

Abstract
A theoretical approach for estimating association free energies of α-helices in nonpolar media has been developed. The parameters of energy functions have been derived from ΔΔG values of mutants in water-soluble proteins and partitioning of organic solutes between water and nonpolar solvents. The proposed approach was verified successfully against three sets of published data: (1) dissociation constants of α-helical oligomers formed by 27 hydrophobic peptides; (2) stabilities of 22 bacteriorhodopsin mutants, and (3) protein-ligand binding affinities in aqueous solution. It has been found that coalescence of helices is driven exclusively by van der Waals interactions and H-bonds, whereas the principal destabilizing contributions are represented by side-chain conformational entropy and transfer energy of atoms from a detergent or lipid to the protein interior. Electrostatic interactions of α-helices were relatively weak but important for reproducing the experimental data. Immobilization free energy, which originates from restricting rotational and translational rigid-body movements of molecules during their association, was found to be less than 1 kcal/mole. The energetics of amino acid substitutions in bacteriorhodopsin was complicated by specific binding of lipid and water molecules to cavities created in certain mutants.
Keywords: binding, thermodynamics, potentials, solvation, membranes, micelles, α-helix, entropy, peptides, bacteriorhodopsin, glycophorin A
Association of α-helices plays an important role during folding, oligomerization, and conformational transitions of membrane proteins (Booth et al. 2001; Arkin 2002; Chin et al. 2002; Engelman et al. 2003; Jiang et al. 2003; Perozo and Rees 2003). Therefore, the physical forces that drive assembly of α-helices in nonpolar media have been a subject of significant interest (Gil et al. 1998; White and Wimley 1999; Popot and Engelman 2000). Nevertheless, there have been relatively few quantitative studies in this area, because of the difficulties with conducting equilibrium unfolding and binding experiments for membrane proteins (Haltia and Freire 1995; Booth et al. 2001). The accumulated data include stabilities of bacteriorhodopsin mutants (Krebs and Isenbarger 2000; Faham et al. 2004; Yohannan et al. 2004) and equilibrium dissociation constants of transmembrane oligomers in bilayers and micelles (Fisher et al. 1999, 2003; Mall et al. 2001; Fleming 2002; Yano et al. 2002; Cristian et al. 2003; DeGrado et al. 2003; Lew et al. 2003). The oligomeric complexes were formed by single-spanning α-helical peptides, such as glycophorin A, designed hydrophobic coiled coils, M2 channel, integrin, synaptobrevin, and others. Unfortunately, except for an NMR structure of the glycophorin A (GpA) dimer, there are no atomic resolution structures of these complexes. Theoretical models of M2 channels, synaptobrevin, and several other oligomers have been calculated by global energy minimization, with a few experimental restraints obtained from mutagenesis and solid-state NMR studies (Fleming and Engelman 2001b; Gottschalk et al. 2002; Nishimura et al. 2002). However, such ab initio models are probably imprecise and differ from each other when developed by independent research teams, as in the case of the M2 tetrameric channel (Wang et al. 2001). More precise homology models can be generated for designed transmembrane coiled coils using crystal structures of their parent water-soluble proteins.
These experimental studies suggest that assembly of transmembrane α-helices is driven mostly by van der Waals (vdW) interactions and H-bonds, whereas hydrophobic forces are insignificant within the nonpolar bilayers or micelles (MacKenzie and Engelman 1998; DeGrado et al. 2003; Lee 2003). The energy of a single H-bond was found to be ~−2 kcal/mole (Lear et al. 2003), whereas the average contribution of vdW interactions to the stability of bacteriorhodopsin mutants was estimated as 26 cal/moleÅ2 (Faham et al. 2004). These values are nearly the same as in interiors of water-soluble proteins, that is, −1.5 kcal/mole to −2.0 kcal/mole for buried H-bonds (Myers and Pace 1996), and 22 cal/mole, 26 cal/mole, and 29 cal/moleÅ2 for vdW interactions of aliphatic carbon, aromatic carbon, and sulfur, respectively (Table 5 in Lomize et al. 2002). It was also found that stability of GpA dimer depends on losses of side-chain conformational entropy (MacKenzie and Engelman 1998). These findings can be better understood by considering the aggregation of α-helices as a process similar to crystallization of small organic molecules, an analogy that has often been applied to protein folding (Shakhnovich and Finkelstein 1989; Murphy and Gill 1991; Graziano et al. 1996; Zhou et al. 1999). The crystallization is also driven by the energy of vdW interactions and H-bonds, which is known as enthalpy of fusion, and it is opposed by an entropic contribution originating from immobilization of the molecules in space and restraining of their torsion angles (Dunitz 1994; Sternberg and Chickos 1994). However, the aggregation in bilayers or micelles is complicated by protein–lipid interactions.
Table 5.
Solvation parameters determined for different types of atoms (see Materials and Methods)
| σ (cal/mole Å2) | ||||||
| Combination of media | Nsolutea | C-aliph | C-arom | N | O | S |
| Water–decadiene | 16 | 13 ± 3b | 19 ± 2 | −53 ± 3 | −57 ± 3 | |
| Water–hexadecene | 16 | 13 ± 4b | 19 ± 2 | −55 ± 4 | −63 ± 4 | |
| Water–hydrocarbons | 34 | 22 ± 1 | 18 ± 1 | −66 ± 2 | −72 ± 2 | −13 ± 7 |
| Water–lipidc | 25 | 19 | −55 | −63 | −13 | |
| Water–proteind | 19 | 7 | −21 | −66 | −1 | |
| Lipid–proteine | −6 | −12 | 34 | −3 | 12 | |
a Number of solutes applied for fitting.
b These values must be disregarded, because the solutes, whose transfer energies were applied for fitting, had few aliphatic carbons.
c Water–lipid parameters were selected as described in the text.
d Parameters obtained from protein engineering data (Lomize et al. 2002).
e The differences of values from two upper rows.
In the present work we propose a simplified theoretical approach for estimation of helix–helix binding free energies as consisting of the following components: (1) transfer energy of protein atoms from a solvent (lipid, detergent, or water) to the interior of the protein complex; (2) H-bond, vdW, and electrostatics interactions within the protein interior; and (3) side-chain conformational entropy losses calculated using the discrete rotamer approximation (Pickett and Sternberg 1993). The transfer energy is estimated using an implicit solvation model (Richards 1977), as a sum of buried surface areas of certain atoms multiplied by their empirical solvation parameters (transfer energies per Å2); this contribution depends on protein–lipid interactions. The H-bond, vdW, and electrostatic interactions between the helices are described by pairwise interatomic potentials. Because the interactions occur in condensed medium rather than in vacuum, the parameters of potentials are different from those in molecular mechanics (see Discussion).
The parameters of energy functions, which are required for calculations using this model, were derived previously from mutagenesis data for water-soluble proteins (Lomize et al. 2002). However, it was necessary to address several additional problems that are specific for binding in nonpolar media. First, the lipid–protein solvation parameters were determined by combining the corresponding water–protein values from our previous study (Lomize et al. 2002) and transfer energies of model organic compounds from water to nonpolar solvents (see Materials and Methods). Second, we selected a dielectric constant for calculations of helix–helix electrostatic interactions in bilayers and micelles. Finally, the association of peptide molecules restricts their rotational and translational movements (Murphy et al. 1994; Holtzer 1995; Gilson et al. 1997). The corresponding immobilization free energy has been found to be small (Yu et al. 2001). Therefore it was simply omitted in the calculations. If present, it would appear as a systematic deviation of the experimental and calculated energies. The proposed computational approach (see Materials and Methods) was tested using all appropriate stability data for transmembrane protein complexes whose three-dimensional structures have been experimentally determined or could be modeled by homology with high precision.
Results
Protein–ligand binding in aqueous solution
Because our energy functions were derived from unfolding ΔΔG values, their feasibility for calculation of binding affinities was not clear. Therefore, they were tested for several protein–ligand complexes in aqueous solution. The corresponding association free energies were calculated for nine small ligands that bind to a small nonpolar cavity in L99A lysozyme (Morton and Matthews 1995). Importantly, this cavity does not contain bound water molecules (Eriksson et al. 1993; Morton et al. 1995). Only complexes with available crystal structures have been considered here to take into account perturbation of protein structures during the binding.
The calculated energies of ligand binding agree reasonably well with experiment, but they are systematically overestimated. The obtained discrepancies (last column of Table 1) were less for four smaller ligands in the set, whose volumes match the size of the binding cavity. The average deviation in this case was only ~0.7 kcal/mole and can be attributed to immobilization free energy of the ligands. However, the additional deviation for five larger ligands (an average of ~2.3 kcal/mole) is probably due to the accumulation of conformational strain. Indeed, the binding of all five larger ligands except ethylbenzene leads to formation of a sterically forbidden side-chain conformer of Val 111 in lysozyme, with χ1 ~+60° in α-helix (Morton et al. 1995). It is also worth noticing that all five complexes with larger ligands have increased B-values of atoms in the area of binding pocket. The interactions of the atoms with higher B-values may be weakened due to dynamic averaging, which might produce the smaller than expected experimental binding energies. Thus, the results obtained suggest that our energy functions are appropriate for calculations of ligand-binding affinities in the absence of significant strain, and that immobilization energies of the ligands are probably less than 1 kcal/mole.
Table 1.
Comparison of experimental and calculated binding free energies (ΔGexpand ΔGclc) for ligands of L99A lysozyme
| Ligand | PDBa | ΔGexpb | ΔGclc | ΔGexp − ΔGclc |
| Smaller ligands | ||||
| Benzene | 1811 | −5.2 | −5.5 | 0.3 |
| Benzofuran | 1821 | −5.5 | −5.0 | −0.5 |
| Indene | 1831 | −5.2 | −7.0 | 1.8 |
| Indole | 1851 | −4.9 | −5.9 | 1.0 |
| Larger ligands | ||||
| Ethylbenzene | lnhb | −5.8 | −8.5 | 2.7 |
| Izobutylbenzene | 1841 | −6.5 | −8.8 | 2.3 |
| n-butylbenzene | 1861 | −6.7 | −9.9 | 3.2 |
| p-xylene | 1871 | −4.7 | −8.7 | 4.0 |
| o-xylene | 1881 | −4.6 | −7.3 | 2.7 |
a Ligand-free structure, 1190.
b From Morton and Matthews (1995).
