

Abstract
Since its introduction into clinical practice, more than 20 years ago, percutaneous transluminal coronary angioplasty (PTCA) has proven to be an effective, minimally invasive alternative to coronary artery bypass grafting (CABG). During this time there have been great improvements in the design of balloon catheters, operative procedures and adjuvant drug therapy, and this has resulted in low rates of primary failure and short-term complications. However, the potential benefits of angioplasty are diminished by the high rate of recurrent disease. Up to 40% of patients undergoing angioplasty develop clinically significant restenosis within a year of the procedure. Although the deployment of endovascular stents at the time of angioplasty improves the short-term outcome, ‘in-stent’ stenosis remains an enduring problem. In order to gain an insight into the mechanisms of restenosis, several experimental models of angioplasty have been developed. These have been used together with the tools provided by recent advances in molecular biology and catheter design to investigate restenosis in detail. It is now possible to deliver highly specific molecular antagonists, such as antisense gene sequences, to the site of injury. The knowledge provided by these studies may ultimately lead to novel forms of intervention. The present review is a synopsis of our current understanding of the pathological mechanisms of restenosis.
Keywords: balloon angioplasty, smooth muscle cells, endothelium, proliferation, migration, rat, rabbit, pig
The first clinical application of coronary angioplasty using a balloon catheter was performed by Gruentzig 1978, however, it was originally used experimentally several years earlier (Baumgartner 1963). Since its introduction, balloon angioplasty has been shown to be safe and effective. In comparisons with CABG, PTCA appears to be an ‘acceptable alternative’ with similar outcomes (CABRI 1995; Pocock et al. 1995, 1996; BARI 1996) After a 6.5-years follow up of the RITA cohort (Henderson et al. 1999), report that patients undergoing PTCA and CABG have similar outcomes with regard to the primary clinical end-points of death or non-fatal MI. However patients undergoing PTCA had a significantly higher requirement for repeat procedures, and suffered a higher prevalence of angina. PTCA is currently used in more than 500 000 patients per annum, world-wide. Acute closure, and primary failure rates are low, however, restenosis remains a major long-term complication (Meier 1989).
Experimental models of restenosis
Restenosis is a narrowing at the site of balloon dilatation (Waller et al. 1991). It develops rapidly following the procedure and may be accompanied by recurrent symptoms (Glazier et al. 1989). The precise mechanisms for restenosis probably vary between patients, but is likely to be a combination of artery wall remodeling and intimal hyperplasia (Austin et al. 1985). Several animal models of restenosis have been developed and these have led to an improvement in our understanding of the cellular mechanisms involved.
Cellular events following angioplasty
Detailed analysis of the cell kinetics following balloon catheter injury has been available since the late 1970s and early 1980s. This knowledge has relied almost entirely on the use of experimental animal models.
Platelet activation
Very rapidly after balloon injury, circulating platelets and leucocytes cover the de-endothelialized surface of the blood vessel (Steele et al. 1985; Ferns et al. 1991c; Groves et al. 1995). The platelets have a flattened appearance, and are clearly involved in the polar release of their contents into the injured artery wall (Winocour et al. 1989). Platelet aggregation is stimulated by the release of subendothelial elements, such as collagen. The process is self-sustaining due to the release of platelet associated pro-aggregatory substances, such as ADP and thromboxane A2. However, within 24 h of balloon injury, the arterial surface becomes significantly less thrombogenic (Kinlough-Rathbone et al. 1983), and the turnover of platelets diminishes (Winocour et al. 1989).
Studies using antiplatelet treatment (aspirin plus dipyridamole) have indicated the importance of platelets to restenosis in the atherosclerotic rabbit (Faxon et al. 1984), although this has not be confirmed by more recent studies (Sun et al. 1993). However, recent clinical trials using glycoprotein IIb/IIIa receptor blockers have reinforced the importance of platelets in restenosis (Lincoff et al. 1999).
Leucocyte infiltration
Inflammatory cells are involved in the acute response to arterial injury. In the subtle injury associated with the placement of a silastic ‘collar’ around the rabbit carotid artery, neutrophils were found to be an important early, though transient, participant (Booth et al. 1989). However, following balloon injury, neutrophils do not form a major constituent of the cellular infiltrate, particularly in the rat (Jonasson et al. 1988; Verheyen et al. 1988; Ferns et al. 1991c). In this model, monocyte-derived macrophages comprise approximately 1% of the neo-intimal cell population (Ferns et al. 1991c). In the cholesterol-fed rabbit these macrophages are transformed into macrophage-derived foam cells, and constitute a substantial part of the neo-intima (Ferns et al. 1992b). The importance of leucocytes to the response to injury has recently been demonstrated by blocking their entry into the artery wall using monoclonal antibodies to cell adhesion molecules (Liao et al. 1997). Mac-1 blockade reduces experimental neointimal thickening, suggesting that leucocyte recruitment and infiltration of injured arteries may be a useful target for preventing intimal hyperplasia (Rogers et al. 1998), and activated circulating monocytes expressing CD11b/CD18 are a good marker of restenosis risk in patients undergoing angioplasty. The role of T lymphocytes in neo-intimal development is unclear, and their importance may vary with the animal model used. Jonasson et al. 1988 have reported that T cells constitute 0.3% of the neo-intimal cell population. Their importance to neo-intimal thickening has been assessed in several ways, but the results of these experiments is inconsistent. The use of cyclosporin A at high doses inhibited neo-intimal thickening in the rat (Jonasson et al. 1988), however, this was not found to be the case in the rabbit when cyclosporin A was used within the therapeutic range (Ferns et al. 1990), nor did the absence of functional T cells affect the cell kinetics of smooth muscle cell proliferation (Ferns et al. 1991c). However Hansson 1994 has reported that T cell depletion is associated with enhanced neo-intimal thickening in the rat.
Smooth muscle kinetics
The characteristic histological feature of restenosis in man and experimental animal models is smooth muscle cell accumulation within the neointima (Austin et al. 1985; Nobuyoshi et al. 1989; Waller et al. 1991). Balloon injury is initially associated with medial smooth muscle cell death (Clowes et al. 1983). There follows several phases of cellular activity (Figure 1). These include:
Figure 1.
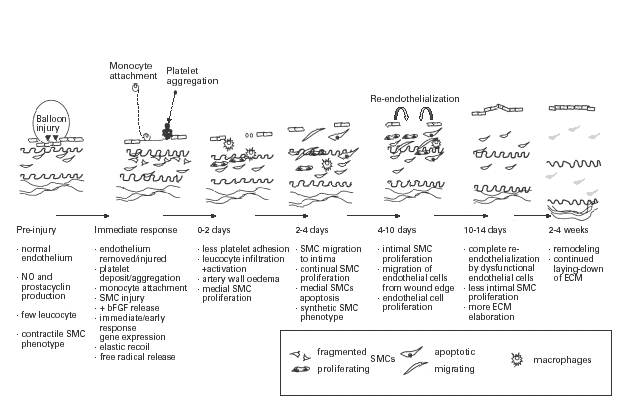
Cellular changes and time course after balloon angioplasty.
• the co-ordinated proliferation of medial smooth muscle cells, driven by growth factors released from platelets and cell lysis;
• migration of a subpopulation of medial smooth muscle cells into the intima in response to the release of chemo-attractants such as platelet-derived growth factor (PDGF) (Ferns et al. 1991c; Rutherford et al. 1997);
• proliferation of intimal smooth muscle cells;
• elaboration of extracellular matrix molecules, such as hyaluronan and chondroitin sulphate by smooth muscle cells of a synthetic phenotype;
• vascular remodeling, including the removal of excess extracellular matrix by matrix metalloproteases (MMPs).
It is a combination of these events that ultimately lead to luminal narrowing, and an understanding of the factors that orchestrate each stage in this process may provide new targets for therapeutic intervention. It is apparent that medial smooth muscle cells are heterogeneous with respect to their responsiveness to chemo-attractants and mitogens released following balloon injury (Clowes et al. 1983; Haudenschild & Grunwald 1985). Smooth muscle cell (SMC) proliferation and phenotypic modulation are important aspects of the arterial response to injury in experimental models of restenosis.
Changes in smooth muscle cell phenotype
SMC undergo profound changes following balloon injury. These changes include: a decrement in myofilament density (Manderson et al. 1989); altered desmin and vimentin positivity (Kocher et al. 1984); an increase in β–actin expression (Kocher et al. 1984; Barja et al. 1986); and changes in extracellular matrix elaboration (Snow et al. 1990). These changes appear to be partially reversible (Grunwald et al. 1987). Smooth muscle cells are also induced to express MHC class II molecules following injury (Jonasson et al. 1988). It has been hypothesized that this is due to the local release of γ–interferon by activated T lymphocytes within the neo-intima. It is unclear to what extent this may be important in restenosis, although, the cells expressing MHC class II molecules were nondividing cells.
In vivo and in vitro observations strongly suggest that marked differences exist in the growth, and matrix-producing capabilities of phenotypically distinct SMC subpopulations within the artery wall (Frid et al. 1997).
SMC migration from the tunica media to the intima in the ballooned rat carotid artery occurs intensively between the second and fifth days after balloon injury. About 80% of the migrating cells are reported to be in the G1 and S phases of the cell cycle. Positive staining for PDGF-B, elastase III B, MMP-I, and MMP-9 of these cells was detected on the fifth day after injury (Yoshida et al. 1997).
Smooth muscle cell apoptosis
Acute apoptotic cell death after vascular injury is a highly regulated process governed by cellular redox state and the relative expression of antiapoptotic genes. Apoptotic cell death is a feature of human and experimental angioplasty. (Perlman et al. 1997) found that up to 70% of medial cells appeared apoptotic within 30 min of injury using the TUNEL assay technique. This was associated with a marked down-regulation of bcl-X expression, particularly in the cells nearest the lumen. Fas ligand induces apoptosis in Fas-bearing cells and (Sata et al. 1998) showed that Fas-ligand gene transfer to the balloon injured rat carotid artery inhibited intimal hyperplasia.
Modifying the vascular redox state by the administration of antioxidants, markedly attenuated both stress-activated protein kinase activation and the induction of apoptosis (Pollman et al. 1999). It has been suggested that the induction of apoptosis in vascular smooth muscle cells is modulated by cell phenotype. In contrast to medial smooth muscle cells, neointimal cells were found to be relatively resistant to apoptotic death induced by balloon injury (Kollum et al. 1997). This resistance to balloon injury-induced death was associated with an upregulation of the antiapoptotic mediator bcl-xL in neointimal smooth muscle cells.
Extra cellular matrix elaboration
The elaboration of extracellular matrix molecules by intimal smooth muscle cells contributes substantially to intimal thickening, hence intimal cross-sectional area continues to increase for some time after cell proliferation has ceased (reviewed by Wight 1995). The size of the neointima reaches a plateau between 1 and 4 months and gradually diminishes in size through the removal of extracellular matrix.
