

Abstract
RNA viruses are responsible for a variety of human diseases, and the pathogenicity of RNA viruses is often attributed to a high rate of mutation. Self-cleavage activity of the wild-type hepatitis delta virus (HDV) ribozyme as measured in standard divalent ion renaturation assays is biphasic and mostly slow and can be improved by multiple rational changes to ribozyme sequence or by addition of chemical denaturants. This is unusual in the sense that wild type is the most catalytically active sequence for the majority of protein enzymes, and RNA viruses are highly mutable. To see whether the ribozyme takes advantage of fast-reacting sequence changes in vivo, we performed alignment of 76 genomic and 269 antigenomic HDV isolates. Paradoxically, the sequence for the ribozyme was found to be essentially invariant in nature. We therefore tested whether three ribozyme sequence changes that improve self-cleavage under standard divalent ion renaturation assays also improve self-cleavage during transcription. Remarkably, wild type was as fast, or faster, than these mutants under cotranscriptional conditions. Slowing the rate of transcription or adding the hepatitis delta antigen protein only further stimulated cotranscriptional self-cleavage activity. Thus, the relative activity of HDV ribozyme mutants depends critically on whether the reaction is assayed under in vivo-like conditions. A model is presented for how wild-type ribozyme sequence and flanking sequence work in concert to promote efficient self-cleavage during transcription. Wild type being the optimal ribozyme sequence under in vivo-like conditions parallels the behavior of most protein enzymes.
Keywords: ribozyme, HDV, cotranscriptional, evolution, catalysis
INTRODUCTION
Small self-cleaving catalytic RNAs, or ribozymes, are critical features of many RNA viruses. The hepatitis delta virus (HDV), a human pathogen, has genomic and antigenomic versions of a small ribozyme embedded in its 1.7-kb RNA genome (Lai 1995; Taylor et al. 1996; Taylor 2006). Other small ribozymes occur in viruses or related species, including the hairpin ribozyme, which is found in viral satellite RNAs (Buzayan et al. 1986), and the hammerhead ribozyme, which is found ubiquitously in viral satellite and viroid RNAs (Forster and Symons 1987). By virtue of their roles in rolling-circle viral replication, these small ribozymes have evolved to function cotranscriptionally, in the presence of flanking RNA sequence and viral proteins and under single turnover conditions.
Replication of hepatitis delta virus RNA is thought to occur without DNA intermediates and to be mediated by host RNA polymerase II (RNAP II). Evidence for RNAP II involvement is threefold: (1) HDV mRNA, which encodes for the hepatitis delta antigen (HDAg) protein, has a 5′ cap and a 3′ poly(A) tail; (2) HDV mRNA production is α-amanitin sensitive; and (3) RNAP II can be associated with HDV RNAs in vivo by cross-linking and coimmunoprecipitation (Taylor 2006; Greco-Stewart et al. 2007). It also appears that HDAg acts as an elongation factor for RNAP II (Yamaguchi et al. 2001; Yamaguchi and Handa 2006). Despite the in vivo relevance of these observations, the vast majority of biochemical studies on HDV and other ribozymes have been conducted on Mg2+-renatured full-length transcripts, conditions referred to herein as “standard” kinetics assays.
During rolling-circle replication of HDV, nascent concatemeric transcripts are cotranscriptionally processed into unit-length monomers by self-cleavage of closely related 85-nt genomic and antigenomic ribozymes (Fig. 1A; Lai 1995; Taylor 2006). After ribozyme self-cleavage, the HDV genome folds into an inactive rod-shaped structure by base-pairing with a highly complementary downstream sequence termed the attenuator (Fig. 1B). The hepatitis delta virus also encodes a single protein, the HDAg, which is essential for replication and has been implicated as an RNA chaperone (Huang and Wu 1998; Huang et al. 2003, 2004; Wang et al. 2003; Lai 2006). It appears that HDAg has a variable effect on self-cleavage under standard assay conditions, which depends on RNA length. For example, the HDAg strongly stimulates −64/112 RNA,1 shows slight stimulation of −30/112, inactivates −8/87, and does not substantially affect −106/112 and −64/172 RNAs (Huang and Wu 1998).
FIGURE 1.

Secondary structures present in HDV ribozyme-containing transcripts. The structures are color coded by native pairings, which are based on the crystal structure of the cleaved form of the ribozyme (Ferre-D'Amare et al. 1998) and extensive mutagenesis experiments (Wadkins et al. 1999; Chadalavada et al. 2000, 2002; Diegelman-Parente and Bevilacqua 2002; Brown et al. 2004). Black arrowheads denote 5′ to 3′ directionality. (A,B) Native secondary structures of constructs studied. These transcripts begin at −54, which allows formation of the P(−1) pairing that facilitates native secondary structure (Chadalavada et al. 2000). P(−1) has the L(−1) hairpin loop at its apex and is joined to the ribozyme by the J(−1/1) single-stranded joining region. Pairings P1, P2, P3, P4, and P1.1 are contained within the ribozyme, and the site of cleavage between −1 and 1 is denoted with an arrow. For the transcript in B, increasing length of downstream sequence is present, which results in formation of P5 and the attenuator. Pairing register of the attenuator, which is mutually exclusive with much of the ribozyme secondary structure, is denoted by an extension (dashed line). (C) Secondary structure of the −30/99 RNA showing some of the known alternative pairings. Alt 1 and Alt 2 involve interactions of upstream flanking sequence with the ribozyme, while Alt 3 involves ribozyme–ribozyme pairing (Chadalavada et al. 2000, 2002). Other alternative pairings not shown include Alt X and Alt Y, which disfavor native RNA folding (Brown et al. 2004). (D) Sequence of wild-type ribozyme (Chadalavada et al. 2002).
In its catalytic fold, the HDV ribozyme assumes a complex double-pseudoknot topology (Fig. 1A; Ferre-D'Amare et al. 1998; Ke et al. 2004). The ribozyme fold consists of five pairings, P1, P2, P3, P4, and P1.1, with P2 and P1.1 comprising pseudoknot pairings, and self-cleaves at the base of P1. Flanking sequence upstream and downstream of the ribozyme folds into pairings termed P(−1) and P5,2 which are compatible with ribozyme self-cleavage, although they do not accelerate it (Fig. 1B; Chadalavada et al. 2000; Diegelman-Parente and Bevilacqua 2002).
Early in vitro studies on wild-type HDV ribozymes revealed that self-cleavage is multiphasic and exceptionally slow (∼90 h half-life), being enhanced by strongly denaturing conditions such as 12.5 M formamide (Perrotta and Been 1990, 1991; Rosenstein and Been 1990), as well as addition of antisense (AS) oligonucleotides that disrupt the Alt 1 pairing (Fig. 1C) and provide rate enhancements of nearly 3000-fold (Chadalavada et al. 2000). These observations instigated studies to determine the structural basis of this effect. Like many functional RNAs, the HDV ribozyme can assume nonnative conformations. Probing the secondary structure of these conformations by enzymatic digestion led to the identification of numerous nonnative, or alternative (Alt), helices, including Alt 1, Alt 2, Alt 3, Alt P1, and a series of Alt X and Alt Y pairings, some of which are depicted in Figure 1C (Chadalavada et al. 2000, 2002; Diegelman-Parente and Bevilacqua 2002; Brown et al. 2004).
These alternative pairings were categorized on the basis of whether they involve interactions with flanking nucleotides: Alt 1, Alt 2, and the attenuator involve ribozyme-flanking nucleotide pairings, while Alt 3, Alt P1, Alt X, and Alt Y involve primarily ribozyme-ribozyme pairings. Disruption of P(−1) can lead to formation of Alt 1 (Fig. 1C; Chadalavada et al. 2000), while extension of P5 facilitates formation of the attenuator (Fig. 1B; Diegelman-Parente and Bevilacqua 2002), both of which provide nonfunctional ribozyme folds. Alt P1 occurs by misalignment of P1 (not shown) and inhibits ribozyme function (Chadalavada et al. 2002).
