

Abstract
Phagocytosis is crucial for host defense against microbial pathogens and for obtaining nutrients in Dictyostelium discoideum. Phagocytosed particles are delivered via a complex route from phagosomes to lysosomes for degradation, but the molecular mechanisms involved in the phagosome maturation process are not well understood. Here, we identify a novel vesicle-associated receptor tyrosine kinase-like protein, VSK3, in D. discoideum. We demonstrate how VSK3 is involved in phagosome maturation. VSK3 resides on the membrane of late endosomes/lysosomes with its C-terminal kinase domain facing the cytoplasm. Inactivation of VSK3 by gene disruption reduces the rate of phagocytosis in cells, which is rescued by re-expression of VSK3. We found that the in vivo function of VSK3 depends on the presence of the kinase domain and vesicle localization. Furthermore, VSK3 is not essential for engulfment, but instead, is required for the fusion of phagosomes with late endosomes/lysosomes. Our findings suggest that localized tyrosine kinase signaling on the surface of endosome/lysosomes represents a control mechanism for phagosome maturation.
Introduction
The internalization and digestion of particulate materials by eukaryotic cells is a complex process that plays important roles in host defense against infection, development, and tissue homeostasis (Aderem and Underhill, 1999; May and Machesky, 2001; Segal, 2005; Stuart and Ezekowitz, 2005). In mammals, phagocytosis of microbial pathogens by so-called professional phagocytes such as macrophages and neutrophils is one of the earliest responses in host defense and is critical for controlling inflammation (Aderem and Underhill, 1999; May and Machesky, 2001; Segal, 2005; Stuart and Ezekowitz, 2005). Phagocytosis begins with the attachment of particles to cell surface receptors, triggering a reorganization of the plasma membrane and actin cytoskeletal components to facilitate particle internalization and the formation of a phagosome. The phagosome matures over time by sequentially interacting with early, then late endocytic vesicles and finally lysosomes, culminating in the formation of a phagolysosome in which the ingested material is degraded (May and Machesky, 2001; Segal, 2005; Stuart and Ezekowitz, 2005). The mechanisms that underlie the early steps of particle internalization and phagosome formation are relatively well understood; however, the molecular machinery regulating phagosome maturation, particularly the process of phagolysosome formation, remains unclear.
Delineating the mechanism of phagosome maturation is critical, as certain intracellular pathogens evade phagolysosomal fusion in macrophages by using a variety of strategies to alter macrophage signaling (Rosenberger and Finlay, 2003). In the case of Mycobacterium tuberculosis (Pieters and Gatfield, 2002; Rosenberger and Finlay, 2003), a number of mechanisms have been proposed for lysosome evasion, including the effects of ammonia production (Gordon et al., 1980), the close apposition between the bacterium and the vacuole membrane (de Chastellier and Thilo, 1998), the ability of surface lipids and cord factor to inhibit vesicular fusion (Fratti et al., 2001; Vergne et al., 2003), and the ability of a eukaryotic-like serine/threonine protein kinase G to inhibit phagosome/lysosome fusion in infected macrophages (Walburger et al., 2004). Other bacterial pathogens are able to alter phagosomal maturation to survive in macrophages by residing in various organelles, such as late endosomes (S. typhimurium), lysosomes (Coxiella burnetti), and rough endoplasmic reticulum (Legionella pneumophila) (Rosenberger and Finlay, 2003). The array of interfering strategies that are used by pathogens suggest that the molecular mechanisms regulating phagosome maturation is complex; this point is underscored by recent proteomic analyses of phagosome-associated proteins in mouse macrophages where more than 160 proteins were identified (Garin et al., 2001).
Cellular signaling proteins have been implicated in phagosome maturation. For example, activation of the toll-like receptor (TLR) signaling pathway by bacteria regulates phagocytosis at multiple steps including internalization and phagosome maturation (Blander and Medzhitov, 2004). In addition, lipid-modulating enzymes such as PI3-kinase and sphingosine kinase, which alter the lipid composition of phagosomal membranes, as well as the lysosome-associated tyrosine kinase Hck (hematopoietic cell kinase) have been shown to regulate phagosome maturation (Cougoule et al., 2006; Yeung et al., 2006). Extensive studies suggest that newly formed phagosomes carry signaling molecules that direct them to specifically interact with endosomes and then lysosomes. In addition to the cell surface receptors (TLRs), small GTPases such as Rab5 (Duclos et al., 2000; Fratti et al., 2001) and Rab7 (Rupper et al., 2001), and LAMP1 and LAMP2 (lysosome-associated membrane proteins) (Chen et al., 1985) play roles in inter-compartment communication, mediating the fusion process during phagosome maturation. It is likely that the surface of endosomes and lysosomes also carry signaling proteins in order to selectively fuse with incoming phagosomes. However, such proteins have yet to be determined.
The single-cell, soil amoeba Dictyostelium discoideum is a professional phagocyte that has proven to be an excellent model system for phagocytosis. D. discoideum is a genetically tractable organism with mutants often displaying clear phagocytosis phenotypes that can be easily screened (Peracino et al., 1998) and has phagocytosis rates severalfold higher than those observed in mammalian macrophages or neutrophils (Thilo, 1985). D. discoideum and mammalian phagocytes share many common molecular components that regulate engulfment and phagosome maturation. Like mammalian phagocytes, F-actin mediates the formation of the phagocytic cup and the internalization of particles, and the WASP family of actin-regulating proteins also plays important roles in regulating phagocytosis. Also in common is the localization of small Rab GTPases and LAMP proteins in phagosomes and a requirement for Rho and Ras family GTPases for regulating phagocytosis (Maniak et al., 1995; Peracino et al., 1998; Muller-Taubenberger et al., 2001; Rupper and Cardelli, 2001). Finally, pathogens that evade death in mammalian phagocytes are also able to escape killing by D. discoideum, suggesting the existence of common targets in both host cell types (Steinert and Heuner, 2005).
Tyrosine phosphorylation plays a major role in the early signaling events of phagocytosis in mammalian phagocytes (Aderem and Underhill, 1999; Stuart and Ezekowitz, 2005) and, as suggested by recent work (Cougoule et al., 2006), may function in later steps as well. In general, tyrosine kinases constitute a large family of signaling molecules and are key regulators of many cellular processes, functioning to transduce signals within and between eukaryotic cells. It is possible that certain tyrosine kinases also regulate the phagosome maturation process. In the present study, we used D. discoideum as a model system to elucidate the physiological functions of a novel receptor tyrosine kinase (RTK)–like protein we termed vesicle-associated kinase (VSK) 3. VSK3 consists of a signal peptide, a single transmembrane domain, a C-terminal kinase domain, and one N-terminal TIG (immunoglobulin-like fold) domain that is found in the MET (HGF receptor tyrosine kinase) kinase family of higher eukaryotes (Goldberg et al., 2006). This report is the first to show that an RTK-like protein localizes to the surface of late endosomes/lysosomes and may serve to mediate vesicle fusion and phagosome maturation.
Results
Identification of a receptor-like tyrosine kinase, VSK3, in D. discoideum
Using a genomic approach to identify receptor-like tyrosine kinases and study their functions in D. discoideum, we searched the D. discoideum genomic database (www.dictybase.org) and found 242 genes that encode proteins containing one of the catalytic domains characteristic of eukaryotic protein kinases. Among them, 46 genes encode the peptide sequence of HRDLXXXN, which is a signature domain in protein tyrosine kinases (Kim et al., 1999). We then analyzed the structure of the 46 putative protein kinase sequences (http://smart.embl-heidelberg.de), and found three previously uncharacterized proteins we termed VSK1, 2, and 3 (see Discussion), which possess a classic receptor kinase domain architecture of a signal peptide, a single trans-membrane domain, and a C-terminal kinase domain. In this study, we focused on the function of VSK3 (Fig. 1 A). Recently, a comprehensive genomic analysis of the protein kinases in D. discoideum also identified the same three receptor-like kinases, which were named receptor kinases (rk) 1, 2, and 3, respectively (Goldberg et al., 2006). We propose that VSK is a more suitable name for these proteins to reflect their subcellular localization and potential function (see Fig. 4 and Discussion).
Figure 1.

