

Abstract
Topology has been shown to be an important determinant of many features of protein folding; however, the delineation of sequence effects on folding remains obscure. Furthermore, differentiation between the two influences proves difficult due to their intimate relationship. To investigate the effect of sequence in the absence of significant topological differences, we examined the folding mechanisms of segment B1 peptostreptococcal protein L and segment B1 of streptococcal protein G. These proteins share the same highly symmetrical topology. Despite this symmetry, neither protein folds through a symmetrical transition state. We analyzed the origins of this difference using theoretical models. We found that the strength of the interactions present in the N-terminal hairpin of protein L causes this hairpin to form ahead of the C-terminal hairpin. The difference in chain entropy associated with the formation of the hairpins of protein G proves sufficient to beget initiation of folding at the shorter C-terminal hairpin. Our findings suggest that the mechanism of folding may be understood by examination of the free energy associated with the formation of partially folded microstates.
Keywords: Protein folding, symmetry breaking, protein L, protein G, folding pathway, transition state
Most proteins perform their biological function in a compact, native state that is reached through folding from a wide array of unfolded states. Despite considerable progress made in the past several decades through experimental and theoretical approaches (Dobson and Karplus 1999), much remains to be learned about the folding process. A complete knowledge of the mechanism by which this process takes place will have broad reaching implications, ranging from understanding the formation and propagation of prion and amyloid diseases (Kelly 1998) to protein design and the prediction of structure from sequence (Jackson 1998).
In recent years, topology has been shown to be a key determinant of many attributes in folding. Simplified protein models which capture little but the topology of the native protein are in some cases able to correctly reproduce many important features of folding (Alm and Baker 1999; Galzitskaya and Finkelstein 1999; Munoz and Eaton 1999; Shea et al. 1999). Furthermore, folding rates of two-state proteins have been shown to correlate very well with contact order, a quantity linked to topology (Plaxco et al. 1998). This correlation has been used to argue that topology may also be a main determinant of transition state structure (Plaxco et al. 2000), and hence the mechanism of protein folding. While this is not true for all proteins, it has been shown to hold in a comparison of two SH3 domains (Martinez et al. 1998; Riddle et al. 1999) and a comparison of acylphosphatase with human procarboxypeptidase A2 activation domain (Villegas et al. 1998; Chiti et al. 1999).
One of the next challenges facing the protein folding community is to push our understanding of protein folding beyond topological effects. While topology can explain many of the gross features of folding, there remain many details which are modulated by energetic factors, and which therefore depend on the protein sequence (Koga and Takada 2001). The importance of sequence effects has also been recognized by other investigators, leading to the incorporation of sequence effects into a simple one-dimensional model known as FOLD-X (Guerois and Serrano 2000). Examples of the importance of energetic factors may be seen in the folding of the B1 segment of the IgG binding domain of peptostreptococcal protein L and the B1 segment of the IgG binding domain of streptococcal protein G.
Fragment B1 of protein L is a 62-residue protein which has been shown to fold via two-state kinetics (Scalley et al. 1997). The structure of protein L is comprised of a four-stranded β-sheet platform made up of two antiparallel β-hairpins connected by an α-helix (Fig. 1a ▶; Wikstrom et al. 1993). The two hairpins are of similar length. Despite the obvious topological symmetry of this protein, the mechanism of folding does not obey the same symmetry. Analysis of the effect of mutations on folding kinetics shows that the N-terminal hairpin (residues 17–38) is predominantly formed in the rate-limiting step of folding, whereas the C-terminal hairpin (residues 58–78) is not formed (Gu et al. 1997; Kim et al. 2000).
Fig. 1.

A comparison of the structures of (a) protein L and (b) protein G. The PDB accession codes are 2PTL and 1PGB, respectively. The figures were generated using MOLSCRIPT (Kraulis 1991).
The B1 segment of the IgG binding domain of streptococcal protein G has a topology which is analogous to that of protein L (Fig. 1b ▶; Gronenborn et al. 1991), and the same symmetry is evident. This fragment is only 56 residues long, and although the two hairpins are again of similar length, both are shorter than the corresponding hairpins in protein L. Like protein L, the folding of protein G under fixed conditions may be coarsely described as a two-state reaction (Alexander et al. 1992). Unlike protein L, however, the C-terminal hairpin of protein G (residues 42–55) has been shown to fold ahead of the N-terminal hairpin (residues 1–20) (Kuszewski et al. 1994; Frank et al. 1995; Sheinerman and Brooks 1998a; Sari et al. 2000). Peptide studies (Blanco and Serrano 1995) also indicate that the unfolded state of protein G in the absence of denaturant shows a considerable degree of native-like residual structure in the C-terminal hairpin: The formation of this residual structure upon dilution of denaturant may explain the observation of an additional fast phase in ultrarapid mixing experiments (Park et al. 1999).
To explore the origin of the symmetry breaking in each protein, we make use of simplified model proteins based on the sequences and native-state structures of both proteins. It has been observed that potential functions containing terms which preferentially stabilize interactions present in the native state lead to model proteins which qualitatively reproduce the folding of small proteins significantly better than their more generic counterparts (Chan and Dill 1998; Nymeyer et al. 1998; Koga and Takada 2001). Such potentials, known as Go models, were first used in discretized simulations on lattices (Taketomi et al. 1975), but have since been extended to minimalist off lattice models (Shea et al. 1999; Zhou and Karplus 1999; Dokholyan et al. 2000). Using these models as a starting point, additional features have been added which permit the balance of forces in the present model to be better matched to those occurring in proteins. We have developed these models using an algorithmic approach that yields protein-specific potentials from a procedure that is not otherwise tuned for each particular target structure and sequence. Upon completion of the study described here, this procedure has subsequently been used without modification in the characterization of folding mechanisms of a protein "test set" consisting of a large number of experimentally characterized proteins, showing excellent overall agreement (J. Karanicolas and C. Brooks, in prep.).
Results and Discussion
Two-state thermodynamics
As noted above, both protein L and protein G are known to fold via apparent two-state kinetics. An important first step in the thermodynamic characterization of these model proteins is the demonstration that they as well fold via two-state mechanisms.
The heat capacity plotted as function of temperature (Fig. 2a ▶) reveals a single sharp peak, which is consistent with a single cooperative first-order-like transition. The temperature at which the peak occurs reflects the transition temperature. The temperature dependence of the average fraction of native contacts (interactions) formed (Fig. 2b ▶) shows that both proteins arrive at conformations with a large amount of native structure from conformations with a small amount of native structure over a small temperature range (∼30°K). In both cases the transition is sharp, which is indicative of the high degree of cooperativity with which folding occurs.
Fig. 2.
