

Abstract
A design principle for a two-photon photochemically removable protecting group based on sequential one-photon processes has been established. The expected performance of such groups in spatially directed photoactivation/photodeprotection has been shown by a kinetic analysis. One particular molecular class fitting into this design, the nitrobenzyl ethers of o-hydroxycinnamates, has been presented. An initial demonstration of two-photon deprotection of one such group prompted further optimization with respect to photochemical deprotection rate. This was accomplished by the preparation and screening of a 135-member indexed combinatorial library. Optimum performance for λ >350 nm deprotection in organic solvent was found with 4,5-dialkoxy and α-cyano substitution in the nitrobenzyl group and 4-methoxy substitution in the cinnamate.
Multiphoton processes are pervasive in science and technology, yet rare in one area, synthetic chemistry. Multiphoton, usually two-photon, excitation provides a means of activating chemical or physical processes with high spatial resolution. It has applications including optical data storage, 3D fluorescence imaging, and stereolithography (1, 2) and takes advantage of the fact that the two-photon absorption probability depends nonlinearly (quadratically) on the light intensity. Absorption is thereby confined to a volume element at the light focus, so that subsequent processes, such as fluorescence or light-induced chemical reactions, are also localized in this volume. In this work, two-photon protecting groups useful for photoactivation have been discovered by using a combinatorial chemistry approach.
Two-photon processes are well known, often exploited in chemistry and biology, and generally involve excitation of a normal UV-absorbing chromophore by using two IR photons. Two-photon phototherapy with psoralen chromophores has been investigated (3), as has the spatial localization of a psoralen-DNA crosslink (4). Two-photon fluorescence microscopy has provided a new avenue to high spatial resolution in 3D visualization of biological structures (5–7). New fluorophores for two-photon fluorescence microscopy have recently been developed (8). New fluorophores have been applied to the study of interactions of therapeutic agents with target cells by using two-photon microscopy (9). Two photon uncaging photochemical microscopy has also been developed (10, 11). In this method, two spatially directed 640-nm photons excite a traditional UV-absorbing nitrobenzyl-caged receptor agonist (carbamyl choline), whose release causes a change in cell current that is mapped to the location of the light. New protecting groups (6-bromo-7-hydroxycoumarinyl, bromohydroxyquinoline) with high two-photon cross sections for IR absorption have been developed for caging applications (12, 13). Another approach to two-photon uncaging is the addition of two conventional photochemically removable protecting groups to a biological effector, either by necessity (14) or design (15).
Materials are another area of application for two-photon processes. Two-photon-initiated polymerization can be used for 3D data storage and microfabrication (16). Superior initiators with much greater two-photon absorption cross sections and therefore higher efficiency in stereolithography, inter alia, have recently been developed (17, 18). Two-photon photochromic materials have also been studied (19).
A technological area in which spatial control is paramount is photolithography, whose impact on advances in electronics and computing cannot be overstated. The ability to write finer and finer lines in resist coatings to fabricate more and more components per unit area has depended on a wide range of advances in physics, chemistry, and engineering (20). One recent advance involves the development of polymeric amplified photoacid resists (21–23). Resists used in semiconductor fabrication generally have the useful (and essential) property of a nonlinear dependence of development rate on photon dose. That is, there is a “threshold” effect in the dissolution of light-sensitive, polymer-based resist materials: below a certain light dose, they are stable, and above it they can be dissolved to permit subsequent processing. In other words, the dissolution of resists is a nonlinear and perhaps noncontinuous function. This enables fine features, comparable to those of the mask, to be created even when the aerial image of the mask has much lower resolution as a result of diffraction (24).
The applications of photolithography have been extended into other areas, such as stereolithography, molecular patterning (25–31), and light-directed combinatorial synthesis (32, 33). The size of the features that can be written and their contrast compared with regions where no features are intended are limited, however. For example, the smallest single molecular feature that has been fabricated photolithographically is 0.15 μm, and degradation of performance is seen in some methods when step-and-repeat processing is necessary (34). This limitation is related to the lack of a nonlinear development stage in these methods, unlike polymer resist-based microfabrication. Rather, chemical processes, generally bond making or breaking, arising directly from irradiation lead to the creation of a desired physical or chemical feature.