Re-endothelialization
The major stimuli for intimal and medial smooth muscle cell proliferation have not been identified, however, factors released by endothelial cells appear to be critical in modulating this effect. Re-endothelialized segments of artery are often associated with less neo-intimal thickening. However this is partially dependent on the extent of the original injury (Walker et al. 1983; Reidy & Silver 1985). Deep and wide injuries may be associated with incomplete re-endothelialization and substantial neo-intimal thickening, whereas superficial, narrow lesions may heal without intimal hyperplasia (Reidy & Silver 1985). Arteries that have sustained extensive endothelial damage may not become fully re-endothelialized but instead become lined by phenotypically modified smooth muscle cells (Stemerman et al. 1977; Reidy 1985). Rapid endothelial cell recoverage of Fogarty balloon-injured arteries in the rabbit may not limit intimal hyperplasia in the centre of the lesion. It is possible that the inability of regenerated endothelium to inhibit intimal hyperplasia at this site is due to its initially dedifferentiated and possibly dysfunctional phenotype (Doornekamp et al. 1997).
Tanaka et al. 1993 found that balloon injury was associated with prolonged expression of activation markers on the regenerating endothelial cells (ICAM-1 and VCAM-1), SMCs (ICAM-1 and class II MHC antigens) and leucocytes. Cell adhesion molecules were upregulated for approximately one week following balloon injury on surviving endothelium and circulating leucocytes. If the selectins were blocked, lumen size remained larger, and intimal size was reduced (Barron et al. 1997). However (Guzman et al. 1995) reported that blocking leucocyte adhesion with a monoclonal antibody to CD18/CD11 did not significantly affect lumen diameter, or percentage stenosis in the rabbit. By contrast (Rogers et al. 1998) found that a monoclonal antibody to Mac-1 (CD11b/CD18) immediately before injury and for 14 days thereafter significantly reduced leucocyte infiltration and intimal hyperplasia.
La Veau et al. 1990 reported that vascular wall reactivity responses were lost after balloon angioplasty, and that the duration of impaired responses was related to the extent of injury (Weidinger et al. 1990). Bosmans et al. 1996 investigated the changes in expression of nitric oxide synthase following balloon injury. This was studied using immunocytochemistry and functionally using isolated arterial segments in organ culture. They reported that endothelium dependent responses of the injured arteries remained impaired for 10 weeks after injury, although re-endothelialization was complete by 2 weeks. Balloon injury was associated with an early reduction in the release of prostacyclin and thromboxane, although this was not sustained (Mattsson et al. 1992, 1993b). The early vasospasm often associated with angioplasty did not appear to be due to endothelin (Mattsson et al. 1993a).
Putative mediators of restenosis
Several growth factors and cytokines probably orchestrate the complex sequence of events described above. They are likely to be derived from a number of sources including platelets, mononuclear cells, endothelial cells and smooth muscle cells. Platelets in particular are a rich repository of growth factors, such as PDGF, IGF, TGF and EGF (Assoian et al. 1984) and have been shown to be important in driving the early cellular events (Fingerle et al. 1990). Activated monocytes are also a rich source of growth factors and cytokines (Nathan 1987). Although they are not present in great abundance in normo-cholesterolaemic animals (Ferns et al. 1991a), they are found in substantial numbers in diseased human arteries and in some hyperlipidaemic animal models (Rosenfeld & Ross 1990; Ferns et al. 1993a). Mononuclear cells remain within the injured artery wall for a considerable time, and hence may contribute to the sustained expansion of the neo-intima. Smooth muscle cells can also be stimulated to elaborate growth factors, including PDGF and IGF that have the potential to act in a paracrine, or autocrine manner. Smooth muscle cell necrosis may also lead to the release of intracellular growth factors such as bFGF. Although the endothelium also has the potential to release growth factors, including PDGF, IGF and bFGF, it appears to exert an overall inhibitory effect on neo-intimal thickening. This may be related to its release of nitric oxide. Other endothelium-derived factors promote endothelial cell re-growth, enhancing re-endothelialization and thereby indirectly inhibiting neo-intimal thickening. Activated T cells are a source of γ–interferon, a putative inhibitor of SMC proliferation Hansson et al. 1988).
The biochemical sequelae of balloon injury
Growth factors and cytokines
The effects of growth factors are likely to be synergistic or at least additive. In their review of clinical atherosclerosis Fuster et al. 1993 drew attention to the complexity of growth factor/cytokine interactions in the development of arterial lesions. Most animal models of restenosis are a gross simplification of this complexity. They are usually single injury models, in arteries with no pre-existing stenotic lesion. They are often in normo-cholesterolaemic animals in whom the restenotic lesion is almost entirely composed of vascular smooth muscle cells. And they are usually models in which other risk factors (e.g. diabetes and hypertension) are not present.
The studies of Ross & Glomset 1976 identified platelet-derived growth factor (PDGF) as a potent stimulator of the VSMC proliferation. However several other growth factors, cytokines and factors involved in the coagulation and fibrinolytic cascades have subsequently been shown to be VSMC mitogens and chemo attractants.
Growth factors are rapidly released from platelets and dying cells after balloon injury. Ligand binding to cell surface receptors results in transient activation and signal transduction. The expression of early response genes is induced within minutes of balloon injury. Among these are c-fos, c-jun, which are upregulated within 30 min, closely followed by c-myc, the expression of which peaks by 2 h (Bauters et al. 1992). Activation of the immediate-early genes results in Go/G1 traverse and entry into the cell cycle. This is co-ordinated by a number of cyclin-dependent kinases, and is associated with the inactivation of several tumour suppressor genes (reviewed by Muller 1997).
Increased polyamine synthesis appears to play an important role in the response to balloon injury, with a transient increase in the expression of ornithine decarboxylase, preceding medial smooth muscle cell proliferation (Thyberg & Fredholm 1987; Majesky et al. 1990; Nishida et al. 1990).
Balloon injury to rat carotid arteries results in temporally related changes in the expression of PDGF receptors (Cercek et al. 1990; Majesky et al. 1990) and their state of tyrosine phosphorylation. Furthermore, tyrosine phosphorylation of PDGF beta receptors is associated with PI-3 kinase activation (Panek et al. 1997).
The profound changes in growth factor and growth factor receptor expression may partially reflect feedback autoregulation, but it is also likely that cross-modulation occurs. For example at low concentrations TGFβ is known to induce PDGF and PDGF receptor expression by smooth muscle cells (Battegay et al. 1990). VSMC exposed simultaneously to both TGF-β1 and PDGF exhibited diminished migration (50%) when compared to cells treated only with PDGF. TGF-β1 can partially reverse the stimulatory effect of PDGF on VSMC migration in vitro possibly by modifying the actin cytoskeleton and the distribution of the αVβ3/β5 integrins (Engel & Ryan 1997).
The importance of PDGF as a SMC chemoattractant has been demonstrated in several animal models, including the rat, rabbit and nonhuman primates, by using specific inhibitors of PDGF. In these models, the BB-isoform of PDGF has emerged as being particularly important in restenosis. Although PDGF is a potent mitogen for mesenchymal cells, a neutralizing antibody was found to inhibit intimal cell accumulation without having a significant effect on SMC proliferation (measured as thymidine uptake) in the balloon rat carotid (Ferns et al. 1991b). This was more recently confirmed using a PDGF B-chain specific antibody (Rutherford et al. 1997). Administering PDGF-BB to the injured rat carotid was also found to stimulate SMC migration rather than proliferation (Jawien et al. 1992). Rat neo-intimal SMC also express PDGF-BB but are heterogeneous in this respect (Lindner et al. 1995).
The interaction between the growth factors has been investigated by (Rutherford et al. 1997) who have demonstrated that PDGF-BB and bFGF are major determinants of the initial response to balloon injury in the rat. Basic FGF is widely expressed in cells of the artery wall and it is upregulated following balloon injury (Lawrence et al. 1995). It appears to have a major role in early medial SMC proliferation, endothelial cell regrowth and possibly SMC migration (Lindner & Reidy 1991; Lindner et al. 1992; Jackson & Reidy 1993; Lawrence et al. 1995; Rutherford et al. 1997) and extracellular matrix elaboration.
Vascular remodeling has emerged as a major component of luminal narrowing (Pasterkamp et al. 1997). Vascular imaging has shown that intimal thickening is not the sole determinant of lumen size. The overall cross-sectional area of an artery may increase, due to the effects of haemodynamic forces, partially compensating for the encroachment of the neo-intima. This process is probably mediated by growth factors and matrix metalloproteinases (MMPs) acting on adventitial cell and extracellular matrix proteins. However, there has been some debate and disagreement about the relative importance of remodeling in the rabbit model (Gertz et al. 1994; Kakuta et al. 1994; Post et al. 1994, 1995; Lafont et al. 1995).
Transforming growth factor-beta-1 (TGFβ1) plays a central role in tissue repair owing to its effects on cell growth and extracellular matrix synthesis. TGFβ1 stimulates adventitial fibroblast differentiation into myofibroblasts after injury, a process that may affect arterial remodelling (Shi et al. 1996). TGFβ also causes an upregulation of lysyl oxidase, an enzyme involved in collagen and elastin cross-linking, and which may, therefore contribute to arterial remodeling after vascular injury (Shanley et al. 1997).
The local inflammatory response elicited by balloon injury is associated with leucocyte activation and the release of inflammatory cytokines such as IL-1 and TNFα. IL-1 is a pleotropic cytokine which has been shown to stimulate SMC proliferation via an autocrine loop (Raines et al. 1989). IL-1 is also a T cell chemoattractant, and stimulates cell adhesion molecule expression on endothelial and VSMC (Wang et al. 1994). Replication of arterial SMCs after balloon injury in rabbits occurs in regions of TNF-α but not IL-1α expression and correlates inversely with the presence of bFGF. Therefore, SMC-derived TNF-α serves as a marker of a modulated SMC phenotype after acute vascular injury and may contribute to local cellular activation and proliferation of SMCs at sites of arterial injury (Tanaka et al. 1996). IL-1β and TNF-α separately induce and synergistically increase P-selectin-dependent leucocyte rolling and firm adhesion (Thorlacius et al. 1997), enhancing leucocyte entry into the artery wall.
IL-1 also has potential to stimulate IGF-1 production by various cells (Glazebrook et al. 1998). IGF-1 is a SMC mitogen that exerts its effects via specific cell surface receptors (reviewed by Ferns et al. 1991a). IGF-1 expression is upregulated by balloon injury (Bornfeldt et al. 1990) as is its receptor. However the role of IGF-1 in restenosis is unclear. Motani et al. 1995 have reported that IGF-1 enhances platelet aggregation, a process that may promote the delivery of platelet associated factors to the injured artery wall. Although the mitogenic effects of IGF-1 could be inhibited in vitro by IGF-BP1, the administration of IGF-BP1 did not significantly affect the intimal response to injury in the rat carotid (Motani et al. 1995). These data suggest that IGF-1 is probably not a major contributor to intimal thickening in the rat model.
Transcription factors
The binding of cytokines and growth factors to their respective receptors stimulate mitogen-activated protein (MAP) kinases. MAP kinase are involved in nuclear signalling. Phosphatases down-regulate MAP kinase activity. Lai et al. 1996 found that MAP kinase phosphatase-1 (MKP-1) expression fell after balloon injury, whereas MAP kinase expression rose. In vitro, over-expression of MKP-1 was associated with growth arrest. Hu et al. (1997) have presented data demonstrating the importance of MAP kinases in mediating the SMC proliferative response. Extracellular signal-regulated kinases-2 (ERK-2) and Jun N-terminal protein kinase (JNK) were rapidly upregulated. Kinase activation is followed by increased c-fos and c-jun expression and activator protein-1 (AP-1) DNA binding activity. Balloon injury rapidly causes the activation of the MAP kinases, including extracellular signal-regulated kinases (ERK), stress activated protein kinases (SAPK) and c-Jun N-terminal protein kinases (JNK) in the rat carotid artery (Hu et al. 1997).
