

Abstract
To understand the evolution of human mental activity, we performed population genetic analyses of nucleotide sequences (∼11 kb) from a worldwide sample of 60 chromosomes of the N-acylsphingosine amidohydrolase (ASAH1) gene. ASAH1 hydrolyzes ceramides and regulates neuronal development, and its deficiency often results in mental retardation. In the region (∼4.4 kb) encompassing exons 3 and 4 of this gene, two distinct lineages (V and M) have been segregating in the human population for 2.4 ± 0.4 million years (MY). The persistence of these two lineages is attributed to ancient population structure of humans in Africa. However, all haplotypes belonging to the V lineage exhibit strong linkage disequilibrium, a high frequency (62%), and small nucleotide diversity (π = 0.05%). These features indicate a signature of positive Darwinian selection for the V lineage. Compared with the orthologs in mammals and birds, it is only Val at amino acid site 72 that is found exclusively in the V lineage in humans, suggesting that this Val is a likely target of positive selection. Computer simulation confirms that demographic models of modern humans except for the ancient population structure cannot explain the presence of two distinct lineages, and neutrality is incompatible with the observed small genetic variation of the V lineage at ASAH1. On the basis of the above observations, it is argued that positive selection is possibly operating on ASAH1 in the modern human population.
HOMO sapiens has evolved to adapt to new and diverse environments, showing rapid population expansion since the exodus from Africa, 60–80 thousand years (KY) ago (Watson et al. 1997; Macaulay et al. 2005; Kivisild et al. 2006; Mellars 2006). At the same time, modern humans have acquired specific mental activity (e.g., language, symbols, culture, arts, etc.) (Henshilwood et al. 2002; Mellars 2006; Bouzouggar et al. 2007), a possible driving force for subsequent dispersal around the world (Klein 1999; Mellars 2006). In this process of modern human evolution, it is likely that some genes, especially those related to mental activity, have evolved under natural selection (Nei 1983). Recent studies have reported positively selected genes for mental activity and/or brain development in the human lineage: Abnormal spindle-like microcephaly associated and Microcephalin in relation to brain size (ASPM, Zhang 2003; MCPH1, Evans et al. 2004; Wang and Su 2004, but see Currat et al. 2006; Yu et al. 2007); Dopamine receptor D4 and Monoamine oxidase A in relation to emotional activity (DRD4, Ding et al. 2002; MAOA, Gilad et al. 2002); Forkhead box P2 in relation to language (FOXP2, Enard et al. 2002; Zhang et al. 2002); and Spinocerebellar ataxia type 2 and Pituitary adenylate cyclase-activating polypeptide in relation to neurodegenerative disorders (SCA2, Yu et al. 2005; PACAP, Wang et al. 2005). In addition, many causal genes for several types of mental retardation, possibly related to brain and cognitive development, have recently been reported (Inlow and Restifo 2004; Mervis and Becerra 2007; Schumacher et al. 2007). These genes are further thought to influence mental activity.
Our interest lies in lipid storage diseases (LSDs) such as Gaucher, Tay-Sachs, Farber, and Niemann-Pick diseases, all of which are the result of inherited deficiency of genes whose products are related to sphingolipid metabolism. Deficiency causes intralysosomal accumulation of unmetabolized sphingolipids, ubiquitous components of eukaryotic cell membranes that play important roles in intracellular signaling and membrane structure (Futerman and Hannun 2004; Futerman and Riezman 2005). Sphingolipids regulate neuronal growth rates, differentiation, and death of neurons. This regulation depends on the concentration of sphingolipids in their metabolism pathway (Buccoliero et al. 2002; Buccoliero and Futerman 2003).
Ceramides are at the hub of sphingolipid metabolism and serve as the first point of significant sphingolipid accumulation in the de novo pathway (Hannun and Obeid 2002; Merrill 2002). This pathway involves acid ceramidase (ASAH1) (also known as N-acylsphingosine amidohydrolase, ASAH) (AC, MIM 228000, EC 3.5.1.23) (Rother et al. 1992), which hydrolyzes ceramides into sphingosines and free fatty acids (Gatt 1963). Catalysis of ASAH1 is highly related to neuronal development (Schwarz and Futerman 1997; Ruvolo 2003), and inherited deficiency leads to accumulation of ceramides in various tissues, resulting in Farber disease also known as Farber lipogranulomatosis (Sugita et al. 1972). Farber disease is a rare disorder with an autosomal recessive mode of inheritance. Typical symptoms include painful swelling of joints, hoarseness, and premature death, and depending on the tissues affected by the storage of ceramides, severe nervous system dysfunction (Moser 1995). Several mutations for Farber disease have been reported; a single nucleotide deletion (V96del, Muramatsu et al. 2002) and nine single nonsynonymous mutations (Y36C, V97E, E138V, L182V, T222K, G235R, R254G, N320D, and P362R, Koch et al. 1996; Li et al. 1999; Bär et al. 2001; Muramatsu et al. 2002; Devi et al. 2006). The gene is located on the short arm of chromosome 8 (8p22–p21.3), is ∼28.5 kb long, appears to be a single-copy gene, and encodes 14 exons. Depending on the splicing pattern, ASAH1 is translated into either 395 or 411 amino acids (NP_808592 and NP_004306).
In this article, we applied the long-range haplotype (LRH) test to eight genes associated with the sphingolipid metabolism and found a probable signature of selective sweep in ASAH1. We therefore examined the tempo and mode of ASAH1 evolution in human populations in more detail. Fifteen African and 15 non-African samples were used to analyze genetic variation. The region sequenced is ∼11 kb long and encompasses exons 3–10, where strong linkage disequilibrium (LD) is manifested. The results of LD and polymorphism analyses suggest that a particular group of haplotypes in ASAH1 represents a signature of recent positive Darwinian selection.