Association of α-helical peptides in micelles and bilayers
In the next series of tests, we considered formation of α-helical dimers and trimers in bilayers and micelles (Tables 2, 3). This was necessary to verify atomic solvation parameters (see Materials and Methods) and dielectric constant, which varies from 3.5 to 7 in biological membranes (Radu et al. 1996; Polevaya et al. 1999; Ermolina et al. 2001; Hughes et al. 2002). The dielectric constant affects electrostatic interactions of α-helix “macrodipoles” (Hol et al. 1981; Ben-Tal and Honig 1996).
Table 2.
Association energies of α-helical peptides in micelles and bilayers (kcal/mole)
| Components of ΔGclc (kcal/mole) | |||||||
| Model peptides | ΔGexp | ΔGclca | ΔHvdW | ΔHHb | ΔGtrnsf | −TΔSsch | ΔHelectr |
| Dimers in micelles | |||||||
| GpA | −6.4b | −6.5 (−6.3) | −13.3 | 0 | 4.3 | 0.9 | 1.6 |
| MS1 | −4.0b | −4.1 (−4.4) | −15.5 | −2.2 | 6.2 | 4.7 | 2.6 |
| Trimers in micelles | |||||||
| MS1 | −5.5b | −6.6 (−6.8) | −24.9 | −4.3 | 12.0 | 5.1 | 5.5 |
| V7N MS1 | −12.4b | −12.7 (−12.9) | −25.9 | −8.3 | 10.3 | 5.3 | 5.5 |
| Dimers in lipid bilayers | |||||||
| L16 + Y16 | −1.5– −3c | −1.1 (−1.4) | −14.6 | 0 | 6.2 | 4.7 | 2.6 |
| L22 + Y22 | −1.5– −3c | −2.7 (−4.2) | −18.8 | 0 | 7.7 | 5.4 | 3.0 |
| Leu-GpA | <−6.4d | −7.4 (−7.2) | −14.5 | 0 | 4.7 | 0.9 | 1.5 |
| L7NL11 | <−6.4d | −6.6 (−7.1) | −19.8 | −2.2 | 6.3 | 6.2 | 2.9 |
| L19 | <−3.4d | −3.7 (−4.2) | −20.3 | 0 | 7.4 | 6.2 | 2.9 |
a These energies were calculated using ɛ = 4 and ΔGimm = 0. Values in parentheses are obtained with ɛ = 8 and ΔGimm = 1 kcal/mole. Only transmembrane segments of α-helices were included in the calculations: 73–95 for GpA and LeuGpA, 3–24 for MS1 and related peptides, 3–25 for L7NL11 and L19, 4–20 for L16 + Y16, and 6–25 for L22 + Y22).
b Experimental energies were recalculated using data from the original publications (Fisher et al. 1999; Fleming 2002; Gratkowski et al. 2002; Lear et al. 2003), as described in Materials and Methods and using partial volumes of detergents from Maire et al. (2000). c Energies varied between −1.5 and −3 kcal/mole, depending on the length of phosphatidylcholine fatty acyl chains (Mall et al. 2001). d An estimation based on results of the TOXCAT assay, as described in the text.
Table 3.
ΔΔGassocvalues (kcal/mole) of GpA and MS1 mutants in micelles
| Glycophorin A dimer | MS1 trimer | ||||
| Mutant | ΔΔGexpa | ΔΔGclc | Mutant | ΔΔGexpb | ΔΔGclc |
| L75A | 1.3 | 0.9 | N14V/V7N | −0.6 | −0.8 |
| I76A | 1.8 | 1.1 | N14Q | −1.8 | −0.6 |
| I77A | 0.1 | 0.1 | N14E | 0.0 | 0.4 |
| F78A | −0.1 | 0.0 | N14L | >1.8 | 4.2 |
| V80A | 0.4 | 0.7 | N14V | >2.1 | 4.2 |
| M81A | −0.2 | 0.0 | N14T | >2.4 | 4.5 |
| V84A | 1.0 | 0.5 | N14S | >2.4 | 6.6 |
| I85A | −0.4 | 0.0 | N14A | >3.0 | 6.8 |
| G86A | −0.1 | −0.1 | |||
| T87A | 0.9 | 1.5 | |||
ΔΔGassoc = ΔΔGassoc (mutant) − ΔΔGassoc (wild type); the values are negative for stabilizing replacements.
a Data of Fleming and Engelman (2001a).
b Based on data of Gratkowski et al. (2001) and Lear et al. (2003). N14D mutant was not included, because it forms a mixture of monomers, trimers, and hexamers (Gratkowski et al. 2001).
Our test set included the following peptides (Table 2): glycophorin A (GpA), glycophorin A with all interfacial residues simultaneously replaced by Leu (Leu-GpA), a redesigned version of two-stranded coiled coil (MS1 peptide), and several poly-Leu peptides with incorporated polar or aromatic residues (L19, L7NL11, L16 + Y16, and L22 + Y22). These peptides form dimers with a crossing angle of ~−30° (GpA and Leu-GpA) or coiled coil dimers and trimers with a crossing angle of ~+20°. The structure of GpA dimer was taken from the Protein Data Bank (1afo; MacKenzie et al. 1997), whereas homology models of other dimers and trimers were generated from crystal structures of the corresponding water-soluble coiled coils, that is, PDB files 2zta and 1zim, respectively. In the models of L16 + Y16 and L22 + Y22 heterodimers, two aromatic side chains from different monomers interact with each other but do not interfere with packing of central Leu residues. The models of MS1 mutants (Table 3) were generated from structures of the most closely related water-soluble coiled coils: the 1zim PDB file (Gonzalez et al. 1996b) for N14L, N14D, N14E, and N14Q mutants, 1ij2 (Akey et al. 2001) for N14T and N14V, and 1zij (Gonzalez et al. 1996a) for N14S and N14A. All side-chain conformers were “traced” as in the experimental templates and adjusted to maximize the number of interhelical H-bonds. Small-to-large mutants, such as Gly → Ala in GpA, were not included, because this requires energy optimization of the original experimental structures to relax the arising overlaps, and the resulting models would be insufficiently precise.
Self-association of GpA and MS1 peptides was studied in the presence of micelles formed by different nonionic or zwitterionic detergents, including C8E5, DPC, and C14-betaine (Fig. 1 ▶). Under such conditions, all experimental energies must be defined using mole concentrations of peptides in detergent rather than peptide concentrations in water or mole fractions. Therefore, the energies from original publications were recalculated, as described in Materials and Methods. The obtained values depend on detergent. For MS1 dimer, they were −4.0 and −2.0 kcal/mole in DPC and C12E8, respectively (Gratkowski et al. 2002). For GpA dimer, they were −6.4, −6.4, −6.8, and −5.2 kcal/mole in C8E5, DPC, DDMAB, and C14-betaine, respectively (Fisher et al. 1999; Fleming 2002; Fleming et al. 2004). Dimerization energies of two other peptides, L16 + Y16 and L22 + Y22, were determined in bilayers (Mall et al. 2001). They varied from −1.5 to −3 kcal/mole, depending on the length of phospatidylcholine fatty acyl chains. Table 2 also includes three peptides whose dimerization propensities were estimated qualitatively in biological membranes using a two-hybrid approach. In the TOXCAT assay, L7NL11 and Leu-GpA dimers were slightly more stable than GpA, whereas L19 was slightly more stable than G83I GpA (Russ and Engelman 1999; Zhou et al. 2001). This can be compared with stabilities of the corresponding GpA and G83I GpA dimers in micelles, that is, −6.4 kcal/mole and −3.4 kcal/mole, respectively, based on data of Fisher et al. (1999) and Bu and Engelman (1999).
Figure 1.

Dimerization of α-helical peptides in micelles. The peptide–detergent ratio is sufficiently low to have less than 1 peptide molecule per micelle.
The calculated stabilities of α-helical dimers and trimers were consistent with the experimental values (Table 2), with an average deviation of 0.4 kcal/mole for oligomers in micelles. The agreement was best with dielectric constant ɛ = 4. However, the value of ɛ could not be defined precisely, because of the uncertain immobilization energy, ΔGimm. For example, the association energies calculated with ɛ = 8 and ΔGimm = 1 kcal/mole were very close to ones with ɛ = 4 and ΔGimm = 0 (Table 2). In the subsequent calculations, we used ɛ = 4. It is worth noting that ΔΔG values in Tables 3 and 4 do not depend on ɛ, because the contribution of backbone electrostatics is identical in wild-type and mutant proteins.
Table 4.
ΔΔGunfoldvalues (kcal/mole) of bacteriorhodopsin mutants
| Mutant | ΔΔGexpa | ΔΔGclc | Mutant | ΔΔGexpa | ΔΔGclc |
| T24S | −0.3 | −0.6 | P50A | −0.1 | 0.6 |
| T24V | 0.2 | 1.4 | I52A | 1.4 | 0.6 |
| T24A | −0.6 | 1.9 | F54A | 1.0 | 0.4 |
| F42A | 1.6 | −0.3 (1.2)b | T55A | 0.1 | 0.0 |
| Y43A | 1.3 | 1.5 | M56A | −1.4 | 0.2 |
| I45A | 1.9 | 0.9 (1.8)b | Y57A | 3.7 | 3.8 |
| T46A | 2.2 | 3.3 | L58A | −0.3 | 0.2 |
| T46S | −0.1 | 1.2 | S59A | −0.1 | 0.0 |
| T47A | 1.0 | 0.8 | M60A | 0.8 | 0.8 |
| L48A | 0.1 | 0.3 | L61A | −0.7 | 0.3 |
| V49A | −0.7 | 1.3 | L62A | −0.5 | 0.0 |
Large-to-small replacements with ΔΔGclc overestimated by >0.5 kcal/mole are indicated by bold.
a Data from Faham et al. (2004) and Yohannan et al. (2004).
b Value in parentheses were calculated assuming that the residue is exposed to water in the SDS-unfolded state.