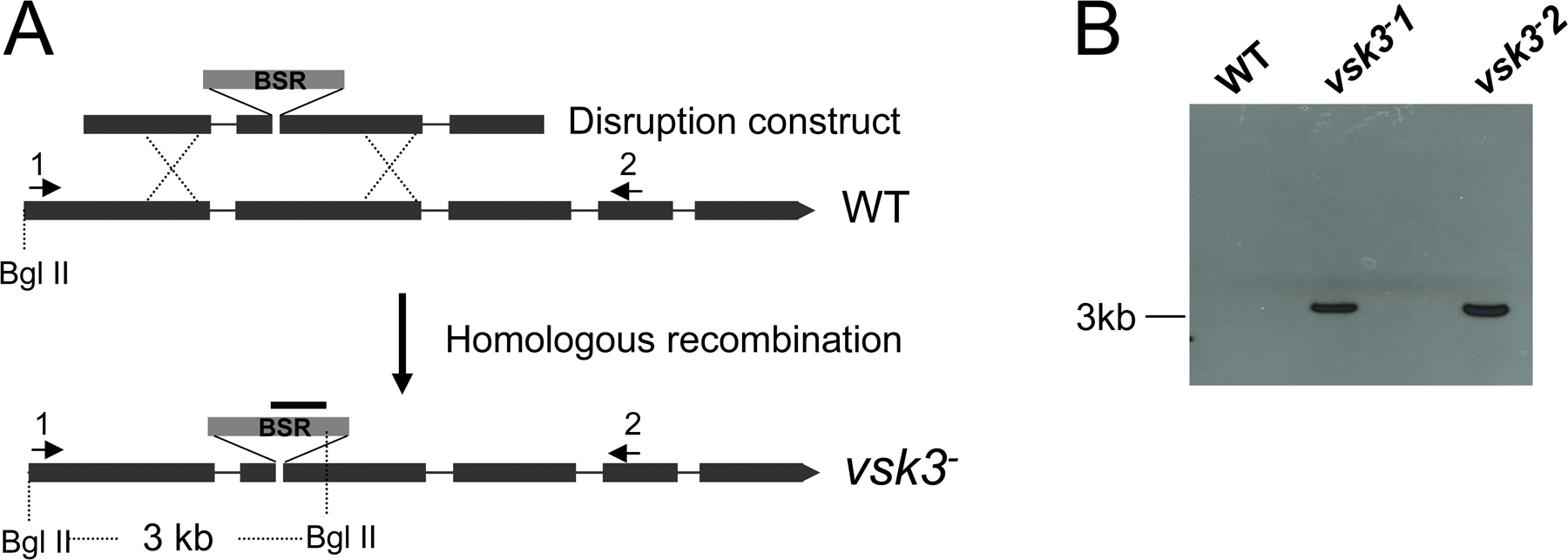
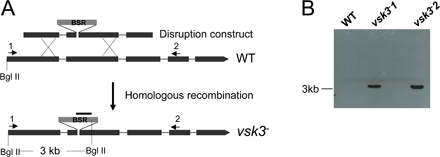
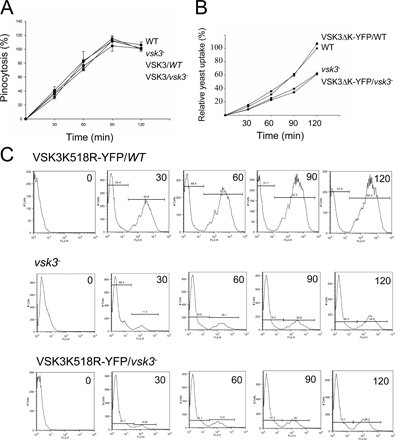
The predicted structure of VSK3, its expression pattern, and disruption are shown. (A) A schematic depiction of the VSK3 protein is shown, highlighting a signal sequence, SS, an N-terminal domain, a transmembrane domain, TM, and a C-terminal kinase domain. (B) The vsk3 mRNA levels decrease during development. Total mRNA was harvested from growing cells (0) and from cells developed in nonnutrient DB buffer with cAMP pulses for 2, 4, and 6 h and vsk3 mRNA levels were assessed by real-time PCR. (C) The predicted molecular weight increase of PCR products from genomic DNA of two vsk3 − clones relative to WT is shown. A marker lane (M) shows the 1-Kb DNA ladder. (D) Clonal plaques of WT and vsk3 − cells on bacterial lawns are shown. Bars: 5 cm. (E) The diameter of individual plaques formed by WT and vsk3 − cells were measured 6 d after cells were plated on bacterial lawns. Means and SD are shown (n = 20). A t-test indicated a statistically significant difference (*, P < 0.01). (F) Growth rates of WT and vsk3 − cells in D3T media are shown. (G) Cells fed TRITC-labeled, heated killed yeast were treated with Trypan blue and processed for fluorometric analysis at the indicated time points to measure phagocytosis rates. Fluorescence intensities were normalized to the maximal value of WT cells. Each measurement was done in triplicate and the experiment was performed three times. The mean and SD are shown from one experiment. (H) WT and the two indicated vsk3 − strains were allowed to take up FITC-dextran by pinocytosis. After washing, FITC fluorescence was measured in aliquots of total cells at the indicated time points by fluorometry.
Figure 4.

Cellular localization and topology of the VSK3 protein. Bar is 5 μm. (A) Cells expressing VSK3-YFP, VSK3ΔK-YFP, VSK3K518R-YFP, and K-YFP were incubated with TRITC-dextran to label lysosomes (Lyso) and were plated in 1-well chambers. Shown are representative cells imaged simultaneously for YFP, TRITC, and DIC by confocal microscopy. Merged images show colocalization of the YFP and TRITC signal. (B) Cells expressing VatM-GFP or VSK3-YFP were briefly incubated in the presence of 2.5 mM neutral red in DB buffer, washed, and visualized by confocal microscopy. Arrow indicates contractile vacuole. (C) The fluorescence protease protection (FFP) assay reveals the topology of the lysosomal membrane protein VSK3. Cells expressing VSK3ΔK-YFP (green) and labeled with TRITC-dextran (red) were treated with digitonin and trypsin at time 0 and were imaged simultaneously for YFP and TRITC-dextran over time. Time in seconds after addition of digitonin and trypsin is indicated at the top right corner of each image. (D) Mean and SD (n = 8) of fluorescence intensity change for YFP and TRITC-dextran after digitonin and trypsin treatment within individual cells are shown. (E) Cartoon of the FPP assay. Enlarged on the far left, is a lysosome (black circle) filled with TRITC-dextran (red) illustrating the deduced topology of VSK3-YFP with an inwardly facing N-terminal domain, and an outwardly facing C-terminal kinase domain (filled gray circle), and YFP moiety (filled green circle). The time course illustrates the selective permeabilization of the plasma membrane (gray circle) by digitonin (D) and the destruction of the YFP moiety (green circle) by the protease (P) trypsin, which leaves lysosomal membranes intact.
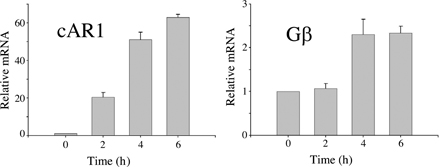
D. discoideum displays various biological behaviors during its life cycle. As free-living amoebae, D. discoideum are professional phagocytes capable of internalizing and digesting bacteria and yeast (Duhon and Cardelli, 2002). Upon starvation, amoebae enter a developmental program during which they aggregate via cAMP-mediated chemotaxis (Mahadeo and Parent, 2006). To evaluate functions of the VSK3 protein, we determined the expression profile of the vsk3 gene during differentiation using real-time PCR. The mRNA level of the vsk3 gene was relatively high in vegetative cells in rich medium, and gradually declined upon starvation (Fig. 1 B), suggesting a requirement for VSK3 in growing cells. Both cAR1 and Gβ mRNA showed the expected expression profile in control amplification experiments (Fig. S1, available at http://www.jcb.org/cgi/content/full/jcb.200701023/DC1).