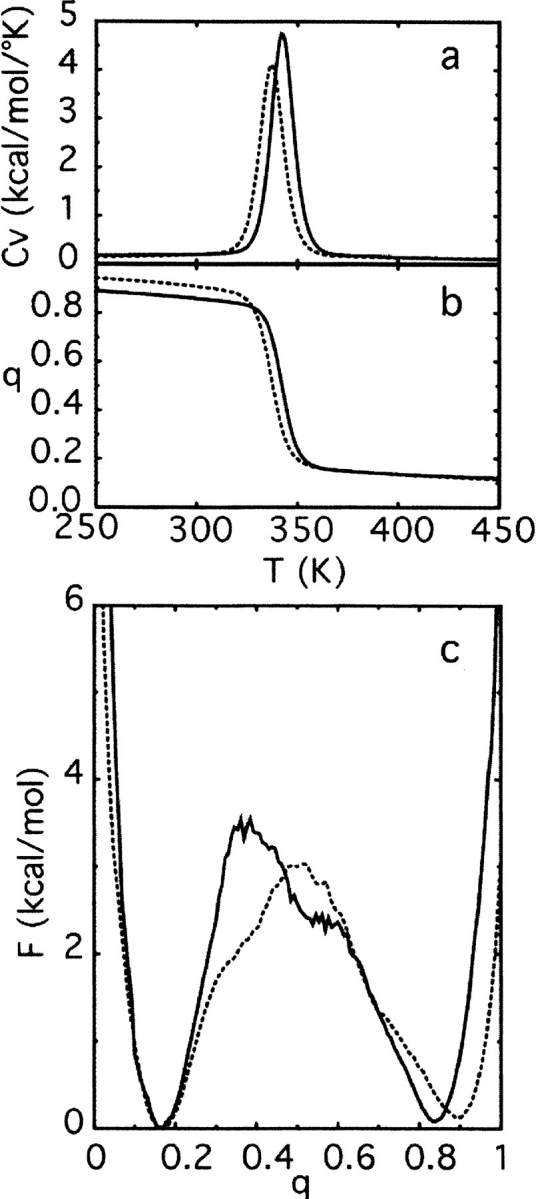
Thermodynamic functions used in the initial characterization of protein L (solid line) and protein G (dotted line) folding. (a) The temperature dependence of the heat capacity, Cv. (b) The temperature dependence of the fraction of native contacts formed, q. (c) The free energy, F, as a function of q at the transition temperature.
The free energy as a function of the fraction of native contacts present is shown at the transition temperature determined from the heat capacity curve (Fig. 2c ▶). At this temperature two equally populated free energy minima are clearly present, which indicates that the transition temperature coincides with the folding temperature. The presence of a free energy barrier separating the two states (each with a height of about 3 kcal/mol, corresponding to more than 4 kBTf) suggests a two-state folding mechanism. Furthermore, the clear separation of the minima along with the presence of the barrier shows that this progress variable is a suitable coordinate for studying the transition between the folded and unfolded states.
These proteins exhibit behavior consistent with a two-state folding mechanism, suggesting that the balance of forces present in the models constructed by the procedure we outline in the Materials and Methods section of this paper provides a reasonable description for the overall folding of these proteins.
Mechanism of folding
To determine the order of events in folding, it is useful to directly examine the dependence of the free energy at the transition temperature on several structural properties. As described in the previous section, the fraction of native contacts formed clearly differentiates between the folded and unfolded states.
The free energy at the transition temperature was computed as a function of the radius of gyration and the fraction of native contacts formed. This surface for both proteins showed excellent agreement with the corresponding free-energy profile constructed from all-atom calculations of protein G (results not shown; Sheinerman and Brooks 1998a), suggesting further that our model captures the key features that determine the folding free energy landscape.
To gain further mechanistic insights into how these proteins fold, free energy surfaces can be computed along a range of different folding progress coordinates (Shea and Brooks 2001; Brooks 2002). Figure 3 ▶ shows the free energy at the transition temperature as a function of the fraction of total native contacts formed and the fraction of native contacts formed in a particular secondary structural element. Examining first protein L, the free energy profile of the N-terminal hairpin (Fig. 3a ▶) shows a "diagonal" profile in these coordinates. Conformations in which this hairpin is formed generally have a high fraction of total native contacts, whereas conformations in which this hairpin is not formed generally have a low fraction of total native contacts. In other words, the degree to which the N-terminal hairpin is folded is a suitable reaction coordinate in describing the folding of the protein as a whole. In contrast, the profile of the C-terminal hairpin (Fig. 3c ▶) shows this hairpin to be formed late in folding. This is inferred from the fact that the fraction of native contacts formed in the C-terminal hairpin is generally low even in conformations for which the fraction of total native contacts is relatively high, suggesting that a considerable population of conformations exist in which the C-terminal hairpin is not formed yet the remainder of the protein is quite native-like. The free energy profile in these coordinates therefore has a backward "L-shape" and occupies the lower right portion of the plot. It is of course impossible for the lower right corner of such an L-shaped plot to be occupied using these progress variables, since the fraction of total native contacts formed cannot be unity unless the fraction of C-terminal native contacts formed is also unity. Finally, the corresponding profile for the helical native contacts shows the helix to be predominantly formed in both the unfolded state and the transition state at this temperature (Fig. 3e ▶). For this reason it is difficult to estimate the degree of formation of the helix in the transition state relative to the unfolded state.
Fig. 3.
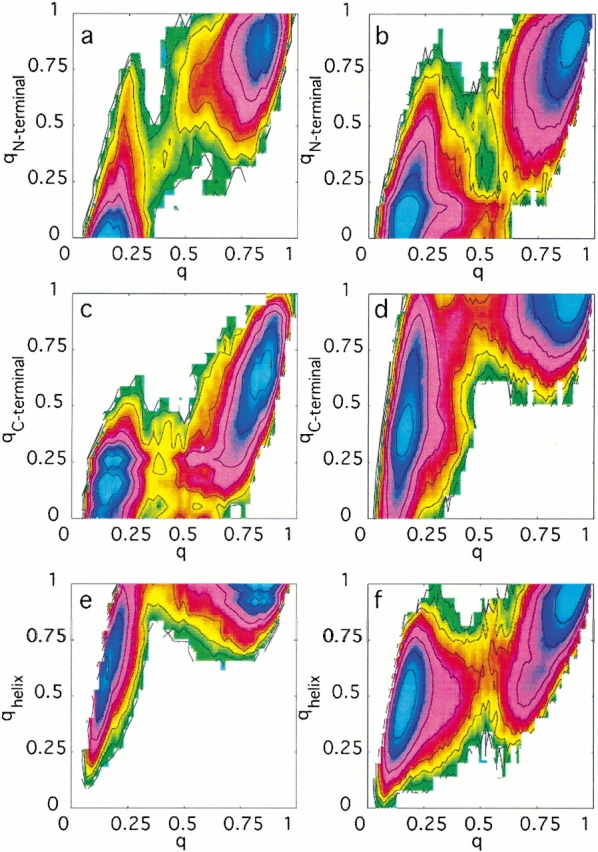
The free energy at the transition temperature as a function of several progress variables for protein L (a,c,e) and protein G (b,d,f). (a,b) The free energy as a function of the fraction of native contacts formed in the N-terminal hairpin (qN-terminal) and the fraction of total native contacts formed (q). (c,d) The free energy as a function of the fraction of native contacts formed in the C-terminal hairpin (qC-terminal) and the fraction of total native contacts formed (q). (e,f) The free energy as a function of the fraction of helical native contacts formed (qhelix) and the fraction of total native contacts formed (q). The free energy difference between adjacent contour lines corresponds to kBTf.
