tails in sla2Δ COF1 cells (top) compared with actin comet tails in sla2Δ cof1-22 cells (bottom). Prebleached cells are shown on the left, and time points are shown that illustrate the progression of the bleached region (indicated by arrows) through the actin comet tails. (C) Quantification of the actin comet tail length and the actin filament flux rates. 20 and 22 cells were quantified for sla2Δ COF1 and sla2Δ cof1-22, respectively. Error bars represent SD. Bars, 2 μm.
Defects in cofilin function perturb the dynamics of the endocytic machinery and actin at endocytic sites
Having established that cof1-22 cells are defective in actin network dynamics in vivo, we wanted to determine how these defects affect endocytosis. In wild-type and cof1-22 cells, we simultaneously imaged the dynamics of Sla1p, an endocytic coat protein, and Abp1p, a marker for actin in endocytic patches, respectively. In wild-type cells, Sla1p assembles at the cell cortex before actin. Subsequent actin assembly, which is marked by Abp1p, drives internalization and triggers disassembly of the endocytic coat (Fig. 4 A, left; and Video 5A, available at http://www.jcb.org/cgi/content/full/jcb.200703092/DC1). Maximum intensity projections of 2-min medial focal plane videos show that assembly and disassembly of the endocytic and actin complexes are restricted to the vicinity of the cell cortex in wild-type cells. In contrast, cof1-22 cells have a dramatic redistribution of F-actin to the cell interior (Fig. 4 A, right; and Video 5 B). However, the endocytic coat marker Sla1p was not abnormally redistributed to the cell interior.
Figure 4.

Cofilin contribution to endocytic machinery and actin dynamics. (A) Single frames from simultaneous two-color imaging of Abp1-mRFP and Sla1-GFP as markers for patch-localized F-actin and the endocytic coat, respectively, in medial focal planes. Maximum intensity projections of 2-min medial focal plane videos are presented as labeled. Bars, 4 μm. Kymographs of representative patches from wild-type and cof1-22 cells are shown on the left and right, respectively. Bars in both kymographs are 5 s on the x axis and 400 nm on the y axis. dfc, distance from cortex. (B, 1 and 2) Traces showing the fluorescence intensity change of Abp1-mRFP (red curves) and the distance from point of origin (blue curves) in wild-type cells and cof1-22 mutant cells, respectively. (3 and 4) Traces showing the fluorescence intensity change of Sla1-GFP (red curves) and the distance from point of origin (blue curves) in wild-type cells and cof1-22 mutant cells, respectively. Each curve is the mean of three representative measurements.
Examination of the dynamics of the endocytic coat and actin markers at the cell cortex in cof1-22 cells by kymograph revealed unexpected behaviors. First, as in wild-type cells, endocytic coat components assembled at the cell cortex and moved inward concurrent with the burst of actin assembly (Fig. 4 A, right kymograph). This observation suggests that endocytic internalization occurs in this mutant, which was not expected because of previously reported defects in fluid-phase endocytosis for this mutant (Lappalainen et al., 1997). These behaviors were quantified, and 90% (n = 71) of Sla1-GFP patches internalized in cof1-22 cells versus 93% (n = 76) of Sla1-GFP patches in wild-type cells. Quantification of Abp1p-mRFP and Sla1p-GFP dynamics in cof1-22 cells indicates that these proteins still assemble at endocytic sites, but with slowed kinetics (Fig. 4 B, panels 2 and 4). After reaching a peak in fluorescence intensity, Sla1p internalized and rapidly disassembled, whereas Abp1p internalized but did not efficiently disassemble, indicating that the F-actin clumps in the cell interior are derived from structures assembled at the cell cortex. The observation that Sla1p is internalized in cof1-22 mutant cells suggests that perturbations to cofilin-mediated actin filament turnover do not block endocytic internalization from the plasma membrane. In addition, the delay in actin assembly at endocytic sites suggests that cofilin function is important for rapid actin assembly.
Endocytic and plasma membrane internalization occurs in cofilin mutants
The observation that endocytic coat complexes internalize concomitant with actin assembly in cof1-22 cells (Fig. 4) was intriguing given the observed defects in the accumulation of fluid-phase endocytic markers in the vacuole (Lappalainen and Drubin, 1997). We tested whether the endocytic coat internalization events reflect receptor-mediated endocytic internalization by quantifying the internalization of radiolabeled yeast mating pheromone. Indeed, in cof1-22 cells, radiolabeled α factor is internalized, albeit with delayed kinetics (Fig. 5 A). The delay in internalization could be reflected by a decrease in the steady-state number of endocytic events in cof1-22 cells. We tested this possibility by quantifying the total number of Sla1-GFP patches per unit area in wild-type and cof1-22 mutant cells and found that there is a slight increase in the number of steady-state endocytic sites in cof1-22 mutant cells (1.7 ± 0.6 endocytic sites per 10 μm2 for wild-type cells [n = 25] vs. 2.5 ± 0.5 endocytic sites per 10 μm2 for cof1-22 mutants [n = 25]). Therefore, we suspect that the delay in α-factor internalization reflects the longer lifetime of the endocytic machinery at the plasma membrane seen in cof1-22 cells (Fig. 4).
Figure 5.

Cofilin defects do not block endocytic internalization but lead to Abp1 association with internal membranes. (A) Internalization of 35S-labeled α factor was measured at 25°C and displayed as percent internalized. Error bars represent the SD from two experiments performed in duplicate (four total measurements per time point). (B) Single medial focal plane image of cof1-22 cells expressing Abp1-GFP 5 min after the addition of FM4-64. Images were captured simultaneously. (C) Kymograph of a cof1-22 cell expressing Abp1-GFP 5 min after the addition of FM4-64. Kymographs represent fluorescent signals in the center of the cells over time. Bars in both kymographs are 5 s on the x axis and 500 nm on the y axis. dfc, distance from cortex. (D) Subcellular fractionation of wild-type and cof1-22 cell extracts using sequential centrifugation steps was used to generate low speed supernatant (S300), medium speed supernatant and pellet (S13,000 and P13,000), and high speed supernatant and pellet (S100,000 and P100,000) fractions. The subcellular distribution of Abp1p and Act1p was detected using immunoblotting with polyclonal anti-Abp1p and Act1p antibodies. Bars, 4 μm.
