

Abstract
The position and role of the unique N-terminal transmembrane (TM) helix, S0, in large-conductance, voltage- and calcium-activated potassium (BK) channels are undetermined. From the extents of intra-subunit, endogenous disulfide bond formation between cysteines substituted for the residues just outside the membrane domain, we infer that the extracellular flank of S0 is surrounded on three sides by the extracellular flanks of TM helices S1 and S2 and the four-residue extracellular loop between S3 and S4. Eight different double cysteine–substituted alphas, each with one cysteine in the S0 flank and one in the S3–S4 loop, were at least 90% disulfide cross-linked. Two of these alphas formed channels in which 90% cross-linking had no effect on the V50 or on the activation and deactivation rate constants. This implies that the extracellular ends of S0, S3, and S4 are close in the resting state and move in concert during voltage sensor activation. The association of S0 with the gating charge bearing S3 and S4 could contribute to the considerably larger electrostatic energy required to activate the BK channel compared with typical voltage-gated potassium channels with six TM helices.
INTRODUCTION
The large-conductance, voltage and calcium (Ca2+)–activated K+ channel (BK, maxi-K, slo, KCNMA1) is expressed in many cell types, including smooth muscle and neurons. BK channel is opened by a rise in intracellular Ca2+ and by depolarization. The outward K+ flux conducted by the BK channel moves the membrane potential in the hyperpolarizing direction, suppressing activation of voltage-dependent channels. To the extent that an increase in intracellular Ca2+ concentration is due to the activation of voltage-gated Ca2+ channels, the BK channel is a negative feedback regulator of cytoplasmic Ca2+ concentration.
The BK channel pore is formed by a tetramer of α subunits. BK α is homologous to the pore-forming subunit of other voltage-gated K+ channels, as can be seen, for example, in its alignment with Kv1.2 and KvAP (Fig. 1 A), the structures of which have been determined to high resolution (Jiang et al., 2003; Long et al., 2005). All voltage-dependent K+ channels have six transmembrane (TM) helices, S1–S6. The BK α subunit has an additional TM helix, S0, which is N terminal to S1–S6. (Wallner et al., 1996; Meera et al., 1997). Where S0 is located relative to S1–S6 in the folded structure of BK α has not previously been examined.
Figure 1.

Mouse BK α. (A) The N-terminal 333 residues of BK α are shown in bold letters. The predicted seven TM helices, S0–S6, and the pore helix are underlined and labeled. Predicted extracellular residues are shown in upper case and the rest in lower case. The extracellular residues that we mutated to Cys are shown in reverse contrast. These Cys substitutions were made in a pseudo-wild-type (pWT) construct, the sequence of which was altered as indicated in boxes above the wild-type sequence (see Materials and methods). We used ClustalW to align BK α residues 106–333, which contain S1–S6, with KvAP residues 31–260 and Kv1.2 residues 97–387. (B) Scheme of the threading of BK α through the membrane. The locations of the changes we made in wild-type α to generate pseudo-wild-type α with a HRV-3C protease cleavage site are shown. The extracellular regions flanking S0–S6, in which Cys were substituted, are indicated by filled rectangles.
Previous work has implicated S0 in the expression and function of the channel. Transfection of cells with BK lacking S0 yielded neither functional (Wallner et al., 1996; Morrow et al., 2006) nor biochemically detectable channels on the cell surface (unpublished data). The α mutants F25W, L26W, and S29W, which are on the same side of the predicted S0 helix, yielded channels with rightward shifted conductance–voltage (G-V) curves compared with wild-type (WT) α (Koval et al., 2007). In addition, replacement of S0 and the preceding N-terminal residues of human BK α with the comparable residues in Drosophila BK α interfered with the functional modulation of this chimera by human β1 subunit (Wallner et al., 1996).
We used disulfide cross-linking to determine the relative proximities of S0 to the other TM helices in α and to determine the effects on channel function of tethering S0 to its neighboring TM helices. We substituted cysteine (Cys) for the first four residues we predicted would just flank the extracellular end of the membrane-embedded portions of the TM helices and expressed 47 double-Cys α mutants containing one Cys flanking S0 and another Cys flanking one of the other TM helices. We determined both the extent of endogenous disulfide bond formation (in the absence of added oxidizing agents) in α heterologously expressed in HEK293 cells and trafficked to the cell surface and the functional effects of the cross-links. We conclude that the extracellular end of S0 is centrally positioned among the extracellular ends of the voltage sensor domain helices, S1–S4, and is particularly close to the extracellular ends of S3 and S4 in both the resting and active states of the voltage sensor.
MATERIALS AND METHODS
Cross-linker
In most experiments, no oxidizing agent was added to the cells. In experiments to test the susceptibility of two Cys on the cell surface to reform a disulfide after reduction, we used the doubly charged oxidant, 4,4′-(azodicarbonyl)-bis-[1,1-dimethylpiperazinium, diiodide] (quaternary piperazinium diamide [QPD]) (Kosower et al., 1974), the synthesis of which was based on that of 4,4′-(azodicarbonyl)-bis-[1-methylpiperazide] (Bose et al., 1984). In brief, N-methylpiperazine was added slowly to a solution of 0.5 equivalent of diethyl diazenedicarboxylate in petroleum ether:ether (3:1) at 0°C. After 4 h stirring at room temperature, the product was filtered and crystallized from hexane/benzene as golden needles. It was quaternized by treatment with excess methyl iodide in acetonitrile at room temperature to yield QPD as a yellow-orange solid. The structure was confirmed by NMR (1H NMR[DMSO-d6, 400 MHz] δ [ppm] 3.97 [m, 4H], 3.83 [m, 4H], 3.57 [m, 4H], 3.52 [m, 4H], 3.19 [s, 12H]); the purity was >98%.
Constructs
Mutants of the BK α subunit (mSlo1, KCNMA1, GenBank/EMBL/DDBJ accession no. NM_010610) were generated by site-directed mutagenesis using QuikChange XL (Stratagene). The β1 (KCNMB1) subunit was cloned by RT-PCR from murine smooth muscle RNA (CLONTECH Laboratories, Inc.). Cys substitutions were made in a pseudo-wild-type background (pWTα) with the following mutations (Fig. 1 A). The FLAG epitope (DYKDDDDK) and a linker (SPGDS) were added to the N terminus. Cys 14 and Cys 141 were each mutated to Ala. The human rhinovirus (HRV-3C) protease consensus cleavage site, LEVLFQGP, which is cleaved between the Q and G, was inserted by mutating Ala89 to Leu and by inserting LFQGP between Val91 and Gly92 (site indicated in Fig. 1 A by *). This construct is called pWT-HRV α.
Expression, Surface Biotinylation, Cell Lysis
HEK293 cells were cultured and transfected as before (Morrow et al., 2006). After 48 h, the cells were collected, washed, and biotinylated with 2 mM sulfoNHS-LC-biotin (Pierce Chemical Co.) in DPBS (Karlin et al., 2004). The cells were solubilized for 20 min at 4°C in lysis buffer (1% Triton-X100 and, in mM, 150 NaCl, 50 Tris, 1 EDTA, 2 NEM, and Roche complete protease inhibitors). Insoluble material was removed by centrifugation.