One factor governing the fidelity of the replication of the mask pattern in light-directed synthesis is the rate of the deprotection reaction, as contrasted with resist systems where the relevant factor is dissolution kinetics. Conventional kinetics analysis of photochemical surface deprotection reactions can be performed (35). The rate of a deprotection reaction is expressed in the integrated form of the rate equation as 1 – e–kIt, where k is a constant that incorporates parameters unique to the particular protecting group used, such as quantum yield, absorption cross section, etc., I is the intensity of the light, and t is time. That is, the deprotection reaction proceeds exponentially with irradiation time, asymptotically approaching 100%. The difference in rates in the unmasked and masked areas, and therefore the contrast between areas intended to be illuminated and those not, is simply reflected in the I term. High optical density masks may provide differential intensity at the mask as large as 105; however, the aerial image of the mask at the work piece will have significantly lower contrast. Furthermore, kinetic analysis using the above model of reactions taken to 99% conversion shows that the aerial contrast ratio is decreased four to five times in unmasked and masked areas. So, even with high-contrast masks, there will be significant deprotection in the masked areas if the reaction is conducted to 100% conversion in the unmasked areas. This is an important consideration for light-directed synthesis because solid-phase chemistry requires essentially quantitative yields in both deprotection and coupling reactions; no purification of intermediate polymer sequences can be performed. Therefore, there will always be “stowaways” (residues incorporated at sites that were not intended to be illuminated) in a light-directed synthesis (32), and polymer sequences made by the method can never be as pure as those prepared by conventional one-at-a-time processing. However, if the photochemistry used in light-directed synthesis were nonlinear, then reactions could be carried to completion with minimal competing deprotection in masked areas, resulting in a higher quality of the sequences prepared.
Previous work in two-photon photoactivation has aimed only to exploit high IR fluences to excite conventional UV-absorbing chromophores with two IR photons, such as the caging groups mentioned earlier and the generation of singlet oxygen (36). Some principles for design of two-photon absorbing chromophores have been advanced (37), but no general strategies are available for the design of a photoreactive chemical group with a high two-photon cross section. Despite the advantages of the two-photon process for spatial control, the long wavelength of IR photons limits the achievable spatial resolution. The technological drive in conventional, one-photon photolithography is toward shorter and shorter wavelengths because of the direct relationship between diffraction-limited feature size and wavelength. Therefore, a UV-excited two-photon photoremovable protecting group would be highly desirable. Few examples of UV two-photon reactions in organic chemistry are known (38), such as a photochromic system (39). An elementary chemical reaction that involves bond formation or cleavage and is nonlinear in photon dose is very rare, limiting the spatial resolution that can be achieved in molecular lithography and calling for the further development of novel nonlinear photochemical processes and groups.
Materials and Methods
Preparation and Irradiation of Ethyl 2-(2′-nitrobenzyloxy)cinnamate (13). A mixture of 0.611 g (5 mmol) of salicylaldehyde and 3.65 g (10.5 mmol) of ethyl (triphenylphosphoranylidene)acetate in 100 ml of benzene was heated under reflux for 12 h. The reaction was cooled and concentrated to afford a semisolid. This residue was loaded onto a small plug of silica gel and eluted with 25% ether in hexane. Evaporation of the solvent afforded a colorless solid (12) that was directly used for the next step. The alkylation of the above solid with 2-nitrobenzylbromide (2.16 g, 10 mmol) was accomplished with potassium fluoride (8 eq) on alumina (40) in acetonitrile for 24 h. The mixture was filtered through a pad of Celite, and the solvent was removed. Repeated crystallization from diethyl ether/pentane afforded colorless crystals of 13 (1.02 g, 62%): mp 81°C; UV λmax 273 nm (ε 31,000); IR (thin film) 2,990, 1,698, 1,522, 1,488, 1,204, 1,149 cm–1; 1H NMR (CDCl3, 300 MHz) δ 8.20 (dd, J = 8.4, 1.2 Hz, 1 H), 8.15 (d, J = 16.2 Hz, 1 H), 7.87 (dd, J = 7.8, 1.2 Hz, 1 H), 7.7 (dt, J = 7.5, 1.2 Hz, 1 H), 7.58 (dd, J = 7.5, 1.8 Hz, 1 H), 7.50 (dt, J = 8.7, 1.2 Hz, 1 H), 7.33 (dt, J = 7.5, 1.8 Hz, 1 H), 7.00 (t, J = 7.8 Hz, 1 H), 6.91 (d, J = 8.7 Hz, 1 H), 6.52 (d, J = 16.2 Hz, 1H), 5.57 (s, 2H), 4.27 (q, J = 6.9 Hz, 2 H), 1.35 (t, J = 6.9 Hz, 3H); 13C NMR (CDCl3, 75 MHz) δ 167.0, 156.3, 146.6, 139.1, 134.1, 133.4, 131.4, 128.3, 128.1, 124.9, 123.7, 121.5, 118.9, 112.6, 67.2, 60.4, 14.4; high-resolution MS calculated for C18H17NO5 327.1107, found 327.1122.