The observation that actin patches have elongated postinternalization lifetimes spurred us to investigate the association of these actin patches with internalized plasma membrane. We used simultaneous imaging of the lipophilic dye FM4-64 and Abp1-GFP to analyze the dynamic interrelationship between plasma membrane internalization and actin in cof1-22 cells. Consistent with the observation that radiolabeled α factor is internalized in cof1-22 cells, 5 min after the addition of FM4-64, labeled internal membranes were observed, indicating that plasma membrane internalization occurs in cof1-22 cells (Fig. 5 B). Strikingly, we observed partial colocalization between cytoplasmic Abp1-GFP and internalized FM4-64 in cof1-22 cells (Fig. 5 B and Video 8, available at http://www.jcb.org/cgi/content/full/jcb.200703092/DC1) but not in wild-type cells (Fig. 5 B and Video 7). The internalized membranes moved rapidly within the cytoplasm. The duration of the colocalization of FM4-64 and Abp1-GFP is evident in kymographs, which demonstrate prolonged colocalization compared with wild-type cells (Fig. 5 C).
We biochemically compared the association of Abp1p with membranes in cof1-22 and wild-type cells by fractionating cell extracts and examining the subcellular distribution of Abp1p. We found that Abp1p and actin are enriched in the heavy membrane fraction (P13,000) from cof1-22 cells (Fig. 5 D). Further fractionation experiments were unsuccessful in determining the exact identity of these Abp1p/actin-associated membranes. These data coupled with the in vivo live cell imaging results suggest that cofilin-mediated actin filament turnover is required for the timely disassembly of actin from endocytic membranes after internalization from the plasma membrane.
Cells defective in cofilin function have abnormal endocytic compartment morphology and secrete CPY
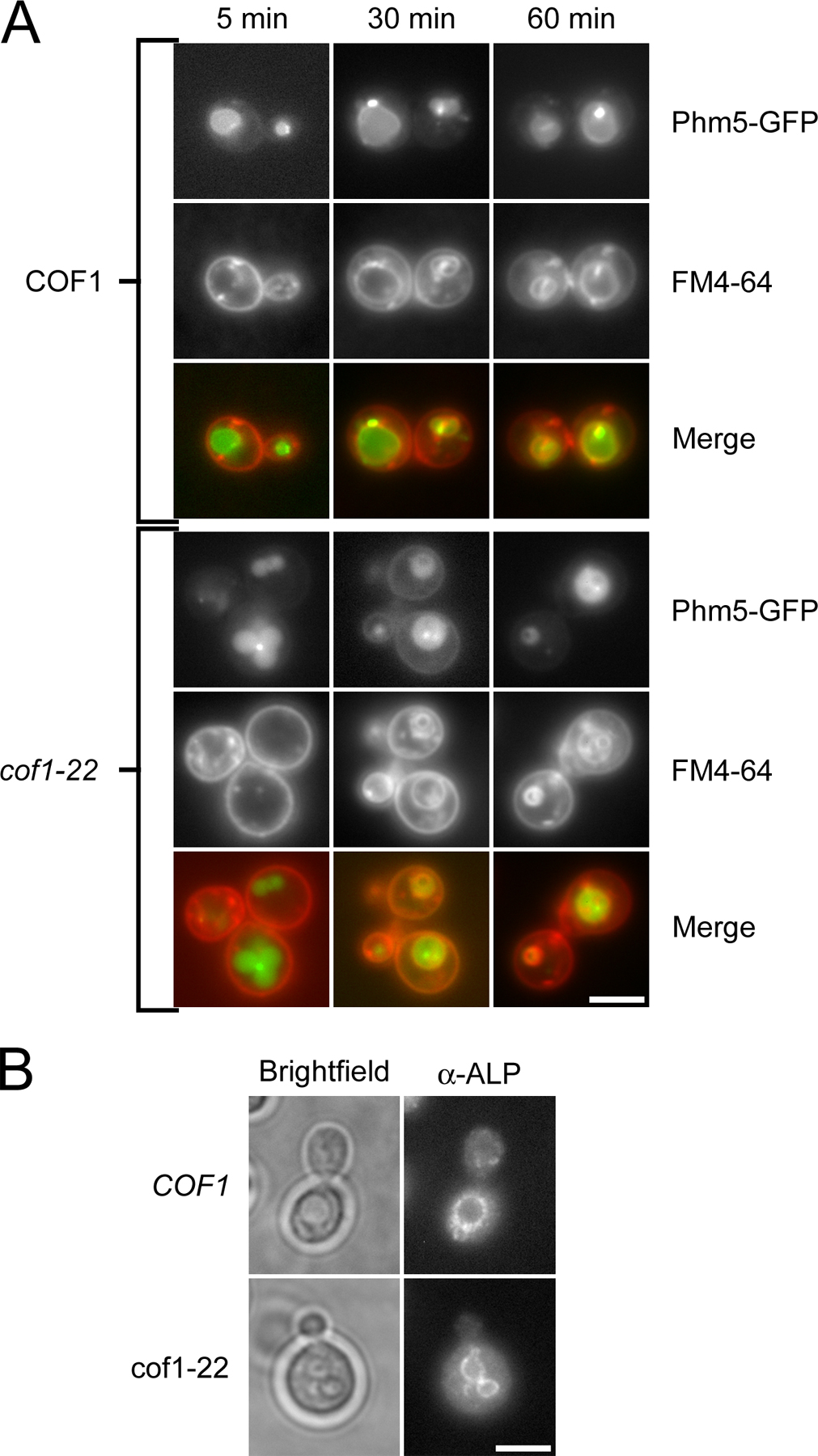
Two observations in cof1-22 cells, the internalization of plasma membrane and endocytic proteins and the lack of accumulation of endocytic cargo in the vacuole, seemed contradictory. We sought to determine the basis for this contradiction, reasoning that the membrane marker FM4-64 would provide better resolution of membrane-trafficking events between the plasma membrane and the vacuole than fluid-phase endocytosis markers. In wild-type cells, FM4-64 initially labeled the plasma membrane (Fig. 6 A). After 15 min, it labeled small, intensely stained endocytic intermediates. After ∼45 min, vacuolar labeling was observed, and, at 60 min, the vacuole was strongly stained. In cof1-22 cells, a dramatic alteration in the morphology of endocytic compartments was observed (Fig. 6 A). After 15 min, large membranous structures were labeled, which persisted throughout the course of the analysis (60 min). These membranous structures were observed in 75% of cof1-22 cells (n = 20) and were never seen in wild-type cells (n = 15). To better identify these structures, we used the vacuolar endopolyphosphatase Phm5p, which was tagged with GFP, as a steady-state marker of vacuolar morphology and followed the internalization of FM4-64 over time (Fig. S4 A, available at http://www.jcb.org/cgi/content/full/jcb.200703092/DC1). In cof1-22 mutant cells, FM4-64 colocalized with Phm5-GFP in the multilobed and fragmented vacuoles after 30 min of internalization of the dye. Multilobed and fragmented vacuoles were observed in 68% of cof1-22 cells (n = 65) versus 7% of wild-type cells (n = 25). These observations indicate that productive cofilin-mediated actin filament turnover is required for the normal morphology of endocytic compartments.
Figure 6.

Defects in endocytic compartment morphology and secretion of the vacuolar hydrolase CPY in a cofilin mutant. (A) Representative medial focal plane images of endocytic internalization of the lipophilic dye FM4-64 in wild-type and cof1-22 mutant cells. Vacuoles identified by brightfield microscopy are marked with red asterisks. (B) Colony blot assay for CPY secretion in wild-type cells and cof1-22, vps4Δ, and vps27Δ mutants grown at 25°C. Bar, 4 μm.