However, not all alternative pairings are inhibitory toward function. In particular, Alt 2 and Alt 3 (Fig. 1C) have been shown both computationally (Isambert and Siggia 2000) and experimentally (Brown et al. 2004) to facilitate HDV ribozyme folding, acting as so-called folding guides, which prevent the formation of more deleterious Alt X and Alt Y pairing registers. In addition, inhibitory effects of certain alternative pairings, in particular of downstream attenuator, can be suppressed by slowing the rate of transcription (Diegelman-Parente and Bevilacqua 2002).
Once the structural nature of these nonfunctional states was established, it was relatively straightforward to enhance self-cleavage activity in standard kinetic assays by a rational, negative-design strategy, similar to that described for proteins (Nautiyal et al. 1995), wherein alternative pairings were weakened by site-directed mutagenesis but native ribozyme pairings were maintained (Chadalavada et al. 2000, 2002; Brown et al. 2004). We generated more than 20 single and double site-directed mutants of noncatalytic nucleotides in an effort to destabilize alternative pairings. Sizeable gains of up to 350-fold in self-cleavage activity were found for standard AS oligonucleotide-containing assays, as was simplification of multiphasic kinetics (Chadalavada et al. 2002; Brown et al. 2004).
The relative ease with which ribozyme function could be enhanced by rational disruption of alternative folds raised the question as to whether the virus takes advantage of such mutations in vivo, and, if not, what other mechanisms for adaptation might be possible. Herein, we present the functional consequences of several ribozyme mutations on cotranscriptional self-cleavage assays. The results suggest that wild type, which is nonoptimal for the self-cleavage of full-length ribozymes under standard conditions, is the optimal sequence for activity of the HDV ribozyme under cotranscriptional conditions.
RESULTS
Conservation of HDV RNA sequence
In an effort to understand the role of sequence variability in ribozyme function, a comparison of 76 HDV genomic sequences encompassing the ribozyme and flanking regions was made. We carried out a sequence comparison on just the ribozyme and upstream regions earlier (Chadalavada et al. 2000); however, at that time, only 21 isolates were available in GenBank. In addition, our knowledge of the structure and function of the flanking regions was relatively limited. Thus, it was critical to reevaluate and extend that initial comparison.
The −54 to 140 region of the genomic HDV RNA was aligned for 76 hepatitis delta isolates using blastn (Altschul et al. 1990). This region encompasses 54 nucleotides (nt) upstream of the cleavage site and 56 nt downstream from the 3′ boundary of the ribozyme. The information content R sequence (L) (in bits per base) at a given position in the sequence, L, was calculated according to Equation 1 (in Materials and Methods) (Schneider et al. 1986; Carothers et al. 2004) and as described in the Materials and Methods. For this analysis, an R sequence (L) value of 2 indicates complete conservation across the 76 isolates (high information content), while a value of 0 indicates no conservation (no information content). A value of 1 indicates that two of the 4 nt are preferred and is consistent with Watson–Crick base-pairing at that position (Schneider et al. 1986; Carothers et al. 2004). Information content (Fig. 2, top) was plotted as a function of genomic sequence for the 5′− and 3′-flanking regions (Fig. 2A,C) and for the ribozyme (Fig. 2B), as well as for the antigenomic ribozyme (Fig. 2D). Also provided (lower portion of Fig. 2) are the actual nucleotide substitutions, in which the size of the font is proportional to the fraction conservation.
FIGURE 2.

Nucleotide conservation of the HDV ribozyme and flanking RNA. Numbering is relative to the cleavage site between nucleotides −1 and +1. The −54 to 140 region of 76 genomic HDV RNA isolates was aligned using nucleotide–nucleotide BLAST (blastn). Information content, R sequence(L), in bits per base at various positions (L) was determined as described in Materials and Methods. To enhance visual clarity, data for genomic sequence have been divided into upstream flanking sequence (A), ribozyme sequence (B), and downstream flanking sequence (C). Panel D shows data for the antigenomic ribozyme sequence. A score of 0–2 was given for the information content in bits. Positions with variability in R sequence(L) values are emphasized by the placement of open circles on the plot. Regions with helical nucleotides are shaded gray, while regions with nonpaired nucleotides are white. The ribozyme and downstream sequences have a few pairing regions that are contiguous; in these cases, a dashed line was used to partition the two helices. The lower half of each panel enumerates the actual nucleotide sequence, with the letter height proportional to the nucleotide frequency. Nucleotides with an occurrence of less than 3% are not depicted.
The −54/−1 upstream flanking sequence includes the P(−1), L(−1), and J(−1/1) elements, as shown in Figure 1A,B. Sequence comprising the P(−1) pairing was found to be well conserved (Fig. 2A). Nonetheless, 57 changes were found in the P(−1) region. In nearly every instance, these mutations occurred adjacent to helical defects and, according to free energy minimization calculations (Mathews et al. 1999; Zuker et al. 1999), allowed a P(−1) of stability within +2.9 to −4.6 kcal/mol of the wild type (ΔG°37 of −18.4) to form, sometimes with migration of the helical defect (Supplemental Fig. S1). These data provide strong support for the presence of an upstream P(−1) pairing in all isolates.
Other upstream elements had somewhat more changes than P(−1) (Fig. 2A; Supplemental Fig. S1). The L(−1) loop was quite variable at its 3′ side, consistent with absence of pairing in this stretch. The other upstream sequence element, J(−1/1), was susceptible to variation at its 5′ and 3′ sides, especially at the −2 and −3 positions, but the central portion of this element, which is pyrimidine rich, was invariant. A similar pyrimidine-rich element was present upstream of the antigenomic ribozyme. It is also noted that nucleotides involved in the Alt 1 pairing between P(−1)3′ and P23′ (Fig. 1C) are well conserved, barring the 3′-most nucleotide of Alt 15′, suggesting a possible functional role for this alternative pairing. In all instances, RNA folding using mfold v3.2 (Mathews et al. 1999; Zuker 2003) suggested that P(−1), L(−1), and J(−1/1) form despite the variation in their sequences, providing strong support for their conservation, which was independently verified experimentally for the wild type (Chadalavada et al. 2000).
Variability in the ribozyme sequence was restricted. Sequence changes were found primarily in three regions: J(1.1/3), P4, and the base of P1. The J(1.1/3) joining region showed significant changes at positions 23, 26, and 27 (Fig. 2B). These three nucleotides were disordered to varying extents in the crystal structure of the ribozyme (Ferre-D'Amare et al. 1998), consistent with absence of critical functional roles. Another variable region was P4, which is dispensable for self-cleavage (Been et al. 1992; Thill et al. 1993). Even here the changes were restricted to relatively few positions, primarily 61 and 64, where deletions occurred with little change to the structure and stability of P4. Conservation of the catalytically dispensable P4 and L4 suggests they may serve some noncatalytic function such as acting as a folding guide (see Discussion).
One additional position of variability within the ribozyme is residue 37, which partakes in the first base pair in P1. In the majority of the genomic isolates (61/76), this residue is a U and forms a GU wobble with G1. However, in the other 15 isolates, it is a C, which can form a GC base pair with G1. Genomic ribozymes with a GC base pair at the base of P1 have nearly identical metal ion and pH kinetic profiles under standard conditions as wild type (A.L. Cerrone-Szakal and P.C. Bevilacqua, in prep.), indicating that this change does not have a functional consequence to ribozyme catalysis. The antigenomic ribozyme is also well conserved, as seen by comparing 269 antigenomic variants (Fig. 2D). Curiously, in all 269 antigenomic isolates, the first base pair in P1 is a GU wobble. Perrotta and Been (2007) recently demonstrated that the −1 position, which is U in the genomic ribozyme and C in the antigenomic, is associated with a change in metal specificity from favoring Mg2+ to favoring Ca2+. Perhaps there is a functional link between the identity of the −1 nt and the first base pair in P1. In summary, variability within the ribozyme is restricted to relatively few nucleotides and occurs in such a way as to allow catalytically important structural elements to remain intact, as expected from the biological function of the ribozyme. Remarkably, none of the ribozyme variants found in nature displayed nucleotide changes known to enhance self-cleavage under standard conditions.
