

Abstract
Glutamate is the major excitatory neurotransmitter in the mammalian CNS. The spatiotemporal profile of the glutamate concentration in the synapse is critical for excitatory synaptic signalling. The control of this spatiotemporal concentration profile requires the presence of large numbers of synaptically localized glutamate transporters that remove pre-synaptically released glutamate by uptake into neurons and adjacent glia cells. These glutamate transporters are electrogenic and utilize energy stored in the transmembrane potential and the Na+/K+-ion concentration gradients to accumulate glutamate in the cell. This review focuses on the kinetic and electrogenic properties of glutamate transporters, as well as on the molecular mechanism of transport. Recent results are discussed that demonstrate the multistep nature of the transporter reaction cycle. Results from pre-steady-state kinetic experiments suggest that at least four of the individual transporter reaction steps are electrogenic, including reactions associated with the glutamate-dependent transporter halfcycle. Furthermore, the kinetic similarities and differences between some of the glutamate transporter subtypes and splice variants are discussed. A molecular mechanism of glutamate transport is presented that accounts for most of the available kinetic data. Finally, we discuss how synaptic glutamate transporters impact on glutamate receptor activity and how transporters may shape excitatory synaptic transmission.
Glutamate in the CNS
In the mammalian central nervous system (CNS), L-glutamate is the chemical transmitter of excitatory signals (Curtis & Johnston, 1974; Curtis & Watkins, 1960; Fonnum, 1984). Glutamate is synthesized and stored in glutamatergic neurons and is released upon specific stimuli (e.g. action potentials) into the synaptic cleft. The cycle of neurotransmission is initiated by the fusion of neurotransmitter-loaded synaptic vesicles with the plasma membrane of the pre-synaptic neuron. Within less than a millisecond, glutamate diffuses across the synaptic cleft and binds to different classes of glutamate receptors which are localized pre- and post-synaptically as well as on astroglial processes (Backus, Kettenmann & Schachner, 1989; Glaum, Holzwarth & Miller, 1990; Nakanishi & Masu, 1994; Sontheimer et al., 1988). The chemical signal is converted into an electrical signal by the transient opening of ligand-gated ion channels. This is achieved either directly by ionotropic glutamate receptors or indirectly by metabotropic glutamate receptors which modulate ion channel activity by second messenger systems (Nakanishi & Masu, 1994). As a result, the chemical signal is integrated and transformed into an electrical signal which results in the transmission of information from one neuron to another. These steps are summarized as glutamatergic neurotransmission of the CNS. Interestingly, the hypothesis of glutamatergic transmission was a subject of extended discussions in the 1960’s which became initially accepted in the 1970’s following the finding of some specific glutamate antagonists and, most importantly, by the discovery of glutamate uptake in the CNS (Balcar & Johnston, 1972; Biscoe et al., 1977; Evans & Watkins, 1978; Logan & Snyder, 1971; Neal & White, 1973; Wofsey, Kuhar & Snyder, 1971). Rapid removal of glutamate from the synaptic cleft is nowadays accepted as an essential step in neurotransmission. The purpose of glutamate uptake is not only to avoid uncontrolled and persistent activation, and thus, damage to secondary neurons, but also to achieve physiological conditions for sustained neuronal signal transmission. Transmitter accumulated in the cytoplasm of pre-synaptic cells is sequestered in synaptic vesicles, ready for a new cycle of neurotransmission (Fig 1). In contrast, glutamate taken up by glial cells passes through the glutamate-glutamine cycle (Hertz, 1979; Derouiche & Rauen, 1995; Rauen & Wiessner, 2000). In this case, glutamate is converted into glutamine by the glia-specific enzyme glutamine-synthetase. Glutamine is then released into the extracellular space and subsequently taken up by glutamatergic neurons for a new cycle of transmitter synthesis (Fig. 1). Both neuronal and glial glutamate uptake systems are capable of transporting glutamate against a several thousand-fold concentration gradient into the cell (Gegelashvili et al., 2001).
Figure 1.
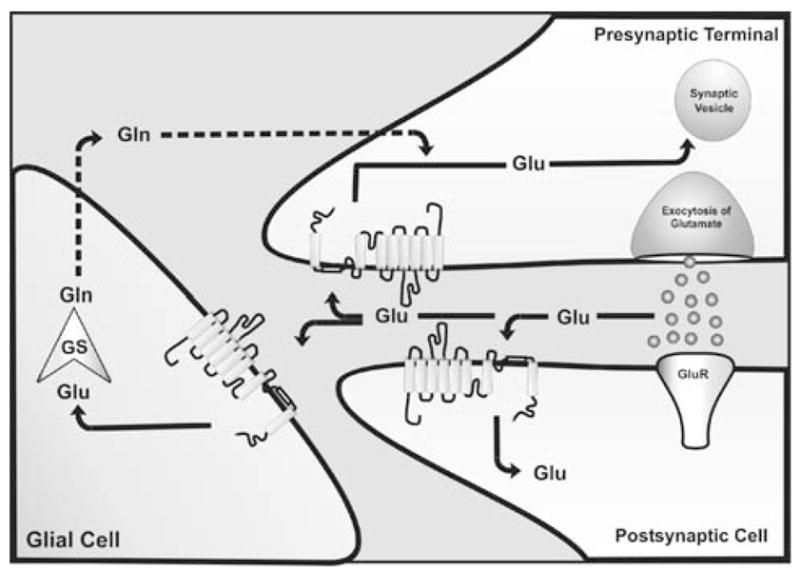
Distribution and function of EAA transporters at the synapse during a synaptic event. Glutamate (circles) binding to glutamate receptors (GluR) mediates the signal to postsynaptic neurons. Rapid removal of glutamate from the synaptic cleft is achieved by glutamate transporters which are located in glial cells and neurons. Once glutamate is transported into the cell (indicated by arrows), it can be sequestered in synaptic vesicles, ready for a new cycle of neurotransmission (indicated by arrows in the presynapse) or is degraded in glial cells by the glia-specific enzyme glutamine-synthetase (GS) to glutamine (Gln), which is released into the extracellular space, and subsequently taken up by glutamatergic neurons for a new cycle of transmitter synthesis (indicated by dotted lines) for a new transmitter synthesis.
Considering the ubiquitous presence of glutamate in the CNS, it is not surprising that malfunction of glutamatergic transmission is related to numerous neurological diseases and psychiatric disorders. At sustained elevated extracellular concentrations (downregulated glutamate uptake activity), glutamate acts as a powerful neurotoxin capable of inducing severe excitotoxic damage to target neurons, a mechanism possibly implicated in several neurodegenerative diseases (e.g., amyotrophic lateral sclerosis (ALS), Huntington’s disease, and Alzheimer’s disease) and brain insults (e.g., ischemia, hypoxia, hypoglycemia, epilepsy). In contrast, low concentrations (upregulated glutamate uptake activity) are implicated in schizophrenia (Behrens et al., 2002; Carlsson & Carlsson, 1990; Choi, 1992; Ingram et al., 2000; Rothstein, 1995; Su et al., 2003). Traditionally, the targets for drugs that interfere with neurotransmission are the receptors that are responsible for the generation of the electrical signal in post-synaptic neurons. In this review, we are aiming at a new and different target in the system; the neurotransmitter transporters that are responsible for the (re)-uptake of the neurotransmitter from the synapse into neuronal cells or surrounding glial cells. The activity of these transporters is assumed to be essential for the temporal and spatial regulation of the neurotransmitter concentration in the synaptic cleft and, thus, is crucial for proper neuronal signalling.
The sodium and potassium dependent glutamate transporter gene family
The biology of glutamate transporters experienced a tremendous boost in the early nineties by the almost simultaneous cloning of the first three eukaryotic glutamate transporters: GLAST1 (Glutamate Aspartate Tansporter 1, (Storck et al., 1992) and GLT1 (Glutamate Transporter 1, (Pines et al., 1992) from rat brain, and EAAC1 (Excitatory Amino Acid Carrier 1, (Kanai & Hediger, 1992) from rabbit small intestine. With the cloning of the human homologues of these three glutamate transporter subtypes, (Arriza et al., 1994) suggested a standardized nomenclature using the acronym EAAT (Excitatory Amino Acid Transporter). Based on sequence similarities, EAAT1 is the human homologue of GLAST1, EAAT2 of GLT1, and EAAT3 of EAAC1. Using homology-screening-strategies resulted not only in the identification of two additional human high affinity glutamate transporters (EAAT4 from cerebellum and EAAT5 from the retina (Arriza et al., 1997; Fairman et al., 1995)), but, remarkably, also in the isolation of two neutral amino acid transporters, namely ASCT1 und ASCT2 (Alanine Serine Cysteine Transporter 1 and 2; (Arriza et al., 1993; Broer et al., 1999; Shafqat et al., 1993)). The five different glutamate transporter subtypes show a high degree of interspecies conservation (for reviews see: Danbolt, 2001; Slotboom, Konings & Lolkema, 1999a), but share no significant homology with the family of sodium- and chloride-dependent neurotransmitter transporters (such as for GABA, glycine, dopamine, serotonin, noradrenalin, proline and taurine), nor do they exhibit any significant homology with any other known protein family. Thus, the diverse glutamate transporter subtypes together with the carriers for neutral amino acids (ASCT1 and ASCT2), as well as the proton-dependent bacterial glutamate and dicarboxylate transporters belong to the same gene-family (reviewed in: Danbolt, 2001; Slotboom et al., 1999a). In addition, there is evidence for additional variants of these glutamate transporter subtypes that arise through alternate mRNA splicing. Huggett et al. (Huggett, Vaughan-Thomas & Mason, 2000) recently discovered a splice variant of GLAST1 (termed GLAST1a) which lacks exon 3. This splice variant appears to be expressed in rat bone and brain. GLT1 (EAAT2), however, appears to exist as a number of distinct splice variants. The original splicing was described in rat by Pines et al. (Pines et al., 1992). Subsequently, Utsunomiya-Tate and colleagues (Utsunomiya-Tate, Endou & Kanai, 1997) described in mouse not only the murine mGLT1 sequence homologous to EAAT2, but also two additional splice variants, mGLT1A and mGLT1B (Fig. 2). These differed from the originally described versions in that the amino-(N) terminus of mGLT1 contained the amino acid sequence MASTEGA, whereas both mGLT1A and mGLT1B contained a short alternately spliced amino-(N)-terminal region (MVSA). In addition, mGLT1B contained an alternately spliced carboxyl-(C)-terminal region (Fig. 2). Recently, Schmitt et al. (Schmitt et al., 2002) described a hybrid GLT1 splice variant in rat which possessed the C-terminal splicing of mGLT1B, but retained the original GLT1 N-terminal region (Fig. 2). They identified this as GLT1v (variant). A similar splice variant was identified by Chen and colleagues (Chen et al., 2002) in rat and referred to as GLT1b. Sullivan et al. (Sullivan et al., 2004) concluded in their study that GLT1v, in contrast to GLT1, may not be involved in shaping the kinetics of synaptic signalling in the brain, but may be critical in preventing spillover of glutamate between adjacent synapses thereby regulating intersynaptic glutamatergic and GABAergic transmission. In a recent study, Rauen et al. have identified a novel GLT1 splicing variant called GLT1c which is expressed pre-synaptically at photoreceptor terminals and has been implied to be the “missing” pre-synaptic photoreceptor glutamate transporter (Rauen et al., 2004). GLT1c is found predominately in rat and human retinal tissue, faintly in cortex and cerebellum, while no product is found in liver, suggesting expression of GLT1c mainly in CNS tissue. The final 23 C-terminal amino acids encoding for GLT1 (Pines et al., 1992) are replaced by 11 amino acids for GLT1c, indicating that 561 amino acids encode GLT1c with a theoretical molecular mass of 60,5 kDa, while 573 amino acids encode for GLT1 (Fig. 2). The carboxy terminal amino acid sequence of GLT1c – SWV – suggests a class I PDZ binding motif (Rauen et al., 2004). A class I PDZ binding motif requires the carboxy terminal sequence (T/S)XV where either serine or threonine are permitted in position 1, any other amino acid residue could substitute in position 2 (X), and valine or a hydrophobic side chain must be present at the carboxy-terminal position (Fanning & Anderson, 1999). Interestingly, EAAT5 (Arriza et al., 1997) and GLT1v (Schmitt et al., 2002) contain similar putative C-terminal PDZ binding motifs. The most general function of PDZ domains may be in spatially clustering and anchoring their ligands within specific subcellular domains. Although no direct experimental evidence currently indicate protein-protein interaction of glutamate transporters with PDZ-domains, the observations that EAAC1, GLT1, GLAST1 and EAAT4 form clusters and are capable of rapid trafficking to and from the plasma membrane further supports the concept that they are connected to other proteins (Davis et al., 1998; Duan et al., 1999; Gegelashvili et al., 2000; Poitry-Yamate, Vutskits & Rauen, 2002).
Figure 2.

Schematic illustration of the known GLT1 splice variants, showing the GLT1 nomenclature according to the original reference, the N-terminal amino acid sequence, reference and protein identity number/GenBank accession number, and the C-terminal amino acid sequence. The prefix “m” denotes mouse; “r” denotes rat.