One hallmark of a subset of postinternalization endocytosis mutants, particularly vacuolar protein sorting (vps) mutants, is the missorting and subsequent secretion of soluble vacuolar hydrolases such as carboxypeptidase Y (CPY). We tested for the secretion of CPY in cof1-22 cells using a colony blot assay (Fig. 6 B). The cof1-22 cells showed markedly elevated CPY secretion compared with wild-type cells. Secretion of CPY from cof1-22 mutant cells was similar to levels observed for two vps mutants, vps4Δ and vps27Δ. To control for the deposition of CPY by cell lysis rather than secretion, log-phase liquid cultures of wild-type and cof1-22 mutant cells were compared for release of the soluble cytosolic marker 3-phosphoglycerate kinase (Pgk1p). No difference in Pgk1p levels was observed in the culture media under these conditions (unpublished data). To further understand the nature of the trafficking defects in cof1-22 mutant cells, we examined the distribution of alkaline phosphatase (Pho8p), a protein that is trafficked to the vacuole via a different biosynthetic route from the one used by CPY (Stepp et al., 1997). We found that Pho8p localizes properly to fragmented/multilobed vacuoles (Fig. S4 B). This result establishes that although certain aspects of vacuole sorting and morphology are abnormal in cof1-22 mutant cells, others remain intact. Collectively, these results indicate that cofilin-mediated actin filament turnover is required for the timely delivery of endocytic material to the vacuole after internalization from the plasma membrane, for certain vacuole sorting processes, and for normal vacuole morphology.
Discussion
We investigated cofilin's dynamics and role in actin subunit flux through filament networks in living yeast cells and obtained in vivo evidence for the dependence of cofilin recruitment on the actin filament nucleotide state. This insight into cofilin- dependent actin filament turnover in yeast allowed us to ask how actin subunit flux contributes to endocytic internalization and to subsequent steps in transport to the vacuole. A cofilin allele with defects in cofilin-dependent actin filament turnover strongly blocked accumulation in the vacuole of a marker for fluid-phase endocytosis but only partially blocked plasma membrane and receptor-mediated cargo internalization. Consistent with these observations, endocytic patches were able to internalize from the plasma membrane concomitantly with a slowed burst of actin polymerization. Defects in actin disassembly did not block disassembly of the other components of the endocytic machinery, including coat proteins subsequent to internalization, but resulted in the prolonged association of actin with internal membranous compartments. F-actin disassembly defects resulted in vacuolar morphology defects and the improper sorting of at least one type of soluble cellular cargo destined for the vacuole.
Nucleotide dependence of cofilin localization in vivo
A role for actin nucleotide state in cofilin affinity for actin filaments has been established biochemically (Maciver et al., 1991; Blanchoin and Pollard, 1999), providing a potential framework for understanding how cells control the timely disassembly of actin filaments. However, a role for actin nucleotide state in cofilin function in vivo had not been established. Using a variety of experimental strategies, our results support a role for Pi release in gating cofilin recruitment to actin filaments in S. cerevisiae. In vertebrate cells, several biochemical and biological observations suggest that additional mechanisms may also regulate cofilin recruitment to actin filaments. However, yeast cofilin appears to have fewer regulatory inputs. For example, yeast cofilin severing activity is pH insensitive and is not regulated by phosphorylation (Moseley and Goode, 2006; Pavlov et al., 2006). Therefore, its localization may be more reliant on the actin nucleotide state. This possibility is consistent with the nature of actin assembly and disassembly at the yeast cell cortex and actin's role in endocytic internalization. Rather than a steady-state cycle of actin assembly and disassembly to generate a treadmilling array of filaments that drives the regulated protrusion of a leading edge, during endocytosis, a transient burst of actin assembly followed almost immediately by complete disassembly of the actin filaments drives plasma membrane invagination and the formation of endocytic vesicles (Kaksonen et al., 2006). Our results support the conclusion that nucleotide hydrolysis and Pi release act as an intrinsic timer for actin disassembly, providing a simple yet effective mechanism for regulating actin disassembly at endocytic sites.