Irradiation of 13 (244 mg) in 12 ml of argon-flushed acetonitrile for 27 h at 350 nm in a Rayonet photochemical reactor (Southern New England Ultraviolet Company, Bransford, CT) yielded coumarin (98.9%) and recovered 13 (2.5 mg, 1.0%). Four other unknown photoproducts were isolated in 1.6- to 12.8-mg amounts. These trace products are presumably derived from photochemistry of nitrosobenzaldehyde, which is formed in the initial photoreaction.
General Procedure for the Bromination of Nitrobenzylic Alcohols. This reaction was performed in the dark as described (41). The alcohol (10 mmol) was dissolved in 30 ml of dichloromethane, and CBr4 (14 mmol) was added. The solution was cooled to 0°C, and solid triphenylphosphine (14 mmol) was added. After 30 min, the cooling bath was removed, and the mixture was stirred at room temperature overnight. The solvent was evaporated, and the residue was purified by chromatography using pentane/ethyl acetate. Select spectral data are given in Supporting Methods, which is published as supporting information on the PNAS web site, www.pnas.org. Structures of benzaldehydes and cyanohydrins are given in Fig. 9, which is published as supporting information on the PNAS web site.
Synthesis of Three A Libraries. Phosphorane A1 (250 mg, 0.5 mmol) was dissolved in 12 ml of dry benzene in a 50-ml round-bottom flask, and equimolar amounts (0.1 mmol each) of the salicaldehydes B1, B2, B3, B4, and B5 were added. The reaction mixture was refluxed for 5 h, the benzene was evaporated, and the residue was freed of triphenylphosphine oxide by passing over a short column (10 g of silica, 450 ml of 4:1 pentane/ethyl acetate). All eluant was collected and evaporated, yielding 183 mg (95%) of a yellow oil. Likewise, for the A2 library, 257 mg (5 mmol) A2 was used, yielding 188 mg of olefination products (94%). For the A3 library, 265 mg (5 mmol) A3 was used, yielding 192 mg of olefination products (92%).
The three AB pools were alkylated with equimolar amounts of nine nitrobenzylbromides C1-C9 in an analogous fashion, creating three libraries containing 45 compounds each. Stock solutions of C1-C9 were prepared in dry acetonitrile. The amounts used were calculated based on 95% conversion in the Wittig reaction such that little excess was present. KF/alumina was added (8× excess), the reactions were protected from light, and they were stirred for 18 h. The reaction mixture was filtered, the cake was washed with 20 ml of acetonitrile, and the solvent was evaporated. Each library was passed through a short silica column (10 g silica, 450 ml of 4:1 pentane/ethyl acetate). The libraries were isolated in the following amounts: A1, 193 mg; A2, 188 mg; A3, 190 mg. Proton NMR spectra of each library show peaks expected for each component.
Synthesis of Five B Libraries. In five reaction vessels salicylaldehydes, B1-B5, were treated with phosphoranes A1, A2, and A3 in benzene. Stock solutions of A1, A2, and A3 were prepared to ensure equimolar representation in each reaction. In each flask 0.09 mmol of a B was treated with 0.03 mmol of A1, 0.03 mmol of A2, and 0.03 mmol of A3. The five reaction mixtures were refluxed for 5 h, the benzene was evaporated, and the residues were freed of triphenylphosphine oxide by passing over a short column (2 g of silica, 150 ml of 4:1 pentane/ethyl acetate) All eluent was collected and evaporated, yielding yellow oils.
The five AB pools were alkylated with nine nitrobenzylbromides, C1-C9, in an analogous fashion, creating five libraries containing 27 compounds each. The amounts used were calculated based on 95% conversion in the Wittig reaction such that little excess was present. Stock solutions of C1-C9 were prepared in dry dimethoxyethane. Dimethoxyethane was used here instead of acetonitrile because it was found to be equally efficient for the alkylation reaction, and the reactants were more soluble. For each of the five AB pools 0.09 mmol of total C was used, so from each of the nine C stock solutions 0.01 mmol was taken. KF/alumina was added (112 mg, 8× excess), the reactions were protected from light, and they were stirred for 18 h. The reaction mixture was filtered, the cake was washed with 20 ml of acetonitrile, and the solvent was evaporated. Each library was passed through a short silica column (2 g of silica, 150 ml of 4:1 pentane/ethyl acetate). The libraries were isolated in the following amounts: B1, 17.1 mg; B2, 28.6 mg; B3, 28.9 mg; B4, 25.9 mg; B5, 23.7 mg. Proton NMR spectra of each library show peaks expected for each component.