MATERIALS AND METHODS
The LRH test:
The LRH test (Sabeti et al. 2002) for eight LSD-associated genes was conducted using the HapMap Project data, which was released in June 2006 (http://www.hapmap.org) (International HapMap Consortium 2005). The eight genes are ASAH1, Glucosidase acid beta (GBA), β-Galactosidase 1 (GLB1), Ganglioside GM2-activator (GM2A), Hexosaminidase A (HEXA), Hexosaminidase B (HEXB), Niemann-Pick disease type C1 (NPC1), and Niemann-Pick disease type C2 (NPC2). For each of Yoruba in Ibadan from Nigeria (YRI) and Northern and Western Europeans in Utah (CEU), the HapMap data of 60 unrelated individuals were analyzed. For Han Chinese from Beijing (CHB) and Japanese from Tokyo (JPT), similar data of 45 and 44 unrelated individuals were used, respectively. To estimate haplotype phases accurately, chromosomes with ≥10% missing genotypes (undetermined genotypes) were excluded. The haplotype phase of these trimmed HapMap data was estimated using the program PHASE v2.1 (Stephens et al. 2001; Stephens and Donnelly 2003). After the phase estimation, haplotypes with a low probability were further excluded. Thus the total number of SNPs and chromosomes used in the LRH test ranges from 83 to 100 and 82 to 120, respectively, as given in supplemental Table 1 at http://www.genetics.org/supplemental/. The extended haplotype homozygosity (EHH) and relative EHH (REHH) in ∼200 kb surrounding specified genomic regions of interest were measured using the software Sweep 1.0 (Sabeti et al. 2002). The significance of the LRH test results was examined with the simulation program ms (Hudson 2002). In the simulation, neutral polymorphism data in a sample of 120 DNA sequences, each of which ∼200 kb in length, were generated without recombination to make the test conservative. One hundred segregating sites were randomly sampled so as to imitate the actual data for the eight LSD-associated genes (supplemental Table 1). One thousand replications were carried out for each of the eight genes. For each gene in each population, the observed and simulated EHH and REHH were compared within the bin that contained haplotypes of the same frequency. The standard deviations of the observed values from the mean in their bin were calculated using the EHH significance calculator option of Sweep 1.0.
DNA samples, PCR, and sequencing:
The 30 human genomic DNA samples used in this study come from 15 Africans (10 Pygmies, 2 African Americans, and 3 Yoruba) and 15 non-Africans (4 Amerinds, 5 Europeans, and 6 Asians). The repository numbers of these samples in the Coriell Cell Repositories are NA10470–10473, 10492–10496, 10469, 10965, 10970, 10975, 11197, 11322, 11324, 11373, 11521, 11587, 13597, 13607, 13617–13618, 13820, 13838, 14537, 14661, 18523, 18853, and 19208.
PCR was used to amplify the part of the ASAH1 gene (∼12.5 kb), ranging from chromosome position 17969623 to 17982155 on chromosome 8 (NCBI build 36.2). The primers were designed using the program Primer3 (Rozen and Skaletsky 2000) and are given in supplemental Table 2 at http://www.genetics.org/supplemental/. PCR was performed with 4 pmol of each primer, 150 ng of human genomic DNA, 0.2 mm dNTPs, 0.7 μl of Elongase enzyme mix (Invitrogen, Carlsbad, CA), and 4 μl of PCR buffer containing 1.9 mm MgCl2 in a total volume of 20 μl. A RoboCycler Gradient 96 (Stratagene, La Jolla, CA) and TGradient (Whatman Biometra, Goettingen, Germany) were used under the following conditions depending on primer pairs: denaturation at 94° for 2 min followed by 40 amplification cycles of 94° for 30 sec, 55°–59° for 30 sec, and 68° for 10 min, and ending with an extension at 68° for 20 min. The amplified products were purified using ExoSAP-IT (United States Biochemical, Cleveland) and sequenced directly. Except for repeated sequences and nucleotides with low quality peaks, the ∼11-kb region was used for subsequent analyses. Sequencing reactions were performed using BigDye Terminator v1.1 and v3.1 cycle sequencing kits (Applied Biosystems, Foster City, CA) and analyzed on an ABI PRISM 377, 3100, and 3730 DNA sequencer (Applied Biosystems). To avoid sequencing errors, the PCR products were read at least twice in both directions. The accession numbers of sequences determined in this study are as follows: AB371370–AB371406.
Population analyses:
From the DNA sequence data of the ∼11-kb region of ASAH1, haplotype phases were inferred using the program PHASE v.2.1 (Stephens et al. 2001; Stephens and Donnelly 2003) and fastPHASE 1.0.1 (Scheet and Stephens 2006). All estimated haplotypes were used for further analyses. The nucleotide diversity (π) (Nei and Li 1979) and Tajima's D (Tajima 1989) were computed, and the HKA test (Hudson et al. 1987) was applied using the program DnaSP 4.10 (Rozas et al. 2003). A gene tree for the strong LD region (after excluding two possible recombinants) was constructed and the time to the most recent common ancestor (TMRCA) was estimated using the software Genetree (Griffiths and Tavaré 1995), assuming the effective population size (Ne) of 104 and the generation time (g) of 20 years (Takahata 1993; Klein and Takahata 2002). To estimate the TMRCA in an alternative way, the average p distance between the V and M lineages (pB) was used. The chimpanzee sequence was downloaded from the Ensemble database (ENSPTRT00000037110). The average per-site pairwise distance (d = 0.0174 ± 0.0003) between 60 human and one chimpanzee chromosomes was calculated in the strong LD region of ∼4.4 kb using the program MEGA 3.1 (Kumar et al. 2004). Under the assumption that humans and chimpanzees diverged 5–7 MYA, the between-lineage TMRCA was estimated as (pB/d) × 5–7 MY. The average nucleotide difference between the root haplotype and every individual in the sample was calculated in each of the V and M lineages. The ratio of these differences to d was then used to estimate the within-lineage TMRCA.
Simulations under various demographic models:
We assumed a model of a panmictic population with bottleneck or expansion as well as that of both recent and ancient structured populations. Under the assumption of selective neutrality, the software ms (Hudson 2002) efficiently evaluated the extent of neutral variation such as the π-value. Twenty-two different sets of demographic parameters were examined with 50,000 replications for each (supplemental Figure 1 and supplemental Table 3 at http://www.genetics.org/supplemental/) by commonly specifying the following parameters: the number of chromosomes, 60; the number of segregating sites, 49 (the observed value in the region of strong LD); and the generation time (g), 20 years. The other parameters for models followed the previous studies (Takahata 1995; Marth et al. 2004; Voight et al. 2005; Williamson et al. 2005).
To evaluate the effect of an advantageous mutation on the pattern of polymorphism in comparison with neutral cases, a forward simulation was also carried out (supplemental Figure 2 at http://www.genetics.org/supplemental/). An ancestral sequence of L = 1000 bp length was generated at random. Two hundred copies (2N0 = 200) of the sequences evolved according to a finite-site model of neutral mutations and random sampling, and this process was repeated till equilibrium. Each of the two subpopulations (popS and popN) was composed of these 100 sequences (N0 = 2N1 = 100, where N1 is the size of subpopulation). To trace a particular lineage (N lineage) in a subpopulation, a single mutation for labeling is introduced into a single gene in the popN. It was assumed that neutral mutations scaled by N0 occur at a rate of N0Lμ = 0.4 per generation, where μ is the neutral mutation rate per site per generation, followed by migration (at a rate of N0m = 0.1, 0.01, and 0.001 where m is the migration rate per gene) and random sampling of N0 sequences for the next generation. No recombination was assumed. Repeating this process for an additional 10N0 generations, two subpopulations admix with each other and the two become a single panmictic one. At 0.25N0 generations before this unification, a single advantageous mutation of N0s = 50 was introduced into a non-N lineage in the popS and a new lineage (S lineage) is generated. The time of 0.25N0 generations is long enough for the mutation to fix in the popS. The fixation time of a single advantageous mutation with N0s = 50 within a subpopulation is ∼0.2N0 generations (Takahata 1991). Simulation was terminated when the frequency of the S lineage in the entire population reached ∼0.7. Then the π-values within the S and N lineages and in the entire population were measured in each replication and ∼100 such π-values were collected.