The observation that Xenopus keratocyte velocity does not affect cofilin localization at the leading edge (Svitkina and Borisy, 1999) implies that cofilin recruitment is not tightly coupled to Pi release rates in keratocytes because the zone of cofilin-free actin filaments would be predicted to increase or decrease in size with increases and decreases in cell velocity, respectively. Moreover, during the assembly of vertebrate actin, there is a delay on the order of several minutes between ATP hydrolysis and Pi release (Carlier and Pantaloni, 1986). It has been suggested that cofilin may accelerate Pi release, which is consistent with its cooperative binding to actin filaments (Blanchoin and Pollard, 1999).
In contrast and consistent with our observations of rapid cofilin recruitment to actin structures in living yeast cells, a delay in Pi release subsequent to actin assembly and ATP hydrolysis is not observed with yeast actin in vitro (Yao and Rubenstein, 2001). Consistent with this result, yeast cofilin binding to yeast actin is noncooperative (Bobkov et al., 2002). Although these observations do not exclude a role for yeast cofilin in accelerating Pi release, they do suggest that ATP hydrolysis and subsequent Pi release gate cofilin recruitment to actin filaments at endocytic sites.
The markedly reduced and uniform association of cofilin with V159N actin filaments, which we have argued resemble filaments in the ADP-Pi state based on biochemical and structural studies (Belmont and Drubin, 1998; Belmont et al., 1999), supports a role for Pi release in gating the association of cofilin with actin filaments. An additional contribution to the delay in cofilin's association with actin filaments in vivo subsequent to assembly might be made by slow binding of cofilin to actin filaments. Although cofilins have been reported to bind somewhat slowly to actin filaments in vitro, we note that yeast cofilin binds to rabbit skeletal muscle ADP–F-actin >80 times faster than amoeba actophorin binds to amoeba ADP–F-actin (2.5 μM−1 s−1 and 0.03 μM−1 s−1, respectively; Blanchoin and Pollard, 1999; Bobkov et al., 2002). A detailed kinetic analysis of yeast cofilin binding to yeast ATP- and ADP–F-actin will be invaluable for fully dissecting the mechanisms that dictate the kinetics of cofilin binding at endocytic sites.
Cofilin's effects on actin subunit flux in actin networks in vivo
A sensitive test of cofilin's in vivo function is a detailed analysis of the effects of cofilin mutations on actin subunit flux in cells. We found that cofilin influences the morphology of actin comet tails and rates of actin flux through filament meshworks in vivo. The cof1-22 allele does not change total cellular actin levels (Lappalainen and Drubin, 1997) but alters the balance of actin in the cell toward assembled F-actin structures at the expense of the G-actin pool, leading to reduced rates of actin assembly proximal to the plasma membrane. The actin comet tails maintain a constant but elongated length. Therefore, an alternate steady state is reached with slowed but balanced rates of actin assembly and disassembly.
Cofilin may contribute to actin assembly and flux rates by generating new barbed ends via filament severing. However, during normal cycles of cortical actin assembly at endocytic sites, cofilin is absent from endocytic sites when actin is assembling and arrives concomitant with the onset of disassembly. In addition, several observations suggest that barbed ends are not limiting for actin assembly associated with endocytic proteins. First, mutations in Arp2/3 complex subunits that perturb actin filament nucleation and branching activity in vitro did not affect actin flux rates in actin comet tails in sla2Δ cells (Martin et al., 2005). Second, low levels of lat A can mimic the defects in actin patch assembly seen in cof1-22 mutant cells (Fig. S5 and Video 6, available at http://www.jcb.org/cgi/content/full/jcb.200703092/DC1). Together, these data suggest that actin filament barbed end concentration is not limiting for actin filament flux in vivo and suggest that slowed actin assembly in vivo in cofilin mutants arises as a result of sequestration of the assembly-competent actin monomer pool in F-actin. This hypothesis is further strengthened by the evidence that cofilin-generated actin filament barbed ends are capped by Aip1p in vitro (Okada et al., 2002; Balcer et al., 2003). Aip1p and cofilin colocalize in cortical patches and distal to the cell cortex in actin comet tails, and they show identical temporal association with actin during endocytic internalization (unpublished data). Collectively, these observations provide evidence that the rate-limiting step for the assembly/disassembly cycle of actin networks in vivo is the cofilin-mediated disassembly of actin filaments required for maintenance of the actin monomer pool.
Aip1p has been proposed to function in association with cofilin to control assembly dynamics at barbed filament ends generated by filament severing (Rodal et al., 1999; Okada et al., 2002; Balcer et al., 2003). The observation that aip1Δ sla2Δ cells have no drastic alterations in actin tail morphology or flux rates raises questions about the role of Aip1p and cofilin in actin filament capping and disassembly. On one hand, the lack of a phenotype could be the result of a compensatory up-regulation of actin disassembly as mediated by cofilin or other factors. Alternatively, this result may reflect the possibility that free barbed ends are not limiting for actin flux rates in vivo (Martin et al., 2005). Utilization of novel assays to describe Aip1p's contribution to actin filament turnover in Arp2/3-generated fluxing arrays of actin filaments in living cells will be required to define its function more clearly.