In contrast to the ribozyme region, the 86–140 downstream flanking sequence was found to have a combination of variable and well-conserved regions (Fig. 2C). The variable region localizes to P5 and L5, which are formed at the tip of the rod (Figs. 1B, 3). As shown in Figure 1B, the experimentally determined P5 pairing (Diegelman-Parente and Bevilacqua 2002) is compatible with the native HDV ribozyme fold. Interestingly, even though the sequence in P5 is variable, changes in the base-pairing region covary. For example, base pair 88:126 was found to be C:G, G:C, and G:G in 13, 62, and 1 isolates, respectively (Fig. 3). (A G:G is the most stable single mismatch thermodynamically; Kierzek et al. 1999.) Similar base-pairing maintenance was observed at positions 96:118, 97:117, and 101:115 (Fig. 3). Subsequent analysis of the 80–140 region alone resulted in a total of 276 genomic isolates, and similar trends were observed (data not shown). Apparently, the secondary structure of P5 is important, although the sequence of certain base pairs is not. This region has been shown to coimmunoprecipitate with RNAP II, which could account for the importance of its secondary structure (Greco-Stewart et al. 2007). It can be noted that the 5′ strand of P5 is particularly pyrimidine rich, much like the J(−1/1) stretch (see Discussion). The L5 loop shows variability at its 5′ side as well, consistent with no known role in catalysis.
FIGURE 3.

Covariation in the P5 helix and flanking regions. Nucleotide sequence corresponding to the 75–140 region of the genomic HDV RNA was folded using mfold v3.2 (Mathews et al. 1999; Zuker 2003). Numbering is relative to the cleavage site between nucleotides −1 and +1. Sequence variability information was obtained from blastn analysis of the 76 genomic HDV RNA isolates. Positions that vary are shaded gray and boxed for base pairs and circled for single-stranded nucleotides. Nonconserved nucleotides that base-pair were observed to covary so as to retain base-pairing. The number of isolates for a given base pair is provided below the figure. Nucleotides with an occurrence of less than 3% are not depicted. In this figure, residue 85 is a C because this is the most common sequence; however, it was G in the isolate used for wild-type, which was isolated from a patient with acute HDV (Makino et al. 1987) (accession M28267). The pairing elements aJ4/2, aP23′, P5, and L5 are denoted in the figure.
Lastly, the well-conserved region of the downstream flanking sequence localizes to regions of the attenuator complementary to the ribozyme, aP23′ 3 and aJ4/2, which are the complements of P23′ and J4/2, respectively (Figs. 2C, 3). Since the ribozyme sequence is conserved in this region, observation that its largely complementary sequence is conserved as well suggests that the attenuator pairing serves an important functional role for the virus.
Ribozyme mutants react faster than wild type under standard divalent ion renaturation conditions
Observation that the sequence of the ribozyme is largely invariant in nature was surprising because prior studies showed that numerous rationally designed single and double mutations to the ribozyme sequence could dramatically improve self-cleavage activity under standard assay conditions (Chadalavada et al. 2000, 2002; Brown et al. 2004). These mutants generally operate by allowing escape from alternative pairings and/or strengthening native pairings. One possible explanation for the absence of these changes in nature is that the effect of mutations on self-cleavage efficiency is specific to the manner in which the assay is conducted. We therefore investigated effects of the wild type and three single mutants under several assay conditions beginning with standard divalent ion renaturation conditions.
The three mutants chosen for study were G10C, G11C, and G85C. Two of these, G11C and G85C, have been previously studied under standard assay conditions (Chadalavada et al. 2002), while G10C is presented here for the first time. These three mutants affect the fold of the ribozyme in a simple and well-understood manner (Nakano et al. 2000; Chadalavada et al. 2002; Brown et al. 2004). G11C acts to destabilize Alt P1 while maintaining P2, albeit with a shifted register having a bulged C84, while G85C stabilizes P2 (Chadalavada et al. 2002). The G10C mutant, on the other hand, is predicted to both destabilize Alt P1 and stabilize P2. Because P25′ competes with the attenuator for the last 5 nt in the ribozyme, these mutants allow this folding process to be probed in different ways. In addition, the G10C and G11C mutants were not found in any of the naturally occurring isolates, while G85C was very common; thus, with these three mutants we explore variants that both occur and do not occur in nature.
Kinetic profiles under standard assay conditions in the presence of 10 mM Mg2+ are shown in Figure 4A, with values provided in Table 1. Previous kinetics studies under standard assay conditions revealed that wild type reacts with a 30% burst phase with a rate of 12.0 min−1, and a 60% slow phase with a rate of 0.17 min−1 (Chadalavada et al. 2002). Thus, wild type is biphasic and largely slow reacting. G85C, on the other hand, reacts in a monophasic fashion with a k obs of 2.2 min−1, while G11C is monophasic with a k obs of 7.1 min−1. As expected, the G10C mutant, which favors the native state by both destabilizing nonnative interactions (Alt P1) and strengthening native state interactions (P2), reacted the fastest under standard assay conditions, with a k obs of 14.4 min−1 (Fig. 4A). In fact, the k obs for G10C is approximately equal to the product of the k obs values for G11C and G85C, suggesting additivity of effects. These findings are consistent with the previously reported observation that G10A, which destabilizes Alt P1 without stabilizing P2, reacts similarly to G11C (Chadalavada et al. 2002).
FIGURE 4.

Effect of ribozyme sequence on full-length transcript self-cleavage and cotranscriptional self-cleavage reactions. (A) Self-cleavage of full-length ribozymes (standard assay) in which the reaction was initiated by addition of MgCl2. The three sequences are wild-type (•), G10C (Δ), and G11C (○). Fits are to Equation 6 and parameters are provided in Table 1. (B,C) Cotranscriptional self-cleavage for −54/99 (B) and −54/140 (C) transcripts. Points are an average of at least two trials. Time points range from 15 sec to 10 min. The four sequences are wild type (•), G10C (Δ), G11C (○), and G85C (▴). Fits for G10C, G85C, and wild type are to Equation 5, while the fit for G11C in the −54/99 background is to Equation 4; fits for closed symbols are solid lines, while fits for open symbols are dashed lines. Parameters are provided in Table 1.
TABLE 1.
Effect of ribozyme sequence on full-length transcript self-cleavage and cotranscriptional self-cleavage reactions

Wild type reacts faster than the mutants under cotranscriptional conditions
Although many ribozymes can be readily engineered to undergo multiple-turnover catalysis as typified by most protein enzymes (Zaug and Cech 1986; Been et al. 1992), small ribozymes and certain large ribozymes are unusual in that the reactions they carry out in nature are single turnover. These ribozymes have therefore evolved to function cotranscriptionally. As such, RNA sequence flanking the ribozyme could be involved in regulating catalysis. We reasoned that one explanation as to why the HDV ribozyme is largely resistant to mutation in nature is that sequence changes that improve self-cleavage under standard assay conditions may not have such effects when assayed cotranscriptionally. To test this possibility, we measured self-cleavage kinetics on the same four ribozyme variants under cotranscriptional assays with various flanking sequences.
Single mutants of the ribozyme were prepared in −54/99 and −54/140 flanking sequence backgrounds, and cotranscriptional self-cleavage assays were carried out as previously described (Diegelman-Parente and Bevilacqua 2002). These two flanking sequence backgrounds were designed to explore downstream effects of P2, P5, and the conserved aP23′ and aJ4/2 regions of the attenuator (Fig. 3) on cleavage efficiency. The upstream flanking sequence was chosen to encompass −54 to −1 because this region forms the P(−1) pairing and J(−1/1) joining region (Fig. 1A), which are compatible with efficient self-cleavage under cotranscriptional conditions (Chadalavada et al. 2000; Diegelman-Parente and Bevilacqua 2002).
