

Abstract
Skeletal muscle expresses high levels of integrin-linked kinase (ILK), predominantly at myotendinous junctions (MTJs) and costameres. ILK binds the cytoplasmic domain of β1 integrin and mediates phosphorylation of protein kinase B (PKB)/Akt, which in turn plays a central role during skeletal muscle regeneration. We show that mice with a skeletal muscle–restricted deletion of ILK develop a mild progressive muscular dystrophy mainly restricted to the MTJs with detachment of basement membranes and accumulation of extracellular matrix. Endurance exercise training enhances the defects at MTJs, leads to disturbed subsarcolemmal myofiber architecture, and abrogates phosphorylation of Ser473 as well as phosphorylation of Thr308 of PKB/Akt. The reduction in PKB/Akt activation is accompanied by an impaired insulin-like growth factor 1 receptor (IGF-1R) activation. Coimmunoprecipitation experiments reveal that the β1 integrin subunit is associated with the IGF-1R in muscle cells. Our data identify the β1 integrin–ILK complex as an important component of IGF-1R/insulin receptor substrate signaling to PKB/Akt during mechanical stress in skeletal muscle.
Introduction
ECM of skeletal muscle consists of a basement membrane (BM) surrounding each myofiber and interstitial connective tissue (endomysium) between the myofibers. The attachment of myofibers to the BM is mainly mediated by integrins and the dystrophin–glycoprotein complex (DGC; Mayer, 2003; Michele and Campbell, 2003). Integrins are expressed throughout the sarcolemma of myofibers but are highly enriched at two force-transducing and force-regulating structures, the myotendinous junctions (MTJs), which connect myofibers to tendons, and the costameres, which are focal adhesion–like structures that connect the sarcomeric z bands with the sarcolemma.
Integrins are a large family of α/β heterodimeric adhesion receptors (Bouvard et al., 2001; Hynes, 2002). Several β1 integrins were shown to play essential roles during myogenesis and muscle homeostasis (Mayer, 2003). Antibody perturbation studies and β1 integrin gene ablations in flies and mice demonstrated that β1 integrins regulate proliferation and fusion of myoblasts and the assembly and maintenance of sarcomeres (Menko and Boettiger, 1987; Volk et al., 1990; Sastry et al., 1996; Hirsch et al., 1998; Schwander et al., 2003). The α7β1 and, until the first postnatal days, the α5β1 integrins are expressed at the MTJs, where they implement and maintain the linkage of the myofiber to the tendon matrix. α5 Integrin–deficient chimeric mice develop a muscle dystrophy associated with reduced adhesion and proliferation of myoblasts (Taverna et al., 1998). α7 Integrin–deficient mice suffer from a progressive muscular dystrophy with disrupted MTJs (Mayer et al., 1997; Miosge et al., 1999).
Integrins transduce important signals. They control actin dynamic and link the actin cytoskeleton with the ECM, and they transduce biochemical signals in cooperation with growth factor receptors, including receptors for insulin-like growth factor (IGF; Goel et al., 2004), PDGF (Schneller et al., 1997; Baron et al., 2002), VEGF (Soldi et al., 1999), and epithelial growth factor (EGF; Moro et al., 1998; Moro et al., 2002). An important and still largely unanswered question is how integrins execute their functions in myoblasts and adult skeletal muscle. Integrin cytoplasmic domains lack actin binding sites and enzymatic activities. Therefore, integrin signals are transduced through accessory molecules such as talin, α-actinin, and integrin-linked kinase (ILK; Brakebusch and Fässler, 2003).
ILK is composed of ankyrin repeats at the N terminus, a pleckstrin homology–like domain, and a putative kinase domain at the C terminus, which binds the cytoplasmic tail of β1 and 3 integrins (Grashoff et al., 2004, Legate et al., 2006). A major function of ILK is to organize the actin cytoskeleton by recruiting actin binding and actin-regulatory proteins, such as PINCH, parvin, paxillin, and kindlin (Legate et al., 2006), and to phosphorylate several proteins, including GSK-3β and PKB/Akt (Delcommenne et al., 1998; Novak et al., 1998; Persad et al., 2000), both of which are important for homeostasis and regeneration of muscle (Glass, 2003; Hoffman and Nader, 2004).
ILK is ubiquitously expressed and essential for the development of vertebrates and invertebrates. Mice lacking ILK die during the periimplantation stage because of abnormal F-actin reorganization and polarity of the epiblast (Sakai et al., 2003). In Drosophila melanogaster and Caenorhabditis elegans, the deletion of ILK leads to muscle detachment resembling the β integrin loss-of-function phenotype (Zervas et al., 2001; Mackinnon et al., 2002). Interestingly, the severe phenotypes both in flies and nematodes can be fully rescued with kinase-dead versions of ILK, suggesting that in invertebrates the kinase activity is dispensable for development and physiology (Zervas et al., 2001; Mackinnon et al., 2002).
Similarly, as in flies and nematodes, mammalian myoblasts and myofibers express high levels of ILK. In myofibers ILK is found at MTJs and costameres. The costameric location makes ILK perfectly suited to transduce contractile forces from the sarcomeres across the sarcolemma to the ECM. Consistent with such a function, mice and zebrafish that lack ILK function in cardiomyocytes exhibit severe defects in mechanotransduction resulting in lethal heart dilation, fibrosis, and disaggregation of cardiomyocytes (Bendig et al., 2006; White et al., 2006). The defects in mouse cardiomyocytes are associated with reduced Ser473 phosphorylation of PKB/Akt. Because PKB/Akt activity is crucial for cardiomyocyte growth and contractility (Condorelli et al., 2002; DeBosch et al., 2006) and ILK phosphorylates Ser473 of PKB/Akt (Delcommenne et al., 1998;Persad et al., 2000), it was concluded that mechanical stress–mediated activation of ILK supports cardiomyocyte homeostasis via PKB/Akt activation.
The role of ILK functions in skeletal muscle is obscure. Overexpression of ILK in C2C12 myoblasts was shown to inhibit myoblast fusion by sustained phosphorylated Erk1/2 activation, thus preventing cell cycle exit and myogenic determination (Huang et al., 2000). However, ILK overexpression in L6 myoblasts was shown to promote fusion and myogenin expression (Miller et al., 2003a). Finally, genetic studies in mice showed that ILK is dispensable for the development and homeostasis of skeletal muscle (White et al., 2006). The latter finding was unexpected and could potentially be because of the severe heart abnormalities and the early death of the mice, or it could alternatively result from incomplete Cre-mediated ILK gene deletion in skeletal muscle.
To test ILK functions in the skeletal muscle of mice without affecting cardiac function, we conditionally ablated the ILK gene using human skeletal α-actin (HSA) promoter–driven Cre expression. We found that loss of ILK triggered a mild, progressive muscular dystrophy, mainly restricted to MTJ areas, which was dramatically aggravated after exercise and accompanied by an impaired phosphorylation of IGF–1 receptor (IGF-1R) and PKB/Akt at the Thr308 and Ser473 residues, respectively.
Results
Skeletal muscle-specific deletion of the ILK gene
Because ILK-null (ILKlacZ/lacZ) mice die shortly after implantation (Sakai et al., 2003), we used the Cre/loxP system to disrupt the ILK gene specifically in skeletal muscle. To obtain mice with the genotype HSAcre+ /ILKflox/flox (called HSACre-ILK), ILKflox/flox mice (Grashoff et al., 2003) were intercrossed with a transgenic mouse strain expressing the Cre recombinase under the control of the HSA promoter (Schwander et al., 2003).
The efficiency of the Cre-mediated deletion of the ILK gene in vivo was tested by Southern blotting using genomic DNA and Western blotting using protein extracts from gastrocnemius (GC) muscle of 3-mo-old control and HSACre-ILK mice. The Southern blots revealed a recombination efficiency of ∼80% (Fig. 1 A) and the Western blots an ∼70% reduction of ILK protein level (Fig. 1 B). Similar experiments with muscle tissue from 4-wk-, 3-mo-, and 1-yr-old HSACre-ILK mice revealed that the ILK gene deletion and ILK protein reduction remained stable (Fig. 1 C and not depicted).
Figure 1.

ILK expression in HSACre-ILK mice. (A) Southern blot analysis of ILK in control (HSAcre− /ILKfl/+) and HSACre-ILK (HSAcre+ /ILKfl/−) GC muscles from two 4-wk-old mice. fl, flox; con, control; rec., recombined allele. (B) Western blot analysis of ILK expression in muscle used for Southern blot assay. GAPDH was used as a loading control. (C) Quantification of the ILK protein content of control and HSACre-ILK muscle by densitometric measurement of the Western blot signals. Data are expressed as the mean ± SD. (D) Immunofluorescence of ILK (red) in control and HSACre-ILK in E16.5 forelimbs, postnatal day 1 (P1) forelimbs, and 3-mo-old GC muscles. ILK is highly expressed at the MTJ (arrowheads). Lower levels of ILK are detected at the sarcolemma. No ILK signal is detected at the MTJ of HSACre-ILK mice. Nuclei are stained with DAPI (blue). Bars: (E16.5) 4 μm; (P1 and 3-mo) 50 μm.