The observed folding mechanism for protein G has both differences and similarities compared to that of protein L. The free energy profile of the protein G N-terminal hairpin (Fig. 3b ▶) is more L-shaped (towards the lower right corner of the plot) than the corresponding profile in protein L. The C-terminal hairpin (Fig. 3d ▶) meanwhile shows a highly L-shaped profile that occupies the upper left corner of the plot, representing a population of conformations in which the C-terminal hairpin is formed yet the remainder of the protein is predominantly not native-like. Finally, the helix in protein G is partially formed in the unfolded state, and shows a similar degree of formation in the transition state (Fig. 3f ▶).
The observed differences between the hairpins in proteins L and G are further highlighted by examination of the free energy as a function of the fraction of N-terminal hairpin native contacts formed and the fraction of C-terminal hairpin native contacts formed (Fig. 4 ▶). This combination of progress coordinates emphasizes the "competition" for symmetry-broken conformational states early in folding. The free energy surface of protein L comprises regions of low free energy in which the N-terminal hairpin is formed yet the C-terminal hairpin is not formed, and has a very high free energy in regions in which the C-terminal hairpin is formed but the N-terminal hairpin is not formed (Fig. 4a ▶). Formation of the C-terminal hairpin therefore requires the presence of the N-terminal hairpin in a native-like conformation. The converse is observed for protein G (Fig. 4b ▶).
Fig. 4.

The free energy at the transition temperature as a function of the fraction of C-terminal hairpin native contacts formed (qC-terminal) and the fraction of N-terminal hairpin native contacts formed (qN-terminal), for (a) protein L and (b) protein G. The free energy difference between adjacent contour lines corresponds to kBTf.
The observed mechanisms of folding show remarkable agreement with other studies. The order of formation of the hairpins concurs with results of other studies on both protein L (Gu et al. 1997; Scalley et al. 1999; Kim et al. 2000) and protein G (Kuszewski et al. 1994; Frank et al. 1995; Sheinerman and Brooks 1998b; McCallister et al. 2000; Sari et al. 2000). Furthermore, a considerable degree of native-like residual structure in the C-terminal hairpin is found in the unfolded state of protein G at the transition temperature (Fig. 3d ▶). This observation is also consistent with previous observations (Blanco and Serrano 1995; Sheinerman and Brooks 1998b; Park et al. 1999).
Studies on protein G conclude that the helix is predominantly formed at an early stage in folding (Kuszewski et al. 1994; Blanco and Serrano 1995; Frank et al. 1995; Sheinerman and Brooks 1998b; Sari et al. 2000), whereas measurement of φ values indicate that the degree of formation of the helix in the transition state (McCallister et al. 2000) resembles the unfolded state more than the folded state. The results obtained in the present study are consistent with both observations.
Like the helix in protein G, the helix in protein L has been shown to exhibit partial formation in the unfolded state (Ramirez-Alvarado et al. 1997; Scalley et al. 1997, 1999; Yi et al. 1997, 2000) and low φ values (Kim et al. 1998, 2000). Although the observed results are in qualitative agreement regarding the presence of residual helical structure in the unfolded state, a comparison with the degree of helical structure in the transition state is difficult due to the small change observed between the unfolded state and the folded state. Nevertheless, it appears that the formation of the helix in the transition state may be slightly overestimated relative to the degree of formation of β-sheet structure. The free energy of formation of a single hydrogen bond in the context of a β-sheet has been shown to be considerably more favorable than the formation of a single hydrogen bond in the context of an α-helix (Tobias et al. 1991). For this reason, it is possible that a model such as this in which the interaction energy is uniform for hydrogen bonds across different secondary structural elements leads to overestimates of the degree of helical structure relative to the degree of β-sheet structure in the transition state.
Origins of the symmetry breaking in hairpin formation
Since the model protein accurately reproduces the basic folding mechanism for both proteins, we may explore the energetic contributions to the folding of both hairpins to delineate the origin of the symmetry breaking in the mechanism of folding. Functional constraints may lead to one of several scenarios, which could in principle explain the symmetry breaking. The energetics of formation of one hairpin may dominate over the formation of the other hairpin, due to a large difference in the intrinsic propensities of the sequences towards β-strand formation. This would be reflected in the dihedral component of the potential functions used in our study. Alternatively, differences in the combination of hydrogen bonding and side-chain contacts within each hairpin may lead to the observed differences, which would be reflected in the nonbonded self-energy terms of these potentials. A third possibility may be that the helix forms stronger interactions with one of the hairpins, which would be reflected in the helix-hairpin interaction energy. To distinguish between these scenarios, we have computed the change upon folding of each of these energetic contributions at the observed folding temperature. The folded and unfolded states were defined in the fraction of native contacts formed, based on the fraction of native contacts formed at the highest point of the barrier separating the two states (Fig. 2c ▶). The mean value of each energetic component in each state at the folding temperature was then computed from the density of states using the weighted histogram method (WHAM) (see Materials and Methods section). The difference between each of these values in the folded state and the unfolded state gives the energetic contribution to folding from each component. The results are presented in Table 1.
Table 1.
A breakdown of the energetic contributions to the folding of the protein L and protein G models
| Component energy differences | Protein L | Protein G |
| Dihedral energy of the N-terminal hairpin | −0.8 | −0.5 |
| Nonbonded self-energy of the N-terminal hairpin | −11.9 | −10.3 |
| N-terminal hairpin - helix interaction energy | −12.0 | −8.1 |
| Dihedral energy of the C-terminal hairpin | 0.2 | −0.8 |
| Nonbonded self-energy of the C-terminal hairpin | −8.3 | −7.9 |
| C-terminal hairpin - helix interaction energy | −8.5 | −5.0 |
| N-terminal hairpin - C-terminal hairpin interaction energy | −14.6 | −14.7 |
All values are in units of kcal/mol, and are taken at the transition temperature.
In the folding of protein L, all three of the energetic contributions described above favor formation of the N-terminal hairpin. Although no single energetic contribution is the cause of the early formation of this hairpin, a large difference between the two hairpins occurs in the nonbonded self-energy terms. This term combines the energetic contribution of both hydrogen bond- and side chain-based native contacts. There are, interestingly, eight hydrogen bond native contacts in each of the hairpins. This suggests that a large contribution to the symmetry breaking of folding in protein L is the difference in the side-chain contacts within each hairpin, which reflects the sequence difference between the two hairpins. The other large contribution is in the hairpin-helix interaction energy, which also reflects the sequence differences between the two hairpins.
Examination of the energetic contributions in protein G shows a very different situation. While the dihedral energy slightly favors the C-terminal hairpin, which folds first, this is more than compensated for by the preferential stabilization of the N-terminal hairpin by nonbonded self-energy term and the interaction with the helix (Table 1). In fact, the difference in the total of these contributions shows that formation of the N-terminal hairpin is energetically much more favorable than formation of the C-terminal hairpin (Table 2). To understand why the C-terminal hairpin forms ahead of the N-terminal hairpin, the entropy difference in forming each hairpin must be considered. Using the difference in the average total energy between the folded state and the unfolded state at the transition temperature, one may estimate the entropy cost per residue associated with folding at the transition temperature. Using the difference in the length of the hairpins, the difference in entropy associated with the formation of each hairpin was estimated (Table 2).