Topology: Structural characteristics of electrogenic glutamate transporters
Our present structural understanding of the glutamate transporter is restricted to topological models. Two such models have recently been proposed, one by S. Amara and colleagues (Seal & Amara, 1998; Seal, Leighton & Amara, 2000) and one by the Kanner and Lolkema groups (Grunewald & Kanner, 2000; Slotboom, Konings & Lolkema, 2001; Slotboom et al., 1999b). The latter model is shown in Fig. 3A. Both models predict the N-terminus on the intracellular side, six N-terminal transmembrane domains (TMD), and an intracellular C-terminus. The two proposals disagree on the topology of the C-terminal part of the polypeptide sequence. In the model shown in Fig. 3A, there are two pore loop-like structures included (RL = re-entrant loop), one which dips into the membrane from the intracellular side (RL1, between TMD6 and TMD7, see Fig. 3A) and one from the extracellular side (RL2, between TMD7 and TMD8). It has been proposed that these re-entrant loop sequences, in analogy with ion channels of known structure (Doyle et al., 1998; Dutzler et al., 2002), are an essential structural element for the permeation of the neurotransmitter and/or the co-transported cations. The model proposed by the Amara group assigns an extracellular re-entrant loop to TMD7 and a transmembrane helix to RL1 (Leighton et al., 2002; Seal & Amara, 1998; Seal et al., 2000). Another similarity to ion channels is the proposed multimeric structure of the transporter. The functional EAAC1 protein was demonstrated to be a pentamer (Eskandari et al., 2000).
Figure 3.
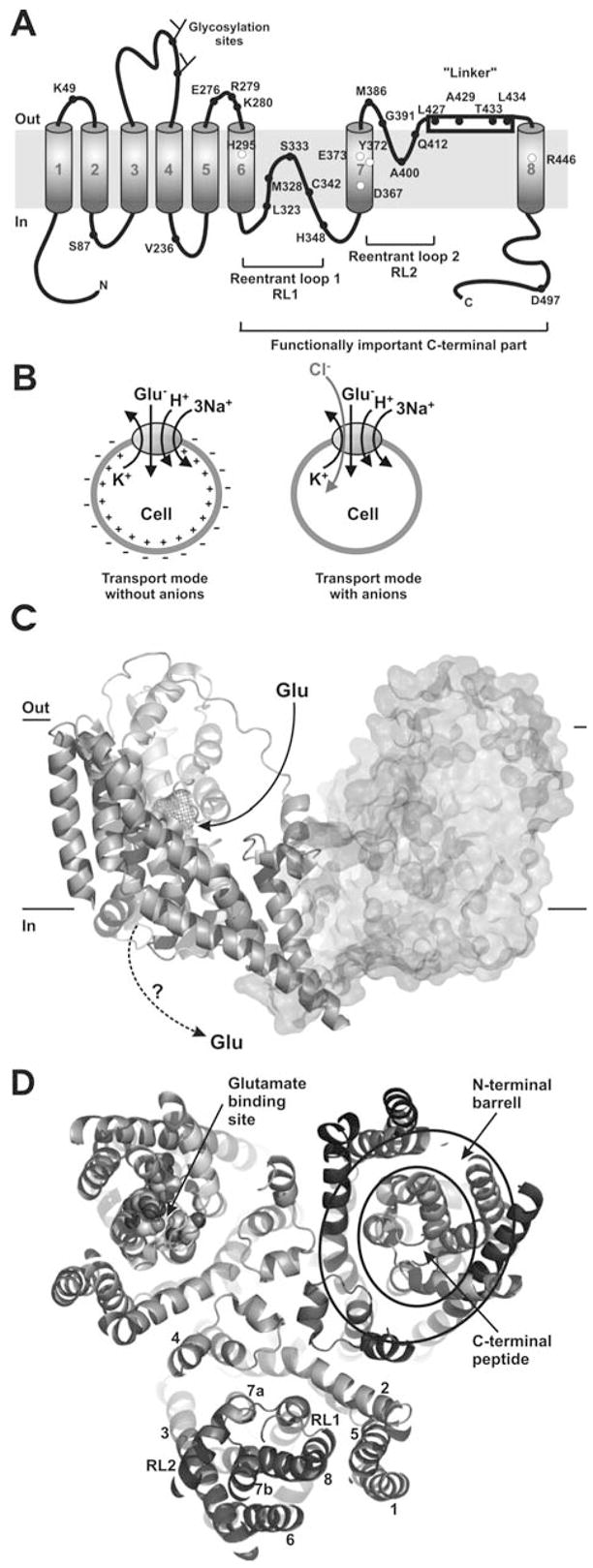
(A) A transmembrane topology model of the GLT-1 according to (Grunewald & Kanner, 2000). The open circles depict some of the functionally important amino acid residues that have been identified by using site-directed mutagenesis. The closed circles represent amino acids that have been used for topology determination (Grunewald & Kanner, 1995; Grunewald & Kanner, 2000; Slotboom et al., 2001; Slotboom et al., 1999b). (B) Illustration of the stoichiometry of glutamate transporters (left panel): Glutamate transport is coupled to the co-transport of three Na+-, one H+- and the counter-transport of one K+-ion. The plus sign (+) inside the cell indicates depolarization of the cell induced by inward glutamate transport. The right panel shows the glutamate-induced anion conductance which might prevent the depolarizing action of glutamate transport by neutralizing the effect of the inwardly-transported positively-charged cations. (C) Side-view of the structure of GltP from Pyrococcus horikoshii (Yernool et al., 2004), PDB ID 1XFH. The third subunit of the trimer pointing towards the viewer was omitted for better clarity. The glutamate binding site in the left subunit is shown as the wire mesh. The right subunit is shown in surface representation. A putative pathway for glutamate binding from the extracellular side is shown by the solid arrow. It is unknown how glutamate dissociates to the intracellular side (dotted arrow). (D) Top view of the trimeric assembly of GltP. The numbering of the TMDs is shown in the bottom subunit. Residues that contribute to glutamate binding are highlighted as spheres in the left subunit. The general subunit architecture is composed of a barrel, which shields the C-terminal part from the membrane and from interaction with the other subunits, as illustrated by the circles in the right subunit. This barrel is generated by the 6 N-terminal TMDs. The functionally important C-terminal part, which is composed of the two re-entrant loops and TMDs 7 and 8, is inserted into this barrel. For clarity, a slab view is shown with the extracellular domain of the transporter cut away approximately at the level of the extracellular face of the membrane. Figures (C) and (D) were created with PyMol (www.pymol.org).
While this manuscript was under review the three-dimensional structure of a glutamate transporter homologue from the thermophilic bacterium Pyrococcus horikoshii was reported (Yernool et al., 2004). The transporter is assembled as a trimer of identical subunits. A side view of two of the three subunits is shown in Fig. 3C. The structure confirms one of the two topological models with two re-entrant loops proposed by the Kanner and Lolkema groups as shown in Fig. 3A (Grunewald & Kanner, 2000; Slotboom, Konings & Lolkema, 2001; Slotboom et al., 1999b). It also confirms that the functionally important part of the glutamate transporter is localized in the C-terminal part starting from RL1. The structure provides no information on the locations of the binding sites for the co- and counter-transported ions. Although the transported substrate, glutamate, is not well resolved at the resolution of 3.8 Å, electron density that probably accounts for bound glutamate was found between RL1 and RL2 and close to TMD8 (wire mesh shown in the left subunit of Fig. 3C). This glutamate binding position agrees well with data obtained from previous mutagenesis studies (Bendahan et al., 2000), and is, surprisingly, localized very close to the extracellular surface of the transporter, and not buried deep within the membrane. Although Yernool et al. (2004) propose an alternating access model in which the two re-entrant loops form the extracellular and intracellular gates that can open alternately to expose the glutamate binding site either to the extracellular side or to the cytoplasm, it is not yet clear how glutamate might dissociate from its binding site close to the extracellular side of the protein into the cytoplasm (Fig. 3C, dashed arrow). This dissociation would require diffusion of glutamate through the major part of the hydrophobic transmembrane interior of the protein. Another possible mechanism that would deviate from the classical alternating access hypothesis would be that the C-terminal part of the protein, which is shielded from the lipid bilayer by a barrel formed by the N-terminus (shown in Fig. 3D, right subunit), could act as a hydrophobic ion that moves glutamate across the membrane in a carrier-type motion. To differentiate between these possibilities, clearly requires further experimentation.
Stoichiometry and electrophysiological properties of glutamate transporters
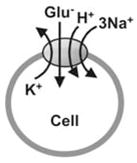
The glutamate transporter belongs to the class of secondary-active, Na+-dependent transporters (Amara, 1998; Danbolt, 2001; Kanner & Schuldiner, 1987). In contrast to primary-active transporters which use metabolic energy sources such as ATP directly for “uphill” transport (Läuger et al., 1981), secondary-active Na+-dependent transporters are energized by the coupling of “uphill” substrate transport to the co-transport of Na+ down its own transmembrane concentration gradient (Crane, 1962; Kanner & Sharon, 1978; Kanai & Hediger, 1992) which is maintained by the Na+/K+-ATPase. In the case of the glutamate transporter, the stoichiometry of this coupling is movement of 1 glutamate−, 3 Na+, and 1 H+ into the cell, along with 1 K+ out of the cell (Billups & Attwell, 1996; Kanner & Bendahan, 1982; Levy, Warr & Attwell, 1998; Zerangue & Kavanaugh, 1996a), as illustrated in Fig. 3B. According to this stoichiometry, glutamate transport is electrogenic (Kanai et al., 1995), as two net positive charges are moved into the cell for each completed transport cycle. Electrogenic transport is associated with the flow of current (Brew & Attwell, 1987) which will be referred to here as transport current (see Table 1). This current is also sometimes termed coupled transport- or uptake current to indicate that it is associated with the charge movement that is stoichiometrically coupled to glutamate transport. Recording of the transport current represents a highly sensitive functional assay for glutamate transporter function, although it is technically not a trivial task. Because of the small turnover number of the glutamate transporter (30 s−1 at 0 mV), about 105 transporter molecules are necessary to generate the same steady-state current as a typical ion channel with a single-channel current of 1 pA.
Table 1.
Current components associated with glutamate transporters.
| Current component | Description | Permeating ions | Reference | |
|---|---|---|---|---|
| Transport Current (TC) | TC is stoichiometrically coupled to glutamate transport. TC is evoked the inward movement of two net by positive charges per transported glutamate. | Glutamate−, Na+, H+, K+ |
|
(Kanai et al., 1995; Zerangue & Kavanaugh, 1996a) |
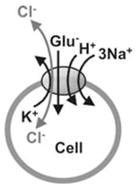
| Anion Current (AC) | AC is activated by glutamate, but is not thermodynamically coupled to glutamate transport. Anion flux follows strictly its own transmembrane electrochemical potential gradient | Cl− |
|
(Billups et al., 1996; Eliasof & Jahr, 1996; Fairman et al., 1995) |
| Leak Currents (LC) | LCs are carried by anions and Na+. Anion LC can be inhibited by non- transportable inhibitors, such as TBOA. | Na+, Cl− |
|
(Grewer et al., 2000; Kanai et al., 1995; Otis & Jahr, 1998; Sonders & Amara, 1996; Vandenberg et al., 1995; Wadiche & Kavanaugh, 1998) |
In addition to this transport current, the transporters also facilitate anion flux across the membrane (Eliasof & Jahr, 1996; Wadiche, Amara & Kavanaugh, 1995a), as shown in Fig. 3B. Anion flux is associated with a current that will be referred to as anion current. This anion current is kinetically, but not stoichiometrically coupled to glutamate transport (Billups, Rossi & Attwell, 1996; Grewer et al., 2000; Otis & Jahr, 1998). Therefore, it is sometimes referred to as uncoupled anion current. This anion current is stimulated by the binding of transported substrates and/or Na+-ions to the transporter. The mechanistic basis of this anion current is described in more detail below (section Glutamate transporter associated anion conductance).
Finally, glutamate transporters catalyze the leak flux of ions across the membrane (Borre & Kanner, 2004; Kanai et al., 1995; Melzer, Biela & Fahlke, 2003; Otis & Jahr, 1998; Watzke & Grewer, 2001). Leak flux occurs already in the absence of the transported substrate. Two components of the leak ion flux have been identified, one being specific for monovalent cations (Kanai et al., 1995; Vandenberg et al., 1995), and the other being specific for anions. The anion component of the leak conductance can be inhibited by applying non-transportable inhibitors to the glutamate transporter (Otis & Jahr, 1998; Watzke & Grewer, 2001). In other secondary transporters, Na+ leaks are caused by uniporter activity (Eskandari et al., 1997). Although it is not clear whether the cation leak of glutamate transporters may be caused by a channel activity, it is more likely that Na+ is imported into the cell via a Na+/K+ exchange mechanism of the transporter. Table 1 lists the major components of glutamate transporter-mediated currents.
Dissecting electrogenic and electroneutral reaction steps of the transport cycle
Secondary transporters are usually studied by uptake of radiolabeled substrate or voltage-clamp current-recording measurements under steady-state conditions. However, under steady-state conditions the properties of the transporter are mainly determined by the rate limiting reaction step in the transport cycle. Therefore, information about other, non-rate-limiting partial reaction steps of the transporter is not easily available from such measurements. In order to overcome this limitation, methods were developed that are based on rapidly perturbing the steady-state and subsequently following the kinetics of the relaxation to a new steady state (pre-steady-state kinetics). One widely used method to perturb the steady state of a secondary transporter is to jump the transmembrane potential to a new value. A subsequent change of the population of state connected by voltage-sensitive reactions can then be monitored by measuring transient membrane currents (Forster et al., 1998; Loo et al., 1993; Lu & Hilgemann, 1999; Mager et al., 1993). The principle of the method is illustrated in Fig. 4A. When such voltage jumps are applied to glutamate transporter-containing membranes, large capacitive transient currents are observed in the absence of the transported substrate, whereas in the presence of extracellular glutamate or non-transportable inhibitors, such as kainate, the transient currents are inhibited (Kanai et al., 1995; Mennerick et al., 1999; Wadiche et al., 1995b; Watzke, Bamberg & Grewer, 2001; Zhang et al., 1998). The simplest explanation for these observations is that an individual reaction step of the empty transporter with no glutamate bound is electrogenic, whereas reactions of the glutamate-bound transporter forms are electroneutral (Kanai et al., 1995; Kanai et al., 1994). Because the magnitude of the voltage-jump-induced transient currents is strongly dependent on the extracellular [Na+], it was further proposed that the glutamate-independent electrogenic reaction is related to Na+ binding to its extracellular binding site on the transporter (Wadiche et al., 1995b). Detailed analysis of the voltage dependence of this Na+ binding reaction permits the estimation of the total charge moved in this reaction step, which is about 40 – 45% of one unitary charge. Thus, electrogenic binding of Na+ contributes only about 20% to the total electrogenicity of the glutamate transporter (inward movement of 2 positive charges). Therefore, other electrogenic partial reactions must exist. Na+ binding is believed to be a source of electrogenicity not only in glutamate transporters, but also in other secondary transporters (Forlani et al., 2001; Forster et al., 1998; Hilgemann & Lu, 1999; Loo et al., 1993; Lu & Hilgemann, 1999; Mackenzie et al., 2003; Mager et al., 1993) and primary ion pumps (Holmgren et al., 2000; Lu et al., 1995). The Na+ binding reaction senses 30% of the electric field in the sugar transporter SGLT1 (Hazama, Loo & Wright, 1997) and 15% of the electric field in the phosphate transporter NaPi-2 (Forster et al., 1998; Murer et al., 2000). Therefore, in analogy to the glutamate transporter, Na+ binding represents only a fraction of the total charge moved during the transport cycle for these transporters. In contrast, Na+ binding is the major electrogenic step in GABA transporters, where it contributes about 90% to the total electrogenicity (Binda et al., 2002; Eckstein-Ludwig, Fei & Schwarz, 1999; Loo et al., 2000; Lu & Hilgemann, 1999; Mager et al., 1996).