It has been reported that the HSA-Cre transgene induces DNA recombination as early as embryonic day (E) 9.5 (Schwander et al., 2003). Despite this early Cre activity, we observed robust ILK immunostaining in all hindlimb muscle of E14.5 and 16.5 HSACre-ILK embryos (Fig. 1 D and not depicted). The intensity and the distribution of ILK immunostaining were comparable to muscle tissue from control mice with a strong signal at the MTJs and a weaker sarcolemmal staining (Fig. 1 D). Peri- and postnatally, ILK was not detected in HSACre-ILK muscle by immunostaining (Fig. 1 D). Quantification of Western blots from muscle tissue lysates showed a 60–70% reduction in ILK protein levels at this stage (Fig. 1 C). These results show that the ILK gene is efficiently deleted by the HSA-Cre transgene and that the ILK mRNA and/or protein have a long half-life in skeletal muscle cell precursors and muscle fibers.
HSACre-ILK mice develop a muscular dystrophy
Intercrosses of HSAcre+ /ILK+/− males with ILKfl/fl females revealed a normal Mendelian distribution of the four possible genotypes among 231 offspring tested: HSAcre+ /ILKfl/−; HSAcre+ /ILKfl/+; HSAcre− /ILKfl/−; and HSAcre− /ILKfl/+ = 24.4; 23.7; 25.2; and 26.8%. HSACre-ILK mice did not show an overt phenotype at birth. They were viable, fertile, and showed normal growth rates with normal weight and body length. At 3 wk of age, when control mice began to securely walk, HSACre-ILK mice still shambled and showed an abnormal walking pattern. To visualize the defect, we painted the front pad of control and HSACre-ILK mice with red ink and the hind pad with blue ink and let them walk on blotting paper. HSACre-ILK mice had an abnormal footprint pattern (Fig. 2 A) with a significantly shorter stride length, and this abnormality was maintained with age (Fig. 2 B).
Figure 2.

Footprint analysis. Footprints of 4-wk-, 3-mo-, and 12-mo-old control and HSACre-ILK mice. (A) Front and back pads were inked in red and blue colors, respectively. (B) The distances between each two footprints were measured by pixel, and to diminish the influence of body length, the distances were divided by body length. The stride length of front and back feet of 4-wk-, 3-mo-, and 12-mo-old mutant mice are significantly shorter in HSACre-ILK mice. Data are expressed as mean ± SD (n = 3; ***, P < 0.001).
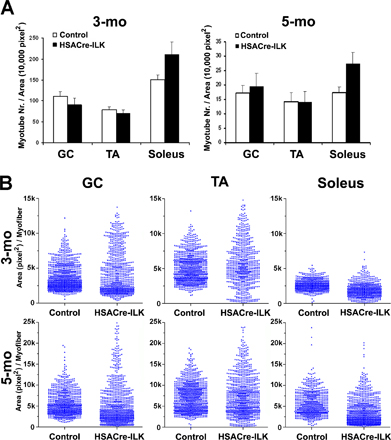
Adult mammalian skeletal muscle is differentiated into distinct fiber types, which are characterized by a unique combination of functional, biochemical, and metabolic properties. To exclude the possibility that loss of ILK affected only specific muscle fiber types, we analyzed the histology of muscles with predominantly fast fibers (tibialis anterior muscle), slow fibers (soleus muscle), and a mixture of slow and fast fibers (GC muscle) from control and HSACre-ILK mice. Samples of all three muscle groups derived from control mice exhibited myofibers with regular diameter and peripherally located nuclei. In contrast, all three muscle types analyzed from HSACre-ILK mice contained myofibers with variable fiber size and centralized nuclei (Fig. 3, A and B; Fig. S1 A; and Fig. S2, A and B, available at http://www.jcb.org/cgi/content/full/jcb.200707175/DC1). Staining of tissue sections for ATPase and NADH activity revealed that both fast and slow fiber types showed centralized nuclei (Fig. 3 B). The irregular fiber size with centralized nuclei could be observed as early as 10 d after birth and were aggravated with age (Fig. 3 A and Fig. S1 B). The number of myofibers with central nuclei increased from 11.6 ± 2.9% in 3-mo-old mice to 22.2 ± 5.1% in 12-mo-old mice, whereas the number in control mice was ∼2% at all ages analyzed. Furthermore, we frequently observed loosened intercellular space filled with fibrotic material and mononuclear cell infiltrates, which were particularly prominent at MTJs and in regions near tendons (Fig. 3 A).
Figure 3.

HSACre-ILK muscle displays signs of a mild dystrophy. (A) Hematoxylin/eosin-stained paraffin sections of the GC muscle of 10-d-, 5-mo-, and 12-mo-old control and HSACre-ILK mice. Note that the myofibers of HSACre-ILK mice show irregular diameter, centrally located nuclei (arrowheads), mononuclear cell infiltrates (asterisk), and fibrosis in 12-mo-old mutant muscle. Bars: (10-d) 40 μm; (5-mo) 50 μm; (12-mo) 60 μm. (B) Myosin ATPase, pH 4.6, and NADH-stained cryosections of the GC muscle of 3-mo-old HSACre-ILK muscles. Myofiber with asterisks indicate type II fibers with centralized nuclei. Arrowheads indicate type I fibers with centralized nuclei. Bar, 80 μm.
The extent of the fibrosis was assessed in more detail by analyzing collagen deposition using trichrome staining. In the GC of 4-wk-old mice, no obvious difference between control and HSACre-ILK was observed. However, at the age of 5 mo, fibrotic regions were observed in the endomysial space around myofibers and at the MTJs of HSACre-ILK mice. The fibrosis became more pronounced in 12-mo-old muscle (Fig. 4).
Figure 4.

Altered collagen deposition in HSACre-ILK muscles. Trichrome staining of paraffin sections of GC muscle of 4-wk-, 5-mo-, and 12-mo-old control and HSACre-ILK mice. Collagen-containing fibrotic regions display blue color signals (arrowheads). Bar, 50 μm.
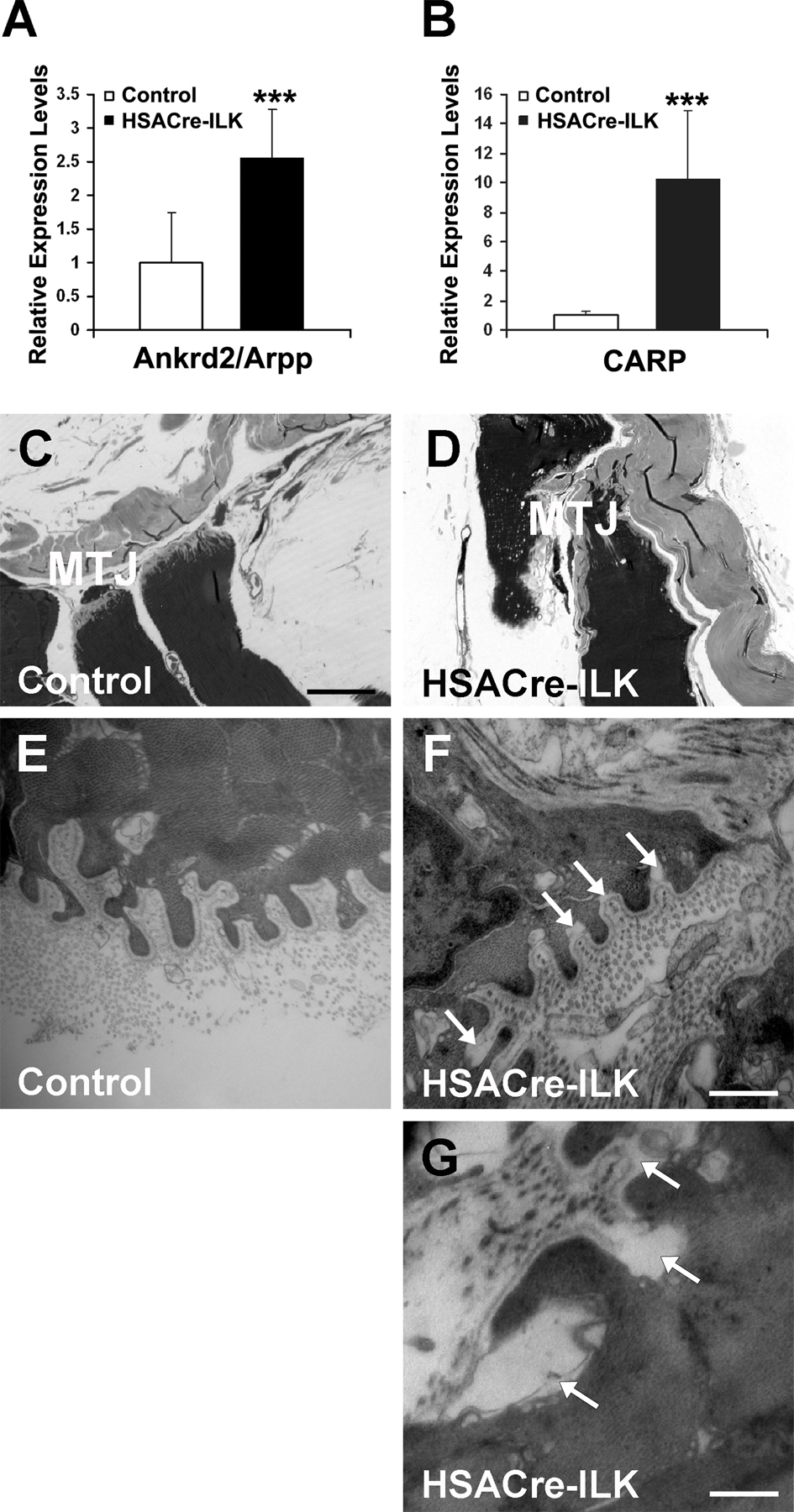
Because ILK-deficient MTJs displayed abnormalities, we further analyzed them at the ultrastructural level. The MTJs from control mice were extensively folded, forming digit-like protuberances of regular size and width that were covered with a well-structured BM and extended from the muscle cells into the collagen-rich tendon matrix (Fig. 5, A and C). The MTJs of 5-mo-old HSACre-ILK mice had folds with irregular size and width (Fig. 5, B and D). We frequently observed that the BM was detached from the sarcolemma at the base of the digit-like protrusions (Fig. 5 D, arrowheads). Interestingly, sarcomeres of HSACre-ILK myofibrils that contained central nuclei and, hence, had regenerated, appeared similar to control mice (Fig. 5, E and F), indicating that regeneration occurs normally in the absence of ILK expression.