Table 2.
Thermodynamics leading to the observed symmetry breaking in the mechanism of folding
| Protein L | Protein G | |
| ΔEfold (N-terminal hairpin) | −39.3 kcal/mol | −33.6 kcal/mol |
| ΔEfold (C-terminal hairpin) | −31.2 kcal/mol | −28.4 kcal/mol |
| ΔΔEfold(N-term − C-term) | −8.1 kcal/mol | −5.2 kcal/mol |
| ΔEfold (total) | −64.7 kcal/mol | −58.8 kcal/mol |
| ΔSfold (per residue) | −0.003 kcal/mol•K | −0.003 kcal/mol•K |
| Length of N-terminal hairpin | 21 residues | 20 residues |
| Length of C-terminal hairpin | 21 residues | 14 residues |
| ΔΔSfold (N-term − C-term) | 0 kcal/mol•K | −0.019 kcal/mol•K |
| -TfΔΔSfold(N-term − C-term) | 0 kcal/mol | 6.3 kcal/mol |
| ΔΔGfold(N-term − C-term) | −8.1 kcal/mol | 1.1 kcal/mol |
The inclusion of entropy effects accounts fully for the observed symmetry breaking in the folding of both protein L and protein G (Table 2). Because the N-terminal hairpin in protein L is of the same length as the C-terminal hairpin, the additional favorable energetic interactions causes the N-terminal hairpin to form first in this system. The entropic contribution to hairpin stability arising from the difference in the lengths of the hairpins in protein G outweighs the energetic contributions, such that the C-terminal hairpin forms first. This is fully consistent with the hypothesis that the hairpin which forms first in these proteins is that which has the lower free energy of formation (McCallister et al. 2000).
Comparison to other simple models
Support for the claim that the incorporation of sequence effects is indeed the basis for the correct differentiation between the folding mechanism of these two proteins can be taken from a recent study aimed at determining the level of accuracy in folding mechanisms which may be expected from a "topology-based" model (Koga and Takada 2001). This model, which differed from the one presented here primarily in its failure to represent sequence effects, predicted an identical folding mechanism for protein L and protein G (Koga and Takada 2001).
To begin to assess the differences between the model presented here and more simple models which also make use of sequence effects, we compare these results to those obtained using the FOLD-X algorithm (Guerois and Serrano 2000). This procedure defines a "configuration" as a series of residues which are either "ordered" or "disordered." Connected paths leading to the native state are enumerated in configuration space, and the free energy of each configuration in the path is evaluated using estimates of entropy. Models such as this may also be classified as Go models, as only interactions present in the native state contribute to the energy of the system. The differences in behavior between one-dimensional (contact order-based) models such as FOLD-X and (topology-based) models which explicitly represent the protein chain in space are examined in detail elsewhere (J. Karanicolas and C. Brooks, in prep.). The FOLD-X algorithm is an improvement over earlier one-dimensional models, in that sequence effects are taken into account.
As shown in Figure 5 ▶, the FOLD-X algorithm predicts initiation of folding at the C-terminal hairpin for both protein L (2ptl) and protein G (1pgb). To verify the robustness of these results, alternate starting structures were used in the characterization for each of these proteins: 1HZ6 for protein L, 1igd for protein G. Regardless of the starting structure, FOLD-X predicts a mechanism of folding involving initiation at the C-terminus for both protein L and protein G or is ambiguous. In contrast, the model building procedure described in the Materials and Methods section resulted in initiation of folding at the N-terminal hairpin for all structures of protein L and initiation of folding at the C-terminal hairpin for all structures of protein G (results not shown), although some ambiguity was present in the results from 1HZ6, due to an exaggerated kink in the C-terminal hairpin of this structure.
Fig. 5.

The φ-values predicted by the FOLD-X algorithm. The C-terminal hairpin is found to fold ahead of the N-terminal hairpin for structures of both protein L (2ptl, 1HZ6) and protein G (1pgb), but the results are ambiguous for the protein G structure 1igd. The φ values were scaled by 100.
Finally, we note that the correct delineation of folding mechanisms of these two proteins has been reproduced in a one-dimensional model by the addition of a specialized potential which favors the hairpin targeted to fold first (Grantcharova et al. 2001). This has been used as further support that elements of structure with the most favorable free energy of formation are formed early in folding.
Conclusions
The set of potentials described here, which employ a barrier corresponding to "desolvation," have led to a simple model which folds with a high degree of cooperativity. The use of such a potential may also allow for a connection to the role of water in the folding of detailed protein models. Expulsion of water from the core has been shown to be one of the final barriers which must be overcome by protein G en route to its native state (Sheinerman and Brooks 1998b). The use of the potential described here results in a free energy surface in agreement with the free energy surface described through all-atom calculations.
The sequence effects incorporated into this simplified model have allowed the discrimination of the roles of the hairpins in the folding of protein L and protein G. The roles of the hairpins appear to be determined by the overall balance of the sequence effects rather than atomic details. This is reinforced by the observations that the relative roles of the hairpins in protein G did not emerge in all-atom models, in which the balance of the forces may have been affected by the addition of potentials which favor the native state (Shakhnovich et al. 2001).
Due to the highly symmetrical native structure of protein L and protein G, topological constraints cannot affect the observed preferential hairpin formation. This reduces the preferred sequence of hairpin formation to a simple competition between the relative stability of formation of the hairpins in the context of the protein. In protein L, the hairpins are of the same length, and the difference in the strength of side-chain interactions within each hairpin leads the N-terminal hairpin to form first. In protein G, the considerable difference in the relative lengths of the hairpins leads the shorter of the two, the C-terminal hairpin, to form first. Using these principles, changes to the relative stability of the hairpins introduced via mutations can be used in a predictable manner to alter the balance between the formation of the hairpins, thus altering the mechanism of folding.
Additional support for this idea derives from a recent study in which the N-terminal hairpin of protein G was redesigned to increase its stability, resulting in folding of this hairpin ahead of the C-terminal hairpin (Nauli et al. 2001). Modification of the native contact strengths in our model due to side chain–side chain contacts to reflect the sequences of these two variants resulted in additional stabilization of −4.1 and −4.9 kcal/mol in the N-terminal native contact strengths for the two modified sequences, respectively. Interestingly, this value is larger than the free-energy difference between the hairpins of 1.1 kcal/mol observed in our model (Table 2), suggesting that the N-terminal hairpin is likely to fold first in models built from these variant sequences. This is in agreement with the experimentally observed results (Nauli et al. 2001) and suggests that this redesign primarily stabilized the N-terminal hairpin microstates to overcome the entropy cost associated with the longer peptide fragment comprising this hairpin. A more difficult and interesting challenge might be to redesign the N-terminal hairpin to equal the C-terminal and explore the effect this "symmetry reestablishment" has on folding stability and kinetics.