Figure 4.
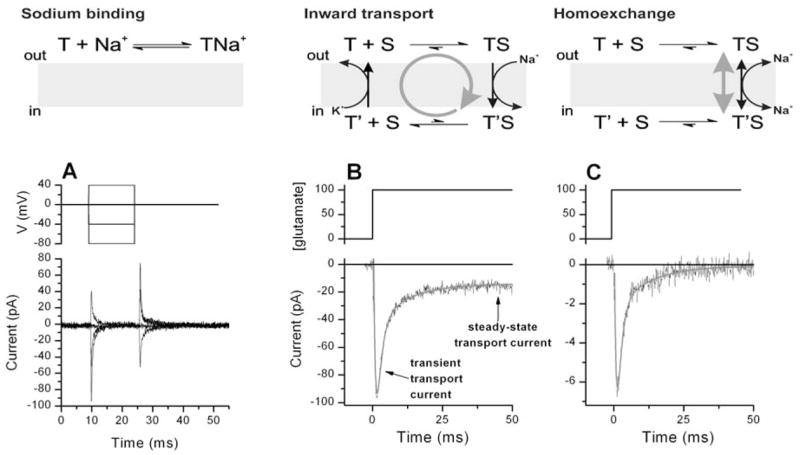
Upper panel: Graphic illustration of Na+ binding to the glutamate transporter (left panel) the inward transport mode (middle panel) and the homoexchange mode (right panel) employed in the experiments shown in A, B and C. The grey arrows indicate the general direction of the transporter reaction. Lower panel: (A) Transient currents induced by voltage jumps in the absence of transported substrate. The voltage jump protocol is indicated in the middle panel. The transient currents are caused by Na+ binding to the glutamate-free form of the transporter. (B) Transient and steady-state transport current induced in glutamate transporter-expressing cells by rapid extracellular application of 100 μM glutamate at time 0 as illustrated in the middle panel (Vm = 0 mV, inward transport mode). (C) Identical experiment as in (B), but glutamate transport is restricted to the homoexchange mode (see grey arrow in the reaction scheme shown in the top panel). Note the absence of a steady-state current. In the homoexchange mode the glutamate-induced transient current is caused by a voltage dependent redistribution of the glutamate-bound states of the transporter.
An alternative approach to determine the pre-steady-state kinetics of glutamate transporters is to rapidly apply glutamate to the transporter-containing membrane, thus perturbing the pre-existing steady-state while keeping the membrane potential constant (Grewer et al., 2001; Grewer et al., 2000). The subsequent relaxation to a new steady-state in the presence of glutamate can then be measured by recording the transport current. As shown in Fig. 4B, glutamate concentration jumps applied to the transporter are followed by a large, but rapidly-decaying inward transport current under inward transport conditions, indicating the existence of a rapid electrogenic process that is induced by glutamate binding to the transport protein (Watzke et al., 2001). The transient current consists of two components that decay with time constants about 10-fold different from each other, suggesting that two electrogenic reactions are involved. Which electrogenic reactions in the transport cycle generate these transient transport currents? First, Na+ binding to the empty transporter can be ruled out since the currents decrease when the extracellular sodium concentration is lowered (in the presence of saturating glutamate concentrations). The opposite effect would be expected if rapid, electrogenic Na+ binding to the transporter in the absence of glutamate would generate the current (Watzke et al., 2001). Secondly, the transient component of the transport current is unaffected by the presence or absence of intracellular K+ (Grewer et al., 2000) which is required for the relocation of the glutamate-free form of EAAC1 (Kanner & Bendahan, 1982). In this relocation reaction the binding sites for glutamate and Na+ ions are recycled from being exposed to the cytoplasm to face the extracellular side of the membrane. The lack of effect by K+ suggests that K+-dependent individual reaction steps of the transporter do not contribute to the transient transport current. Third, glutamate binding can be ruled out since glutamate binds to the transporter in its negatively charged form which would generate an outward current (Bendahan et al., 2000; Watzke et al., 2000). Interestingly, the transient current does not rise instantaneously, suggesting that the electrogenic reaction is preceded by an electroneutral reaction step which is, most likely, glutamate binding. This interpretation is in line with results from steady-state kinetic experiments that indicate voltage independence of the Km of the transporter for glutamate and non-transportable blockers (Grewer et al., 2000; Wadiche et al., 1995b). Together, these results suggest that the glutamate-induced transient transport current is caused by electrogenic reactions of the fully loaded transporter and, possibly, by glutamate translocation.
To test this hypothesis, the glutamate concentration jumps can be applied to the transporter in the homoexchange transport mode (Fig. 4C). In the homoexchange transport mode glutamate, as well as cotransported Na+ ions must be present on both sides of the membrane at saturating concentrations, whereas K+ is absent. This leads to an exchange of glutamate across the membrane in the absence of net transport. Under these conditions, the population of the transporter states is limited to those states that are associated with glutamate translocation. Unexpectedly, the transient transport current is also present under homoexchange conditions (Fig. 4C and (Watzke et al., 2001). Therefore, these results directly demonstrate that the electrogenicity is associated with individual reaction steps of the glutamate-bound forms of EAAC1. Similar to the forward transport mode, the transient transport current consists of two components which is indicative of two electrogenic processes. Each of these two electrogenic processes contributes about 25–30% to the total transporter electrogenicity. Therefore, taken together with voltage-dependent Na+ binding, these glutamate-dependent electrogenic reactions can still not account for the total electrogenicity of the glutamate transporter leaving other voltage-dependent reactions to be identified. Although the results indicate that the glutamate translocation reaction may be electrogenic, it can not be ruled out that rapid intracellular Na+ dissociation is the actual electrogenic step which follows the slower, electroneutral glutamate translocation. This problem in interpretation arises because the affinity of the transporter for intracellular Na+ is not known. Therefore, intracellular Na+ dissociation may not have been prevented in the homoexchange experiments since the internal sodium binding site(s) may not have been saturated. In any case, the finding that reactions of the glutamate-dependent half cycle of the carrier are associated with electrogenicity is in contrast with the observation that electrogenicity of many other sodium-coupled transporters is restricted to reactions of the empty transporter or the sodium-bound form for the transporter that has not yet bound substrate (Forlani et al., 2001; Forster et al., 1998; Hilgemann & Lu, 1999; Loo et al., 1993; Lu & Hilgemann, 1999; Mackenzie et al., 2003; Mager et al., 1993). Whether this difference in transport mechanism reflects a general difference between the glutamate transporters and other sodium coupled transporters, or if other transporters also display electrogenicity associated with the substrate translocation half cycle remains to be determined. Interestingly, it appears that the electrogenicity is distributed over many partial reactions of the glutamate transporter (Watzke et al., 2001). This interpretation is consistent with current models of the Na+/K+-ATPase in which four partial reactions in the Albers-Post cycle were identified to be electrogenic (Bamberg, Clarke & Fendler, 2001).
Rate limitation of glutamate transport and voltage dependence at steady state
It was initially proposed by Hediger and colleagues in 1994 that glutamate translocation is the rate limiting step in the transport cycle (Kanai et al., 1994). In contrast to this proposal, recent evidence based on pre-steady-state kinetic experiments supports an interpretation in which the reaction steps associated with glutamate binding and translocation are rapid (Grewer et al., 2000; Auger & Attwell, 2000; Otis & Kavanaugh, 2000). The time constants of these reactions are in the range of 1–8 ms, in contrast to the 30 ms cycle time of EAAC1 (Grewer et al., 2000), indicating that these reactions do not limit steady-state turnover. One other possibility is that reactions of the potassium-dependent branch of the transport cycle are rate limiting. This hypothesis can be tested by performing pre-steady-state experiments after reducing the intracellular [K+] from its physiological value of 140 mM to 5 mM, which should dramatically slow any K+-dependent process. Such a reduction in the intracellular K+ concentration results in a 10-fold reduction of the steady-state transport rate, whereas rate constants associated with the glutamate-dependent half-cycle of the transporter are virtually unaffected. Therefore, it now appears likely that the K+-induced relocation of the glutamate-free form of the transporter is mainly responsible for limiting the steady-state turnover rate (Bergles, Tzingounis & Jahr, 2002; Grewer et al., 2000; Larsson et al., 2004). This interpretation is consistent with the fact that in the presence of intracellular Cs+ instead of K+ the steady-state transport current is reduced, whereas the pre-steady-state component of the current is not affected (Bergles et al., 2002). Thus, Cs+ can substitute for K+ and slows the turnover by slowing the relocation reaction of the transporter.
The rate limiting step in a transport cycle is generally the main determinant for the properties of the transporter at steady state. How is it then possible that the stationary transport rate is exponentially accelerated by negative membrane potentials (Arriza et al., 1994; Barbour, Brew & Attwell, 1988; Grewer et al., 2000), when outward movement of the positively-charged K+ ion should be inhibited by negative voltages? The answer to this question is most likely that the positive charge of the outward moving K+ in the relocation step is over-compensated by intrinsic negative charges of the transport protein that move across the electric field of the membrane in the same direction and in concert with K+. In this interpretation, the intrinsic charge of the transporter must be more negative than −1. It can be speculated that this negative charge serves to counterbalance the positive charge of cations bound to the transporter, and, therefore, increases their mobility within the membrane. Consistently, negatively charged cation binding sites have been postulated for other Na+-coupled secondary transporters (Loo et al., 1993; Murer et al., 2000) and cation pumps (Koenderink et al., 2003; MacLennan, Rice & Green, 1997; Toyoshima et al., 2000). Thus, negatively charged cation binding sites appear to be a general feature of cation-transporting membrane proteins.
The charge compensation model discussed in the preceding paragraph predicts that the inwardly-directed current should be associated with the K+-induced relocation of the glutamate transporter. So far, this current has only been observed at steady-state. However, it should be possible to isolate this current by performing pre-steady-state voltage jump experiments in the presence of K+ on both sides of the membrane in order to lock the transporter in the K+-exchange mode. Such experiments have yet to be performed in order to obtain direct evidence for the charge compensation mechanism.
Kinetics of different glutamate transporter subtypes and splice-variants
Only limited information is available in the literature about the kinetics of the various glutamate transporter subtypes and splice variants. So far, pre-steady-state experiments have been carried out on the recombinantly expressed subtypes EAAT1, EAAT2 (GLT-1), EAAC1, the splice variant GLT1v, and some transporters natively expressed in neurons and astrocytes. From these data it appears that EAAT2 is the fastest of the three transporter subtypes. At physiological membrane potentials (−80 to −90 mV) the turnover rate of EAAT2 was estimated to be 100 s−1 (Bergles & Jahr, 1998; Bergles et al., 2002) which is about 1.5-times faster than the turnover rate determined for EAAC1 (Grewer et al., 2000) and 6-times faster than that determined for EAAT1 (Wadiche & Kavanaugh, 1998) under similar conditions. The decay of glutamate-induced pre-steady-state anion currents is also fastest for EAAT2 and occurs with a time constant of 1– 1.5 ms (Bergles & Jahr, 1997; Bergles & Jahr, 1998; Bergles et al., 2002; Otis & Kavanaugh, 2000), whereas the time constants for EAAC1 have been determined to be 2.5 ms (Grewer et al., 2000; Watzke et al., 2001) and 14 ms for EAAT1 (Wadiche & Kavanaugh, 1998). A splice variant of GLT1, GLT1v (Fig. 2), however, exhibits kinetics indistinguishable of those of GLT1 suggesting that alternate C-terminal splicing of GLT1 does not have significant effects on its glutamate transport properties (Sullivan et al., 2004). This result is entirely consistent with the earlier work of Utsunomiya-Tate et al. (1997) and the subsequent data from Chen et al. (2002). Instead of showing different functional properties, both proteins, GLT1 and GLT1v, exhibited distinct cellular and subcellular distribution patterns in retinal neurons as well as in brain astrocytes. Thus, the purpose of differential targeting of GLT-1 and GLT1v most likely is not to create different glutamate clearance properties in different CNS microdomains, such as in areas immediately around synapses, compared with areas distal to such. Instead, Sullivan et al. (2004) suggested that differential splicing and the subsequent differential targeting of GLT1 in distinct membrane domains may be a mechanism that facilitates independent regulation of the immediate perisynaptic glutamate level and the level of glutamate in the extracellular space in general.
