

Abstract
Tumor necrosis factor (TNF) is key to the pathogenesis of various arthritic diseases and inflammatory bowel disease (IBD). Anti-TNF therapies have proved successful in the clinical treatment of these diseases, but a mechanistic understanding of TNF function is still lacking. We have investigated early cellular mechanisms of TNF function in these diseases using an established TNF transgenic model, which develops a spondyloarthritis-like disease characterized by peripheral joint arthritis, sacroiliitis, enthesitis, and Crohn's-like IBD. Bone marrow grafting experiments demonstrated that development of arthritis requires TNF receptor I (TNFRI) expression in the radiation-resistant compartment, which is also known to be a sufficient target of TNF in the development of Crohn's-like IBD in the same model. Early activation of synovial fibroblasts and intestinal myofibroblasts could also be demonstrated by perturbed expression of matrix metalloproteases and their inhibitors. Notably, selective Cre/loxP-mediated TNFRI expression in mesenchymal cells resulted in a fully arthritic–spondyloarthritic and intestinal phenotype, indicating that mesenchymal cells are primary and sufficient targets of TNF in these pathologies. Our results offer a novel mechanistic perspective for TNF function in gut and joint pathologies and indicate early common cellular pathways that may also explain the often observed synovial–gut axis in human disease.
Chronic inflammatory joint and intestinal diseases are often coexpressed in individual patients, indicating common underlying pathogenic mechanisms. Epidemiological studies revealed that a substantial group of patients suffering from spondyloarthritis shows asymptomatic gut inflammation, and some of them (7%) eventually develop inflammatory bowel disease (IBD), particularly Crohn's disease (CD) (1, 2). Conversely, spondyloarthritis is not uncommonly associated with IBD (3). The coexistence of these diseases is usually classified in the group of spondyloarthropathies (SpAs) and is defined as interrelated and overlapping chronic inflammatory diseases that mainly include ankylosing spondylitis, reactive and psoriatic arthritis, and CD (4).
Spondyloarthritis, rheumatoid arthritis, and CD are diseases showing major clinical responses to anti-TNF treatment (5, 6). An important finding validating the role of TNF in specifically driving inflammatory arthritis in vivo was provided by the generation of transgenic mice overexpressing human TNF (Tg197 mice) and developing chronic inflammatory polyarthritis, which was reversible by anti–human TNF antibody administration (7). More recently, it was recognized that Tg197 mice also develop spondyloarthritis (8). Importantly, a previously established TNF-overexpressing mouse model (TnfΔARE mice) characterized by the development of both chronic inflammatory polyarthritis and Crohn's-like IBD (9) is shown in this report to also develop extraintestinal features of spondyloarthritis such as sacroiliitis and enthesitis. These results establish TnfΔARE mice as a new model for SpAs uniquely associated with CD (similarly to human SpAs), offering an appropriate system in which further mechanistic studies, including a potential common basis of TNF function in these pathologies, can be explored.
Mechanistic hypotheses addressing the coincidence of gut–joint axis diseases in humans favor an aetiopathogenic role for common molecular determinants on effector lymphocytes, which render them capable of homing equally efficiently to the intestinal mucosa and joints (10). An experimental model in support of this hypothesis, the HLA-B27 transgenic rat, a spontaneous model for SpAs, develops colitis, gastritis, and arthritis (11). Although adaptive immune responses and, more specifically, CD8+ effector function are required for the development of Crohn's-like IBD in the TnfΔARE model (12), arthritis develops independently of the adaptive immune compartment (9), indicating that T cell responses may not be commonly required in the pathogenesis of these two diseases. Therefore, alternative mechanisms are likely to be responsive for TNF-mediated pathology in gut–joint diseases. Current hypotheses suggest that TNF delivers innate activation and proinflammatory signals through its action on various cell types, including myeloid/monocytic, lymphocytic, endothelial, mesenchymal, or epithelial cell types (5, 13). Using TNF-driven disease models, we demonstrate for the first time that mesenchymal cells, such as the fibroblasts/myofibroblasts of the joint and the intestine, are primary responder cells sufficient for full pathogenic TNF/TNFRI signaling in arthritis, sacroiliitis, and Crohn's-like IBD. Our findings provide novel mechanistic insights into the cellular and molecular events underlying TNF function in joint–gut axis diseases and establish an early dominant role for mesenchymal cell responses in their pathogenesis.
RESULTS AND DISCUSSION
TnfΔARE mice as a model for SpAs
TnfΔARE/+ mutant mice have been described previously (9, 12) to develop chronic polyarthritis starting at weeks 5–6 and Crohn's-like IBD starting at week 6. Prompted by the coincidence of both joint and intestinal disease in these mice, we have examined whether additional features of spondyloarthritic disease are developing in this model. Histological analysis of sacroiliac joints from TnfΔARE/+ mice revealed bilateral inflammation of the sacroiliac joints, a prominent feature of ankylosing spondylitis. Invasion of inflammatory tissue was detected in the subchondral bone and iliac BM of TnfΔARE/+ mice (Fig. 1). Initial signs of enthesitis were evident by week 4 (not depicted). No gender bias was apparent. These data establish TnfΔARE mutant mice as a disease model resembling human SpAs. Interestingly, the TnfΔARE model is characterized by the combined presence of arthritis–spondyloarthritis with a CD-like pathology localized primarily in the small intestine, as typically occurs in patients suffering from SpAs (2).
Figure 1.