Synthesis of Nine C Libraries. In a single reaction vessel were refluxed together A1 (135 mg, 0.27 mmol), A2 (139 mg, 0.27 mmol) and A3 (143 mg, 0.27 mmol), with B1 (19.8 mg, 0.162 mmol), B2 (29.5 mg, 0.162 mmol), B3 (26.9 mg, 0.162 mmol), B4 (31.3 mg, 0.162 mmol), and B5 (24.6 mg, 0.162 mmol) in dry benzene for 20 h. After evaporation of the solvent, the mixture was filtered through a short column (2 g of silica, 150 ml of 4:1 pentane/ethyl acetate), yielding 268.4 mg (0.67 mmol) of an AB pool containing 15 different cinnamates. A stock solution of this mixture in dimethoxyethane was split into nine portions and alkylated in separate vessels with one each of the nitrobenzylbromides C1-C9 (0.074 mmol). The following amounts of nitrobenzylbromide were used for each library: C1, 16.1 mg; C2, 17.1 mg; C3, 17.9 mg; C4, 20.5 mg; C5, 21.6 mg; C6, 22.4 mg; C7, 18.3 mg; C8, 20.4 mg; C9, 21.2 mg. Each vessel contained 1 ml of the AB stock solution, a nitrobenzylbromide C, and 1 ml of dry dimethoxyethane. KF/alumina was added (93 mg each, 8× xs), the reactions were protected from light, and they were stirred for 18 h. The reaction mixture was filtered, the cake was washed with 20 ml of acetonitrile, and the solvent was evaporated. Each library was passed through a short silica column (2 g of silica, 150 ml of 4:1 pentane/ethyl acetate). The libraries were isolated in the following amounts: C1, 19.0 mg; C2, 17.8 mg; C3, 28.0 mg; C4, 35.2 mg; C5, 31.3 mg; C6, 25.3 mg; C7, 28.4 mg; C8, 33.9 mg; C9, 30.3 mg. Proton NMR spectra of each library show peaks expected for each component.
Gas Chromatography. We used a HP-5890 gas chromatograph, J & W Scientific (Folsom, CA) capillary column (DB-5.625, 30 m × 0.25 mm id, 0.25 μm film); injector, 230°C; detector, 260°C; temperature program, start 130°C, end 230°C, rate 15°C/min. A stock solution of biphenyl was prepared as internal standard (59 mg/25 ml of benzene).
Library Screening. A stock solution of each library was prepared in 5 ml of benzene. For each photolysis 1 ml of the library stock solution was mixed with 0.5 ml of the internal standard and 0.5 ml of benzene, making up a total volume of 2 ml. This solution was irradiated in an NMR tube positioned 2 cm from a UV lamp in a Rayonet photoreactor. Samples were taken and injected after 0, 60, 180, 300, 480, and 660 s.
Results
A photochemical bond-cleavage process equivalent to two-photon photochemistry can be achieved by consecutive one-photon processes (Eq. 1) provided that the first step is reversible, either thermally or photochemically. The basic concept is that a preirradiation step prepares the group for a second irradiation that actually does the removal. It is easiest to conceive of the process by assuming that the two irradiations are conducted at different wavelengths and in series, but the principle applies equally if the wavelengths are the same and the processes therefore occur in parallel. Although not necessarily intuitive, this is merely an application of the Curtin–Hammett principle (42).
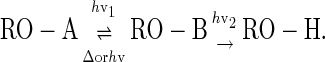 |
[1] |
The performance of such a group is most easily seen by examining a scenario in molecular photolithography (Fig. 1). Ideally, irradiation at λ1 produces B groups in the intended zone, which are removed as irradiation is performed at λ2. Because the contrast between the masked and unmasked areas is expressed in both steps, the overall contrast ratio should be their square. The small fraction of light (related to the optical density of the mask and diffraction) that falls in the unintended zone in step 1 slightly converts A to B in a masked area. This is an error waiting to be deprotected in a subsequent irradiation cycle when this zone is also unintended. This problem can be overcome if the possibility exists for reversion of the “photoactivated” photoremovable group B to the inert group A, producing the idealized work piece.
Fig. 1.

Expected performance of a two-photon photoremovable protecting group in molecular photolithography. In the irradiation at λ1, all A groups in the unmasked region are converted to B groups. In the masked region, a few A groups are converted to B groups, their ratio determined by kinetics and the (lower) light intensity. In the irradiation at λ2, all of the B groups in the unmasked region are deprotected. The contrast between the masked region and the unmasked region minimizes the amount of B deprotection in the masked region in this step, but B groups still remain that might be deprotected in a subsequent step. The conversion of B to A resets the substrate for a subsequent two-photon deprotection cycle.
Kinetics analysis of the behavior of such a group is readily performed based on earlier-established methods (21–23, 35). The solutions of the rate equations are given in Eqs. 2–4, where k (Eq. 5) includes terms for light intensity, quantum yield, and extinction coefficient. Simulations of these functions show the expected behavior, i.e., B accumulates when kb < ka. If ka = 100 kb, photochemical deprotection can be easily conducted in a stepwise manner (with change of wavelength, if desired/needed) to five half-lives ([B] ≥ 100 [ROH]).