VSK3 is required for efficient phagocytosis
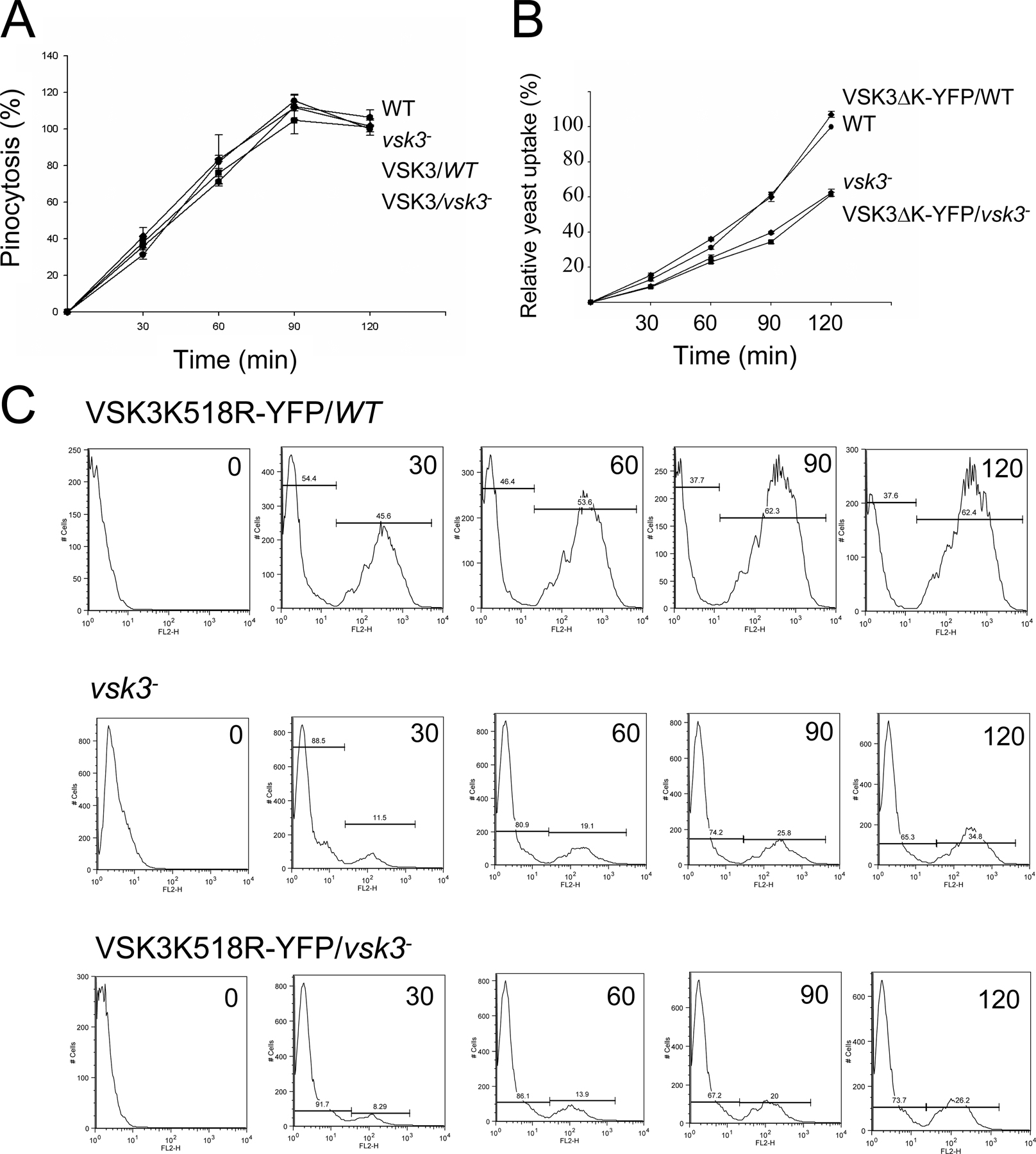
vsk3-null (vsk3 −) cells were generated through homologous recombination of the vsk3 genomic sequence flanking the blasticidin resistance cassette into the vsk3 open reading frame of wild-type (AX2) cells. Disruption mutants were identified by PCR and Southern blot analyses (Fig. 1 C; Fig. S2, A and B, available at http://www.jcb.org/cgi/content/full/jcb.200701023/DC1). To analyze potential growth and developmental phenotypes, wild-type and vsk3 − cells were grown on bacterial lawns supported on an agar substrate. The bacteria provide a food source for growing D. discoideum. As bacteria are consumed and cleared, circular plaques form on the bacterial lawns, which reflect growth and the phagocytic capacity of D. discoideum cells. As a response to the loss of nutrients in the center of plaques, cells chemotax into multicellular aggregates and ultimately differentiate into fruiting bodies consisting of dormant spore cells supported by stalk cells. When grown on bacterial lawns, we observed that vsk3 − cells form significantly smaller plaques (Fig. 1, D and E) relative to wild-type cells, suggesting a defect in growth or phagocytosis. We examined the growth rate of vsk3 − and wild-type cells in shaking, rich, liquid medium. Under these conditions, cells obtain liquid nutrients through pinocytosis (Hacker et al., 1997). We found no significant difference in either the growth rate (Fig. 1 F) or the rate of pinocytosis between strains (Fig. S3 A, available at http://www.jcb.org/cgi/content/full/jcb.200701023/DC1), suggesting that the defect in plaque size on bacterial lawns is due to impaired phagocytosis. To test for potential phagocytic defects, phagocytosis was measured as the cell-associated fluorescence intensity of TRITC-labeled yeast after washing (Fig. 1 G). Cells lacking VSK3 exhibit a clear decrease in yeast uptake in comparison to wild-type cells, indicating that the protein is required for normal phagocytosis.
To determine if VSK3 affects the acidification of endosomal vesicles, we pulse labeled wild-type and vsk3 − cells with FITC-dextran and measured the change in FITC fluorescence over time. FITC fluorescence is sensitive to pH change and FITC-dextran has been used as a probe to measure endosomal pH along the fluid-phase endocytosis pathway (Yamashiro and Maxfield, 1988). After the cells were loaded with FITC-dextran and then washed with phosphate buffer (pH 6.6) the endosomal pH dropped rapidly, reaching a minimal value of ∼pH 5.0 by 20 min and then returning to the extracellular value of pH 6.6 (Fig. 1 H), as previously reported (Aubry et al., 1993). We found no substantial difference in the kinetics of pH change as measured by FITC fluorescence in two independent vsk3 − clones (Fig. 1 H); these data indicate that VSK3 does not affect endosomal acidification.
Finally, vsk3 − cells did not display considerable developmental phenotypes. The vsk3 − cells form wild-type–like fruiting bodies in plaques on bacterial lawns and on nonnutrient agar. In addition, we found that the vsk3 − cells exhibit relatively normal chemotaxis responses to cAMP using a micropipette assay (unpublished data).
Quantitative analyses of phagocytosis in vsk3 mutants
To further analyze the phagocytic abilities of vsk3 mutants, we used a flow cytometry assay to measure uptake of heat-killed yeast fluorescently labeled with TRITC (Fig. 2). In this assay, the fluorescence of ingested yeast particles was determined after quenching the signal from uningested yeast by the addition of Trypan blue. Fig. 2 A shows representative flow cytometry results of cells incubated with TRITC-labeled yeast over a 2-h time course at 22°C. The first peak shows the background autofluorescence of cells having no internalized TRITC-labeled yeast and the appearance of the second peak over time indicates the rise in the level of ingested fluorescent yeast. Relative to wild-type, vsk3 − cells display a considerable decrease in yeast uptake during the time course with only a fraction (35.5%) of vsk3 − cells containing internalized yeast compared with 82.6% for wild-type cells by 2 h (Fig. 2 A).
Figure 2.

Quantitative analyses of phagocytosis. (A) Phagocytosis of TRITC labeled yeast of various cells was measured by FACS. Wild-type (WT) and vsk3 − cells were incubated with TRITC-labeled, heated killed yeast at room temperature to allow internalization. Samples were collected at the indicated times (min) and treated with Trypan blue to quench fluorescence of the noninternalized yeast. The left peak represents the background autofluorescence of cells and the right peak represents cells that have internalized TRITC labeled yeast particles. (B) vsk3-YFP mRNA levels were assessed by real-time PCR for the transformants and normalized to the vsk3 mRNA level in wild-type cells. Each measurement was done in triplicate and the mean and SD are shown. (C) VSK3-YFP protein expression in vsk3 − cells selected at the indicated levels of G418 was determined by probing Western blots of total cellular protein with anti-GFP antibody. (D) Expressing VSK3-YFP at the proper level rescues the phagocytosis defect of vsk3 − cells. Cells were incubated with TRITC-labeled yeast for 120 min and measured by FACS. vsk3 − and wild-type cells expressing VSK3-YFP were selected after transformation with the indicated concentration of G418 (top right corner). Each measurement was done in triplicate and all experiments were performed three times.
To rescue the vsk3-null phenotype, we transformed null cells with a nonintegrating, extrachromasomal plasmid that constitutively expresses a VSK3-YFP fusion protein (see Fig. 3 A) under the control of the actin 15 promoter. Transformants were selected with various levels of G418 to control the level of VSK3-YFP (Comer et al., 2005). As expected, increasing selective drug pressure increased the level of VSK3-YFP mRNA and VSK3-YFP protein as detected by real-time PCR and Western blot analysis, respectively (Fig. 2, B and C). The phagocytosis defect observed in vsk3 − cells (Fig. 2 A) was substantially rescued in transformants selected with the lower concentration of drug (Fig. 2 D). Interestingly, selection of transformed vsk3 − or wild-type cells with a concentration of G418 at 10 μg/ml caused a reduction in phagocytic capacity (Fig. 2, B–D). Similar negative effects on phagocytosis have been observed for both loss- and gain-of-function mutants of RacB (Lee et al., 2003), suggesting that a balance of signaling activity is required for optimal phagocytosis.
Figure 3.

VSK3 contains a functional tyrosine kinase domain. (A) A schematic depiction of VSK3, VSK3ΔK, the VSK3 kinase domain, K, and VSK3K518R tagged with YFP is shown (SS, signal sequence; TM, transmembrane domain). (B) Lysates of cells expressing VSK3-YFP, VSK3ΔK-YFP, K-YFP, and VSK3K518R were mixed with protein G beads that were coupled with anti-GFP antibodies (rabbit), and bound proteins were analyzed by immunoblotting with anti-GFP antibodies (mouse). The lysate of parental cells was used as control. (C) The relative levels of VSK3-YFP, VSK3ΔK-YFP, and K-YFP from equivalent cell lysates were quantified from band intensities on immunoblots. The measurement was done three times and the mean and SD are shown. (D) Relative tyrosine kinase activity (counts per min, cpm) were measured from immunoprecipitates of equal lysates from cells expressing VSK3-YFP, VSK3ΔK-YFP, VSK3K518R, and K-YFP compared with positive (Src) and negative kinase controls provided by the SignaTECT kit (see Materials and methods). (E) Relative Ser/Thr kinase activity was measured from immunoprecipitates of equal lysates from cells expressing VSK3-YFP, VSK3ΔK-YFP, VSK3K518R, and K-YFP compared with positive (MAPK) and negative kinase controls provided by the KinEASE FP kit (see Materials and methods).
The VSK3 tyrosine kinase domain is required for phagocytosis
To measure protein kinase activity and to determine the localization of VSK3 in vivo, we created a VSK3 fusion protein tagged at its C terminus with YFP (VSK3-YFP). For controls, we deleted the kinase domain from VSK3-YFP (VSK3ΔK-YFP), deleted the N terminus and transmembrane domain to fused the kinase domain to YFP (K-YFP) and mutated a conserved lysine to arginine in the presumptive ATP binding site (VSK3K518R-YFP; Fig. 3 A). The K518R point mutation was based on previous studies that demonstrated this conversion abrogates kinase activity (Kamps and Sefton, 1986; Weinmaster et al., 1986; Ebina et al., 1987). As shown above, VSK3-YFP rescues the phagocytosis defect of vsk3 − cells (Fig. 2 A), indicating that YFP moiety does not interfere with VSK3 function in vivo. In contrast, expressing VSK3ΔK-YFP or VSK3K518R-YFP in vsk3 − cells does not rescue the defect (Fig. S3, B and C), showing that the kinase domain is required for VSK3 function.