For a neutral case, the population size of N0 was set to be 20, because the simulation of the same scheme takes an enormously long time to collect sufficient data. The simulation was ceased at 0.1N0 generations after the unification of two subpopulations. At any segregating site, the lineage with a nucleotide of its frequency ∼0.7 is defined as the S lineage, while if the frequency is ∼0.3, the lineage with the nucleotide is defined as the N lineage. The π-values within the S or N lineage and in the entire population were calculated in the same way for the case of selection, and ∼1000 π-values within each lineage were collected.
Phylogenetic analysis:
ASAH1 orthologous DNA sequences in mammals and birds were retrieved from the NCBI and Ensembl databases. The accession numbers of the sequences used are as follows: humans (NM_177924), chimpanzees (ENSPTRT00000037110), orangutans (CR859721), rhesus monkeys (ENSMMUT00000021738), mice (NM_019734), rats (NM_053407), cows (ENSBTAT00000014960), dogs (ENSCAFT00000039154), hedgehogs (ENSETET00000012263), opossums (ENSMODT00000024178), and chickens (NM_001006453). These nucleotide sequences were aligned by Clustal X (Thompson et al. 1997) and the alignment was further checked manually. A neighbor-joining (NJ) tree (Saitou and Nei 1987) was constructed by MEGA 3.1 (Kumar et al. 2004). The number of nonsynonymous sites was counted by a modified Nei–Gojobori method (Nei and Gojobori 1986; Ina 1995; Zhang et al. 1998) with the complete-deletion option.
RESULTS AND DISCUSSION
Linkage disequilibrium of LSD-associated genes:
To identify target genes that showed a plausible signature of positive selection, we focused on eight LSD-associated genes whose deficiency clearly results in symptoms of mental retardation. The LRH test (Sabeti et al. 2002) was applied to these genes using the HapMap data of four populations, YRI, CEU, CHB, and JPT (International HapMap Consortium 2005). The test is intended to detect the signature of positive selection, i.e., selective sweep. Positive selection results in such rapid spread of a selected site within a population that recombination does not take place frequently to break down LD at nearby sites. A haplotype containing the selected site becomes dominant while maintaining long-range LD (Sabeti et al. 2006) and resulting in significantly higher EHH and REHH than under neutrality (Aquadro et al. 1994; Sabeti et al. 2002).
The core region of each of the eight LSD-associated genes is determined separately in YRI, CEU, CHB, and JPT. For each gene, the core region turns out to be almost the same in the four populations, or the pattern of LD does not differ among populations. Both the EHH and the REHH of each haplotype in the core region (core haplotype) were measured in either the ∼200-kb region upstream or downstream from the core region and compared with simulation data under a neutral model without recombination (materials and methods). Among these 16 surrounding regions of the eight genes, a particular core haplotype including ASAH1 shows a significantly high EHH and REHH in the ∼200-kb downstream region for all four populations (P < 0.05; Figures 1 and 2, and supplemental Table 5 at http://www.genetics.org/supplemental/). The EHH and REHH associated with the ASAH1 core haplotype are kept high throughout (Figure 2A), whereas no other core regions chosen in the ∼200-kb region exhibit such a tendency. The most parsimonious explanation for the sharing of this pattern across all four populations is that the sweep occurred prior to the radiation of modern humans out of Africa.
Figure 1.—

The relative extended haplotype homozygosity (REHH) of ASAH1 genes. The REHH values (y-axis) are plotted against the core haplotype frequency (x-axis). Observed REHHs were compared with simulated data with 1000 replications. Brown dots represent simulation results, and blue dots represent the observations of each of the four populations.
Figure 2.—

A map of genes and SNPs, REHH by distance, and a LD plot of ASAH1. (A) Blue rectangles indicate genes in the ∼200-kb region, with the official symbol of the gene shown on the rectangles. Under the genes, blue bars indicate the SNPs used for the LRH test and dark blue bars represent SNPs in the core region. The x-axis indicates the distance from the core region and the y-axis, the REHH values of each haplotype in the region. Core haplotype sequences are shown in supplemental Figure 3A at http://www.genetics.org/supplemental/. The data presented is based on the CEU panel from the HapMap. (B) A LD plot was drawn using the software Haploview 3.32 (Barrett et al. 2005) and analyzed on the basis of HapMap genotype data in which SNPs were shared in all four populations. The black line expresses the gene structure of ASAH1, and on the line, the dark blue rectangle represents the region sequenced in this study. The black bar in the light blue rectangle shows the location of the SNPs used for the LD analysis, with the reference SNP identification number indicated under each SNP. The triangle represents the LD plot calculated from these SNPs. Numbers in squares represent D′ values (×100, Lewontin 1964) and the color of each square expresses the extent of LD: bright red, LOD (Morton 1955) ≥ 2 and D′ = 1; red and shades of pink, LOD ≥ 2 and D′ < 1; blue, LOD < 2 and D′ = 1; and white, LOD < 2 and D′ < 1. The triangle surrounded by a thick black line shows the LD block according to the definition of Gabriel et al. (2002).
Coalescence analysis of ASAH1 sequences:
About an 11-kb region of the ASAH1 gene was sequenced for 15 Africans and 15 non-Africans. The region includes exons 3–10, which are part of the functional domain, and covers the strongest LD block with significantly high EHH and REHH (Figure 2B). There are 106 segregating sites and 37 haplotypes within this region (Figure 3). The small number of haplotypes relative to the large number of segregating sites directly indicates fairly strong LD in this region. Nonetheless, we classified the region into two in terms of LD, a ∼4.4-kb subregion under strong LD (SL) and the remaining ∼6.6-kb subregion under moderately strong LD (ML). With an ancestor sequence being inferred from a chimpanzee sequence, the gene tree constructed for the SL subregion (Figure 4) reveals two distinct lineages. They are distinguished at two nonsynonymous polymorphic sites, M72V and I93V. One lineage possesses Val (derived type) at amino acid site 72 and Ile (ancestral type) at amino acid site 93, whereas the other possesses Met (ancestral type) and Val (derived type) at these sites, respectively. The two lineages are named M and V with respect to this M72V dimorphism and are found in both the African and the non-African samples (supplemental Table 6 at http://www.genetics.org/supplemental/). The TMRCA between the V and M lineages is estimated as 2.4 ± 0.4 MY from the average pairwise nucleotide differences (pB) (materials and methods). This is relatively old compared to the average TMRCA of 0.8 MY, if Ne = 104 and g = 20 years (Takahata 1993; Klein and Takahata 2002). However, there are several reports about genes with TMRCA >2 MY (Harris and Hey 1999; Barreiro et al. 2005; Garrigan et al. 2005a; Stefansson et al. 2005; Garrigan and Hammer 2006; Hayakawa et al. 2006). Except for one gene (Garrigan et al. 2005b), the ancient TMRCA of these genes is attributed to the presence of two distinct lineages in Africa (Satta and Takahata 2004). In particular, Hayakawa et al. (2006) show that the TMRCA at the CMP-N-acetylneuraminic acid hydroxylase (CMAH) locus is 2.9 MY and suggest that this rather ancient TMRCA may result from partially isolated populations in the Pleistocene period in Africa. To compare the TMRCA at ASAH1 with that at other loci, we applied the HKA test to the nucleotide diversity and divergence at 10 loci including CMAH (Hayakawa et al. 2006) and those at ASAH1, but no significant differences are detected in the test. This suggests that the TMRCA at ASAH1 is not exceptional (supplemental Table 7 at http://www.genetics.org/supplemental/).