Actin flux is essential for multiple steps in the endocytic pathway
The effects of decreasing the actin monomer pool and actin subunit flux rates fivefold are manifested in vivo by the inability of cells to efficiently transport fluid-phase cargo to the vacuole even though plasma membrane internalization and receptor- mediated endocytosis still occur. Examination of the dynamics of endocytic coat proteins at the cell cortex revealed that endocytic components still assemble at the cell cortex and are internalized. Surprisingly, disassembly of the endocytic coat was not delayed, although disassembly of actin was markedly perturbed, resulting in the prolonged association of actin and actin-binding proteins with internal membranes. This phenotype is partially similar to that caused by mutations of the Ark/Prk protein kinases, which lead to the accumulation of vesicular membranes in the cytoplasm that are associated with actin filaments (Sekiya-Kawasaki et al., 2003). However, the internal membranes in ark1 prk1 mutants also accumulate endocytic coat components, such as Sla1p and Pan1p, and the WASP (Wiskott-Aldrich syndrome protein)-related protein Las17p. Ark/Prk protein kinases are important for attenuating actin filament polymerization through the Arp2/3 activator Pan1p and for disassembly of the endocytic coat module (Zeng et al., 2001; Sekiya-Kawasaki et al., 2003; Toshima et al., 2005). These observations demonstrate that cofilin-mediated actin disassembly can be uncoupled from endocytic coat disassembly, which implies that endocytic coat disassembly mechanisms are independent of actin disassembly mechanisms.
We suspected that later events in the endocytic pathway are perturbed by the decrease in actin subunit flux rates in cof1-22 cells. A previous study examining the defects in endocytic internalization in this mutant used the fluid-phase endocytic marker Lucifer yellow to show defects in the vacuolar accumulation of fluid-phase cargo (Lappalainen and Drubin, 1997). However, this marker does not provide information regarding the state of membranes between the plasma membrane and the vacuole. Therefore, we used the observation that cof1-22 cells are able to internalize FM4-64 to examine the morphology of the endocytic pathway. Consistent with defects in late trafficking events, drastic morphological alterations of the endocytic pathway were observed in cof1-22 cells. Accordingly, these cells were unable to properly traffic the soluble vacuolar hydrolase CPY to the lumen of the vacuole, resulting in its secretion. However, cof1-22 cells were able to properly traffic alkaline phosphatase to the vacuole. These data suggest that trafficking to the vacuole is not completely blocked by defects in actin subunit flux but is slowed, which leads to defects in the timely delivery of soluble vacuolar resident proteins. The prolonged association of Abp1/actin with internalized membranes raises the possibility that cofilin-mediated actin filament disassembly is important to disassemble actin filaments associated with internalized endocytic membranes, which is important for steps in fluid-phase endocytosis such as vesicle–endosome fusion. Alternatively, regeneration of the actin monomer pool may be important for late vacuolar delivery dynamics, which is consistent with in vitro work implicating actin dynamics in vacuolar fusion events (Eitzen et al., 2002).
Our observations lead us to propose a model (Fig. 7) for the timing of actin-associated events during clathrin- and actin- dependent endocytosis in yeast, wherein actin nucleotide dynamics modulate cofilin association with dynamic actin networks. Cofilin associates preferentially with aged F-actin and facilitates its rapid disassembly. This rapid disassembly is essential for removing actin from internalized endocytic compartments and replenishment of the assembly-competent G-actin pool, the dynamics of which are important for new rounds of actin filament polymerization at the cell cortex and later trafficking events to the vacuole.
Figure 7.

Model for cofilin function during endocytosis. Recruitment of the endocytic coat complex and Arp2/3 activators/myosin stimulates actin filament assembly, which drives invagination of the plasma membrane (Kaksonen et al., 2005; Sun et al., 2006). Actin nucleotide dynamics control cofilin association with F-actin and facilitate rapid turnover of aged filaments. The rapid turnover of F-actin from postinternalization endocytic membranes is essential for replenishment of the assembly-competent G-actin pool, the dynamics of which are important for new rounds of assembly at the cell cortex and for downstream membrane-trafficking events to the vacuole.
Materials and methods
Media, plasmids, and strains
Yeast strains used in this study are listed in Table S1 (available at http://www.jcb.org/cgi/content/full/jcb.200703092/DC1). Yeast strains were grown in standard rich media (YPD) or synthetic dextrose media (SD) supplemented with the appropriate amino acids. CasAA media is selective for uracil prototrophy and is composed of 1% casamino acids (Difco), 0.7% yeast nitrogen base (Difco), and 2% dextrose. All strains were cultured at 25°C unless otherwise noted. T. Doyle (Stanford University, Stanford, CA) provided the ACT1-GFP plasmid, and H. Pelham (Medical Research Council Laboratory of Molecular Biology, Cambridge, UK) provided the PHM5-GFP plasmid. The GFP-cofilin construct, which has S65T-GFP and a 12–amino acid linker inserted in between amino acids N74 and G75 of cofilin, was made as follows. Genomic DNA from DDY426 was used as a template for PCR to generate a COF1 DNA fragment corresponding to 750 bp upstream of the translational start site and 400 bp downstream of the translational start flanked by BamHI and SpeI restriction sites at the 5′ and 3′ ends, respectively, which were introduced into the primers. A second COF1 fragment was generated corresponding to the region 401 bp downstream from the translational start and 350 bp downstream of the translational stop flanked by NotI and SacII sites at the 5′ and 3′ ends, respectively, which were introduced into the primers. pFA6a-GFP(S65T)-kanMX6 was used as a template to generate by PCR a fragment of DNA corresponding to GFP(S65T) flanked by a linker sequence encoding the amino acids GHGTGSTGSGSS and flanked by SpeI and NotI sites at the 5′ and 3′ ends, respectively. These constructs were cloned into pRS426 to generate pDD2090, which was used in Fig. S1, and pRS316 to generate pDD2091, which was used in the rest of this study. The entire construct was sequenced, and the only error introduced into the relevant portion of the coding sequence was a missense mutation converting the third glycine in the 5′ GFP linker into an arginine. Primer sequences used for generating this construct are provided in Table S2.