Table 2 shows the different components of free energy. The major stabilizing contribution comes from dispersion attraction of the α-helices (ΔHvdW). This was expected, because the interactions of uncharged molecules are usually dominated by dispersion forces (Israelachvili 1992). Some of the oligomers are also greatly stabilized by hydrogen bonds (−8.3 kcal/mole in V7N MS1 trimer). The largest destabilizing component was represented by the transfer energy (ΔGtrnsf). This contribution is unfavorable, because surfaces of the α-helices consist mostly of aliphatic and aromatic groups that are more “soluble” in the nonpolar hydrocarbon cores of bilayers or micelles than in the protein interior, judging from the corresponding solvation parameters (bottom row in Table 5). However, the solid-state vdW interactions outweigh this nonspecific solvation effect (|ΔHvdW | > ΔGtrnsf in Table 2). The contribution of side-chain entropy is also destabilizing, but it strongly depends on flexibility of residues affected by interaction of α-helices. The entropy changes were close to zero for GpA dimer, because its binding interface consists mostly of Val, Gly, and Thr residues, whose side chains are already restrained in isolated α-helices before the association. This contribution is higher for other complexes that contain a number of Leu residues, whose conformations are restrained during formation of the oligomers. However, the entropy changes are still relatively small, because Leu has only two allowed rotamers in an isolated α-helix. Electrostatic interactions of α-helices are also destabilizing, because the helices are roughly parallel in the oligomers.
In the next test, we estimated association energies of GpA and MS1 mutants (Table 3). The calculated ΔΔGassoc values were consistent with experiment: The average deviation was 0.3 kcal/mole for mutants of GpA. The results obtained for MS1 trimer were more difficult to compare. Some of the ΔΔG values are indicated by inequalities in Table 3, because they were not precisely determined (Lear et al. 2003). The corresponding MS1 mutants form trimers only at relatively high peptide–detergent mole ratios, close to 1:100 (Gratkowski et al. 2001). Such ratios may correspond to more than one peptide molecule per micelle, because the aggregation number of C14-betaine micelles is ~83–130 (Maire et al. 2000). Thus, two or more peptide molecules might be forced to occupy the same micelle, which differs from the situation shown in Figure 1 ▶.
In summary, the results of calculations for 27 peptides (Tables 2, 3) indicate that the implicit solvation model works successfully with parameters derived from transfer energies of model compounds. The experimental binding affinities were satisfied using ɛ of 4 to 8 and immobilization energy <1 kcal/mole.
Stabilities of bacteriorhodopsin mutants
In the last test, we reproduced ΔΔG values of 22 bacteriorhodopsin mutants (Table 4). The protein was dissolved in mixed DMPC/CHAPSO micelles and reversibly unfolded by gradually increasing the concentration of SDS (Faham et al. 2004; Yohannan et al. 2004). Under such conditions, the folded protein exists in a monomeric form that can be crystallized from bicelles (Faham and Bowie 2002). The “unfolded” state of bacteriorhodopsin in SDS micelles probably represents a mixture of four or five nativelike α-helices (A to E), nonnative β-structure, and coil (Hunt et al. 1997; Marti 1998; Booth et al. 2001).
The substituted residues (Table 4) were located in trans-membrane helices A and B of bacteriorhodopsin. These helices are individually stable in SDS, as demonstrated by two-dimensional NMR studies of the corresponding peptides, 1–36, 34–65, and 1–71 (Lomize et al. 1992; Pervushin and Arseniev 1992; Pervushin et al. 1994; Orekhov et al. 1995). Therefore, the unfolding ΔΔG values of residues in these helices were calculated as the differences of free energies describing dissociation of the entire 7α-helical domain into individual α-helices in the wild-type and mutant proteins. The crystal structures of mutants T24S, T24V, T24A, T46S, P50A, and M56A were taken from the PDB (1s51, 1s52, 1s54, 1s53, 1pxr, and 1pxs files, respectively; Faham et al. 2004; Yohannan et al. 2004). Models of other mutants, without available crystal structures, were generated by substituting the corresponding residues by Ala in wild-type monomer (1py6). Substitutions of residues 37–41 were not included, because this region is disordered in SDS (Lomize et al. 1992).
The results of calculations indicate that stabilities of the mutants were reproduced in most but not all cases (Table 4). For a majority of mutants, the average deviation of ΔΔG values was 0.3 kcal/mole, just as for GpA. However, Phe 42 and Ile 45 residues are probably exposed to water in the denatured state, because their ΔΔG values were reproduced much better using water–protein instead of lipid–protein solvation parameters (the default assumption was that all residues from the hydrophobic segments are buried from water). These two residues are situated at the edge of a hydrophobic segment and separated by a proline kink from the rest of helix B.
Discrepancies were also found for eight large-to-small replacements, whose calculated stabilities were systematically underestimated (indicated by bold in Table 4). These replacements usually improve stability of bacteriorhodopsin, despite the loss of vdW interactions within the protein structure. The observed stabilization can be attributed to tight specific binding of water or lipid molecules to cavities created in these mutants. All the corresponding residues are either buried within the transmembrane α-bundle (46, 49, and 50) or situated at the protein–lipid interface (24, 56, and 61). All crystal structures of mutants from the first group (1s53 and 1pxr) include a bound-water molecule, which spatially substitutes for the removed side chain and forms H-bonds with the protein (the water molecules were not included in the calculations). All residues from the second group (Thr 24, Met 56, and Leu 61) are involved in formation of lipid-binding sites, because they form contacts with crystallized lipids in bacteriorhodopsin trimer (1c3w) or monomer (1kme). The lipid-binding cavities become deeper after substituting these residues by Ala, which would strengthen the lipid–protein association and stabilize the structures of the mutants (however, lipid molecules were not included in PDB entries of the mutants). It is worth noting that no similar deviations of ΔΔG values were observed for mutants of GpA, except possibly I85A (Table 3). The 7α-helical bundle of bacteriorhodopsin is probably more prone to formation of lipid-binding cavities than the small GpA dimer, because it has a more complicated surface geometry.
The obtained results suggest that stabilities of mutants in transmembrane proteins can be theoretically predicted. However, there are certain complications here. The folded protein structure may be stabilized by tight specific binding of water and lipid molecules. In addition, the denatured state may be unfolded in some regions of the polypeptide chain, but form nativelike α-helices in others. These independently stable α-helices are probably dissociated or form a molten globule-like state in SDS micelles.
Discussion
The experimental binding affinities were reproduced successfully in all systems considered, including protein–ligand complexes in aqueous solution, α-helical bundles in micelles, and bacteriorhodopsin mutants (Tables 1–4). Thus, the described theoretical approach can be applied hereafter. For example, the dimerization free energies can now be estimated for any transmembrane oligomers deposited in the PDB (these energies are typically −2 to −8 kcal/ mole according to our preliminary results). In addition, an analysis of helix–helix binding energies within individual transmembrane subunits in the PDB would be important for understanding of protein folding in biological membranes. Finally, an automated membrane protein docking procedure can be developed by combining our method for evaluation of energy with previously developed optimization software (Tuffery et al. 1994; Adams et al. 1995; Pappu et al. 1999; Torres et al. 2001; Kim et al. 2003).
The stability of transmembrane oligomers depends on several factors, most of which were known previously: (1) tight packing of α-helices; (2) formation of hydrogen bonds; (3) formation of aliphatic, aromatic, and polar clusters, in accordance to the “like dissolves like” rule that applies for vdW forces in media (see below); (4) a preferential solvation of aliphatic and aromatic groups by detergents and lipids, as reflected in their protein–lipid transfer energies; (5) content of conformationally constrained Val, Gly, Ala, Thr, or Pro residues; and (6) mutual orientations of helices, which affects electrostatic energy. The corresponding energy contributions are discussed below in more detail.
Protein–protein interactions
All α-helical bundles are stabilized exclusively by helix–helix vdW interactions and H-bonds (ΔHvdW and ΔHHB in Table 2). This is a purely enthalpic contribution. Surprisingly, even in aqueous solution, the binding of nonpolar ligands to proteins is usually enthalpy driven (Morton et al. 1995; Klotz 1997; Weber and Salemme 2003), which means it depends more on interactions within the complexes than on hydrophobic effect. Importantly, the corresponding helix–helix interaction energies, for example −13 kcal/mole in GpA dimer, are several times smaller than would be calculated with molecular mechanics potentials, such as CHARMM or OPLS (Torres et al. 2001; Im et al. 2003; Lazaridis 2003). This discrepancy has a simple explanation. The molecular mechanics potentials are derived from enthalpies of sublimation or vaporization describing transfer of organic molecules from molecular crystals or liquids to the gas phase (Jorgensen et al. 1996; Gavezzotti and Filippini 1997; Ewig et al. 1999), or at least they reproduce these enthalpies, when derived from different sources (Momany et al. 1974). Therefore, such potentials are usually regarded as specifying interactions in vacuum (Lazaridis et al. 1995). However, vdW forces are much weaker in condensed media than in vacuum, as evident from theory and experiment, because these forces are of electrostatic origin (Israelachvili 1992; Leckband and Israelachvili 2001). Furthermore, the aggregation and even folding of protein molecules is similar to crystallization or liquid–solid transition (Murphy and Gill 1991; Zhou et al. 1999). Therefore, the corresponding energy gain must be related to enthalpy of fusion (Shakhnovich and Finkelstein 1989; Graziano et al. 1996), which is smaller than the enthalpies of sublimation or vaporization, consistent with the weaker interactions in media. Further, the vdW interactions in media are expected to follow the “like dissolves like” pattern, in which the same types of atoms interact more strongly than different types (Israelachvili 1992). In contrast, all empirical force fields are based on the Slater–Kirkwood equation or “combinatorial rules,” which assume that interaction energy of different types of atoms, A and B, is intermediate between A–A and B–B energies.