Cotranscriptional self-cleavage assays on wild-type and single-mutant ribozymes were first tested in the −54/99 background (Fig. 4B), which is sufficiently short to prevent formation of P5 or the competing attenuator (Diegelman-Parente and Bevilacqua 2002). Only data for G11C in the −54/99 background could be fit to a simple one-term equation (Equation 4) for cotranscriptional cleavage (Long and Uhlenbeck 1994). Fitting of wild-type and other single mutants required an equation with an additional term (Equation 5), in which the early portion of the data was treated as a burst fraction (Diegelman-Parente and Bevilacqua 2002). As suggested by previously published simulations, this added complexity can be caused by the presence of either an off-path channel or an on-path intermediate that has a rate of breaking down similar to the rate of chemistry (Diegelman-Parente and Bevilacqua 2002). The kinetic parameters from these fits are presented in Table 1. Because of the complex behavior of the mutants, the reaction is treated largely qualitatively in terms of overall self-cleavage efficiency.
In the −54/99 background, wild type, G10C, and G85C have similar cotranscriptional behavior. At early times, the G11C mutant reacted slightly slower than the other ribozymes, but reached similar fraction cleaved at intermediate times. Thus, unlike standard assay conditions, cotranscriptional kinetics revealed that the wild type reacts similarly to the mutants.
Next, cotranscriptional experiments were conducted in the −54/140 background, which contains the conserved portions of the attenuator aP23′ and aJ4/2, which are complementary to P23′ and J4/2. Mutants in the −54/140 background had a larger range of effects, with kinetic profiles spread out across the plot (Fig. 4C). The wild type was more efficient overall at self-cleavage than the mutants. G85C and G10C, which behaved similarly in the −54/99 background, had self-cleavage efficiency similar to the wild type at early times, but were both substantially less efficient than wild type at later times. The G11C mutant, on the other hand, was less efficient at early times, but reacted similarly to the wild type at later times.
Slowing the rate of transcription and adding delta antigen protein further improves self-cleavage activity
Having found that wild type is the most efficient sequence under cotranscriptional conditions, we tested whether other factors that mimic in vivo conditions might further improve self-cleavage efficiency. The rate of transcription for phage T7 RNA polymerase, which was used in cotranscriptional experiments, is particularly fast (∼200–400 nt/sec) (Uptain et al. 1997; Pan et al. 1999; Pan and Sosnick 2006). On the other hand, the rate of elongation for RNA polymerase II, which is thought to replicate HDV (Taylor 2006; Yamaguchi and Handa 2006), is much slower, with an estimate of ∼12–20 nt/sec (Kugel and Goodrich 1998), although the actual rate for replication of HDV is approximately two- to fourfold faster (see Discussion) (Yamaguchi et al. 2007). The rate of transcription by T7 RNA polymerase was slowed by limiting the concentrations of NTPs in the reaction (Materials and Methods). Controls showed that the rate of transcription was constant over the 10-min time course of the experiments for both low and high NTP concentrations, with transcription reactions in 10 μM NTP going ∼100-fold slower than in 600 μM NTPs (Supplemental Fig. S2). In addition, we introduced a construct with a 3′ end that extends to position 155, which is complementary to the 3′ strand of P4, in order to more fully evaluate the importance of folding and cleaving during transcription.
As shown in Figure 5A, lowering the rate of transcription improved self-cleavage efficiency of the longest construct (−54/155) the most. In general, this effect is most likely due to the additional window of time afforded for self-cleavage prior to synthesis of the attenuator. Supporting this interpretation, lowering the rate of transcription had a minimal effect on promoting self-cleavage in the −54/99 and −54/140 backgrounds, which have no or less extensive ribozyme complementarity, respectively (Fig. 5A). Apparently, the kinetics of partitioning between self-cleavage and attenuator synthesis are critical (see Discussion).
FIGURE 5.

Effect of lowering the rate of transcription and of HDAg on wild-type cotranscriptional self-cleavage reactions. (A) Cotranscriptional self-cleavage in which the rate of transcription was decreased by lowering NTP concentrations. The rate of transcription was slowed by lowering the concentrations of all NTPs other than GTP in the transcription reaction to 10 μM. The three backgrounds are −54/99 (•, ○), −54/140 (■, □), and −54/155 (▲, △). Closed symbols are at 600 μM NTPs, while open symbols are at 10 μM NTPs (600 μM GTP). (B) Cotranscriptional self-cleavage in which the HDAg was added. The delta antigen was added to a final concentration of 3 μM in the background of 600 μM each NTP. The three backgrounds are −54/99 (•, ○), −54/140 (■, □), and −54/155 (▲, △). Closed symbols are without HDAg, while open symbols include 3 μM HDAg. Data for lower NTP concentrations and HDAg could not be fit to Equations 4 or 5. In these cases the lines are present only to help visualize data trends. Lines for closed symbols are solid, while fits for open symbols are dashed.
The results presented above show that self-cleavage efficiency of the HDV ribozyme is sensitive to a number of factors, including whether the reaction is carried out cotranscriptionally, ribozyme sequence, flanking regions, and transcription rate. One other factor that has been shown to influence the cleavage rate of full-length HDV ribozymes (both positively and negatively) in standard reaction assays is the HDAg (Huang and Wu 1998; Huang et al. 2003, 2004; Wang et al. 2003), which is the only protein that HDV encodes. However, the effects of the HDAg on ribozyme kinetics during transcription have not been reported to our knowledge.
The effects of the HDAg on self-cleavage efficiency during transcription were tested on the wild-type sequence in the −54/99, −54/140, and −54/155 backgrounds (Fig. 5B). Addition of HDAg to the wild type strongly stimulated self-cleavage for all length constructs. The cotranscriptional effect of HDAg could be due to direct effects on folding of full-length HDV transcripts, especially given the literature precedent in standard assays (Huang and Wu 1998; Huang et al. 2003, 2004; Wang et al. 2003), or to ribozyme-indirect effects including decreasing the rate of transcription, since this also favors cotranscriptional cleavage efficiency as shown in Figure 5A.
The effect of HDAg on the rate of transcription was monitored at 600 μM each NTP, as described in the Materials and Methods. It was found that 1 or 3 μM HDAg slows the rate of transcription at 600 μM NTPs approximately sevenfold (Supplemental Fig. S2). Thus, part of the effect of the HDAg in stimulating wild-type activity cotranscriptionally could be due to slowing the rate of transcription (see Discussion). However, we note that HDAg decreases the rate of transcription by only sevenfold, which is much less than the 100-fold effect of lowering the NTP concentrations to 10 μM (Supplemental Fig. S2). This suggests that effects of HDAg on ribozyme self-cleavage efficiency may not be due only to slowing transcription.
Potential effects of HDAg on cleavage of full-length transcripts, referred to as “basal effects,” were determined for wild type in standard (i.e., noncotranscriptional) assays in the −30/99 + AS1 background (Supplemental Fig. S3). It is seen that HDAg increases both the rate of self-cleavage of the major phase and the amplitude of the burst phase approximately twofold. Apparently, HDAg stimulates HDV ribozyme cleavage by two distinct kinetic mechanisms: slowing the rate of T7 transcription and improving basal folding and cleavage rates.
The basal effect can also be inferred from cotranscriptional assays for the wild type in the −54/99 background. Kinetics of cleavage of −54/99 are substantially faster in the presence of the HDAg (Fig. 5B) even though slowing the rate of transcription does not affect cleavage efficiency of this construct (Fig. 5A). Similar conclusions can be reached from the −54/140 wild-type data. In sum, under cotranscriptional conditions with native flanking sequence, wild type is no longer slow reacting. It has self-cleavage efficiency equal to or greater than the three mutants, and wild-type self-cleavage becomes even more efficient when the rate of transcription is attenuated and HDAg protein is included.