To summarize, diverse glutamate transporter phenotypes are expressed in the CNS including different subtypes and various splicing variants, a situation which might be reminiscent of that of structural and functional heterogeneous glutamate receptor expression in neuronal tissue (reviewed in: Dev, Nakanishi & Henley, 2001; Nakanishi & Masu, 1994; Perez-Otano & Ehlers, 2004). Thus, different subtypes and splicing variants of glutamate transporters expressed specifically in different subsets of cells with differential subcellular distribution pattern might reflect different functional roles of glutamate transporters in the CNS: glial glutamate uptake for clearance of extracellular glutamate, neuronal reuptake of synaptically released glutamate, transporter inherent feedback systems to control cell excitability, uptake for metabolic reasons, and light-dependent uptake in the retina might all require different transporter characteristics.
The mechanism of proton transport by glutamate transporters
Inwardly-directed glutamate transport is accompanied by intracellular acidification (Zerangue & Kavanaugh, 1996b). Several models were proposed to explain this acidification effect. It was proposed that either protons are co-transported together with glutamate (Erecinska et al., 1986; Zhang, Pines & Kanner, 1994), or OH− ions are counter-transported together with K+ (Bouvier et al., 1992). In a third model, it was suggested that H+ is counter-exchanged with K+ in the relocation reaction of the glutamate-free transporter which should also result in net inward movement of protons (Auger & Attwell, 2000). However, the latter model can not explain the presence of a modulatory effect of protons on the affinity of the transporter for glutamate in the absolute absence of potassium ions. Taken together, the recent data on EAAT proton transport support the model of proton co-transport. Specifically, the distinct kinetic isotope effect in the presence of D2O on the partial reactions linked to glutamate translocation is in agreement with the co-transport model, but it rules out a model in which OH− is counter-transported (Watzke et al., 2000). Since the mass ratio between OH− and OD− is close to 1, a deuterium effect should not be observed if hydroxide is the transported species.
Since protons are the transported species, the question arises as to how they are co-transported with glutamate. It was initially proposed that glutamate is transported by EAATs in its acidic, protonated form (Zerangue & Kavanaugh, 1996b), or that H+ protonates the γ-carboxylate of glutamate once it is bound to the transporter (Slotboom et al., 1999a). In contrast to this interpretation, Kanner and collegues (Bendahan et al., 2000) found that glutamate transporters can be converted into transporters for neutral amino acids by replacing an arginine residue in position 446 (EAAC1 nomenclature) with cysteine. This result strongly supports a model in which the γ-carboxylate of glutamate associates with the guanidinium group of the arginine 446. This is only possible when the γ-carboxylate is present in its negatively charged form. Furthermore, the structurally related neutral amino acid transporters (ASCTs) which possess a cysteine in the homologous position, but not an arginine, transport acidic amino acids only when the pH is lowered (Vadgama & Christensen, 1984). This suggests that in the absence of a positive charge in position 446 glutamate can not bind as the negatively charged species, but it can only bind as the uncharged zwitterion. Instead, proton co-transport is most likely mediated by protonation of a titratable group intrinsic to the glutamate transporter protein. It has been recently suggested that a glutamate residue in position 373 (EAAC1) forms part of this protonation site. When glutamate (E373) was replaced by glutamine (E373Q) the mutant transporter is still able to operate in the homoexchange mode (Grewer et al., 2003; Pines, Zhang & Kanner, 1995), indicating that negative charge on E373 is not required for the glutamate translocation reaction. However, glutamate transport by the E373Q mutant transporter was found to be insensitive to extracellular pH (Grewer et al., 2003). Interestingly, ASCT1 and ASCT2 contain a glutamine residue in the comparable position (Q392 in ASCT2). In contrast to EAAC1, ASCT does not cotransport protons (Broer et al., 2000; Grewer et al., 2003). Moreover, pH sensitivity could be engineered into ASCT2 by introducing the reverse mutation, Q392E (Grewer et al., 2003). Together, these results suggest that E373 has to be protonated (neutral) to allow glutamate translocation. Another potential amino acid residue to be involved in proton transport is the histidine 295 (EAAC1 nomenclature) (Zhang et al., 1994) which is conserved among the EAAT family. Because of its pKa value near the physiological pH range, histidine is generally thought to be a likely mediator of proton transport in general.
Glutamate transporter associated anion conductance
Glutamate transport is associated with an anion conductance. This anion conductance is activated by glutamate binding to the transporter. The anion conductance is not stoichiometrically coupled to the transport of glutamate, but it is activated by glutamate only under ionic conditions that also favor glutamate translocation (e.g., external sodium) (Eliasof & Jahr, 1996; Picaud et al., 1995; Wadiche et al., 1995a). Hence, glutamate uptake can take place already in the absence of chloride ions, but no kinetically-coupled anion flux occurs without the activation of the glutamate transporter by substrate and ion binding (Wadiche et al., 1995a). Thus, glutamate transporters have mixed characteristics that are, in part, carrier-like and, in part, similar to ligand-gated chloride channels. In analogy to ligand-gated ion channels it was proposed that anion permeation may be through a pore formed in the center of the multimeric subunit complex (Eskandari et al., 2000). The anion conductance is selective for hydrophobic anions. The anion permeability follows the sequence PSCN− > PNO3− > PI− > PBr− > PCl− > PF− ≫ PGluconate− (Melzer et al., 2003; Wadiche et al., 1995a; Wadiche & Kavanaugh, 1998).
The mechanism of anion permeation is not yet fully understood. Although activation of a secondary anion-conducting protein by the glutamate transporter can not be totally ruled out at present, substantial evidence supports the idea that the permeation pathway is an intrinsic part of the glutamate transporter protein. Initially, it was suggested that glutamate, once it is bound to the transporter, provides part of the permeation pathway for anions (Wadiche et al., 1995a). However, recent findings do not favor this interpretation. First, it is possible to knock out transport by site-directed mutagenesis without affecting the properties of the anion conductance (Ryan & Vandenberg, 2002; Seal et al., 2001). Second, the permeation properties of the anion conductance are altered by mutations in the TMD2-region of the transporters without affecting the rate of glutamate transport (Ryan, Mitrovic & Vandenberg, 2004). Thus, it appears that glutamate and anions move through the transporter by taking different pathways that can be modulated independently of each other. Third, activation of the anion conductance is delayed with respect to glutamate binding to the transporter (Fig. 5A), suggesting that glutamate binding alone is not sufficient to provide the anion permeation pathway (Watzke et al., 2001). Glutamate binding, however, triggers the binding of Na+ to the transporter and the Na+-bound form is anion conducting. Therefore, it appears that the anion conductance is not a glutamate-gated anion channel, but instead a sodium-gated anion channel (Watzke et al., 2001). This interpretation is supported by the finding that Na+ binding to the transporter in the absence of glutamate can already activate a leak anion conducting pathway (Watzke et al., 2001). A hypothetical model of the anion conductance activation is shown in Fig. 5B.
Figure 5.
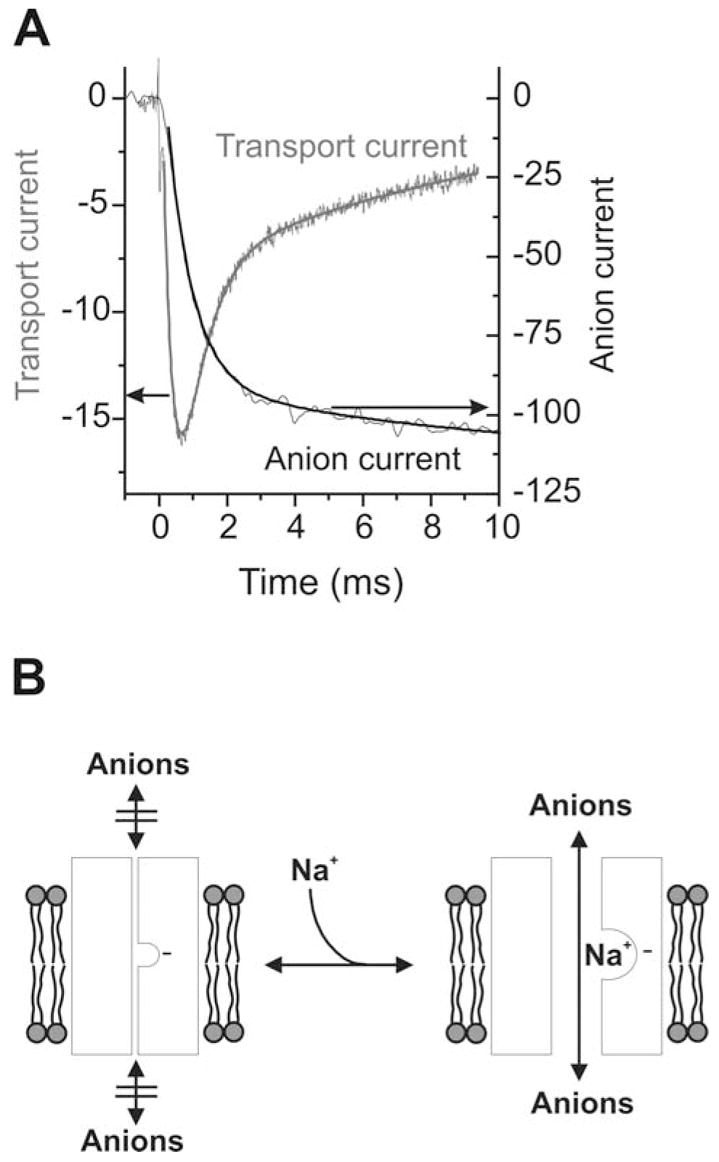
Mechanism of the anion conductance catalyzed by the glutamate transporter EAAC1 (Watzke et al., 2001). (A) Transport and anion currents generated by EAAC1 after rapid application of glutamate at time 0. Currents were recorded in the absence (transport current) and presence (anion current) of intracellular SCN−. Both current components show biphasic behaviour with similar time constants of the two phases (Watzke et al., 2001). The rapidly-decaying phase of the transport current (left y-axis), which reflects Na+ binding to the transporter (Watzke et al., 2001), precedes the activation of the anion current (right y-axis). Although the experimentally measured anion current represents a sum of transport current and pure anion current, the transport component can be neglected because of its comparatively small magnitude. (B) A model of the Na+-dependent gating of the glutamate transporter associated anion conductance. The anion conducting conformation is indicated by I. The empty Na+ binding site is negatively charged, indicated by the – sign.
One possible physiological function of the anion conductance would be that the inwardly-directed anion flux (at the resting potential of neurons) balances the inflow of positively-charged sodium ions during glutamate inward transport (Fig. 3B), and thus, keeps the membrane potential at a hyperpolarized level that favors Na+ entry and glutamate uptake (Arriza et al., 1997; Sonders & Amara, 1996). In other words, the anion conductance keeps glutamate transport more elektroneutral. A second physiological role might be related to a direct effect on synaptic signaling, as the glutamate-regulated chloride permeability can affect the excitability of the membrane. In fact, the size of the chloride conductance relative to transport capacity can vary dramatically between different transporter subtypes (Arriza et al., 1997; Arriza et al., 1994; Fairman et al., 1995). For example, the glutamate-elicited current in EAAT5 is carried largely by chloride ions, in contrast to the rather small but measurable chloride conductance associated with GLAST1, GLT1 and EAAC1 activity. Comparing the uptake rate (the amount of transported glutamate molecules per minute) with the magnitude of the chloride conductance of each transporter subtype, the following reverse pattern arises: Uptake systems with a high transport rate, like GLT1, GLAST1 or EAAC1, usually show a relative low chloride conductance, whereas the transport systems EAAT4 and EAAT5 with a low transport rate, or in particular the photoreceptor glutamate transporters, are associated with a high chloride conductance (Eliasof et al., 1998; Larsson et al., 1996; Picaud et al., 1995). Thus, the glutamate transport rate is in a reversed relationship to the chloride conductance of the respective transporter subtypes suggesting that GLT1, GLAST1 and EAAC1 act as classical glutamate uptake systems while EAAT4, EAAT5 and especially the photoreceptor glutamate transporter behave like Na+/glutamate-gated chloride channels (inhibitory glutamate receptor).
General mechanism for glutamate transport
The currently available data on glutamate transport are summarized in a kinetic mechanism shown in Fig. 6 (Watzke et al., 2001). This mechanism represents the minimum number of reaction steps necessary to describe the experimental observations. The mechanism incorporates sequential binding of Na+ and glutamate to the transporter, where Na+ binds in different reaction steps to both the glutamate-free and glutamate-bound conformations of the excitatory amino acid transporters (in the study of Watzke et al., 2001, EAAC1 was used). Glutamate translocation is possible only if the transporter is fully loaded with both glutamate and three sodium ions. Protonation of the transporter takes place before glutamate has bound. In the model, protonation is placed after binding of the first sodium ion to the transporter, according to Larsson et al. (2004), although the precise sequence of Na+ and H+ binding is not yet known. Low pH increases the affinity of the transporter for Na+ in the first sodium binding step, and, thus, shifts the Q-V curve for electrogenic Na+ binding to the right (Larsson et al., 2004). This could be explained by proton binding either before or after Na+ binding. Potassium ion transport occurs in steps separate from the glutamate translocation reaction, to be precise, in the relocation halfcycle of the transporter (Kanner & Bendahan, 1982). Four conformational states of the glutamate transporter are included in the model. Three of these states have been directly observed by voltage-clamp fluorimetry (Larsson et al., 2004). A fourth state was included because of kinetic evidence for a conformational change that is induced by glutamate binding which precedes glutamate translocation (Watzke et al., 2001). Most likely, even more conformational states will be identified in the future.
Figure 6.

A kinetic model of glutamate transport including 4 anion conducting states (indicated by I and II). The electrogenic individual reaction steps are marked by the shaded boxes. Time constants for individual reactions in the forward transport direction are given for physiological membrane potentials (−70 to −80 mV) typical for neurons (Kandel, Schwartz & Jessell, 1995). Positive and negative charges of the transporter binding sites are indicated by the + and – signs, respectively. G = glutamate, N = Na+, H = H+, K = K+.