As follows from this discussion, the in vacuo molecular mechanics potentials are not appropriate for calculations of interatomic forces and energies in media. For example, all protein–protein and protein–lipid attractive forces are strongly overestimated and conceptually incorrect (they do not follow the “like dissolves like” rule) in molecular dynamics simulations with explicit lipids and water. Better energy functions can be derived from thermodynamic parameters of protein folding, ligand binding, crystal dissolution, or melting (the processes that occur in condensed media), rather than from heats of sublimation or vaporization describing transitions of condensed phases to gases (Murphy and Gill 1991; Eriksson et al. 1992; Robertson and Murphy 1997; Funahashi et al. 2001; Guerois et al. 2002). However, the parameters of distance-dependent (r−6) potentials have only recently been estimated from such thermodynamic data (Lomize et al. 2002). The depths of the potentials, specifically in the protein interior, were smaller than in molecular mechanics and followed the “like dissolves like” rule, as expected. Apparently, these potentials can also be applied for protein complexes in bilayers and micelles, as follows from results of this work. However, interactions across water would be weaker (Lomize et al. 2002).
We have found that electrostatic interactions of α-helices are relatively weak. Surprisingly, any value of dielectric constant between 4 and 8 could satisfy our limited set of data. These values of ɛ are consistent with experimental measurements of dielectric constant in different biological membranes, which vary from 3.5 to 7 (Radu et al. 1996; Polevaya et al. 1999; Ermolina et al. 2001; Hughes et al. 2002) and represent an average value for the protein and lipid components (Hianik and Passechnik 1995). At first glance, ɛ ~4–6 seems too high, because ɛ is ~2 in pure “black” bilayers (Benz et al. 1975) and often assumed to be between 3 and 4 in proteins (Gilson and Honig 1986). However, the experimental values of ɛ in the protein core can actually vary from 5 to 20, depending on local packing density of the polypeptide chain and penetration of single molecules of water (Ramesh et al. 1997; Dwyer et al. 2000; Mertz and Krishtalik 2000; Cohen et al. 2002). At lower packing densities or higher temperatures, the polypeptide chain becomes more flexible, which facilitates relaxation of the peptide dipoles in the electric field (Sham et al. 1998), thus increasing ɛ of dry synthetic polypeptides up to 20 (Tredgold and Hole 1976). The value of 4 can be considered as a lower limit for proteins that was observed in tightly packed crystals of acetamide and silk-like polypeptides (Tredgold and Hole 1976; Lide 2003).
Protein–lipid interactions
Association of α-helices is also mediated by peptide–lipid interactions. These interactions were treated here using an implicit solvation model, which operates with empirical transfer free energies (per squared Å) for different types of atoms determined from partition coefficients of organic compounds (Table 5). The transfer energies (atomic solvation parameters) include enthalpic and entropic components that may show up in various ways (Seelig 1997; Heerklotz and Epand 2001). For example, the GpA dimer is partially stabilized by a small entropic contribution (Fisher et al. 1999) that may originate from the gain of conformational freedom by detergent molecules that are released during association of the peptides, similar to annular lipids (Marsh and Horvath 1998; Lee 2003). Such entropic effects are difficult to reproduce in simulations with explicit lipids. Therefore, we prefer the implicit solvation model. Importantly, this model yields attractive protein–lipid interactions that destabilize the complexes (ΔGtrnsf > 0 in Table 2).
The implicit solvation model has certain limitations. First of all, the atomic solvation parameters may depend on a specific experimental system. For example, the transition of lipid bilayers to gel phase or addition of cholesterol decreases water-membrane transfer energies of hexane and benzene (DeYoung and Dill 1988, 1990). This can be interpreted as reduced solvation parameters of aliphatic and aromatic carbons, which would promote helix–helix association in the presence of cholesterol, as has actually been observed for several peptides (Mall et al. 2001; Cristian et al. 2003). It has also been found that aggregation of α-helical peptides can be modulated by replacing a detergent or lipid, although the corresponding energetic effects are relatively small, ~1.5 kcal/mole for dimers (Mall et al. 2001; Fleming 2002; Gratkowski et al. 2002), and ~2.7 kcal/mole for M2 tetramer (Cristian et al. 2003), that is, ~0.7 kcal/ mole per α-helix. Such effects may be explained by different factors that were not considered here, such as lateral pressure, hydrophobic mismatch, or specific interactions with detergent head groups.
Furthermore, the implicit solvation model can be applied only in liquid solvents whose interactions with a solute have been reduced and averaged because of the high mobility and lower density of the liquid state. The tight specific binding of a solvent molecule to a protein cavity is an entirely different situation that was observed for certain large-to-small mutants of bacteriorhodopsin (see Results). The stabilities of such mutants are probably improved by solid-state interactions with the water or lipid molecules attached to the rigid protein core, a situation that can be treated only using explicit interatomic potentials.
It is the binding of solvent molecules that probably explains the unusually high proportion of stability-enhancing mutations in membrane proteins (Bowie 2001; Howard et al. 2002). The specific binding of solvent may be more typical for membrane than water-soluble proteins for two reasons. First, the interiors of transmembrane channels, receptors, and transporters have a higher content of polar residues, which facilitate formation of H-bonds with bound water. Second, the lipid molecules can stick to shallow clefts in proteins, because they have a reduced mobility in the liquid crystalline state. The corresponding stability gain associated with tight solvent binding can be as high as 2 kcal/mole, judging from deviations of ΔΔG values in Table 4.
Other energy contributions
The results of our work suggest that immobilization of molecules during their binding costs very little (<1 kcal/mole for a dimer), no matter whether this is protein–ligand binding in water or association of α-helices in micelles (Tables 1, 2). This is in agreement with the earlier cross-linking studies of protein dimers (Yu et al. 2001). The small value of immobilization free energy can be explained by considering it as a sum of the corresponding enthalpic and entropic components ΔHimm and -TΔSimm (Yu et al. 2001). The value of -TΔSimm should not exceed fusion entropy of rigid asymmetric molecules, which can be approximated by a constant of ~3.9 kcal/mole at 300 K, according to Walden’s rule (Chickos et al. 1999). Indeed, the corresponding entropic contributions were found to be ~3.0 kcal/mole and ~2 kcal/mole for dimerization of proteins and crystallization of water, respectively (Dunitz 1994; Tamura and Privalov 1997; Yu et al. 2001). The enthalpy of immobilization, ΔHimm, originates from loss of kinetic energy that has been transformed to heat during the nonelastic collision of two molecules. The kinetic energy of a molecule is 1/2NkT (where N is the number of degrees of freedom; Wannier 1966), that is, ~−1.8 kcal/mole at 300 K (N = 6). Finally, the ΔHimm - TΔSimm value must be less than 2 kcal/mole for dimerization of asymmetric molecules in three-dimensional space, and even smaller in two-dimensional membranes.
Importantly, the transmembrane oligomers with larger buried surfaces are not necessarily more stable, just as with complexes of water-soluble proteins (Brooijmans et al. 2002). The energy of vdW interactions and H-bonds increases with the number of interatomic interactions: It is −18 kcal/mole in MS1 dimer and −29 kcal/mole in MS1 trimer (Table 2). However, this energy gain is almost cancelled by the growing destabilizing contributions, such as transfer energy (peptide–lipid interactions) and side-chain conformational entropy. Indeed, all stabilizing and destabilizing energy terms are expected to increase simultaneously with the number of atoms, and their balance depends on fine details of the structure, such as close packing, formation of specific H-bonds, or clustering of aliphatic, aromatic, and polar side chains. The total energy can be zero or positive. Thus, only complementary domains, with negative binding energy, will aggregate in biological membranes.
Materials and methods
Experimental binding free energies
The noncovalent binding of two molecules, A and B, can be considered as a generalized chemical reaction, A + B = AB. The condition for equilibrium is that
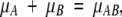 |
(1) |
or
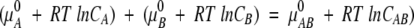 |
(2) |
Thus
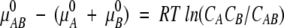 |
(3) |
where each CX is the equilibrium concentration of species X = A, B, or AB, μX = μX0 + RTlnCX is the chemical potential of X, and μX0 is the standard chemical potential of X.
The theoretical energy of binding represents the difference of standard chemical potentials, ΔGclc = μAB0 − (μA0 + μB0), and it should be compared with the corresponding experimental energy determined from the equilibrium concentrations, CA, CB, and CAB:
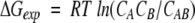 |
(4) |
Importantly, all concentrations in equations 2–4 must actually be defined as CA/CA0, CB/CB0, and CAB/CAB0, where CA0 = CB0 = CAB0 = 1 mole (Alberty 1983; Gilson et al. 1997). Therefore, the equilibrium constant, KAB = CAB/CACB, is a dimensionless quantity.
Experimental binding energy depends on concentration units applied in equation 4. For example, ΔGexp describing self-association of a peptide in the presence of detergent can be defined using: (1) mole peptide concentrations in water, (2) mole fractions (moles of peptide per 1 mole of detergent), or (3) moles of peptide in 1 L of detergent. This yields three different values, ΔGaq, ΔGMF, and ΔGdeterg, respectively, which are related as follows:
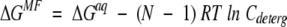 |
(5) |
where Cdeterg is the concentration of detergent, N is the number of molecules in the complex (N = 2 for dimer, N = 3 for trimer, etc.), and
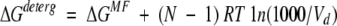 |
(6) |
where Vd is the partial volume of the solvent (detergent or lipid), in units of centimeters cubed per mole. For example, the ΔGaq, ΔGMF, and ΔGdeterg values for dimerization of glycophorin A in C8E5 micelles are −9 kcal/mole (Fleming et al. 1997), −7 kcal/mole, and −6.4 kcal/mole, respectively. The applicability of equation 5 has been justified previously (Fleming 2002; Fleming et al. 2004).