Farnesylation appears to be an important process in activating ras-proteins. Chemla et al. (1997) inhibited the expression of the enzyme responsible for catalysing this reaction using antisense oligonucleotides and also inhibited intimal hyperplasia. Farnesylation is also affected by the HMGCoA reductase inhibitor, lovastatin, which was found to inhibit luminal narrowing in a double-injury model of the hypercholesterolaemic rabbits (Gellman et al. 1991).
NF-kB and its inhibitory proteins (IK-B) form an autoregulatory system that has a potential role in vascular disease. Immediately after balloon catheter injury, the levels of IKBα, IKBβ and p105 were found to be dramatically reduced whereas the expression of the NFKB regulated genes, vascular cell adhesion molecule (VCAM)-1 and monocyte chemotactic protein (MCP)-1, were increased in VSMCs. In chronically denuded vessels, SMCs on the luminal surface continued to express high levels of VCAM-1 and MCP-1, which may account for the continuing presence of macrophages. These findings provide a link between the activation of NFKB and intimal lesion formation (Landry et al. 1997).
The myocyte enhancer binding factor-2 (MEF2) family of transcription factors are known to regulate the differentiation of skeletal and cardiac muscle. In the rat model of restenosis, the expression of MEF2A,B and D mRNAs were found to be upregulated in the neointima, with the highest levels in the layer of cells nearest to the lumen. These data suggest that the MEF2s are associated with the activated smooth muscle phenotype (Firulli et al. 1996). It was also shown that after balloon injury in the rat there was an increased immuno-reactivity of ETS-1 transcription factor that activates expression of matrix-degrading proteinases such as collagenase and stromelysin (Hultgardh-Nilsson et al. 1996)
The coagulation cascade
Plasmin is a fibrinolytic enzyme derived from plasminogen by proteolytic activation. Balloon injury is associated with altered interactions between plasminogen and the artery wall (Hatton et al. 1988), and changes in plasminogen activator expression (Clowes et al. 1990). In uninjured vessels the expression of both urokinase-type plasminogen activator (uPA) and tissue-type plasminogen activator (tPA) is low. Following injury uPA expression rises to an initial peak within 24 h. Subsequently mRNA levels for both tPA and uPA rise to a maximum at approximately 7 days after injury, hence uPA is expressed during the early mitogenic phase, whilst tPA is present during the phase of medial smooth muscle cell migration (Reidy et al. 1996).
von Willebrand factor
von Willebrand factor (vWF) is a well-characterized multimeric glycoprotein present in platelets and plasma and synthesized by vascular endothelial cells and megakaryocytes.
The deposition of vWF in the media of the injured precedes vascular smooth muscle proliferation and endothelial regrowth (Giddings et al. 1997). However its absence in the von Willebrand disease pig was not associated with a significant effect on lesion size or SMC proliferation.
Renin-angiotensin system
The renin-angiotensin paracrine/autocrine networks are responsible for the local generation and receptor binding of angiotensin II within the vessel wall (Nakamura et al. 1988). There is also evidence that angiotensin II may modulate smooth muscle growth in vitro and in vivo (Dzau et al. 1989). Enalaprilat, an ACE inhibitor reduced intimal hyperplasia (Law et al. 1994). Neo-intimal thickening was shown to be enhanced by aldosterone and inhibited by spironolactone, an aldosterone antagonist (Van Belle et al. 1995).
Renin mRNA is upregulated in the balloon-injured rat carotid artery and this precedes neointima formation (Iwai et al. 1997). The renin-angiotensin system is in a heightened state of activation in the aortae of spontaneously hypertensive Sprague-Dawley rats SHR both before and after balloon injury compared with normotensive rats (Jandeleit-Dahm et al. 1997).
Huckle et al. (1996) have demonstrated that that balloon injury enhances the expression of angiotensin II receptor subtypes following balloon injury. PAI-1 regulates fibrinolysis and extracellular matrix turnover. It has been suggested that PAI-1 activity is modulated by angiotensin II. Hamdan et al. (1996) used the ACE inhibitor Captopril to investigate this possible interaction. After balloon injury levels of PAI-1 mRNA were increased by 3 h, falling to baseline at 2 days and rising again between 4 and 7 days. Captopril inhibited the induction of PAI-1 at the 7 day time point, but not the early phase of induction. Cilazapril, another ACE inhibitor reduced the expression of bFGF in medial SMCs after injury (Iwata et al. 1998). Powell et al. (1991) have found that the benefits of Cilazapril treatment were greater when treatment was started before angioplasty, and inhibition was enhanced by combining treatment with heparin. Miyauchi et al. (1998) used Cilazapril in the lathryritic rat model. In this animal, connective tissue is abnormal and the internal elastic lamina is fractured following balloon injury. The ACE inhibitor was administered for 1 week before injury, and for 3 weeks afterwards. The authors found that Cilazapril inhibited intimal hyperplasia in animals receiving a mild injury, but not when the injury was sufficiently severe to rupture the internal elastic lamina. ATII receptor type 1 blockade with a selective non peptide antagonist, TCV-116, inhibited the induction of immediate/early genes, such as c-fos, c-jun. Egr-1, ornithine decarboxylase and fibronectin (Kim & Iwao 1997). There was a corresponding reduction in intimal hyperplasia, and the content of extracellular matrix in the neo-intima.
Reactive oxygen and nitrogen species
The benefits of antioxidant treatment in clinical trials, and experimental animal models of restenosis, support the notion that free radicals play an important role in restenosis (Ferns et al. 1992a, b; Konneh et al. 1995; Tardif et al. 1997). The local release of free radicals can be detected directly using ‘spin-traps’ combined with mass spectrometry (McCormick et al. 1998). These free radical species lead to the formation of lipid peroxides (Blann et al. 1993) and isoprostanes, products of arachidonic acid oxidation (Reilly et al. 1997; Witztum & Berliner 1998). Golino et al. (1996) have also demonstrated that free radicals may enhance tissue factor expression by endothelial cells, potentially causing activation of the extrinsic coagulation pathway.
Endothelial cells in areas adjacent to the site of injury are likely to be dysfunctional. Short-term exposure of endothelial cells to low oxygen tension results in the elaboration of predominantly vasoconstricting effectors, while longer-term and more severe hypoxic exposure generates factors that can induce smooth muscle proliferation and remodeling (Faller 1999).
Peroxynitrite (derived by reaction of superoxide with nitric oxide) and transition metal ions (perhaps released by injury to the vessel wall) may contribute to lipid peroxidation in atherosclerotic lesions (Darley-Usmar & Halliwell 1996; Van et al. 1997).
Nitric oxide (NO) may be involved in the response to balloon injury in several ways. NO, which is normally derived from a healthy endothelium, inhibits platelet and leucocyte adhesion and platelet aggregation (Gonzalez-Fernandez et al. 1998) and may thereby limit thrombosis and inflammation. NO also stimulates the differentiation of SMCs and inhibits their proliferation and migration (Sarkar & Webb 1998). Groves et al. (1993) have reported that the NO donor SIN-1 (3-morpholino-sydnonimine) inhibits platelet deposition on the balloon-injured artery, and work from the same group subsequently reported that the orally active nitric oxide donor, molsidomine, inhibited intimal and medial SMC proliferation in arteries where the injury was moderate but not when rupture of the internal elastic lamina was present (Groves et al. 1995). More recently, Provost et al. (1997) have reported that SIN-1 inhibited platelet and neutrophil adhesion, and attenuated mural thrombosis and post angioplasty vasoconstriction. There is functional evidence suggesting that endothelial denudation stimulates inducible nitric oxide synthase (iNOS) activity in the vascular wall (Gonzalez-Fernandez et al. 1998). In vitro studies have shown that iNOS expression in smooth muscle cells is reduced by endothelial cells. It was shown that platelets prevent iNOS protein expression early after endothelial balloon damage, an effect that can be avoided with GP IIb/IIIa blocking agents (Gonzalez-Fernandez et al. 1998). Adenovirus-mediated transfer of the gene encoding NO synthase (NOS) in balloon-injured arteries may restore NO production and inhibit neointima formation. Human endothelial constitutive NOS cDNA (AdCMVceNOS) gene transfer to balloon-injured rat carotid arteries restored vascular NO production and reduced neointima formation, at least in part because of an antiproliferative effect on medial SMCs (Janssens et al. 1998).
l-arginine, a precursor of NO, inhibits intimal hyperplasia after experimental balloon angioplasty in the hypercholesterolemic rabbit whereas the NOS inhibitor L-NAME enhances lesion formation (Le Tourneau et al. 1999). However, McNamara et al. (1993) and Greenlees et al. (1997) found that although l-arginine reduced intimal thickening in the cholesterol-fed rabbit it appeared to adversely affect endothelial function at 4 weeks after injury. This may be related to the effects of lipids on nitric oxide synthase activity. Local, intramural delivery of l-arginine enhanced NO generation and reduced intimal hyperplasia (Schwarzacher et al. 1997).
Endothelin
Endothelin-1 (ET-1) is a SMC mitogen and its circulating levels are elevated after PTCA. ET-1 may be derived from dysfunctional endothelial cells and possibly from activated macrophages. Exogenous ET-1 administration promotes neointimal formation, suggesting that ET-1 is involved in the pathogenesis of balloon-induced lesion formation in the rat (Douglas et al. 1994).
Endothelin-converting enzyme-1 (ECE-1) is responsible for the conversion of pro-ET-1 to biologically active ET-1 and is expressed in neointimal smooth muscle cells in rat balloon-injured arteries, and in both smooth muscle cells and macrophages in human coronary atherosclerotic lesions. Blockade of ECE-1 was effective in reducing neointima formation after balloon injury. Thus, ECE-1 activity may contribute to the process of injury-induced neointimal formation and atherosclerosis through the autocrine/paracrine effects of endothelin-1 (Minamino et al. 1997).
Using quantitative radioligand binding (Viswanathan et al. 1997) investigated the expression of endothelin receptor subtypes ET(A) and ET(B) in the rat carotid artery after balloon-catheter injury. These studies and the use of selective ET receptor antagonists suggest that the ET(B) receptor may have a significant role in injury-induced vascular smooth muscle proliferation and neointima formation. Similarly, Azuma et al. (1995) have examined the effects of balloon injury on the expression of subtypes of the endothelin-1 receptor, in the rabbit and showed that the relative expression was greater for ET(B) than ET(A) receptor, with ET(B) being mainly expressed within the neo-intima, and ET(A) in the tunica media.
Lipoprotein interactions
Arterial injury is associated with a marked increase in permeability for plasma constituents. Lipoprotein influx increases dramatically and this results in a 10-fold increase in cholesterol ester accumulation within 5 days (Schwenke & Zilversmit 1987). This is probably also related to changes in the synthesis of lipoprotein-binding proteoglycan by smooth muscle cells (Alavi et al. 1989), alterations in smooth muscle cell phenotype (Parlavecchia et al. 1989), and vessel wall cholesterol esterase activity (Hajjar et al. 1981).