Understanding the role of competing energetics in a protein subject to strong topological constraints, such as the SH3 domain, presents an additional future challenge. A complete comprehension of the "competition" between topology and sequence effects will be important both in protein design and in understanding diseases related to improper protein folding.
Materials and methods
The model we employ makes use of a minimalist representation of the protein where each amino acid residue is described by one "bead," located at the α-carbon position. The mass of each bead is taken to be the mass of the amino acid residue that the bead represents. The beads are connected via "virtual bonds" along the protein backbone.
Given the structure of the native state, a fixed set of encoded "rules" was used to build a set of potentials that yield the potential energy landscape on which we study the folding behavior of these two proteins. Our model potentials extend earlier models by introducing different energy scales to describe nonbonded interactions between side chains, hydrogen bonding in regular secondary structure, and sequence-dependent virtual dihedral potentials to represent sequence modulated flexibility and conformational preferences. The resulting free energy surface should mimic that of the real protein more closely than earlier models, which employed fewer energy scales or targeted specific encoding of the backbone structure with virtual dihedral potentials. Although the set of potentials we develop here are specific to the protein of interest, the "rules" used to derive the potentials were developed based on a wide range of protein topologies.
The energy terms employed for this purpose include: a favorable interaction between residues that are in contact in the native state, a repulsive interaction between all other pairs of residues, harmonic potentials applied to each of the bonds and angles, and a dihedral term reflecting the preferences of the backbone dihedral angles of the residues involved but otherwise not tuned to the specific protein topology.
For the interaction energy of residues separated in sequence by three or more bonds, we use the following functional form:
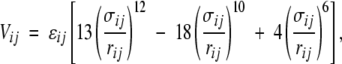 |
(1) |
where rij is the distance between residues i and j, σij is the distance between i and j at which the interaction energy is a minimum, and -ɛij is the strength of the interaction at this distance.
It is worth noting the differences between this potential and a typical Lennard-Jones potential. In addition to an increase in the curvature of the potential around the minimum, this function includes a small energy barrier. We physically rationalize this barrier as corresponding to a "desolvation penalty," which any pair of residues must pay before a favorable contact can be formed (Jernigan and Bahar 1996; Sheinerman and Brooks 1998b; Bilsel and Matthews 2000; Cheung et al. 2002). The height of this barrier for a given contact scales as the minimum energy for that contact. The use of this potential leads to enhanced cooperativity of the folding transition for a number of proteins (results not shown).
As discussed earlier, a G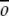 -like model is one in which residues that are in contact in the native state are defined to have a favorable interaction energy, while those not in contact in the native state have a less favorable interaction energy: repulsive, neutral, or less attractive. To build a G
-like model is one in which residues that are in contact in the native state are defined to have a favorable interaction energy, while those not in contact in the native state have a less favorable interaction energy: repulsive, neutral, or less attractive. To build a G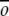 -like model from a protein's native-state structure, a set of native contacts must be defined. Native contacts were defined to represent as accurately as possible the determinants of a given protein structure, within the framework of the simplified model. Native contacts are defined from hydrogen bonding between backbone atoms and side chain–side chain interactions.
-like model from a protein's native-state structure, a set of native contacts must be defined. Native contacts were defined to represent as accurately as possible the determinants of a given protein structure, within the framework of the simplified model. Native contacts are defined from hydrogen bonding between backbone atoms and side chain–side chain interactions.
A list of hydrogen bonds was generated using the method of Kabsch and Sander (1983). This method estimates the hydrogen bond energy based on electrostatics, which is a function of both the hydrogen bond distance and the alignment of donor, acceptor, and attendant N and C atoms. A hydrogen bond is present when this energy lies below a threshold value. Any pair of residues that are hydrogen-bonded interact favorably in our model via the potential shown in Eq. 1, with ɛij set to unity and σij set to the α-carbon separation distance of this pair in the native-state structure. Native contacts separated by less than two residues in sequence (i,i+2 contacts) were excluded.
It should be noted that the simple representation of this model does not consider the relative orientation of the residues involved in hydrogen bonding directly. Residues in β-sheets or hairpins may be in contact via two hydrogen bonds, which imposes an additional requirement on the orientation of the backbone. In cases where a hydrogen bond was assigned to a pair in which a native contact had already been defined, four additional weak native contacts were defined. If residues i and j interact via either two hydrogen bonds or a hydrogen bond and a side-chain contact, then the pairs (i−1,j), (i,j−1), (i,j+1) and (i+1,j) are also defined as native contacts. These contacts interact favorably via the potential shown in Eq. 1, with ɛij set to 0.25 times the strength of the hydrogen bond and σij set to the α-carbon separation distance of each pair in the native-state structure. The spirit of this approach is similar to a previous model for hydrogen bonding networks, in which an additional favorable energetic term is applied when adjacent hydrogen bonds are formed (Kolinski and Skolnick 1994).
Side chain–side chain native contacts were assigned by collecting all pairs of residues with nonhydrogen side chain atoms within 4.5 Å. The distance dependence of the interaction energy was the same as discussed above (Eq. 1), but due to the large difference in the chemical natures of amino-acid side chains, the interaction strength was not fixed for all pairs of side chain–side chain native contacts. The relative value of ɛij was instead scaled in proportion to the contact energy reported by Miyazawa and Jernigan (1996) for this pair. Due to the introduction of a new energy scale relative to the hydrogen bond native contact energy scale, it was necessary to rescale these contact potentials. Thus, interactions were renormalized by dividing each Miyazawa-Jernigan contact energy by one-half the average value of all 210 contact energies. The fundamental interaction length parameter σij was again set to the α-carbon separation distance of this pair in the native structure. Both sequence and structure for specific proteins thus influence the density of favorable interactions along the chain.
To determine an overall energy scale for interactions in our model, we assume the protein has two dominant states, the native state and the unfolded state. The temperature at which both states are equally populated is dependent on the magnitude of the interactions present in the native state relative to those of the unfolded state. At the temperature where both states are equally populated, Tf, the free energy of both states must be equal, where F represents the free energy of the state, E represents the internal energy of the state, S represents the entropy of the state, and T is the temperature.
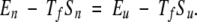 |
(2) |
If we assume that all native contacts are fully formed in the native manifold of states, and none are present in the unfolded manifold of states, and furthermore, that this difference exclusively accounts for the energy difference between the folded and unfolded states, that is,
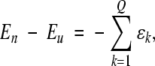 |
(3) |
where k is index over the Q native contacts. Then if the entropy difference between the native and unfolded states of an average residue is approximately constant for all proteins,
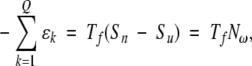 |
(4) |
where N is the number of residues and ω is a constant, which is equal to the Boltzmann constant multiplied by the natural logarithm of the ratio of the densities of states of the unfolded and native states. By determining the folding temperature for a variety of proteins built with a fixed value of 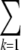 ɛk (results not shown), ω k=1 has been found to have a value of 0.0054 kcal/mol•K for the α-carbon based models employed here. Using Eq. 4, it is possible to specify the sum of the native contact strengths that will give rise to the desired folding temperature. This relationship may be used to scale the interaction energies of each of the assigned native contacts, so that the model will fold at a predetermined temperature.