Fluorescence microscopy
All cells were grown at 25°C to early log phase in SD media lacking tryptophan (TRP [SD−TRP]) to minimize background fluorescence. Simultaneous two-color imaging and fluorescence recovery after photobleaching experiments were performed as described previously (Kaksonen et al., 2003). All images were acquired with MetaMorph software (Molecular Devices). P-values were determined using a two-tailed t test in Excel (Microsoft).
For the visualization of cofilin redistribution upon treatment with jasplakinolide and lat A, flow cells were constructed using two stacked pieces of Scotch brand double-sided tape along two edges of a concanavalin A–coated (0.1 μg/ml) coverslip (24 × 50 mm). Cells were allowed to attach to the concanavalin A–coated coverslip, and a second coverslip of the same size was placed perpendicular to the first. A 24 × 10-mm piece of Whatman filter paper (to act as a sink to allow for rapid flow through the chamber) was then attached at one end of the flow cell with vacuum grease. Cells were imaged continuously as 400 μM lat A (Sigma-Aldrich) or 50 μM jasplakinolide (Sigma-Aldrich) in 2% dextrose was flowed through the chamber on a fluorescence microscope (IX71; Olympus) equipped with a 100× 1.4 NA objective and a CCD camera (Orca II; Hamamatsu).
For simultaneous imaging of Abp1-GFP and FM4-64 internalization, FM4-64 (Invitrogen) at 8 μM in SD–TRP was perfused over cells in flow chambers as described above for visualization of cofilin redistribution upon treatment with jasplakinolide and lat A. Labeling of endocytic intermediates and the vacuole was performed essentially as described previously (Vida and Emr, 1995) with the following modifications. Cells were grown in SD–TRP at 25°C to early log phase. A final concentration of 25 μM FM4-64 was added to the cells, and aliquots were harvested at 0, 15, 30, 45, and 60 min after dye addition. The aliquots of cells were washed into ice-cold SD–TRP containing 15 mM sodium azide and 15 mM sodium fluoride and were then imaged on a microscope (IX71; Olympus) equipped as described in the previous paragraph.
For immunofluorescence of alkaline phosphatase (Pho8p), wild-type and cof1-22 cells were grown to early log phase in YPD. Cells were fixed with 4% formaldehyde for 30 min at 25°C in YPD and overnight in 4% formaldehyde, 50 mM potassium phosphate, pH 7.4, and 1 mM MgCl2. The fixed cells were treated with TEB (200 mM Tris-HCl, pH 8.0, 20 mM EDTA, and 144 mM β-mercaptoethanol) for 30 min at 30°C. Cells were spheroplasted with 0.75 mg/ml Zymolyase 20T (Seikagaku Corp.) in 1.2 M sorbitol, 50 mM potassium phosphate, pH 7.4, and 1 mM MgCl2 for 45 min at 30°C. Cells were washed twice with 1.2 M sorbitol and were treated for 2 min with 1.2 M sorbitol with 2% SDS. Cells were washed twice with 1.2 M sorbitol, allowed to settle on a chambered slide, and processed for immunofluorescence using a 1:10 dilution of monoclonal α-ALP antibodies (Invitrogen) and a 1:50 dilution of FITC-conjugated donkey α-mouse antibodies (Jackson ImmunoResearch Laboratories).
α-Factor internalization
35S-labeled α factor was prepared as described previously (Sekiya-Kawasaki et al., 2003). A continuous presence protocol was used. Cells were grown in YPD media at 25°C, harvested by centrifugation at 1,500 g for 4 min, and resuspended in internalization media (YPD media with 0.5% casamino acids and 1% BSA). Aliquots were taken at the indicated time points and diluted in ice-cold 50 mM potassium phosphate buffer at pH 6.0 (total bound α factor) or pH 1.1 (internalized α factor). The cells were collected by filtration, and counts per minute were determined in a scintillation counter (LS 6500; Beckman Coulter). Percent uptake is represented by multiplying 100 times the ratio of counts per minute at pH 1.1/pH 6.0 for each time point.
Subcellular fractionation
1.5 liters of cells were grown at 25°C to log phase (A600 = 0.5/ml) in YPD. Cells were pelleted by centrifugation at 1,500 g for 5 min and washed once with water. The cell wall was partially destabilized by treating cells at 20 OD600/ml with 100 mM Tris-HCl, pH 9.4, and 50 mM β-mercaptoethanol for 15 min at 30°C. Cells were washed once with 100 ml spheroplast buffer (1.2 M sorbitol, 20 mM potassium phosphate, pH 7.5, and 5 mM MgCl2) and resuspended to 50 OD600/ml in spheroplast buffer with 3 mg/ml Zymolyase 20T (Seikagaku Corp.). Cell walls were digested for 1–1.5 h at 30°C, whereas spheroplast formation was assayed using phase-contrast light microscopy. Cells were pelleted at 1,500 g for 5 min at 4°C, washed once with 100 ml of ice-cold spheroplast buffer, and resuspended in ice-cold membrane isolation buffer (0.6 M sorbitol, 20 mM Hepes-KOH, pH 7.5, 1 mM PMSF, and 3 mM benzamidine) to 50 OD600/ml. Cells were lysed in a Dounce homogenizer (Wheaton Scientific) with 15 strokes using the tight pestle. The resulting cell lysate was centrifuged at 300 g for 5 min at 4°C to pellet unlysed cells and other dense cell debris. Half of the supernatant (S300) was harvested and spun at 13,000 g for 10 min at 4°C, yielding the S13,000 and P13,000 fractions. Three quarters of the S13,000 was spun at 100,000 g for 1 h at 4°C to yield the S100,000 and P100,00 fractions. Fractions were solubilized in Tris urea buffer (50 mM Tris-Cl, pH 6.8, 3 M urea, 1% SDS, and 5% β-mercaptoethanol) and analyzed by SDS-PAGE followed by immunoblotting with polyclonal Abp1p and Act1p antibodies.