Quantitating Intrasubunit Cross-linking
The cell lysate was mixed with Neutravidin-Sepharose beads (Pierce Chemical Co.) for 90 min at room temperature. In a typical sample, lysate of ∼25% of the cells from a 100-mm plate in 240-μl lysis buffer was mixed with 30 μl of a 50% slurry of Neutravidin beads, previously washed and suspended in PBS. The beads were washed seven times with lysis buffer (without NEM and protease inhibitors) and two additional times with HRV buffer (in mM, 150 NaCl, 50 Tris, pH 7.5, 1 EDTA, 0.5% Triton X-100). The beads were mixed with 5 U of HRV-3C protease (Novagen) in 50 μl HRV-3C buffer and nutated overnight at 4°C. To elute bound protein and fragments released by cleavage, we added 100 μl 8 M urea, 4% SDS, 200 mM Tris, pH 8, and heated the mixture to 100°C for 2 min. After a short spin, the supernatant was collected. A 75-μl portion was treated with 2 μl of 400 mM DTT to reduce disulfides. Both the unreduced and the reduced portions were electrophoresed, transferred to nitrocellulose, and immunoblotted by sequential treatment with anti-BK-α-C-terminus antibody (BD Bioscience), horseradish-peroxidase-conjugated secondary antibody, and ECL reagent (Pierce Super Signal West). We quantitated the chemiluminescence from each band with a CCD camera (Alpha Innotech Corporation) and ImageQuant software (Molecular Dynamics).
Part of each sample was exhaustively reduced, and the reduced and unreduced portions were electrophoresed in adjacent lanes. We calculated the extent of cross-linking, uncorrected for the efficiency of cleavage, from the lane with the unreduced portion and the efficiency of protease cleavage from the lane with the reduced portion. The calculation of the corrected extent of cross-linking is as follows (Fig. 2). In the reduced lane, the density of the 110-kD band divided by the sum of the densities of the 110-kD band and the 125-kD band is the fraction of α cleaved by protease; i.e., the efficiency of cleavage. In the unreduced lane, the same quotient is the uncorrected fraction of α that was not cross-linked, and hence ran as 110-kD after cleavage. This fraction was corrected to 100% cleavage by dividing it by the efficiency of protease cleavage, yielding the corrected fraction of α that was not cross-linked. The corrected fraction of α that was cross-linked equals one minus the corrected fraction of uncrosslinked α.
Figure 2.

Determination of disulfide cross-linking. (A) Scheme of the method, in which cleavage at the HRV-3C protease site in the S0–S1 loop of α produces an ∼15-kD N-terminal fragment and an ∼110-kD C-terminal fragment. Shown schematically are the species generated from different starting α forms after treatment with protease and then treatment with DTT. (B) Western blots illustrating cross-linking of Cys pairs and controls. The blots were developed with an antibody against a C-terminal epitope, reacting with both the 125-kD full-length α and with the 110-kD C-terminal fragment generated by HRV-3C protease cleavage. (i) Control showing that the mobility of pWT α is not affected either by DTT (lane 1) or by HRV-3C protease followed by DTT (lane 2). (ii) α with an HRV site is cleaved by HRV-3C protease and is unaffected by DTT. (iii) α in which the native pair, Cys14 and Cys141, is endogenously cross-linked, as shown by the preponderance of the 125-kD band after protease treatment (lane 1) and the loss of this band after protease and DTT (lane 2). In panels iv, v, and vi are shown the endogenous cross-linking of three additional pairs of Cys, one Cys in each case in S0 and one in S1 (iv), S2 (v), or S3–S4 (vi). At the bottom of each gel is the fraction of α cross-linked corrected for the efficiency of cleavage (see Materials and methods).
All double-Cys mutants were independently expressed and tested twice if there was little cross-linking and three or more times if there was substantial cross-linking. In each experiment, duplicate reduced and unreduced lanes were run. Finally, the means of the individual means were calculated.
Quantitating Intersubunit Cross-linking
The amounts of DNA required to yield equivalent coexpression of a single-Cys α mutant in S0 and a single-Cys α mutant in S5 or S6 were determined based on quantitative immunoblotting. The single-Cys mutants were coexpressed in HEK293 cells. After 48 h, the cell surface proteins were biotinylated, and the cells were lysed as above. Biotinylated proteins were captured on Neutravidin beads, which were washed and eluted, also as above. There was no cleavage step. Samples were size fractionated on SDS-PAGE, transferred to nitrocellulose, and immunoblotted with anti-BK-α antibody. The extents of S0 to S5 and S0 to S6 cross-linking were determined by the density of the dimer band at 250 kD divided by the sum of densities of the bands at 125 kD and 250 kD. Similar results were obtained by including the densities of the lanes around the expected positions of trimer (375 kD) and of tetramer (500 kD).
Comparing the Functional Effects of Cross-linking
G-V curves and the activation and deactivation kinetics were determined with inside-out and outside-out macropatch experiments, as previously described (Zakharov et al., 2005; Morrow et al., 2006). The bath solution for inside-out patches and the pipette solution for outside-out patches contained (in mM): 158 KCl, 5 TES, 5 EGTA, pH 7.0, with appropriate [Ca2+] calculated using the program Max Chelator (maxchelator.stanford.edu). The pipette solution for inside-out patches and the bath solution for outside-out patches contained (in mM): 158 KCl, 5 TES, 1 MgCl2, pH 7.0, except during reduction, when the pH was 8.0.
We focused on two types of functional effects of mutations and cross-linking. The effect on V50 was calculated as ΔV50 = V50,MUT − V50,pWT-HRV. The effects on the rate constants of opening and closing were calculated as log(kMUT/kpWT-HRV). The rationale is that V50 is proportional to the free energy between ground states and log(k) is proportional to the free energy between a ground state and a transition state, and therefore V50s can be added and subtracted, and the same applies to log(k)s.
Normalization of Functional Effects
Because the double Cys mutants were cross-linked to different extents, the observed functional characteristics were a composite of the characteristics of channels composed of uncrosslinked and of cross-linked α subunits in varying proportions. To estimate the effects of the disulfide cross-links themselves, we made some simplifying assumptions: (a) the effect of cross-linking increases linearly with the extent of cross-linking; (b) the effect of two uncrosslinked Cys is the sum of the effects of each single Cys mutation; and (c) in cross-linked Cys, the effect of the disulfide cross-link predominates over the effect of the replacement of the native residues. Based on these assumptions, we calculated what the functional effects would be if the Cys were 100% cross-linked.
Let δ be an effect, either ΔV50 or log(kMUT/kpWT-HRV). We define δ1 = the observed effect of the first single Cys mutation; δ2 = the observed effect of the second single Cys mutation; δ12 = the effect of the double Cys mutation before disulfide bond formation; δSS = the effect of the double Cys mutation, with the two Cys completely disulfide cross-linked, which is to be calculated; ε = the observed effect of the double Cys mutation, cross-linked to the fractional extent, x, determined by Western blotting.
Assuming linear addition of the effect on each α in the tetrameric channel complex, the observed effect of the double Cys mutation is the sum of the effect of the cross-linked Cys and the effect of the noncross-linked (i.e., reduced) Cys:
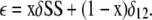 |
1 |
Assuming that δ12 = δ1 + δ2, we get
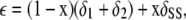 |
2 |
and rearranging, we get
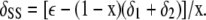 |
3 |
Online Supplemental Material
The supplemental material (available at http://www.jgp.org/cgi/content/full/jgp.200809968/DC1) consists of Table S1, which contains the mean extents and standard errors of disulfide bond formation of all mutants pairs in which mutant α alone was expressed and in which mutant α was coexpressed with wild-type β1 subunit. Fig. S1 shows additional Western blots of mutant α, and Fig. S2 shows activation and deactivation recordings for representative mutant α.