DISCUSSION
RNA viruses mutate at an exceptionally high rate. For instance, RNA polymerases that lack proofreading functions make mistakes every 1 in 103 to 105 bases (Domingo and Holland 1997). The remarkable evolvability of RNA viruses is often attributed to this elevated rate of mutation. We compared 76 genomic HDV sequences and found that while flanking regions often undergo mutation, almost no change occurs in the ribozyme region. Absence of variation of the ribozyme sequence was initially surprising because mutants react faster than wild type under standard kinetics assays and can be rationally designed with relative ease. The main finding from the present study is that the wild type is the optimal sequence of the HDV ribozyme when assays are conducted under in vivo-like conditions of cotranscriptional cleavage, presence of extensive native flanking sequence, slower transcription rate, and presence of HDAg protein. These observations suggest that wild-type ribozyme sequences encode catalytic information that is optimized for cellular-like conditions.
Under standard assay conditions, wild type is substantially less efficient at self-cleavage than the mutants. For example, at 15 sec the G10C and G11C mutants cleaved to completion, while only 30% of the wild type self-cleaved (Fig. 4A). However, under cotranscriptional folding conditions, wild type is within 10% cleavage efficiency of the mutants for the −54/99 background (Fig. 4B) and as fast or faster than these mutants for the more extensive −54/140 flanking background (Fig. 4C). Lowering the NTP concentration or adding HDAg increased the self-cleavage efficiency of wild type even further, with an additional 25%–35% increase at early time points (Fig. 5).
The effect of the HDAg is due to effects on the rate of transcription and basal effects on full-length ribozyme. The overall rate of T7 transcription, as judged by monitoring total RNA produced with time, decreased approximately sevenfold in the presence of HDAg (Supplemental Fig. S2). The stimulatory basal effects of HDAg on the wild-type are revealed by approximately twofold increases in burst amplitude and main phase rate under standard conditions. Together, these findings indicate that that HDAg promotes cleavage of the wild-type ribozyme under in vivo-like conditions.
A recent report indicates that HDAg stimulates the rate of elongation of RNAP II, with approximately two- to fourfold increases (Yamaguchi et al. 2007). Although HDAg affects rates of T7 and RNAP II transcription in opposite directions, the end effect is similar rates of transcription. For example, using rates of transcription for T7 of ∼200–400 nt/sec (Uptain et al. 1997; Pan et al. 1999; Pan and Sosnick 2006) and RNAP II of ∼12–20 nt/sec (Kugel and Goodrich 1998) and correcting for the sevenfold attenuation on T7 and the two- to fourfold increase on RNAP II, similar final rates of transcription are found. It is not clear whether this similarity is simply fortuitous or reflects some common mechanism; nonetheless, the rate of T7 transcription in the presence of HDAg may be similar to that for RNAP II in the presence of HDAg in vivo.
Given the similar rates of transcription for T7 and RNAP II in the presence of HDAg, it is of interest to compare the rate of ribozyme self-cleavage to the rate of attenuator RNA synthesis. Using the rates for RNAP II in the presence of HDAg given above, synthesis of the RNA from position 84 (3′ end of the ribozyme; Fig. 1) to 155 (position of attenuator where significant wild-type inhibition is observed; Fig. 5) should occur in less than 3 sec. The wild-type cleaves with a burst phase of 42% (where the burst is faster than 15 sec), while optimal sequences in vitro cleave with rates of 1 sec−1 (Brown et al. 2004). Although this analysis does not consider the potential importance of transcriptional pausing events, which have not been characterized yet for HDV in vivo, it appears likely that ribozymes have a selective pressure to cleave on the seconds time scale in vivo.
Of the various mutants, G85C was the least efficient in promoting self-cleavage of −54/140 cotranscriptionally, especially at longer time points (Fig. 4C; Table 1). This is likely due to the G85C change strengthening the aP23′ portion of the attenuator (Fig. 3), which competes with P2 formation in the ribozyme. Consistent with observations, this effect does not manifest itself under standard reaction assays or cotranscriptional assays with −54/99, which do not contain enough nucleotides to form aP23′.
G10C is the next least efficient in cotranscriptional cleavage assays on −54/140, followed by G11C (Fig. 4C). Both G10C and G11C are involved in formation of the P2 pseudoknot pairing (Fig. 1D). The effects on G10C and G11C are unlikely to be due to less efficient self-cleavage of the native state itself since these mutants were the fastest self-cleavers under standard assay conditions (Fig. 4A). One possibility is that the G10C and G11C changes interfere with the formation of transient folding guides. Isambert and Siggia (2000) proposed an 8-bp guide helix termed “P8” that has G10 and G11 engaged in Watson–Crick pairs with the L4 nucleotides C58 and C57, respectively. Interestingly, our sequence alignments show that all 8 bp in P8 are conserved. The G to C mutations at positions 10 and 11 could interfere with formation of the P8 guides. Observation that G10C and G11C mutations only manifest themselves negatively under cotranscriptional cleavage with an attenuator present implies that their roles in accelerating folding only matters when challenged with a competing RNA folding event.
It was previously proposed that formation of the pseudoknot portions of the genomic HDV ribozyme is the slow step in overall folding (Chadalavada et al. 2002), while it is known that the local folding of hairpins such as P5 is rapid (Turner et al. 1990). Thus, it is reasonable that the attenuator could compete with P2 pseudoknot pairing, even though the former is synthesized later. This model would also explain why slowing the rate of transcription leads to enhanced cleavage efficiency (Fig. 5A).
The sequence comparison of isolates conducted herein suggests that HDV has evolved several solutions to promote efficient cotranscriptional ribozyme folding and cleavage. An RNA folding model is presented in Figure 6 that captures some of the salient features for cotranscriptional folding. The major theme in this model is avoidance of noncatalytic pairings. Beginning at the 5′ end of the transcript, the first 38 nt are sequestered in the P(−1) stem–loop, which prevents them from interacting with either the J(−1/1) region (blocked pairing 1) or the ribozyme (blocked pairing 2) (Fig. 6). The presence of a stable P(−1) pairing is strongly supported by folding of sequence variants (Supplemental Fig. S1). Next to be synthesized is the pyrimidine-rich J(−1/1) joining region. Being pyrimidine rich, this region avoids interacting with the (soon to be synthesized) self-folding purine–pyrimidine mixed ribozyme (blocked pairing 3) or with the downstream pyrimidine-rich P55′ (blocked pairing 4), which exists transiently as unpaired during replication. The purine–pyrimidine mixed ribozyme folds with itself, which prevents folding with pyrimidine-rich P55′ (blocked pairing 5) or with the folded P5 pairing (blocked pairing 6). Interestingly, by being pyrimidine rich, J(−1/1) and P55′ not only avoid interacting with each other, but also avoid self-structure that could inhibit their ability to later interact with their purine-rich complements in the attenuator. Similarly, the purine-rich nature of P53′ prevents its interacting with the purine–pyrimidine mixed attenuator (blocked pairing 7) or the purine-rich anti J(−1/1) region (blocked pairing 8).
FIGURE 6.

Flanking sequence promotes efficient cotranscriptional ribozyme folding. (A) Rod model for the genomic HDV RNA, starting at −54. Ribozyme is base-paired to its complementary attenuator sequence. The site of cleavage between −1 and 1 is denoted with an arrow. The 5′-upstream nucleotides are shown forming the P(−1) pairing element since the attenuator sequence complementary to this region is not depicted. The 5′ boundary of the attenuator is defined as the region that begins base-pairing with the ribozyme (Fig. 3). The pyrimidine-rich nucleotide sequence for the upstream J(−1/1) element and P55′ are highlighted in yellow; these regions are complementary to the purine-rich downstream P53′ and anti J(−1/1) respectively, highlighted in blue. There is a small stretch of nucleotides between P23′ and P55′ that is composed of both R and Y residues (see Fig. 3). The actual nucleotide sequences for the two R-rich and Y-rich regions are shown in panel A. Ways in which flanking sequence conspires to avoid noncatalytic ribozyme pairings are illustrated in panel B and described in the text.