To assay VSK3 kinase activity, VSK3-YFP, VSK3ΔK-YFP, K-YFP, and VSK3K518R-YFP were partially purified by immunoprecipitation from equal numbers of transformed D. discoideum cells selected with 10 μg/ml of G418. Western blot analyses of immunoprecipitates showed that the level of VSK3-YFP protein was consistently fourfold lower than that of K-YFP and VSK3ΔK-YFP expressed in the same parental cells (Fig. 3, B and C). Both VSK3-YFP and K-YFP were able to phosphorylate the tyrosine kinase–specific peptide substrate 60% as well as the control mammalian Src tyrosine kinase (Fig. 3 D), but did not show any detectable activity in a similar Ser/Thr kinase assay (Fig. 3 E). We are unable to offer a simple explanation as to why immunoprecipitates from cells expressing K-YFP did not yield more kinase activity relative to the amount of input protein when compared with the full-length VSK3-YFP; synergism from the N-terminal domain or associated proteins may account for the relative higher level of kinase activity from VSK3-YFP immunoprecipitates. As expected, neither VSK3ΔK-YFP nor VSK3K518R-YFP showed detectable protein kinase activity in either assay (Fig. 3, D and E; Fig. S4, available at http://www.jcb.org/cgi/content/full/jcb.200701023/DC1). Together, these results demonstrate that VSK3 contains an active tyrosine kinase domain that is required for its normal function.
VSK3 resides in late endosomal/lysosomal membranes with its kinase domain facing the cytoplasm
To study the subcellular distribution of VSK3 in live cells, we separately expressed VSK3-YFP, VSK3ΔK-YFP, VSK3K518R-YFP, and K-YFP in both wild-type and vsk3 − cells, and examined fluorescence in growing cells using confocal laser scanning microscopy (Fig. 4). VSK3-YFP, VSK3ΔK-YFP, and VSK3-K518R-YFP were observed at the periphery of intracellular vesicles, while K-YFP was seen in the entire cytosol. These findings indicate that sequence determinants outside of the kinase domain dictate VSK3 localization. We incubated these cells with TRITC-dextran, which is taken into cells by pinocytosis and selectively labels late endosomes and lysosomes (Padh et al., 1993; Rodriguez-Paris et al., 1993), and found that VSK3-YFP, VSK3ΔK-YFP, and VSK3-K518R-YFP localize to the surface of dextran-labeled vesicles (Fig. 4 A). Together, these data suggest that both VSK3 localization and kinase activity are required together for its normal functions (Fig. 3 and Fig. 4 A; Fig. S3, B and C; unpublished data).
To further characterize VSK3-containing vesicles, we loaded wild-type cells expressing VSK3-YFP and, as a control, cells expressing VatM-GFP with neutral red, which accumulates in acidic vesicles (Clarke et al., 2002). VatM is the 100-kD transmembrane subunit of the vacuolar H(+)-ATPase and localizes to both contractile vacuoles and acidic endosomal vesicles that fuse with phagosomes (Clarke et al., 2002). As expected, neutral red colocalizes with VatM-GFP in endosomal vesicles (Fig. 4 B), with the exception of contractile vacuoles (Fig. 4 B, arrow). We found that neutral red also accumulates in VSK3-YFP labeled vesicles, indicating that VSK3 resides in acidic compartments (Fig. 4 B).
It seemed likely that VSK3′s N terminus would be oriented toward the interior of vesicles given the existence of a putative N-terminal signal peptide and a single transmembrane domain, thus enabling the kinase domain to reside in the cytoplasm where ATP is plentiful. To test this prediction, we used a recently established fluorescence protease protection (FPP) assay used to study the topology of transmembrane proteins within live cells (Lorenz et al., 2006). Cells expressing VSK3-YFP and labeled with TRITC-dextran were exposed to digitonin, which specifically permeablizes the plasma membrane but not membranes of intracellular organelles, and were then treated with trypsin to degrade unprotected proteins and protein moieties within the cytosol (Lorenz et al., 2006). After digitonin and trypsin treatment, TRITC-dextran fluorescence persisted (Fig. 4, C and D), indicating that lysosomes remained intact. If VSK3-YFP were oriented in the membrane such that the YFP moiety was inside the vesicle, it would be protected from trypsin digestion and its fluorescence would persist. However, we observed that the YFP fluorescence was rapidly lost after digitonin and trypsin treatment (Fig. 4, C and D), showing that VSK3 is oriented with its N terminus within late endosomes/lysosomes and its C-terminal tyrosine kinase domain facing the cytosol (Fig. 4 E).
To determine if loss of VSK3 incurs ultrastructural changes in cells, we imaged wild-type and vsk3 − cells expressing VSK3ΔK-YFP by transmission electron microscopy (TEM). VSK3ΔK-YFP could be used as a marker for VSK3 containing vesicles in both wild-type and vsk3 − cells as our data indicate it does not rescue the null phenotype (see Fig. S3 B; unpublished data). The YFP epitope was detected with an anti-GFP primary and a gold-conjugated secondary antibody. In control experiments, some nonspecific nucleation of the enhancement reagent occurred on material other than gold during the particle enhancement step (unpublished data) and nonspecific staining was observed throughout wild-type cells not expressing VSK3ΔK-YFP when both primary and secondary antibodies were used (Fig. 5). In cells expressing the VSK3 marker, gold particles were found in high density around intracellular vesicles in bold wild-type and null cells well above background staining (Fig. 5). These data are consistent with VSK3ΔK-YFP localization as determined by epifluorescence microscopy (see Fig. 4 A). The morphology of positively staining vesicles varied in shape and size, ranging from 0.1 to 1 μm, but no striking differences were apparent between vesicles observed in wild-type and vsk3 − cells. Finally, in support of our FPP assay results (see Fig. 4 C), high magnification images of individual vesicles revealed that gold particles accumulated on the outside of vesicles (Fig. 5), confirming that the C terminus of VSK3 is positioned toward the cytoplasm.
Figure 5.

Loss of vsk3 doss not incur ultrastructural change in vesicles. Vegetative wild-type cells (WT-control), WT cells expressing VSK3ΔK-YFP (VSK3ΔK/WT), and vsk3 − cells expressing VSK3ΔK-YFP (VSK3ΔK/vsk3 −), which does not rescue the null phenotype, were fixed, probed with a rabbit anti-GFP antibody followed by a gold-conjugated secondary antibody to reveal VSK3-containing vesicles in electron micrographs. Arrows indicates the vesicles. Low and high magnifications are shown.
Formation of the actin-dependent phagocytic cup is normal in vsk3 − cells
To determine whether VSK3 is involved in actin-mediated internalization during the early steps of phagocytosis, we visualized the formation of the phagocytic cup around TRITC-labeled yeast particles in wild-type and vsk3 − cells expressing coronin-GFP, which is known to colocalize with F-actin during particle engulfment (Maniak et al., 1995). At the point of attachment, the formation of phagocytic cup can be seen as an accumulation of coronin-GFP around the yeast. Once internalized, coronin-GFP rapidly dissociates from the newly formed phagosome. In vsk3 − cells, as in wild-type cells, coronin-GFP rapidly accumulates at the site of attachment of yeast and remains in association throughout the engulfment process, and then disappears once the yeast is internalized in the newly formed phagosome (Fig. 6). We recorded and quantified the time from the formation of phagocytic cup to the formation of the phagosome in both wild-type (107 s, SD = 14.8 s; n = 10) and vsk3 − (102 s, SD = 10.3 s, n = 10) cells, and found that there was no considerable difference between them. Based on these observations, we conclude that VSK3 is not essential for this step of phagocytosis.
Figure 6.

Dynamic formation of actin-dependent phagocytic cups in wild-type (WT) and vsk3− cells. WT and vsk3 − cells expressing coronin-GFP were fed with TRITC-labeled, heat-killed yeast. Individual cells were imaged simultaneously for GFP and TRITC fluorescence during a time course by confocal microscopy shown left to right at 30-s intervals. See Videos 1 and 2 (available at http://www.jcb.org/cgi/content/full/jcb.200701023/DC1).
VSK3 plays a role in fusion between early phagosomes and lysosomes
To further explore VSK3′s function in phagocytosis, we tracked the dynamic localization of VSK3-containing vesicles as cells were fed heat-killed, Texas red–labeled yeast with confocal microscopy. We used VSK3ΔK-YFP as a marker because the expression of full-length VSK3-YFP to detectable levels for epifluorescent microscopy exerts a dominant-negative effect on phagocytosis (see Fig. 2 D), while VSK3ΔK-YFP has no effect on phagocytosis and localizes to the same vesicles (see Fig. S3 A and Fig. 4 A). VSK3ΔK-YFP is not detected around yeast particles during the early phase of phagocytic cup formation, but within a minute of full engulfment, VSK3ΔK-associated late endosomes/lysosomes began to surround the yeast-containing phagosome and by five minutes complete fusion occurred (Fig. 7 A). The kinetics of vesicle fusion we observed is consistent with previously reported rates of phagosome–lysosome fusion in D. discoideum (Souza et al., 1997). These results suggest that VSK3 may be involved in the fusion process between phagosomes and late endosomes/lysosomes. Finally, images captured 20 min after feeding show that VSK3ΔK-YFP still surrounds yeast containing phagosomes, again consistent with late stage localization (Fig. 7 B).