RESULTS
Mutant α Subunits
Our approach was to mutate to Cys at least the first four residues just extracellular to the membrane domain of each TM helix. The TM helices in BK α were predicted originally by Wallner et al. (1996); the PHDhtm algorithm (Rost et al., 1995) predicted similar boundaries of the TM helices. Furthermore, the predicted BK S1–S6 helices align with the homologous S1–S6 of KvAP (Jiang et al., 2003) and Kv1.2 (Long et al., 2005) (Fig. 1 A), although the boundaries of the membrane-embedded domains are not precisely defined in these channels crystallized from detergent solution.
BK α contains two extracellular Cys, Cys14 and Cys141, which we mutated to Ala to generate our background construct, pseudo-wild-type (pWT) α. In addition, we inserted a human rhinovirus-3C (HRV-3C) protease cleavage site between S0 and S1 (Fig. 1) to permit the detection of intrasubunit cross-links (Wolin and Kaback, 2000). The construct with the HRV-3C site we call pWT-HRV, the functional characteristics of which are identical to WT (see below). In this background, we substituted Cys for the four residues flanking S0, S1, and S2, and two of the four residues in the loop between S3 and S4. In addition, in the pWT α background, we substituted Cys for eight residues flanking S5 and six residues flanking S6. Among all the possible combinations, we tested 47 double-Cys mutants, in which one Cys was in the flank of S0 and one Cys was in flank of one of the other helices. We focused our analysis on cell surface channels, assuming that only well-folded and assembled α tetramers were trafficked to the surface membrane. We selected these channels by biotinylating the surface proteins in intact cells with the relatively impermeant sulfoNHS-LC-biotin and by capturing the biotinylated channels on avidin beads after cell lysis. The efficiencies of transfection and cell surface expression of the double-Cys α mutants involving S0–S4 and of pWT α were similar.
Quantitating Intrasubunit Cross-links
The α subunit has an apparent molecular mass of ∼125 kD (Knaus et al., 1995). Intersubunit disulfide cross-links would generate dimers, trimers, or tetramers of 250, 375, and 500 kD, respectively. By contrast, α with an intrasubunit disulfide cross-link between S0 and another helix had no change in mass and no detectable change in electrophoretic mobility. In the presence of a disulfide between S0 and another helix, HRV-3C cleavage between S0 and S1 also did not change the mass or the mobility (Fig. 2 B). After both HRV-3C cleavage and reduction of the disulfide, however, two fragments are obtained, a 15-kD N-terminal fragment and a 110-kD C-terminal fragment, as shown schematically (Fig. 2 A) and experimentally (Fig. 2 B). The extent of intrasubunit cross-linking was quantitated as described in the Materials and methods.
To quantitate the extent of intrasubunit cross-linking, we cleaved the biotinylated and Neutravidin-bound α with HRV-3C protease. The α subunit and fragments of α were eluted from the Neutravidin beads in SDS, and a portion of the sample was reduced with DTT. Unreduced and reduced samples were electrophoresed and immunoblotted with an antibody against the C terminus of α. We quantitated the relative amounts of the 125-kD and the 110-kD species by measuring the chemiluminescence generated by a secondary HRP-tagged antibody (Fig. 2 B). We adjusted the conditions of cleavage by HRV-3C protease so that there was no cleavage of pWT α, which does not contain a HRV-3C cleavage site (Fig. 2 B, i). Under these conditions, pWT-HRV was cleaved by HRV-3C protease ∼70% (Fig. 2 B, ii). In each experiment, the efficiency of cleavage was determined and was used in the calculation of the extent of cross-linking (see Materials and methods).
An Extracellular Disulfide in Native α
We tested in wild-type α modified only with the HRV-3C site whether the two extracellular, wild-type Cys, namely Cys14, seven residues N terminal to S0, and Cys141, seven residues N terminal to S2, form a disulfide bond. There was extensive endogenous, intrasubunit disulfide-bond formation (Fig. 2 B, iii). We conclude that in WT BK α, the extracellular, N-terminal tail preceding S0 is tethered to the loop between S1 and S2 by a disulfide between Cys14 and Cys141.
Disulfides Formed in Double-Cys Mutants of α
Cys in the S0 flank readily cross-linked to Cys in the S1 flank (Fig. 2 B, iv), in the S2 flank (Fig. 2 B, v), and in the S3–S4 loop (Fig. 2 B, vi) (see Fig. S1 for more examples and Table S1 for complete quantitation, available at http://www.jgp.org/cgi/content/full/jgp.200809968/DC1). Among the Cys flanking S1 and S2, the Cys substituted four residues out from the membrane domain, P137C flanking S1, and F144C flanking S2 showed the greatest extents of cross-linking to S0 (Fig. 3, A and B). All four Cys flanking S0 were cross-linked to P137C (S1) and to F144C (S2) to the extent of 40% or more.
Figure 3.

Mean extents of endogenous disulfide-bond formation between Cys substituted in the extracellular flank of S0 and Cys substituted in the extracellular flanks of S1–S6. (A) S0 flank and S1 flank; (B) S0 flank and S2 flank; (C) S0 flank and S3–S4 loop; (D) S0 flank and S5 flank; and (E) S0 flank and S6 flank. The four residues substituted by Cys in the S0 flank are indicated along the front edge of the base, and the residues substituted by Cys in the paired flank are indicated along the right edge of the base. The extent of disulfide bond formation is represented by bars in the z direction, placed at the intersection of the lines from the paired Cys-substituted residues. In the cases in which the mean extent of disulfide bond formation was zero, the value 0.5% was plotted to identify these pairs as tested. Pairs that were not tested lead to empty intersections. Except for S3–S4, the residues closest to the membrane are closest to the front, right corner of the base. For the S3–S4 loop, L199 is closest to the predicted membrane domain of S3, and S202 is closest to the predicted membrane domain of S4. (F) Proposed location of the extracellular end of S0 relative to the extracellular ends of S1–S6, consistent with the extents of disulfide bond formation. We have taken a model of the structure of Kv1.2 in the closed state (Yarov-Yarovoy et al., 2006) as a model for the structure of BK α S1–S6. In this en face view, each of the four α subunits is rendered in a distinct color. We have marked the extracellular end of each TM helix with a numbered circle in the same color as the subunit. The extracellular end of S0 is indicated as a circle marked with a zero. Its position is consistent with the cross-linking results in A-E. We have taken the means of the top three extents of cross-linking in each of the panels A–E as a measure of the relative proximity between the segments: the mean extent of cross-linking from S0 to S1 was 66 ± 20%; from S0 to S2, 52 ± 11%; from S0 to S3–S4, 96 ± 0%; from S0 to S5, 8 ± 1%; and from S0 to S6, 2 ± 1%.
All eight pairs of Cys tested for cross-linking between the S0 flank and the S3–S4 loop were cross-linked 88% or more (Fig. 3 C). These results are consistent with close and extensive contact between the residues immediately flanking S0 and the short loop between S3 and S4.
The patterns of intrahelix cross-linking between S0 and S1, S2 and S3–S4 in α expressed alone (Fig. 3, A–C) were nearly the same in α coexpressed with WT β1 (Table S1). In six double-Cys mutants that we tested functionally, we found that coexpressed β1 associated with the cross-linked α, based on β1 modulation of the conductance–voltage (G-V) curve (see below), but we do not know whether that association preceded disulfide bond formation in α.