Voltage dependence is assigned to four individual reaction steps in the transport cycle, as marked by the shaded areas in Fig. 6: Na+ binding to the empty transporter, Na+ binding to the glutamate-bound transporter, translocation of the fully loaded transporter form, and relocation of the K+-bound transporter. The sequence of dissociation of glutamate and Na+ on the intracellular side is not yet known so it is not specified in this mechanism. Only two of the three Na+ binding reactions are shown in Fig. 6, because it is unknown whether the third sodium ion binds to the transporter before or after glutamate. However, binding before glutamate appears to be preferred in recently proposed mechanisms (Bergles et al., 2002; Larsson et al., 2004).
The anion conductance is assigned to states of the transporter that have either bound Na+, or are fully loaded with Na+ and the neurotransmitter (Grewer et al., 2000; Otis & Kavanaugh, 2000; Wadiche & Kavanaugh, 1998). In the model shown here, the anion conducting states are only populated when the glutamate binding site faces the extracellular side of the membrane. However, considerable evidence is available that some inwardly facing conformations are also anion conducting (Billups et al., 1996; Watzke & Grewer, 2001). Specifically, blockers of glutamate transport inhibit a leak anion current when applied to the intracellular side, suggesting that the glutamate-free transporter with the binding site facing the cytoplasm is also anion conducting.
Impact of glutamate transporters on synaptic glutamate receptor activation
The glial and neuronal localization of glutamate transporters at the synapse and the transport processes active during a synaptic event are schematically shown in Fig. 1. Since it was first discovered that the amplitude and/or the decay time of excitatory postsynaptic currents (EPSCs) is increased after blocking synaptic glutamate transporters in some types of excitatory synapses, it is clear that the transporters must play a crucial role in the glutamatergic signal transmission process other than just maintaining the steady-state glutamate concentration in the extracellular space at a low level (Barbour et al., 1994; Diamond & Jahr, 1997; Mennerick et al., 1999; Mennerick & Zorumski, 1994; Overstreet et al., 1999; Tong & Jahr, 1994; Turecek & Trussell, 2000). Although it is probable that the function of the transporters is to shape the spatiotemporal profile of the glutamate concentration in the synaptic cleft, the exact physiological role of the glutamate transporters in synaptic transmission is still unclear. Recently, progress towards understanding this role was made mainly by being able to record evoked synaptic transport currents (Bergles & Jahr, 1997; Diamond, Bergles & Jahr, 1998; Mennerick & Zorumski, 1994; Otis, Kavanaugh & Jahr, 1997) in combination with the better understanding of the microscopic kinetics of the glutamate transporter subtypes involved (Grewer et al., 2000; Otis & Jahr, 1998; Otis & Kavanaugh, 2000; Wadiche & Kavanaugh, 1998; Watzke et al., 2001). One hypothesis put forward suggests that the transporters affect the concentration of glutamate in the synaptic cleft by buffering glutamate through rapid binding to the large number of transporter binding sites present within the synaptic cleft (up to 10,000 per μm2) (Diamond & Jahr, 1997; Lehre & Danbolt, 1998; Tong & Jahr, 1994). Consistent with this hypothesis, binding of glutamate to the transporters has been shown to be rapid, occurring with a rate constant of 1–2.107 M−1s−1 (Grewer et al., 2000; Otis & Jahr, 1998; Wadiche & Kavanaugh, 1998). Therefore, glutamate will bind to the transporters at the synapse within 10–100 μs at typical released concentrations (approximately 1 mM, (Clements et al., 1992). This glutamate-buffering hypothesis solved the problem that steady-state glutamate transport was, by far, too slow to account for the experimentally observed rapid removal of glutamate from the synaptic cleft. However, it should be noted that turnover rates for glutamate transport have been typically determined at room temperature. Given the high temperature dependence of glutamate transport with a Q10 of about 3 (Wadiche and Kavanaugh, 1998) compared to the weak temperature dependence of diffusion, removal of glutamate from the synapse by steady-state turnover of the transporters may be more favourable at the physiological temperature of 37 °C.
A second, less well defined parameter important for the buffering hypothesis is the rate of glutamate dissociation from the transporter binding site. Together with the rate of translocation, this value determines whether free glutamate in the synaptic cleft is in rapid equilibrium with the transporter-bound form or if it is permanently trapped by the transporter once it is bound. Pre-steady-state kinetic experiments indicate that glutamate dissociation from EAAC1 occurs with a time constant of about 500 μs (Grewer et al., 2000) which is about 10-fold faster compared to glutamate translocation. Therefore, it appears likely that glutamate can bind to and dissociate from the transporter binding sites multiple times before it is either translocated across the membrane or diffuses out of the synaptic cleft. In terms of buffering properties, the transporters would be considered to be a rapid buffer which is comparable to, for example, BAPTA as a rapid buffer for calcium ions (Neher, 1998). In contrast, EGTA, is a slow buffer for Ca2+ because of its slow rates of binding to calcium and dissociation of calcium (Naraghi, 1997). Analogous to calcium buffers, the glutamate transporters in the synapse are predicted to slow diffusion of glutamate out of the synaptic cleft. Although a topic of debate (Barbour, 2001), slowed glutamate diffusion is an interesting effect because it may lead to prolonged activation of synaptic glutamate receptors at low concentrations of glutamate (Rusakov & Kullmann, 1998a; Rusakov & Kullmann, 1998b). The temporal profile of the synaptic glutamate concentration in the presence and absence of glutamate transporters predicted by numerical simulations is shown in Fig. 7A. The presence of glutamate transporter binding sites at the synapse leads to a marked biphasic behaviour in the decay profile. The fast phase is dominated by glutamate transporter-mediated glutamate buffering, whereas the slow phase is dominated by diffusion and glutamate trapping by the transporters.
Figure 7.
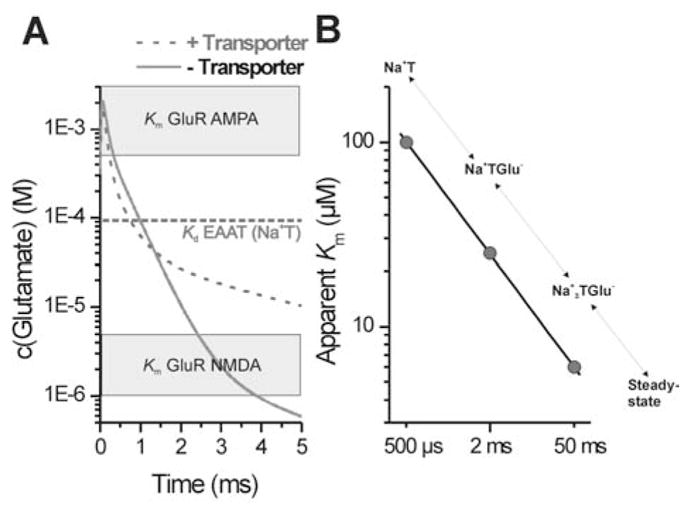
(A) The time dependence of the synaptic glutamate concentration in the presence (dashed line) or absence (solid line) of glutamate transport as calculated by numerical simulations using a formalism for diffusive initial value problems with radial symmetry as described in (Smith, 1965) with a diffusion coefficient for glutamate of 10−6 cm2/s. The diameter of the synapse was assumed to be 0.5 μm. The glutamate transporter concentration was set to 1 mM and the concentration of released glutamate was 10 mM. The binding rate of the transporter was 2.107 M−1s−1 and the dissociation rate 2000 s−1. The translocation rate was set to 400 s−1. Apparent Km values for AMPA receptors (0.5 – 5 mM) and NMDA receptors (1 – 5 μM) are indicated by grey boxes. The intrinsic Kd value for EAATs of about 100 μM which reflects the intrinsic affinity of the transporters for glutamate immediately after pre-synaptic release is indicated by the dotted line. (B) The apparent Km values of the glutamate transporter subtype EAAC1 for glutamate are plotted as a function of time after glutamate release from the pre-synaptic cell (circles). The conformational states of EAAC1 corresponding to the respective point in time after glutamate release are shown on top of the data points. The apparent affinity for glutamate increases from 100 μM immediately after pre-synaptic glutamate release to 5 μM at steady state.
In addition to the buffering kinetics, a second critical parameter defining the buffer properties is the concentration of the maximum buffering power which depends on the Km of the buffer for its substrate. For glutamate transporters, this Km seems to be time dependent, as shown in Fig. 7B. Initially, glutamate binds to the transporters with relatively low intrinsic affinity (Kd = 50 – 100 μM for EAAC1) (Bergles et al., 2002; Grewer et al., 2000). As the glutamate:transporter complex moves along the transport pathway, binding becomes tighter with a final affinity of about 5 μM at steady-state (EAAC1) (Grewer et al., 2000). This reflects the trapping of glutamate by the transporter as additional states following the initial glutamate:transporter complex are occupied. Thus, the buffering properties of the transporter are time dependent (Grewer et al., 2000). Interestingly, the intrinsic Kd of the transporters for glutamate is in-between the relatively high apparent Km for AMPA (DL-α-amino-3-hydroxy-5-methyl-4-isoxazole-4-propionic acid) receptors (0.5 – 5 mM) (Dingledine et al., 1999 ; Hausser & Roth, 1997; Li, Oswald & Niu, 2003; Mosbacher et al., 1994) and the low Km of the high-affinity NMDA (N-Methyl-D-aspartic acid) receptors (1 – 5 μM) (Anson et al., 1998; Laube et al., 1997; Lester & Jahr, 1992). Thus, rapid buffering of glutamate by the transporters may serve as a discriminating factor in shaping the temporal profile of the glutamate concentration such that the low-affinity AMPA receptors experience only a very short burst of glutamate concentration. This is in contrast to the high-affinity NMDA receptors which are activated by a prolonged low glutamate concentration within the synaptic cleft (Fig. 7A and B). Third, the effect of glutamate translocation has to be considered. Recent kinetic evidence indicates that glutamate translocation can be as rapid as a 1–3 milliseconds in GLT1 (Otis & Kavanaugh, 2000) and EAAC1 (Grewer et al., 2000). Thus, buffering may only play a role on very short, submillisecond timescales, whereas the transporters then rapidly remove glutamate from the synapse by translocation. Since the concentration of transporters at the synapse is sufficiently high to capture most of the released transmitter molecules, each transporter completes only one halfcycle during a synaptic event. Thus, a high turnover number is not necessary for glutamate transport because there is enough time in-between synaptic events to regenerate the transporter binding sites to be exposed to the extracellular side.
Concluding remarks
This review has summarized data available in the literature on the kinetic and mechanistic properties of glutamate transporters. It is becoming clear that glutamate transporters are highly complex transmembrane proteins that move glutamate across membranes by mechanism that we have only begun to understand. This complexity is exemplified by many studies on structure-function, kinetics, regulation, and physiological function of these transporters. The mechanism of transport, for example, consists of multiple reaction steps, of which at least four are electrogenic. These individual reaction steps occur on time scales ranging from 100 μs to 100 ms. The challenge for the future will be to determine the structural basis of this complex functional behavior. Although structure-function studies based on site-directed mutagenesis will be invaluable in this respect, much will also depend on the determination of the three-dimensional structure of the transporter. Fortunately, high-resolution structures of secondary transporters are becoming available now (Abramson et al., 2003), and a structure of a bacterial glutamate transporter was published recently (Yernool at eal., 2004), while this article was under review. Although this structure answers many questions regarding transmembrane topology, assembly of subunits, and arrangement of TMDs, a transport pathway for glutamate and the co-transported ions is not immediately obvious from the structure. Thus, more functional work will need to be done to obtain a clear picture of how glutamate is moved across the membrane. The structure, however, will serve as a valuable scaffold for more specific site-directed mutagenesis to determine the transport pathway.
Complexity is also reflected by the large number of transporter subtypes and splice variants that have been identified, along with their differential expression in different cell types and sub-cellular structures. At present, it is not well understood why an organism would need such a variety of transport proteins for one substrate. Although the current understanding is that the various subtypes and splice variants are important for targeting of expression in different cell types, it will be also necessary to determine the possible differences in function and regulation of these subtypes. Thus, it is likely that glutamate transporters will remain an important target for future biophysical, cell-biological, and pharmacological research.
Acknowledgments
The authors thank Dr. Georg Nagel for critical reading of the manuscript and helpful comments. This work was supported by the National Institutes of Health Grant R01-NS0493 to CG and by the Deutsche Forschungsgemeinschaft Grants GR 1393/2-2,3 to CG and RA 753/1-1,2 to TR. CG and TR are also grateful for support by the Max-Planck-Institute for Biophysics and Brain Research, respectively.