Figure 3.—

Segregating sites in 37 haplotypes and their frequencies in a worldwide sample of 60 chromosomes. Dots indicate nucleotides that are identical to those in the top line sequence. The number above the sequence indicates the base pair position of each site, and sites in the coding region are represented by the larger font size. Nucleotides with an asterisk (*) indicate a nonsynonymous polymorphic site; the amino acid substitutions at these sites are shown above the top line sequence. Sequences in the rectangle are in complete linkage disequilibrium except for two haplotypes (M090 and M100). The numbers in the far right-hand column indicate the frequency of each haplotype.
Figure 4.—

A gene tree of 13 human ASAH1 haplotypes in the SL subregion. The chimpanzee sequence was used to root the tree. Solid circles represent nucleotide substitutions. The haplotype names at the tip of the tree are provided in supplemental Table 6. The numbers below the haplotype names represent the frequency of each haplotype on the 58 examined chromosomes, excluding two recombinant sequences. Since the nucleotide at site 32 (Figure 3) in M080 is the same as that in the chimpanzee, parallel substitution was thought to have occurred at this site; the site is incompatible with others and therefore excluded from the tree. After exclusion of this site, M080 in the SL subregion is identical to M2. For the method of TMRCA estimation, see materials and methods.
The π-value (Nei and Li 1979) in the SL subregion of 60 chromosomes is high (0.37 ± 0.02%; Table 1), more than four times higher than the average value in the human genome (0.08%; Sachidanandam et al. 2001). It was examined whether this large π is compatible with demographic models that have been proposed for human evolution so far by computer simulation (see materials and methods, and supplemental Figure 1). The probability of π > 0.37% from simulated polymorphism data was estimated under neutrality. The results of 50,000 replications showed that the probability was <0.03 except for the ancient population-structure model (supplemental Table 4 at http://www.genetics.org/supplemental/). It should be noted that this ancient population-structure model (supplemental Figure 1H) is consistent with π-values at other loci, i.e., more than half of the 10 loci used for the HKA test (data not shown).
TABLE 1.
Genetic diversity at ASAH1 in a worldwide sample of 60 chromosomes
| Lineagea | Nb | Sc | Hd | πe | |
|---|---|---|---|---|---|
| Worldwide | All | 60 | 49 | 17 | 0.37 (0.024) |
| V | 37 | 9 | 6 | 0.05 (0.006) | |
| M | 23 | 19 | 11 | 0.13 (0.017) | |
| African | All | 30 | 44 | 11 | 0.36 (0.043) |
| V | 20 | 8 | 5 | 0.06 (0.010) | |
| M | 10 | 12 | 6 | 0.13 (0.016) | |
| Non-African | All | 30 | 41 | 9 | 0.39 (0.026) |
| V | 17 | 4 | 3 | 0.04 (0.005) | |
| M | 13 | 16 | 6 | 0.11 (0.034) |
Defined by V and M at site 72.
Number of sequences.
Number of segregating sites.
Number of haplotypes.
Percentage of nucleotide diversity per site (standard deviation).
The frequency of V and M in the total sample is 0.62 and 0.38, respectively (supplemental Table 6). Under the assumption of Hardy–Weinberg equilibrium, the expected heterozygosity with V and M is 0.47. However, the observed value in the sample is 0.23, which is significantly lower than the expectation (P < 0.005, chi-square test). This heterozygosity deficiency is also observed in both the African and non-African samples. Under overdominance selection, we may also expect an excess of heterozygotes over Hardy–Weinberg equilibrium, although the extent is not necessarily large if mating occurs at random every generation. The deficiency is inconsistent with the possibility of overdominance that might maintain two allelic lineages for a long time (Takahata 1990). Alternatively, this deficiency may suggest that the V and M lineages have been maintained in a partially isolated subpopulation until the exodus from Africa. When migration is limited, it is likely that genes within each subpopulation coalesce to a common ancestor and form a single cluster in a tree (Takahata 1991). Subpopulation-specific lineages tend to be reciprocally monophyletic and independent mutations can accumulate in a lineage-specific manner. This pattern is consistent with the observed tree topology. In this context, it should be noted that the pattern of genetic diversity of ASAH1 and other loci is compatible with the proposal that the human population was once geographically structured and genetically differentiated in Africa (Takahata 1995; Satta and Takahata 2004).
ASAH1 polymorphism:
The value of nucleotide diversity (π) within the V lineage (πVSL = 0.05 ± 0.01%; Table 1) is smaller than the overall π-value on chromosome 8 (0.08%) (Sachidanandam et al. 2001) on which ASAH1 is located. More importantly, the πVSL value is significantly smaller than the nucleotide diversity within the M lineage (πMSL = 0.13 ± 0.02%). Furthermore, the number of haplotypes in the V lineage is only 6, yet it is 11 in the M lineage. These hold true in both African and non-African samples (Table 1). We can therefore expect that the TMRCA within the V lineage is younger than that within the M lineage as far as the SL subregion is concerned (Figure 4). Indeed, the TMRCA of the V lineage is estimated as 200 ± 50 KY from Genetree analysis (Griffiths and Tavaré 1995) and 340 ± 80 KY on the basis of the average nucleotide diversity (materials and methods). On the other hand, the TMRCA of the M lineage is 320 ± 70 KY from the Genetree analysis and 680 ± 180 KY from the nucleotide diversity. Compared with the M lineage, the relatively recent origin of the predominant V lineage implies that it has been rapidly increasing in frequency. In accordance with this, Tajima's D value is negative (−0.11) for the V lineage and positive (0.27) for the M lineage, although both are not statistically different from zero (P > 0.1) (Tajima 1989).