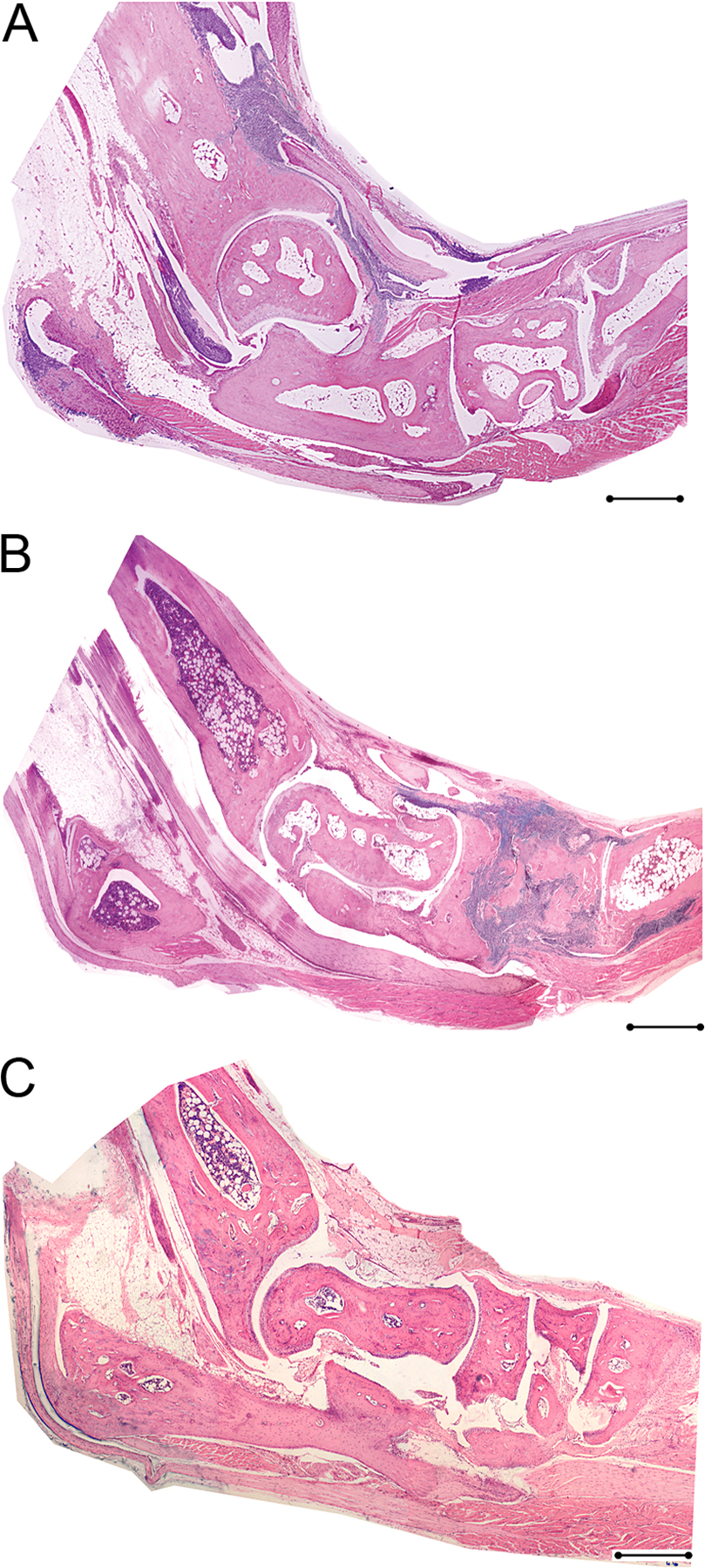
Bilateral inflammation in sacroiliac joints of TnfΔARE mice. (A–C) Sacroiliac joint from a healthy C57BL/6 control. The lower two thirds of the joint consists of fibrocartilagenous tissue, and it is well defined and separated from the periost of the iliac bone. (D–F) Sacroiliac joint from a TnfΔARE/+ mouse on the C57BL/6 background. The presence of a mononuclear infiltrate within the sacroiliac joint with an increased number of blood vessels is shown. The fibrocartilagenous portion of the joint is invaded by mononuclear cells that protrude from the periost of the iliac bone into the BM. Paraffin sections are stained with hematoxylin and eosin. The boxes in B and E indicate the high-magnification areas shown in C and F, respectively. Bars: (A and D) 500 μm; (B and E) 200 μm; (C and F) 50 μm.
TNFRI expression on radiation-resistant, tissue stroma–residing cells is necessary and sufficient for the induction of arthritis
The dominant role of TNFRI in mediating TNF pathogenic signals in modeled arthritis and IBD has been previously established (7, 9). To gain further insight into the cellular specificity of TNFRI-mediated signaling, we used BM engraftment experiments into lethally irradiated recipients, using TNFRI-deficient mice (14) as either recipients or donors, to restrict TNFRI expression in either hematopoietic or radiation-resistant tissue stromal cells. Using this system, we have previously reported the capacity of BM-derived cells from the TnfΔARE mice to induce IBD and, more importantly, that radiation-resistant, tissue stroma–residing cells are sufficient TNF targets for the induction of IBD (12). We have now extended these previous findings by demonstrating that at 12 wk after transplantation, lethally irradiated WT mice transplanted with TnfΔARE/+ BM cells also develop arthritis (Table I; and Fig. S1 A, available at http://www.jem.org/cgi/content/full/jem.20070906/DC1), indicating that a source of pathogenic TNF production in this model resides within the hematopoietic compartment. Transfer of TnfΔARE/ΔARE TnfRI−/− BM into WT irradiated recipients resulted in overt arthritis, similar to the TnfΔARE/+-reconstituted WT mice, as indicated by histopathological analysis of joints performed 12 wk after engraftment (Table I and Fig. S1 B), suggesting that radiation-resistant, stroma–residing cells are sufficient TNF targets for the induction of arthritis. Reciprocal engraftment of TnfΔARE/+ BM into TnfRI−/− recipients did not result in any arthritic manifestation 12 wk after transplantation (Table I and Fig. S1 C), indicating that radiation-resistant, tissue stroma–residing cells are also required TNF targets for the induction of arthritis. Similar results were obtained using the huTNF transgenic Tg197 model of arthritis (unpublished data). Collectively, these results demonstrate that in TNF-mediated arthritis, TNFRI expression on radiation-resistant cells is a necessary and sufficient condition for the development of disease.
Table I.
Arthritis development following TnfΔARE BM reconstitution of lethally irradiated recipients
| Donor genotype | Recipient genotype | Arthritis development |
|---|---|---|
| WT | WT | 0/20 |
| TnfΔARE/+ | WT | 19/21 |
| TnfΔARE/ΔARE TnfRI−/− | WT | 25/25 |
| TnfΔARE/+ | TnfRI−/− | 0/8 |
BM isolated from 2-mo-old female B6,129 mice (donor genotype) was engrafted into 6–8-wk-old syngeneic, lethally irradiated, female mice (recipient genotype). Assessment of arthritis development was based on histopathological evaluation of joint samples at 12 wk after transfer.