Absence of Disulfide Formation between the Extracellular Flanks of S0 and S5 or S6
We also tested the susceptibility to disulfide formation of Cys flanking S0 with Cys flanking S5 and S6. Given the structure of the homologous Kv1.2 (Fig. 3 F), any cross-linking between S0 and S5 or S6 was likely to be between different subunits and to result in dimers or higher oligomers. Nevertheless, we first tested double-Cys mutants of α, one Cys flanking S0 and one Cys flanking S5 or S6, in the same subunit. These resulted in no significant cross-linking. These constructs, however, yielded poor surface expression, and the mutants tested showed no function, even after reduction with DTT. In a second approach, we coexpressed equal amounts of two single-Cys α mutants, an α with a Cys substituted in the S0 flank and an α with a Cys substituted in the flank of S5 or S6. All single mutants with a Cys just in S5 or S6 expressed well and resulted in functional channels. The hybrid channels resulting from transfection with both an α S0 mutant and α S5 or S6 mutant also expressed well. Because there was no detectable cross-linking between S0 and any Cys within four residues of the membrane domain of either S5 or S6, we made Cys mutations further out from the membrane. We tested Cys substituted for the first eight residues flanking S5 and for four residues among six flanking S6. There was <8% cross-linking among any of the 32 different combinations of S0 and S5 Cys mutants (Fig. 3 D) and among any of the 16 combinations of S0 and S6 Cys mutants (Fig. 3 E; also Table S1). To control for possible interference with the cross-linking by Cys277 in the pore helix (Fig. 1 A), we also retested all of these combinations in a background in which Cys277 was mutated to Ala (Savalli et al., 2006), none of which showed any more cross-linking than previously seen (unpublished data). It is likely that the extracellular end of S0 is not close to the extracellular ends of S5 and S6.
Functional Effects of the Single-Cys and Double-Cys Mutations
We examined the function of the cross-linked channels to determine whether or not they were in a near-native conformation. We assume that a near-native structure would have near-native function. We note first that the background constructs, pWT-HRV α and pWT α, and WT α have the same activation and deactivation kinetics (Fig. 4, A–C), and the same G-V curves and sensitivity to Ca2+ (Fig. 4 D). In addition, all three were identically modulated by coexpressed β1 (unpublished data). We infer that the native cross-link between Cys14 and Cys141 in WT α, but not present in pWT α and pWT-HRV α, does not influence these electrophysiological properties.
Figure 4.

Function of background constructs and single-Cys mutants. Macroscopic currents conducted by (A) wild-type (WT) α, (B) pseudo-wild type (pWT) α with the native extracellular Cys mutated to Ala (i.e., C14A, C141A), and (C) pWT α with the HRV consensus site inserted between S0–S1 transmembrane segments (pWT-HRV α). Each construct was expressed in HEK293 cells, and the currents shown were in response to step depolarizations to 0, 40, and 80 mV, in 10 μM Ca2+. (D) For WT, pWT, and pWT-HRV in inside-out macropatches, normalized conductance is plotted against the activating voltage. The data were from the maximum of the tail currents obtained after stepping the voltage from the plotted voltage to −80 mV in nominally zero (open symbols) or 10 μM (filled symbols) Ca2+. The data were fit by 1/{1 + exp[−zF(V − V50)/RT)] }. (E) For each of the single-Cys mutants indicated on the abscissa, V50 was determined as in D in nominally 0 and 10 μM Ca2+. From the mean V50 (n > 3) was subtracted the mean V50 for pWT-HRV to obtain ΔV50. (F) For each single-Cys mutant, the time course of activation at 240 mV was fit with an exponential function to estimate the rate constant for activation; the mean rate constants (n > 4) were divided by the mean rate constant for activation of pWT-HRV, and the log of this ratio is plotted. Similarly, the mean rate constants for deactivation at −80 mV were determined and divided by the mean deactivation rate constant for pWT-HRV. The log of this ratio is shown. The errors in the differences and ratios are propagated from the standard errors of the means of the arguments, and the errors in the logs are from the approximation log(x ± err) = log(x) + log(1 ± err/x) ≈ log(x) ± 0.434(err/x), where err is the standard error of the mean x.
We characterized the function of the double-Cys mutants that are representative of the different pairs of segments and that showed the highest extents of disulfide bond formation: P137C (S1), F144C (S2), L199C (S3–S4), and R201 (S3–S4) paired with each of the four S0 Cys mutants. To determine the functional effects of cross-linking, it was necessary to separate its effects from the effects of mutating native residues to Cys. We determined the functional effects of mutating each of the eight residues, one at a time, to Cys (Fig. 4, E and F). Except for F144C, none of the single-Cys mutants showed |ΔV50| greater than 20 mV or more than a twofold change in the rate constants for activation and deactivation.
Each of the 16 double-Cys mutant αs was functional (Fig. 5). Each was activated by depolarization and modulated by Ca2+. Six of these double mutants (R17C-P137C, R17C-F144C, Q19C-F144C, G18C-L199C, R17C-R201C, R20C-R201C) were also tested for modulation by coexpressed β1, which shifted the G-V curve of five of the six double mutants to the left by 50 mV or more (unpublished data). β1 had a similar effect on WT α. Coexpression of β1 with the sixth double mutant, Q19C-F144C, shifted the G-V curve by ∼25 mV.
Figure 5.

Functional effects of endogenous cross-linking of double-Cys mutants. In pWT-HRV, Cys was substituted for each of the four residues flanking S0 (R17C, G18C, Q19C, and R20C, as indicated on the abscissas). These were paired with Cys substituted for residues flanking (A and B) S1 (P137C), (C and D) S2 (F144C), and (E–H) S3–S4 (L199C and R201C), as indicated in the top right of each panel. The conditions were as in Fig. 4. ΔV50 and kMUT/kpWT-HRV were corrected for the effects of the single Cys mutations and were extrapolated to 100% cross-linking, taking into account the actual extent of cross-linking, as described in Materials and methods. The errors were propagated as in Fig. 4 (n > 4 in all cases). (I) Binned effects of cross-linking on V50, represented by color-coded lines connecting the cross-linked residues, superimposed on the extracellular ends of S0–S4, taken from Fig. 3 F. (J) Binned effects of cross-linking on kACT,MUT/kACT,pWT-HRV or kDEACT,MUT/kDEACT,pWT-HRV, whichever was greater.
Because the double Cys mutants were endogenously cross-linked to different extents, as we determined above, the observed functional characteristics were a composite of the characteristics of channels composed of uncrosslinked and of cross-linked α subunits in varying proportions. To compare the functional effects of the different cross-links, we normalized (see Materials and methods) the functional effects to 100% cross-linking (Fig. 5).
It was notable that two cross-links between S0 and S3–S4, R20C to L199C and R17C to R201C, had almost no effect on the V50 or on the rate constants for activation and deactivation (Fig. 5, E–H; Fig. S2), implying that these residues are close in the resting, activated, and transitions states. Other cross-links between S0 and S3–S4 did have significant effects on V50 or gating kinetics. The cross-links between R17C, G18C, and Q19C, in the S0 flank, and L199C, in the S3–S4 loop, all caused a rightward shift in the G-V curves (Fig. 5 E), indicating that more electrostatic energy was needed to activate the channel; i.e., the closed state was stabilized relative to the open state. By contrast, cross-links between Q19C or R20C and R201C caused a leftward shift in the G-V curve and hence stabilized the open state relative to the closed state (Fig. 5 G). The shifts were similar in 0 and 10 μM Ca2+, implying that the cross-links did not affect the dependence of V50 on Ca2+.