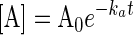 |
[2] |
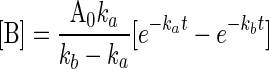 |
[3] |
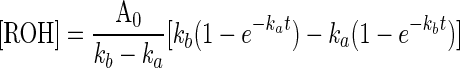 |
[4] |
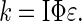 |
[5] |
From these equations may be derived the rate of deprotection in adjacent sites (only I differs between them, scaled by A, the contrast ratio of the aerial image of the mask). The contrast ratio S for the one-photon case is given by Eq. 6, and is 215 at 99% conversion assuming A = 1,000. The contrast ratio S for the two-photon case is given by Eq. 7. For reactions taken to significant conversion, it simplifies to Eq. 8. It has no analytical solution, but can be simulated. An analysis of these equations by mathematica is given in Supporting Text, which is published as supporting information on the PNAS web site. A contrast enhancement of five times (basically recovering the contrast of the aerial image lost with one-photon groups) can be expected for two-photon deprotection for ka = 100 kb.
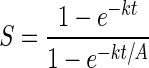 |
[6] |
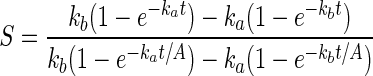 |
[7] |
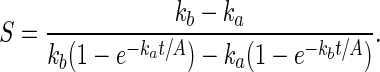 |
[8] |
We have designed, based on the foregoing principles, a two-photon photoremovable protecting group that could be used in photolithographic synthesis of peptides or nucleic acids. It combines the well-known nitrobenzyl protecting group chemistry (43–47) with the hydroxycinnamate group developed by Porter and colleagues (48, 49). An idealized version of its intended operation is shown in Fig. 2: photochemical cis-trans isomerization of 7 leads to 8, which could revert to 7 by thermal or photochemical isomerization, or be converted to the free alcohol 9 by nitrobenzyl deprotection. Lactonization of this material would occur spontaneously, as is already known. Of course, because of possible overlap of electronic absorptions or energy transfer, it is not necessary that the deprotection reaction proceed only through this pathway.
Fig. 2.

Example of a molecule that could function as required by Eq. 1.
Initial experimental investigation of such protecting groups began with 12 (Fig. 3). This known compound (50, 51) was alkylated with nitrobenzylbromide/KF/alumina. Irradiation of 13 (Rayonet photochemical reactor, 350 nm) produces coumarin in quantitative yield. Irradiation of 14 produces nonanol (76% isolated) and coumarin (76% isolated).
Fig. 3.

Prototype reactions for two-photon photodeprotection.
It was clear that optimization of the deprotection rate with >350 nm light of this group, with its (assumed) multiple photon absorptions and multiple chemical and photochemical steps, would be difficult to conduct rationally. Therefore, a 3D indexed combinatorial library (52, 53) of 135 such groups was designed from three phosphoranes A, five salicaldehydes B, and nine benzylic halides C (Fig. 4).
Fig. 4.

Building blocks for the indexed combinatorial library. Group A, phosphoranes; group B, salicaldehydes; group C, nitrobenzylbromides.
The indexed combinatorial library approach involves the preparation of organized pools or sublibraries in which one of the combinatorial sites is held constant while others are varied. This library can be conceived as in Fig. 5, where the preparation and screening is organized around 17 “slices” through this 3D space.
Fig. 5.

Conceptual framework for 3D indexed combinatorial library. Syntheses are conducted and screens are performed on slices through this cube.
The synthesis scheme is shown in Fig. 6. Wittig condensation of a salicaldehyde B with a substituted phosphorane A provides an o-hydroxycinnamate, which is alkylated with a benzylic bromide C. Both reactions are high yielding and can be driven to completion, which is essential to afford equimolar representation of each library member in each pool. Proofing of each of the building blocks in the reactions of this synthesis scheme was conducted with single compounds and on a pool basis before library preparation. For example, phosphorane A1 reacts with salicaldehyde B4 for 7 h in refluxing benzene to yield cinnamate A1B4 in 97% yield. This compound was used to demonstrate the alkylation by nitrobenzyl bromides in groups of three (C1, C4, C7; C2, C5, C8; C3, C6, C9). In each case, NMR proved the presence of all three products in an equimolar ratio. A pool synthesis was performed with 100 μmol of phosphorane A1 and 25 μmol each of salicaldehydes B1, B2, B4, and B5. After 3 h, B1, B4, and B5 were consumed, whereas the slower reacting B2 required an additional 3 h to reach completion. The isolated yield of this pool was 92%, and NMR analysis confirmed the presence of all products in an equimolar ratio, with no impurities or side products. Likewise, these four cinnamates (A1B1, A1B2, A1B4, and A1B5) were alkylated with bromide C1, which was used in a ratio equimolar to their sum. The reaction was complete after overnight stirring, and this pool was isolated in 90% yield. NMR proved the presence of all four products in an equimolar ratio.