Early activation of mesenchymal cells in modeled arthritis and Crohn's-like IBD
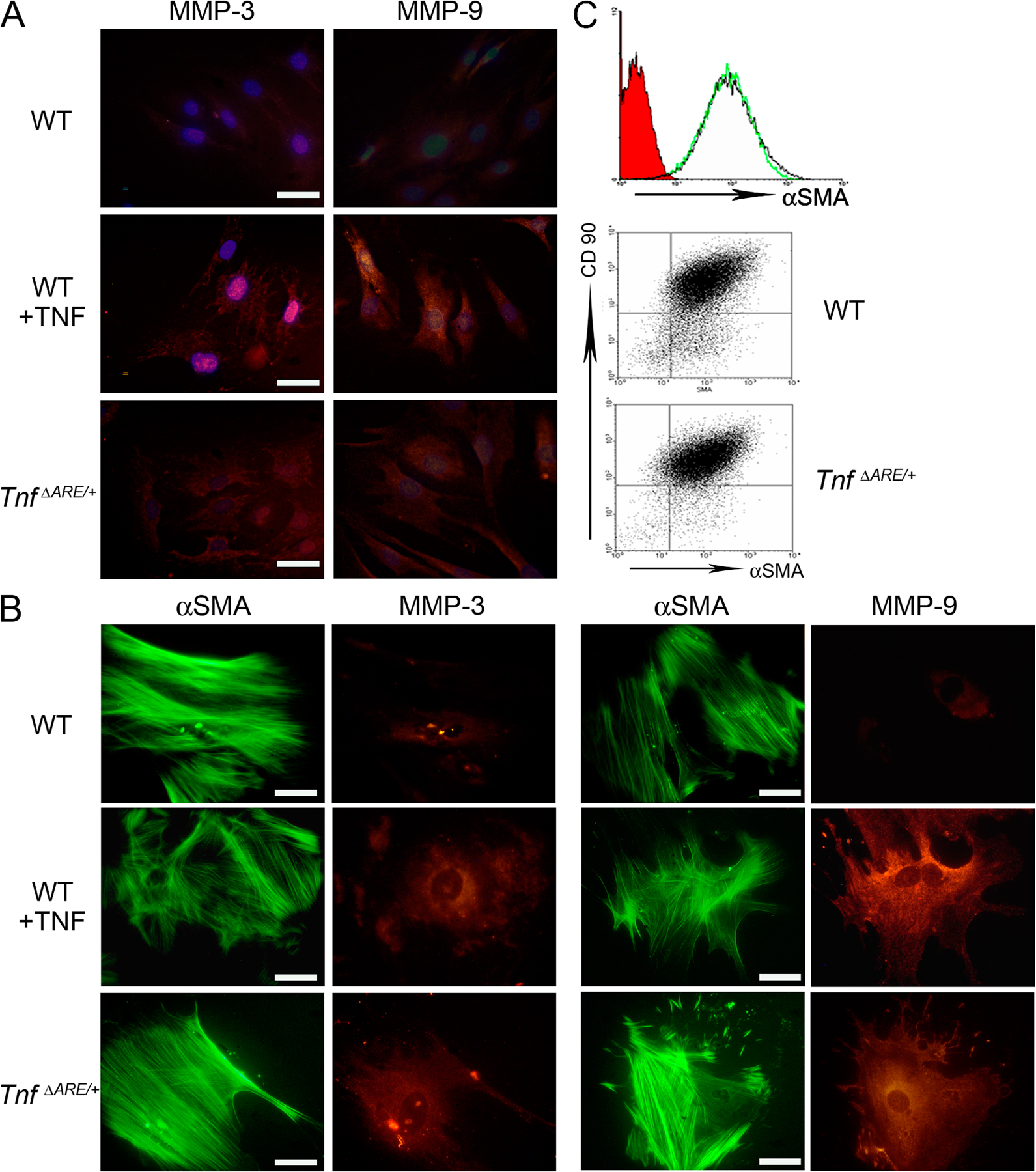
In inflamed tissues, mesenchymal cells become activated and act as both recruiters of lymphocytes, by the increased expression of adhesion molecules (15, 16), and as effectors of tissue destruction, through the production of several matrix degrading enzymes such as matrix metalloproteinases (MMPs) (17, 18). Importantly, TNFRI-mediated mesenchymal MMP secretion in human fetal small intestine explant cultures has been previously associated with intestinal injury (19). To examine whether mesenchymal cells of TnfΔARE mice are activated before the onset of disease, we isolated synovial fibroblasts (SFs) of ankle joints and intestinal myofibroblasts (IMFs) from WT and TnfΔARE/+ mice before the appearance of the inflammatory infiltrate (at 4 wk of age) (9, 12). Increased proteolytic activity against gelatin caused by MMP9 expression was detected in SF and IMF cell extracts in contrast to MMP2 activity, which remained unaltered (Fig. 2 A). Additionally, semiquantitative RT-PCR revealed the up-regulation of MMP3 and the strong down-regulation of tissue inhibitor of MMPs 1 (TIMP1) messenger RNA, leading to severely misbalanced ratios of MMP3 versus TIMP1 expression in both SF and IMF cells of TnfΔARE/+ compared with WT mice (Fig. 2 B). Furthermore, using immunofluorescent staining of ex vivo–derived SFs and IMFs, we detected elevated protein expression of MMP3 and MMP9 in TnfΔARE cultures (Fig. S2, A and B, respectively, available at http://www.jem.org/cgi/content/full/jem.20070906/DC1). Interestingly, we detected α–smooth muscle actin (αSMA) expression by SFs as well as IMFs (Fig. S2 C), suggesting common phenotypic features between these two cell types. These results indicate that mesenchymal/myofibroblast-like cells of TnfΔARE mice may represent the local primary targets of TNF in the induction of joint and gut pathology. Misbalanced production of MMPs and TIMPs by SFs and IMFs confirms early proinflammatory triggering that could potentially promote primary matrix degradation in the respective tissues.
Figure 2.

Mesenchymal cells from TnfΔARE mice are activated before disease onset. (A) Representative gelatin zymogram showing MMP9 and MMP2 secretion either from SFs (left) or IMFs (right) derived from WT and TnfΔARE/+ 4-wk-old mice. (B) Representative semiquantitative RT-PCR for MMP3 and TIMP1 expression in SFs (left) or IMFs (right) derived from WT and TnfΔARE/+ 4-wk-old mice. WT cells treated with TNF were used as a positive control in A and B.