ɛk (results not shown), ω k=1 has been found to have a value of 0.0054 kcal/mol•K for the α-carbon based models employed here. Using Eq. 4, it is possible to specify the sum of the native contact strengths that will give rise to the desired folding temperature. This relationship may be used to scale the interaction energies of each of the assigned native contacts, so that the model will fold at a predetermined temperature.
The native contact energy per residue, 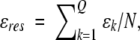 is a more fundamental energy scale than the average native contact energy, as the latter is dependent on the density of native contacts, and hence is sensitive to the definitions used in assigning native contacts. The native contact energy per residue, on the other hand, is equal to Tfω (see Eq. 4), which may be considered fixed for a protein of a given stability. Because this is the energy scale to which the interaction energies of each of the assigned native contacts is set, we use ɛres to set the energy scale to which all remaining terms in the Hamiltonian of the model may be defined.
is a more fundamental energy scale than the average native contact energy, as the latter is dependent on the density of native contacts, and hence is sensitive to the definitions used in assigning native contacts. The native contact energy per residue, on the other hand, is equal to Tfω (see Eq. 4), which may be considered fixed for a protein of a given stability. Because this is the energy scale to which the interaction energies of each of the assigned native contacts is set, we use ɛres to set the energy scale to which all remaining terms in the Hamiltonian of the model may be defined.
Having set the energy scale for the Hamiltonian, we now turn our attention to the remaining terms which comprise the Hamiltonian. Pairs of residues not defined as native contacts were subject to repulsive interactions of the form:
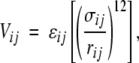 |
(5) |
where ɛij for all pairs of residues not in contact in the native state was set to 1.5×10−3 ɛres. Each residue was assigned a "repulsive radius," and the value of σij for each residue pair was equal to the average of the radii of the residues involved. The repulsive radius of a residue was set to the distance to the closest residue which had not been assigned as a native contact.
A harmonic potential was applied to each of the virtual bonds and angles, with the minimum located at the native state value and force constants of 200 ɛres and 40 ɛres, respectively.
The chirality of the amino acid constituents of a protein impose preferences on the backbone dihedral angles of the protein, as reflected in Ramachandran plots. Furthermore, differences in the sizes and geometry of the side chains mean that the backbone dihedral angle preferences of each amino acid are slightly different. Examination of the protein backbone shows that the virtual dihedral angle, that is, the dihedral angle between four adjacent α-carbons, is dependent only on the backbone dihedral angles of the two middle residues, two φ angles and two ξ angles, provided that the backbone dihedral angles ω, which contain the peptide bond, are held fixed at 180°. This relationship has been observed by Park and Levitt (1995). With this mapping in mind, it was possible to obtain the probability of a given virtual dihedral angle ρ from the probabilities of the backbone dihedrals which define it.
Using Ramachandran plots obtained from a survey (J. Srinivasan and C. Brooks, unpubl.) of the Protein Data Bank (Bernstein et al. 1977), a probability distribution for the virtual dihedral angle was obtained for each of the 400 possible ordered pairs of amino acid residues. The natural logarithm of the probability distribution can be related to the free energy as a function of the virtual dihedral angle. Since entropy is approximately uniform over this degree of freedom, this free energy is proportional to the energy as a function of the virtual dihedral angle. The natural logarithm of the probability distribution was therefore used as a sequence-dependent potential reflecting the relative propensity of ordered pairs of amino acids towards various secondary structural elements, and applied to the virtual dihedral angles in this model. The virtual dihedral potential generally consisted of two minima located at approximately +45° and −135°, corresponding to local α-helical and β-strand geometry, respectively. These "statistical" potentials were modeled as a cosine series consisting of up to four terms. As expected, the balance between these two minima reflects the propensity of the component amino acids towards these elements of secondary structure. Pairs of amino acids containing glycine showed lower barriers between the minima, while pairs containing proline showed a high barrier near 0° (examples of several potentials are available as supplementary material). To allow for the same dependence of the virtual dihedral strength on the total native contact energy, an additional proportionality constant equal to 0.4 ɛres was applied to the virtual dihedral potential.
This dihedral potential is sequence-specific, but does not depend on the specific protein topology. Directly encoding the native state in the backbone dihedral angles may result in locally driven folding, which should be avoided for several reasons. First, this deemphasizes the importance of the formation of native contacts, which reduces the search problem associated with reaching the native state from an ensemble of unfolded states. Furthermore, the cooperativity of the folding transition may be reduced due to the introduction of a population of conformations with energy between that of the native manifold of states and the unfolded manifold of states.
Whereas the native structure used to construct the protein G model was solved via X-ray crystallography, the protein L structure was solved via NMR. To most fully capture the information present in the ensemble of protein L structures, a model was built from each of the 21 members of the ensemble, and these models were subsequently averaged to yield the model used in the thermodynamic characterization.
All simulations were carried out using the CHARMM macromolecular mechanics package (Brooks et al. 1983) (parameter files in the CHARMM format based on the Hamiltonian described above are available for both proteins upon request). The time scale was defined by τ =(m/ɛres)½r0, where m is the mass of the average residue (119 amu) and r0 is the average distance between adjacent (bonded) beads in the native state (3.8 Å). The model was then allowed to evolve following high-friction Langevin dynamics, using friction coefficient β = 0.2/τ and timestep Δτ = 0.0075τ. The virtual bond lengths were kept fixed using the SHAKE algorithm (Ryckaert et al. 1977).
To improve sampling efficiency, each protein was simulated using a two-dimensional extension (Sugita and Okamoto 2002) of the replica exchange algorithm (Sugita and Okamoto 1999). Each replica was assigned one of four temperatures (300 K, 330 K, 363 K, and 400 K) and one of four harmonic potentials applied to the radius of gyration (each of which had a force constant of 0.5 kcal/ mol/Å2 and a minimum at 1.0 Rg0, 1.5 Rg0, 2.0 Rg0, or 2.5 Rg0, where Rg0 represents the radius of gyration in the native state). After an initial equilibration period, each replica was simulated for 2×107 Δτ, testing for exchanges every 2×104 Δτ. Data was collected only every 500 Δτ, which is beyond the conformational correlation time of the model protein. This procedure was carried out twice, to ensure convergence of the results.
In our analysis of the resulting conformational ensembles, a particular native contact was deemed to be formed if the distance between the α-carbons involved in the contact was less than 1.2 times the distance in the native state.
A complete thermodynamic analysis was carried out using the weighted histogram analysis method (Ferrenberg and Swendsen 1989), and has recently been reviewed in the context of both detailed atomic and minimalist models (Shea and Brooks 2001).
Acknowledgments
We thank our colleague Dr. Michael Feig, who provided the computational infrastructure for performing the replica-exchange sampling simulations used in this study. We also acknowledge financial support from the N.I.H. (GM48807, RR12255) and through a fellowship from NSERC (J.K.).