Overall, it appears that HDV has evolved several solutions to achieve efficient cotranscriptional ribozyme folding. These are characterized by the presence of flanking regions that avoid self-structure and long-range pairings by being pyrimidine rich. Pyrimidines may be a better choice than purines for flanking the ribozyme in that the latter are also good stackers and have the potential to form G-quartets, which could interfere with efficient cotranscriptional ribozyme folding. One additional possibility is that the P(−1)/J(−1/1) combination, which has both 5′ self-structure and potentially weak template interactions, facilitates release of the ribozyme from the template, as is the case for factor-independent transcription termination (Martin and Tinoco 1980; Chadalavada et al. 2000).
Consistent with flanking sequence helping the ribozyme avoid noncatalytic pairings (rather than flanking sequence directly forming catalytic pairings with the ribozyme), we have previously shown through structure–function studies that P(−1) and P5, while compatible with self-cleavage, do not accelerate it (Chadalavada et al. 2000; Diegelman-Parente and Bevilacqua 2002). Observation that wild-type reacts with similar reaction parameters in the −54/99 and −54/140 backgrounds (Table 1) is also consistent with absence of direct participation of these regions in catalysis. In addition, inspection of HDV ribozyme crystal structures (Ferre-D'Amare et al. 1998; Ke et al. 2004) does not reveal any obvious ways in which the P(−1) and P5 regions could interact with the ribozyme.
In summary, the optimal sequence for efficient cotranscriptional self-cleavage by the HDV ribozyme is wild type. Mechanistically it appears that efficient self-cleavage is accomplished by the proper combination of flanking sequence that avoids interaction with the ribozyme and internal sequence that allows folding guides to drive efficient formation of pseudoknots prior to downstream sequence interactions. The HDAg protein further aids this process by basal effects and slowing transcription, although establishing the mechanistic basis for basal effects will require further study. The finding that wild-type is the most efficient ribozyme sequence under cellular-like conditions is in line with the behavior of most protein enzymes.
MATERIALS AND METHODS
Cloning and vectors
The HDV sequence was from a human patient with acute delta hepatitis (PDB accession number M28267). The −30/99 genomic HDV RNA was transcribed from pT7–30/99 using phage T7 RNA polymerase, as described (Chadalavada et al. 2000). The wild-type −54/99 and −54/140 plasmids were prepared by downstream extension of a shorter construct containing the −54/−1 sequence and cloning into pUC19 as described earlier (Diegelman-Parente and Bevilacqua 2002). All constructs contain variable lengths of this sequence inserted between EcoRI and BamHI. A phage T7 promoter was introduced at the 5′ end. Mutant plasmids were generated using the QuikChange kit (Stratagene). Sequences were confirmed by the dideoxy method after a miniprep purification (Qiagen). Internal BfaI and MscI sites were used to generate −54/99 and −54/140 run-off transcripts, respectively. According to published nomenclature, transcripts contain HDV RNA sequences 659–787 (−30/99), 635–787 (−54/99), and 635–828 (−54/140), where the ribozyme cleavage site is between positions 688 and 689.
Nucleotide alignment
The −54 to 140 region of the genomic HDV RNA was aligned using nucleotide–nucleotide BLAST (blastn), resulting in 76 sequence hits. The amount of information needed to specify the sequence at any given position, R sequence (L) was determined by subtracting the Shannon uncertainty for that position, H sequence (L) from the maximum uncertainty possible, H genome (Schneider et al. 1986); thus,
The Shannon uncertainty (H) gives the information content in bits and was calculated using the general formula (Shannon and Weaver 1949)
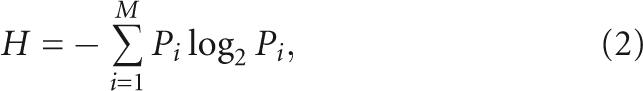 |
in which P i is the intrinsic probability of symbol i, and there are a total of M symbols. Here, M = 4. H genome was calculated using Equation 2 and turned out to be 1.85; this value is sufficiently close to 2 that a random distribution of nucleotides was used in the actual calculations of R sequence (L). Using the information obtained from aligning all of the 76 sequences, H sequence (L) was then calculated for every position in the −54/140 genomic HDV RNA. R sequence (L) was calculated using Equation 1, and the variability in information content at every position in the sequence was plotted.
In vitro cotranscriptional cleavage experiments
Restriction enzymes BfaI or MscI were used to linearize the −54/99 and −54/140 plasmids, respectively, for run-off transcriptions. The linearized plasmid was phenol/chloroform extracted, ethanol precipitated, and resuspended in 1× TE at an approximate concentration of 1 μg/μL. Typical cotranscriptional cleavage assays were performed using 1 μg of linearized plasmid, 600 μM each ATP, CTP, GTP, and UTP, 0.5 μL of 10 μCi/μL [α-32P]GTP, 20 mM Tris-HCl (pH 8.0), 75 mM KCl, 10 mM MgCl2, 1 mM DTT, and 100 μg/mL acetylated bovine serum albumin in a total reaction volume of 30 μL. Note that cotranscriptional cleavage assays and standard divalent ion renaturation cleavage assays contained the same final concentrations of Mg2+. Reactions set up to examine the effect of hepatitis HDAg on cotranscriptional cleavage contained 3 μM small hepatitis HDAg. In some cases where lower rates of transcriptions were desired, 10 μM ATP, CTP, and UTP were used, and the concentration of GTP was retained at 600 μM to help initiate transcription. In either case the reaction was incubated for 2 min at 37°C, followed by initiation with 65 units of T7 RNA polymerase. Two-microliter aliquots were withdrawn at specific time points and quenched by addition to 3 μL of the 95% formamide loading buffer/0.1 M EDTA quench. Time points for the −54/99 and −54/140 reactions ranged from 15 sec to 10 min. The RNA was fractionated by 8% denaturing PAGE, run at 25 W for ∼1 h. The gel was dried and quantified by a PhosphorImager (Molecular Dynamics).
Transcription does not progress evenly with time but instead is largely dependent on initiation and elongation events, interspersed by pause signals, resulting in polymerase being stalled during transcription (Pan and Sosnick 2006). We were thus unable to obtain an absolute value for the rate of transcription. As such, we compared the relative rates of transcription for the 600 μM NTP containing transcription reactions with those carried out in the presence of lowered NTP concentrations. The relative rates of transcription were determined by plotting the total RNA counts in a given lane versus the time course of the reaction. The data were well fit to a straight line, and the resulting slopes were used to determine relative rates (Supplemental Fig. S2).
Ribozyme kinetics and data fitting
Each transcript was labeled internally by [α-32P]GTP as it was transcribed, allowing three bands to be visualized: the full-length uncleaved starting material (A), the upstream cleavage fragment (−54/−1), and the downstream cleavage fragment (B). The fraction of the RNA uncleaved, fun, was determined from Equation 3:
The counts for A, B, and −54/−1 were determined at each time point using a PhosphorImager. The plots of fraction uncleaved versus time were fit (Kaleidagraph, Synergy Software) to Equation 4:
 |
which is derived for the case wherein a full-length transcript is an intermediate in a coupled reaction of transcription and cleavage (Long and Uhlenbeck 1994). This equation holds for any rate of transcription, as long as it is constant. Evidence that the rate of transcription is constant for several different NTP concentrations and in the presence HDAg is presented in Supplemental Figure S2. We chose to plot the fraction uncleaved rather than the fraction cleaved, since this simplifies Equation 4.
In certain cases, the data were biphasic, requiring the two terms in Equation 5:
where k 1 and k 2 are the observed first-order rate constants for the burst and nonburst phases, respectively, t is time, and A is the burst fraction. In all plots, the first nonzero time point was 15 sec. The burst phase, when present, was sufficiently fast that a rate could not be obtained; nonetheless, the two-term equation was required because of the coupled nature of transcription and folding and cleavage.