TNFRI-expressing mesenchymal cells are sufficient TNF targets for the development of SpA-related pathologies
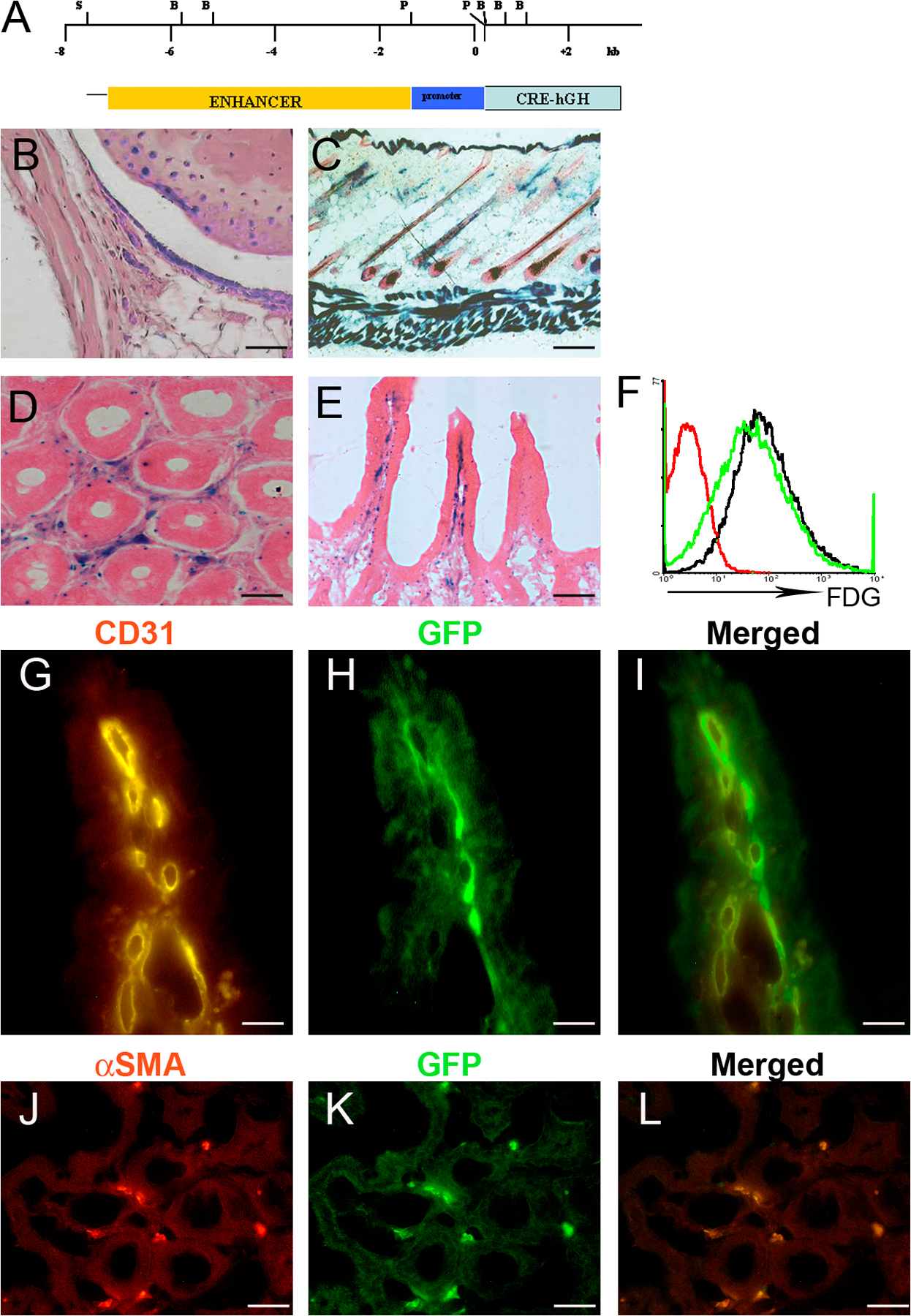
To investigate the specific role of mesenchymal cells in the TnfΔARE and Tg197 pathologies, we generated transgenic mice expressing Cre recombinase under the control of a collagen VI (ColVI) promoter cassette (Fig. S3 A, available at http://www.jem.org/cgi/content/full/jem.20070906/DC1), which is known to drive gene expression in mesenchymal cell types (20). The specificity of Cre-mediated recombination in the ColVI-Cre mice was assessed using the ROSA26 and Z/EG reporter mouse strains (21, 22). X-gal staining in various tissues of ColVI-Cre ROSA26flx/+ mice indicated Cre-mediated recombination in joint SFs, articular chondrocytes and myocytes, skin keratinocytes and dermal fibroblasts, the muscle layer around arteries of the heart (not depicted), and underneath the epithelial layer of the gut (Fig. S3, B–E). Cre activity was not detected in hematopoietic tissues (spleen and thymus; not depicted). Efficiency of Cre-mediated recombination in ex vivo–cultured SFs measured by flow cytometric analysis using fluorodeoxyglucose staining was >80% (Fig. S3 F). The localization of X-gal–positive cells neighboring the epithelium of ColVI-Cre ROSA26flx/+ reporter mice indicated Cre recombinase expression in the subepithelial myofibroblast cell layer (Fig. S3, D and E). Indeed, in ColVI-Cre Z/EG mice (22), GFP-expressing cells stained positive for αSMA (Fig. S3, J–L) but negative for the CD31 endothelial cell marker (Fig. S3, G–I), indicating Cre-mediated recombination in subepithelial myofibroblasts.