To see if the reduced level of polymorphism within the V lineage is restricted to the SL subregion, the π-value is compared with that of the ML subregion defined as moderately strong LD (Figure 5). Possible recombination in the ML subregion does not allow us to make phylogenetic analysis and the concept of lineage per se becomes equivocal. For this reason, we simply defined the V and M lineages in the ML subregion as those that are linked with V and M in the SL subregion. We then calculated the nucleotide diversity as πVML = 0.10 ± 0.02% and πMML = 0.18 ± 0.02%. They are not significantly different from each other (P > 0.05, Z-test), but πVML is significantly larger than πVSL (P < 0.01, Z-test; Table 1).
Figure 5.—

Window analysis of π in the sequenced region of ∼11 kb in length. The π-values were calculated in a window size of 500 bp with a step size of 250 bp. The x-axis indicates the distance in the sequenced region and the y-axis, the π-value of each window. (A) The π-values obtained from a sample of 60 sequences. (B) The π-values within the V lineage. The arrow indicates the location of the M72V polymorphism. (C) The π-values within the M lineage.
As mentioned earlier, the frequency of the V lineage in the total sample is higher (0.62) than that of the M lineage (0.38). The predominance of the V lineage is observed in both Africans and non-Africans (supplemental Table 6). The HapMap data also gives similar results: 0.83 in YRI, 0.56 in CEU, and 0.67 in both CHB and JPT. Despite the relatively high frequency of the V lineage irrespective of data and populations, the π-value is smaller and the within-lineage TMRCA is shorter than the corresponding value in the M lineage. All these features are consistently explained by positive Darwinian selection operating for the V lineage. In addition, it should be noted that this small genetic diversity was limited to the SL subregion, suggesting that the target of the selection is located within the subregion.
Lineage-specific amino acid changes:
We have also attempted to identify a target site of positive selection. In the SL subregion of ASAH1, there are 46 segregating sites in introns, one synonymous and two nonsynonymous segregating sites. The synonymous mutation is observed in only one chromosome (V040), whereas two nonsynonymous mutations (M72V and I93V) are “fixed” within each lineage. Regarding M72V, the Val is a derived and human-specific amino acid, because in chimpanzees, orangutans, and rhesus monkeys the amino acid at this site is exclusively occupied by Met. Regarding I93V, although the Val is shared by rhesus monkeys, but the site is occupied by Ile in chimpanzees and orangutans (Figure 6). It is likely that site 93 is subject to recurrent substitution to Val.
Figure 6.—

Alignment of ASAH1 orthologs from birds and mammals in the SL subregion (A) and the NJ tree obtained on the basis of nonsynonymous substitutions in the entire ASAH1 of these orthologs (B). (A) Dots indicate amino acids that are identical to those in the top line sequence. Dashes represent a deletion. Boldface type indicates amino acid substitutions in the human lineage compared with chimpanzees and birds and other mammals. (B) Numbers near the node indicate the bootstrap probability in 500 replications. The scale bar below the tree represents the number of nonsynonymous substitutions per site. The nonsynonymous sites of 751 bp were compared. The chicken sequence was used as an outgroup. Uppercase letters next to the species name indicate the amino acid at position 72.
The SL subregion contains exon 3 (31 amino acids) and exon 4 (29 amino acids). Although these exons do not encode the active center of ASAH1, mutations in these exons are associated with Faber disease. Three such nonsynonymous mutations are Y36C, V96del, and V97E (Bär et al. 2001; Muramatsu et al. 2002). It thus appears that these exons encode important functional parts of ASAH1. It is interesting to note that the 659-bp region encompassing M72V did not accumulate any mutations within the V lineage (Figure 5). This is again a signature of selective sweep on the region encompassing Val at site 72, suggesting the amino acid might be a likely target of positive selection for the V lineage.
Theoretical considerations of natural selection operating on ASAH1:
The above results show that positive selection is likely to have favored the V lineage commonly sharing Val at amino acid site 72 and showing reduction of genetic diversity in the lineage despite its predominance (the large population size). To examine the relationship between π within a lineage and population size with and without selection, we carried out computer simulations under the ancient population-structure model (materials and methods; supplemental Figure 2). To check the validity of the computer program for the model, we compare simulation results with the theoretical expectation for neutral cases.
To obtain the theoretical formulas for the coalescence time of two neutral genes at a locus, we use Equation 4 for a finite island model in Takahata (1995). The model assumes that the ancestral panmictic population of effective size M has been subdivided into l island populations with effective size N1 for ti generations. Our model is different in that the subpopulations have further admixed to form a panmictic population of effective size N0 for subsequent tm generations to the present. We sample two genes from this panmictic population and derive the formula of the mean coalescence time. Although the formula is complicated for any values of l, M, N1, and N0, it is much simplified for the case of l = 2, M = N0 = 2N1, and this simplified formula is sufficient for our present purpose.
The coalescence can occur in the admixed population during the period of tm generation. This conditional TMRCA, T0, is readily given by
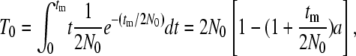 |
(1) |
where a = 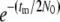 . On the other hand, if two genes do not coalesce during tm, the two ancestral gene lineages reside in the same subpopulations with probability P(0) and in two different populations with probability Q(0) in Takahata's designations. Obviously,
. On the other hand, if two genes do not coalesce during tm, the two ancestral gene lineages reside in the same subpopulations with probability P(0) and in two different populations with probability Q(0) in Takahata's designations. Obviously, 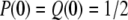 in the present model with l = 2. Equation 4 in Takahata (1995) then reduces to
in the present model with l = 2. Equation 4 in Takahata (1995) then reduces to
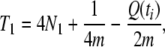 |
(2) |
where
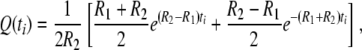 |
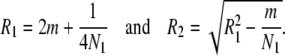 |
Noting that this coalescence is conditional with probability a and from Equations 1 and 2, we obtain the formula of the unconditional coalescence time T = T0 + aT1:
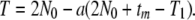 |
(3) |
For 4N0m > 1, T approaches to 2N0 generations, whereas for small 4N0m, T becomes large in proportion to the reciprocal of m.
Finally, to define the effective population size and the expected nucleotide diversity π for the present nonequilibrium demographic model, we set Ne = T/2 as in Nei and Takahata (1993) and obtain
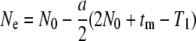 |
(4a) |
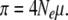 |
(4b) |
Under neutrality, simulation assumes N0Lμ = 0.4, L = 1000, and N0 = 20. In a wide range of N0m, π in Equations 4a and 4b agrees well with that in simulation (Table 2). The same simulation also demonstrates that the π-value within the predominant lineage (S lineage) is significantly larger than that within the other lineage (N lineage), even though with limited gene flow (4N0m = 0.004–0.4) (Table 2; t-test, P < 1E − 130). Thus neutrality cannot explain the observed reduction of nucleotide diversity in the V lineage in the SL subregion. On the other hand, in the presence of selection, the mean and variance of nucleotide diversity in the S lineage are significantly smaller than those in the N lineage with any tested migration rate (Table 2; F-test, P < 1E − 10; t-test, P < 1E − 6). Selective sweep can thus result in reduced nucleotide diversity within a lineage that carries an advantageous mutation. This pattern is exactly what we observed at ASAH1.