Abbreviations
- CNS
Central nervous system
- ALS
Amyotrophic lateral sclerosis
- EAAT
Excitatory amino acid transporter
- EAAC1
Excitatory amino acid carrier 1
- GLT1
Glutamate transporter 1
- GltP
Prokaryotic glutamate transporter
- GLAST
Glutamate aspartate transporter
- ASCT
Alanine Serine Cysteine Transporter
- GluR
Glutamate receptor
- GS
Glutamine synthase
- Gln
Glutamine
- GABA
γ-Aminobutyric acid
- NMDA
N-Methyl-D-aspartic acid
- AMPA
DL-α-amino-3-hydroxy-5-methyl-4-isoxazole-4-propionic acid
- PDZ
PSD-95, Dlg and ZO-1/2 (motif)
- ATP
Adenosinetriphosphate
- EGTA
ethylene glycol-bis-(beta-aminoethyl ether)-N,N,N′,N′-tetracetic acid
- BAPTA
O,O′-Bis(2-aminophenyl)ethyleneglycol-N,N,N′,N′-tetraacetic acid
- RL
Re-entrant loop
- TMD
Transmembrane domain
References
- Abramson J, Smirnova I, Kasho V, Verner G, Kaback HR, Iwata S. Structure and Mechanism of the Lactose Permease of Escherichia coli. Science. 2003;301:610–15. doi: 10.1126/science.1088196. [DOI] [PubMed] [Google Scholar]
- Amara SG. Methods Enzymol. Academic Press; San Diego: 1998. Neurotransmitter transporters. [Google Scholar]
- Anson LC, Chen PE, Wyllie DJ, Colquhoun D, Schoepfer R. Identification of amino acid residues of the NR2A subunit that control glutamate potency in recombinant NR1/NR2A NMDA receptors. J Neurosci. 1998;18:581–9. doi: 10.1523/JNEUROSCI.18-02-00581.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arriza JL, Eliasof S, Kavanaugh MP, Amara SG. Excitatory amino acid transporter 5, a retinal glutamate transporter coupled to a chloride conductance. Proc Natl Acad Sci U S A. 1997;94:4155–160. doi: 10.1073/pnas.94.8.4155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arriza JL, Fairman WA, Wadiche JI, Murdoch GH, Kavanaugh MP, Amara SG. Functional comparisons of three glutamate transporter subtypes cloned from human motor cortex. J Neurosci. 1994;14:5559–5569. doi: 10.1523/JNEUROSCI.14-09-05559.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arriza JL, Kavanaugh MP, Fairman WA, Wu YN, Murdoch GH, North RA, Amara SG. Cloning and expression of a human neutral amino acid transporter with structural similarity to the glutamate transporter gene family. J Biol Chem. 1993;268:15329–32. [PubMed] [Google Scholar]
- Auger C, Attwell D. Fast removal of synaptic glutamate by postsynaptic transporters. Neuron. 2000;28:547–58. doi: 10.1016/s0896-6273(00)00132-x. [DOI] [PubMed] [Google Scholar]
- Backus KH, Kettenmann H, Schachner M. Pharmacological characterization of the glutamate receptor in cultured astrocytes. J Neurosci Res. 1989;22:274–82. doi: 10.1002/jnr.490220307. [DOI] [PubMed] [Google Scholar]
- Balcar VJ, Johnston GA. The structural specificity of the high affinity uptake of L-glutamate and L-aspartate by rat brain slices. J Neurochem. 1972;19:2657–66. doi: 10.1111/j.1471-4159.1972.tb01325.x. [DOI] [PubMed] [Google Scholar]
- Bamberg E, Clarke RJ, Fendler K. Electrogenic Properties of the Na+,K+-ATPase Probed by Presteady State and Relaxation Studies. J Bioenerg Biomembr. 2001;33:401–405. doi: 10.1023/a:1010667407003. [DOI] [PubMed] [Google Scholar]
- Barbour B. An evaluation of synapse independence. J Neurosci. 2001;21:7969–84. doi: 10.1523/JNEUROSCI.21-20-07969.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barbour B, Brew H, Attwell D. Electrogenic glutamate uptake in glial cells is activated by intracellular potassium. Nature. 1988;335:433–5. doi: 10.1038/335433a0. [DOI] [PubMed] [Google Scholar]
- Barbour B, Keller BU, Llano I, Marty A. Prolonged presence of glutamate during excitatory synaptic transmission to cerebellar Purkinje cells. Neuron. 1994;12:1331–43. doi: 10.1016/0896-6273(94)90448-0. [DOI] [PubMed] [Google Scholar]
- Behrens PF, Franz P, Woodman B, Lindenberg KS, Landwehrmeyer GB. Impaired glutamate transport and glutamate-glutamine cycling: downstream effects of the Huntington mutation. Brain. 2002;125:1908–22. doi: 10.1093/brain/awf180. [DOI] [PubMed] [Google Scholar]
- Bendahan A, Armon A, Madani N, Kavanaugh MPBIK. Arginine 447 plays a pivotal role in substrate interactions in a neuronal glutamate transporter. J Biol Chem. 2000;275:37436–42. doi: 10.1074/jbc.M006536200. [DOI] [PubMed] [Google Scholar]
- Bergles DE, Jahr CE. Synaptic activation of glutamate transporters in hippocampal astrocytes. Neuron. 1997;19:1297–308. doi: 10.1016/s0896-6273(00)80420-1. [DOI] [PubMed] [Google Scholar]
- Bergles DE, Jahr CE. Glial contribution to glutamate uptake at Schaffer collateral-commissural synapses in the hippocampus. J Neurosci. 1998;18:7709–16. doi: 10.1523/JNEUROSCI.18-19-07709.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bergles DE, Tzingounis AV, Jahr CE. Comparison of Coupled and Uncoupled Currents during Glutamate Uptake by GLT-1 Transporters. J Neurosci. 2002;22:10153–10162. doi: 10.1523/JNEUROSCI.22-23-10153.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Billups B, Attwell D. Modulation of non-vesicular glutamate release by pH. Nature. 1996;379:171–4. doi: 10.1038/379171a0. [DOI] [PubMed] [Google Scholar]
- Billups B, Rossi D, Attwell D. Anion conductance behavior of the glutamate uptake carrier in salamander retinal glial cells. J Neurosci. 1996;16:6722–31. doi: 10.1523/JNEUROSCI.16-21-06722.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Binda F, Bossi E, Giovannardi S, Forlani G, Peres A. Temperature effects on the presteady-state and transport-associated currents of GABA cotransporter rGAT1. FEBS Lett. 2002;512:303–7. doi: 10.1016/s0014-5793(02)02271-8. [DOI] [PubMed] [Google Scholar]
- Biscoe TJ, Evans RH, Francis AA, Martin MR, Watkins JC, Davies J, Dray A. D-alpha-Aminoadipate as a selective antagonist of amino acid-induced and synaptic excitation of mammalian spinal neurones. Nature. 1977;270:743–5. doi: 10.1038/270743a0. [DOI] [PubMed] [Google Scholar]
- Borre L, Kanner BI. Arginine 445 Controls the Coupling between Glutamate and Cations in the Neuronal Transporter EAAC-1. J Biol Chem. 2004;279:2513–2519. doi: 10.1074/jbc.M311446200. [DOI] [PubMed] [Google Scholar]
- Bouvier M, Szatkowski M, Amato A, Attwell D. The glial cell glutamate uptake carrier countertransports pH-changing anions. Nature. 1992;360:471–4. doi: 10.1038/360471a0. [DOI] [PubMed] [Google Scholar]
- Brew H, Attwell D. Electrogenic glutamate uptake is a major current carrier in the membrane of axolotl retinal glial cells. Nature. 1987;327:707–9. doi: 10.1038/327707a0. [DOI] [PubMed] [Google Scholar]
- Broer A, Brookes N, Ganapathy V, Dimmer KS, Wagner CA, Lang F, Broer S. The astroglial ASCT2 amino acid transporter as a mediator of glutamine efflux. J Neurochem. 1999;73:2184–94. [PubMed] [Google Scholar]
- Broer A, Wagner C, Lang F, Broer S. Neutral amino acid transporter ASCT2 displays substrate-induced Na+ exchange and a substrate-gated anion conductance. Biochem J. 2000;346:705–710. [PMC free article] [PubMed] [Google Scholar]
- Carlsson M, Carlsson A. Interactions between glutamatergic and monoaminergic systems within the basal ganglia--implications for schizophrenia and Parkinson’s disease. [Review] [61 refs] Trends Neurosci. 1990;13:272–276. doi: 10.1016/0166-2236(90)90108-m. [DOI] [PubMed] [Google Scholar]
- Chen W, Aoki C, Mahadomrongkul V, Gruber CE, Wang GJ, Blitzblau R, Irwin N, Rosenberg PA. Expression of a variant form of the glutamate transporter GLT1 in neuronal cultures and in neurons and astrocytes in the rat brain. J Neurosci. 2002;22:2142–52. doi: 10.1523/JNEUROSCI.22-06-02142.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi DW. Excitotoxic cell death. J Neurobiol. 1992;23:1261–1276. doi: 10.1002/neu.480230915. [DOI] [PubMed] [Google Scholar]
- Clements JD, Lester RA, Tong G, Jahr CE, Westbrook GL. The time course of glutamate in the synaptic cleft. Science. 1992;258:1498–501. doi: 10.1126/science.1359647. [DOI] [PubMed] [Google Scholar]
- Crane RK. Hypothesis for mechanism of intestinal active transport of sugars. Fed Proc. 1962;21:891–5. [PubMed] [Google Scholar]
- Curtis DR, Johnston GA. Amino acid transmitters in the mammalian central nervous system. Ergeb Physiol. 1974;69:97–188. doi: 10.1007/3-540-06498-2_3. [DOI] [PubMed] [Google Scholar]
- Curtis DR, Watkins JC. The excitation and depression of spinal neurones by structurally related amino acids. J Neurochem. 1960;6:117–41. doi: 10.1111/j.1471-4159.1960.tb13458.x. [DOI] [PubMed] [Google Scholar]
- Danbolt NC. Glutamate uptake. Prog Neurobiol. 2001;65:1–105. doi: 10.1016/s0301-0082(00)00067-8. [DOI] [PubMed] [Google Scholar]
- Davis KE, Straff DJ, Weinstein EA, Bannerman PG, Correale DM, Rothstein JD, Robinson MB. Multiple signaling pathways regulate cell surface expression and activity of the excitatory amino acid carrier 1 subtype of Glu transporter in C6 glioma. J Neurosci. 1998;18:2475–85. doi: 10.1523/JNEUROSCI.18-07-02475.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Derouiche A, Rauen T. Coincidence of L-glutamate/L-aspartate transporter (GLAST) and glutamine synthetase (GS) immunoreactions in retinal glia: evidence for coupling of GLAST and GS in transmitter clearance. J Neurosci Res. 1995;42:131–43. doi: 10.1002/jnr.490420115. [DOI] [PubMed] [Google Scholar]
- Dev KK, Nakanishi S, Henley JM. Regulation of mglu(7) receptors by proteins that interact with the intracellular C-terminus. Trends Pharmacol Sci. 2001;22:355–61. doi: 10.1016/s0165-6147(00)01684-9. [DOI] [PubMed] [Google Scholar]
- Diamond JS, Bergles DE, Jahr CE. Glutamate release monitored with astrocyte transporter currents during LTP. Neuron. 1998;21:425–33. doi: 10.1016/s0896-6273(00)80551-6. [DOI] [PubMed] [Google Scholar]
- Diamond JS, Jahr CE. Transporters buffer synaptically released glutamate on a submillisecond time scale. J Neurosci. 1997;17:4672–87. doi: 10.1523/JNEUROSCI.17-12-04672.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dingledine R, Borges K, Bowie D, Traynelis SF. The Glutamate Receptor Ion Channels. Pharmacol Rev. 1999;51:7–62. [PubMed] [Google Scholar]
- Doyle DA, Morais Cabral J, Pfuetzner RA, Kuo A, Gulbis JM, Cohen SL, Chait BT, MacKinnon R. The structure of the potassium channel: molecular basis of K+ conduction and selectivity [see comments] Science. 1998;280:69–77. doi: 10.1126/science.280.5360.69. [DOI] [PubMed] [Google Scholar]
- Duan S, Anderson CM, Stein BA, Swanson RA. Glutamate induces rapid upregulation of astrocyte glutamate transport and cell-surface expression of GLAST. J Neurosci. 1999;19:10193–200. doi: 10.1523/JNEUROSCI.19-23-10193.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dutzler R, Campbell EB, Cadene M, Chait BT, MacKinnon R. X-ray structure of a ClC chloride channel at 3.0 Å reveals the molecular basis of anion selectivity. Nature. 2002;415:287–294. doi: 10.1038/415287a. [DOI] [PubMed] [Google Scholar]
- Eckstein-Ludwig U, Fei J, Schwarz W. Inhibition of uptake, steady-state currents, and transient charge movements generated by the neuronal GABA transporter by various anticonvulsant drugs. Br J Pharmacol. 1999;128:92–102. doi: 10.1038/sj.bjp.0702794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eliasof S, Arriza JL, Leighton BH, Amara SG, Kavanaugh MP. Localization and function of five glutamate transporters cloned from the salamander retina. Vision Research. 1998;38:1443–1454. doi: 10.1016/s0042-6989(97)00452-5. [DOI] [PubMed] [Google Scholar]
- Eliasof S, Jahr CE. Retinal glial cell glutamate transporter is coupled to an anionic conductance. Proc Natl Acad Sci U S A. 1996;93:4153–4158. doi: 10.1073/pnas.93.9.4153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Erecinska M, Troeger MB, Wilson DF, Silver IA. The role of glial cells in regulation of neurotransmitter amino acids in the external environment. II Mechanism of aspartate transport. Brain Res. 1986;369:203–14. doi: 10.1016/0006-8993(86)90529-9. [DOI] [PubMed] [Google Scholar]
- Eskandari S, Loo DFD, Dai D, Levy O, Wright EM, Carrasco N. Thyroid Na+/I Symporter. J Biol Chem. 1997;272:27230–27238. doi: 10.1074/jbc.272.43.27230. [DOI] [PubMed] [Google Scholar]