In the rabbit, high levels of β–VLDL are associated with a significantly larger neo-intima following injury (Walker & Bowyer 1984; Ferns et al. 1993a). The entry of β–VLDL into the artery wall has the potential to enhance leucocyte recruitment, due to monocyte chemotactic peptide-1 release (Berliner et al. 1997), and to prolong the duration of endothelial dysfunction (Stemerman 1981). Lp(a) is another lipoprotein that may influence the development of the neo-intima by its inhibition of tPA activation of plasminogen (Miles et al. 1989). Although it is not present in the plasma of most mammals apart from primates, several apo(a) transgenic animal models have been developed that have allowed an investigation of the role of Lp(a) in restenosis (Luoma 1997). The accumulation of Lp(a) is significantly greater in injured vessels than in normal vessels. Moreover, the accumulation of Lp(a) was relatively greater than for LDL (Nielsen et al. 1996). The degradation rate of Lp(a) is markedly increased in atherosclerotic lesions of rabbits, supporting the notion that Lp(a) may contribute to foam cell formation (Nielsen et al. 1998).
Extra cellular matrix molecules and their reciprocal effects on cell function
The extracellular matrix is now recognized as a biologically active constituent of the artery wall. It consists of structural, adhesive, and counteradhesive fibrous proteins embedded in a hydrated ground substance of glycosaminoglycans. Resident arterial cells are able to detect small differences in the specific composition and distribution of matrix components. Following injury there is a perturbation of matrix formation and degradation, that can lead to remodeling. Modifying cell–matrix interactions may provide a novel therapeutic approach in the prevention of unfavourable remodeling that leads to restenosis (Coats & Faxon 1997). After balloon injury, SMC migration is dependent on chemotactic factors and the effects of matrix metalloproteinases that free SMCs from their ECM ‘cage’. In vitro bFGF and PDGF-BB but not PDGF-AA increased the migration of SMC from primate aortic explants. This process was found to be dependent on endogenous MMP2 and MMP9 activity (Kenagy et al. 1997).
Following balloon angioplasty there is a significant increase in collagen, elastin, and proteoglycan synthesis, up to 4–10 times above control. This increase in synthesis is accompanied by a significant increase in collagen and elastin content (by approximately 35%) that coincides with the temporal increase in cross-sectional area (Strauss et al. 1994). Analysis of cultured SMCs from the ballooned rat carotid artery showed that these cells express higher levels of type VIII collagen in response to serum and growth factors, notably platelet-derived growth factor (PDGF)-BB. VSMCs adhere significantly less to type VIII collagen than to type I collagen substrata and showed greater PDGF-BB-stimulated migration (by 2.2-fold) on type VIII collagen than on type I collagen (Sibinga et al. 1997). Degradation of newly synthesized collagen is an important mechanism regulating collagen accumulation and the matrix metalloproteinases MMPs have an integral role in collagen turnover after balloon angioplasty (Strauss et al. 1996).
The accumulation of proteoglycans (PGs), such as decorin, versican and hyaluronan, is regulated by growth factors such as platelet-derived growth factor (PDGF) and transforming growth factor (TGF)-β. PG deposition is localized to areas of intense growth factor immunostaining. Notably, decorin and TGF-β1 are distributed similarly, predominantly in the macrophage-rich core, whereas biglycan is prominent in the smooth muscle cell matrix adjoining TGF-β1-positive macrophages. Versican and hyaluronan are enriched in the extracellular matrix adjacent to both PDGF-and TGF-β1-positive cells. PG accumulation varies with lesion severity, structural characteristics, and in relation to PDGF and TGF-β expression (Evanko et al. 1998). PDGF was associated with a reduction in perlecan expression whilst only the PDGF-AA isoform was associated with increased syndecan-1 mRNA expression. In vitro the migratory response and PG synthesis induced by PDGF-AA was accompanied by changes in the morphology of SMCs. PDGF-AA dramatically induced the spreading of SMCs, whereas heparin lyase treatment of PDGF-AA-stimulated cultures diminished cell spreading. PDGF-AA selectively modifies heparan sulphate PG accumulation on SMCs and thereby potentially influences the interactions of SMCs with heparin-binding ECM proteins. These interactions, in turn, generate signals that may suppress SMC migration (Koyama et al. 1998). Purified intimal-medial proteoglycan (PG) extracts from rat aortae were analysed for protein and glycosaminoglycan (GAG) content and GAG distribution pattern. The neointima of injured aortas synthesized sulphated PG at a significantly higher concentration than the intima of normal aortae. Size exclusion chromatography revealed that neointima synthesized higher molecular weight PG, and in a higher proportion, than PG from normal aortae. PG accumulating in neointima of injured aortas showed a significantly altered GAG distribution pattern (Ismail et al. 1997).
Matrix metalloproteinases
The role of basement membrane-degrading matrix metalloproteinases (MMPs) in enabling vascular smooth muscle cell migration after vascular injury has been demonstrated in several animal models. Vascular SMCs constitutively elaborate the zymogen form of gelatinase A (MMP-2). MMP-2 forms complexes with its inhibitor, the tissue inhibitor of metalloproteinases (TIMP)-2 at sites of vascular injury. Thrombin may activate MMP-2 in situ and thereby facilitate cell migration and proliferation. In the case of complicated atherosclerotic plaques, episodes of intraplaque haemorrhage or plaque disruption may promote plaque instability by increasing local matrix-degrading activity (Galis et al. 1997). Upregulation of metalloproteinase MMP-2 and MMP-9 accompanies neointima formation in rat, pig and cholesterol-fed rabbits models of angioplasty (Bendeck et al. 1996; Jenkins et al. 1998). Similar findings have been reported in organ cultures of human saphenous veins (George et al. 1997). In situ hybridization detected increased expression of TIMP-4 as early as 24 h after injury and a marked induction in neointimal cells 7 days after injury. The temporal relationship between the upregulation of TIMP-4, its accumulation, and the onset of collagen deposition suggest an important role for TIMP-4 in the proteolytic balance of the vasculature controlling both smooth muscle migration and collagen accumulation in the injured arterial wall (Dollery et al. 1999). Secretion of TIMP-1 and TIMP-2 is greatly increased during neointima formation in human saphenous veins in vitro. TIMP-1 is readily released, whereas TIMP-2 remains partially attached and TIMP-3 exclusively attached to the extracellular matrix. Regulation of TIMP expression is therefore an important determinant of net MMP activity during neointima formation, restricting this to the pericellular environment (Kranzhofer et al. 1999). Expression of TIMP-3 mRNAs is increased by PDGF and TGF-β, particularly when used in combination. Divergent regulation of gelatinase and TIMP expression implies that either net synthesis or net degradation of basement membrane can be mediated by appropriate combinations of growth factors and cytokines (Fabunmi et al. 1996). TIMP-2 overexpression reduces neointimal thickening, primarily by inhibiting MMP activity and hence smooth muscle cell migration (George et al. 1998). TIMP-1 is upregulated in the rabbit aorta in response to a balloon-catheter induced de-endothelialization at the mRNA and protein levels (Wang et al. 1996a).
Bone associated genes
Osteopontin is an arginine-glycine-asparatate (RGD) containing adhesive glycoprotein. It is constitutively expressed in rat arteries and is upregulated after balloon injury (Giachelli et al. 1995) with a peak at approximately 1–3 days (Wang et al. 1996b). Osteopontin is chemotactic for VSMCs and may therefore be involved in vascular remodeling. Osteopontin's expression is regulated by PDGF-AB and −BB but not PDGF-AA (Wang et al. 1996b). Shanahan et al. (1997) have identified another bone-associated gene, osteoglycin, that is upregulated following balloon injury, using differential cDNA library screening. In vitro osteoglycin is downregulated by a number of growth factors, including bFGF, PDGF, ATII and TGFβ. In uninjured arteries, cells in the media and adventitia expressed osteoglycin, whereas after balloon injury, neo-intimal cells also expressed osteoglycin.
Animal models
Many of the earlier studies of the mechanisms of restenosis were undertaken in the rat and rabbit carotid artery (Clowes et al. 1983; Reidy et al. 1983; Richardson et al. 1984). Because the calibre of these arteries is smaller than a human coronary, it is not possible to use an angioplasty catheter, and many of these studies used a Fogarty embolectomy catheter. In order to produce complete de-endothelialization, a partially inflated catheter is repeatedly drawn through the artery bed. Hence the forces to which the artery wall is subjected are different in magnitude and nature to those experienced in the clinical setting. A further problem is that coronary angioplasty is used to dilate arteries with pre-existing disease, whereas most of the published data using experimental animal models have been performed in normal arteries. The compliance of human arteries containing advanced atherosclerotic lesions, often with regions of calcification, is poor, and PTCA is consequently accompanied by considerable tissue damage, including intimal tears and splitting of the tunica media (Waller 1989; Brady & Warren 1991). The degree of injury produced by angioplasty has been shown to affect the biological response that ensues, and hence data from animal models must be interpreted with caution. Despite the use of different experimental models, different forms of vascular injury, different methods of analysis and different definitions of arterial remodelling, most animal studies have demonstrated the importance of remodeling in the maintenance of vascular patency in both atherogenesis and in restenosis following angioplasty particularly in the nonhuman primate. Balloon catheterization as usually performed clinically can cause a much deeper injury than simple endothelial denudation. However these models have been useful for an understanding human restenosis, and for testing therapies aimed at preventing restenosis after balloon angioplasty or other coronary interventional procedures.
The rat model
Although the rat is not the ideal animal model for human disease, the ballooned rat carotid model remains the best characterized to date. The rat common carotid artery is easily accessible via the external carotid artery and the common carotid is a relatively long unbranched arterial segment, that permits the harvesting of a reasonable amount of tissue for histological and biochemical analysis. However the rat carotid has a relatively simple structure. The intima consists of a monolayer of endothelial cells apposed to the basement membrane, or internal elastic lamina. For this reason it is not an ideal model of the human coronary artery, that has a well developed intima. The model has proved useful in the study of the kinetics of smooth muscle cell response to injury. Another major difference between the rat carotid model and clinical angioplasty is the extent of injury. The gentle passage of the embolectomy catheter in the rat model does not usually create extensive tears or lacerations in the tunica media whereas this is a consistent finding in man. Indolfi et al. (1995) have found that SMC proliferation after balloon injury is proportional to the extent of injury.