In standard divalent-ion-induced self-cleavage assays, plots of fraction product versus time were fit to the single-exponential equation:
where fc is the fraction of precursor ribozyme cleaved, k obs is the first order rate constant for ribozyme cleavage for the nonburst phase, t is time, A is the fraction of the ribozyme cleaved at completion, and A+B is the burst fraction (present under some of the reaction conditions). The first time point was taken at 15 sec for hand mixing and 0.1 sec for rapid mixing. Kinetic parameters were obtained using nonlinear, least-squares fitting by Kaleidagraph (Synergy Software). Self-cleavage kinetics for the full-length −30/99 transcript under standard divalent ion renaturation assays contained ∼2 nM 5′-end-labeled RNA, 25 mM HEPES (pH 8.0), 10 mM MgCl2, and 10 μM AS1 oligonucleotide (Chadalavada et al. 2000). In certain reactions, HDAg was present at 1 or 3 μM. In all cases, reaction was initiated by the addition of MgCl2. Hand-mixing and rapid-mixing kinetics were carried out for the wild-type RNA, while data for the faster reacting mutants were obtained by rapid mixing alone, as described previously (Chadalavada et al. 2002).
SUPPLEMENTAL DATA
All Supplemental Materials and information can be found at http://research.chem.psu.edu/pcbgroup/Pages/pubs.html.
ACKNOWLEDGMENTS
This work was supported by NIH Grant R01-58709. We thank Dave Gilmour for discussions on RNAP II elongation and the reviewers for helpful suggestions.
Footnotes
Article published online ahead of print. Article and publication date are at http://www.rnajournal.org/cgi/doi/10.1261/rna.778107.
In this notation, the negative number is how many nucleotides upstream from the cleavage site, which is between −1 and +1, the transcript begins. The positive number is how many nucleotides downstream from the cleavage site the transcript ends, where a transcript with a full-length ribozyme is 85 nt.
P5 is defined as that portion of the stem–loop downstream of the ribozyme that does not pair with the ribozyme. For functional reasons, its 3′-end boundary is defined by the last nucleotide before the nucleotide that pairs with the 3′ end of the ribozyme. Specifically, P5 runs from C86 to G128, and the attenuator begins at G129 (see Fig. 3). This definition is imperfect because it is possible that mutations that strengthen P5 can still favor unfolding of the ribozyme because of the physical connection between P5 and the attenuator.
Regions of the attenuator that are complementary to specific secondary structural elements in the ribozyme are denoted with the name of that element preceded with an “a” for attenuator.
REFERENCES
- Altschul, S.F., Gish, W., Miller, W., Myers, E.W., Lipman, D.J. Basic local alignment search tool. J. Mol. Biol. 1990;215:403–410. doi: 10.1016/S0022-2836(05)80360-2. [DOI] [PubMed] [Google Scholar]
- Been, M.D., Perrotta, A.T., Rosenstein, S.P. Secondary structure of the self-cleaving RNA of hepatitis delta virus: Applications to catalytic RNA design. Biochemistry. 1992;31:11843–11852. doi: 10.1021/bi00162a024. [DOI] [PubMed] [Google Scholar]
- Brown, T.S., Chadalavada, D.M., Bevilacqua, P.C. Design of a highly reactive HDV ribozyme sequence uncovers facilitation of RNA folding by alternative pairings and physiological ionic strength. J. Mol. Biol. 2004;341:695–712. doi: 10.1016/j.jmb.2004.05.071. [DOI] [PubMed] [Google Scholar]
- Buzayan, J.M., Gerlach, W.L., Bruening, G. Satellite tobacco ringspot virus RNA: A subset of the RNA sequence is sufficient for autolytic processing. Proc. Natl. Acad. Sci. 1986;83:8859–8862. doi: 10.1073/pnas.83.23.8859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carothers, J.M., Oestreich, S.C., Davis, J.H., Szostak, J.W. Informational complexity and functional activity of RNA structures. J. Am. Chem. Soc. 2004;126:5130–5137. doi: 10.1021/ja031504a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chadalavada, D.M., Knudsen, S.M., Nakano, S., Bevilacqua, P.C. A role for upstream RNA structure in facilitating the catalytic fold of the genomic hepatitis delta virus ribozyme. J. Mol. Biol. 2000;301:349–367. doi: 10.1006/jmbi.2000.3953. [DOI] [PubMed] [Google Scholar]
- Chadalavada, D.M., Senchak, S.E., Bevilacqua, P.C. The folding pathway of the genomic hepatitis delta virus ribozyme is dominated by slow folding of the pseudoknots. J. Mol. Biol. 2002;317:559–575. doi: 10.1006/jmbi.2002.5434. [DOI] [PubMed] [Google Scholar]
- Diegelman-Parente, A., Bevilacqua, P.C. A mechanistic framework for co-transcriptional folding of the HDV genomic ribozyme in the presence of downstream sequence. J. Mol. Biol. 2002;324:1–16. doi: 10.1016/s0022-2836(02)01027-6. [DOI] [PubMed] [Google Scholar]
- Domingo, E., Holland, J.J. RNA virus mutations and fitness for survival. Annu. Rev. Microbiol. 1997;51:151–178. doi: 10.1146/annurev.micro.51.1.151. [DOI] [PubMed] [Google Scholar]
- Ferre-D'Amare, A.R., Zhou, K., Doudna, J.A. Crystal structure of a hepatitis delta virus ribozyme. Nature. 1998;395:567–574. doi: 10.1038/26912. [DOI] [PubMed] [Google Scholar]
- Forster, A.C., Symons, R.H. Self-cleavage of plus and minus RNAs of a virusoid and a structural model for the active sites. Cell. 1987;49:211–220. doi: 10.1016/0092-8674(87)90562-9. [DOI] [PubMed] [Google Scholar]
- Greco-Stewart, V.S., Miron, P., Abrahem, A., Pelchat, M. The human RNA polymerase II interacts with the terminal stem–loop regions of the hepatitis delta virus RNA genome. Virology. 2007;357:68–78. doi: 10.1016/j.virol.2006.08.010. [DOI] [PubMed] [Google Scholar]
- Huang, Z.S., Wu, H.N. Identification and characterization of the RNA chaperone activity of hepatitis delta antigen peptides. J. Biol. Chem. 1998;273:26455–26461. doi: 10.1074/jbc.273.41.26455. [DOI] [PubMed] [Google Scholar]
- Huang, Z.S., Su, W.H., Wang, J.L., Wu, H.N. Selective strand annealing and selective strand exchange promoted by the N-terminal domain of hepatitis delta antigen. J. Biol. Chem. 2003;278:5685–5693. doi: 10.1074/jbc.M207938200. [DOI] [PubMed] [Google Scholar]
- Huang, Z.S., Chen, A.Y., Wu, H.N. Characterization and application of the selective strand annealing activity of the N-terminal domain of hepatitis delta antigen. FEBS Lett. 2004;578:345–350. doi: 10.1016/j.febslet.2004.11.043. [DOI] [PubMed] [Google Scholar]
- Isambert, H., Siggia, E.D. Modeling RNA folding paths with pseudoknots: Application to hepatitis delta virus ribozyme. Proc. Natl. Acad. Sci. 2000;97:6515–6520. doi: 10.1073/pnas.110533697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ke, A., Zhou, K., Ding, F., Cate, J.H., Doudna, J.A. A conformational switch controls hepatitis delta virus ribozyme catalysis. Nature. 2004;429:201–205. doi: 10.1038/nature02522. [DOI] [PubMed] [Google Scholar]
- Kierzek, R., Burkard, M.E., Turner, D.H. Thermodynamics of single mismatches in RNA duplexes. Biochemistry. 1999;38:14214–14223. doi: 10.1021/bi991186l. [DOI] [PubMed] [Google Scholar]