- Eskandari S, Kreman M, Kavanaugh MP, Wright EM, Zampighi GA. Pentameric assembly of a neuronal glutamate transporter. Proc Natl Acad Sci U S A. 2000;97:8641–6. doi: 10.1073/pnas.97.15.8641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Evans RH, Watkins JC. Specific antagonism of excitant amino acids in the isolated spinal cord of the neonatal rat. Eur J Pharmacol. 1978;50:123–9. doi: 10.1016/0014-2999(78)90007-9. [DOI] [PubMed] [Google Scholar]
- Fairman WA, Vandenberg RJ, Arriza JL, Kavanaugh MP, Amara SG. An excitatory amino-acid transporter with properties of a ligand- gated chloride channel. Nature. 1995;375:599–603. doi: 10.1038/375599a0. [DOI] [PubMed] [Google Scholar]
- Fanning AS, Anderson JM. PDZ domains: fundamental building blocks in the organization of protein complexes at the plasma membrane. J Clin Invest. 1999;103:767–72. doi: 10.1172/JCI6509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fonnum F. Glutamate: a neurotransmitter in mammalian brain. J Neurochem. 1984;42:1–11. doi: 10.1111/j.1471-4159.1984.tb09689.x. [DOI] [PubMed] [Google Scholar]
- Forlani G, Bossi E, Ghirardelli R, Giovannardi S, Binda F, Bonadiman L, Ielmini L, Peres A. Mutation K448E in the external loop 5 of rat GABA transporter rGAT1 induces pH sensitivity and alters substrate interactions. J Physiol. 2001;536:479–94. doi: 10.1111/j.1469-7793.2001.0479c.xd. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Forster I, Hernando N, Biber J, Murer H. The voltage dependence of a cloned mammalian renal type II Na+/Pi cotransporter (NaPi-2) J Gen Physiol. 1998;112:1–18. doi: 10.1085/jgp.112.1.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gegelashvili G, Dehnes Y, Danbolt NC, Schousboe A. The high-affinity glutamate transporters GLT1, GLAST, and EAAT4 are regulated via different signalling mechanisms. Neurochem Int. 2000;37:163–70. doi: 10.1016/s0197-0186(00)00019-x. [DOI] [PubMed] [Google Scholar]
- Gegelashvili G, Robinson MB, Trotti D, Rauen T. Regulation of glutamate transporters in health and disease. Prog Brain Res. 2001;132:267–86. doi: 10.1016/S0079-6123(01)32082-4. [DOI] [PubMed] [Google Scholar]
- Glaum SR, Holzwarth JA, Miller RJ. Glutamate receptors activate Ca2+ mobilization and Ca2+ influx into astrocytes. Proc Natl Acad Sci U S A. 1990;87:3454–8. doi: 10.1073/pnas.87.9.3454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grewer C, Madani Mobarekeh SA, Watzke N, Rauen T, Schaper K. Substrate translocation kinetics of excitatory amino acid carrier 1 probed with laser-pulse photolysis of a new photolabile precursor of D-aspartic acid. Biochemistry. 2001;40:232–40. doi: 10.1021/bi0015919. [DOI] [PubMed] [Google Scholar]
- Grewer C, Watzke N, Rauen T, Bicho A. Is the Glutamate Residue Glu-373 the Proton Acceptor of the Excitatory Amino Acid Carrier 1? J Biol Chem. 2003;278:2585–2592. doi: 10.1074/jbc.M207956200. [DOI] [PubMed] [Google Scholar]
- Grewer C, Watzke N, Wiessner M, Rauen T. Glutamate translocation of the neuronal glutamate transporter EAAC1 occurs within milliseconds. Proc Natl Acad Sci U S A. 2000;97:9706–9711. doi: 10.1073/pnas.160170397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grunewald M, Kanner B. Conformational changes monitored on the glutamate transporter GLT-1 indicate the existence of two neurotransmitter-bound states. J Biol Chem. 1995;270:17017–17024. doi: 10.1074/jbc.270.28.17017. [DOI] [PubMed] [Google Scholar]
- Grunewald M, Kanner BI. The accessibility of a novel reentrant loop of the glutamate transporter GLT-1 is restricted by its substrate. J Biol Chem. 2000;275:9684–9. doi: 10.1074/jbc.275.13.9684. [DOI] [PubMed] [Google Scholar]
- Hausser M, Roth A. Dendritic and somatic glutamate receptor channels in rat cerebellar Purkinje cells. J Physiol. 1997;501:77–95. doi: 10.1111/j.1469-7793.1997.077bo.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hazama A, Loo DD, Wright EM. Presteady-state currents of the rabbit Na+/glucose cotransporter (SGLT1) J Membr Biol. 1997;155:175–86. doi: 10.1007/s002329900169. [DOI] [PubMed] [Google Scholar]
- Hertz L. Functional interactions between neurons and astrocytes. I Turnover and metabolism of putative amino acid transmitters. Prog Neurobiol. 1979;13:277–323. doi: 10.1016/0301-0082(79)90018-2. [DOI] [PubMed] [Google Scholar]
- Hilgemann DW, Lu CC. GAT1 (GABA:Na+:Cl-) cotransport function. Database reconstruction with an alternating access model. J Gen Physiol. 1999;114:459–75. doi: 10.1085/jgp.114.3.459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holmgren M, Wagg J, Bezanilla F, Rakowski RF, De Weer P, Gadsby DC. Three distinct and sequential steps in the release of sodium ions by the Na+/K+-ATPase. Nature. 2000;403:898–901. doi: 10.1038/35002599. [DOI] [PubMed] [Google Scholar]
- Huggett J, Vaughan-Thomas A, Mason D. The open reading frame of the Na(+)-dependent glutamate transporter GLAST-1 is expressed in bone and a splice variant of this molecule is expressed in bone and brain. FEBS Lett. 2000;485:13–8. doi: 10.1016/s0014-5793(00)02175-x. [DOI] [PubMed] [Google Scholar]
- Ingram EM, Tessler S, Bowery NG, Emson PC. Glial glutamate transporter mRNAs in the genetically absence epilepsy rat from Strasbourg. Brain Res Mol Brain Res. 2000;75:96–104. doi: 10.1016/s0169-328x(99)00301-0. [DOI] [PubMed] [Google Scholar]
- Kanai Y, Hediger MA. Primary structure and functional characterization of a high- affinity glutamate transporter. Nature. 1992;360:467–471. doi: 10.1038/360467a0. [DOI] [PubMed] [Google Scholar]
- Kanai Y, Nussberger S, Romero MF, Boron WF, Hebert SC, Hediger MA. Electrogenic properties of the epithelial and neuronal high affinity glutamate transporter. J Biol Chem. 1995;270:16561–16568. doi: 10.1074/jbc.270.28.16561. [DOI] [PubMed] [Google Scholar]
- Kanai Y, Stelzner M, Nussberger S, Khawaja S, Hebert SC, Smith CP, Hediger MA. The Neuronal and Epithelial Human High Affinity Glutamate Transporter: Insights into structure and mechanism of transport. J Biol Chem. 1994;269:20599–20606. [PubMed] [Google Scholar]
- Kandel ER, Schwartz JH, Jessell TM. Essentials of Neural Science and Behavior. Appleton & Lange 1995 [Google Scholar]
- Kanner BI, Bendahan A. Binding order of substrates to the sodium and potassium ion coupled L-glutamic acid transporter from rat brain. Biochemistry. 1982;21:6327–30. doi: 10.1021/bi00267a044. [DOI] [PubMed] [Google Scholar]
- Kanner BI, Schuldiner S. Mechansim of transport and storage of neurotransmitters. CRC Crit Rev Biochem. 1987;22:1–38. doi: 10.3109/10409238709082546. [DOI] [PubMed] [Google Scholar]
- Kanner BI, Sharon I. Active transport of L-glutamate by membrane vesicles isolated from rat brain. Biochemistry. 1978;17:3949–53. doi: 10.1021/bi00612a011. [DOI] [PubMed] [Google Scholar]
- Koenderink JB, Geibel S, Grabsch E, De Pont JJHHM, Bamberg E, Friedrich T. Electrophysiological Analysis of the Mutated Na, K-ATPase Cation Binding Pocket. J Biol Chem. 2003;278:51213–51222. doi: 10.1074/jbc.M306384200. [DOI] [PubMed] [Google Scholar]
- Larsson HP, Picaud SA, Werblin FS, Lecar H. Noise analysis of the glutamate-activated current in photoreceptors. Biophys J. 1996;70:733–742. doi: 10.1016/S0006-3495(96)79613-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larsson HP, Tzingounis AV, Koch HP, Kavanaugh MP. Fluorometric measurements of conformational changes in glutamate transporters. Proc Natl Acad Sci U S A. 2004;101:3951–3956. doi: 10.1073/pnas.0306737101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laube B, Hirai H, Sturgess M, Betz H, Kuhse J. Molecular determinants of agonist discrimination by NMDA receptor subunits: analysis of the glutamate binding site on the NR2B subunit. Neuron. 1997;18:493–503. doi: 10.1016/s0896-6273(00)81249-0. [DOI] [PubMed] [Google Scholar]
- Läuger P, Benz R, Stark G, Bamberg E, Jordan PC, Fahr A, Brock W. Relaxation studies of ion transport systems in lipid bilayer membranes. Q Rev Biophys. 1981;14:513–98. doi: 10.1017/s003358350000247x. [DOI] [PubMed] [Google Scholar]
- Lehre KP, Danbolt NC. The number of glutamate transporter subtype molecules at glutamatergic synapses: chemical and stereological quantification in young adult rat brain. J Neurosci. 1998;18:8751–8757. doi: 10.1523/JNEUROSCI.18-21-08751.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leighton BH, Seal RP, Shimamoto K, Amara SG. A hydrophobic domain in glutamate transporters forms an extracellular helix associated with the permeation pathway for substrates. J Biol Chem. 2002;277:29847–29855. doi: 10.1074/jbc.M202508200. [DOI] [PubMed] [Google Scholar]
- Lester RA, Jahr CE. NMDA channel behavior depends on agonist affinity. J Neurosci. 1992;12:635–643. doi: 10.1523/JNEUROSCI.12-02-00635.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levy LM, Warr O, Attwell D. Stoichiometry of the glial glutamate transporter GLT-1 expressed inducibly in a Chinese hamster ovary cell line selected for low endogenous Na+-dependent glutamate uptake. J Neurosci. 1998;18:9620–9628. doi: 10.1523/JNEUROSCI.18-23-09620.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li G, Oswald RE, Niu L. Channel-Opening Kinetics of GluR6 Kainate Receptor. Biochemistry. 2003;42:12367–75. doi: 10.1021/bi034797t. [DOI] [PubMed] [Google Scholar]
- Logan WJ, Snyder SH. Unique high affinity uptake systems for glycine, glutamic and aspartic acids in central nervous tissue of the rat. Nature. 1971;234:297–9. doi: 10.1038/234297b0. [DOI] [PubMed] [Google Scholar]
- Loo DD, Eskandari S, Boorer KJ, Sarkar HK, Wright EM. Role of Cl- in electrogenic Na+-coupled cotransporters GAT1 and SGLT1. J Biol Chem. 2000;275:37414–22. doi: 10.1074/jbc.M007241200. [DOI] [PubMed] [Google Scholar]
- Loo DD, Hazama A, Supplisson S, Turk E, Wright EM. Relaxation kinetics of the Na+/glucose cotransporter. Proc Natl Acad Sci U S A. 1993;90:5767–71. doi: 10.1073/pnas.90.12.5767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu CC, Hilgemann DW. GAT1 (GABA:Na+:Cl-) cotransport function. Kinetic studies in giant Xenopus oocyte membrane patches. J Gen Physiol. 1999;114:445–57. doi: 10.1085/jgp.114.3.445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu CC, Kabakov A, Markin VS, Mager S, Frazier GA, Hilgemann DW. Membrane transport mechanisms probed by capacitance measurements with megahertz voltage clamp. Proc Natl Acad Sci U S A. 1995;92:11220–4. doi: 10.1073/pnas.92.24.11220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mackenzie B, Schafer MK-H, Erickson JD, Hediger MA, Weihe E, Varoqui H. Functional Properties and Cellular Distribution of the System A Glutamine Transporter SNAT1 Support Specialized Roles in Central Neurons. J Biol Chem. 2003;278:23720–23730. doi: 10.1074/jbc.M212718200. [DOI] [PubMed] [Google Scholar]
- MacLennan DH, Rice WJ, Green NM. The Mechanism of Ca2+ Transport by Sarco(Endo)plasmic Reticulum Ca2+-ATPases. J Biol Chem. 1997;272:28815–28818. doi: 10.1074/jbc.272.46.28815. [DOI] [PubMed] [Google Scholar]
- Mager S, Kleinberger-Doron N, Keshet GI, Davidson N, Kanner BI, Lester HA. Ion binding and permeation at the GABA transporter GAT1. J Neurosci. 1996;16:5405–14. doi: 10.1523/JNEUROSCI.16-17-05405.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mager S, Naeve J, Quick M, Labarca C, Davidson N, Lester HA. Steady states, charge movements, and rates for a cloned GABA transporter expressed in Xenopus oocytes. Neuron. 1993;10:177–88. doi: 10.1016/0896-6273(93)90309-f. [DOI] [PubMed] [Google Scholar]
- Melzer N, Biela A, Fahlke C. Glutamate Modifies Ion Conduction and Voltage-dependent Gating of Excitatory Amino Acid Transporter-associated Anion Channels. J Biol Chem. 2003;278:50112–50119. doi: 10.1074/jbc.M307990200. [DOI] [PubMed] [Google Scholar]
- Mennerick S, Shen W, Xu W, Benz A, Tanaka K, Shimamoto K, Isenberg KE, Krause JE, Zorumski CF. Substrate turnover by transporters curtails synaptic glutamate transients. J Neurosci. 1999;19:9242–51. doi: 10.1523/JNEUROSCI.19-21-09242.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mennerick S, Zorumski CF. Glial contributions to excitatory neurotransmission in cultured hippocampal cells. Nature. 1994;368:59–62. doi: 10.1038/368059a0. [DOI] [PubMed] [Google Scholar]
- Mosbacher J, Schoepfer R, Monyer H, Burnashev N, Seeburg PH, Ruppersberg JP. A molecular determinant for submillisecond desensitization in glutamate receptors. Science. 1994;266:1059–62. doi: 10.1126/science.7973663. [DOI] [PubMed] [Google Scholar]