Consistent with the above shifts in V50 were the changes in activation and deactivation kinetics. For all S0 Cys to S3–S4 L199C cross-links that caused a rightward shift in V50, the rate constants for activation were slowed more than the rate constants for deactivation (Fig. 5, E and F). In contrast, for the pairs between S0 and R201C that showed a leftward shift in V50, the rate constants for deactivation were slowed more than the rate constants for activation (Fig. 5, G and H).
All cross-links from the S0 flank to the S1 flank and from the S0 flank to the S2 flank resulted in effects on V50 or on the kinetics of activation and deactivation (Fig. 5, A–D; Fig. S2). These effects could reflect the cross-linking of Cys substituted for residues that in the native structure are in different positions in the different states of the voltage sensor, or the effects could reflect a perturbation of a static part of the structure that in turn affects parts that do move. Again, that these perturbations do not completely disrupt channel function and that the extents of endogenous cross-linking are moderately high indicate that the Cys involved, and the native residues they replaced, are not far apart in either the resting or the activated states of the voltage sensor.
Reducing and Reforming Disulfides on the Cell Surface
In three double-Cys mutants with large ΔV50s, we tested by reduction of the disulfide on the cell surface whether the functional changes were due to disulfide bond formation per se or to the effects of the mutations (Fig. 6). We exposed outside-out patches to 20 mM DTT at pH 8.0 for 5–10 min (Fig. 6, A–D). DTT shifted the G-V curves back toward the G-V curves of the single Cys mutants, leftward in R17C-P137C (Fig. 6, B and E) and rightward in R20C-R201C (Fig. 6, D and E). In R20C-F144C (Fig. 6, C and E), DTT shifted the G-V curve rightward toward the G-V curve of the single mutant, F144C, the only single mutant the function of which was significantly different than that of pWT-HRV (Fig. 4 E). DTT did not affect the G-V curve of pWT-HRV (Fig. 6 A). Also, in no case did DTT increase the maximum tail currents at −80 mV, from which we conclude that there was no pool of channels held in a nonfunctional state by disulfide cross-linking.
Figure 6.

Reversibility by reduction of the functional effects of cross-linking. (A) Normalized conductance as a function of voltage for pWT-HRV before (unfilled circles) and after (filled circles) 20 mM DTT (pH 8.0) for 5 min. The measurements were made in outside-out macropatches with 10 μM Ca2+ inside the pipet. G-V curves were determined before and after DTT on the same patch. Similarly, G-V curves were determined before and after DTT for (B) R17C–P137C, (C) R20C–F144C, and (D) R20C–R201C. (E) V50 (±SD and n) for pWT-HRV and the three double-Cys mutants, before and after DTT.
One criterion of the near-native structure of cross-linked double-Cys mutant was that they were transported to the cell surface. Another criterion would be whether the disulfide cross-link could be reduced and reoxidized at the cell surface. For reoxidation we used a membrane-impermeant, novel bis-quaternary-ammonium diamide derivative (QPD) (Fig. 7 A), which, like diamide, can induce disulfide bond formation. QPD did not induce cross-linking of the single-Cys mutants (unpublished data). We tested the reformation by QPD of the disulfide between G18C and L199C and between R20C and R201C (Fig. 7 B), two pairs that were almost completely cross-linked endogenously and in which the cross-link had measurable functional effects. In both cases, DTT reduced, and QPD reformed, the disulfides.
Figure 7.

Reduction and reformation of disulfides. (A) Structure of 4,4′-(azodicarbonyl)-bis-[1,1-dimethylpiperazinium, diiodide] (QPD). (B) Reduction and reoxidation of the disulfide between G18C and L199C (left) and R20C and R201C (right). The intact cells were surface biotinylated, reduced with DTT, and oxidized for 6 min with 40 μM QPD. The extent of cross-linking was estimated from each pair of lanes. (C) Functional effects of reduction and reoxidation: G-V curve in outside-out macropatches from cells expressing the mutant α G18C-L199C (left) and R20C- R201C (right). The cells were treated with 20 mM DTT, pH 8.0, for 20 min, before pulling the patch. After first G-V curve, the patch was exposed to 40 μM QPD and the effects monitored with 1-s ramps (−100 to +100 mV) from a holding potential of −80 mV, with 5-s intervals at −80 mV. When the reaction was complete, a second G-V curve was determined. The G-V curve from a different cell expressing the same double-Cys mutant that was neither reduced nor reoxidized is shown as a dashed line. The pipet contained 10 μM Ca2+. (D) The mean change in V50 induced by QPD acting on cells expressing the double-Cys mutants, R20C-R201C and G18C-L199C, and each of the single-Cys mutants, from records such as in C. The means, standard errors, and n are shown.
We also determined whether QPD could restore the functional effects of cross-linking G18C-L199C α and R20C-R201C α (Fig. 7 C). In G18C-L199C α, endogenous cross-linking shifted the G-V curve to the right relative to pWT-HRV (Fig. 5 E); DTT shifted it to the left; and QPD shifted it back to the right (Fig. 7 C). In R20C-R201C α endogenous cross-linking shifted the G-V curve to the left relative to pWT-HRV (Fig. 5 G). DTT shifted the G-V curve to the right relative to that of the unreduced mutant, and QPD shifted the curve back toward that of the endogenously cross-linked α (Fig. 7 C). QPD did not affect the V50 of the single mutants (Fig. 7 D). Thus, for these two mutants, the cross-link could be reduced and reformed at the cell surface with similar effects on V50 to those resulting from endogenous cross-linking.
DISCUSSION
Disulfide Cross-linking
Our approach to locating S0 among the other TM helices, S1–S6, was to mutate to Cys, one at a time, at least the first four residues just extracellular to the membrane domain of the TM helices. In the polar juxtamembrane environment, the Cys thiol can ionize to the reactive thiolate and is accessible to oxidizing reagents and oxidizing enzymes. Furthermore, Cys just outside the membrane domain of different TM helices are more or less in the same plane parallel to the membrane. Because at least four consecutive positions are mutated, all directions parallel to the membrane are sampled, whatever the local secondary structure, and in neighboring segments at least some substituted Cys are likely to be pointing toward each other and susceptible to disulfide bond formation.
The induction of disulfide bond formation between substituted Cys has been widely used to infer the structure and structural dynamics of proteins (Bass et al., 2007). In Shaker K+ channels, disulfide bond formation between substituted Cys has provided evidence for the proximity between the extracellular end of S4 of one subunit and the extracellular end of S5 of an adjacent subunit (Laine et al., 2003; Elliott et al., 2004), an arrangement that is compatible with the model in Fig. 3 F. Also in Shaker, cross-linking was obtained between the extracellular end of S4 and positions approximately six residues from the extracellular ends of S1 and S2 (Campos et al., 2007).
Even though the centers of the sulfur atoms in a disulfide are only 3 Å apart, disulfide bond formation has been induced between Cys SH known to be 15 Å apart in the crystal structure of a protein (Butler and Falke, 1996). This is consistent with the fluctuating nature of protein structures, where even some moderately distant residues have finite probability of colliding, the shorter the distance, the greater the probability. Based on this, the relative proximities of cysteines have been inferred from the relative rate constants for disulfide bond formation (Bass et al., 2007). Distance, however, is not the only determinant of the rate constant for disulfide bond formation. Structural flexibility and the likelihood of overcoming steric hindrance and of achieving the correct orientation for reaction may differ in different parts of a protein. The electrostatic environment also plays a role in at least two ways: it affects the pKA of the cysteine thiol (the thiolate is the reactive species), and it affects the collision probability of the charged reactants. At the water-accessible surface of a protein, these effects are modest in the absence of a local, high concentration of positive or negative side chains (Hansen et al., 2005).