Figure 5.

Abnormalities at the MTJs of HSACre-ILK mice. Alteration of MTJs in HSACre-ILK mice. Electron micrographs of the MTJ of 5-mo-old control (A and C) and HSACre-ILK (B and D) mice. (A and B) MTJs with multiple finger-like interdigitations of regular organization and length are observed in control (A) and HSACre-ILK (B) mice. (C) The finger-like folds are covered by a well-organized and closely attached BM interdigitating with collagen fibrils in control mice. (D) HSACre-ILK mice showed folds of slightly different length and width with a partial detachment of the BM (arrowheads). (E and F) The sarcomeric structure was normal in fibers of HSACre-ILK containing centralized nuclei (asterisk). The open white rectangle in E indicates the region shown in F. Bars: (A and B) 2.2 μm; (C and D) 500 nm; (E) 20 μm; (F) 800 nm.
Collectively, these results suggest that the deletion of the ILK gene from skeletal muscles leads to a mild muscular dystrophy characterized by abnormalities at MTJs, variation of myofiber size, and increased fibrosis.
Loss of ILK affects integrin localization at MTJs
ILK forms a ternary complex with PINCH and parvin that is important for the stability of the individual components and the recruitment of the complex into focal adhesions. Similarly, as reported for other cell types and tissues, HSACre-mediated loss of ILK was associated with reduced PINCH1 and β-parvin levels, which are both highly expressed in skeletal muscle (Fig. 6 A). The ILK–PINCH–parvin complex is thought to regulate integrin function and actin reorganization. Interestingly, however, ultrastructural analysis revealed no signs of F-actin detachment from the sarcolemma both at the MTJs and in central areas of the muscle tissue (Fig. S3, available at http://www.jcb.org/cgi/content/full/jcb.200707175/DC1). This indicates that, in contrast with flies and nematodes, ILK is not essential for anchoring actin filaments to the muscle cell membrane.
Figure 6.

Expression and distribution of ILK-associated proteins. (A) ILK, Pinch, and parvin are decreased in HSACre-ILK muscle, whereas β1, β1D, α5, α7A, and α7B integrin and phosphopaxillin (p-paxillin), β-dystroglycan (β-DG), and caveolin-3 are not changed. GAPDH was used as the loading control. (B) Immunofluorescence staining of α7A, α7B, α5, and β1D integrins and β-dystroglycan. Note the prominent α7A integrin signals (red) at MTJ of control muscles and the reduction at MTJs of HSACre-ILK muscles. Although α7B integrin (red) is expressed on the sarcolemma of all myofibers in the control, it shows an irregular staining pattern in HSACre-ILK muscle. In contrast to the control muscle, α5 integrin signals are not restricted to the tendon of HSACre-ILK muscles. β1D Integrin and β-dystroglycan signals are comparable between control and HSACre-ILK muscles. Bars: (α7A integrin, α5 integrin, and β-dystroglycan) 40 μm; (α7B integrin) 35 μm; (β1D integrin) 60 μm.
The predominant integrin of skeletal muscle is the α7β1D integrin. The α7 integrin gene is alternatively spliced, producing the α7B splice variant (α7Bβ1), which is the predominant form in skeletal muscle found at the sarcolemma and the MTJ, and the α7A splice variant (α7Aβ1), which is expressed at MTJs (Nawrotzki et al., 2003). Both α7 subunits associate with a splice variant of the β1 integrin subunit called β1D integrin, which is found at the MTJs and costameres (Belkin et al., 1996; van der Flier et al., 1997). Western blotting and immunostaining revealed strong β1D integrin expression at MTJs and lower levels at the sarcolemma both in control and HSACre-ILK muscle (Fig. 6, A and B). Although similar amounts were detected by immunoblotting, immunostaining for α7Aβ1 and α7Bβ1 integrins revealed irregular staining patterns with reduced signals in some areas of the muscle tissue (Fig. 6 B). Furthermore, unlike in control muscle, the α5 staining was not restricted to the tendon but partly extended into the muscle fibers (Fig. 6 B).
The BM around myofibers is assembled by integrins and the DGC (Mayer et al., 1997; Miosge et al., 1999; Nawrotzki et al., 2003; Guo et al., 2006; Rooney et al., 2006). Immunostaining revealed normal expression of β-dystroglycan, laminin α2, and dystrophin (Fig. 6 B and Fig. S4, available at http://www.jcb.org/cgi/content/full/jcb.200707175/DC1). Collectively, we conclude that ILK deficiency in muscle does not affect F-actin anchorage and BM assembly but affects the distribution of integrins.
Normal myoblast fusion in vitro
Because of the long half-life of the ILK mRNA and/or protein, HSACre-ILK embryos still contained significant levels of ILK in skeletal muscle tissue around the time of myoblast migration and fusion (Fig. 1 D). Therefore, we isolated primary myoblasts from the hindlimbs of 2-d-old control and HSACre-ILK mice and compared their ability to form myotubes in vitro. Western blotting of freshly isolated myoblasts cultured in growth medium (GM) or differentiation medium (DM) revealed faint levels of ILK (Fig. 7 A), which were most likely derived from fibroblast contaminations (desmin-negative cells; not depicted). The switch from GM to DM induced the expression of the myogenic marker myogenin and repressed the expression of MyoD to a similar extent in both control and HSACre-ILK cells. PINCH1 levels decreased concomitantly with ILK, whereas the levels of integrin β1D, phosphorylated PKB/Akt, and phosphorylated Erk1/2 were similar between control and HSACre-ILK myoblasts (Fig. 7 A). When the primary myoblasts were induced to form myotubes, we observed that HSACre-ILK myoblasts were able to efficiently form multinucleated myotubes (Fig. 7, B–D). Neither the number of myotubes nor the kinetic of their formation differed between the cultures of control and HSACre-ILK myoblasts. Furthermore, quantitative analysis revealed similar numbers of nuclei per myotube in control and HSACre-ILK cultures (Fig. 7, C and D). These data indicate that ILK plays no obvious role in myoblast proliferation, differentiation, and fusion.
Figure 7.

Normal fusion of HSACre-ILK myoblasts. (A) Protein levels of ILK and PINCH1 are dramatically decreased in primary HSACre-ILK myoblasts both in GM and DM. Signals of desmin and myogenin increase and MyoD decreases in response to differentiation in both cell types. The levels of β1D integrin, pSer473-Akt, pThr308-Akt, Akt, pErk1/2, and Erk1/2 are comparable. GAPDH was used as the loading control. (B) Myoblast cells were isolated from postnatal day 2 control and HSACre-ILK hindlimbs, incubated in DM, and evaluated microscopically. Differentiated myoblasts and myotubes were stained with sarcomeric α-actinin antibody (red) and DAPI (blue). (C) Quantification of myotube numbers from control and HSACre-ILK myoblasts plated at different cell densities. Data are expressed as mean ± SD (n = 3; 200 and 400 cells/nm2). (D) Determination of the number of nuclei per fused myotube. No difference between control and HSACre-ILK mice was observed (P = 0.253). Bars: (phase) 50 μm; (sarcomeric α-actinin) 80 μm.