Several antisense oligonucleotide strategies have been used to inhibit intimal hyperplasia, targeting different processes. Both Abe et al. (1994) and Morishita et al. (1994) used this approach to inhibit the cell cycle related molecules cdc2 and cdk2. They reported a 47% reduction in intimal cell accumulation when inhibited singly, with a near complete inhibition when cdc2 and cdk2 antisense oligonucleotides were used in combination. Bennett et al. (1997) found that antisense oligonucleotides to c-myc, administered adventitially, suppressed medial SMC replication 2 days after injury, and intimal thickening at 14 days. Luminal area was increased at this time point. Most of the effect of the oligonucleotide was due to suppression of cell migration. Antisense c-myb oligonucleotides were found to have a similar effect (Villa et al. 1995). Various adenovirus vector constructs have also been used to elucidate the mechanisms of restenosis. Chang et al. (1995b) transfected injured arteries with a adenoviral vector encoding nonphosphorylatable retinoblastoma gene product. This reduced SMC proliferation. Over-expression of the cyclin/cyclin-dependent kinase inhibitor, p21, similarly inhibited SMC proliferation and neointima formation (Chang et al. 1995a). p21 acts by inhibiting cyclin dependent kinases required for the initiation of the S phase of the cell cycle and inhibition is associated with inhibition of the phosphorylation of Rb. Gax is a homeobox gene that is expressed by VSMCs and is down regulated by mitogen stimulation. It is also down regulated following balloon injury (Weir et al. 1995). When microinjected into VSMCs gax protein inhibits cell cycle entry into S-phase. This inhibitory effect is associated with an upregulation of p21 and reduced cdk2 activity. Local delivery of gax using an adenoviral vector, inhibited neointima formation and luminal narrowing (Smith et al. 1997). The proto-oncogene c-Harvey-ras is thought to be a key transducer in growth factor signalling. Ueno et al. (1997) inhibited c-Harvey-ras in vivo, transfecting a dominant negative gene construct into the injured rat carotid artery, and inhibited intimal hyperplasia in this way. The local delivery of an adenoviral vector expressing hirudin, an inhibitor of thrombin, was found to reduce neointima formation, supporting the notion that local thrombin activity has a role in its pathogenesis (Rade et al. 1996).
Using differential display Autieri et al. (1995), 1996) have reported a number of novel genes upregulated following balloon injury in the rat. These include protein kinase C regulatory protein 14–3-3 gamma, BART-1 and allograft inflammatory factor-1. The expression of AIF-1 was increased at 1 and 3 days, declining by 7 days. In vitro AIF-1 is expressed by human SMCs stimulated by serum and cytokines. 14–3-3 may play a role in cell proliferation by activating protein kinase RAF-1.
Healthy endothelial cells elaborate nitric oxide due to a constitutively expressed nitric oxide synthase (ecNOS). Douglas et al. (1994) have examined time-dependent changes in vascular reactivity after balloon injury in the rat carotid. Angioplasty was associated with an increased sensitivity to ET-1 induced contractility, which they suggest is due to the loss of ET-1 induced release of nitric oxide. Chen et al. (1998) expressed ecNOS aberrantly in VSMCs that were subsequently seeded onto injured arteries. These cells produced NO locally, inhibited neointima formation, and caused vessel dilatation. Nitric oxide donors are known to inhibit SMC proliferation in vitro. Seki et al. (1995) found that a novel NO donor (FK409) suppressed intimal hyperplasia by 48%, whereas isosorbide mononitrate (ISMN) was ineffective. FK409 inhibited intimal and medial SMC proliferation, and had a significant effect on intimal hyperplasia even when administered on the day of injury and for 4 days subsequently.
The development of an intimal proliferative lesion after balloon catheter de-endothelialization has been studied in congenitally athymic nude rats lacking T lymphocytes. There did not appear to be any significant effect on SMC kinetics in the nude animals (Ferns et al. 1991c). Hancock et al. (1994) investigated the cell types and cytokine expression associated with intimal hyperplasia after balloon injury in the Sprague-Dawley rat. Large numbers of CD4+/ED1 + cells but no T cells were identified by immunocytochemistry. IL-1β, TNFα and IL-8 were widely expressed, together with PDGF and TGFβ. The authors administered an anti CD4 monoclonal antibody to the rats, and this was associated with a reduction in intimal hyperplasia, and reduced monocyte accumulation.
The importance of PDGF in the rat model of restenosis has become evident over recent years. Following balloon injury there are well characterized temporal changes in the expression and phosphorylation state of PDGF receptor subunits (α– and β–subunits) (Majesky et al. 1990; Panek et al. 1997). Abe et al. (1996) have found that injury is associated with tyrosyl phosphorylation of PDGF receptor subunits (αand β) but not EGF receptor, nor insulin receptor substrate-1 (IRS-1). An angiotensin II receptor antagonist was found to inhibit tyrosyl phosphorylation of PDGF receptor subunits and also intimal hyperplasia.
Platelet-derived growth factor accounts for approximately 50% of the mitogenic activity of a rat platelet releasate. PDGF-BB and PDGF-AB were both potent chemotactic agents for the nude rat carotid smooth muscle cells with a peak response at approximately 10 ng/ml. In contrast, PDGF-AA, transforming growth factor beta, and basic fibroblast growth factor were only weak chemoattractants for these cells (Ferns et al. 1991c). In the balloon injury rat model PDGF appears to be a major chemoattractant (Ferns et al. 1991b; Jawien et al. 1992). Lindner et al. (1995) found that a subpopulation of SMCs, comprising approximately 10% of the total express PDGF-B chain. More recently, Rutherford et al. (1997) have reported that the PDGF-BB isoform and bFGF act synergistically and account for the majority of the chemotactic effect in this model. In contrast, Bjornsson et al. (1991) found that administration of acidic FGF intravenously inhibited intimal thickening and promoted endothelial regrowth. The receptors for FGF are upregulated following balloon injury, and this fact has been used to target toxins (such as saporin, using a bFGF conjugate) to proliferating intimal cells (Casscells et al. 1992; Farb et al. 1997). Monocytes adhere preferentially to the migrating cells at the edge of the regenerating endothelium and Wempe et al. (1997) have reported that this is probably due to an induction of monocyte chemotactic peptide-1 (MCP-1) stimulated by bFGF.
The stimuli for intimal hyperplasia are probably derived from the blood and the injured artery wall. West & Hubbell (1996) attempted to clarify the relative importance of factors from these two sources by using a hydrogel barrier that blocked thrombus formation after balloon injury. The authors measured the intimal response to injury, endogenous bFGF mobilization within the artery wall, thrombosis and medial cell re-population. They reported that factors other than PDGF-BB and thrombin contributed to intimal hyperplasia, and that a luminally derived factor was necessary for bFGF mobilization. Platelet adhesion and aggregation occur rapidly after arterial injury, causing the release of several platelet-associated factors. Zahger et al. (1995) used a platelet GP 1b receptor antagonist to inhibit platelet adhesion at the site of injury, a recombinant form of the GP 1b binding domain of von Willebrand factor (vWF) which inhibits platelet binding to vWF. It was found to reduce platelet adhesion by more than 80% and reduced intimal thickening and luminal stenosis, without significantly affecting SMC proliferation.
TGFβ mRNA was found to be increased 5-fold by 24 h after injury, with significant increases being apparent as early as 6 h, and remaining elevated for up to 2 weeks (Majesky et al. 1991). Increased levels of TGFβ immuno-reactivity were associated with increased expression of fibronectin, and collagen, and infusion of TGFβ into injured arteries stimulated SMC DNA synthesis (Majesky et al. 1991). More recently, Ward et al. (1997) investigated the expression of several other TGFβ–related genes following balloon injury. TGFβ–1 and TGFβ–3 were both induced within 48 h. There were also elevations in TGFβ-type-1 (ALK-2 and ALK–5) and -2 receptors. The expression of two integrins (αv and β3), normally responsive to TGFβ were also upregulated. These changes could be inhibited substantially by the tyrosine kinase inhibitor, genistein. A TGFβ antagonist, Tranilast, also attenuated these effects in vivo (Ward et al. 1998).
The role of IGF-I in intimal hyperplasia is unclear (Ferns et al. 1991a; Motani et al. 1995). Yumi et al. (1997) used a specific inhibitor of growth hormone and the IGF-1 axis, octreotide to probe this issue. Octreotide inhibited IGF-I mRNA expression following injury, but had no effect on IGF-BP4, which was upregulated following balloon injury. Treatment with octreotide inhibited neo-intimal thickening by up to 90%.
Endothelin-related proteins are rapidly modulated following balloon injury (Wang et al. 1996c; Viswanathan et al. 1997). Endothelin converting enzyme (ECE-1) expression preceded that of prepro-ET-1 and -3. Expression of the ET(A) and ET(B) receptors were markedly upregulated within 3 days of injury. Douglas & Ohlstein (1993) reported that ET-1 stimulates rat SMC proliferation in vitro, and enhanced intimal hyperplasia when administered in vivo. They also reported that an ET receptor blocker inhibited intimal thickening in the rat. Douglas et al. (1994) compared the effects of an ET(A) receptor antagonist vs. a dual ET(A)/(B) antagonist. They found that the dual antagonist was effective in inhibiting intimal hyperplasia, but the specific ET(A) antagonist was ineffective. These data suggest that the ET(B) receptor subtype is involved in the development of intimal hyperplasia. The possible role of endothelin in intimal hyperplasia has been strengthened by the demonstration that SB 217242, and TAK-044, both mixed ET(A)/(B) receptor antagonist (Tsujino et al. 1995; Chandra et al. 1998) reduced the intima/media ratio by more than 40%. Endothelin converting enzyme (ECE-1) is a key neutral metalloprotease involved in the processing of endothelin-1, and is normally present in endothelial cells. Minamino et al. (1997) examined the expression and localization of ECE-1 following carotid injury. ECE-1 mRNA increased between 2 and 5 days after injury. In the absence of endothelium, ECE-1 was detected in the neointimal smooth muscle cells up to 14 days after injury. The authors concluded that ECE-1 may contribute to neointimal thickening through an autocrine, or paracrine effect on ET-1.
The importance of vascular remodeling has received less attention in the rat model. However, Jenkins et al. (1998) have recently investigated the expression of matrix metalloproteinase following balloon injury. Previous studies demonstrated that MMP-2 and MMP-9 were upregulated in this model, and that MMP-2 is involved in SMC migration. Jenkins et al. (1998) examined the effects on membrane-type MMP-1 that may be involved in the regulation of MMP-2. It may do so by affecting the tissue localization of MMP-2, its activation state, and the expression of an inhibitor, tissue inhibitor of metalloproteinase (TIMP-2). They reported that MT-MMP-1 steady-state mRNA levels were increased 6-fold within 3 days of injury, and that there was a temporal relationship with increased MMP-2 activity, measured by zymography. Collagen type α1 VIII and α2 VIII mRNA are both upregulated after balloon injury (Sibinga et al. 1997). The pattern of expression indicates that collagen type VIII is expressed by activated, dedifferentiated SMCs of the intima and media. In vitro expression could be stimulated by PDGF-BB, and SMCs were less adherent to collagen type VIII than to type I collagen. SMCs were also more responsive to chemotactic agents on a matrix of collagen type VIII vs. collagen type I. Proteoglycan accumulation varies with lesion severity and with local growth factor elaboration (Evanko et al. 1998).
Heat shock proteins (HSPs) are a family of highly conserved proteins, expressed during periods of physiological stress, that are protective during re-exposure to stress. Neschis et al. (1998) induced HSPs in Sprague-Dawley rats by whole body hyperthermia and subsequently these animals were subject to balloon injury. Hyperthermia treatment was associated with a significant reduction in intimal hyperplasia and medial cell proliferation.
The response to balloon injury is sexually dimorphic due to the protective effects of oestrogens. Oparil et al. (1997) found that exogenous oestrogen was ineffective in inhibiting intimal hyperplasia in male rats, although serum 17β–oestradiol concentrations reached physiological female levels. Progestin (medroxyprogesterone acetate) blocks the effects of oestrogen, and enhanced the response to balloon injury in female rats, but the response in male rats was unaffected. The oestrogen, oestradiol, enhanced the rate of functional recovery of the endothelium after balloon injury (Krasinski et al. 1997).