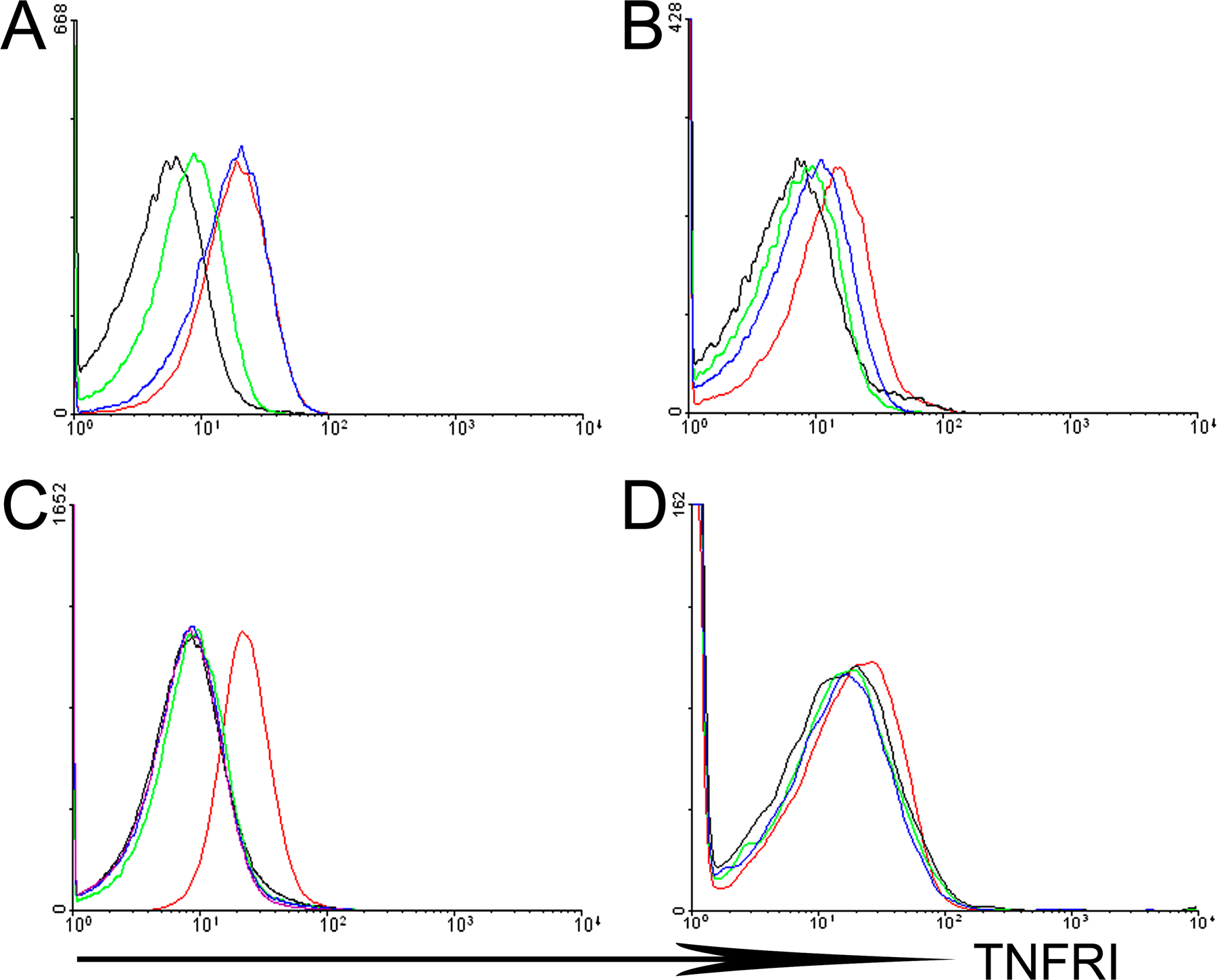
To obtain mesenchymal cell–specific expression of TNFRI, we used previously generated mutant mice carrying a conditional gain-of-function allele for this receptor (TnfRIflxneo mice). In these mice, TNFRI expression is inhibited by the presence of a floxed neomycin cassette but is restored upon Cre-mediated neo excision (23). The specificity of recombination for the floxed neo TNFRI allele was examined in ColVI-Cre TnfRIflxneo/flxneo SFs, IMFs, BM-derived macrophages, and CD11bhi splenic myeloid cells by flow cytometric analysis of TNFRI expression, confirming Cre-mediated TNFRI reactivation in SFs and IMFs but not in other nonmesenchymal cell types such as myeloid cells (Fig. S4, available at http://www.jem.org/cgi/content/full/jem.20070906/DC1). ColVI-Cre–driven TNFRI expression in ColVI-Cre TnfΔARE/+ TnfRIflxneo/flxneo mice was sufficient to induce a course of arthritic pathology similar to the TnfΔARE mice, consisting of pannus formation with evident cartilage and bone erosions (Fig. 3, A and B; and Table II). Flow cytometric analysis for the detection of TNFRI in SFs from individual ColVI-Cre TnfΔARE/+ TnfRIflxneo/flxneo mice confirmed reactivation of the TNFRI allele in these cells, whereas TNFRI expression was undetectable in the same cells from TnfΔARE / + TnfRIflxneo/flxneo mice (Fig. 3 J). Histological examination of the ileum of ColVI-Cre TnfΔARE/+ TnfRIflxneo/flxneo mice revealed profound inflammatory changes consistent with the Crohn's–like disease phenotype of TnfΔARE mice (Table II). Intestinal pathology in the ColVI-Cre TnfΔARE/+ TnfRIflxneo/flxneo mice consisted of mucosal abnormalities with intestinal villous blunting and broadening, and mucosal and submucosal infiltration of inflammatory cells with characteristics typical of transmural inflammation (Fig. 3 C). None of the TnfΔARE/+ TnfRIflxneo/flxneo littermates exhibited signs of disease (Fig. 3 D), whereas TnfΔARE/+ TnfRIflxneo/+ littermates developed intestinal pathology (Table II). Furthermore, the spondyloarthritic characteristic of bilateral sacroiliitis observed in the TnfΔARE model was also evident in the ColVI-Cre TnfΔARE/+ TnfRIflxneo/flxneo mice examined at 40 wk of age but not in TnfΔARE/+ TnfRIflxneo/flxneo control mice (four mice per group; Fig. 3, E and F, respectively). Notably, introduction of the ColVI-Cre transgene in the Tg197 TnfRIflxneo/flxneo background also resulted in the development of progressive inflammatory arthritis starting at 5–6 wk of age and with 100% incidence (10 out of 10 mice), similar to the timing and course of disease seen in Tg197 mice (Fig. 3 G). Littermate control Tg197 TnfRIflxneo/flxneo mice remained healthy even at 50 wk of age (15 out of 15 mice; Fig. 3 H), further confirming the sufficiency of TNFRI signaling in mesenchymal cells for the development of TNF-driven arthritis. Southern blot analysis of tissue DNA from ColVI-Cre Tg197 TnfRIflxneo/flxneo mice confirmed the recombination of the TNFRIflxneo allele in joints, skeletal muscle, skin, gut, and heart (Fig. 3 I). These findings demonstrate that mesenchymal cells are common primary targets for TNF in the development of inflammatory polyarthritis, Crohn's-like IBD, and anklylosing spondylitis, and that selective expression of TNFRI on these cells is sufficient to orchestrate the complete development of such SpA-related pathologies.
Figure 3.