Abnormal mechanical stress response
To test how mechanical stress affects the integrity of HSACre-ILK muscles, we subjected 5-mo-old control and HSACre-ILK mice, respectively, to daily treadmill exercise with an upward inclination of 10° at 18 m/min for 60 min, 5 d/wk. In the first 3 wk, both control and HSACre-ILK mice maintained the required speed of 18 m/min at the 10° inclination during the whole training sessions. Because HSACre-ILK mice were unable to run at this speed without breaks after the 3-wk training period, we terminated the treadmill exercise and analyzed the muscle tissue.
Hematoxylin/eosin and trichrome staining of trained HSACre-ILK muscles revealed an increase in fibrosis which was not detected in control muscles (Fig. 8 A). The myofibers of untrained HSACre-ILK mice showed mild dystrophic changes and were positive for methylene blue staining (Fig. 8 B). In contrast, fibers of trained HSACre-ILK muscle were frequently negative for methylene blue, indicating muscle damage (Fig. 8 B). To evaluate the mechanical stress–induced damage more quantitatively, we investigated the levels of stretch/injury-responsive muscle ankyrin-repeat proteins Ankrd2 and CARP (Miller et al., 2003b; Hentzen et al., 2006). Both Ankrd2 and CARP mRNA were found to be significantly up-regulated in trained HSACre-ILK muscles, further confirming the exercise-induced muscle damage in HSACre-ILK mice (Fig. S5, A and B, available at http://www.jcb.org/cgi/content/full/jcb.200707175/DC1).
Figure 8.

Exercise-induced alterations in myofibers of HSACre-ILK mice. (A) Hematoxylin/eosin (H&E)- and trichrome-stained cryosections of the GC muscle of trained control and HSACre-ILK mice. Arrowheads indicate fibrotic regions. Bar, 50 μm. (B) Methylene blue–stained skeletal muscle fibers of untrained and trained HSACre-ILK mice. Trained HSACre-ILK muscles contain necrotic fibers (asterisk). Bars: (top) 35 μm; (bottom) 900 nm.
Furthermore, we observed profound abnormalities in MTJs of HSACre-ILK muscles at the ultrastructural level. Although control mice had normal MTJs after the exercise program (Fig. S5, C and D), the MTJs from HSACre-ILK mice almost completely lost their digit-like interdigitations and instead formed irregular membrane protrusions and invaginations (Fig. S5, E and F). Concomitantly with these defects, the BM detachment from the sarcolemma was further aggravated (Fig. S5, F and G) when compared with untrained muscle (Fig. 5 F). In addition, in some areas the BM was replaced by an electron-dense material (Fig. S5 F).
Training experiments with 9-mo-old HSACre-ILK mice showed that they were unable to exercise at a speed of 18 m/min. This age-dependent decline in running capacity made it impossible to perform a training intervention comparable to the 5-mo-old mice. Collectively, these data demonstrate that ILK-deficient skeletal muscle is highly susceptible to mechanical stress.
ILK modulates IGF signaling
Growth of skeletal muscle critically depends on the activation of mTOR kinase by PKB/Akt (Glass, 2003; Hoffman and Nader, 2004). ILK phosphorylates Ser473 of PKB/Akt and is thought to be required for full PKB/Akt activation. To test PKB/Akt activity, we isolated muscle tissue and performed Western blotting using phosphospecific antibodies. Untrained HSACre-ILK muscle displayed levels of Ser473 and Thr308 phosphorylation comparable with those of control muscle (Fig. 9 A). Upon training, however, we observed a significant increase in the phosphorylation of Ser473 as well as of Thr308 residues of PKB/Akt in control muscle, whereas the HSACre-ILK muscle showed a strongly attenuated response (Fig. 9 A).
Figure 9.

Altered IGF signaling after endurance exercise training. (A and B) Western blot analysis of untrained and endurance exercise–trained control and HSACre-ILK GC muscle for ILK, pSer473-Akt, pThr308-Akt, total Akt, pTyr397-FAK, and total FAK (A) and pTyr1131/1135/1136-IGF-1R and total IGF-1R (B). GAPDH was determined to show equal loading. (C) Densitometric quantification of pTyr-IGF-1R levels from resting and endurance exercised–trained control and HSACre-ILK muscles. The increase of pTyr-IGF-1R was significantly reduced in muscle from HSACre-ILK mice (n = 4; **, P < 0.01). (D) Real-time PCR analysis of IGF-1R mRNA expression of control and HSACre-ILK muscles. The levels were not significantly different between control and HSACre-ILK mice (n = 4; P = 0.199). (E) Measurement of autocrine IGF-1 levels of control and HSACre-ILK muscles by ELISA. The level of IGF-1 in trained HSACre-ILK was 75% higher than in trained control muscles. Data are expressed as mean ± SD (n = 4; *, P < 0.05). (F) C2C12 mouse myoblasts were lysed before (0) and after 1 and 4 d of differentiation with or without IGF-1 treatment, immunoprecipitated with rabbit anti–β1 integrin antiserum or rabbit IgG (as control), and immunoblotted with rabbit anti–IGF-1R, rabbit anti–β1 integrin, or rabbit anti–EGF-R antiserum. 8 μg of lysates were used as inputs.
In muscle, PKB/Akt can be activated by an intracellular signaling cascade that is triggered through the activation of IGF-1R (Mourkioti and Rosenthal, 2005). Therefore, we tested whether IGF-1R levels and/or activation were altered in exercised HSACre-ILK muscle. Western blotting and real-time PCR revealed that the total protein and RNA levels of IGF-1R were similar before and after exercise in control and HSACre-ILK mice (Fig. 9, B and C). Upon exercise, the phosphorylation of cytoplasmic tyrosines in the activation loop of IGF-1R (Tyr1131/1135/1136), which are known to induce PKB/Akt-activation (Vasilcanu et al., 2004), significantly increased by 57.9 ± 0.19% in exercised control muscle (Fig. 9, B and D). In contrast, there was no increase in phosphorylation of IGF-1R in HSACre-ILK muscle upon exercise (Fig. 9, B and D). Importantly, the failure of increased IGF-1R phosphorylation upon training was not caused by diminished IGF-1 secretion because IGF-1 levels were even higher in trained HSACre-ILK muscle than in that of trained controls (Fig. 9 E). These findings suggest that ILK acts in concert with growth factors to protect muscle from mechanical damage by regulating the IGF-1R–PBK–Akt signaling pathway.
The β1 integrin subunit can associate with the IGF-1R in several cell types (Goel et al., 2006). To test whether a similar association occurs before and/or during the formation of myofibers, we cultured C2C12 cells for different days in fusion medium in the presence or absence of IGF-1, cross-linked and immunoprecipitated β1 integrin subunits, and finally probed the precipitate with antibodies against IGF-1R, EGFR, β-dystroglycan, and β1 integrin. As shown in Fig. 9 F, the β1 integrin associated with the IGF-1R, with or without IGF-1 treatment and before and after myoblast fusion. Interestingly, the amount of IGF-1R–β1 integrin complexes increased in response to IGF-1 treatment. The association was specific because β1 subunits neither coimmunoprecipitated with EGFR (Fig. 9 F) nor with β-dystroglycan (not depicted).
Discussion
In this paper, we report the skeletal muscle–specific ablation of the ILK gene, which leads to a mild muscular dystrophy and increased susceptibility to stress-induced damage. The exercise-induced defects are associated with reduced PKB/Akt activation, which is likely caused by an impaired cross talk between β1 integrin–ILK and the IGF-1R–insulin receptor substrate 1–PI-3K signaling pathway.
ILK maintains MTJs of untrained muscle
Loss of ILK resulted in a very mild phenotype characterized by a persistent shamble without affecting weight or life span and damaged muscle fibers with increased presence of centralized nuclei and large variation of myofiber size. Other alterations were restricted to the MTJs and included increased fibrosis, infiltration of a few inflammatory cells, and detachment of BMs at the base of the interdigitations. Because we observed normal Erk1/2 activation and normal levels of activated PKB/Akt in ILK-deficient muscle, we conclude that muscle regeneration works efficiently in the absence of ILK, and that muscle damage likely results from a mechanical rather than a signaling failure. Furthermore, it seems that ILK is important for the stabilization of adhesion sites exposed to high mechanical forces, such as MTJs, whereas at costameres, where less force is transmitted, ILK seems to be dispensable.
The destabilization of MTJs is caused by a detachment of the BM rather than a detachment of F-actin from the sarcolemma, indicating that loss of ILK primarily affects ligand binding of integrins. This observation differs from the loss-of-function studies of ILK in flies (Zervas et al., 2001), where ILK is primarily required to attach the actin cytoskeleton at the plasma membrane and, to a lesser extent, for integrin binding to the ECM. It is possible that the requirements of ILK are slightly different between invertebrate and vertebrate muscle. Alternatively, it could also be that our mice have subtle actin defects that escaped detection.