Figure 7.

Late endosomes/lysosomes containing VSK3ΔK-YFP fuse with phagosomes containing newly ingested yeast. (A) Wild-type (WT) cells expressing VSK3ΔK-YFP were fed heat killed, TRITC-labeled yeast and were imaged over time by confocal microscopy to capture the engulfment process, the formation of a phagosome, and phagosome-late endosome/lysosome fusion. The first nine panels show a merged YFP (green), TRITC (red), and DIC time series of a cell ingesting a yeast particle; time in seconds is indicated. The last three frames (time points indicated) are from the same time series without the DIC channel. See Video 3 (available at http://www.jcb.org/cgi/content/full/jcb.200701023/DC1). (B) Higher resolution image of cells in (A) 20 min after feeding showing VSK3ΔK-YFP surrounding yeast filled phagosomes.
We then compared the percentage of phagosome–lysosome fusion between wild-type and vsk3 − cells. We monitored the maturation of phagosomes containing Texas red–labeled yeast (red) by their ability to colocalize with lysosomes labeled with FITC-dextran (green) over time (Fig. 8). In wild-type cells, most internalized yeast colocalized with FITC-dextran by 1 h (Fig. 8 A). In vsk3 − cells, fewer yeast were internalized, and those that were rarely colocalized with FITC-dextran (Fig. 8 A). Fig. 8 B shows a quantitative analysis of colocalization of yeast and FITC-dextran images of wild-type and vsk3 − cells as a scatter diagram, demonstrating a high degree of colocalization (linearity) in wild-type but not in vsk3 − cells. We also quantified the percentage of phagosome-lysosome fusion by counting the percentage of colocalized Texas red–yeast and FITC-dextran in wild-type and vsk3 − cells using fluorescence microscopy (Fig. 8 C). Consistently, vsk3 − cells had significantly fewer yeast colocalized with lysosomes, demonstrating that without functional VSK3, cells are defective in the fusion of phagosomes and lysosomes.
Figure 8.

Phagosome–lysosome fusion in wild-type (WT) and vsk3− cells. (A) WT and vsk3 − cells were incubated with FITC-dextran (green) for 3 h to label lysosomes and were fed Texas red–labeled, heated killed yeast (red). After washing away uningested yeast, cells were imaged for lysosomes, yeast and DIC using confocal microscopy. Merged images show colocalization of yeast and FITC-dextran in WT but not in vsk3 − cells. Bars: 10 μm. (B) Quantitative analysis of colocalization of FITC-dextran and Texas red–yeast images as a scatter diagram (LSM 510META software; Carl Zeiss MicroImaging, Inc.). The scatter diagram shows three fluorescence positive regions: region 1: FITC positive and Texas red negative; region 2: FITC negative and Texas red positive; and region 3: FITC and Texas red positive. In region 3, FITC-dextran and Texas red–yeast display a high degree of colocalization in WT but not vsk3 − cells. The color bar shows colocalization frequency between the two channels. (C) Phagosome– lysosome fusion is significantly reduced in vsk3 − cells. Lysosomes were prelabeled with FITC-dextran in WT and vsk3 − cells and cells were fed Texas red–labeled yeast as in (A). 100 cells with internalized yeast were imaged by confocal microscopy and the percentage of intracellular yeast that colocalized with FITC-dextran was calculated and expressed as mean and SD for six independent experiments. Asterisk indicates a significant difference relative to WT (P < 0.01).
Discussion
Although intense study of mammalian phagocytes and D. discoideum has uncovered numerous molecules that regulate the early events of phagocytosis, signaling pathways that mediate the later events of phagosome maturation have yet to be clearly defined. In this study, we have identified and characterized a novel RTK-like kinase, VSK3, that localizes to late endosomes/lysosomes (see Fig. 4). VSK3 has a characteristic RTK topology, spanning the membrane of intracellular vesicles once with a lumenal N-terminal domain and a cytoplasmic C-terminal kinase domain (see Fig. 1 A, Fig. 4, and Fig. 5). The kinase domain actively phosphorylates tyrosine residues (see Fig. 3), consistent with a localized signaling role for VSK3 on the cytosolic face of vesicles. VSK3 is required for normal phagocytosis but the defect lies in the number of cells in a population that can ingest particles (see Fig. 1 G and Fig. 2). It is possible that vsk3 − cells cannot efficiently bind to particles, but once attachment occurs, actin-mediated phagocytic cup and phagosome formation are indistinguishable from that observed in wild-type cells (see Fig. 6). VSK3 does not appear to regulate fluid phase uptake or processing (Fig. 1 H; Fig. S3 A); however and most significantly, in null cells that do ingest particles, it appears that VSK3 is required for phagosome maturation as phagosome and late-endosome/lysosome fusion is significantly impaired (see Fig. 8).
In mammalian cells, tyrosine phosphorylation plays an essential role during the earliest stages of phagocytosis in the clustering of Fcγ receptors during particle recognition and uptake and is accomplished by Src and Syk families of tyrosine kinases (Aderem and Underhill, 1999; Kwiatkowska and Sobota, 1999). In addition to these early functions, another Src-family tyrosine kinase, Hck, was shown to be involved in phagosome maturation in macrophages (Astarie-Dequeker et al., 2002). Hck, although not an RTK-like molecule, localizes to a subset of lysosomal vesicles that specifically fuse with phagosomes containing particles whose entry is mediated by Fcγ receptor signaling, implicating a general role for tyrosine kinase signaling throughout the phagocytic pathway.
Metazoans encode a wide variety of single-pass, transmembrane tyrosine kinases. These prototypic RTKs localize to the plasma membrane and function to transduce extracellular signals to intracellular pathways, and only a few have been implicated in phagocytosis. Distinct from mammalian RTKs, VSK3 localizes to intracellular vesicles, but contains an N-terminal, immunoglobulin-like fold TIG domain (Goldberg et al., 2006), which, according to our work (see Fig. 5), would face the lumen of vesicles. The presence of TIG domain makes VSK3 most similar to the MET family of mammalian RTKs (Goldberg et al., 2006), which includes RON kinase, an RTK expressed in macrophages that functions, among other roles, in attenuating the inflammatory response (Correll et al., 1997). In comparison to higher eukaryotes, D. discoideum encodes a limited number of potential RTK-like molecules (Goldberg et al., 2006). Including this report, functions have been attributed to seven of the nine putative kinases (Zeng et al., 2000; Chibalina et al., 2004). Although the role of two potential single pass RTKs we term VSK1 and 2 (rk1 and rk2 in Goldberg et al., [2006]) remain to be determined, it is interesting to note that when expressed as GFP fusion proteins, VSK2 localizes on the membrane of late endosomes/lysosomes like VSK3, while VSK1 is distributed in membranes of other vesicles (unpublished data), suggesting that tyrosine kinase signaling may play additional roles in vesicle trafficking and phagosome maturation.
It has long been recognized that certain pathogenic organisms escape destruction and grow in both D. discoideum and mammalian phagocytes by undermining phagosome maturation (Steinert and Heuner, 2005). Death avoidance strategies uncovered thus far indicate that different microorganisms have adapted specialized means to subvert the completion of phagocytosis. For example, M. tuberculosis disrupts early endosome autoantigen and PI3K function, thus inhibiting trafficking downstream of Rab5 (Fratti et al., 2001), whereas Salmonella inhibits the trafficking of NADPH oxidase and nitric oxide synthase, two antimicrobial enzymes (Vazquez et al., 2000). Due to its role in phagosome maturation, it is intriguing to speculate that VSK3 may serve as a potential target by an invading pathogen to prevent late endosome/lysosome fusion.
In conclusion, our work shows for the first time that an RTK-like molecule, which resides in late endosomes/lysosomes, functions to mediate phagosome maturation. It has become increasingly clear that receptor signaling occurs not only at the plasma membrane but also along the endocytic pathway (Miaczynska et al., 2004). It may be possible that VSK3 functions like a typical receptor generating specific signals in these cytoplasmic compartments. One of numerous scenarios that can be envisaged is that signals generated in phagosomes by an engulfed particle activate VSK3 in vesicles as they fuse. In turn, activated VSK3 may regulate the downstream fate of the maturing phagosome, similar to the function of TLR9 in the response to bacterial DNA in endosomes of macrophages (Ahmad-Nejad et al., 2002). Although such functions remain to be tested, it will be interesting to determine in the future whether localized tyrosine kinase signaling represents a control mechanism for phagosome maturation in other phagocytes besides D. discoideum.
Materials and methods
Bioinformatic analysis
Tyrosine and serine/threonine kinase domains were verified according to Kim et al. (1999) in putative protein kinases as identified by the Dictyostelium Sequencing Project (Eichinger et al., 2005). Protein structural motifs were identified using DNASTAR and sequence analysis tools found at http://smart.embl-heidelberg.de/ and http://www.cbs.dtu.dk/services/SignalP/.