The rabbit model
The rabbit model of balloon injury was one of the first to be used (Groves et al. 1979). Rabbits are very responsive to atherogenic diets, developing hyperlipidaemia and early lesions soon after initiation. Indeed, Tashiro et al. (1998) found that small amounts of saturated fatty acid (such as lard) promoted the formation of intimal thickening without affecting serum lipid levels. There are also inbred rabbit strains with genetic abnormalities of lipid metabolism, equivalent to human conditions such as Familial Hypercholesterolaemia (the Watanabe Heritable hyperlipidaemic rabbit; Takagi et al. 1997) and Familial combined hyperlipidaemia (Seddon et al. 1987). For this reason it is possible to investigate the combined effects of mechanical injury and dyslipidaemia (Faxon et al. 1982; Alavi et al. 1983). Moreover it has recently been possible to establish transgenic rabbit models. For example a human apolipoprotein A1 transgenic rabbit line has been created (Duverger et al. 1996) which may be useful to explore the role of high density lipoprotein in the response to balloon injury.
More et al. (1994) and Stadius et al. (1994) have presented the detailed cell kinetics in the rabbit after balloon injury. Re-endothelialization was complete by day 14 and could be enhanced by administration of vascular permeability factor (Callow et al. 1994). Cellular proliferation reached a maximum by day 7 for the media and day 14 for the intima, before returning to baseline by one month. Urokinase type plasminogen activator (uPA) increased within 2 h of injury and remained elevated at 24 h but fell below baseline by 7 days, whilst tissue type plasminogen activator (tPA) fell after injury, rising to baseline by 7 days (More et al. 1995). Stadius et al. (1996) also examined the effect of double injury in the rabbit.
The importance of PDGF in the rabbit model of restenosis is also clear. PDGF B-chain is expressed in the balloon-injured artery, with PDGF-B chin mRNA peaking by 7 days, and remain elevated for 21 days, whereas PDGF A-chain is only modestly increased. PDGF receptor alpha and beta subunits are also upregulated (Uchida et al. 1996). Trapidil, a PDGF antagonist, inhibited intimal hyperplasia, and increased luminal area in the atherosclerotic rabbit (Liu et al. 1990).
Using immuno-cytochemical analysis, Lawrence et al. (1995) found that following balloon injury bFGF positive cells were found in the media and in recently migrated cells of the neo-intima for up to 14 days after injury.
The collagen content of ballooned rabbit artery was found to increase over the first four weeks after injury, however, the ratio of collagen subtypes was unchanged (Coats et al. 1996). Coats et al. (1997) subsequently reported an inverse relationship between collagen content and gelatinase activity. Strauss et al. (1996) investigated the turnover of collagen and its regulation by MMP in a double injury model. Peak collagen synthesis and degradation occurred at one week, as did MMP-2 activity. The expression of matrix metalloproteinase-2 is increased in the neo-intima of the rabbit (Wang et al. 1998) and its inhibition results in a reduction in VSMC proliferation and migration. MMP-2 requires activation for full activity. This appears to be due to membrane type MMP-1 (MT-MMP-1) (Wang & Keiser 1998).
In vitro, heparan sulphate PGs (HSPG) maintained VSMCs in a quiescent state by inhibiting phenotypic change and DNA synthesis. Aorta-derived HSPG reduced neo-intimal thickening by 35% when applied peri-adventially (Bingley et al. 1998). Hyaluronic acid, a major constituent of aortic GAG also reduced intimal thickening and monocyte influx in the cholesterol-fed rabbit (Ferns et al. 1995), as did low molecular weight heparin (Currier et al. 1991) which may be partly related to its effect on SMC proliferation (Oberhoff et al. 1998).
Endotoxin-stimulatable tissue-factor expression was increased in circulating mononuclear cells following balloon angioplasty, and this was blunted by treating animals with l-arginine (Corseaux et al. 1998). Brown et al. (1996) and Asada et al. (1998) have investigated the effects of inhibiting fibrin formation using a tissue factor pathway inhibitor administered locally immediately after injury. Jang et al. (1995) investigated the contribution of the coagulation cascade on restenosis in the hyper-cholesterolaemic rabbit using inhibitors for different levels of the coagulation pathway. Factor VIIa binding to tissue factor was blocked with active-site inactivated factor VIIa (DEGR-VIIa). Recombinant tissue factor pathway inhibitor (TFPI), binds to factor Xa and inhibits tissue factor-VIIa complex formation, and recombinant tick anticoagulant peptide (TAP) and hirudin block factor Xa and thrombin, respectively. DEGR-VIIa and TFPI inhibited neo-intimal hyperplasia indicating the importance of the extrinsic coagulation pathway, especially factor VII and tissue factor in the arterial response to injury. Thrombin inhibition, by hirudin, suppressed arterial thrombin activity (Barry et al. 1996), and restenosis (Barry et al. 1997). Hirudin probably operates partly by reducing tissue factor expression. Gertz et al. (1998) have reported that hirudin inhibits tissue factor expression in the injured rabbit femoral artery and porcine coronary artery. Ragosta et al. (1996) found that hirudin did not inhibit cell proliferation. Recombinant tissue plasminogen activator inhibits intimal hyperplasia in the hypercholesterlaemic rabbit (Kanamasa et al. 1996).
The efficiency of adeno-virus transfection is very low (approximately 0.2% of intimal and medial cells) in atherosclerotic rabbit arteries (Feldman et al. 1996). However a vector construct containing the growth arrest homeobox gene (Gax) reduced intimal thickening (Maillard & Walsh 1996; Maillard et al. 1997).
The pig model
The anatomical and physiological similarities between human and porcine cardiovascular systems have led to the use of the pig to model restenosis (Steele et al. 1985). It is possible to use the same balloon catheters and similar inflation pressure as used clinically, and the morphology of the lesion that follows balloon injury is similar to that seen in human postangioplasty restenosis (Schwartz et al. 1990; Karas et al. 1992).
Groves et al. (1995) have provided detailed kinetic data of VSMC proliferation in the pig and examined the possible variations with respect to degree of injury (medial dilatation vs. medial tearing). Although medial enlargement was similar for both degrees of injury, they found that the total thickening of the injured artery was greater when a deep tear breached the media. The effects of balloon injury on SMC phenotype previously shown in the rat have also been demonstrated in the pig (De Leon et al. 1997). Non-muscle myosin heavy chain-B isoform is expressed by proliferating VSMCs, and its expression following balloon injury in the pig is associated with cell proliferation in the adventitia and intima. However, nonmuscle myosin heavy chain-B isoform expression was not limited to proliferating cells.
von Willebrand factor (vWF), is involved in platelet–vessel wall interactions. Giddings et al. (1997) have investigated the sequential changes in tissue distribution of vWF following balloon injury, and report that it is localized to the cells of the tunica media for up to 7 days, and may be involved in medial smooth muscle cell proliferation. There are some significant differences between pig and human platelet function, for example their responsiveness to agonists such as adrenaline and arachidonic acid (reviewed by Softeland et al. 1992).
Following balloon injury, arteries sometimes undergo vasoconstriction. Lam et al. (1987) found that the arterial spasm is related to the degree of platelet deposition, and could be inhibited by aspirin. Although neutrophils are not a major cellular component of the ballooned artery wall in most animal models, neutropenia induced by treatment with cyclophosphamide was associated with reduced platelet aggregation and deposition, and reduced vasoconstrictor response (Merhi et al. 1994). Lam et al. (1992) have reported that cod liver oil at a dose of 1 ml/kg/day reduced platelet deposition after deep carotid injury by balloon injury. This was also associated with reduced vasoconstriction. However, using porcine arteries ex vivo, Fischell et al. (1989) reported that the vasoconstriction may also be endothelium-dependent, particularly in situations where residual endothelial cells remain on the partially denuded artery. Vasoconstriction appeared to be mediated by cyclo-oxygenase-derived products, and could be blocked by indomethacin. The pig model has also been used to show the importance of arterial remodeling in late luminal narrowing; (Post et al. 1994; Andersen et al. 1996). Lamawansa et al. (1997) also present evidence that the sympathetic innervation may influence arterial remodeling and wall thickening.
Studies in the pig model have shown that endothelin stimulates SMC proliferation via the ET(A) receptor. Burke et al. (1997) have reported that an orally active ET(A) receptor antagonist, A127722.5, inhibited intimal thickening and luminal narrowing. It also reduced the collagen content of the tunica media. These data support the role of endothelin in restenosis, although the relative importance of ET(A) and ET(B) receptors may differ between the animal models.
The pig has also been used to test a number of putative intervention strategies which have provided a clearer insight into the mechanisms of restenosis. For example Banai et al. (1998) have reported that a specific blocker of PDGF-receptor tyrosine kinase (tyrphostin AG1295) when delivered locally to the site of balloon injury, significantly reduced intimal thickening, and luminal narrowing. Selective αvβ3 integrin blockade, using a cyclic Arginine-Glycine-Asparagine (RGD) pepidometic antagonist, inhibited neointimal hyperplasia in the pig coronary artery (Srivatsa et al. 1997). Antithrombin III is the major physiological inhibitor of thrombin. Combined with heparin, ATIII significantly inhibited the reduction in minimal luminal diameter at 4 weeks after angioplasty (Ali et al. 1996). There was a modest reduction in neo-intimal proliferation associated with treatment with the ACE inhibitor, trandolapril (Huber et al. 1993). Three Ang II receptor antagonists, including AT1 selective (L-158 809), balanced AT1/AT2 (L-163 082), and AT2 selective (L-164 282) agents, were evaluated for their ability to inhibit vascular intimal thickening in a porcine coronary artery model of vascular injury. It was shown that chronic blockade of Ang II receptors by either site-selective or balanced AT1/AT2 antagonists is insufficient to inhibit intimal hyperplasia after experimental coronary vascular injury in the pig. Unlike in the rat carotid artery, Ang II is not a major mediator of intimal thickening in the pig coronary artery (Huckle et al. 1996).
The calibre of the pig coronary artery has allowed the local delivery of antisense oligonucleotides. Using this model, Gunn et al. (1997) have demonstrated that an antisense oligonucleotides to c-myb, a early gene expressed after vascular injury, reduced c-myb mRNA and protein, and subsequently led to a significant reduction in the intimal response to injury. However this study was confounded by nonspecific effects of the oligonucleotides.
Shi et al. (1997) investigated the cellular origin of type I collagen and elastin after balloon injury. By the second day after injury, type I pro-collagen was expressed by cells of the adventitia. This was followed by enhanced neo-intimal expression of pro-collagen and elastin.
The non-human primate model
The arterial response to angioplasty in atherosclerotic monkeys appears to closely parallel that seen in humans. Plaque fracture, delayed recoil, intimal hyperplasia, and remodeling each appear to be important in determining late lumen calibre. In monkeys that were fed an atherogenic diet and subsequently underwent experimental angioplasty, the pattern of matrix and integrin expression within the injured artery wall is in many ways analogous to that of healing wounds (Geary et al. 1996). The provisional extracellular matrix laid down after injury is invaded by leucocytes and alpha-actin-positive smooth muscle cells. These SMCs express hyaluronan and are associated with versican proteoglycans expression. Versican is expressed throughout the neointima as it enlarges, but expression later diminishes. Procollagen-I expression initially increases in the adventitia and then subsequently within the developing neointima. Procollagen-I expression persists within the adventitia and in the neointima in SMCs nearest the lumen. Elastin staining is prominent within the mature neointima but is not evident at earlier time points. Integrin expression also increases within the injured artery wall. αVβ3 staining characteristic of the fibrin[ogen] receptor, increases in the injured media and is then apparent throughout the newly formed neointima. α2β1 integrin expression (collagen receptor) increased in the neointima in SMCs nearest the lumen (Geary et al. 1998).