ILK is not required for myoblast fusion and sarcomere assembly
Although the β1 integrins were shown to regulate myoblast fusion and sarcomere assembly (Menko and Boettiger, 1987; Hirsch et al., 1998; Schwander et al., 2003), it is still unclear how β1 integrins execute these functions. Two studies reported that ILK is acting downstream of β1 integrin to control fusion and sarcomere formation. The studies overexpressed wild-type and mutant ILK cDNAs in either mouse or rat myoblast cell lines and came to opposite conclusions. Although one study showed that ILK antagonizes myoblast fusion by sustained Erk1/2 activation, preventing cell cycle withdrawal and myogenic determination, another study reported that ILK stimulates fusion and myogenic determination (Huang et al., 2000; Miller et al., 2003a).
Unfortunately, the long half-life time of the ILK mRNA and/or protein did not permit us to study myoblast fusion and sarcomere assembly in vivo. Therefore, we isolated primary myoblasts from newborn control and HSACre-ILK mice and tested whether they fuse and assemble sarcomeres in vitro. We found that primary ILK-deficient myoblasts exhibit normal fusion and sarcomere assembly, indicating that ILK does not play an obvious and very prominent role during myogenic differentiation. These findings are in line with recent zebrafish data showing that loss of ILK function results in severe heart failure but normal development of skeletal muscle.
ILK protects myofibers from force-induced damage
Contrary to the mild defects of ILK-deficient skeletal muscle, disruption of ILK function in zebrafish or the myocardium of mice leads to lethality with progressive contraction defects, heart dilation, and fibrosis (Bendig et al., 2006; White et al., 2006). The strong phenotype suggests that mechanical loading triggers the severe defects. We tested this assumption by exposing control and mutant mice to forced treadmilling and found that mutant mice could not complete the 4-wk training protocol with a treadmilling speed of 18 m/min, demonstrating that the function of HSACre-ILK muscle was profoundly compromised. After training for 3 wk, the MTJs of HSACre-ILK muscle displayed an almost complete loss of interdigitations, extensive BM detachments, and myofiber necrosis.
Although treadmilling profoundly augmented the defects at the MTJs, we still observed normal insertions of actin filaments at the sarcolemma at MTJs. This could have several reasons. It is possible that the training protocol was not vigorous enough to trigger actin filament detachment as observed in flies and nematodes. It is also conceivable that ILK plays no or only a minor role as a mechanical linkage for the terminal sarcomeres at MTJs. Finally, loss of ILK may be compensated by other actin-linked adhesion molecules such as the DGC. Consistent with the latter assumption, it was demonstrated that mdx/α7 integrin−/− double mutant mice develop a more severe muscle dystrophy than dystrophin or α7 integrin single mutant mice (Guo et al., 2006; Rooney et al., 2006).
ILK is required for the mechanical stress-induced activation of PKB/Akt
ILK is believed to phosphorylate and, thereby, modulate the activity of several target proteins, including PKB/Akt, which, in turn, plays a central role during the repair of skeletal muscle by activating the mTOR complex and downstream targets such as S6K and 4E-BP (Bodine et al., 2001; Rommel et al., 2001). Several reports have recently challenged this view and showed that the phosphorylation of Ser473 is mediated by the mTOR kinase rather than ILK, that the phosphorylation of Thr308 is sufficient for activation of PKB/Akt, and that the phosphorylation of Ser473 determines the specificity of PKB/Akt toward FOXO1 and FOXO3 and, to a lesser extent, the absolute activity of PKB/Akt (Frias et al., 2006; Guertin et al., 2006; Jacinto et al., 2006; Shiota et al., 2006). Despite these novel findings, ablation of the ILK gene in cardiomyocytes of mice led to a dramatically reduced Ser473 phosphorylation of PKB/Akt. Because the phosphorylation levels of Thr308 were not determined in ILK-deficient cardiomyocytes, it is unclear whether loss of ILK affected the specificity of PKB/Akt only or also the overall activity of PKB/Akt.
We observed similar phosphorylation levels of Ser473 and Thr308 in control and HSACre-ILK muscle of untrained mice, suggesting that the mild muscular dystrophy upon ILK loss is not a result of diminished ILK-dependent PKB/Akt activation. In sharp contrast, forced treadmilling triggered an increase of Ser473 phosphorylation in the skeletal muscle of control mice, whereas phospho-Ser473 levels failed to rise in HSACre-ILK muscle. Moreover, the reduced PKB/Akt phosphorylation was not restricted to the potential ILK target Ser473 but was also observed on Thr308, indicating that mechanical load–induced activation of both the PDK-1 and Ser473 kinase activities requires ILK.
These findings suggest that loss of ILK likely affects an activator, which is upstream of both the PDK-1–Thr308-PKB–Akt and the Ser473–PKB/Akt pathways in mechanically challenged muscle. A potential candidate for such an upstream activator is IGF-1R, which plays a central role during muscle repair (Musaro et al., 2001), acts through PI-3K/PKB/Akt/mTor signaling (Bodine et al., 2001; Rommel et al., 2001), cross talks with the β1 integrin signaling pathway (Goel et al., 2004, 2006), and was shown to activate ILK (Attwell et al., 2000). We performed Western blot assays with antibodies against the phosphorylated form of IGF-1R and could indeed demonstrate that IGF-1R activation is severely impaired in trained HSACre-ILK muscle. It is well known that physical training triggers IGF-1R phosphorylation and the downstream activation of PKB/Akt and the mTor complexes, which in turn give rise to the formation of new myofibrils. Consistent with the reduced PKB/Akt phosphorylation, the training-induced phosphorylation of IGF-1R at tyrosine residues (Tyr1131/1135/1136) in the activation loop of the kinase domain was almost completely abrogated in HSACre-ILK muscle. Phosphorylation of Tyr1136 was shown to be required for the IGF-1–induced phosphorylation of insulin receptor substrate 1, the interaction of the regulatory p85 subunit of PI-3K with IGF-1R, and the downstream-activation of PKB/Akt (Vasilcanu et al., 2004). Moreover, we observed that the β1 integrin subunit can form a complex with IGF-1R in C2C12 cells before and after fusion into myotubes. Interestingly, IGF-1 treatment increases the amount of β1 integrin–IGF-1R complex, corroborating that the complex formation has a functional role in IGF signaling. Collectively, our data suggest that the β1 integrin–IGF-1R complex is using ILK to activate the IGF-1R signaling machinery, leading to PKB/Akt activation and regeneration of exercise-induced muscle damage.
Materials and methods
Mouse strains
To obtain mice with a skeletal muscle–restricted deletion of the ILK gene, floxed ILK mice (Grashoff et al., 2003) were crossed with transgenic mice expressing the Cre gene under the control of the HSA promoter (Schwander et al., 2003). All animals were fed ad libitum and housed according to the guidelines of the Society of Laboratory Animal Science.
Antibodies
Antibodies used in this study were mouse anti-GAPDH (Millipore), mouse anti–sarcomeric α-actinin (Sigma-Aldrich), rabbit anti-caveolin3 (Abcam), rabbit anti-Erk1/2 (Cell Signaling Technology), rabbit anti–phospho-Erk1/2 (Thr202/Tyr204; Cell Signaling Technology), mouse anti-desmin (BD Biosciences), mouse anti-paxillin (Transduction Laboratories), rabbit anti-phosphopaxillin (Tyr118; Cell Signaling Technology), mouse anti-myogenin (BD Biosciences), rabbit anti-MyoD (Santa Cruz Biotechnology, Inc.), rabbit anti-FAK (Millipore), rabbit anti–phospho-FAK (Tyr397; Invitrogen), mouse anti-dystrophin (Abcam), goat anti–β-dystroglycan (Santa Cruz Biotechnology, Inc.), rabbit anti–EGF-R (Cell Signaling Technology), rabbit anti–phospho–IGF-1R (Tyr1131/1135/1136; Acris Antibodies), rabbit anti–IGF-1R (Cell Signaling Technology), and mouse anti–IGF-1R (Millipore). Fluorescent dye–conjugated secondary antibodies were obtained from Invitrogen. All other antibodies used have been described previously (Nawrotzki et al., 2003; Sakai et al., 2003; Stanchi et al., 2005; Chu et al., 2006; Guo et al., 2006).
Western blotting
Muscle tissue was homogenized in modified RIPA buffer (50 mM Tris-HCl, pH 7.4, 150 mM NaCl, 5 mM EDTA, 0.1% SDS, 1% Triton X-100, 1% sodium deoxycholate, protease inhibitors [Roche], and phosphatase inhibitors [Sigma-Aldrich]). Extracted proteins were gel separated and immunoprobed as previously described (Grashoff et al., 2003).