Fig. 6.

Synthetic scheme for the preparation of sublibraries of the indexed combinatorial library.
The synthesis of the indexed libraries was performed under similar conditions. Stock solutions of salicaldehydes and nitrobenzylbromides were used, and the starting size of the library was 0.5 mmol (≈250 mg phosphorane). All operations were carried out with exclusion of light to prevent premature deprotection of fast-reacting individual library members. Isolated yields in the first stage were 92–95%, and in the second stage they were 92–94%.
This protocol created a total of 17 sublibraries of compounds (three A, five B, and nine C) with generic structure 7. By simultaneously irradiating these sublibraries and quantifying the released (cyclododecane)methanol by gas chromatography, the most efficient two-photon photoremovable protecting group could be determined. Photolyses were carried out in NMR tubes held equidistant from the light source (350-nm UV lamps in a Rayonet photoreactor). Stock solutions of all pools were prepared in dry benzene, and biphenyl was added as an internal standard before photolysis. Alcohol released was plotted versus time, and superior groups were identified from these progress curves (Fig. 7 and Figs. 10–18, which are published as supporting information on the PNAS web site). From this study it was concluded that compound A1B5C6 (the 2-cyano-4,5-dialkoxynitrobenzyl derivative) is superior. Reaction of A1B5C6 is sufficiently rapid that it shows complete deprotection in kinetic runs in which progress curves for all other groups are still increasing.
Fig. 7.

Representative GC progress curve for library irradiation. Alcohol production was determined by digital integration relative to biphenyl.
A variant of the library synthesis (using the cyanohydrin tosylate; see Materials and Methods) was used for the preparation in quantity of ethyl ester 15 (Fig. 8), bearing our superior group, for characterization. Its electronic spectrum is given in Fig. 19, which is published as supporting information on the PNAS web site. Spectra of the isolated chromophores show that each is quite similar to that of 15. Because of the high photochemical reactivity of 15, the ethyl ester 16 of the next lower reactive group, A1B5C9, was also prepared and characterized. In preparative irradiation experiments, 7-methoxycoumarin is isolated from 15 in >80% yield. The deprotection of both of these compounds was evaluated by UV spectroscopy, monitoring production of 7-methoxycoumarin at 315 nm. In a 0.1 mM acetonitrile solution in a 2-mm pathlength cuvette with 365-nm light at a power of 4 mW/cm2, 15 is deprotected with a half-time of 30 s, whereas 16 shows a half-time of 35 s. This compares to deprotection half-times under very similar conditions (but with a power of 14.5 mW/cm2)of18–31 s reported for nucleosides protected with the MeNPOC group (33). Thus, scaling for power, both groups are as reactive or more reactive than standard one-photon groups. Quantum yields for 365-nm irradiation in comparison to nitroveratryl alcohol (Φ = 0.10) were also determined for these two groups (15 = 0.018; 16 = 0.013). These values are comparable to known quantum yields of nitroveratryl ethers and suggest, consistent with their design principles, that the ether deprotection is the rate-limiting step.
Fig. 8.

Optimum two-photon protecting groups in this library.
A virtue of the indexed combinatorial library approach used here is that the sublibraries are enduring resources. If an optimal group is required for different reaction conditions, it can be discovered merely by rescreening the sublibraries.
The grouping discovered in this work should provide superior results in applications involving spatial definition of light, such as stereolithography or directed drug delivery, and, because the contrast ratio directly affects the fidelity of a light-directed synthesis, higher-quality photolithographically generated arrays of nucleic acids and peptides.
Supplementary Material
Acknowledgments
We thank F. Cerrina for assistance with mathematica. Financial support was provided by National Institutes of Health Grant GM46720 and a Deutsche Forschungsgemeinschaft Postdoctoral Fellowship (to W.P.).
This paper was submitted directly (Track II) to the PNAS office.