TABLE 2.
The distribution of nucleotide diversity under an ancient population-structure model with several migration rates
| Total
|
S lineage
|
N lineage
|
|||||
|---|---|---|---|---|---|---|---|
| N0m | Meana | SDb | Mean | SD | Mean | SD | |
| Neutral | 0.1 | 0.33 (0.29) | 0.17 | 0.18 | 0.15 | 0.05 | 0.08 |
| 0.01 | 0.46 (0.47) | 0.17 | 0.25 | 0.19 | 0.05 | 0.06 | |
| 0.001 | 0.51 (0.51) | 0.16 | 0.28 | 0.21 | 0.05 | 0.07 | |
| Selection | 0.1 | 0.37 | 0.18 | 0.01 | 0.01 | 0.12 | 0.07 |
| 0.01 | 0.38 | 0.12 | 0.01 | 0.02 | 0.07 | 0.06 | |
| 0.001 | 0.38 | 0.15 | 0.01 | 0.02 | 0.07 | 0.06 | |
Percentage of mean of nucleotide diversity (theoretical expectation).
Percentage of standard deviation of nucleotide diversity.
Conclusions:
The pattern and level of genetic variability at ASAH1 cannot be explained by demographic causes alone and/or overdominance selection. Rather, they suggest positive Darwinian selection operating on the predominant V lineage of a relatively recent origin. It is likely that the human-specific Val at amino acid site 72 is a possible target for positive Darwinian selection and may have occurred before the most recent common ancestor of all V lineage haplotypes, 200–340 KYA. It is speculated that, when modern humans dispersed from Africa, admixture of the distinct V and M lineages occurred and the V lineage has since spread in the entire population by possible positive selection. Although there is no functional assay of the Val substitution at present, the Val may influence the enzyme activity of ASAH1, thereby affecting catalysis of ceramides and possibly neuronal development as well. Further comprehension of ASAH1 and other genes could shed light on the evolution of mental activity in modern human evolution.
Acknowledgments
We thank Naoyuki Takahata, Tatsuya Ota, and Andrew G. Clark for their critical reading of the manuscript and for their numerous suggestions and three anonymous reviewers for their helpful comments on an early version of the manuscript. This study was supported in part by an intramural grant from the Hayama Center for Advanced Studies.
References
- Aquadro, C. F., D. J. Begun and E. C. Kindahl, 1994. Non-neutral evolution, pp. 46–56 in Selection, Recombination and DNA Polymorphism in Drosophila, edited by B. Golding. Chapman & Hall, London.
- Bär, J., T. Linke, K. Ferlinz, U. Neumann, E. H. Schuchman et al., 2001. Molecular analysis of acid ceramidase deficiency in patients with Farber disease. Hum. Mutat. 17 199–209. [DOI] [PubMed] [Google Scholar]
- Barreiro, L. B., E. Patin, O. Neyrolles, H. M. Cann, B. Gicquel et al., 2005. The heritage of pathogen pressures and ancient demography in the human innate-immunity CD209/CD209L region. Am. J. Hum. Genet. 77 869–886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barrett, J. C., B. Fry, J. Maller and M. J. Daly, 2005. Haploview: analysis and visualization of LD and haplotype maps. Bioinformatics 15 263–265. [DOI] [PubMed] [Google Scholar]
- Bouzouggar, A., N. Barton, M. Vanhaeren, F. d'Errico, S. Collcutt et al., 2007. 82,000-year-old shell beads from North Africa and implications for the origins of modern human behavior. Proc. Natl. Acad. Sci. USA 104 9964–9969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buccoliero, R., and A. H. Futerman, 2003. The roles of ceramide and complex sphingolipids in neuronal cell function. Pharmacol. Res. 47 409–419. [DOI] [PubMed] [Google Scholar]
- Buccoliero, R., J. Bodennec and A. H. Futerman, 2002. The role of sphingolipids in neuronal development: lessons from models of sphingolipid storage diseases. Neurochem. Res. 27 565–574. [DOI] [PubMed] [Google Scholar]
- Currat, M, L. Excoffier, W. Maddison, S. P. Otto, N. Ray et al., 2006. Comment on “Ongoing adaptive evolution of ASPM, a brain size determinant in Homo sapiens” and “Microcephalin, a gene regulating brain size, continues to evolve adaptively in humans.” Science 313 172. [DOI] [PubMed] [Google Scholar]
- Devi, A. R. R., M. Gopikrishna, R. Ratheesh, G. Savithri, G. Swarnalata et al., 2006. Farber lipogranulomatosis: clinical and molecular genetic analysis reveals a novel mutation in an Indian family. J. Hum. Genet. 51 811–814. [DOI] [PubMed] [Google Scholar]
- Ding, Y. C., H. C. Chi, D. L. Grady, A. Morishima, J. R. Kidd et al., 2002. Evidence of positive selection acting at the human dopamine receptor D4 gene locus. Proc. Natl. Acad. Sci. USA 99 309–314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Enard, W., M. Przeworski, S. E. Fisher, C. S. Lai, V. Wiebe et al., 2002. Molecular evolution of FOXP2, a gene involved in speech and language. Nature 418 869–872. [DOI] [PubMed] [Google Scholar]
- Evans, P. D., J. R. Anderson, E. J. Vallender, S. S. Choi and B. T. Lahn, 2004. Reconstructing the evolutionary history of microcephalin, a gene controlling human brain size. Hum. Mol. Genet. 13 1139–1145. [DOI] [PubMed] [Google Scholar]
- Futerman, A. H., and Y. A. Hannun, 2004. The complex life of simple sphingolipids. EMBO Rep. 5 777–782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Futerman, A. H., and H. Riezman, 2005. The ins and outs of sphingolipid synthesis. Trends Cell Biol. 15 312–318. [DOI] [PubMed] [Google Scholar]
- Gabriel, S. B., S. F. Schaffner, H. Nguyen, J. M. Moore, J. Roy et al., 2002. The structure of haplotype blocks in the human genome. Science 296 2225–2229. [DOI] [PubMed] [Google Scholar]
- Garrigan, D., and M. F. Hammer, 2006. Reconstructing human origins in the genomic era. Nat. Rev. Genet. 7 669–680. [DOI] [PubMed] [Google Scholar]