The glycoprotein (GP) IIb/IIIa found on platelets cross-links fibrinogen and vWF, thereby mediating platelet aggregation. Guth et al. (1997) studied the effects of blocking GP IIb/IIIa, using a new nonpeptidic molecule, BIBU52. The inhibition of platelet aggregation by BIBU52 was found to be species specific. BIBU52 inhibited aggregation in platelet rich plasma from rhesus and marmoset but was ineffective in rat plasma.
Recently, Giese et al. (1999) selectively blocked PDGFα and β receptor subunits using monoclonal antibodies and demonstrated that PDGF probably promotes neointima formation via PDGF receptor β subunit.
Lipoprotein(a) (Lp(a)) has been proposed as a risk factor for both restenosis and coronary heart disease. Recently, Ryan et al. (1997) identified Lp(a) in the arterial wall during the early phases of neointimal growth in cynomolgus monkeys. Ryan et al. (1998) also investigated the relationship between circulating Lp(a) levels and the extent of early neointima formation. A four-fold increase in circulating plasma Lp(a) levels did not result in a statistically significant enhancement in neointima formation at 14 days after balloon injury.
The mouse model
Because of its small size, it has not been possible to develop an authentic mouse model of angioplasty. Models of arterial injury have been developed which are associated with endothelial injury and intimal thickening (Lindner et al. 1993; Kumar & Lindner 1997). Carmeliet et al. (1997b) have described a mouse model of restenosis induced by perivascular femoral artery injury using an electric current. Endothelial injury in the mouse femoral artery was also induced by photochemical reaction between localized irradiation by green light and intravenous injection of rose bengal (Miyazawa et al. 1998). Although none of these models is ideal for studying the complete process of restenosis, it is possible that they will provide reliable information when combined with the use of transgenic strains such as those described by Liao et al. (1996) and Leppanen et al. (1998). The use of transgenic mouse models has demonstrated that leucocyte-endothelium adhesion receptors play a significant part in promoting monocyte recruitment and consequently lesion growth (Dong & Wagner 1998). Mechanical and electric vascular injury of plasminogen activator inhibitor-1 (PAI-1) deficient mice showed that an inhibitory role by PAI-1 was played in neointima formation and that the adenoviral gene transfer reduced neointimal formation (Carmeliet et al. 1997a).
Conclusions
Animal models of intimal hyperplasia have provided a profound insight into the cellular and biochemical mechanisms of restenosis following angioplasty. They have been less useful in predicting the efficacy of therapeutic interventions. Nevertheless, these models have provided a test-bed for novel strategies including the new molecular approaches. It is clear that interspecies differences, and the relative simplicity of the animal models have yielded some misleading results that have not been translated into clinical benefit. Nonetheless, they have collectively provided a number of novel therapeutic targets, and as their sophistication increases their reliability as models of human diseases will improve pari passu.
Acknowledgments
Much of the authors' own work in this area has been supported by grants from the British Heart Foundation and Medical Research Council. Dr Tony Avades is supported by a project grant from the BHF.
References
- Abe H, Bandai A, Makuuchi M, et al. Hyperinsulinaemia accelerates accumulation of cholesterol ester in aorta of rats with transplanted pancreas. Diabetologia. 1996;39:1276–1283. doi: 10.1007/s001250050570. [DOI] [PubMed] [Google Scholar]
- Abe J, Zhou W, Taguchi J, et al. Suppression of neointimal smooth muscle cell accumulation in vivo by antisense cdc2 and cdk2 oligonucleotides in rat carotid artery. Biochem. Biopys. Res. Commun. 1994;198:16–24. doi: 10.1006/bbrc.1994.1003. [DOI] [PubMed] [Google Scholar]
- Alavi M, Dunnett CW, Moore S. Lipid composition of rabbit aortic wall following removal of endothelium by balloon catheter. Arteriosclerosis. 1983;3:413–419. doi: 10.1161/01.atv.3.5.413. [DOI] [PubMed] [Google Scholar]
- Alavi MZ, Richardson M, Moore S. The in vitro interactions between serum lipoproteins and proteoglycans of the neointima of rabbit aorta after a single balloon catheter injury. Am. J. Pathol. 1989;134:287–294. [PMC free article] [PubMed] [Google Scholar]
- Ali MN, Mazur W, Kleiman NS, et al. Inhibition of coronary restenosis by antithrombin III in atherosclerotic swine. Coron. Art. Dis. 1996;7:851–861. doi: 10.1097/00019501-199611000-00010. [DOI] [PubMed] [Google Scholar]
- Andersen HR, Maeng M, Thorwest M, Falk E. Remodeling rather than neointimal formation explains luminal narrowing after deep vessel wall injury: insights from a porcine coronary (re) stenosis model. Circulation. 1996;93:1716–1724. doi: 10.1161/01.cir.93.9.1716. [DOI] [PubMed] [Google Scholar]
- Asada Y, Hara S, Tsuneyoshi A, et al. Fibrin-rich and platelet-rich thrombus formation on neointima: recombinant tissue factor pathway inhibitor prevents fibrin formation and neointimal development following repeated balloon injury of rabbit aorta. Thromb. Haemostas. 1998;80:506–511. [PubMed] [Google Scholar]
- Assoian RK, Grotendorst GR, Miller DM, Sporn MB. Cellular transformation by co-ordinated action of three peptide growth factors from human platelets. Nature. 1984;309:804–806. doi: 10.1038/309804a0. [DOI] [PubMed] [Google Scholar]
- Austin GE, Ratliff NB, Hollman J, Tabei S, Phillips DF. Intimal proliferation of smooth muscle cells as an explanation for recurrent coronary artery stenosis after percutaneous transluminal coronary angioplasty. J. Am. Coll. Cardiol. 1985;6:369–375. doi: 10.1016/s0735-1097(85)80174-1. [DOI] [PubMed] [Google Scholar]
- Autieri MV, Feuerstein GZ, Yue T, Ohlstein EH, Douglas SA. Use of differential display to identify differentially expressed mRNAs induced by rat carotid artery balloon angioplasty. Lab. Invest. 1995;72:656–661. [PubMed] [Google Scholar]
- Autieri MV, Haines DS, Romanic AM, Ohlstein EH. Expression of 14–3–3 gamma in injured arteries and growth factor- and cytokine-stimulated human vascular smooth muscle cells. Cell Growth Different. 1996;7:1453–1460. [PubMed] [Google Scholar]
- Azuma H, Hamasaki H, Sato J, Isotani E, Obayashi S, Matsubara O. Different localisation of ETA and ETB receptors in the hyperplastic vascular wall. J. Cardiovasc. Pharmacol. 1995;25:802–809. doi: 10.1097/00005344-199505000-00017. [DOI] [PubMed] [Google Scholar]
- Banai S, Wolf Y, Golomb G, et al. PDGF-receptor tyrosine kinase blocker AG1295 selectively attenuates smooth muscle cell growth in vitro and reduces neointimal formation after balloon angioplasty in swine. Circulation. 1998;97:1960–1969. doi: 10.1161/01.cir.97.19.1960. [DOI] [PubMed] [Google Scholar]
- BARI. BARI Investigators; Comparison of coronary bypass surgery with angioplasty in patients with multivessel disease. N. Engl. J. Med. 1996;335:217–225. doi: 10.1056/NEJM199607253350401. [DOI] [PubMed] [Google Scholar]
- Barja F, Coughlin C, Belin D, Gabbiani G. Actin isoform synthesis and mRNA levels in quiescent and proliferating rat aortic smooth muscle cells in vivo and in vitro. Lab. Invest. 1986;55:226–233. [PubMed] [Google Scholar]
- Barron MK, Lake RS, Buda AJ, Tenaglia AN. Intimal hyperplasia after balloon injury is attenuated by blocking selectins. Circulation. 1997;96:3587–3592. doi: 10.1161/01.cir.96.10.3587. [DOI] [PubMed] [Google Scholar]
- Barry WL, Gimple LW, Humphries JE, et al. Arterial thrombin activity after angioplasty in an atherosclerotic rabbit model: time course and effect of hirudin. Circulation. 1996;94:88–93. doi: 10.1161/01.cir.94.1.88. [DOI] [PubMed] [Google Scholar]
- Barry WL, Wiegman PJ, Gimple LW, et al. A new single-injury model of balloon angioplasty in cholesterol-fed rabbits: beneficial effect of hirudin and comparison with double-injury model. Lab. Invest. 1997;77:109–116. [PubMed] [Google Scholar]
- Battegay EJ, Raines EW, Seifert RA, Bowen-Pope DF, Ross R. TGF-beta induces bimodal proliferation of connective tissue cells via complex control of an autocrine PDGF loop. Cell. 1990;63:515–524. doi: 10.1016/0092-8674(90)90448-n. [DOI] [PubMed] [Google Scholar]
- Baumgartner HR. Eine neue methode zur erzeungung vor thromben durch gesielte uberdehnung der gefässwand. Z. Ges. Exp. Med. 1963;137:227. [PubMed] [Google Scholar]
- Bauters C, De Groote P, Adamantidis M, et al. Proto-oncogene expression in rabbit aorta after wall injury. First marker of the cellular process leading to restenosis after angioplasty. Eur. Heart J. 1992;13:556–559. doi: 10.1093/oxfordjournals.eurheartj.a060213. [DOI] [PubMed] [Google Scholar]
- Bendeck MP, Irvin C, Reidy MA. Inhibition of matrix metalloproteinase activity inhibits smooth muscle cell migration but not neointimal thickening after arterial injury. Circ. Res. 1996;78:38–43. doi: 10.1161/01.res.78.1.38. [DOI] [PubMed] [Google Scholar]
- Bennett MR, Lindner V, Deblois D, Reidy MA, Schwartz SM. Effect of phosphorothioated oligonucleotides on neointima formation in the rat carotid artery. Dissecting the mechanism of action. Arterioscler. Thromb. Vasc. Biol. 1997;17:2326–2332. doi: 10.1161/01.atv.17.11.2326. [DOI] [PubMed] [Google Scholar]
- Berliner J, Leitinger N, Watson A, Huber J, Fogelman A, Navab M. Oxidized lipids in atherogenesis: formation destruction and action. Thromb. Haemostas. 1997;78:195–199. [PubMed] [Google Scholar]