References
- 1.Calvert, P. & Crockett, R. (1997) Chem. Mater. 9 650–663. [Google Scholar]
- 2.Neckers, D. C. (1990) CHEMTECH 20 615–619. [Google Scholar]
- 3.Oh, D. H., Stanley, R. J., Lin, M., Hoeffler, W. K., Boxer, S. G., Berns, M. W. & Bauer, E. A. (1997) Photochem. Photobiol. 65 91–95. [DOI] [PubMed] [Google Scholar]
- 4.Oh, D. H., King, B. A., Boxer, S. G. & Hanawalt, P. C. (2001) Proc. Natl. Acad. Sci. USA 98 11271–11276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Williams, R. M., Piston, D. W. & Webb, W. W. (1994) FASEB J. 8 804–813. [DOI] [PubMed] [Google Scholar]
- 6.Piston, D. W. (1999) Trends Cell Biol. 9 66–69. [DOI] [PubMed] [Google Scholar]
- 7.So, P. T., Konig, K., Berland, K., Dong, C. Y., French, T., Buhler, C., Ragan, T. & Gratton, E. (1998) Cell Mol. Biol. 44 771–793. [PubMed] [Google Scholar]
- 8.Belfield, K. D., Shafer, K. J., Mourad, W. & Reinhardt, B. A. (2000) J. Org. Chem. 65 4475–4481. [DOI] [PubMed] [Google Scholar]
- 9.Wang, X., Krebs, L. J., Al-Nuri, M., Pudavar, H. E., Ghosal, S., Liebow, C., Nagy, A. A., Schally, A. V. & Prasad, P. N. (1999) Proc. Natl. Acad. Sci. USA 96 11081–11084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kubitscheck, U., Tschodrich-Rotter, M., Wedekind, P. & Peters, R. (1996) J. Microsc. 182 225–233. [DOI] [PubMed] [Google Scholar]
- 11.Denk, W. (1994) Proc. Natl. Acad. Sci. USA 91 6629–6633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Furuta, T., Wang, S. S., Dantzker, J. L., Dore, T. M., Bybee, W. J., Callaway, E. M., Denk, W. & Tsien, R. Y. (1999) Proc. Natl. Acad. Sci. USA 96 1193–1200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Fedoryak, O. D. & Dore, T. M. (2002) Org. Lett. 4, 3419–3422. [DOI] [PubMed] [Google Scholar]
- 14.Qiao, L., Kozikowski, A. P., Olivera, A. & Spiegel, S. (1998) Bioorg. Med. Chem. Lett. 8 711–714. [DOI] [PubMed] [Google Scholar]
- 15.Pettit D. L., Wang, S. S., Gee, K. R. & Augustine, G. J. (1997) Neuron 19 465–471. [DOI] [PubMed] [Google Scholar]
- 16.Belfield, K. D., Ren, X., Van Stryland, E. W., Hagan, D. J., Dubikovski, V. & Meisak, E. J. (2000) J. Am. Chem. Soc. 122 1217–1219. [Google Scholar]
- 17.Cumpston, B. H., Ananthavel, S. P., Barlow, S., Dyer, D. L., Ehrlich, J. E., Erskine, L. L., Heikal, A. A., Kuebler, S. M., Lee, I.-Y. S.; McCord-Maughont, D., et al. (1999) Nature 398 51–54. [Google Scholar]
- 18.Albota, M., Beljonne, D., Bredas, J.-L., Ehrlich, J. E., Fu, J.-Y., Heikal, A. A., Hess, S. E., Kogej, T., Levin, M. D., Marder, S. R., et al. (1998) Science 281 1653–1656. [DOI] [PubMed] [Google Scholar]
- 19.Harada, J., Uekusa, H. & Ohashi, Y. (1999) J. Am. Chem. Soc. 121 5809–5810. [Google Scholar]
- 20.Wallraff, G. M. & Hinsberg, W. D. (1999) Chem. Rev. 99 1801–1821. [DOI] [PubMed] [Google Scholar]
- 21.MacDonald, S. A., Willson, C. G. & Frechet, J. M. J. (1994) Acc. Chem. Res. 27 151–158. [Google Scholar]
- 22.Willson, C. G. (1991) Polym. Mater. Sci. Eng. 64 18–20. [Google Scholar]
- 23.Willson, C. G. & Bowden, M. J. (1989) CHEMTECH 19 182–189. [Google Scholar]
- 24.Thompson, L. F., Willson, C. G. & Bowden, M. J., eds. (1994) Introduction to Microlithography (Am. Chem. Soc., Washington, DC).