- Kugel, J.F., Goodrich, J.A. Promoter escape limits the rate of RNA polymerase II transcription and is enhanced by TFIIE, TFIIH, and ATP on negatively supercoiled DNA. Proc. Natl. Acad. Sci. 1998;95:9232–9237. doi: 10.1073/pnas.95.16.9232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lai, M.M. The molecular biology of hepatitis delta virus. Annu. Rev. Biochem. 1995;64:259–286. doi: 10.1146/annurev.bi.64.070195.001355. [DOI] [PubMed] [Google Scholar]
- Lai, M.M. Hepatitis delta antigen: Biochemical properties and functional roles in HDV replication. In: Handa H., Yamaguchi Y., editors. Hepatitis delta virus. Landes Bioscience; Georgetown, TX: 2006. pp. 38–51. [Google Scholar]
- Long, D.M., Uhlenbeck, O.C. Kinetic characterization of intramolecular and intermolecular hammerhead RNAs with stem II deletions. Proc. Natl. Acad. Sci. 1994;91:6977–6981. doi: 10.1073/pnas.91.15.6977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Makino, S., Chang, M.F., Shieh, C.K., Kamahora, T., Vannier, D.M., Govindarajan, S., Lai, M.M. Molecular cloning and sequencing of a human hepatitis delta (δ) virus RNA. Nature. 1987;329:343–346. doi: 10.1038/329343a0. [DOI] [PubMed] [Google Scholar]
- Martin, F.H., Tinoco I., Jr DNA–RNA hybrid duplexes containing oligo(dA:rU) sequences are exceptionally unstable and may facilitate termination of transcription. Nucleic Acids Res. 1980;8:2295–2299. doi: 10.1093/nar/8.10.2295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mathews, D.H., Sabina, J., Zuker, M., Turner, D.H. Expanded sequence dependence of thermodynamic parameters improves prediction of RNA secondary structure. J. Mol. Biol. 1999;288:911–940. doi: 10.1006/jmbi.1999.2700. [DOI] [PubMed] [Google Scholar]
- Nakano, S., Chadalavada, D.M., Bevilacqua, P.C. General acid–base catalysis in the mechanism of a hepatitis delta virus ribozyme. Science. 2000;287:1493–1497. doi: 10.1126/science.287.5457.1493. [DOI] [PubMed] [Google Scholar]
- Nautiyal, S., Woolfson, D.N., King, D.S., Alber, T. A designed heterotrimeric coiled coil. Biochemistry. 1995;34:11645–11651. doi: 10.1021/bi00037a001. [DOI] [PubMed] [Google Scholar]
- Pan, T., Sosnick, T. RNA folding during transcription. Annu. Rev. Biophys. Biomol. Struct. 2006;35:161–175. doi: 10.1146/annurev.biophys.35.040405.102053. [DOI] [PubMed] [Google Scholar]
- Pan, T., Artsimovitch, I., Fang, X.W., Landick, R., Sosnick, T.R. Folding of a large ribozyme during transcription and the effect of the elongation factor NusA. Proc. Natl. Acad. Sci. 1999;96:9545–9550. doi: 10.1073/pnas.96.17.9545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perrotta, A.T., Been, M.D. The self-cleaving domain from the genomic RNA of hepatitis delta virus: Sequence requirements and the effects of denaturant. Nucleic Acids Res. 1990;18:6821–6827. doi: 10.1093/nar/8.10.2295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perrotta, A.T., Been, M.D. A pseudoknot-like structure required for efficient self-cleavage of hepatitis delta virus RNA. Nature. 1991;350:434–436. doi: 10.1038/350434a0. [DOI] [PubMed] [Google Scholar]
- Perrotta, A.T., Been, M.D. A single nucleotide linked to a switch in metal ion reactivity preference in the HDV ribozymes. Biochemistry. 2007;46:5124–5130. doi: 10.1021/bi602569x. [DOI] [PubMed] [Google Scholar]
- Rosenstein, S.P., Been, M.D. Self-cleavage of hepatitis delta virus genomic strand RNA is enhanced under partially denaturing conditions. Biochemistry. 1990;29:8011–8016. doi: 10.1021/bi00487a002. [DOI] [PubMed] [Google Scholar]
- Schneider, T.D., Stormo, G.D., Gold, L., Ehrenfeucht, A. Information content of binding sites on nucleotide sequences. J. Mol. Biol. 1986;188:415–431. doi: 10.1016/0022-2836(86)90165-8. [DOI] [PubMed] [Google Scholar]
- Shannon, C.E., Weaver, W. The mathematical theory of communication. University of Illinois Press; Urbana, IL: 1949. [Google Scholar]
- Taylor, J.M. Structure and replication of hepatitis delta virus RNA. In: Handa H., Yamaguchi Y., editors. Hepatitis delta virus. Landes Bioscience; Georgetown, TX: 2006. pp. 20–37. [Google Scholar]
- Taylor, D.R., Lee, S.B., Romano, P.R., Marshak, D.R., Hinnebusch, A.G., Esteban, M., Mathews, M.B. Autophosphorylation sites participate in the activation of the double-stranded-RNA-activated protein kinase PKR. Mol. Cell. Biol. 1996;16:6295–6302. doi: 10.1128/mcb.16.11.6295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thill, G., Vasseur, M., Tanner, N.K. Structural and sequence elements required for the self-cleaving activity of the hepatitis delta virus ribozyme. Biochemistry. 1993;32:4254–4262. doi: 10.1021/bi00067a013. [DOI] [PubMed] [Google Scholar]
- Turner, D.H., Sugimoto, N., Freier, S.M. Thermodynamics and kinetics of base-pairing and of DNA and RNA self-assembly and helix coil transition. vol. 1C of Landolt–Bornstein Series. In: Saenger W., editor. Nucleic acids. Springer-Verlag; Berlin: 1990. pp. 201–227. [Google Scholar]
- Uptain, S.M., Kane, C.M., Chamberlin, M.J. Basic mechanisms of transcript elongation and its regulation. Annu. Rev. Biochem. 1997;66:117–172. doi: 10.1146/annurev.biochem.66.1.117. [DOI] [PubMed] [Google Scholar]
- Wadkins, T.S., Perrotta, A.T., Ferre-D'Amare, A.R., Doudna, J.A., Been, M.D. A nested double pseudoknot is required for self-cleavage activity of both the genomic and antigenomic hepatitis delta virus ribozymes. RNA. 1999;5:720–727. doi: 10.1017/s1355838299990209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang, C.C., Chang, T.C., Lin, C.W., Tsui, H.L., Chu, P.B., Chen, B.S., Huang, Z.S., Wu, H.N. Nucleic acid binding properties of the nucleic acid chaperone domain of hepatitis delta antigen. Nucleic Acids Res. 2003;31:6481–6492. doi: 10.1093/nar/gkg857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamaguchi, Y., Handa, H. Hepatitis delta antigen and RNA polymerase II. In: Handa H., Yamaguchi Y., editors. Hepatitis delta virus. Landes Bioscience; Georgetown, TX: 2006. pp. 66–75. [Google Scholar]
- Yamaguchi, Y., Filipovska, J., Yano, K., Furuya, A., Inukai, N., Narita, T., Wada, T., Sugimoto, S., Konarska, M.M., Handa, H. Stimulation of RNA polymerase II elongation by hepatitis delta antigen. Science. 2001;293:124–127. doi: 10.1126/science.1057925. [DOI] [PubMed] [Google Scholar]
- Yamaguchi, Y., Mura, T., Chanarat, S., Okamoto, S., Handa, H. Hepatitis delta antigen binds to the clamp of RNA polymerase II and affects transcriptional fidelity. Genes Cells. 2007;12:863–875. doi: 10.1111/j.1365-2443.2007.01094.x. [DOI] [PubMed] [Google Scholar]
- Zaug, A.J., Cech, T.R. The intervening sequence RNA of Tetrahymena is an enzyme. Science. 1986;231:470–475. doi: 10.1126/science.3941911. [DOI] [PubMed] [Google Scholar]
- Zuker, M. Mfold web server for nucleic acid folding and hybridization prediction. Nucleic Acids Res. 2003;31:3406–3415. doi: 10.1093/nar/gkg595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zuker, M., Mathews, D.H., Turner, D.H. Algorithms and thermodynamics for RNA secondary structure prediction: A practical guide. In: Barciszewski J., Clark Brian F.C., editors. RNA biochemistry and biotechnology. NATO ASI Series, Kluwer Academic Publishers; Dordrecht, the Netherlands: 1999. pp. 11–43. [Google Scholar]