The publication costs of this article were defrayed in part by payment of page charges. This article must therefore be hereby marked "advertisement" in accordance with 18 USC section 1734 solely to indicate this fact.
Abbreviations
Tf, folding temperature
WHAM, weighted histogram method
φ-value, phi-value
q, fraction of native contacts
Article and publication are at http://www.proteinscience.org/cgi/doi/10.1110/ps.0205402.
References
- Alexander, P., Orban, J., and Bryan, P. 1992. Kinetic analysis of folding and unfolding the 56 amino acid IgG-binding domain of streptococcal protein G. Biochemistry 31 7243–7248. [DOI] [PubMed] [Google Scholar]
- Alm, E. and Baker, D. 1999. Prediction of protein-folding mechanisms from free-energy landscapes derived from native structures. Proc. Natl. Acad. Sci. 96 11305–11310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bernstein, F., Koetzle, T., Williams, G., Meyer, E., Brice, M., Rodgers, J., Kennard, O., Shimanouchi, T., and Tasumi, M. 1977. The Protein Data Bank: A computer-based archival file for macromolecular structures. J. Mol. Biol. 112 535–542. [DOI] [PubMed] [Google Scholar]
- Bilsel, O. and Matthews, C.R. 2000. Barriers in protein folding reactions. Adv. Prot. Chem. 53 153–207. [DOI] [PubMed] [Google Scholar]
- Blanco, F.J. and Serrano, L. 1995. Folding of protein G B1 domain studied by the conformational characterization of fragments comprising its secondary structure elements. Eur. J. Biochem. 230 634–649. [DOI] [PubMed] [Google Scholar]
- Brooks, B.R., Bruccoleri, R.E., Olafson, B., States, D., Swaminathan, S., and Karplus, M. 1983. CHARMM: A program for the macromolecular energy, minimization, and dynamics calculations. J. Comp. Chem. 4 187–217. [Google Scholar]
- Brooks III, C.L. 2002. Viewing protein folding from many perspectives. Proc. Natl. Acad. Sci. 99 1099–1100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan, H.S. and Dill, K.A. 1998. Protein folding in the landscape perspective: Chevron plots and non-Arrhenius kinetics. Proteins 30 2–33. [DOI] [PubMed] [Google Scholar]
- Cheung, M.S., Garcia, A.E., and Onuchic, J.N. 2002. Protein folding mediated by solvation: Water expulsion and formation of the hydrophobic core occur after structural collapse. Proc. Natl. Acad. Sci. 99 685–690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiti, F., Taddei, N., White, P.M., Bucciantini, M., Magherini, F., Stefani, M., and Dobson, C.M. 1999. Mutational analysis of acylphosphatase suggests the importance of topology and contact order in protein folding. Nat. Struct. Biol. 6 1005–1009. [DOI] [PubMed] [Google Scholar]
- Dobson, C.M. and Karplus, M. 1999. The fundamentals of protein folding: Bringing together theory and experiment. Curr. Opin. Struct. Biol. 9 92–101. [DOI] [PubMed] [Google Scholar]
- Dokholyan, N.V., Buldyrev, S.V., Stanley, H.E., and Shakhnovich, E.I. 2000. Identifying the protein folding nucleus using molecular dynamics. J. Mol. Biol. 296 1183–1188. [DOI] [PubMed] [Google Scholar]
- Ferrenberg, A.M. and Swendsen, R.H. 1989. Optimized Monte Carlo data analysis. Phys. Rev. Lett. 63 1195–1198. [DOI] [PubMed] [Google Scholar]
- Frank, M.K., Clore, G.M., and Gronenborn, A.M. 1995. Structural and dynamic characterization of the urea denatured state of the immunoglobulin binding domain of streptococcal protein G by multidimensional heteronuclear NMR spectroscopy. Protein Sci. 4 2605–2615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galzitskaya, O.V., and Finkelstein, A.V. 1999. A theoretical search for folding/unfolding nuclei in three-dimensional protein structures. Proc. Natl. Acad. Sci. 96 11299–11304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grantcharova, V., Alm, E.J., Baker, D., and Horwich, A.L. 2001. Mechanisms of protein folding. Curr. Opin. Struct. Biol. 11 70–82. [DOI] [PubMed] [Google Scholar]
- Gronenborn, A.M., Filpula, D.R., Essig, N.Z., Achari, A., Whitlow, M., Wingfield, P.T., and Clore, G.M. 1991. A novel, highly stable fold of the immunoglobulin binding domain of streptococcal protein G. Science 253 657–661. [DOI] [PubMed] [Google Scholar]
- Gu, H., Kim, D., and Baker, D. 1997. Contrasting roles for symmetrically disposed β-turns in the folding of a small protein. J. Mol. Biol. 274 588–596. [DOI] [PubMed] [Google Scholar]
- Guerois, R. and Serrano, L. 2000. The SH3-fold family: Experimental evidence and prediction of variations in the folding pathways. J. Mol. Biol. 304 967–982. [DOI] [PubMed] [Google Scholar]
- Jackson, S.E. 1998. How do small single-domain proteins fold? Fold. Des. 3 R81–91. [DOI] [PubMed] [Google Scholar]
- Jernigan, R.L. and Bahar, I. 1996. Structure-derived potentials and protein simulations. Curr. Opin. Struct. Biol. 6 195–209. [DOI] [PubMed] [Google Scholar]
- Kabsch, W. and Sander, C. 1983. Dictionary of protein secondary structure: Pattern recognition of hydrogen-bonded and geometrical features. Biopolymers 22 2577–2637. [DOI] [PubMed] [Google Scholar]
- Kelly, J.W. 1998. The environmental dependency of protein folding best explains prion and amyloid diseases. Proc. Natl. Acad. Sci. 95 930–932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim, D.E., Yi., Q., Gladwin, S.T., Goldberg, J.M., and Baker, D. 1998. The single helix in protein L is largely disrupted at the rate-limiting step in folding. J. Mol. Biol. 284 807–815. [DOI] [PubMed] [Google Scholar]
- Kim, D.E., Fisher, C., and Baker, D. 2000. A breakdown of symmetry in the folding transition state of protein L. J. Mol. Biol. 298 971–984. [DOI] [PubMed] [Google Scholar]
- Koga, N. and Takada, S. 2001. Roles of native topology and chain-length scaling in protein folding: A simulation study with a Go-like model. J. Mol. Biol. 313 171–180. [DOI] [PubMed] [Google Scholar]
- Kolinski, A. and Skolnick, J. 1994. Monte Carlo simulations of protein folding. I. Lattice model and interaction scheme. Proteins 18 338–352. [DOI] [PubMed] [Google Scholar]
- Kraulis, P.J. 1991. Molscript—A program to produce both detailed and schematic plots of protein structures. J. Appl. Crystallog. 24 946–950. [Google Scholar]
- Kuszewski, J., Clore, G.M., and Gronenborn, A.M. 1994. Fast folding of a prototypic polypeptide: The immunoglobulin binding domain of streptococcal protein G. Protein Sci. 3 1945–1952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martinez, J.C., Pisabarro, M.T., and Serrano, L. 1998. Obligatory steps in protein folding and the conformational diversity of the transition state. Nat. Struct. Biol. 5 721–729. [DOI] [PubMed] [Google Scholar]