Cell culturing and development
For development, D. discoideum were grown in D3-T nutrient media (KD-Medical) to log phase (1–3 × 106 cells/ml) and harvested by low speed centrifugation (2000 g) for 3 min. Cells were washed in Development Buffer (DB; 7.4 mM NaH2PO4·H2O; 4 mM Na2HPO4·7H2O; 2 mM MgCl2; 0.2 mM CaCl2; pH 6.5), centrifuged and resuspended in DB to 2 × 107 cells/ml (Xu et al., 2005). For synchronous development in shaking suspension, cells were rotated at 100 rpm on a platform shaker at 22°C in DB and received exogenous 75-nM pulses of cAMP every 6 min. Development and the ability to phagocytose bacteria were assessed on bacterial lawns of Klebsiella aerogenes grown on SM-agar plates.
Real-time PCR
Cells were differentiated by exogenous cAMP pulses in DB and 108 cells were harvested at various times. Total RNA was isolated using TRIzol reagent (Invitrogen) according to manufacturer's instructions. 1 μg of DNase-treated RNA was converted to cDNA using the SuperScript first-strand synthesis system (Invitrogen). A 5% volume of the cDNA reaction was used as template for real-time PCR using a Light-Cycle thermal cycler (Roche Applied Science) and PCR products were detected with SYBR Green I. Real-time PCR conditions were according to the QIAGEN protocol except the extension temperature was 60°C. cDNA copy number was determined by using QuantiTect SYBR Green PCR kit (QIAGEN). The primers used to amplify experimental and control cDNAs are as follows: vsk3: 5′-TGACCCATATACTGAGAAAG-3′ and 5′-GACGTGTGAATGGAGCGATACT-3′; cAR1: 5′-TGGGCATCTGTCACATTTATCT-3′ and 5′-GGAACTACATTGCACATCATCAC-3′; Gβ subunit: 5′-CAGTGGTGCTTGTGATGCTA-3′ and 5′-ATGTTGTCGTGGGTGTATTG-3′.
Plasmid constructs and transformation
The vsk3 disruption construct was generated by inserting the blasticidin resistant (BSR) cassette into the vsk3 cDNA 951 nucleotides from the translation start codon and was cloned into pBluescript KS+. To create vsk3-null strains, the vsk3-BSR construct was cut and purified from the carrier plasmid and transformed into D. discoideum. Transformants were grown and selected in D3-T media containing 10 μg/ml blasticidin in 10-cm cell culture dishes. Individual colonies were picked 5–7 d later and homologous recombinants were identified by PCR and confirmed by Southern blot.
To fuse YFP to the C terminus of VSK3, the vsk3 coding sequence was amplified from cDNA with oligonucleotides that provided 5 BglII and 3 AgeI restriction sites. The amplified product was cloned into the pEYFP-N1 (CLONTECH Laboratories, Inc.) to create pVSK3-YFP. To engineer VSK3ΔK-YFP and K-YFP, phosphorylated primers were designed to anneal at locations flanking the sequence in pVSK3-YFP to be deleted and amplify the remaining plasmid. The PCR product of each reaction was ligated and transformed into TOP10 cells for propagation. For VSK3ΔK-YFP, the following sense and antisense primers were used for the deletion of aa 492–740 from VSK3: aatctctgatatatcaattggttT and gaaatcgttaaaagattgga. For K-YFP, the following primers were used delete the N-terminal domain (aa 1–481), which includes the transmembrane domain, from VSK3: cattttttaataagatctgagtccggtagcgctagc and tttgaaattaaaccaattgatatatcag. PCR fidelity was confirmed by DNA sequencing. The wild-type and mutant VSK3-YFP coding sequences were subsequently cloned into the D. discoideum expression vector pYU20, containing the G418 resistant cassette. Cells were transformed, grown and selected in D3-T media containing 20 μg/ml G418. The predicted molecular weight of the VSK3-YFP fusion protein expressed in cells was confirmed by Western blot with anti-GFP antibodies.
The invariant lysine in the predicted ATP binding site of VSK3 was mutated to arginine by a two-step fusion PCR mutagenesis. Primer 1 anneals to the 5′ end of the VSK3-YFP cDNA and includes a BglII site for subcloning and a ribosome binding site (underscored). Primer 4 anneals to the 3′ end and includes a NotI site for subcloning (underscored). Primers 2 and 3 target the invariant lysine on opposite strands of the cDNA. A single nucleotide was changed from A to G to encode the lysine to arginine conversion and a neighboring alanine (GCA) was mutated to leucine (CTT) to create a HindIII site to screen for positive mutants (underscored). For the first round of PCR, primers 1 and 3 or primers 2 and 4 were mixed with VSK3-YFP cDNA and the resulting amplification products of each reaction were gel purified. For the second, fusion PCR reaction, the purified amplification products were mixed together with the flanking primers 1 and 4. The resulting VSK3-YFP arginine mutant was subcloned into a bacterial shuttle vector and finally placed into the D. discoideum expression vector pCV5 using the BglII and NotI sites. Primer1: ACAGATCTTATTAAAAAATGATAATAATAAATAAATATATACGGATG; primer 2: TATTGTTGCAATTAGAAAGCTTAAATTATTGAATGAAGAC; primer 3: TCAATAATTTAAGCTTTCTAATTGCAACAATAATACC; primer 4: CCGCGGCCGCTTACTTGTACAGCTCGTCC.
Endosomal pH determination
Cells were loaded with FITC-dextran for 10 min at 22°C with 2.5 mg/ml FITC-dextran in phosphate buffer, pH 6.6, and washed with ice-cold phosphate buffer. Cells were then incubated at 22°C and the evolution of pH change in endosomes was monitored in aliquots over time by measuring the change in the ratio of FITC fluorescence at 518 nm when excited with 450-nm light to fluorescence at 518 nm when excited with 494-nm light. Endosomal pH values were calculated from a standard curve generated from the resulting fluorescence of FITC-dextran at differing pH values.
Phagocytosis and pinocytosis assays
Quantitative phagocytosis assays were conducted as described previously (Maniak et al., 1995). In brief, cells grown to log-phase (1–3 × 106 cell/ml) were washed and suspended to 2 × 106 cells/ml in phosphate buffer with magnesium (PM; 7.4 mM NaH2PO4-H20; 4 mM Na2HPO4-7H2O; 2 mM MgCl2) and fed TRITC-labeled, heat-killed yeast in shaking suspension (150 rpm) at 22°C. Samples were taken at indicated times and the fluorescence of noninternalized yeast was quenched by the addition of Trypan blue. Fluorescence was measured with a Perkin-Elmer spectrophotometer using 544-nm excitation and 574-nm emission filters. In addition, a flow-cytometry method was used to determine the phagocytosis rates as previously described (Ramet et al., 2002). In brief, cells grown to log-phase were fed TRITC-labeled, heat-killed yeast in PM in shaking suspension at 22°C over a time course. After incubation, the cells were quenched and flow cytometry was performed on 10,000 cells for each sample. Fluorescence data were analyzed with FlowJo software. Quantitative pinocytosis assays were performed as described previously (Hacker et al., 1997) by incubating log-phase cells with TRITC-dextran in suspension culture at 22°C. At various times, aliquots were obtained and extracellular fluorescence was quenched with Trypan blue. Internalized fluorescence was measured with a spectrophotometer as above.
Quantitation of phagosome–lysosome fusion
Quantitation of phagosome and lysosome fusion was performed as described previously (Gildea et al., 2005) with some modification. Log-phase cells were incubated with FITC-dextran (1 mg/ml) in D3-T medium for 3 h to mark lysosomes. Next, heat-killed, Texas red–labeled yeast were mixed 5:1 with D. discoideum in shaking suspension at 160 rpm for 1 h. Cells were then placed in a 4-well chamber for 20 min and allowed to adhere. The uningested yeast were washed away and cells were incubated for an additional hour. Phagosome–lysosome fusion was evaluated by confocal microscopy as a colocalization of yeast and FITC-dextran and was quantified by determining the percentage of phagosomes to phagolysosomes by randomly counting 100 yeast-containing D. discoideum cells.
Topology determination of VSK3
The membrane topology of VSK3 was determined as previously described (Lorenz et al., 2006) with some modification. Cells expressing VSK3-YFP were incubated with TRITC dextran for 3 h in shaking suspension at 160 rpm in D3-T medium to label lysosomes. Cells were placed in an 8-well Lab-Tek chamber and were imaged by confocal microscopy. A final concentration of 10 μM digitonin, to selectively permeabilize the plasma membrane, and 4 mM trypsin, to digest cytosolic-facing moieties of internal vesicle membrane proteins, were added to the chamber. Changes in the YFP and TRITC signal were recorded over time.
Immunoprecipitation and protein Tyr and Ser/Thr kinase activity assays
For immunoprecipitation of YFP-tagged proteins, 100 μl of Protein G beads were prepared (Invitrogen) by washing four times with ice-cold PBS and once with ice-cold lysis buffer (1% NP-40, 50 mM Tris, pH 7.5, 150 mM NaCl, protease inhibitors, and 1 mM Na3VO4). The beads were resuspended in 1 ml ice-cold lysis buffer and coupled to anti-GFP antibody (CLONTECH Laboratories, Inc.) for 2 h at 4°C and then washed four more times with lysis buffer. To carry out immunoprecipitation, 108 log-phase cells were washed twice with ice-cold PBS containing protease inhibitors and 1 mM Na3VO4. The cells were resuspended in 5 ml of lysis buffer and vortexed briefly every 5 min for 20 min. Insoluble matter was removed from the lysate by centrifugation (20,000 g) for 15 min. The supernatant was precleared by incubation with 100 μl of Protein G beads (no antibody coupling) for 1 h. The beads were removed by centrifugation and the supernatant was incubated with 100 μl anti-GFP-coupled beads for 3 h at 4°C. The supernatant was removed by centrifugation and the beads were washed four times with ice-cold lysis buffer. Proteins were removed from a 10-μl bed volume of beads and for Western blot analysis. The remaining beads were used to for a tyrosine kinase activity assay. The tyrosine kinase activity assay was conducted by using the SignaTECT Protein Tyrosine Kinase Assay System (Promega) according to the manufacturer's instructions. Ser/Thr kinase activity assay was conducted by using the KinEASE FP Fluorescein Green Assay (Upstate Cell Signaling Solutions) according to the manufacturer's instructions.