TNFRI-expressing mesenchymal cells are sufficient targets of pathogenic TNF. (A–D) Histological examination of joint (8-wk-old) and ileal (16-wk-old) sections from ColVI-Cre TnfΔARE/+ TnfRIflxneo/flxneo (A and C) and TnfΔARE/+ TnfRIflxneo/flxneo mice (B and D). (E and F) Histological examination of sacroiliac joint sections from 10-mo-old ColVI-Cre TnfΔARE/+ TnfRIflxneo/flxneo (E) and TnfΔARE/+ TnfRIflxneo/flxneo mice (F). (G and H) Histological examination of joint sections from 7-wk-old ColVI-Cre Tg197 TnfRIflxneo/flxneo (G) and Tg197 TnfRIflxneo/flxneo mice (H). Paraffin sections were stained with hematoxylin and eosin. Bars: (A, B, G, and H) 600 μm; (C–F) 100 μm. (I) Southern blot analysis of BamHI-digested tissue DNA from ColVI-Cre TnfRIflxneo/flxneo mice for detection of the recombined loxP TnfRI allele. (J) Flow cytometric analysis or the detection of TNFRI expression in ColVI-Cre TnfΔARE/+ TnfRIflxneo/flxneo SFs derived from the cultures of individual mice. White lines indicate that intervening lanes have been spliced out.
Table II.
Incidence of arthritic and intestinal pathology in TnfΔARE/+ TnfRIflxneo/flxneo ColVI-Cre mice and littermates
| Genotype | Incidence of pathology
|
|||
|---|---|---|---|---|
| Arthritic
|
Intestinal
|
|||
| wk 8 | wk 16 | wk 8 | wk 16 | |
| TnfΔARE/+TnfRIflxneo/flxneo | 0/7 | 0/10 | 0/7 | 0/10 |
| 0% | 0% | 0% | 0% | |
|
ColVI-Cre TnfΔARE/
+
TnfRIflxneo/flxneo |
5/5 | 21/21 | 5/5 | 21/21 |
| 100% | 100% | 100% | 100% | |
| TnfΔARE/+ TnfRIflxneo/+ | 5/6 | 4/4 | 4/6 | 4/4 |
| 83% | 100% | 66% | 100% | |
Collectively, the data in this study demonstrate that mesenchymal cell targeting by TNF represents a common pathogenic mechanism in chronic inflammatory joint and intestinal diseases. Deregulated MMP expression was detected as an early marker of SF and intestinal subepithelial fibroblast activation by TNF. Increased levels of MMP3 and other MMPs have been reported in CD (24) and arthritic patients (25). Sustained perturbations on MMP expression are now strongly implicated in various processes central to inflammation and immunity, such as cytokine and chemokine production, leukocyte recruitment, and tissue repair (26). It is thus possible that, in susceptible hosts, dysfunctional responses to tissue injury, infections, or other environmental stimuli may lead to sustained TNF/TNFRI-dependent activation of mesenchymal cells. Failure to regulate these proinflammatory and tissue-repair activities may be responsible for driving the transition of acute-to-chronic tissue inflammation and fibrosis (15). SFs do play a critical role in these processes, as they have been suggested to be capable of propagating both inflammation and joint destruction in rheumatoid arthritis (27, 28). An equally important role may be played by the intestinal subepithelial myofibroblast, which is also considered important in maintaining tissue architecture, regulating inflammation, and repair in the gut (29). Intestinal inflammation has also been prominently linked to epithelial barrier dysfunction (13). It was recently demonstrated that NF-κB-deficiency sensitizes epithelial cells to TNFRI-dependent apoptosis, linking innate immunity to intestinal inflammation (30). It will also be interesting to investigate whether TNFRI-mediated signaling on IMFs may contribute to epithelial barrier dysfunction in CD.
The identification of mesenchymal cells as a cellular subset linking joint and intestinal pathologies provides an alternative cellular basis to understand the common occurrences of these pathologies in humans, as well as the remarkable response of a significant number of patients to anti-TNF therapies (6). Mesenchymal cell replacement by stem cell or BM transplantation may offer future therapeutic approaches for the resolution of chronic inflammatory processes. The identification of TNF-responsive mesenchymal cells as dominant contributors to joint and gut inflammatory diseases suggests that attenuation of the TNFRI pathway in these cells or their precursors may hold promise for more rational and effective therapeutic interventions.
MATERIALS AND METHODS
Mice.
The generation of Tg197 (7), TnfΔARE (9), and mutant mice carrying a conditional gain-of-function allele for TnfRI (TnfRIflxneo mice) have been previously described (23). TnfRI−/− (14) and Z/EG (22) mice were purchased from the Jackson Laboratory. ROSA26flx/+ (21) mice were provided by P. Soriano (Fred Hutchinson Cancer Research Center, Seattle, WA). The ColVI promoter cassette used for the generation of ColVI-Cre mice was provided by G. Bressan (University of Milano, Milano, Italy). All mice were bred and maintained on C57BL/6J and on mixed C57BL/6J × 129S6 or C57BL/6 × CBA genetic backgrounds in the animal facilities of the BSRC “Alexander Fleming” under specific pathogen-free conditions. All mice were used in accordance with the guidance of the Institutional Animal Care and Use Committee of BSRC “Alexander Fleming.”
BM transplantation.
BM transplantation experiments were performed as previously described (12).
Cell culture.
SF cultures were established by enzymatic treatment as previously described (1). IMF cultures were established by enzymatic treatment of terminal ilea (5 cm). In brief, the intestinal pieces were flushed extensively with HBSS (Sigma-Aldrich)/2% FBS (Biochrome), and cut longitudinally. The epithelial layer was removed by incubation in DMEM/10% FBS-containing collagenase XI (300 U/ml)/dispase (0.1 mg/ml; Sigma-Aldrich) for 10 min at 30°C. The digested tissues were washed with HBSS/2% FBS and subjected to a second incubation with collagenase/dispase for 20 min at 30°C. The mucosal samples were then washed and cultured at 37°C in a humidified CO2 incubator in DMEM (Invitrogen) supplemented with 10% FBS, 1% nonessential amino acids, 100 U/ml penicillin, 100 μg/ml streptomycin (Invitrogen), and 1 μg/ml amphotericin B (Sigma-Aldrich). Cells were used after three passages. The purity of myofibroblast cultures was determined by flow cytometric detection of CD90 (BD Biosciences) and αSMA (Sigma-Aldrich) expression. The generation of BM-derived macrophages was performed as previously described (9).