It has been emphasized that only mole concentrations, not mole fractions, can be applied for extracting the standard free energies (Ben-Naim 1980; Holtzer 1994, 1995). Thus, we used molarity units in all cases, including protein–ligand binding in water (Table 1), partitioning of model compounds (Table 5), and oligomerization of α-helical peptides (Table 2). However, because the peptides are highly hydrophobic, they are dissolved in detergent or lipid pseudo-phases, not in water. Therefore we used ΔGdeterg. Nevertheless, the corresponding mole fraction-based energies, ΔGMF, could also serve as a reasonable approximation (Lear et al. 2001; Mall et al. 2001; Fleming 2002), because they are very close to ΔGdeterg, with a difference of only 0.6 kcal/mole for binding of two molecules in detergents (N = 2, Vd ~350 cm3) and 0.2 kcal/ mole in lipids (Vd ~750cm3), as follows from equation 6.
To summarize, we assumed that hydrophobic peptides are dissolved in the detergent or lipid phases, and that law of mass action can be applied to determine their experimental self-association free energies (Fleming 2002), as long as the following conditions are satisfied: (1) The peptide–detergent molar ratio is sufficiently low to have less than one peptide molecule per micelle (Fig. 1 ▶); (2) Cdeterg is significantly higher than the critical micelle concentration (the presence of nonmicellar detergent was neglected); and (3) peptide concentrations are expressed in proper units. All these conditions were satisfied for data in Tables 2 and 3.
Theoretical association free energies
The theoretical binding energies, ΔGassoc, were calculated using our program Assembl and three-dimensional models of the complexes as input. They represented a difference of the energies calculated for the complex and individual molecules (subunits):
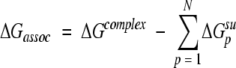 |
(7) |
The energies of the complex and subunits were represented as follows:
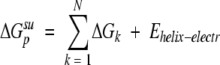 |
(8) |
where Ehelix-electr is the energy of electrostatic interactions of TM α-helices, ΔGk is the contribution of residue k, and N is number of residues. The contribution of residue k included transfer and interaction energies of its atoms, averaged over all side-chain conformers of residue k, and conformational entropy of this residue:
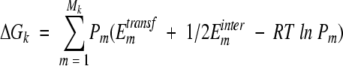 |
(9) |
where Pm, Emtransf, and Eminter are occupancy, transfer energy, and interaction energy of conformer m, respectively, and Mk is number of side-chain conformers of residue k. The 1/2 multiplier appears in equation 9 because the interaction energy of residues k and l will be summed twice. The contributions ΔGk were included only for “active” residues, which form the interaction interface between the subunits in a complex. A residue was considered as active if it had at least one contact (the distance between closest atoms <7 Å) with another subunit. Operating with only active residues served to reduce the errors associated with summation over a large set of pairwise energies dependent on imprecisely determined atomic coordinates. The set of active residues was further limited during calculations of ΔΔG values for bacteriorhodopsin mutants by including only α-helical segments around the substituted residue.
Transfer energy of residue k in conformer m from the external environment (water or lipid) to protein interior was defined as follows:
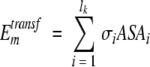 |
(10) |
where σ are the corresponding water–protein or lipid–protein atomic solvation parameters from Table 5, and ASAi is accessible surface area of atom i.
Interaction energy of conformer m of residue k with other active residues was defined as follows:
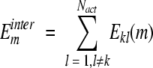 |
(11) |
where interaction energy of residues k and l is defined as follows:
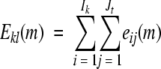 |
(12) |
where eij is the interaction energy of atoms i and j that belong to residues k and l, respectively, and Ik and Jl are numbers of atoms in residues k and l. During the calculations, side chain of residue k occupies conformer m, whereas residue l occupies its conformer present in the experimental structure or model of the complex. Thus, the averaging of side-chain conformers was done individually for each residue k, rather than for clusters of interacting amino acid residues. We have not included here torsion energy and side-chain–backbone vdW interactions within the same α-helix, which were assumed to be unchanged during binding.
The conformer occupancies, Pm in equation 9 were defined as follows:
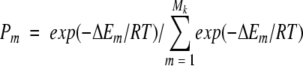 |
(13) |
where ΔEm is the relative energy of conformer m:
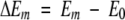 |
(14) |
E0 is the lowest energy, and
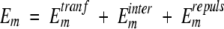 |
(15) |
The potentials for vdW interactions and H-bonds were used as 6–12 and 10–12 functions, respectively, with softened repulsions (Kuhlman and Baker 2000; Lomize et al. 2002):
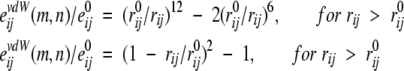 |
(16) |
for vdW interactions, and
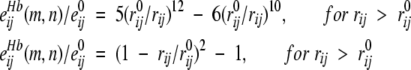 |
(17) |
for H-bonds, where rij is the distance between atoms i and j, e0 ij is energy at the minimum of the potential, and the equilibrium distances, r0ij, were taken from ECEPP/2 (Momany et al. 1974). The repulsions were softened, because the experimental coordinates of atoms are determined with a precision of ~0.3 Å, which would produce significant errors in the calculated energies.
The electrostatic interactions between large systems of oriented peptide dipoles in α-helices were calculated with partial atomic charges from CHARMM:
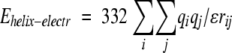 |
(18) |
where atoms i and j belong to backbones of different α-helices (including polar hydrogens).
The repulsion energy, Emrepuls, was added in equation 15 to exclude all sterically forbidden side-chain conformers. It was calculated similar to Eminter (equations 11 and 12), except that interaction energy of atoms i and j was defined differently:
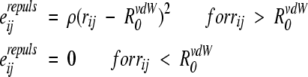 |
(19) |
where R0vdW are radii of Chothia reduced by 0.1 Å, and the weight factor, ρ, was chosen to be 3 kcal/moleÅ2. We have also assumed that repulsion energy does not change during peptide–peptide association. Therefore, Emrepuls was not included directly in binding energy (equation 9) and could only affect conformer occupancies.
The calculations for ligand–lysozyme complexes were accomplished as described above, using water–protein atomic solvation parameters. The distance cutoff defining the set of active residues was reduced to include only 19 lysozyme residues around the ligands. These residues were mostly inaccessible to water; their side chains were considered as rigid during calculations. The ligand binding energy included interactions between all active lysozyme residues in the bound and ligand-free protein structures, in addition to the protein–ligand interactions, as follows from equations 7 and 8. Conformational entropies of ligands were estimated simply from the number of their conformers, that is, 3 and 9 (0.7 kcal/mole and 1.1 kcal/mole) for izobutylbenzene and n-butylbenzene, respectively. Torsion energy was included for χ angles of active residues, as described previously for protein mutants (Lomize et al. 2002). Total contribution of torsion energy did not exceed 0.5 kcal/mole.
Determination of solvation parameters in membranelike environments
The required σlipid → protein parameters were defined as differences of the corresponding σwater → protein and σwater → lipid energies, where σwater → protein values were obtained previously from mutagenesis data for water-soluble proteins (Lomize et al. 2002), and σwater → lipid parameters were determined here based on experimental transfer energies of model compounds from water to non-polar solvents. Importantly, all transfer energies had to be obtained based on mole concentrations of the solutes rather than mole fractions (Ben-Naim 1980; Radzicka and Wolfenden 1988; Holtzer 1994).
The required σwater → lipid value was estimated first for aliphatic carbon. Transfer energies of aliphatic compounds from water to nonpolar solvents, micelles, and bilayers are slightly different. For example, transfer energies of hexane from water to hexadecane or DMPC vesicles are −6.1 or −5.4 kcal/mole, respectively, based on data of Abraham et al. (1994) and DeYoung and Dill (1990). The most reliable estimate here can be obtained using the “consensus” increment value of a CH2 group, which is 0.7 kcal/mole (Heerklotz and Epand 2001). This yields σ = 25 cal/moleÅ2 for aliphatic carbon with radii of Chothia (ASA of CH2 group is 28 Å2), consistently with 20–26 cal/moleÅ2, which is usually applied for hydrocarbons (Richards 1977). The σ of aromatic carbon was nearly the same in all considered water–solvent systems, ~19 cal/moleÅ2 (Table 5). This was expected, because transfer energies of benzene (as a representative aromatic solute) from water to hexadecane, SDS micelles, and DMPC vesicles are almost identical, −2.9, −2.8, and −2.8 kcal/mole at 300 K, respectively, judging from the corresponding partition coefficients (DeYoung and Dill 1988; Abraham et al. 1994; Hussam et al. 1995).
Solvation parameters of oxygen and nitrogen are more difficult to determine than for carbon. They cannot be derived directly from the partition coefficients of polar compounds between water and bilayers or micelles, because such compounds occupy a lipid–water interface rather than the hydrophobic interior of the bilayer (Wimley and White 1993). A possible solution here comes from studies of permeability barriers across lipid bilayers. These studies show that the membrane core is much less polar than octanol (Walter and Gutknecht 1986) and can be best approximated by nonpolar solvents with one or two double bonds, such as hexadecene or decadiene (Xiang and Anderson 1994a,b). Therefore, we used partition coefficients of 16 uncharged solutes containing mostly polar and aromatic groups, which have been determined for water/1,9-decadiene and water/hexadecene systems by Xiang and Anderson (1994a,b). The set included water, acetamide, adenine, 2′3′-dideoxyadenosine, and formic, acetic, benzoic, butyric, p-toluic, α-hydroxy-p-toluic, α-methoxy-p-toluic, α-carboxy-p-toluic, α-carbamido-p-toluic, α-naphtoic, β-naphtoic, and 9-an-throic acids. The atomic solvation parameters were determined by the least square fitting of the corresponding transfer energies, using ASA in the most extended conformers of the solutes generated using QUANTA. ASA were calculated using the subroutine SOLVA from NACCESS (Hubbard and Thornton 1993) with atomic radii of Chothia (1975), probe radius of 1.4 Å, and without hydrogens. The parameters obtained for polar atoms decrease with the number of double bonds in a solvent (zero in decane, one in hexadecene, and two in decadiene; Table 5). The final parameters were taken as for hexadecene.