Confocal studies
Confocal fluorescent observations were made using a laser-scanning microscope (LSM 510 META; Carl Zeiss MicroImaging, Inc.). Either a 40×/1.3 NA or 63×/1.4 NA oil immersion objective lens was used to image cells (Xu et al., 2005). An argon 488-nm laser was used to excite GFP, YFP, and FITC fluorescence and a HeNe 543-nm laser was used for neutral red and TRITC excitation.
EM studies
Vegetative wild-type cells, wild-type cells expressing VSK3ΔK-YFP, and vsk3 − cells expressing VSK3ΔK-YFP were washed from growth media and shaken at 100 rpm for 30 min in PBS (pH 7.4). Cells were plated at a density of 75 × 105 cells/cm2 in PBS on to a circular plastic coverslip (Thermanox) placed in a well of a 24-well plate and allowed to adhere for 10 min. Cells were fixed with 1% formaldehyde, 0.1% glutaraldehyde, 0.01% digitonin, in Pipes/EGTA buffer (15 mM Pipes, pH 7.4, and 1 mM EGTA) for 15 min and were washed 2 × 2 min then 1 × 15 min in 1% formaldehyde in Pipes/EGTA. Next, cells were washed 3 × 5 min with Pipes/EGTA and then incubated in block solution (50 mM NH4Cl, 0.1% digitonin, and 1% BSA in PBS) for 30 min. Cells were incubated overnight at 4°C with rabbit anti-GFP antibody (ABCAM 290) diluted to 1:500 or 1:1,000 in block solution on a gently gyrating platform. Cells were washed 6 × 2 min then 1 × 5 min in PBS and incubated with goat anti–rabbit Nanogold 1.4-nm particle Fab fragment (Nanoprobes) diluted 1:50 in block solution for 3.5 h at 22°C on a gently gyrating platform. Cells were washed 6 × 2 min in PBS then 3 × 5 min in distilled water. Gold particle enhancer was prepared per manufacturer's protocol (Nanoprobes GEEM GoldEnhance kit) and incubated with cells for 8 min. The reaction was stopped by washing with distilled water 4 × 2 min. Cells were stored in 1% glutaraldehyde in PBS. For TEM analysis, the cells were washed 3× with 0.1 M cacodylate buffer and incubated in 0.5% OsO4, 0.8% K4Fe (CN)6 in 0.1m cacodylate buffer for 30 min. This was followed by 1 wash with 0.1 M cacodylate buffer and 2 washes with distilled water. The cells were dehydrated in ethanol series of 50, 75, 95, and 100% for 10 min, respectively. The last ethanol concentration was repeated two more times after which the cells were infiltrated with 3:1 ethanol/Spurrs resin for 1 h, 1:1 ethanol/Spurrs for 2 h, and overnight in 1:3 ethanol/Spurrs. The next day the cells were infiltrated with 100% Spurrs resin for 4 h and embedded in beam capsules at 68°C for 24 h. Ultrathin sections were obtained using the MT-7000 ultra microtome and stained with UA. The images were obtained using Philips C10 TEM.
Online supplemental materials
Fig. S1 shows the mRNA levels of the cAR1 and Gβ genes during development as detected by RT-PCR. Fig. S2. shows the cloning strategy and analysis of vsk3 disruptant mutants. Fig. S3 A shows that wild-type and vsk3 mutant strains have similar rates of pinocytosis. Fig. S3 (B and C) shows the results of phagocytosis assays of cells expressing VSK3 kinase-dead mutants. Fig. S4 shows that the VSK3K518R-YFP point mutant does not have detectable kinase activity. Videos 1 and 2 show the dynamic formation of actin-dependent phagocytic cups in wild-type (WT) (Video 1) and vsk3 − (Video 2) cells. Video 3 shows late endosomes/lysosomes containing VSK3ΔK-YFP fusing with phagosomes containing newly ingested yeast. Online supplemental material is available at http://www.jcb.org/cgi/content/full/jcb.200701023/DC1.
Supplementary Material
Acknowledgments
We thank A. Kimmel, D. Hereld, M. Dykstra, and X. Xiang for discussions and critical reading the manuscript. We thank M. Clarke for kindly providing the VatM-GFP plasmid and J. Papakrivos and T.E. Wellems for their technical assistance with pH determination. We also thank Y. Wang, T. Khurana, W. Song, A. Bagorda, C. Parent, X. Xu, L. Yi, and J. Yan for their helpful suggestions.
This research was supported by the Intramural Research Program of NIH, NIAID.
J. Fang and J.A. Brzostowski contributed equally to this paper.
Abbreviations used in this paper: FPP, fluorescence protease protection; LAMP, lysosome-associated membrane protein; RTK, receptor tyrosine kinase; TLR, toll-like receptor; VSK, vesicle-associated kinase.
References
- Aderem, A., and D.M. Underhill. 1999. Mechanisms of phagocytosis in macrophages. Annu. Rev. Immunol. 17:593–623. [DOI] [PubMed] [Google Scholar]
- Ahmad-Nejad, P., H. Hacker, M. Rutz, S. Bauer, R.M. Vabulas, and H. Wagner. 2002. Bacterial CpG-DNA and lipopolysaccharides activate Toll-like receptors at distinct cellular compartments. Eur. J. Immunol. 32:1958–1968. [DOI] [PubMed] [Google Scholar]
- Astarie-Dequeker, C., S. Carreno, C. Cougoule, and I. Maridonneau-Parini. 2002. The protein tyrosine kinase Hck is located on lysosomal vesicles that are physically and functionally distinct from CD63-positive lysosomes in human macrophages. J. Cell Sci. 115:81–89. [DOI] [PubMed] [Google Scholar]
- Aubry, L., G. Klein, J.L. Martiel, and M. Satre. 1993. Kinetics of endosomal pH evolution in Dictyostelium discoideum amoebae. Study by fluorescence spectroscopy. J. Cell Sci. 105:861–866. [DOI] [PubMed] [Google Scholar]
- Blander, J.M., and R. Medzhitov. 2004. Regulation of phagosome maturation by signals from toll-like receptors. Science. 304:1014–1018. [DOI] [PubMed] [Google Scholar]
- Chen, J.W., T.L. Murphy, M.C. Willingham, I. Pastan, and J.T. August. 1985. Identification of two lysosomal membrane glycoproteins. J. Cell Biol. 101:85–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chibalina, M.V., C. Anjard, and R.H. Insall. 2004. Gdt2 regulates the transition of Dictyostelium cells from growth to differentiation. BMC Dev. Biol. 4:8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clarke, M., J. Köhler, Q. Arana, T. Liu, J. Heuser, and G. Gerisch. 2002. Dynamics of the vacuolar H(+)-ATPase in the contractile vacuole complex and the endosomal pathway of Dictyostelium cells. J. Cell. Sci. 115:704–708 [DOI] [PubMed] [Google Scholar]
- Comer, F.I., C.K. Lippincott, J.J. Masbad, and C.A. Parent. 2005. The PI3K-mediated activation of CRAC independently regulates adenylyl cyclase activation and chemotaxis. Curr. Biol. 15:134–139. [DOI] [PubMed] [Google Scholar]
- Correll, P.H., A. Iwama, S. Tondat, G. Mayrhofer, T. Suda, and A. Bernstein. 1997. Deregulated inflammatory response in mice lacking the STK/RON receptor tyrosine kinase. Genes Funct. 1:69–83. [DOI] [PubMed] [Google Scholar]
- Cougoule, C., S. Hoshino, A. Dart, J. Lim, and E. Caron. 2006. Dissociation of recruitment and activation of the small G-protein Rac during Fcgamma receptor-mediated phagocytosis. J. Biol. Chem. 281:8756–8764. [DOI] [PubMed] [Google Scholar]
- de Chastellier, C., and L. Thilo. 1998. Modulation of phagosome processing as a key strategy for Mycobacterium avium survival within macrophages. Res. Immunol. 149:699–702. [DOI] [PubMed] [Google Scholar]
- Duclos, S., R. Diez, J. Garin, B. Papadopoulou, A. Descoteaux, H. Stenmark, and M. Desjardins. 2000. Rab5 regulates the kiss and run fusion between phagosomes and endosomes and the acquisition of phagosome leishmanicidal properties in RAW 264.7 macrophages. J. Cell Sci. 113:3531–3541. [DOI] [PubMed] [Google Scholar]
- Duhon, D., and J. Cardelli. 2002. The regulation of phagosome maturation in Dictyostelium. J. Muscle Res. Cell Motil. 23:803–808. [DOI] [PubMed] [Google Scholar]