Histology, immunofluorescence, and electron microscopy
Muscle tissues from embryos or newborn or adult mice was excised and either frozen in liquid nitrogen–cooled isopentane or dehydrated and embedded in paraffin. 8 μm of transverse sections were cut and collected onto SuperFrost Plus (Menzel-Gläser) slides. The area of myofibers was determined on hematoxylin/eosin-stained paraffin sections using the Axiovision software (Version 4.6.3.0; Carl Zeiss, Inc.).
Immunofluorescence was done on cryosections as described in Mayer et al. (1997). Images were collected by confocal microscopy (DMIRE2; Leica) using Leica Confocal Software (version 2.5, build 1227) with 40 or 63× oil objectives, by fluorescence microscopy (DMRA2; Leica) using SimplePCI software (version 5.1.0.0110; GTI Microsystems) with 20, 40, or 63× oil objectives, or by bright field microscopy (Axiovert; Carl Zeiss, Inc.) using IM50 software (Leica) with 10 or 40× objectives. All images were collected at RT. Digital images were manipulated and arranged using Photoshop CS2 (Adobe). Transmission electron microscopy was performed using an electron microscope (902A; Carl Zeiss, Inc.) as described in Hirsch et al. (1998).
In brief, muscle biopsies were fixed in 4% buffered PFA, rinsed three times in cacodylate buffer, and then treated with 1% uranyl acetate in 70% ethanol for 8 h. The biopsies were subsequently dehydrated in a graded series of ethanol and then embedded in Araldite (SERVA). Semithin sections (500 μm) were cut with a glass knife on an ultramicrotome (Reichert) and stained with Methylene blue. Ultrathin sections (30–60 nm) for electron microscopic observation were processed on the same microtome with a diamond knife and placed on copper grids.
Isolation and differentiation of primary myoblasts
Primary myoblasts were isolated as described by Rando and Blau (1994). In brief, hindlimbs were dissected from 1–2-d-old mice, placed in PBS, minced with a razor blade, and enzymatically dissociated with a mixture of collagenase II (0.1%; Worthington Biochemical) and dispase (2.4 U/ml; grade II; Roche). The slurry, maintained at 37°C for 30–45 min, was triturated every 15 min with a 5-ml plastic pipette. After centrifugation at 350 g for 10 min, the pellet was resuspended in DME containing 20% FCS, 2 mM glutamin, and 1% Pen/Strep (Invitrogen) and preplated into noncoated tissue culture dishes for 20 min for attaching fibroblasts to the dish surface. The nonadherent cells were then transferred into 0.2% gelatin-coated 6-well plates (approximately two limbs for one well). Differentiation was induced with 5% horse serum (Invitrogen) in DME for 2–4 d. A myotube was defined as having three or more nuclei.
Endurance exercise training
Experiments were performed with 5-mo-old control (seven mice; weight: 27.0 ± 0.7 g) and HSACre-ILK (six mice; weight: 25.1 ± 0.9 g) mice. The treadmill (Exer3/6; Columbus Instruments) endurance training consisted of a 60-min treadmill exercise 5 d a week at a velocity of 18 m/min at an angle of 10°. Mice were elicited to run by touching their back with a pencil. Mice were accommodated to the situation for 1 wk before starting the experiments. The velocity of 18 m/min was chosen because in these preexperiments, control and HSACre-ILK mice were able to constantly run for 1 h at this velocity. An angle of 10° was chosen to increase the muscle load during the training. The training was performed for 4 wk. At the end of this period, animals were killed and the vastus lateralis and GC muscles were isolated, fixed in 4% buffered PFA for 6 h, and prepared for ultrastructural analysis.
An additional test was initiated with 9-mo-old control (n = 6) and HSACre-ILK (n = 6) mice under the conditions described in the previous paragraph. Because HSACre-ILK mice were unable to abide the exercise, the experiment was terminated. The local Animal Care Committee approved all experimental procedures.
Real-time PCR
Muscle RNA was isolated with the RNeasy Mini kit (QIAGEN). 1 μl cDNA generated from 200 ng RNA with the iScript cDNA Synthesis kit (Bio-Rad Laboratories) was subjected to real-time PCR using the iQ SYBR Green Supermix (Bio-Rad Laboratories) and the iCycler (Bio-Rad Laboratories). The following primers (Mouse Genome Informatics number1204415) were used for detecting IGF-1R: forward, 5′-TGGCACCTACAGGTTCGAG-3′; and reverse, 5′-TGATGGACACACCTGCATG-3′. The following primers were used for CARP: forward, 5′-GAGAAGTTAATGGAGGCTGG-3′; and reverse, 5′-GTTCAGCAACAGTTTCAGGAC-3′. The following primers were used for Ankrd2: forward, 5′-CCACAGAGCTCATCGAGCAG-3′; and reverse, 5′-CTAGCACTAGCATGTCCATGG-3′. Gene expression was quantified using the Gene Expression Analysis Program for the iCycler iQ Real-Time PCR Detection system (Bio-Rad Laboratories) and normalized to GAPDH levels.
Cross-linking and immunoprecipitation
C2C12 mouse myoblasts were maintained in GM and differentiation was induced with DM. Treatment with 100 ng/ml mIGF-1 (R&D Systems) in DME was performed 14 h after starvation.
Cross-linking reaction was performed in 1 mM DSP (Thermo Fisher Scientific) in PBS for 20 min on ice. Cells were lysed in IP buffer containing 1% Triton X-100, 0.05% sodium deoxycholate, 150 mM NaCl, and 50 mM Tris-HCl, pH 8, with protease inhibitors and phosphatase inhibitors (cocktails 1 and 2). For coimmunoprecipitation of β1 integrin, 700 μg of lysates were incubated with anti–β1 integrin antiserum for 30 min at 4°C and then with 35 μl of protein A–Agarose for another 1 h. Protein complexes were washed three times in wash buffer (0.1% Triton X-100, 0.005% sodium deoxycholate, 150 mM NaCl, and 50 mM Tris-HCl, pH 8) and subsequently extracted with 5× SDS loading buffer for 5 min at 95°C.
Myosin ATPase, pH 4.6, staining
Unfixed cryosections were preincubated for 10 min at RT in incubation buffer (0.1 M NaOAc and 1 mM EDTA adjusted to pH 4.6). Slides were dipped in incubation buffer, pH 9.6, and then immediately incubated in ATP solution (10 mg ATP in 10 ml incubation buffer, pH 9.6) for 10 min at 37°C. After washing with double-distilled H2O (ddH2O), slides were immersed in 2% CoCl2 for 5 min. After another wash with ddH2O, slides were immersed in 0.1% ammonium sulfide solution for 30 s. Finally, the slides were washed under running water for 5 min, dehydrated, and mounted in glycerine jelly.
NADH staining
Unfixed cryosections were incubated for 30 min in 0.2M Tris-HCl, pH 7.4, 1.5 mM NADH, and 1.5 mM Nitroblue tetrazolium (Sigma-Aldrich) at 37°C. After incubation, slides were rinsed three times with ddH2O and mounted in Evanol.
Masson's trichrome staining
Slides were mordant in preheated Bouin's solution (saturated picric acid/formaldehyde/glacial acetic acid = 15:5:1) for 15 min at 56°C. After cooling to RT, slides were washed under running water to remove the yellow color and stained in Weigert's iron hematoxylin solution for 5 min. Slides were then washed for 5 min under running water, rinsed in ddH2O, and stained in Biebrich scarlet-acid Fuchsin for 5 min. After rinsing in ddH2O, slides were transferred to Aniline blue Solution for 5 min and, subsequently, to 1% acetic acid for 2 min. Finally, the slides were rinsed, dehydrated through alcohol, cleared in xylene, and mounted. All chemicals were from Sigma-Aldrich.
IGF-1 measurement
Muscle tissue was homogenized in PBS, pH 7.4. After two freeze–thaw cycles, the homogenates were centrifuged for 5 min at 5,000 g. The supernatant was then removed and stored at −80°C. IGF-1 measurement was performed as described in the manual of the Quantikine Mouse IGF-1 kit (R&D Systems).
Statistical analysis
Statistical evaluation was performed with GraphPad Prism software (GraphPad, Inc.). Statistical significance between data groups was determined by the Mann-Whitney test and subdivided into three groups (*, P < 0.05; **, P < 0.01; ***, P < 0.001).
Online supplemental material
Fig. S1 shows the centralized nuclei in soleus, extensor digitorum longus, and tibialis anterior muscle of HSACre-ILK mice. Fig. S2 shows the measurements of myofiber density and size in HSACre-ILK and control mice. Fig. S3 shows that the sarcolemmal F-actin is unaffected in HSACre-ILK muscle. Fig. S4 shows the normal distribution and expression of β1 integrin, vinculin, laminin α2, and dystrophin in HSACre-ILK muscle. Fig. S5 shows the analysis of Ankrd2 and CARP levels as markers of muscle damage in trained mice as well as ultrastructural analysis showing detachment of the BM in trained HSACre-ILK muscle. Online supplemental material is available at http://www.jcb.org/cgi/content/full/jcb.200707175/DC1.