The solvation parameter of sulfur was estimated using the “water–hydrocarbons” data set, which combined transfer energies from water to decane (16 polar solutes) and from water to hexadecane (18 nonpolar solutes: pentane, hexane, heptane, octane, nonane, benzene, butylbenzene, pentylbenzene, naphthalene, biphenyl, anthracene, phenantrene, pyrene, perylene, phenylmethyl sulfide, buthylthiol, diethyl disulfide, and di-n-propyl sulfide) (data from Abraham et al. 1994; Xiang and Anderson 1994a).
Acknowledgments
We thank Dr. Simon Hubbard for providing the program NACCESS. This work was supported by NIH grant GM061299 and the Upjohn Research Award from the College of Pharmacy, University of Michigan.
The publication costs of this article were defrayed in part by payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 USC section 1734 solely to indicate this fact.
Abbreviations
vdW, van der Waals interactions
ASA, accessible surface area
PDB, Protein Data Bank
SDS, sodium dodecylsulfate
DPC, N-dodecylphosphocholine
DDMAB, N-dodecyl-N,N-(dimethylammonio) butyrate
DMPC, 1,2 Dimytristoyl-sn-glycerol-3-phosphocholine
GpA, glycophorin A
MS1, QLLIAVLLLIAVNLILLIAVARLRYLVG
L7NL11, RARL7NL11GILIN
L19, RARL19GILIN
L16, KKGL7WL9KKA
Y16, KKGL7YL9KKA
L22, KKGL10WL12KKA
Y22, KKGL10YL12KK.
Article published online ahead of print. Article and publication date are at http://www.proteinscience.org/cgi/doi/10.1110/ps.04850804.
References
- Abraham, M.H., Chadha, H.S., Whiting, G.S., and Mitchell, R.C. 1994. Hydrogen bonding. 32. An analysis of water-octanol and water-alkane partitioning and the ΔlogP parameter of Seiler. J. Pharmaceut. Sci. 83 1085–1100. [DOI] [PubMed] [Google Scholar]
- Adams, P.D., Arkin, I.T., Engelman, D.M., and Brunger, A.T. 1995. Computational searching and mutagenesis suggest a structure for the pentameric transmembrane domain of phospholamban. Nat. Struct. Biol. 2 154–162. [DOI] [PubMed] [Google Scholar]
- Akey, D.L., Malashkevich, V.N., and Kim, P.S. 2001. Buried polar residues in coiled-coil interfaces. Biochemistry 40 6352–6360. [DOI] [PubMed] [Google Scholar]
- Alberty, R.A. 1983. Physical chemistry, 6th ed. Wiley, New York.
- Arkin, I.T. 2002. Structural aspects of oligomerization taking place between the transmembrane α-helices of bitopic membrane proteins. Biochim. Biophys. Acta 1565 347–363. [DOI] [PubMed] [Google Scholar]
- Ben-Naim, A. 1980. Hydrophobic interactions. Plenum Press, New York.
- Ben-Tal, N. and Honig, B. 1996. Helix–helix interactions in lipid bilayers. Biophys. J. 71 3046–3050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benz, R., Frohlich, O., Lauger, P., and Montal, M. 1975. Electrical capacity of black lipid films and of lipid bilayers made from monolayers. Biochim. Biophys. Acta. 394 323–334. [DOI] [PubMed] [Google Scholar]
- Booth, P.J., Templer, R.H., Meijberg, W., Allen, S.J., Curran, A.R., and Lorch, M. 2001. In vitro studies of membrane protein folding. Crit. Rev. Biochem. Mol. Biol. 36 501–603. [DOI] [PubMed] [Google Scholar]
- Bowie, J.U. 2001. Stabilizing membrane proteins. Curr. Op. Struct. Biol. 11 397–402. [DOI] [PubMed] [Google Scholar]
- Brooijmans, N., Sharp, K.A., and Kuntz, I.D. 2002. Stability of macromolecular complexes. Proteins 48 645–653. [DOI] [PubMed] [Google Scholar]
- Bu, Z. and Engelman, D.M. 1999. A method for determining transmembrane helix association and orientation in detergent micelles using small angle X-ray scattering. Biophys. J. 77 1064–1073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chickos, J.S., Acree, W.E., and Liebman, J.F. 1999. Estimating solid–liquid phase change enthalpies and entropies. J. Phys. Chem. Ref. Data 28 1535–1673. [Google Scholar]
- Chin, C.N., vonHeijne, G., and Gier, J.-W.L. 2002. Membrane proteins: Shaping up. Trends Biochem. Sci. 27 231–234. [DOI] [PubMed] [Google Scholar]
- Chothia, C. 1975. Structural invariants in protein folding. Nature 254 304–308. [DOI] [PubMed] [Google Scholar]
- Cohen, B.E., McAnaney, T.B., Park, E.S., Jan, Y.N., Boxer, S.G., and Jan, L.Y. 2002. Probing protein electrostatics with a synthetic fluorescent amino acid. Science 296 1700–1703. [DOI] [PubMed] [Google Scholar]
- Cristian, L., Lear, J.D., and DeGrado, W.F. 2003. Use of thioldisulfide equilibria to measure the energetics of assembly of transmembrane helices in phospholipid bilayers. Proc. Natl. Acad. Sci. 100 14772–14777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeGrado, W.F., Gratkowski, H., and Lear, J.D. 2003. How do helix–helix interactions help determine the folds of membrane proteins? Perspectives from the study of homo-oligomeric helical bundles. Protein Sci. 12 647–665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeYoung, L.R. and Dill, K.A. 1988. Solute partitioning into lipid bilayer membranes. Biochemistry 27 5281–5289. [DOI] [PubMed] [Google Scholar]
- ———. 1990. Partitioning of nonpolar solutes into bilayers and amorphous n-alkanes. J. Phys. Chem. 94 801–809. [Google Scholar]
- Dunitz, J.D. 1994. The entropic cost of bound water in crystals and biomolecules. Science 264 670. [DOI] [PubMed] [Google Scholar]
- Dwyer, J.J., Gittis, A.G., Karp, D.A., Lattman, E.E., Spencer, D.S., Stites, W.E., and Garcia-Moreno, B.E. 2000. High apparent dielectric constants in the interior of a protein reflect water penetration. Biophys. J. 79 1610–1620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Engelman, D.M., Chen, Y., Chin, C.N., Curran, A.R., Dixon, A.M., Dupuy, A.D., Lee, A.S., Lehnert, U., Matthews, E.E., Reshetnyak, Y.K., et al. 2003. Membrane protein folding: Beyond the two stage model. FEBS Lett. 555 122–125. [DOI] [PubMed] [Google Scholar]
- Eriksson, A.E., Baase, W.A., Zhang, X.J., Heinz, D.W., Blaber, M., Baldwin, E.P., and Matthews, B.W. 1992. Response of a protein structure to cavity-creating mutations and its relation to the hydrophobic effect. Science 255 178–183. [DOI] [PubMed] [Google Scholar]
- Eriksson, A.E., Baase, W.A., and Matthews, B.W. 1993. Similar hydrophobic replacements of Leu99 and Phe153 within the core of T4-lysozyme have different structural and thermodynamic consequences. J. Mol. Biol. 229 747–769. [DOI] [PubMed] [Google Scholar]
- Ermolina, I., Strinkovski, A., Lewis, A., and Feldman, Y. 2001. Observation of liquid-crystal-like ferroelectric behavior in a biological membrane. J. Phys. Chem. B 105 2673–2676. [Google Scholar]
- Ewig, C.S., Thacher, T.S., and Hagler, A.H. 1999. Derivation of class II force fields. 7. Nonbonded force field parameters for organic compounds. J. Phys. Chem. B 103 6998–7014. [Google Scholar]
- Faham, S. and Bowie, J.U. 2002. Bicelle crystallization: A new method for crystallizing membrane proteins yields a monomeric bacteriorhodopsin structure. J. Mol. Biol. 316 1–6. [DOI] [PubMed] [Google Scholar]
- Faham, S., Yang, D., Bare, E., Yohannan, S., Whitelegge, J.P., and Bowie, J.U. 2004. Side-chain contributions to membrane protein structure and stability. J. Mol. Biol. 335 297–305. [DOI] [PubMed] [Google Scholar]
- Fisher, L.E., Engelman, D.M., and Sturgis, J.N. 1999. Detergents modulate dimerization, but not helicity, of the glycophorin A transmembrane domain. J. Mol. Biol. 293 639–651. [DOI] [PubMed] [Google Scholar]
- ———. 2003. Effect of detergents on the association of the glycophorin A transmembrane helix. Biophys. J. 85 3097–3105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fleming, K.G. 2002. Standardizing the free energy change of transmembrane helix–helix interactions. J. Mol. Biol. 323 563–571. [DOI] [PubMed] [Google Scholar]
- Fleming, K.G. and Engelman, D.M. 2001a. Specificity in transmembrane helix–helix interactions can define a hierarchy of stability for sequence variants. Proc. Natl. Acad. Sci. 98 14340–14344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ———. 2001b. Computation and mutagenesis suggest a right-handed structure for the synaptobrevin transmembrane dimer. Proteins 45 313–317. [DOI] [PubMed] [Google Scholar]
- Fleming, K.G., Ackerman, A.L., and Engelman, D.M. 1997. The effect of point mutations on the free energy of transmembrane α-helix dimerization. J. Mol. Biol. 272 266–275. [DOI] [PubMed] [Google Scholar]
- Fleming, K.G., Ren, C.C., Doura, A.K., Eisley, M.E., Kobus, F.J., and Stanley, A.M. 2004. Thermodynamics of glycophorin A transmembrane helix dimerization in C14 betaine micelles. Biophys. Chem. 108 43–49. [DOI] [PubMed] [Google Scholar]
- Funahashi, J., Takano, K., and Yutani, K. 2001. Are the parameters of various stabilization factors estimated from mutant human lysozymes compatible with other proteins? Protein Eng. 14 127–134. [DOI] [PubMed] [Google Scholar]
- Gavezzotti, A. and Filippini, G. 1997. Energetic aspects of crystal packing: Experiment and computer simulations. In Theoretical aspects and computer modeling. The molecular solid state (ed. A. Gavezzotti), Vol. 1., pp. 62–97. Wiley, New York.