- Garrigan, D., Z. Mobasher, S. B. Kingan, J. A. Wilder and M. F. Hammer, 2005. a Deep haplotype divergence and long-range linkage disequilibrium at xp21.1 provide evidence that humans descend from a structured ancestral population. Genetics 170 1849–1856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garrigan, D., Z. Mobasher, T. Severson, J. A. Wilder and M. F. Hammer, 2005. b Evidence for archaic Asian ancestry on the human X chromosome. Mol. Biol. Evol. 22 189–192. [DOI] [PubMed] [Google Scholar]
- Gatt, S., 1963. Enzymic hydrolysis and synthesis of ceramides. J. Biol. Chem. 238 3131–3133. [PubMed] [Google Scholar]
- Gilad, Y., S. Rosenberg, M. Przeworski, D. Lancet and K. Skorecki, 2002. Evidence for positive selection and population structure at the human MAO-A gene. Proc. Natl. Acad. Sci. USA 99 862–867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Griffiths, R. C., and S. Tavaré, 1995. Unrooted genealogical tree probabilities in the infinitely-many-sites model. Math. Biosci. 127 77–98. [DOI] [PubMed] [Google Scholar]
- Hannun, Y. A., and L. M. Obeid, 2002. The ceramide-centric universe of lipid-mediated cell regulation: stress encounters of the lipid kind. J. Biol. Chem. 277 25847–25850. [DOI] [PubMed] [Google Scholar]
- Harris, E. E., and J. Hey, 1999. X chromosome evidence for ancient human histories. Proc. Natl. Acad. Sci. USA 96 3320–3324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayakawa, T., I. Aki, A. Varki, Y. Satta and N. Takahata, 2006. Fixation of the human-specific CMP-N-acetylneuraminic acid hydroxylase pseudogene and implications of haplotype diversity for human evolution. Genetics 172 1139–1146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henshilwood, C. S., F. d'Errico, R. Yates, Z. Jacobs, C. Tribolo et al., 2002. Emergence of modern human behavior: Middle Stone Age engravings from South Africa. Science 295 1278–1280. [DOI] [PubMed] [Google Scholar]
- Hudson, R. R., 2002. Generating samples under a Wright-Fisher neutral model of genetic variation. Bioinformatics 18 337–338. [DOI] [PubMed] [Google Scholar]
- Hudson, R. R., M. Kreitman and M. Aguadé, 1987. A test of neutral molecular evolution based on nucleotide data. Genetics 116 153–159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ina, Y., 1995. New methods for estimating the numbers of synonymous and nonsynonymous substitutions. J. Mol. Evol. 40 190–226. [DOI] [PubMed] [Google Scholar]
- Inlow, J. K., and L. L. Restifo, 2004. Molecular and comparative genetics of mental retardation. Genetics 166 835–881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- International HapMap Consortium, 2005. A haplotype map of the human genome. Nature 437 1299–1320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kivisild, T., P. Shen, D. P. Wall, B. Do, R. Sung et al., 2006. The role of selection in the evolution of human mitochondrial genomes. Genetics 172 373–387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klein, R. G., 1999. Anatomically modern humans, pp. 495–573 in The Human Career: Human Biological and Cultural Origins, Ed. 2. The University of Chicago Press, Chicago.
- Klein, J., and N. Takahata, 2002. Where Do We Come From? The Molecular Evidence for Human Descent. Springer-Verlag, Berlin.
- Koch, J., S. Gärtner, C. M. Li, L. E. Quintern, L. Bernardo et al., 1996. Molecular cloning and characterization of a full-length complementary DNA encoding human acid ceramidase. Identification of the first molecular lesion causing Farber disease. J. Biol. Chem. 271 33110–33115. [DOI] [PubMed] [Google Scholar]
- Kumar, S., K. Tamura and M. Nei, 2004. MEGA3: integrated software for molecular evolutionary genetics analysis and sequence alignment. Brief. Bioinform. 5 150–163. [DOI] [PubMed] [Google Scholar]
- Lewontin, R. C., 1964. The interaction of selection and linkage. I. General considerations; heterotic models. Genetics 49 49–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li, C. M., J. H. Park, X. He, B. Levy, F. Chen et al., 1999. The human acid ceramidase gene (ASAH): structure, chromosomal location, mutation analysis, and expression. Genomics 62 223–231. [DOI] [PubMed] [Google Scholar]
- Macaulay, V., C. Hill, A. Achilli, C. Rengo, D. Clarke et al., 2005. Single, rapid coastal settlement of Asia revealed by analysis of complete mitochondrial genomes. Science 308 1034–1036. [DOI] [PubMed] [Google Scholar]
- Marth, G. T., E. Czabarka, J. Murvai and S. T. Sherry, 2004. The allele frequency spectrum in genome-wide human variation data reveals signals of differential demographic history in three large world populations. Genetics 166 351–372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mellars, P., 2006. Why did modern human populations disperse from Africa ca. 60,000 years ago? A new model. Proc. Natl. Acad. Sci. USA 103 9381–9386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Merrill, Jr., A. H., 2002. De novo sphingolipid biosynthesis: a necessary, but dangerous, pathway. J. Biol. Chem. 277 25843–25846. [DOI] [PubMed] [Google Scholar]
- Mervis, C. B., and A. M. Becerra, 2007. Language and communicative development in Williams syndrome. MRDD Res. Rev. 13 3–15. [DOI] [PubMed] [Google Scholar]
- Morton, N. E., 1955. Sequential tests for the detection of linkage. Am. J. Hum. Genet. 7 277–318. [PMC free article] [PubMed] [Google Scholar]
- Moser, H. W., 1995. Ceramidase deficiency: Farber lipogranulomatosis, pp. 2589–2599 in The Metabolic Basis of Inherited Disease, Ed. 7, edited by C. R. Scriver, A. L. Beaudet, W. S. Sly and D. Valle. McGraw-Hill, New York.
- Muramatsu, T., N. Sakai, I. Yanagihara, M. Yamada, T. Nishigaki et al., 2002. Mutation analysis of the acid ceramidase gene in Japanese patients with Farber disease. J. Inherit. Metab. Dis. 25 585–592. [DOI] [PubMed] [Google Scholar]
- Nei, M., 1983. Genetic polymorphism and the role of mutation in evolution, pp. 165–190 in Evolution of Genes and Protein, edited by M. Nei and R. K. Koehn. Sinauer Associates, Sunderland, MA.