- Ebina, Y., E. Araki, M. Taira, F. Shimada, M. Mori, C.S. Craik, K. Siddle, S.B. Pierce, R.A. Roth, and W.J. Rutter. 1987. Replacement of lysine residue 1030 in the putative ATP-binding region of the insulin receptor abolishes insulin- and antibody-stimulated glucose uptake and receptor kinase activity. Proc. Natl. Acad. Sci. USA. 84:704–708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eichinger, L., J.A. Pachebat, G. Glockner, M.A. Rajandream, R. Sucgang, M. Berriman, J. Song, R. Olsen, K. Szafranski, Q. Xu, et al. 2005. The genome of the social amoeba Dictyostelium discoideum. Nature. 435:43–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fratti, R.A., J.M. Backer, J. Gruenberg, S. Corvera, and V. Deretic. 2001. Role of phosphatidylinositol 3-kinase and Rab5 effectors in phagosomal biogenesis and mycobacterial phagosome maturation arrest. J. Cell Biol. 154:631–644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garin, J., R. Diez, S. Kieffer, J.F. Dermine, S. Duclos, E. Gagnon, R. Sadoul, C. Rondeau, and M. Desjardins. 2001. The phagosome proteome: insight into phagosome functions. J. Cell Biol. 152:165–180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gildea, L.A., G.M. Ciraolo, R.E. Morris, and S.L. Newman. 2005. Human dendritic cell activity against Histoplasma capsulatum is mediated via phagolysosomal fusion. Infect. Immun. 73:6803–6811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldberg, J.M., G. Manning, A. Liu, P. Fey, K.E. Pilcher, Y. Xu, and J.L. Smith. 2006. The Dictyostelium kinome—analysis of the protein kinases from a simple model organism. PLoS. Genet. 2:e38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gordon, A.H., P.D. Hart, and M.R. Young. 1980. Ammonia inhibits phagosome-lysosome fusion in macrophages. Nature. 286:79–80. [DOI] [PubMed] [Google Scholar]
- Hacker, U., R. Albrecht, and M. Maniak. 1997. Fluid-phase uptake by macropinocytosis in Dictyostelium. J. Cell Sci. 110:105–112. [DOI] [PubMed] [Google Scholar]
- Kamps, M.P., and B.M. Sefton. 1986. Neither arginine nor histidine can carry out the function of lysine-295 in the ATP-binding site of p60src. Mol. Cell. Biol. 6:751–757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim, L., J. Liu, and A.R. Kimmel. 1999. The novel tyrosine kinase ZAK1 activates GSK3 to direct cell fate specification. Cell. 99:399–408. [DOI] [PubMed] [Google Scholar]
- Kwiatkowska, K., and A. Sobota. 1999. Signaling pathways in phagocytosis. Bioessays. 21:422–431. [DOI] [PubMed] [Google Scholar]
- Lee, E., D.J. Seastone, E. Harris, J.A. Cardelli, and D.A. Knecht. 2003. RacB regulates cytoskeletal function in Dictyostelium spp. Eukaryot. Cell. 2:474–485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lorenz, H., D.W. Hailey, and J. Lippincott-Schwartz. 2006. Fluorescence protease protection of GFP chimeras to reveal protein topology and subcellular localization. Nat. Methods. 3:205–210. [DOI] [PubMed] [Google Scholar]
- Mahadeo, D.C., and C.A. Parent. 2006. Signal relay during the life cycle of Dictyostelium. Curr. Top. Dev. Biol. 73:115–140. [DOI] [PubMed] [Google Scholar]
- Maniak, M., R. Rauchenberger, R. Albrecht, J. Murphy, and G. Gerisch. 1995. Coronin involved in phagocytosis: dynamics of particle-induced relocalization visualized by a green fluorescent protein tag. Cell. 83:915–924. [DOI] [PubMed] [Google Scholar]
- May, R.C., and L.M. Machesky. 2001. Plagiarism and pathogenesis: common themes in actin remodeling. Dev. Cell. 1:317–318. [DOI] [PubMed] [Google Scholar]
- Miaczynska, M., L. Pelkmans, and M. Zerial. 2004. Not just a sink: endosomes in control of signal transduction. Curr. Opin. Cell Biol. 16:400–406. [DOI] [PubMed] [Google Scholar]
- Muller-Taubenberger, A., A.N. Lupas, H. Li, M. Ecke, E. Simmeth, and G. Gerisch. 2001. Calreticulin and calnexin in the endoplasmic reticulum are important for phagocytosis. EMBO J. 20:6772–6782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Padh, H., J. Ha, M. Lavasa, and T.L. Steck. 1993. A post-lysosomal compartment in Dictyostelium discoideum. J. Biol. Chem. 268:6742–6747. [PubMed] [Google Scholar]
- Peracino, B., J. Borleis, T. Jin, M. Westphal, J.M. Schwartz, L. Wu, E. Bracco, G. Gerisch, P. Devreotes, and S. Bozzaro. 1998. G protein beta subunit-null mutants are impaired in phagocytosis and chemotaxis due to inappropriate regulation of the actin cytoskeleton. J. Cell Biol. 141:1529–1537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pieters, J., and J. Gatfield. 2002. Hijacking the host: survival of pathogenic mycobacteria inside macrophages. Trends Microbiol. 10:142–146. [DOI] [PubMed] [Google Scholar]
- Ramet, M., P. Manfruelli, A. Pearson, B. Mathey-Prevot, and R.A. Ezekowitz. 2002. Functional genomic analysis of phagocytosis and identification of a Drosophila receptor for E coli. Nature. 416:644–648. [DOI] [PubMed] [Google Scholar]
- Rodriguez-Paris, J.M., K.V. Nolta, and T.L. Steck. 1993. Characterization of lysosomes isolated from Dictyostelium discoideum by magnetic fractionation. J. Biol. Chem. 268:9110–9116. [PubMed] [Google Scholar]
- Rosenberger, C.M., and B.B. Finlay. 2003. Phagocyte sabotage: disruption of macrophage signalling by bacterial pathogens. Nat. Rev. Mol. Cell Biol. 4:385–396. [DOI] [PubMed] [Google Scholar]
- Rupper, A., and J. Cardelli. 2001. Regulation of phagocytosis and endo-phagosomal trafficking pathways in Dictyostelium discoideum. Biochim. Biophys. Acta. 1525:205–216. [DOI] [PubMed] [Google Scholar]
- Rupper, A., B. Grove, and J. Cardelli. 2001. Rab7 regulates phagosome maturation in Dictyostelium. J. Cell Sci. 114:2449–2460. [DOI] [PubMed] [Google Scholar]
- Segal, A.W. 2005. How neutrophils kill microbes. Annu. Rev. Immunol. 23:197–223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Souza, G.M., D.P. Mehta, M. Lammertz, J. Rodriguez-Paris, R. Wu, J.A. Cardelli, and H.H. Freeze. 1997. Dictyostelium lysosomal proteins with different sugar modifications sort to functionally distinct compartments. J. Cell Sci. 110:2239–2248. [DOI] [PubMed] [Google Scholar]
- Steinert, M., and K. Heuner. 2005. Dictyostelium as host model for pathogenesis. Cell. Microbiol. 7:307–314. [DOI] [PubMed] [Google Scholar]
- Stuart, L.M., and R.A. Ezekowitz. 2005. Phagocytosis: elegant complexity. Immunity. 22:539–550. [DOI] [PubMed] [Google Scholar]
- Thilo, L. 1985. Quantification of endocytosis-derived membrane traffic. Biochim. Biophys. Acta. 822:243–266. [DOI] [PubMed] [Google Scholar]
- Vazquez, S., B. Garner, M.M. Sheil, and R.J. Truscott. 2000. Characterisation of the major autoxidation products of 3-hydroxykynurenine under physiological conditions. Free Radic. Res. 32:11–23. [DOI] [PubMed] [Google Scholar]
- Vergne, I., J. Chua, and V. Deretic. 2003. Tuberculosis toxin blocking phagosome maturation inhibits a novel Ca2+/calmodulin-PI3K hVPS34 cascade. J. Exp. Med. 198:653–659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walburger, A., A. Koul, G. Ferrari, L. Nguyen, C. Prescianotto-Baschong, K. Huygen, B. Klebl, C. Thompson, G. Bacher, and J. Pieters. 2004. Protein kinase G from pathogenic mycobacteria promotes survival within macrophages. Science. 304:1800–1804. [DOI] [PubMed] [Google Scholar]
- Weinmaster, G., M.J. Zoller, and T. Pawson. 1986. A lysine in the ATP-binding site of P130gag-fps is essential for protein-tyrosine kinase activity. EMBO J. 5:69–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu, X., M. Meier-Schellersheim, X. Jiao, L.E. Nelson, and T. Jin. 2005. Quantitative imaging of single live cells reveals spatiotemporal dynamics of multistep signaling events of chemoattractant gradient sensing in Dictyostelium. Mol. Biol. Cell. 16:676–688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamashiro, D.J., and F.R. Maxfield. 1998. Regulation of endocytic processes by pH. Trends Pharmacol. Sci. 9:190–193. [DOI] [PubMed] [Google Scholar]
- Yeung, T., B. Ozdamar, P. Paroutis, and S. Grinstein. 2006. Lipid metabolism and dynamics during phagocytosis. Curr. Opin. Cell Biol. 18:429–437. [DOI] [PubMed] [Google Scholar]
- Zeng, G., F.H. Nystrom, L.V. Ravichandran, L.N. Cong, M. Kirby, H. Mostowski, and M.J. Quon. 2000. Roles for insulin receptor, PI3-kinase, and Akt in insulin-signaling pathways related to production of nitric oxide in human vascular endothelial cells. Circulation. 101:1539–1545. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}