We determined the extent of endogenous disulfide bond formation in intact cells. No oxidizing agent was added. Endogenous disulfide formation of a protein targeted to the cell membrane can be due to uncatalyzed Cys thiol oxidation by dissolved O2 (i.e., spontaneous disulfide formation) or to catalytic oxidation by protein disulfide isomerase (PDI) homologues in the ER(Wilkinson and Gilbert, 2004), by secreted PDI homologues (Jiang et al., 1999), or by secreted oxidases (Becker et al., 2006). Endogenous disulfide bond formation between juxtamembrane Cys in membrane proteins has been observed (Whitley et al., 1993; Jansen and Akabas, 2006; Iscla et al., 2007). For membrane-spanning proteins, only Cys in the extracytoplasmic domain or close to the exofacial lipid–water interface are substrates for PDI homologues (Luo et al., 2004; Lu et al., 2006; Wilkinson and Gilbert, 2004).
The mechanism of PDI and its homologues provides a rationale for relating the extent of disulfide bond formation to the proximity of the Cys involved. These enzymes make and break disulfides repeatedly in the context of chaperone-enabled refolding, so that at the end of the process the disulfides formed are the most stable ones possible (Wilkinson and Gilbert, 2004). It seems likely that if there are only two extracellular Cys accessible in the target protein, then the extent of disulfide formation should depend on the stability of this target disulfide relative to the stability of the disulfide between the two Cys in the catalytic site of the PDI homologue. Thus, the stability of the PDI catalytic site disulfide provides a baseline. Like the rate constant for disulfide bond formation discussed above, the stability of the target disulfide depends on the proximity of the two Cys in a stable structure of the protein. The stability of the disulfide-bonded structure will depend on the differences in both electrostatic and nonelectrostatic terms in the free energy difference between the disulfide bonded structure and the structures with free cysteines. We sought to determine the average proximity of short segments of the BK α by testing multiple Cys pairs to average out the effects of relative orientation and local electrostatics.
We quantitated the extent of endogenous disulfide bond formation in α trafficked to the plasmalemma in intact cells. We assume that 48 h after transfection when the cells were harvested, the surface densities at the cell surface of reduced and oxidized α species were in a steady state. In the steady state, the extent of cross-linking, xEX, of α on the cell surface is given by
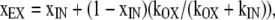 |
4 |
in which xIN is the extent of cross-linking of α in the intracellular pool feeding the cell surface, kOX is the overall rate constant for both spontaneous and catalyzed disulfide-bond formation of α on the cell surface, and kIN is the rate constant for endocytosis of α (see Appendix). xIN is determined by the activity of the PDI homologues in the ER and by the argument above is a measure of proximity of the Cys. kOX also depends on the proximity of the Cys thiols in the native structure of the protein in the plasmalemma. Thus, xEX is a measure of proximity. Where xIN is close to 1, or kOX << kIN (see Appendix),
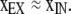 |
5 |
Placing the Extracellular End of S0 in a Model of Kv1.2
As a measure of the relative proximity of the extracellular flank of S0 to the flanks of S1–S6, we averaged the top three extents of cross-linking for each group (Fig. 3, A–E). In a model of BK α based on a Rosetta model of Kv1.2 in the closed state (Yarov-Yarovoy et al., 2006), we placed a circle representing the extracellular end of S0 in a location that satisfies qualitatively the relative proximities estimated from the mean extents of cross-linking and is consistent with the lack of cross-linking to S5 and S6 (Fig. 3 F). The extracellular end of S0 is in a cove three-quarters surrounded by the extracellular ends of S1–S4, closest to the short loop between S3 and S4.
Our inference is based not only on the high extent of endogenous cross-linking of the S0 flank to the S1 and S2 flanks and to the S3–S4 loop but also on the lack of significant cross-linking to the flanks of S5 and S6. We guarded against the possibility that this lack of cross-linking could be due to a misalignment normal to the membrane plane by testing Cys substituted in positions six (S6) and eight (S5) residues from the predicted extracellular side of the membrane (Fig. 3, D and E). There was a small extent of cross-linking of S0 to Cys five and eight residues from the membrane domain of S5 (Fig. 3 D), most likely a reflection of the flexibility of the S0 flank and the increased flexibility of the S5 flank some distance from the membrane. Could the meager extent of cross-linking of S0 to S5 and S6 be due to the suppression of the reactivity of the Cys due to local negatively charged residues? Although we do not know the exact three-dimensional structure of these regions in BK α, Asp and Glu are not concentrated in the sequences around the residues mutated to Cys (Fig. 1 A). There are as many negatively charged residues in or around the flanks of S1 and S2, which did cross-link to S0, as around the flank of S5, which did not cross-link to S0. Furthermore, the flank of S0 is positively charged, which should more than compensate for the effect of negatively charged residues around a Cys in another flank (Hansen et al., 2005).
Functional Effects of Cross-linking
Our initial purpose of looking at the functional effects of the cross-linking was to determine whether or not a cross-linked α was in a near-native conformation. Our assumption was that near-normal function implies near-normal structure. We found that each of the 16 double-Cys mutants that we tested for function were expressed at the cell surface and were functional. Thus, the most highly cross-linked double-Cys mutants were functionally competent and had near-native structures.
Of course, the combination of the mutation of two native residues to Cys and the cross-linking of the Cys by a disulfide bond must perturb the native structure to some extent. What can we infer about the structural dynamics of the cross-linked segments from the functional effects of cross-linking? This question is easiest to answer for cross-links that form endogenously to a high extent and have no functional effects. Two cross-links between S0 and S3–S4, R20C to L199C and R17C to R201C, had almost no effect on the V50 or on the rate constants for activation and deactivation (Fig. 5, E–H; Fig. S2). Because the extents of endogenous cross-linking of these pairs were 96% and 88%, respectively, we conclude that the two Cys in each of these pairs, and the native residues that they replaced, are near neighbors in the resting state of the voltage sensor in which disulfide bond formation takes place. But they are also likely to be closely apposed in the activated state of the voltage sensor, because the cross-linked structures favor neither the open nor the closed state. If the cross-link stabilized either state, ΔV50 would have been significantly different than zero, which it was not. Furthermore, the two Cys (and the native residues that they replaced) are unlikely to move relative to one another during the transitions between states, because the cross-linking of the substituted Cys has little effect on the kinetics of opening and closing. We conclude that the two Cys, and the residues that they replaced, are close neighbors in the closed, open, and transition states of the channel.
The native residues, R20 and L199, are predicted to be the first residues just flanking the membrane domains of S0 and S3, respectively. Thus, the adjacent membrane domains of S0 and S3 in both the deactivated/closed and activated/open states and in the transition states are also close. Similarly, R17C and R201C formed a cross-link between the S0 flank and the S3–S4 loop, two residues from the membrane domain of S4. It seems likely that the four residues flanking the extracellular end of S0 and the four-residue loop between S3 and S4 follow close, parallel trajectories, because all of the double-Cys mutants in these segments cross-linked nearly completely (Fig. 3 C; Table S1). Thus, to the extent that the extracellular ends of S3 and S4 move during activation and deactivation, the extracellular end of S0 moves with them. Some of the cross-links between the S0 flank and the S3–S4 loop do result in changes in V50 or in the rate constants of activation and deactivation (Fig. 5, E–H), which is consistent with some differences in the interface between these segments in the resting, transition, or activated states.