- Nei, M., and T. Gojobori, 1986. Simple methods for estimating the numbers of synonymous and nonsynonymous nucleotide substitutions. Mol. Biol. Evol. 3 418–426. [DOI] [PubMed] [Google Scholar]
- Nei, M., and W. H. Li, 1979. Mathematical model for studying genetic variation in terms of restriction endonucleases. Proc. Natl. Acad. Sci. USA 76 5269–5273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nei, M., and N. Takahata, 1993. Effective population size, genetic diversity, and coalescence time in subdivided populations. J. Mol. Evol. 37 240–244. [DOI] [PubMed] [Google Scholar]
- Rother, J., G. van Echten, G. Schwarzmann and K. Sandhoff, 1992. Biosynthesis of sphingolipids: dihydroceramide and not sphinganine is desaturated by cultured cells. Biochem. Biophys. Res. Commun. 189 14–20. [DOI] [PubMed] [Google Scholar]
- Rozas, J., J. C. Sanchez-DelBarrio, X. Messeguer and R. Rozas, 2003. DnaSP, DNA polymorphism analyses by the coalescent and other methods. Bioinformatics 19 2496–2497. [DOI] [PubMed] [Google Scholar]
- Rozen, S., and H. Skaletsky, 2000. Primer3 on the WWW for general users and for biologist programmers, pp. 365–386 in Bioinformatics Methods and Protocols: Methods in Molecular Biology, edited by S. Krawetz and S. Misener. Humana Press, Totowa, NY. [DOI] [PubMed]
- Ruvolo, P. P., 2003. Intracellular signal transduction pathways activated by ceramide and its metabolites. Pharmacol. Res. 47 383–392. [DOI] [PubMed] [Google Scholar]
- Sabeti, P. C., D. E. Reich, J. M. Higgins, H. Z. Levine, D. J. Richter et al., 2002. Detecting recent positive selection in the human genome from haplotype structure. Nature 419 832–837. [DOI] [PubMed] [Google Scholar]
- Sabeti, P. C., S. F. Schaffner, B. Fry, J. Lohmueller, P. Varilly et al., 2006. Positive natural selection in the human lineage. Science 312 1614–1620. [DOI] [PubMed] [Google Scholar]
- Sachidanandam, R., D. Weissman, S. C. Schmidt, J. M. Kakol, L. D. Stein et al., 2001. A map of human genome sequence variation containing 1.42 million single nucleotide polymorphisms. Nature 409 928–933. [DOI] [PubMed] [Google Scholar]
- Saitou, N., and M. Nei, 1987. The neighbor-joining method: a new method for reconstructing phylogenetic trees. Mol. Biol. Evol. 4 406–425. [DOI] [PubMed] [Google Scholar]
- Satta, Y., and N. Takahata, 2004. The distribution of the ancestral haplotype in finite stepping-stone models with population expansion. Mol. Ecol. 13 877–886. [DOI] [PubMed] [Google Scholar]
- Scheet, P., and M. Stephens, 2006. A fast and flexible statistical model for large-scale population genotype data: applications to inferring missing genotypes and haplotypic phase. Am. J. Hum. Genet. 78 629–644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schumacher, J., P. Hoffmann, C. Schmäl, G. Schulte-Körne and M. M. Nöthen, 2007. Genetics of dyslexia: the evolving landscape. J. Med. Genet. 44 289–297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwarz, A., and A. H. Futerman, 1997. Distinct roles for ceramide and glucosylceramide at different stages of neuronal growth. J. Neurosci. 17 2929–2938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stefansson, H., A. Helgason, G. Thorleifsson, V. Steinthorsdottir, G. Masson et al., 2005. A common inversion under selection in Europeans. Nat. Genet. 37 129–137. [DOI] [PubMed] [Google Scholar]
- Stephens, M., and P. Donnelly, 2003. A comparison of Bayesian methods for haplotype reconstruction from population genotype data. Am. J. Hum. Genet. 73 1162–1169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stephens, M., N. J. Smith and P. Donnelly, 2001. A new statistical method for haplotype reconstruction from population data. Am. J. Hum. Genet. 68 978–989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sugita, M., J. T. Dulaney and H. W. Moser, 1972. Ceramidase deficiency in Farber's disease (lipogranulomatosis). Science 178 1100–1102. [DOI] [PubMed] [Google Scholar]
- Tajima, F., 1989. Statistical method for testing the neutral mutation hypothesis by DNA polymorphism. Genetics 123 585–595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takahata, N., 1990. A simple genealogical structure of strongly balanced allelic lines and trans-species evolution of polymorphism. Proc. Natl. Acad. Sci. USA 87 2419–2423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takahata, N., 1991. Genealogy of neutral genes and spreading of selected mutations in a geographically structured population. Genetics 129 585–595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takahata, N., 1993. Allelic genealogy and human evolution. Mol. Biol. Evol. 10 2–22. [DOI] [PubMed] [Google Scholar]
- Takahata, N., 1995. A genetic perspective on the origin and history of humans. Annu. Rev. Ecol. Syst. 26 343–372. [Google Scholar]
- Thompson, J. D., T. J. Gibson, F. Plewniak, F. Jeanmougin and D. G. Higgins, 1997. The CLUSTAL_X windows interface: flexible strategies for multiple sequence alignment aided by quality analysis tools. Nucleic Acids Res. 25 4876–4882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Voight, B. F., A. M. Adams, L. A. Frisse, Y. Qian, R. R. Hudson et al., 2005. Interrogation multiple aspects of variation in a full resequencing data set to infer human population size changes. Proc. Natl. Acad. Sci. USA 102 18508–18513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang, Y. Q. and B. Su, 2004. Molecular evolution of microcephalin, a gene determining human brain size. Hum. Mol. Genet. 13 1131–1137. [DOI] [PubMed] [Google Scholar]
- Wang, Y. Q., Y. P. Qian, S. Yang, H. Shi, C. H. Liao et al., 2005. Accelerated evolution of the pituitary adenylate cyclase-activation polypeptide precursor gene during human origin. Genetics 170 801–806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watson, E., P. Forster, M. Richards and H. J. Bandelt, 1997. Mitochondrial footprints of human expansions in Africa. Am. J. Hum. Genet. 61 691–704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williamson, S. H., R. Hernandez, A. Fledel-Alon, L. Zhu, R. Nielsen et al., 2005. Simultaneous inference of selection and population growth from patterns of variation in the human genome. Proc. Natl. Acad. Sci. USA 102 7882–7887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu, F., P. C. Sabeti, P. Hardenbol, Q. Fu, B. Fry et al., 2005. Positive selection of a pre-expansion CAG repeat of the human SCA2 gene. PLoS Genet. 1 e41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu, F., R. S. Hill, S. F. Schaffner, P. C. Sabeti, E. T. Wang et al., 2007. Comment on “Ongoing adaptive evolution of ASPM, a brain size determinant in Homo sapiens.” Science 316 370. [DOI] [PubMed] [Google Scholar]
- Zhang, J., 2003. Evolution of the human ASPM gene, a major determinant of brain size. Genetics 165 2063–2070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang, J., H. F. Rosenberg and M. Nei, 1998. Positive Darwinian selection after gene duplication in primate ribonuclease genes. Proc. Natl. Acad. Sci. USA 95 3708–3713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang, J., D. M. Webb and O. Podlaha, 2002. Accelerated protein evolution and origins of human-specific features: Foxp2 as an example. Genetics 162 1825–1835. [DOI] [PMC free article] [PubMed] [Google Scholar]