Gelatin zymography and Western blotting.
In brief, synchronized cells were grown in confluence on 10-cm plates in the presence or absence of 20 ng/ml of mouse TNF. Cell extracts (in 50 mM Tris-HCl [pH 7.4], 150 mM NaCl, 0.1% SDS, 1% Triton X-100, 2 μg/ml leupeptin, 2 μg/ml aprotinin, 1mM PMSF) were collected at the time points indicated in the figures. The samples were sonicated and stored at −70°C until use. 20 μg of total protein per sample was analyzed on 8% SDS-PAGE containing 1 mg/ml of gelatin under nonreduced conditions, at 4°C, and at a constant voltage (100 V). The gel was rinsed for 60 min in 2.5% Triton X-100 and incubated in MMP activation buffer (50 mM Tris-HCl [pH 7.5], 5 mM CaCl2, 0.02% NaN3, 1 μM ZnCl2) for 36 h at 37°C with constant shaking. Gels were stained for 30 min with 0.5% Coomassie blue R250 and destained in 40% methanol/10% acetic acid. The gels were stored either dried or in water/20% glycerol. The specificity of reaction was tested with the use of specific inhibitors that were added to the activation buffer (10 mM EDTA and 0.1 mM phenantroline). For detection of actin, 20 mg of total protein per sample was analyzed on 8% SDS-PAGE, electrotransfered in nitrocellulose membrane, and detected with Western blot analysis using polyclonal antibodies against actin (Santa Cruz Biotechnology, Inc.).
RNA isolation and RT.
RNA was isolated with TRIzol (Invitrogen). RT to complementary DNA was performed with 12 μg RNA by MMLV-RT (Promega) at a final volume of 20 μl, according to manufacturer's instructions. The volume of the reaction was increased to 50 μl and the dilutions used in reactions were always performed in accordance with the housekeeping gene content. The sequences of the primers are as follows: MMP3 sense, 5′-CCACAGACTTGTCCCGTTTCC-3′; MMP3 antisense, 5′-GAGTCCTGAGAGATTTGCGCC-3′; TIMP1 sense, 5′-ACAAGTCCCAGAACCGCAGTA-3′; TIMP1 antisense, 5′-GGACCTGATCCGTCCACAAAC-3′; β2 microglobulin (β2M) sense, 5′-TTCTGGTGCTTGTCTCACTGA-3′; and β2M antisense, 5′-CAGTATGTTCGGCTTCCCATTC-3′. The annealing temperature for all primers was 53°C for 30 cycles. Product sizes were as follows: MMP3, 237 bp; TIMP1, 234 bp; and β2M, 104 bp.
Online supplemental material.
Fig. S1 shows the histopathological manifestations of BM chimeras. Fig. S2 shows the immunohistochemical detection of MMPs in mesenchymal cells of TnfΔARE mice before disease onset. Fig. S3 describes the generation and characterization of ColVI-Cre mice. Fig. S4 demonstrates the flow cytometric analysis of TNFRI expression in mesenchymal and nonmesenchymal cells of ColVI-Cre TnfRIflxneo/flxneo mice. Online supplemental material is available at http://www.jem.org/cgi/content/full/jem.20070906/DC1.
Supplemental Material
Acknowledgments
We thank M. Papamichael for allowing access to the γ irradiator at the Agios Savvas Hospital. We also thank S. Lalos for excellent technical assistance in histopathology, and A. Giannakopoulou, P. Athanasakis, and G. Alexakos for animal breeding.
This work was supported by a grant from the European Commission program MUGEN (LSHG-CT-2005–005203), and by grants from the Fund for Scientific Research–Flanders and the Research Fund of Ghent University. P. Jacques is a research assistant with the Fund for Scientific Research–Flanders.
The authors have no conflicting financial interests.
References
- 1.Mielants, H., E.M. Veys, C. Cuvelier, and M. de Vos. 1988. Ileocolonoscopic findings in seronegative spondylarthropathies. Br. J. Rheumatol. 27(Suppl. 2):95–105. [DOI] [PubMed] [Google Scholar]
- 2.Mielants, H., E.M. Veys, C. Cuvelier, M. De Vos, S. Goemaere, L. De Clercq, L. Schatteman, and D. Elewaut. 1995. The evolution of spondyloarthropathies in relation to gut histology. II. Histological aspects. J. Rheumatol. 22:2273–2278. [PubMed] [Google Scholar]
- 3.de Vlam, K., H. Mielants, C. Cuvelier, F. De Keyser, E.M. Veys, and M. De Vos. 2000. Spondyloarthropathy is underestimated in inflammatory bowel disease: prevalence and HLA association. J. Rheumatol. 27:2860–2865. [PubMed] [Google Scholar]
- 4.Khan, M.A. 2002. Update on spondyloarthropathies. Ann. Intern. Med. 136:896–907. [DOI] [PubMed] [Google Scholar]
- 5.Feldmann, M., and L. Steinman. 2005. Design of effective immunotherapy for human autoimmunity. Nature. 435:612–619. [DOI] [PubMed] [Google Scholar]
- 6.Strand, V., R. Kimberly, and J.D. Isaacs. 2007. Biologic therapies in rheumatology: lessons learned, future directions. Nat. Rev. Drug Discov. 6:75–92. [DOI] [PubMed] [Google Scholar]
- 7.Keffer, J., L. Probert, H. Cazlaris, S. Georgopoulos, E. Kaslaris, D. Kioussis, and G. Kollias. 1991. Transgenic mice expressing human tumour necrosis factor: a predictive genetic model of arthritis. EMBO J. 10:4025–4031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Redlich, K., B. Gortz, S. Hayer, J. Zwerina, G. Kollias, G. Steiner, J.S. Smolen, and G. Schett. 2004. Overexpression of tumor necrosis factor causes bilateral sacroiliitis. Arthritis Rheum. 50:1001–1005. [DOI] [PubMed] [Google Scholar]
- 9.Kontoyiannis, D., M. Pasparakis, T.T. Pizarro, F. Cominelli, and G. Kollias. 1999. Impaired on/off regulation of TNF biosynthesis in mice lacking TNF AU-rich elements: implications for joint and gut-associated immunopathologies. Immunity. 10:387–398. [DOI] [PubMed] [Google Scholar]