- Murer H, Hernando N, Forster I, Biber J. Proximal Tubular Phosphate Reabsorption: Molecular Mechanisms. Physiol Rev. 2000;80:1373–1409. doi: 10.1152/physrev.2000.80.4.1373. [DOI] [PubMed] [Google Scholar]
- Nakanishi S, Masu M. Molecular diversity and functions of glutamate receptors. Annu Rev Biophys Biomol Struct. 1994;23:319–48. doi: 10.1146/annurev.bb.23.060194.001535. [DOI] [PubMed] [Google Scholar]
- Naraghi M. T-jump study of calcium binding kinetics of calcium chelators. Cell Calcium. 1997;22:255–68. doi: 10.1016/s0143-4160(97)90064-6. [DOI] [PubMed] [Google Scholar]
- Neal MJ, White RD. Proceedings: Multiplicity of transport systems of L-glutamate and L-aspartate in the retina. Br J Pharmacol. 1973;49:172P–173P. [PMC free article] [PubMed] [Google Scholar]
- Neher E. Usefulness and limitations of linear approximations to the understanding of Ca++ signals. Cell Calcium. 1998;24:345–357. doi: 10.1016/s0143-4160(98)90058-6. [DOI] [PubMed] [Google Scholar]
- Otis TS, Jahr CE. Anion currents and predicted glutamate flux through a neuronal glutamate transporter. J Neurosci. 1998;18:7099–110. doi: 10.1523/JNEUROSCI.18-18-07099.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Otis TS, Kavanaugh MP. Isolation of current components and partial reaction cycles in the glial glutamate transporter EAAT2. J Neurosci. 2000;20:2749–57. doi: 10.1523/JNEUROSCI.20-08-02749.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Otis TS, Kavanaugh MP, Jahr CE. Postsynaptic glutamate transport at the climbing fiber-Purkinje cell synapse. Science. 1997;277:1515–1518. doi: 10.1126/science.277.5331.1515. [DOI] [PubMed] [Google Scholar]
- Overstreet LS, Kinney GA, Liu YB, Billups D, Slater NT. Glutamate transporters contribute to the time course of synaptic transmission in cerebellar granule cells. J Neurosci. 1999;19:9663–73. doi: 10.1523/JNEUROSCI.19-21-09663.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perez-Otano I, Ehlers MD. Learning from NMDA Receptor Trafficking: Clues to the Development and Maturation of Glutamatergic Synapses. Neurosignals. 2004;13:175–89. doi: 10.1159/000077524. [DOI] [PubMed] [Google Scholar]
- Picaud SA, Larsson HP, Grant GB, Lecar H, Werblin FS. Glutamate-gated chloride channel with glutamate-transporter-like properties in cone photoreceptors of the tiger salamander. J Neurophysiol. 1995;74:1760–1771. doi: 10.1152/jn.1995.74.4.1760. [DOI] [PubMed] [Google Scholar]
- Pines G, Danbolt NC, Bjoras M, Zhang Y, Bendahan A, Eide L, Koepsell H, Storm Mathisen J, Seeberg E, Kanner BI. Cloning and Expression of a Rat Brain L Glutamate Transporter. Nature. 1992;360:464–467. doi: 10.1038/360464a0. [DOI] [PubMed] [Google Scholar]
- Pines G, Zhang Y, Kanner BI. Glutamate 404 is involved in the substrate discrimination of GLT-1, a (Na+ + K+)-coupled glutamate transporter from rat brain. J Biol Chem. 1995;270:17093–17097. doi: 10.1074/jbc.270.29.17093. [DOI] [PubMed] [Google Scholar]
- Poitry-Yamate CL, Vutskits L, Rauen T. Neuronal-induced and glutamate-dependent activation of glial glutamate transporter function. J Neurochem. 2002;82:987–97. doi: 10.1046/j.1471-4159.2002.01075.x. [DOI] [PubMed] [Google Scholar]
- Rauen T, Wiessner M. Fine tuning of glutamate uptake and degradation in glial cells: common transcriptional regulation of GLAST1 and GS. Neurochem Int. 2000;37:179–89. doi: 10.1016/s0197-0186(00)00021-8. [DOI] [PubMed] [Google Scholar]
- Rauen T, Wiessner M, Sullivan R, Lee A, Pow DV. A new GLT-1 splice variant: Cloning and immunolocalization of GLT1c in the mammalian retina and brain. Neurochem Int. 2004;104:1095–106. doi: 10.1016/j.neuint.2004.04.006. [DOI] [PubMed] [Google Scholar]
- Rothstein JD. Excitotoxicity and neurodegeneration in amyotrophic lateral sclerosis. Clinical Neuroscience. 1995;3:348–359. [PubMed] [Google Scholar]
- Rusakov DA, Kullmann DM. Extrasynaptic glutamate diffusion in the hippocampus: ultrastructural constraints, uptake, and receptor activation. J Neurosci. 1998a;18:3158–70. doi: 10.1523/JNEUROSCI.18-09-03158.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rusakov DA, Kullmann DM. A tortuous and viscous route to understanding diffusion in the brain. Trends Neurosci. 1998b;21:469–470. doi: 10.1016/s0166-2236(98)01309-5. [DOI] [PubMed] [Google Scholar]
- Ryan RM, Mitrovic AD, Vandenberg RJ. The chloride permeation pathway of a glutamate transporter and its proximity to the glutamate translocation pathway. J Biol Chem. 2004;24:24. doi: 10.1074/jbc.M304433200. [DOI] [PubMed] [Google Scholar]
- Ryan RM, Vandenberg RJ. Distinct Conformational States Mediate the Transport and Anion Channel Properties of the Glutamate Transporter EAAT-1. J Biol Chem. 2002;277:13494–13500. doi: 10.1074/jbc.M109970200. [DOI] [PubMed] [Google Scholar]
- Schmitt A, Asan E, Lesch KP, Kugler P. A splice variant of glutamate transporter GLT1/EAAT2 expressed in neurons: cloning and localization in rat nervous system. Neuroscience. 2002;109:45–61. doi: 10.1016/s0306-4522(01)00451-1. [DOI] [PubMed] [Google Scholar]
- Seal RP, Amara SG. A reentrant loop domain in the glutamate carrier EAAT1 participates in substrate binding and translocation. Neuron. 1998;21:1487–98. doi: 10.1016/s0896-6273(00)80666-2. [DOI] [PubMed] [Google Scholar]
- Seal RP, Leighton BH, Amara SG. A model for the topology of excitatory amino acid transporters determined by the extracellular accessibility of substituted cysteines. Neuron. 2000;25:695–706. doi: 10.1016/s0896-6273(00)81071-5. [DOI] [PubMed] [Google Scholar]
- Seal RP, Shigeri Y, Eliasof S, Leighton BH, Amara SG. Sulfhydryl modification of V449C in the glutamate transporter EAAT1 abolishes substrate transport but not the substrate-gated anion conductance. Proc Natl Acad Sci U S A. 2001;98:15324–15329. doi: 10.1073/pnas.011400198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shafqat S, Tamarappoo K, Kilberg MS, Puranam RS, McNamara JO, Guadano-Ferraz A, Fremeau RTJ. Cloning and expression of a novel Na+-dependent neutral amino acid transporter structurally related to mammalian Na+/glutamate cotransporter. J Biol Chem. 1993;268(21):15351–15355. [PubMed] [Google Scholar]
- Slotboom DJ, Konings WN, Lolkema JS. Structural features of the glutamate transporter family. Microbiol Mol Biol Rev. 1999a;63:293–307. doi: 10.1128/mmbr.63.2.293-307.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Slotboom DJ, Konings WN, Lolkema JS. Glutamate transporters combine transporter- and channel-like features. Trends in Biochem Sci. 2001;26:534–539. doi: 10.1016/s0968-0004(01)01925-9. [DOI] [PubMed] [Google Scholar]
- Slotboom DJ, Sobczak I, Konings WN, Lolkema JS. A conserved serine-rich stretch in the glutamate transporter family forms a substrate-sensitive reentrant loop. Proc Natl Acad Sci U S A. 1999b;96:14282–7. doi: 10.1073/pnas.96.25.14282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith GD. Numerical Solution of Partial Differential Equations. Oxford University Press; London: 1965. [Google Scholar]
- Sonders MS, Amara SG. Channels in transporters. Curr Op Neurobiol. 1996;6:294–302. doi: 10.1016/s0959-4388(96)80111-5. [DOI] [PubMed] [Google Scholar]
- Sontheimer H, Kettenmann H, Backus KH, Schachner M. Glutamate opens Na+/K+ channels in cultured astrocytes. Glia. 1988;1:328–36. doi: 10.1002/glia.440010505. [DOI] [PubMed] [Google Scholar]
- Storck T, Schulte S, Hofmann K, Stoffel W. Structure, expression, and functional analysis of a Na(+)- dependent glutamate/aspartate transporter from rat brain. Proc Natl Acad Sci USA. 1992;89:10955–10959. doi: 10.1073/pnas.89.22.10955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Su ZZ, Leszczyniecka M, Kang DC, Sarkar D, Chao W, Volsky DJ, Fisher PB. Insights into glutamate transport regulation in human astrocytes: cloning of the promoter for excitatory amino acid transporter 2 (EAAT2) Proc Natl Acad Sci U S A. 2003;100:1955–60. doi: 10.1073/pnas.0136555100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sullivan R, Rauen T, Fischer F, Wiessner M, Grewer C, Bicho A, Pow DV. Cloning, transport properties, and differential localization of two splice variants of GLT-1 in the rat CNS: Implications for CNS glutamate homeostasis. Glia. 2004;45:155–69. doi: 10.1002/glia.10317. [DOI] [PubMed] [Google Scholar]
- Tong G, Jahr CE. Block of glutamate transporters potentiates postsynaptic excitation. Neuron. 1994;13:1195–203. doi: 10.1016/0896-6273(94)90057-4. [DOI] [PubMed] [Google Scholar]
- Toyoshima C, Nakasako M, Nomura H, Ogawa H. Crystal structure of the calcium pump of sarcoplasmic reticulum at 2.6 A resolution. Nature. 2000;405:647–55. doi: 10.1038/35015017. [DOI] [PubMed] [Google Scholar]
- Turecek R, Trussell LO. Control of synaptic depression by glutamate transporters. J Neurosci. 2000;20:2054–63. doi: 10.1523/JNEUROSCI.20-05-02054.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Utsunomiya-Tate N, Endou H, Kanai Y. Tissue specific variants of glutamate transporter GLT-1. FEBS Lett. 1997;416:312–316. doi: 10.1016/s0014-5793(97)01232-5. [DOI] [PubMed] [Google Scholar]
- Vadgama JV, Christensen HN. Wide distribution of pH-dependent service of transport system ASC for both anionic and zwitterionic amino acids. J Biol Chem. 1984;259:3648–52. [PubMed] [Google Scholar]
- Vandenberg RJ, Arriza JL, Amara SG, Kavanaugh MP. Constitutive Ion Fluxes and Substrate Binding Domains of Human Glutamate Transporters. J Biol Chem. 1995;270:17668–17671. doi: 10.1074/jbc.270.30.17668. [DOI] [PubMed] [Google Scholar]
- Wadiche JI, Amara SG, Kavanaugh MP. Ion fluxes associated with excitatory amino acid transport. Neuron. 1995a;15:721–728. doi: 10.1016/0896-6273(95)90159-0. [DOI] [PubMed] [Google Scholar]
- Wadiche JI, Arriza JL, Amara SG, Kavanaugh MP. Kinetics of a human glutamate transporter. Neuron. 1995b;14:1019–1027. doi: 10.1016/0896-6273(95)90340-2. [DOI] [PubMed] [Google Scholar]
- Wadiche JI, Kavanaugh MP. Macroscopic and microscopic properties of a cloned glutamate transporter/chloride channel. J Neurosci. 1998;18:7650–7661. doi: 10.1523/JNEUROSCI.18-19-07650.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watzke N, Bamberg E, Grewer C. Early intermediates in the transport cycle of the neuronal excitatory amino acid carrier EAAC1. J Gen Physiol. 2001;117:547–62. doi: 10.1085/jgp.117.6.547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watzke N, Grewer C. The anion conductance of the glutamate transporter EAAC1 depends on the direction of glutamate transport. FEBS Lett. 2001;503:121–5. doi: 10.1016/s0014-5793(01)02715-6. [DOI] [PubMed] [Google Scholar]
- Watzke N, Rauen T, Bamberg E, Grewer C. On the mechanism of proton transport by the neuronal excitatory amino acid carrier 1. J Gen Physiol. 2000;116:609–22. doi: 10.1085/jgp.116.5.609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wofsey AR, Kuhar MJ, Snyder SH. A unique synaptosomal fraction, which accumulates glutamic and aspartic acids, in brain tissue. Proc Natl Acad Sci U S A. 1971;68:1102–6. doi: 10.1073/pnas.68.6.1102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yernool D, Boudker O, Jin Y, Gouaux E. Structure of a glutamate transporter homologue from Pyrococcus horikoshii. Nature. 2004;431:811–8. doi: 10.1038/nature03018. [DOI] [PubMed] [Google Scholar]
- Zerangue N, Kavanaugh MP. Flux coupling in a neuronal glutamate transporter. Nature. 1996a;383:634–637. doi: 10.1038/383634a0. [DOI] [PubMed] [Google Scholar]
- Zerangue N, Kavanaugh MP. Interaction of L-cysteine with a human excitatory amino acid transporter. J Physiol (Lond) 1996b;493:419–23. doi: 10.1113/jphysiol.1996.sp021393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Bendahan A, Zarbiv R, Kavanaugh MP, Kanner BI. Molecular determinant of ion selectivity of a (Na+ plus K+)-coupled rat brain glutamate transporter. Proc Natl Acad Sci U S A. 1998;95:751–755. doi: 10.1073/pnas.95.2.751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Pines G, Kanner BI. Histidine 326 is critical for the function of GLT-1, a (Na+ + K+)-coupled glutamate transporter from rat brain. J Biol Chem. 1994;269:19573–19577. [PubMed] [Google Scholar]