- 25.Rozsnyai, L. F., Benson, D. R., Fodor, S. P. A. & Schultz, P. G. (1992) Angew. Chem. Int. Ed. 31 759–761. [Google Scholar]
- 26.Pritchard, D. J., Morgan, H. & Cooper, J. M. (1995) Angew. Chem. Int. Ed. 34 91–93. [Google Scholar]
- 27.Yan, M., Cai, S. X., Wybourne, M. N. & Keana, J. F. W. (1993) J. Am. Chem. Soc. 115 814–816. [Google Scholar]
- 28.Bhatia, S. K., Hickman, J. J. & Ligler, F. S. (1992) J. Am. Chem. Soc. 114 4432–4433. [Google Scholar]
- 29.Bhatia, S. K., Shriver-Lake, L. C., Prior, K. J., Georger, J. H., Calvert, J. M., Bredehosrst, R., Hickman, J. J. & Ligler, F. S. (1989) Anal. Biochem. 178 408–413. [DOI] [PubMed] [Google Scholar]
- 30.Sundberg, S. A., Barrett, R. W., Stryer, L., Pirrung, M. C., Liu, A. T., Kiangsoontra, B. & Holmes, C. P. (1995) J. Am. Chem. Soc. I 117 12050–12057. [Google Scholar]
- 31.Pirrung, M. C. & Huang, C.-Y. (1996) Bioconjugate Chem., 7 317–321. [DOI] [PubMed] [Google Scholar]
- 32.Fodor, S. P. A., Read, J. L., Pirrung, M. C., Stryer, L., Liu, A. T. & Solas, D. (1991) Science 251 767–773. [DOI] [PubMed] [Google Scholar]
- 33.Pease, A. C., Solas, D., Sullivan, E. J., Cronin, M. T., Holmes, C. P. & Fodor, S. P. A. (1994) Proc. Natl. Acad. Sci. USA 91 5022–5026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Blawas, A. S., Oliver, T. F., Pirrung, M. C. & Reichert, W. M. (1998) Langmuir 14 4243–4250. [Google Scholar]
- 35.McGall, G. H., Barone, A. D., Diggelmann, M., Fodor, S. P. A., Gentalen, E. & Ngo, N. (1997) J. Am. Chem. Soc. 119 5081–5090. [Google Scholar]
- 36.Frederiksen, P. K., Jorgensen, M. & Ogilby, P. R. (2001) J. Am. Chem. Soc. 123 1215–1221. [DOI] [PubMed] [Google Scholar]
- 37.Pati, S. K., Marks, T. J. & Ratner, M. A. (2001) J. Am. Chem. Soc. 123 7287–7291. [DOI] [PubMed] [Google Scholar]
- 38.Pérez-Prieto, J., Miranda, M. A., García, H., Kónya, K. & Scaiano, J. C. (1996) J. Org. Chem. 61 3773–3777. [DOI] [PubMed] [Google Scholar]
- 39.Uchida, M. & Irie, M. (1993) J. Am. Chem. Soc. 115 6442–6443. [Google Scholar]
- 40.Ando, T., Yamawaki, J., Kawate, T., Sumi, S. & Hanafusa, T. (1982) Bull. Chem. Soc. Jpn. 55 2504–2507. [Google Scholar]
- 41.Hayashi, H., Nakanishi, K., Brandon, C. & Marmur, J. (1973) J. Am. Chem. Soc. 95 8749–8757. [DOI] [PubMed] [Google Scholar]
- 42.Curtin, D. Y. (1954) Rec. Chem. Prog. 15 111. [Google Scholar]
- 43.Patchornik, A., Amit, B. & Woodward, R. B. (1970) J. Am. Chem. Soc. 92 6333–6335. [Google Scholar]
- 44.Amit, B., Zehavi, U. & Patchornik, A. (1974) J. Org. Chem. 39 192–196. [Google Scholar]
- 45.Zehavi, U., Amit, B. & Patchornik, A. (1972) J. Org. Chem. 37 2281–2284. [Google Scholar]
- 46.Zehavi, U. & Patchornik, A. (1972) J. Org. Chem. 37 2285–2288. [Google Scholar]
- 47.Amit, B., Zehavi, U. & Patchornik, A. (1974) J. Org. Chem. 39 192–195. [Google Scholar]
- 48.Turner, A. D., Pizzo, S. V., Rozakis, G. W. & Porter, N. A. (1987) J. Am. Chem. Soc. 109 1274–1275. [Google Scholar]
- 49.Turner, A. D., Pizzo, S. V., Rozakis, G. W. & Porter, N. A. (1988) J. Am. Chem. Soc. 110 244–250. [Google Scholar]
- 50.Liu, Y. Y., Thom, E. & Liebmann, A. A. (1979) J. Heterocyclic Chem. 16 799–801. [Google Scholar]
- 51.Black, M., Cadogan, J. I. G., Cartwright, G. A., McNab, H. & MacPherson, A. (1993) J. Chem. Soc. Chem. Commun. 959–960.
- 52.Pirrung M. C. & Chen, J. (1995) J. Am. Chem. Soc. 117 1240–1245. [Google Scholar]
- 53.Pirrung, M. C., Chau, J. H.-L. & Chen, J. (1996) in Combinatorial Chemistry: A High-Tech Search for New Drug Candidates, eds. Wilson, S. R. & Murphy, R. (Wiley, New York).
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}