- 10.Salmi, M., D.P. Andrew, E.C. Butcher, and S. Jalkanen. 1995. Dual binding capacity of mucosal immunoblasts to mucosal and synovial endothelium in humans: dissection of the molecular mechanisms. J. Exp. Med. 181:137–149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hammer, R.E., S.D. Maika, J.A. Richardson, J.P. Tang, and J.D. Taurog. 1990. Spontaneous inflammatory disease in transgenic rats expressing HLA-B27 and human beta 2m: an animal model of HLA-B27-associated human disorders. Cell. 63:1099–1112. [DOI] [PubMed] [Google Scholar]
- 12.Kontoyiannis, D., G. Boulougouris, M. Manoloukos, M. Armaka, M. Apostolaki, T. Pizarro, A. Kotlyarov, I. Forster, R. Flavell, M. Gaestel, et al. 2002. Genetic dissection of the cellular pathways and signaling mechanisms in modeled tumor necrosis factor–induced Crohn's-like inflammatory bowel disease. J. Exp. Med. 196:1563–1574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Korzenik, J.R., and D.K. Podolsky. 2006. Evolving knowledge and therapy of inflammatory bowel disease. Nat. Rev. Drug Discov. 5:197–209. [DOI] [PubMed] [Google Scholar]
- 14.Pfeffer, K., T. Matsuyama, T.M. Kundig, A. Wakeham, K. Kishihara, A. Shahinian, K. Wiegmann, P.S. Ohashi, M. Kronke, and T.W. Mak. 1993. Mice deficient for the 55 kd tumor necrosis factor receptor are resistant to endotoxic shock, yet succumb to L. monocytogenes infection. Cell. 73:457–467. [DOI] [PubMed] [Google Scholar]
- 15.Buckley, C.D., D. Pilling, J.M. Lord, A.N. Akbar, D. Scheel-Toellner, and M. Salmon. 2001. Fibroblasts regulate the switch from acute resolving to chronic persistent inflammation. Trends Immunol. 22:199–204. [DOI] [PubMed] [Google Scholar]
- 16.Saada, J.I., I.V. Pinchuk, C.A. Barrera, P.A. Adegboyega, G. Suarez, R.C. Mifflin, J.F. Di Mari, V.E. Reyes, and D.W. Powell. 2006. Subepithelial myofibroblasts are novel nonprofessional APCs in the human colonic mucosa. J. Immunol. 177:5968–5979. [DOI] [PubMed] [Google Scholar]
- 17.Pender, S.L., and T.T. MacDonald. 2004. Matrix metalloproteinases and the gut–new roles for old enzymes. Curr. Opin. Pharmacol. 4:546–550. [DOI] [PubMed] [Google Scholar]
- 18.Zhu, J., and D.T. Yu. 2006. Matrix metalloproteinase expression in the spondyloarthropathies. Curr. Opin. Rheumatol. 18:364–368. [DOI] [PubMed] [Google Scholar]
- 19.Pender, S.L., J.M. Fell, S.M. Chamow, A. Ashkenazi, and T.T. MacDonald. 1998. A p55 TNF receptor immunoadhesin prevents T cell-mediated intestinal injury by inhibiting matrix metalloproteinase production. J. Immunol. 160:4098–4103. [PubMed] [Google Scholar]
- 20.Braghetta, P., C. Fabbro, S. Piccolo, D. Marvulli, P. Bonaldo, D. Volpin, and G.M. Bressan. 1996. Distinct regions control transcriptional activation of the alpha1(VI) collagen promoter in different tissues of transgenic mice. J. Cell Biol. 135:1163–1177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Soriano, P. 1999. Generalized lacZ expression with the ROSA26 Cre reporter strain. Nat. Genet. 21:70–71. [DOI] [PubMed] [Google Scholar]
- 22.Novak, A., C. Guo, W. Yang, A. Nagy, and C.G. Lobe. 2000. Z/EG, a double reporter mouse line that expresses enhanced green fluorescent protein upon Cre-mediated excision. Genesis. 28:147–155. [PubMed] [Google Scholar]
- 23.Victoratos, P., J. Lagnel, S. Tzima, M.B. Alimzhanov, K. Rajewsky, M. Pasparakis, and G. Kollias. 2006. FDC-specific functions of p55TNFR and IKK2 in the development of FDC networks and of antibody responses. Immunity. 24:65–77. [DOI] [PubMed] [Google Scholar]
- 24.Bailey, C.J., R.M. Hembry, A. Alexander, M.H. Irving, M.E. Grant, and C.A. Shuttleworth. 1994. Distribution of the matrix metalloproteinases stromelysin, gelatinases A and B, and collagenase in Crohn's disease and normal intestine. J. Clin. Pathol. 47:113–116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Tchetverikov, I., L.R. Lard, J. DeGroot, N. Verzijl, J.M. TeKoppele, F.C. Breedveld, T.W. Huizinga, and R. Hanemaaijer. 2003. Matrix metalloproteinases-3, -8, -9 as markers of disease activity and joint damage progression in early rheumatoid arthritis. Ann. Rheum. Dis. 62:1094–1099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Parks, W.C., C.L. Wilson, and Y.S. Lopez-Boado. 2004. Matrix metalloproteinases as modulators of inflammation and innate immunity. Nat. Rev. Immunol. 4:617–629. [DOI] [PubMed] [Google Scholar]
- 27.Firestein, G.S., and N.J. Zvaifler. 2002. How important are T cells in chronic rheumatoid synovitis?: II. T cell-independent mechanisms from beginning to end. Arthritis Rheum. 46:298–308. [DOI] [PubMed] [Google Scholar]
- 28.Muller-Ladner, U., J. Kriegsmann, B.N. Franklin, S. Matsumoto, T. Geiler, R.E. Gay, and S. Gay. 1996. Synovial fibroblasts of patients with rheumatoid arthritis attach to and invade normal human cartilage when engrafted into SCID mice. Am. J. Pathol. 149:1607–1615. [PMC free article] [PubMed] [Google Scholar]
- 29.Powell, D.W., R.C. Mifflin, J.D. Valentich, S.E. Crowe, J.I. Saada, and A.B. West. 1999. Myofibroblasts. II. Intestinal subepithelial myofibroblasts. Am. J. Physiol. 277:C183–C201. [DOI] [PubMed] [Google Scholar]
- 30.Nenci, A., C. Becker, A. Wullaert, R. Gareus, G. van Loo, S. Danese, M. Huth, A. Nikolaev, C. Neufert, B. Madison, et al. 2007. Epithelial NEMO links innate immunity to chronic intestinal inflammation. Nature. 446:557–561. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}