Whatever the interactions are between the S0 flank and the S3–S4 loop, their residues are not irreplaceable for the function of α alone, because deletion of the N-terminal extracellular residues, starting with R20, did not perturb the function of α; this deletion did, however, prevent the modulation of the V50 by by β1(Morrow et al., 2006). Therefore, the effect of S0 on the properties of the BK voltage sensor must depend on interactions either with the S0 membrane domain, for which there is evidence (Koval et al., 2007), and/or with the intracellular S0–S1 loop.
Possible Effect of S0 on Voltage Sensor Properties
Compared with other voltage-gated K+ channels, BK channel V50s for both gating current and for opening are shifted to much more positive voltages. Although the BK channel has a smaller gating charge, the electrostatic energy for activating the voltage sensors is greater for the BK channel than that for other voltage-gated K+ channels. F × qgating × V50,gating is a measure of the standard free energy difference per mole between the resting and the activated states. For the BK channel composed of α alone, qgating is 0.55 charges per subunit and V50 is 156 mV (Stefani et al., 1997; Horrigan and Aldrich, 1999; Bao and Cox, 2005); thus, the standard free energy difference is 8.5 kJ/mole-subunit. For Kv1.2 (Scholle et al., 2004), Kv2.1 (Scholle et al., 2004), and Shaker (Schoppa et al., 1992), the standard free energy differences range from −3.6 to −15 kJ/mole-subunit. Thus, the standard free energy difference, activated state minus resting state, is 12 to 23 kJ per mole-subunit (2.9 to 5.5 kcal/mol-subunit) more positive in BK channel than in Kv1.2, Kv2.1, or Shaker channels. Niu et al. (2004) suggested that the cytoplasmic Ca2+-dependent linker–gating ring complex exerts drag on the BK channel gate and the voltage sensor. Given the position of S0 in the midst of S1–S4 and its close proximity to S3 and S4, it seems possible that S0 also contributes to the stabilization of the resting state of the BK channel relative to other voltage-gated K+ channels.
Acknowledgments
We thank Vladimir Yarov-Yarovoy and William Catterall for supplying the Kv1.2 model used in Fig. 3 F and Jonathan Javitch and Xiao-Wei Niu for comments on the manuscript.
This work was supported in part by National Institutes of Health research grant awards P01 HL081172 from the National Heart, Lung, and Blood Institute, R01 NS054946 from National Institute of Neurological Disorders and Stroke, and UL1 RR024156 from National Center for Research Resources and the Arlene and Arnold Goldstein Family Foundation. S.O. Marx is an Established Investigator of the American Heart Association.
Lawrence G. Palmer served as editor.
APPENDIX
Steady-State Extent of Disulfide Bond Formation
The rate of change of reduced α in the plasmalemma is
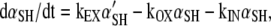 |
(A1) |
where αSH is the concentration of reduced α in the cell surface membrane, α'SH is the concentration of well-folded, reduced α in the intracellular membranes, kEX is the rate constant for exocytosis of both α'SH and of α'SS (oxidized α), kIN is the rate constant for endocytosis of both αSH and αSS, and kOX is the rate constant for oxidation of αSH on the cell surface.
The rate of change of oxidized α in the plasmalemma is
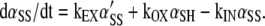 |
(A2) |
In the steady-state both derivatives equal zero, implying
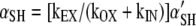 |
(A3) |
and
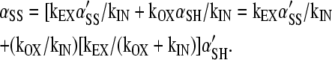 |
(A4) |
By definition, the fraction xEX of α on the surface that is oxidized is
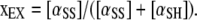 |
(A5) |
Substituting Eq. A3 and A4 in A5, we obtain xEX = {kEXα'SS/kIN + (kOX/kIN)[kEX/(kOX + kIN)]α'SH }/{kEXα'SS/kIN + (kOX/kIN) [kEX/(kOX + kIN)]α'SH + [kEX/(kOX + kIN)]α'SH }. This is simplified by the following operations on both the numerator and denominator: factor out kEX, multiply by kIN, divide by α'SH, and simplify the numerator, to obtain:
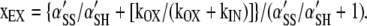 |
(A6) |
By definition,
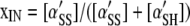 |
(A7) |
or
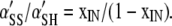 |
(A8) |
Combining A6 and A8 yields
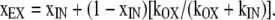 |
(A9) |
If either kOX << kIN or xIN ∼1,
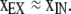 |
(A10) |
Spontaneous oxidation of protein Cys is very slow in the absence of metal catalysts (Wilkinson and Gilbert, 2004; Becker et al., 2006) and is unlikely to be faster than the rate of spontaneous intramolecular disulfide bond formation in dithiothreitol, which has a k ≤ 0.05/h at neutral pH and room temperature (unpublished data). The half-time for internalization of the K+ channel, Kv1.4, transiently transfected in HEK293 cells, was 87 min (i.e., kIN = ∼0.5/h) (Jugloff et al., 2000), and kIN = 0.4/h to 0.8/h for an N-type Ca2+ channel, also transiently transfected in HEK293 cells (Bernstein and Jones, 2007). Assuming a similar kIN for BK α and that catalytic disulfide bond formation is taking place mainly in the ER, kOX ≤ 0.05/h, kOX/kIN < 0.1, and Eq. A10 applies.
G. Liu and S.I. Zakharov contributed equally to this work.
Abbreviations used in this paper: BK, large-conductance, voltage- and calcium-activated potassium; HRV, human rhinovirus; PDI, protein disulfide isomerase; pWT, pseudo-wild-type; QPD, quaternary piperazinium diamide; TM, transmembrane; WT, wild-type.