CPY secretion
Sterile 0.45-μm nitrocellulose filter disks were placed on YPD plates, and cells were streaked on top and allowed to grow for 10 h. The nitrocellulose disks were then washed and subjected to standard immunoblotting techniques using polyclonal CPY antibodies (provided by R. Schekman, University of California, Berkeley, Berkeley, CA).
Online supplemental material
Fig. S1 shows complementation of a cof1Δ strain by a high-copy GFP- cofilin plasmid, the inability of this strain to grow in the absence of the plasmid, and the relative amounts of GFP-cofilin in vivo when it is expressed from low or high-copy plasmids. Fig. S2 shows that native untagged cofilin is restricted from regions of actin tails engaged in active actin filament assembly. Fig. S3 shows GFP-cofilin relocalization in actin tails in sla2Δ cells upon treatment with lat A. Fig. S4 shows vacuolar morphology defects in cof1-22 cells using Phm5-GFP and FM4-64 and shows that alkaline phosphatase is properly trafficked to fragmented vacuoles in cof1-22 mutant cells. Fig. S5 shows low levels of lat A slow actin assembly at endocytic sites. Video 1 shows GFP-cofilin and Abp1-mRFP dynamics in vivo using two-color real-time image acquisition. Videos 2 and 3 show GFP-cofilin relocalization in Abp1-mRFP–labeled actin tails in sla2Δ cells treated with jasplakinolide (Video 2) and lat A or DMSO (Video 3) using two-color real-time image acquisition. Video 4 shows fluorescence recovery after photobleaching analysis of GFP-actin within actin comet tails in sla2Δ and sla2Δ cof1-22 cells using real-time acquisition. Video 5 shows Sla1-GFP and Abp1-mRFP dynamics in wild-type cells and cof1-22 mutant cells using two-color real-time image acquisition. Video 6 shows Abp1-GFP dynamics in wild-type cells treated with low levels of lat A using real-time image acquisition. Videos 7 and 8 show FM4-64 and Abp1-GFP dynamics in wild-type (Video 7) and cof1-22 mutant (Video 8) cells using two-color real-time image acquisition. Table S1 presents the yeast strains used in this study. Table S2 presents the primer sequences used to generate GFP-cofilin. Online supplemental material is available at http://www.jcb.org/cgi/content/full/jcb.200703092/DC1.
Supplementary Material
Acknowledgments
We thank Tim Doyle, Hugh Pelham, and Randy Schekman for providing valuable reagents. We are grateful to I. Le Blanc, Y. Sun, B. Young, and M. Kaksonen for their critical reading of the manuscript and to the rest of the Drubin/Barnes laboratory for helpful discussion. Confocal imaging data were acquired at the Cancer Research Laboratory Molecular Imaging Center at the University of California, Berkeley.
This work was supported by National Institutes of Health RO1 grants GM42759 and GM50399 to D.G. Drubin.
Abbreviations used in this paper: CPY, carboxypeptidase Y; F-actin, filamentous actin; lat A, latrunculin A; mRFP, monomeric RFP; SD, synthetic dextrose; TRP, tryptophan; vps, vacuolar protein sorting.
References
- Andrianantoandro, E., and T.D. Pollard. 2006. Mechanism of actin filament turnover by severing and nucleation at different concentrations of ADF/cofilin. Mol. Cell. 24:13–23. [DOI] [PubMed] [Google Scholar]
- Ayscough, K.R. 2000. Endocytosis and the development of cell polarity in yeast require a dynamic F-actin cytoskeleton. Curr. Biol. 10:1587–1590. [DOI] [PubMed] [Google Scholar]
- Balcer, H.I., A.L. Goodman, A.A. Rodal, E. Smith, J. Kugler, J.E. Heuser, and B.L. Goode. 2003. Coordinated regulation of actin filament turnover by a high-molecular-weight Srv2/CAP complex, cofilin, profilin, and Aip1. Curr. Biol. 13:2159–2169. [DOI] [PubMed] [Google Scholar]
- Belmont, L.D., and D.G. Drubin. 1998. The yeast V159N actin mutant reveals roles for actin dynamics in vivo. J. Cell Biol. 142:1289–1299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Belmont, L.D., A. Orlova, D.G. Drubin, and E.H. Egelman. 1999. A change in actin conformation associated with filament instability after Pi release. Proc. Natl. Acad. Sci. USA. 96:29–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blanchoin, L., and T.D. Pollard. 1999. Mechanism of interaction of Acanthamoeba actophorin (ADF/Cofilin) with actin filaments. J. Biol. Chem. 274:15538–15546. [DOI] [PubMed] [Google Scholar]
- Bobkov, A.A., A. Muhlrad, K. Kokabi, S. Vorobiev, S.C. Almo, and E. Reisler. 2002. Structural effects of cofilin on longitudinal contacts in F-actin. J. Mol. Biol. 323:739–750. [DOI] [PubMed] [Google Scholar]
- Carlier, M.F., and D. Pantaloni. 1986. Direct evidence for ADP-Pi-F-actin as the major intermediate in ATP-actin polymerization. Rate of dissociation of Pi from actin filaments. Biochemistry. 25:7789–7792. [DOI] [PubMed] [Google Scholar]
- Eitzen, G., L. Wang, N. Thorngren, and W. Wickner. 2002. Remodeling of organelle-bound actin is required for yeast vacuole fusion. J. Cell Biol. 158:669–679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Idrissi, F.Z., B.L. Wolf, and M.I. Geli. 2002. Cofilin, but not profilin, is required for myosin-I-induced actin polymerization and the endocytic uptake in yeast. Mol. Biol. Cell. 13:4074–4087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaksonen, M., Y. Sun, and D.G. Drubin. 2003. A pathway for association of receptors, adaptors, and actin during endocytic internalization. Cell. 115:475–487. [DOI] [PubMed] [Google Scholar]