- Gil, T., Ipsen, J.H., Mouritsen, O.G., Sabra, M.C., Sperotto, M.M., and Zuckermann, M.J. 1998. Theoretical analysis of protein organization in lipid membranes. Biochem. Biophys. Acta 1376 245–266. [DOI] [PubMed] [Google Scholar]
- Gilson, M.K. and Honig, B.H. 1986. The dielectric constant of a folded protein. Biopolymers 25 2097–2119. [DOI] [PubMed] [Google Scholar]
- Gilson, M.K., Given, J.A., Bush, B.L., and McCammon, J.A. 1997. The statistical-thermodynamic basis for computation of binding affinities: A critical review. Biophys. J. 72 1047–1069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gonzalez, L., Brown, R.A., Richardson, D., and Alber, T. 1996a. Crystal structures of a single coiled-coil peptide in two oligomeric states reveal the basis for structural polymorphism. Nat. Struct. Biol. 3 1002–1010. [DOI] [PubMed] [Google Scholar]
- Gonzalez, L., Wolfson, D.N., and Alber, T. 1996b. Buried polar residues and structural specificity in the GCH4 leucine zipper. Nat. Struct. Biol. 3 1011–1018. [DOI] [PubMed] [Google Scholar]
- Gottschalk, K.E., Adams, P.D., Brunger, A.T., and Kessler, H. 2002. Transmembrane signal transduction of the α (IIb)β (3) integrin. Protein Sci. 11 1800–1812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gratkowski, H., Lear, J.D., and DeGrado, W.F. 2001. Polar side chains drive the association of model transmembrane peptides. Proc. Natl. Acad. Sci. 98 880–885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gratkowski, H., Dai, Q., Wand, A.J., DeGrado, W.F., and Lear, J.D. 2002. Cooperativity and specificity of association of a designed transmembrane peptide. Biophys. J. 83 1613–1619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Graziano, G., Catanzano, F., Del Vecchio, P., Giancola, C., and Barone, G. 1996. Thermodynamic stability of globular proteins: A reliable model from small molecule studies. Gazetta Chim. Italiana 126 559–567. [Google Scholar]
- Guerois, R., Nielsen, J.E., and Serrano, L. 2002. Predicting changes in the stability of proteins and protein complexes: A study of more than 1000 mutations. J. Mol. Biol. 320 369–387. [DOI] [PubMed] [Google Scholar]
- Haltia, T. and Freire, E. 1995. Forces and factors that contribute to the structural stability of membrane proteins. Biochim. Biophys. Acta 1228 1–27. [DOI] [PubMed] [Google Scholar]
- Heerklotz, H. and Epand, R.M. 2001. The enthalpy of acyl chain packing and the apparent water-accessible apolar surface area of phospholipids. Biophys. J. 80 271–279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hianik, T. and Passechnik, V.I. 1995. Bilayer lipid membranes: Structure and mechanical properties. Kluwer, Dordrecht, The Netherlands.
- Hol, W.G.J., Halie, L.M., and Sander, C. 1981. Dipoles of the α-helix and β-sheet: Their role in protein folding. Nature 294 532–536. [DOI] [PubMed] [Google Scholar]
- Holtzer, A. 1994. Does Flory–Huggins theory help in interpreting solute partitioning experiments? Biopolymers 34 315–320. [DOI] [PubMed] [Google Scholar]
- ———. 1995. The “cratic correction” and related fallacies. Biopolymers 35 595–602. [DOI] [PubMed] [Google Scholar]
- Howard, K.P., Lear, J.D., and DeGrado, W.F. 2002. Sequence determinants of the energetics of folding of a transmembrane four-helix-bundle protein. Proc. Natl. Acad. Sci. 99 8568–8572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hubbard, S.J. and Thornton, J.M. 1993. ‘NACCESS’ computer program. Department of Biochemistry and Molecular Biology, University College London, London.
- Hughes, M.P., Morgan, H., and Rison, F.J. 2002. Measuring the dielectric properties of herpes simplex virus type 1 virions with dielectrophoresis. Biochem. Biophys. Acta 1571 1–8. [DOI] [PubMed] [Google Scholar]
- Hunt, J.F., Earnest, T.N., Bousche, O., Kalghatgi, K., Reilly, K., Horvath, C., Rothschild, K.J., and Engelman, D.M. 1997. A biophysical study of integral membrane protein folding. Biochemistry 36 15156–15176. [DOI] [PubMed] [Google Scholar]
- Hussam, A., Basu, S.C., Hixon, M., and Olumee, Z. 1995. General method for the study of solute-surfactant association equilibria of volatile solutes by headspace gas chromatography. Anal. Chem. 67 1459–1464. [Google Scholar]
- Im, W., Feig, M., and Brooks, C.L. 2003. An implicit membrane generalized Born theory for the study of structure, stability, and interactions of membrane proteins. Biophys. J. 85 2900–2918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Israelachvili, J.N. 1992. Intermolecular and surface forces. Academic Press, San Diego, CA.
- Jiang, Y.X., Ruta, V., Chen, J.Y., Lee, A., and MacKinnon, R. 2003. The principle of gating charge movement in a voltage-dependent K+ channel. Nature 423 42–48. [DOI] [PubMed] [Google Scholar]
- Jorgensen, W.L., Maxwell, D.S., and Tirado-Rives, J. 1996. Development and testing of the OPLS all-atom force field on conformational energetics and properties of organic liquids. J. Am. Chem. Soc. 118 11225–11236. [Google Scholar]
- Kim, S., Chamberlain, A.K., and Bowie, J.U. 2003. A simple method for modeling transmembrane helix oligomers. J. Mol. Biol. 329 831–840. [DOI] [PubMed] [Google Scholar]
- Klotz, I.M. 1997. Ligand-receptor energetics. A guide for the perplexed. Wiley, New York.
- Krebs, M.P. and Isenbarger, T.A. 2000. Structural determinants of purple membrane assembly. Biochem. Biophys. Acta 1460 15–26. [DOI] [PubMed] [Google Scholar]
- Kuhlman, B. and Baker, D. 2000. Native protein sequences are close to optimal for their structures. Proc. Natl. Acad. Sci. 97 10383–10388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lazaridis, T. 2003. Effective energy function for proteins in lipid membranes. Proteins 52 176–192. [DOI] [PubMed] [Google Scholar]
- Lazaridis, T., Archontis, G., and Karplus, M. 1995. Enthalpic contribution to protein stability: Insights from atom-based calculations and statistical mechanics. Adv. Prot. Chem. 47 231–307. [DOI] [PubMed] [Google Scholar]
- Lear, J.D., Gratkowski, H., and DeGrado, W.F. 2001. Membrane active peptides. Biochem. Soc. Trans. 29 559–564. [DOI] [PubMed] [Google Scholar]
- Lear, J.D., Gratkowski, H., Adamian, L., Liang, J., and DeGrado, W.F. 2003. Position-dependence of stabilizing polar interactions of asparagines in trans-membrane helical bundles. Biochemistry 42 6400–6407. [DOI] [PubMed] [Google Scholar]
- Leckband, D. and Israelachvili, J. 2001. Intermolecular forces in biology. Quart. Rev. Biophys. 34 105–267. [DOI] [PubMed] [Google Scholar]
- Lee, A.G. 2003. Lipid–protein interactions in biological membranes: A structural perspective. Biochim. Biophys. Acta 1612 1–40. [DOI] [PubMed] [Google Scholar]
- Lew, S., Caputo, G.A., and London, E. 2003. The effect of interactions involving ionizable residues flanking membrane-inserted hydrophobic helices upon helix–helix interaction. Biochemistry 42 10833–10842. [DOI] [PubMed] [Google Scholar]
- Lide, D.R., ed. 2003. CRC handbook of chemistry and physics. CRC Press, Boca Raton, FL.