References
- Bao, L., and D.H. Cox. 2005. Gating and ionic currents reveal how the BKCa channel's Ca2+ sensitivity is enhanced by its β1 subunit. J. Gen. Physiol. 126:393–412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bass, R.B., A.S. Miller, S.L. Gloor, and J.J. Falke. 2007. The PICM chemical scanning method for identifying domain-domain and protein-protein interfaces: applications to the core signaling complex of E. coli chemotaxis. Methods Enzymol. 423:1–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Becker, L., M.E. Nesheim, and M.L. Koschinsky. 2006. Catalysis of covalent Lp(a) assembly: evidence for an extracellular enzyme activity that enhances disulfide bond formation. Biochemistry. 45:9919–9928. [DOI] [PubMed] [Google Scholar]
- Bernstein, G.M., and O.T. Jones. 2007. Kinetics of internalization and degradation of N-type voltage-gated calcium channels: role of the α2/δ subunit. Cell Calcium. 41:27–40. [DOI] [PubMed] [Google Scholar]
- Bose, A.K., M.S. Manhas, D.P. Sahu, and V.R. Hedge. 1984. Stereospecific cyclization of β-hydroxyl aryl amides to β-lactams. Can. J. Chem. 62:2498–2505. [Google Scholar]
- Butler, S.L., and J.J. Falke. 1996. Effects of protein stabilizing agents on thermal backbone motions: a disulfide trapping study. Biochemistry. 35:10595–10600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campos, F.V., B. Chanda, B. Roux, and F. Bezanilla. 2007. Two atomic constraints unambiguously position the S4 segment relative to S1 and S2 segments in the closed state of Shaker K channel. Proc. Natl. Acad. Sci. USA. 104:7904–7909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elliott, D.J., E.J. Neale, Q. Aziz, J.P. Dunham, T.S. Munsey, M. Hunter, and A. Sivaprasadarao. 2004. Molecular mechanism of voltage sensor movements in a potassium channel. EMBO J. 23:4717–4726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hansen, R.E., H. Ostergaard, and J.R. Winther. 2005. Increasing the reactivity of an artificial dithiol-disulfide pair through modification of the electrostatic milieu. Biochemistry. 44:5899–5906. [DOI] [PubMed] [Google Scholar]
- Horrigan, F.T., and R.W. Aldrich. 1999. Allosteric voltage gating of potassium channels II. Mslo channel gating charge movement in the absence of Ca2+. J. Gen. Physiol. 114:305–336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iscla, I., G. Levin, R. Wray, and P. Blount. 2007. Disulfide trapping the mechanosensitive channel MscL into a gating-transition state. Biophys. J. 92:1224–1232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jansen, M., and M.H. Akabas. 2006. State-dependent cross-linking of the M2 and M3 segments: functional basis for the alignment of GABAA and acetylcholine receptor M3 segments. J. Neurosci. 26:4492–4499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang, X.M., M. Fitzgerald, C.M. Grant, and P.J. Hogg. 1999. Redox control of exofacial protein thiols/disulfides by protein disulfide isomerase. J. Biol. Chem. 274:2416–2423. [DOI] [PubMed] [Google Scholar]
- Jiang, Y., A. Lee, J. Chen, V. Ruta, M. Cadene, B.T. Chait, and R. MacKinnon. 2003. X-ray structure of a voltage-dependent K+ channel. Nature. 423:33–41. [DOI] [PubMed] [Google Scholar]
- Jugloff, D.G., R. Khanna, L.C. Schlichter, and O.T. Jones. 2000. Internalization of the Kv1.4 potassium channel is suppressed by clustering interactions with PSD-95. J. Biol. Chem. 275:1357–1364. [DOI] [PubMed] [Google Scholar]
- Karlin, A., C. Wang, J. Li, and Q. Xu. 2004. Transfer in SDS of biotinylated proteins from acrylamide gels to an avidin-coated membrane filter. Biotechniques. 36:1010–1016. [DOI] [PubMed] [Google Scholar]
- Knaus, H.G., A. Eberhart, R.O. Koch, P. Munujos, W.A. Schmalhofer, J.W. Warmke, G.J. Kaczorowski, and M.L. Garcia. 1995. Characterization of tissue-expressed α subunits of the high conductance Ca2+-activated K+ channel. J. Biol. Chem. 270:22434–22439. [DOI] [PubMed] [Google Scholar]
- Kosower, E.M., N.S. Kosower, H. Kenety-Londner, and L. Levy. 1974. Glutathione. IX. New thiol-oxidizing agents: DIP, DIP+1, DIP+2. Biochem. Biophys. Res. Commun. 59:347–351. [DOI] [PubMed] [Google Scholar]
- Koval, O.M., Y. Fan, and B.S. Rothberg. 2007. A role for the S0 transmembrane segment in voltage-dependent gating of BK channels. J. Gen. Physiol. 129:209–220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laine, M., M.C. Lin, J.P. Bannister, W.R. Silverman, A.F. Mock, B. Roux, and D.M. Papazian. 2003. Atomic proximity between S4 segment and pore domain in Shaker potassium channels. Neuron. 39:467–481. [DOI] [PubMed] [Google Scholar]
- Long, S.B., E.B. Campbell, and R. Mackinnon. 2005. Crystal structure of a mammalian voltage-dependent Shaker family K+ channel. Science. 309:897–903. [DOI] [PubMed] [Google Scholar]
- Lu, X., A.W. Gross, and H.F. Lodish. 2006. Active conformation of the erythropoietin receptor: random and cysteine-scanning mutagenesis of the extracellular juxtamembrane and transmembrane domains. J. Biol. Chem. 281:7002–7011. [DOI] [PubMed] [Google Scholar]
- Luo, B.H., T.A. Springer, and J. Takagi. 2004. A specific interface between integrin transmembrane helices and affinity for ligand. PLoS Biol. 2:e153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meera, P., M. Wallner, M. Song, and L. Toro. 1997. Large conductance voltage- and calcium-dependent K+ channel, a distinct member of voltage-dependent ion channels with seven N-terminal transmembrane segments (S0-S6), an extracellular N terminus, and an intracellular (S9-S10) C terminus. Proc. Natl. Acad. Sci. USA. 94:14066–14071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morrow, J.P., S.I. Zakharov, G. Liu, L. Yang, A.J. Sok, and S.O. Marx. 2006. Defining the BK channel domains required for β1-subunit modulation. Proc. Natl. Acad. Sci. USA. 103:5096–5101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niu, X., X. Qian, and K.L. Magleby. 2004. Linker-gating ring complex as passive spring and Ca2+-dependent machine for a voltage- and Ca2+-activated potassium channel. Neuron. 42:745–756. [DOI] [PubMed] [Google Scholar]
- Rost, B., R. Casadio, P. Fariselli, and C. Sander. 1995. Transmembrane helices predicted at 95% accuracy. Protein Sci. 4:521–533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Savalli, N., A. Kondratiev, L. Toro, and R. Olcese. 2006. Voltage-dependent conformational changes in human Ca2+- and voltage-activated K+ channel, revealed by voltage-clamp fluorometry. Proc. Natl. Acad. Sci. USA. 103:12619–12624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scholle, A., S. Dugarmaa, T. Zimmer, M. Leonhardt, R. Koopmann, B. Engeland, O. Pongs, and K. Benndorf. 2004. Rate-limiting reactions determining different activation kinetics of Kv1.2 and Kv2.1 channels. J. Membr. Biol. 198:103–112. [DOI] [PubMed] [Google Scholar]
- Schoppa, N.E., K. McCormack, M.A. Tanouye, and F.J. Sigworth. 1992. The size of gating charge in wild-type and mutant Shaker potassium channels. Science. 255:1712–1715. [DOI] [PubMed] [Google Scholar]
- Stefani, E., M. Ottolia, F. Noceti, R. Olcese, M. Wallner, R. Latorre, and L. Toro. 1997. Voltage-controlled gating in a large conductance Ca2+-sensitive K+ channel (hslo). Proc. Natl. Acad. Sci. USA. 94:5427–5431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wallner, M., P. Meera, and L. Toro. 1996. Determinant for β-subunit regulation in high-conductance voltage-activated and Ca2+-sensitive K+ channels: an additional transmembrane region at the N terminus. Proc. Natl. Acad. Sci. USA. 93:14922–14927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whitley, P., L. Nilsson, and G. von Heijne. 1993. Three-dimensional model for the membrane domain of Escherichia coli leader peptidase based on disulfide mapping. Biochemistry. 32:8534–8539. [DOI] [PubMed] [Google Scholar]
- Wilkinson, B., and H.F. Gilbert. 2004. Protein disulfide isomerase. Biochim. Biophys. Acta. 1699:35–44. [DOI] [PubMed] [Google Scholar]
- Wolin, C.D., and H.R. Kaback. 2000. Thiol cross-linking of transmembrane domains IV and V in the lactose permease of Escherichia coli. Biochemistry. 39:6130–6135. [DOI] [PubMed] [Google Scholar]
- Yarov-Yarovoy, V., D. Baker, and W.A. Catterall. 2006. Voltage sensor conformations in the open and closed states in ROSETTA structural models of K+ channels. Proc. Natl. Acad. Sci. USA. 103:7292–7297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zakharov, S.I., J.P. Morrow, G. Liu, L. Yang, and S.O. Marx. 2005. Activation of the BK (SLO1) potassium channel by mallotoxin. J. Biol. Chem. 280:30882–30887. [DOI] [PubMed] [Google Scholar]