Supplemental Material
Acknowledgments
We thank the members of the Fässler laboratory for lively discussions and careful reading of the manuscript.
S.A. Wickström is supported by the Sigrid Juselius Foundation and the Finnish Cultural Foundation. This work was funded by the Welcome Trust (grant 060549 to U. Mayer), the Austrian Science Fund (grant SFB021), and the Max Planck Society (to R. Fässler).
Abbreviations used in this paper: BM, basement membrane; ddH2O, double-distilled H2O; DGC, dystrophin–glycoprotein complex; DM, differentiation medium; E, embryonic day; EGF, epithelial growth factor; GC, gastrocnemius; GM, growth medium; HSA, human skeletal α-actin; IGF, insulin-like growth factor; IGF-1R, IGF receptor 1; ILK, integrin-linked kinase; MTJ, myotendinous junction.
References
- Attwell, S., C. Roskelley, and S. Dedhar. 2000. The integrin-linked kinase (ILK) suppresses anoikis. Oncogene. 19:3811–3815. [DOI] [PubMed] [Google Scholar]
- Baron, W., S.J. Shattil, and C. ffrench-Constant. 2002. The oligodendrocyte precursor mitogen PDGF stimulates proliferation by activation of αvβ3 integrins. EMBO J. 21:1957–1966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Belkin, A.M., N.I. Zhidkova, F. Balzac, F. Altruda, D. Tomatis, A. Maier, G. Tarone, V.E. Koteliansky, and K. Burridge. 1996. β1D integrin displaces the β1A isoform in striated muscles: localization at junctional structures and signaling potential in nonmuscle cells. J. Cell Biol. 132:211–226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bendig, G., M. Grimmler, I.G. Huttner, G. Wessels, T. Dahme, S. Just, N. Trano, H.A. Katus, M.C. Fishman, and W. Rottbauer. 2006. Integrin-linked kinase, a novel component of the cardiac mechanical stretch sensor, controls contractility in the zebrafish heart. Genes Dev. 20:2361–2372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bodine, S.C., T.N. Stitt, M. Gonzalez, W.O. Kline, G.L. Stover, R. Bauerlein, E. Zlotchenko, A. Scrimgeour, J.C. Lawrence, D.J. Glass, and G.D. Yancopoulos. 2001. Akt/mTOR pathway is a crucial regulator of skeletal muscle hypertrophy and can prevent muscle atrophy in vivo. Nat. Cell Biol. 3:1014–1019. [DOI] [PubMed] [Google Scholar]
- Bouvard, D., C. Brakebusch, E. Gustafsson, A. Aszodi, T. Bengtsson, A. Berna, and R. Fässler. 2001. Functional consequences of integrin gene mutations in mice. Circ. Res. 89:211–223. [DOI] [PubMed] [Google Scholar]
- Brakebusch, C., and R. Fässler. 2003. The integrin-actin connection, an eternal love affair. EMBO J. 22:2324–2333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chu, H., I. Thievessen, M. Sixt, T. Lammermann, A. Waisman, A. Braun, A.A. Noegel, and R. Fässler. 2006. γ-Parvin is dispensable for hematopoiesis, leukocyte trafficking, and T-cell-dependent antibody response. Mol. Cell. Biol. 26:1817–1825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Condorelli, G., A. Drusco, G. Stassi, A. Bellacosa, R. Roncarati, G. Iaccarino, M.A. Russo, Y. Gu, N. Dalton, C. Chung, et al. 2002. Akt induces enhanced myocardial contractility and cell size in vivo in transgenic mice. Proc. Natl. Acad. Sci. USA. 99:12333–12338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeBosch, B., I. Treskov, T.S. Lupu, C. Weinheimer, A. Kovacs, M. Courtois, and A.J. Muslin. 2006. Akt1 is required for physiological cardiac growth. Circulation. 113:2097–2104. [DOI] [PubMed] [Google Scholar]
- Delcommenne, M., C. Tan, V. Gray, L. Rue, J. Woodgett, and S. Dedhar. 1998. Phosphoinositide-3-OH kinase-dependent regulation of glycogen synthase kinase 3 and protein kinase B/AKT by the integrin-linked kinase. Proc. Natl. Acad. Sci. USA. 95:11211–11216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frias, M.A., C.C. Thoreen, J.D. Jaffe, W. Schroder, T. Sculley, S.A. Carr, and D.M. Sabatini. 2006. mSin1 is necessary for Akt/PKB phosphorylation, and its isoforms define three distinct mTORC2s. Curr. Biol. 16:1865–1870. [DOI] [PubMed] [Google Scholar]
- Glass, D.J. 2003. Signalling pathways that mediate skeletal muscle hypertrophy and atrophy. Nat. Cell Biol. 5:87–90. [DOI] [PubMed] [Google Scholar]
- Goel, H.L., M. Fornaro, L. Moro, N. Teider, J.S. Rhim, M. King, and L.R. Languino. 2004. Selective modulation of type 1 insulin-like growth factor receptor signaling and functions by β1 integrins. J. Cell Biol. 166:407–418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goel, H.L., L. Moro, M. King, N. Teider, M. Centrella, T.L. McCarthy, M. Holgado-Madruga, A.J. Wong, E. Marra, and L.R. Languino. 2006. Beta1 integrins modulate cell adhesion by regulating insulin-like growth factor-II levels in the microenvironment. Cancer Res. 66:331–342. [DOI] [PubMed] [Google Scholar]
- Grashoff, C., A. Aszodi, T. Sakai, E.B. Hunziker, and R. Fässler. 2003. Integrin-linked kinase regulates chondrocyte shape and proliferation. EMBO Rep. 4:432–438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grashoff, C., I. Thievessen, K. Lorenz, S. Ussar, and R. Fässler. 2004. Integrin-linked kinase: integrin's mysterious partner. Curr. Opin. Cell Biol. 16:565–571. [DOI] [PubMed] [Google Scholar]
- Guertin, D.A., D.M. Stevens, C.C. Thoreen, A.A. Burds, N.Y. Kalaany, J. Moffat, M. Brown, K.J. Fitzgerald, and D.M. Sabatini. 2006. Ablation in mice of the mTORC components raptor, rictor, or mLST8 reveals that mTORC2 is required for signaling to Akt-FOXO and PKCalpha, but not S6K1. Dev. Cell. 11:859–871. [DOI] [PubMed] [Google Scholar]
- Guo, C., M. Willem, A. Werner, G. Raivich, M. Emerson, L. Neyses, and U. Mayer. 2006. Absence of α7 integrin in dystrophin-deficient mice causes a myopathy similar to Duchenne muscular dystrophy. Hum. Mol. Genet. 15:989–998. [DOI] [PubMed] [Google Scholar]
- Hoffman, E.P., and G.A. Nader. 2004. Balancing muscle hypertrophy and atrophy. Nat. Med. 10:584–585. [DOI] [PubMed] [Google Scholar]
- Hentzen, E.R., M. Lahey, D. Peters, L. Mathew, I.A. Barash, J. Fridén, and R.L. Lieber. 2006. Stress-dependent and -independent expression of the myogenic regulatory factors and the MARP genes after eccentric contractions in rats. J. Physiol. 570:157–167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirsch, E., L. Lohikangas, D. Gullberg, S. Johansson, and R. Fässler. 1998. Mouse myoblasts can fuse and form a normal sarcomere in the absence of β1 integrin expression. J. Cell Sci. 111:2397–2409. [DOI] [PubMed] [Google Scholar]
- Huang, Y., J. Li, Y. Zhang, and C. Wu. 2000. The roles of integrin-linked kinase in the regulation of myogenic differentiation. J. Cell Biol. 150:861–872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hynes, R.O. 2002. Integrins: bidirectional, allosteric signaling machines. Cell. 110:673–687. [DOI] [PubMed] [Google Scholar]
- Jacinto, E., V. Facchinetti, D. Liu, N. Soto, S. Wei, S.Y. Jung, Q. Huang, J. Qin, and B. Su. 2006. SIN1/MIP1 maintains rictor-mTor complex integrity and regulates Akt phosphorylation and substrate specificity. Cell. 127:125–137. [DOI] [PubMed] [Google Scholar]
- Legate, K.R., E. Montanez, O. Kudlacek, and R. Fässler. 2006. ILK, PINCH and parvin: the tIPP of integrin signalling. Nat. Rev. Mol. Cell Biol. 7:20–31. [DOI] [PubMed] [Google Scholar]
- Mackinnon, A.C., H. Qadota, K.R. Norman, D.G. Moerman, and B.D. Williams. 2002. C. elegans PAT-4/ILK functions as an adaptor protein within integrin adhesion complexes. Curr. Biol. 12:787–797. [DOI] [PubMed] [Google Scholar]
- Mayer, U. 2003. Integrins: redundant or important players in skeletal muscle? J. Biol. Chem. 278:14587–14590. [DOI] [PubMed] [Google Scholar]