- Kaksonen, M., C.P. Toret, and D.G. Drubin. 2005. A modular design for the clathrin- and actin-mediated endocytosis machinery. Cell. 123:305–320. [DOI] [PubMed] [Google Scholar]
- Kaksonen, M., C.P. Toret, and D.G. Drubin. 2006. Harnessing actin dynamics for clathrin-mediated endocytosis. Nat. Rev. Mol. Cell Biol. 7:404–414. [DOI] [PubMed] [Google Scholar]
- Lappalainen, P., and D.G. Drubin. 1997. Cofilin promotes rapid actin filament turnover in vivo. Nature. 388:78–82. [DOI] [PubMed] [Google Scholar]
- Lappalainen, P., E.V. Fedorov, A.A. Fedorov, S.C. Almo, and D.G. Drubin. 1997. Essential functions and actin-binding surfaces of yeast cofilin revealed by systematic mutagenesis. EMBO J. 16:5520–5530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maciver, S.K., H.G. Zot, and T.D. Pollard. 1991. Characterization of actin filament severing by actophorin from Acanthamoeba castellanii. J. Cell Biol. 115:1611–1620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin, A.C., X.P. Xu, I. Rouiller, M. Kaksonen, Y. Sun, L. Belmont, N. Volkmann, D. Hanein, M. Welch, and D.G. Drubin. 2005. Effects of Arp2 and Arp3 nucleotide-binding pocket mutations on Arp2/3 complex function. J. Cell Biol. 168:315–328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moon, A.L., P.A. Janmey, K.A. Louie, and D.G. Drubin. 1993. Cofilin is an essential component of the yeast cortical cytoskeleton. J. Cell Biol. 120:421–435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moseley, J.B., and B.L. Goode. 2006. The yeast actin cytoskeleton: from cellular function to biochemical mechanism. Microbiol. Mol. Biol. Rev. 70:605–645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okada, K., L. Blanchoin, H. Abe, H. Chen, T.D. Pollard, and J.R. Bamburg. 2002. Xenopus actin-interacting protein 1 (XAip1) enhances cofilin fragmentation of filaments by capping filament ends. J. Biol. Chem. 277:43011–43016. [DOI] [PubMed] [Google Scholar]
- Pavlov, D., A. Muhlrad, J. Cooper, M. Wear, and E. Reisler. 2006. Severing of F-actin by yeast cofilin is pH-independent. Cell Motil. Cytoskeleton. 63:533–542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perrais, D., and C.J. Merrifield. 2005. Dynamics of endocytic vesicle creation. Dev. Cell. 9:581–592. [DOI] [PubMed] [Google Scholar]
- Pollard, T.D., and G.G. Borisy. 2003. Cellular motility driven by assembly and disassembly of actin filaments. Cell. 112:453–465. [DOI] [PubMed] [Google Scholar]
- Rodal, A.A., J.W. Tetreault, P. Lappalainen, D.G. Drubin, and D.C. Amberg. 1999. Aip1p interacts with cofilin to disassemble actin filaments. J. Cell Biol. 145:1251–1264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodal, A.A., L. Kozubowski, B.L. Goode, D.G. Drubin, and J.H. Hartwig. 2005. Actin and septin ultrastructures at the budding yeast cell cortex. Mol. Biol. Cell. 16:372–384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sekiya-Kawasaki, M., A.C. Groen, M.J. Cope, M. Kaksonen, H.A. Watson, C. Zhang, K.M. Shokat, B. Wendland, K.L. McDonald, J.M. McCaffery, and D.G. Drubin. 2003. Dynamic phosphoregulation of the cortical actin cytoskeleton and endocytic machinery revealed by real-time chemical genetic analysis. J. Cell Biol. 162:765–772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stepp, J.D., K. Huang, and S.K. Lemmon. 1997. The yeast adaptor protein complex, AP-3, is essential for the efficient delivery of alkaline phosphatase by the alternate pathway to the vacuole. J. Cell Biol. 139:1761–1774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun, Y., A.C. Martin, and D.G. Drubin. 2006. Endocytic internalization in budding yeast requires coordinated actin nucleation and myosin motor activity. Dev. Cell. 11:33–46. [DOI] [PubMed] [Google Scholar]
- Svitkina, T.M., and G.G. Borisy. 1999. Arp2/3 complex and actin depolymerizing factor/cofilin in dendritic organization and treadmilling of actin filament array in lamellipodia. J. Cell Biol. 145:1009–1026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toshima, J., J.Y. Toshima, A.C. Martin, and D.G. Drubin. 2005. Phosphoregulation of Arp2/3-dependent actin assembly during receptor-mediated endocytosis. Nat. Cell Biol. 7:246–254. [DOI] [PubMed] [Google Scholar]
- Vida, T.A., and S.D. Emr. 1995. A new vital stain for visualizing vacuolar membrane dynamics and endocytosis in yeast. J. Cell Biol. 128:779–792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yao, X., and P.A. Rubenstein. 2001. F-actin-like ATPase activity in a polymerization-defective mutant yeast actin (V266G/L267G). J. Biol. Chem. 276:25598–25604. [DOI] [PubMed] [Google Scholar]
- Yarar, D., C.M. Waterman-Storer, and S.L. Schmid. 2005. A dynamic actin cytoskeleton functions at multiple stages of clathrin-mediated endocytosis. Mol. Biol. Cell. 16:964–975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeng, G., X. Yu, and M. Cai. 2001. Regulation of yeast actin cytoskeleton-regulatory complex Pan1p/Sla1p/End3p by serine/threonine kinase Prk1p. Mol. Biol. Cell. 12:3759–3772. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}