- Lomize, A.L., Pervushin, K.V., and Arseniev, A.S. 1992. Spatial structure of (34–65)bacterioopsin polypeptide in SDS micelles determined from nuclear magnetic resonance data. J. Biomol. NMR 2 361–372. [DOI] [PubMed] [Google Scholar]
- Lomize, A.L., Reibarkh, M.Y, and Pogozheva, I.D. 2002. Interatomic potentials and solvation parameters from protein engineering data for buried residues. Protein Sci. 11 1984–2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacKenzie, K.R. and Engelman, D.M. 1998. Structure-based prediction of the stability of transmembrane helix–helix interactions: The sequence dependence of glycophorin A dimerization. Proc. Natl. Acad. Sci. 95 3583–3590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacKenzie, K.R., Prestgard, J.H., and Engelman, D.M. 1997. A transmembrane helix dimer: Structure and implications. Science 276 131–135. [DOI] [PubMed] [Google Scholar]
- Maire, M., Champeil, P., and Moller, J.V. 2000. Interactions of membrane proteins and lipids with solubilizing detergents. Biochim. Biophys. Acta 1508 86–111. [DOI] [PubMed] [Google Scholar]
- Mall, S., Broadbridge, R., Sharma, R.P., East, J.M., and Lee, A.G. 2001. Self-association of model transmembrane α-helices is modulated by lipid structure. Biochemistry 40 12379–12386. [DOI] [PubMed] [Google Scholar]
- Marsh, D. and Horvath, L.I. 1998. Structure, dynamics and composition of the lipid–protein interface. Perspectives from spin-labeling. Biochim. Biophys. Acta 1376 267–296. [DOI] [PubMed] [Google Scholar]
- Marti, T. 1998. Refolding of bacteriorhodopsin from expressed polypeptide fragments. J. Biol. Chem. 273 9312–9322. [DOI] [PubMed] [Google Scholar]
- Mertz, E.L. and Krishtalik, L.I. 2000. Low dielectric response in enzyme active site. Proc. Natl. Acad. Sci. 97 2081–2086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Momany, F.A., Carruthers, L.M., McGuire, R.F., and Scheraga, H.A. 1974. Intermolecular potentials from crystal data. III. Determination of empirical potentials and application to the packing configurations and lattice energies in crystals of hydrocarbons, carboxylic acids, amines, and amides. J. Phys. Chem. 78 1595–1620. [Google Scholar]
- Morton, A. and Matthews, B.W. 1995. Energetic origins of ligand binding in an interior nonpolar cavity of T4 lysozyme. Biochemistry 34 8564–8575. [DOI] [PubMed] [Google Scholar]
- Morton, A., Baase, W.A., and Matthews, B.W. 1995. Specificity of ligand binding in a buried nonpolar cavity of T4 lysozyme: Linkage of dynamics and structural plasticity. Biochemistry 34 8576–8588. [DOI] [PubMed] [Google Scholar]
- Murphy, K.P. and Gill, S.J. 1991. Solid model compounds and the thermodynamics of protein unfolding. J. Mol. Biol. 222 699–709. [DOI] [PubMed] [Google Scholar]
- Murphy, K.P., Xie, D. Thompson, K.S., Amzel, L.M., and Freire, E. 1994. Entropy in biological binding processes: Estimation of translational entropy loss. Proteins 18 63–67. [DOI] [PubMed] [Google Scholar]
- Myers, J.K. and Pace, C.N. 1996. Hydrogen bonding stabilizes globular proteins. Biophys. J. 71 2033–2039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishimura, K., Kim, S.G., Zhang, L., and Cross, T.A. 2002. The closed state of a H+ channel helical bundle combining precise orientational and distance restraints from solid state NMR-1. Biochemistry 41 13170–13177. [DOI] [PubMed] [Google Scholar]
- Orekhov, V.Y., Pervushin, K.V., Korzhnev, D.M., and Arseniev, A.S. 1995. Backbone dynamics of (1–71)bacterioopsin and (1–31)bacterioopsin studied by two-dimensional 1H-15N NMR spectroscopy. J. Biomol. NMR 6 113–122. [DOI] [PubMed] [Google Scholar]
- Pappu, R.V., Marshall, G.R., and Ponder, J.W. 1999. A potential smoothing algorithm accurately predicts transmembrane helix packing. Nat. Struct. Biol. 6 50–55. [DOI] [PubMed] [Google Scholar]
- Perozo, E. and Rees, D.C. 2003. Structure and mechanism in prokaryotic mechanosensitive channels. Curr. Opin. Struct. Biol. 13 432–442. [DOI] [PubMed] [Google Scholar]
- Pervushin, K.V. and Arseniev, A.S. 1992. Three-dimensional structure of (1–36)bacterioopsin in methanol chloroform mixture and SDS micelles determined by 2D 1H NMR. FEBS Lett. 308 190–196. [DOI] [PubMed] [Google Scholar]
- Pervushin, K.V., Orekhov, V.Yu., Popov, A.I., Musina, L.Yu., and Arseniev, A.S. 1994. Three-dimensional strucutre of (1–71)bacterioopsin solubilized in methanol/chloroform and SDS micelles determined by 15N-1H hetero-nuclear NMR spectroscopy. Eur. J. Biochem. 219 571–583. [DOI] [PubMed] [Google Scholar]
- Pickett, S.D. and Sternberg, M.J. 1993. Empirical scale of side-chain conformational entropy in protein folding. J. Mol. Biol. 231 825–839. [DOI] [PubMed] [Google Scholar]
- Polevaya, Y., Ermolina, I., Schlesinger, M., Ginzburg, B.Z., and Feldman, Y. 1999. Time domain dielectric spectroscopy study of human cells. II. Normal and malignant white blood cells. Biochim. Biophys. Acta 1419 257–271. [DOI] [PubMed] [Google Scholar]
- Popot, J.L. and Engelman, D.M. 2000. Helical membrane protein folding, stability, and evolution. Annu. Rev. Biochem. 69 881–922. [DOI] [PubMed] [Google Scholar]
- Radu, M., Petcu, I., Sommer, A., and Avram, D. 1996. Changes in membrane electrical parameters of yeast following chemical treatment for protoplast isolation. Biochem. Bioenerg. 40 159–166. [Google Scholar]
- Radzicka, A. and Wolfenden, R. 1988. Comparing the polarities of the amino acids: Side-chain distribution coefficients between the vapor phase, cyclohexane, 1-octanol, and neutral aqueous solution. Biochemistry 27 1664–1670. [Google Scholar]
- Ramesh, C.V., Jayakumar, R., and Puvanakrishnan, R. 1997. Physicochemical characterization of a novel surfactant peptide containing an arginine cation and laurate anion. Colloid Polym. Sci. 275 1162–1168. [Google Scholar]
- Richards, F.M. 1977. Areas, volumes, packing, and protein structure. Annu. Rev. Biophys. Bioeng. 6 151–176. [DOI] [PubMed] [Google Scholar]
- Robertson, A.D. and Murphy, K.P. 1997. Protein structure and energetics of protein stability. Chem. Rev. 97 1251–1267. [DOI] [PubMed] [Google Scholar]
- Russ, W.P. and Engelman, D.M. 1999. TOXCAT: A measure of transmembrane helix association in a biological membrane. Proc. Natl. Acad. Sci. 96 863–868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seelig, J. 1997. Titration calorimetry of lipid–peptide interactions. Biochim. Biophys. Acta 1331 103–116. [DOI] [PubMed] [Google Scholar]
- Shakhnovich, E.I. and Finkelstein, A.V. 1989. Theory of cooperative transitions in protein molecules. I. Why denaturation of globular proteins is a first-order phase transition. Biopolymers 28 1667–1680. [DOI] [PubMed] [Google Scholar]
- Sham, Y.Y., Muegge, I., and Warshel, A. 1998. The effect of protein relaxation on charge–charge interactions and dielectric constants of proteins. Biophys. J. 74 1744–1753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sternberg, M.J.E. and Chickos, J.S. 1994. Protein side-chain conformational entropy derived from fusion data—comparison with other empirical scales. Protein Eng. 7 149–155. [DOI] [PubMed] [Google Scholar]
- Tamura, A. and Privalov, P.L. 1997. The entropy cost of protein association. J. Mol. Biol. 273 1048–1060. [DOI] [PubMed] [Google Scholar]
- Torres, J., Kukol, A. and Arkin, I.T. 2001. Mapping the energy surface of transmembrane helix–helix interactions. Biophys. J. 81 2681–2692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tredgold, R.H. and Hole, P.N. 1976. Dielectric behavior of dry synthetic polypeptides. Biochim. Biophys. Acta 443 137–142. [DOI] [PubMed] [Google Scholar]
- Tuffery, P., Etchebest, C., Popot, J.L., and Lavery, R. 1994. Prediction of the positioning of the seven transmembrane α-helices of bacteriorhodopsin. A molecular simulation study. J. Mol. Biol. 236 1105–1122. [DOI] [PubMed] [Google Scholar]
- Walter, A. and Gutknecht, J. 1986. Permeability of small nonelectrolytes through lipid bilayer membranes. Membr. Biol. 90 207–217. [DOI] [PubMed] [Google Scholar]
- Wang, J.F., Kim, S., Kovacs, F., and Cross, T.A. 2001. Structure of the transmembrane region of the M2 protein H+ channel. Protein Sci. 10 2241–2250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wannier, G.H. 1966. Statistical physics. Wiley, New York.
- Weber, P.C. and Salemme, F.R. 2003. Application of calorimetric methods to drug discovery and the study of protein interactions. Curr. Opin. Struct. Biol. 13 115–121. [DOI] [PubMed] [Google Scholar]
- White, S.H. and Wimley, W.C. 1999. Membrane protein folding and stability: Physical principles. Annu. Rev. Biophys. Biomol. Struct. 28 319–365. [DOI] [PubMed] [Google Scholar]
- Wimley, W.C. and White, S.H. 1993. Membrane partitioning: Distinguishing bilayer effects from the hydrophobic effect. Biochemistry 32 6307–6312. [DOI] [PubMed] [Google Scholar]
- Xiang, T.X. and Anderson, B.D. 1994a. The relationship between permeant size and permeability in lipid bilayer membranes. J. Membr. Biol. 140 111–122. [DOI] [PubMed] [Google Scholar]
- ———. 1994b. Substituent contributions to the transport of substituted p-toluic acids across lipid bilayer membranes. J. Pharm. Sci. 83 1511–1518. [DOI] [PubMed] [Google Scholar]
- Yano, Y., Takemoto, T., Kobayashi, S., Yasui, H., Sakurai, H., Ohashi, W., Niwa, M., Futaki, S., Sugiura, Y., and Matsuzaki, K. 2002. Topological stability and self-association of a completely hydrophobic model transmembrane helix in lipid bilayers. Biochemistry 41 3073–3080. [DOI] [PubMed] [Google Scholar]
- Yohannan, S., Faham, S., Yang, D., Grosfeld, D., Chamberlain, A.K., and Bowie, J.U. 2004. Cα-H. . .O hydrogen bond in a membrane protein is not stabilizing. J. Amer. Chem. Soc. 126 2284–2285. [DOI] [PubMed] [Google Scholar]
- Yu, B.Y., Privalov, P.L, and Hodges, R.S. 2001. Contribution of translational and rotational motions to molecular association in aqueous solution. Biophys. J. 81 1632–1642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou, Y., Vitkup, D., and Karplus, M. 1999. Native proteins are surface-molten solids: Application of the Lindermann criterion for the solid versus liquid state. J. Mol. Biol. 285 1371–1375. [DOI] [PubMed] [Google Scholar]
- Zhou, F.X., Merianos, H.J., Brunger, A.T., and Engelman, D.M. 2001. Polar residues drive association of polyleucine transmembrane helices. Proc. Natl. Acad. Sci. 98 2250–2255. [DOI] [PMC free article] [PubMed] [Google Scholar]