- Mayer, U., G. Saher, R. Fässler, A. Bornemann, F. Echtermeyer, H. von der Mark, N. Miosge, E. Poschl, and K. von der Mark. 1997. Absence of integrin α7 causes a novel form of muscular dystrophy. Nat. Genet. 17:318–323. [DOI] [PubMed] [Google Scholar]
- Menko, A.S., and D. Boettiger. 1987. Occupation of the extracellular matrix receptor, integrin, is a control point for myogenic differentiation. Cell. 51:51–57. [DOI] [PubMed] [Google Scholar]
- Michele, D.E., and K.P. Campbell. 2003. Dystrophin-glycoprotein complex: post-translational processing and dystroglycan function. J. Biol. Chem. 278:15457–15460. [DOI] [PubMed] [Google Scholar]
- Miller, M.G., I. Naruszewicz, A.S. Kumar, T. Ramlal, and G.E. Hannigan. 2003. a. Integrin-linked kinase is a positive mediator of L6 myoblast differentiation. Biochem. Biophys. Res. Commun. 310:796–803. [DOI] [PubMed] [Google Scholar]
- Miller, M.K., M.L. Bang, C.C. Witt, D. Labeit, C. Trombitas, K. Watanabe, H. Granzier, A.S. McElhinny, C.C. Gregorio, and S. Labeit. 2003. b. The muscle ankyrin repeat proteins: CARP, ankrd2/Arpp and DARP as a family of titin filament-based stress response molecules. J. Mol. Biol. 333:951–964. [DOI] [PubMed] [Google Scholar]
- Miosge, N., C. Klenczar, R. Herken, M. Willem, and U. Mayer. 1999. Organization of the myotendinous junction is dependent on the presence of α7β1 integrin. Lab. Invest. 79:1591–1599. [PubMed] [Google Scholar]
- Moro, L., M. Venturino, C. Bozzo, L. Silengo, F. Altruda, L. Beguinot, G. Tarone, and P. Defilippi. 1998. Integrins induce activation of the EGF receptor: role in MAP kinase induction and adhesion-dependent cell survival. EMBO J. 17:6622–6632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moro, L., L. Dolce, S. Cabodi, E. Bergatto, E. Boeri Erba, M. Smeriglio, E. Turco, S.F. Retta, M.G. Giuffrida, M. Venturino, et al. 2002. Integrin induced epidermal growth factor (EGF) receptor activation requires c-src and p130Cas and leads to phosphorylation of specific EGF receptor tyrosines. J. Biol. Chem. 277:9405–9414. [DOI] [PubMed] [Google Scholar]
- Mourkioti, F., and N. Rosenthal. 2005. IGF-1, inflammation and stem cells. Interactions during muscle regeneration. Trends Immunol. 26:535–542. [DOI] [PubMed] [Google Scholar]
- Musaro, A., K. McCullagh, A. Paul, L. Houghton, G. Dobrowolny, M. Molinaro, E.R. Barton, H.L. Sweeney, and N. Rosenthal. 2001. Localized Igf-1 transgene expression sustains hypertrophy and regeneration in senescent skeletal muscle. Nat. Genet. 27:195–200. [DOI] [PubMed] [Google Scholar]
- Nawrotzki, R., M. Willem, N. Miosge, H. Brinkmeier, and U. Mayer. 2003. Defective integrin switch and matrix composition at α7-deficient myotendinous junctions precede the onset of muscular dystrophy in mice. Hum. Mol. Genet. 12:483–495. [DOI] [PubMed] [Google Scholar]
- Novak, A., S.C. Hsu, C. Leung-Hagesteijn, G. Radeva, J. Papkoff, R. Montesano, C. Roskelley, R. Grosschedl, and S. Dedhar. 1998. Cell adhesion and the integrin-linked kinase regulate the LEF-1 and beta-catenin signaling pathways. Proc. Natl. Acad. Sci. USA. 95:4374–4379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Persad, S., S. Attwell, V. Gray, M. Delcommenne, A. Troussard, J. Sanghera, and S. Dedhar. 2000. Inhibition of integrin-linked kinase (ILK) suppresses activation of protein kinase B/Akt and induces cell cycle arrest and apoptosis of PTEN-mutant prostate cancer cells. Proc. Natl. Acad. Sci. USA. 97:3207–3212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rando, T.A., and H.M. Blau. 1994. Primary mouse myoblast purification, characterization, and transplantation for cell-mediated gene therapy. J. Cell Biol. 125:1275–1287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rommel, C., S.C. Bodine, B.A. Clarke, R. Rossman, L. Nunez, T.N. Stitt, G.D. Yancopoulos, and D.J. Glass. 2001. Mediation of IGF-1-induced skeletal myotube hypertrophy by PI(3)K/Akt/mTOR and PI(3)K/Akt/GSK3 pathways. Nat. Cell Biol. 3:1009–1013. [DOI] [PubMed] [Google Scholar]
- Rooney, J.E., J.V. Welser, M.A. Dechert, N.L. Flintoff-Dye, S.J. Kaufman, and D.J. Burkin. 2006. Severe muscular dystrophy in mice that lack dystrophin and α7 integrin. J. Cell Sci. 119:2185–2195. [DOI] [PubMed] [Google Scholar]
- Sakai, T., S. Li, D. Docheva, C. Grashoff, K. Sakai, G. Kostka, A. Braun, A. Pfeifer, P.D. Yurchenco, and R. Fässler. 2003. Integrin-linked kinase (ILK) is required for polarizing the epiblast, cell adhesion, and controlling actin accumulation. Genes Dev. 17:926–940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sastry, S.K., M. Lakonishok, D.A. Thomas, J. Muschler, and A.F. Horwitz. 1996. Integrin α subunit ratios, cytoplasmic domains, and growth factor synergy regulate muscle proliferation and differentiation. J. Cell Biol. 133:169–184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schneller, M., K. Vuori, and E. Ruoslahti. 1997. αvβ3 integrin associates with activated insulin and PDGFbeta receptors and potentiates the biological activity of PDGF. EMBO J. 16:5600–5607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwander, M., M. Leu, M. Stumm, O.M. Dorchies, U.T. Ruegg, J. Schittny, and U. Muller. 2003. β1 integrins regulate myoblast fusion and sarcomere assembly. Dev. Cell. 4:673–685. [DOI] [PubMed] [Google Scholar]
- Shiota, C., J.T. Woo, J. Lindner, K.D. Shelton, and M.A. Magnuson. 2006. Multiallelic disruption of the rictor gene in mice reveals that mTOR complex 2 is essential for fetal growth and viability. Dev. Cell. 11:583–589. [DOI] [PubMed] [Google Scholar]
- Soldi, R., S. Mitola, M. Strasly, P. Defilippi, G. Tarone, and F. Bussolino. 1999. Role of alphavbeta3 integrin in the activation of vascular endothelial growth factor receptor-2. EMBO J. 18:882–892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stanchi, F., R. Bordoy, O. Kudlacek, A. Braun, A. Pfeifer, M. Moser, and R. Fässler. 2005. Consequences of loss of PINCH2 expression in mice. J. Cell Sci. 118:5899–5910. [DOI] [PubMed] [Google Scholar]
- Taverna, D., M.H. Disatnik, H. Rayburn, R.T. Bronson, J. Yang, T.A. Rando, and R.O. Hynes. 1998. Dystrophic muscle in mice chimeric for expression of α5 integrin. J. Cell Biol. 143:849–859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van der Flier, A., A.C. Gaspar, S. Thorsteinsdottir, C. Baun, E. Groeneveld, C.L. Mummery, and A. Sonnenberg. 1997. Spatial and temporal expression of the beta1D integrin during mouse development. Dev. Dyn. 210:472–486. [DOI] [PubMed] [Google Scholar]
- Vasilcanu, D., A. Girnita, L. Girnita, R. Vasilcanu, M. Axelson, and O. Larsson. 2004. The cyclolignan PPP induces activation loop-specific inhibition of tyrosine phosphorylation of the insulin-like growth factor-1 receptor. Link to the phosphatidyl inositol-3 kinase/Akt apoptotic pathway. Oncogene. 23:7854–7862. [DOI] [PubMed] [Google Scholar]
- Volk, T., L.I. Fessler, and J.H. Fessler. 1990. A role for integrin in the formation of sarcomeric cytoarchitecture. Cell. 63:525–536. [DOI] [PubMed] [Google Scholar]
- White, D.E., P. Coutu, Y.F. Shi, J.C. Tardif, S. Nattel, R. St Arnaud, S. Dedhar, and W.J. Muller. 2006. Targeted ablation of ILK from the murine heart results in dilated cardiomyopathy and spontaneous heart failure. Genes Dev. 20:2355–2360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zervas, C.G., S.L. Gregory, and N.H. Brown. 2001. Drosophila integrin-linked kinase is required at sites of integrin adhesion to link the cytoskeleton to the plasma membrane. J. Cell Biol. 152:1007–1018. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}