- McCallister, E.L., Alm, E., and Baker, D. 2000. Critical role of β-hairpin formation in protein G folding. Nat. Struct. Biol. 7 669–673. [DOI] [PubMed] [Google Scholar]
- Miyazawa, S. and Jernigan, R.L. 1996. Residue-residue potentials with a favorable contact pair term and an unfavorable high packing density term, for simulation and threading. J. Mol. Biol. 256 623–644. [DOI] [PubMed] [Google Scholar]
- Munoz, V. and Eaton, W.A. 1999. A simple model for calculating the kinetics of protein folding from three-dimensional structures. Proc. Natl. Acad. Sci. 96 11311–11316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nauli, S., Kuhlman, B., and Baker, D. 2001. Computer-based redesign of a protein folding pathway. Nat. Struct. Biol. 8 602–605. [DOI] [PubMed] [Google Scholar]
- Nymeyer, H., Garcia, A.E., and Onuchic, J.N. 1998. Folding funnels and frustration in off-lattice minimalist protein landscapes. Proc. Natl. Acad. Sci. 95 5921–5928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park, B.H. and Levitt, M. 1995. The complexity and accuracy of discrete state models of protein structure. J. Mol. Biol. 249 493–507. [DOI] [PubMed] [Google Scholar]
- Park, S.H., Shastry, M.C., and Roder, H. 1999. Folding dynamics of the B1 domain of protein G explored by ultrarapid mixing. Nat. Struct. Biol. 6 943–947. [DOI] [PubMed] [Google Scholar]
- Plaxco, K.W., Simons, K.T., and Baker, D. 1998. Contact order, transition state placement and the refolding rates of single domain proteins. J. Mol. Biol. 277 985–945. [DOI] [PubMed] [Google Scholar]
- Plaxco, K.W., Simons, K.T., Ruczinski, I., and David, B. 2000. Topology, stability, sequence, and length: Defining the determinants of two-state protein folding kinetics. Biochemistry 39 11177–11183. [DOI] [PubMed] [Google Scholar]
- Ramirez-Alvarado, M., Serrano, L., and Blanco, F.J. 1997. Conformational analysis of peptides corresponding to all the secondary structure elements of protein L B1 domain: Secondary structure propensities are not conserved in proteins with the same fold. Protein Sci. 6 162–174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riddle, D.S., Grantcharova, V.P., Santiago, J.V., Alm, E., Ruczinski, I and Baker, D. 1999. Experiment and theory highlight role of native state topology in SH3 folding. Nat. Struct. Biol. 6 1016–1024. [DOI] [PubMed] [Google Scholar]
- Ryckaert, J-P, Ciccotti, G and Berendsen, H.J.C. 1977. Numerical integration of the cartesian equations of motion of a system with constraints: Molecular dynamics of n-alkanes. J. Comp. Phys. 23 327–341. [Google Scholar]
- Sari, N., Alexander, P., Bryan, P.N., and Orban, J. 2000. Structure and dynamics of an acid-denatured protein G mutant. Biochemistry 39 965–977. [DOI] [PubMed] [Google Scholar]
- Scalley, M.L., Yi, Q., Gu, H., McCormack, A., Yates 3rd, J.R., and Baker, D. 1997. Kinetics of folding of the IgG binding domain of peptostreptococcal protein L. Biochemistry 36 3373–3382. [DOI] [PubMed] [Google Scholar]
- Scalley, M.L., Nauli, S., Gladwin, S.T., and Baker, D. 1999. Structural transitions in the protein L denatured state ensemble. Biochemistry 38 15927–15935. [DOI] [PubMed] [Google Scholar]
- Shakhnovich, E.I., Shimada, J., and Kussell, E. 2001. Folding thermodynamics and kinetics: Insights from all-atom simulations. Abstr. Pap. Am. Chem. Soc. 220.
- Shea, J.E. and Brooks III, C.L. 2001. From folding theories to folding proteins: A review and assessment of simulation studies of protein folding and unfolding. Annu. Rev. Phys. Chem. 52 499–535. [DOI] [PubMed] [Google Scholar]
- Shea, J.E., Onuchic, J.N., and Brooks III, C.L. 1999. Exploring the origins of topological frustration: Design of a minimally frustrated model of fragment B of protein A. Proc. Natl. Acad. Sci. 96 12512–12517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheinerman, F.B., and Brooks III, C.L. 1998a. Calculations on folding of segment B1 of streptococcal protein G. J. Mol. Biol. 278 439–456. [DOI] [PubMed] [Google Scholar]
- Sheinerman, F.B. and Brooks III, C.L. 1998b. Molecular picture of folding of a small alpha/beta protein. Proc. Natl. Acad. Sci. 95 1562–1567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sugita, Y. and Okamoto, Y. 1999. Replica-exchange molecular dynamics method for protein folding. Chem. Phys. Lett. 314 141–151. [Google Scholar]
- Sugita, Y. and Okamoto, Y. 2002. Free-energy calculations in protein folding by generalized-ensemble algorithms. In Lecture notes in computational science and engineering, (eds. H. H. Gan and T. Schlick), pp. 303–331. Springer-Verlag, Berlin.
- Taketomi, H., Ueda, Y., and Go, N. 1975. Studies on protein folding, unfolding and fluctuations by computer simulation. I. The effect of specific amino acid sequence represented by specific inter-unit interactions. Int. J. Pept. Protein Res. 7 445–459. [PubMed] [Google Scholar]
- Tobias, D.J., Sneddon, S.F., and Brooks III, C.L. 1991. The stability of protein secondary structures in aqueous solution. Adv. Biomolecular Simul. (AIP Conf. Proc.) 239 174–199. [Google Scholar]
- Villegas, V., Martinez, J.C., Aviles, F.X., and Serrano, L. 1998. Structure of the transition state in the folding process of human procarboxypeptidase A2 activation domain. J. Mol. Biol. 283 1027–1036. [DOI] [PubMed] [Google Scholar]
- Wikstrom, M., Sjobring, U., Kastern, W., Bjorck, L., Drakenberg, T., and Forsen, S. 1993. Proton nuclear magnetic resonance sequential assignments and secondary structure of an immunoglobulin light chain-binding domain of protein L. Biochemistry 32 3381–3386. [DOI] [PubMed] [Google Scholar]
- Yi, Q., Scalley, M.L., Simons, K.T., Gladwin, S.T., and Baker, D. 1997. Characterization of the free energy spectrum of peptostreptococcal protein L. Fold. Des. 2 271–280. [DOI] [PubMed] [Google Scholar]
- Yi, Q., Scalley-Kim, M.L., Alm, E.J., and Baker, D. 2000. NMR characterization of residual structure in the denatured state of protein L. J. Mol. Biol. 299 1341–1351. [DOI] [PubMed] [Google Scholar]
- Zhou, Y. and Karplus, M. 1999. Interpreting the folding kinetics of helical proteins. Nature 401 400–403. [DOI] [PubMed] [Google Scholar]