- Bingley JA, Hayward IP, Campbell JH, Campbell GR. Arterial heparan sulfate proteoglycans inhibit vascular smooth muscle cell proliferation and phenotype change in vitro and neointimal formation in vivo. J. Vasc. Surg. 1998;28:308–318. doi: 10.1016/s0741-5214(98)70167-3. [DOI] [PubMed] [Google Scholar]
- Bjornsson TD, Dryjski M, Tluczek J, et al. Acidic fibroblast growth factor promotes vascular repair. Proc. Natl. Acad. Sci. USA. 1991;88:8651–8655. doi: 10.1073/pnas.88.19.8651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blann A, Midgley H, Burrows G, et al. Free radicals, antioxidants, and endothelial cell damage after percutaneous transluminal coronary angioplasty. Coron. Art. Dis. 1993;4:905–910. doi: 10.1097/00019501-199310000-00010. [DOI] [PubMed] [Google Scholar]
- Booth RF, Martin JF, Honey AC, Hassall DG, Beesley JE, Moncada S. Rapid development of atherosclerotic lesions in the rabbit carotid artery induced by perivascular manipulation. Atheroscler. 1989;76:257–268. doi: 10.1016/0021-9150(89)90109-3. [DOI] [PubMed] [Google Scholar]
- Bornfeldt KE, Arnqvist HJ, Norstedt G. Regulation of insulin-like growth factor-I gene expression by growth factors in cultured vascular smooth muscle cells. J. Endocrinol. 1990;125:381–386. doi: 10.1677/joe.0.1250381. [DOI] [PubMed] [Google Scholar]
- Bosmans JM, Bult H, Vrints CJ, Kockx MM, Herman AG. Balloon angioplasty and induction of non-endothelial nitric oxide synthase in rabbit carotid arteries. Eur. J. Pharmacol. 1996;310:163–174. doi: 10.1016/0014-2999(96)00377-9. [DOI] [PubMed] [Google Scholar]
- Brady AJB, Warren JB. Angioplasty and restenosis. B.M.J. 1991;303:729–730. doi: 10.1136/bmj.303.6805.729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown DM, Kania NM, Choi ET, et al. Local irrigation with tissue factor pathway inhibitor inhibits intimal hyperplasia induced by arterial interventions. Arch. Surg. 1996;131:1086–1090. doi: 10.1001/archsurg.1996.01430220080018. [DOI] [PubMed] [Google Scholar]
- Burke SE, Lubbers NL, Gagne GD, et al. Selective antagonism of the ET (A) receptor reduces neointimal hyperplasia after balloon-induced vascular injury in pigs. J. Cardiovasc. Pharmacol. 1997;30:33–41. doi: 10.1097/00005344-199707000-00006. [DOI] [PubMed] [Google Scholar]
- CAbri. CABRI Trial Participants; First-year results of CABRI (Coronary Angioplasty versus Bypass Revascularisation Investigation) Lancet. 1995;346:1179–1184. [PubMed] [Google Scholar]
- Callow AD, Choi ET, Trachtenberg JD, et al. Vascular permeability factor accelerates endothelial regrowth following balloon angioplasty. Growth Factors. 1994;10:223–228. doi: 10.3109/08977199409000240. [DOI] [PubMed] [Google Scholar]
- Carmeliet P, Moons L, Lijnen R, et al. Inhibitory role of plasminogen activator inhibitor-1 in arterial wound healing and neointima formation: a gene targeting and gene transfer study in mice. Circulation. 1997a;96:3180–3191. doi: 10.1161/01.cir.96.9.3180. [DOI] [PubMed] [Google Scholar]
- Carmeliet P, Moons L, Stassen JM, et al. Vascular wound healing and neointima formation induced by perivascular electric injury in mice. Am. J. Pathol. 1997b;150:761–776. [PMC free article] [PubMed] [Google Scholar]
- Casscells W, Lappi DA, Olwin BB, et al. Elimination of smooth muscle cells in experimental restenosis: targeting of fibroblast growth factor receptors. Proc. Natl. Acad. Sci. USA. 1992;89:7159–7163. doi: 10.1073/pnas.89.15.7159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cercek B, Fishbein C, Forrester JS, Helfant RH, Fagin JA. Induction of insulin-like growth factor 1 mRNA in rat aorta after balloon denudation. Circ. Res. 1990;66:1755–1760. doi: 10.1161/01.res.66.6.1755. [DOI] [PubMed] [Google Scholar]
- Chandra S, Sarkar S, Elliot JD, Ohlstein EH. Application of magnetic resonance imaging for evaluation of the efficacy of SB 217242 in neointimal formation. J. Cardiovasc. Pharmacol. 1998;31:S317–S319. doi: 10.1097/00005344-199800001-00088. [DOI] [PubMed] [Google Scholar]
- Chang MW, Barr E, Lu MM, Barton K, Leiden JM. Adenovirus-mediated over-expression of the cyclin/cyclin-dependent kinase inhibitor, p21 inhibits vascular smooth muscle cell proliferation and neointima formation in the rat carotid artery model of balloon angioplasty. J. Clin. Invest. 1995a;96:2260–2268. doi: 10.1172/JCI118281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang MW, Barr E, Seltzer J, et al. Cytostatic gene therapy for vascular proliferative disorders with a constitutively active form of the retinoblastoma gene product. Science. 1995b;267:518–522. doi: 10.1126/science.7824950. [DOI] [PubMed] [Google Scholar]
- Chemla E, Castier Y, Julia P, Pirotski E, Carpentier A, Fabiani JN. Inhibition of intimal hyperplasia by an antisense oligonucleotide of farnesyl transferase delivered endoluminally during iliac angioplasty in a rabbit model. Ann. Vascular Surgery. 1997;11:581–587. doi: 10.1007/s100169900095. [DOI] [PubMed] [Google Scholar]
- Chen L, Daum G, Forough R, Clowes M, Walter U, Clowes AW. Overexpression of human endothelial nitric oxide synthase in rat vascular smooth muscle cells and in balloon-injured carotid artery. Circ. Res. 1998;82:862–870. doi: 10.1161/01.res.82.8.862. [DOI] [PubMed] [Google Scholar]
- Clowes AW, Reidy MA, Clowes MM. Kinetics of cellular proliferation after arterial injury. I. Smooth muscle cell growth in the absence of endothelium. Lab. Invest. 1983;49:327–333. [PubMed] [Google Scholar]
- Clowes AW, Clowes MM, Au YPT, Reidy MA, Belin D. Smooth muscle cells express urokinase during mitogenesis and tissue type plasminogen activator during migration in injured rat carotid artery. Circ. Res. 1990;67:61–67. doi: 10.1161/01.res.67.1.61. [DOI] [PubMed] [Google Scholar]
- Coats WDJ, Cheung DT, Han B, Currier JW, Faxon DP. Balloon angioplasty significantly increases collagen content but does not alter collagen subtype I/III ratios in the atherosclerotic rabbit iliac model. J. Moll. Cell. Cardiol. 1996;28:441–446. doi: 10.1006/jmcc.1996.0040. [DOI] [PubMed] [Google Scholar]
- Coats WDJ, Faxon DP. The role of the extracellular matrix in arterial remodeling. Semin. Intervent. Cardiol. 1997;2:167–176. [PubMed] [Google Scholar]
- Coats WDJ, Whittaker P, Cheung DT, Currier JW, Han B, Faxon DP. Collagen content is significantly lower in restenotic versus nonrestenotic vessels after balloon angioplasty in the atherosclerotic rabbit model. Circulation. 1997;95:1293–1300. doi: 10.1161/01.cir.95.5.1293. [DOI] [PubMed] [Google Scholar]
- Corseaux D, Le Tourneau T, Six I, et al. Enhanced monocyte tissue factor response after experimental balloon angioplasty in hypercholesterolemic rabbit: inhibition with dietary L-arginine. Circulation. 1998;98:1776–1782. doi: 10.1161/01.cir.98.17.1776. [DOI] [PubMed] [Google Scholar]
- Currier JW, Pow TK, Haudenschild CC, Minihan AC, Faxon DP. Low molecular weight heparin (enoxaparin) reduces restenosis after iliac angioplasty in the hypercholesterolemic rabbit. J. Am. Coll. Cardiol. 1991;17:118B–118B. doi: 10.1016/0735-1097(91)90947-8. [DOI] [PubMed] [Google Scholar]
- Darley-Usmar V, Halliwell B. Blood radicals: reactive nitrogen species, reactive oxygen species, transition metal ions, and the vascular system. Pharmaceut Res. 1996;13:649–662. doi: 10.1023/a:1016079012214. [DOI] [PubMed] [Google Scholar]
- De Leon H, Scott NA, Martin F, Simonet L, Bernstein KE, Wilcox JN. Expression of nonmuscle myosin heavy chain-B isoform in the vessel wall of porcine coronary arteries after balloon angioplasty. Circ. Res. 1997;80:514–519. doi: 10.1161/01.res.80.4.514. [DOI] [PubMed] [Google Scholar]
- Dollery CM, McEwan JR, Wang M, Sang QA, Liu YE, Shi YE. TIMP-4 is regulated by vascular injury in rats. Circ. Res. 1999;84:498–504. doi: 10.1161/01.res.84.5.498. [DOI] [PubMed] [Google Scholar]
- Dong ZM, Wagner DD. Leukocyte-endothelium adhesion molecules in atherosclerosis. J. Lab. Clin. Med. 1998;132:369–375. doi: 10.1016/s0022-2143(98)90107-x. [DOI] [PubMed] [Google Scholar]
- Doornekamp FN, Borst C, Post MJ. The influence of lesion length on intimal hyperplasia after Fogarty balloon injury in the rabbit carotid artery: role of endothelium. J. Vasc. Res. 1997;34:260–266. doi: 10.1159/000159232. [DOI] [PubMed] [Google Scholar]
- Douglas SA, Ohlstein EH. Endothelin-1 promotes neointima formation after balloon angioplasty in the rat. J. Cardiovasc. Pharmacol. 1993;22(Suppl.):S371–S373. doi: 10.1097/00005344-199322008-00097. [DOI] [PubMed] [Google Scholar]
- Douglas SA, Louden C, Vickery-Clark LM, et al. A role for endogenous endothelin-1 in neointimal formation after rat carotid artery balloon angioplasty. Protective effects of the novel nonpeptide endothelin receptor antagonist SB 209670. Circ. Res. 1994;75:190–197. doi: 10.1161/01.res.75.1.190. [DOI] [PubMed] [Google Scholar]
- Duverger N, Viglietta C, Berthou L, et al. Transgenic rabbits expressing human apolipoprotein A-I in the liver. Arterioscler. Thromb. Vasc. Biol. 1996;16:1424–1429. doi: 10.1161/01.atv.16.12.1424. [DOI] [PubMed] [Google Scholar]
- Dzau VJ, Paul M, Nakamura N, Pratt RE, Ingelfinger JR. Role of molecular biology in hypertension research. State of the Art lecture. Hypertension. 1989;13:731–740. doi: 10.1161/01.hyp.13.6.731. [DOI] [PubMed] [Google Scholar]
- Engel L, Ryan U. TGF-beta 1 reverses PDGF-stimulated migration of human aortic smooth muscle cells in vitro. In Vitro Cell. Dev. Biol. 1997;33:443–451. doi: 10.1007/s11626-997-0062-x. [DOI] [PubMed] [Google Scholar]
- Evanko SP, Raines EW, Ross R, Gold LI, Wight TN. Proteoglycan distribution in lesions of atherosclerosis depends on lesion severity, structural characteristics and the proximity of platelet-derived growth factor and transforming growth factor-beta. Am. J. Pathol. 1998;152:533–546. [PMC free article] [PubMed] [Google Scholar]
- Fabunmi RP, Baker AH, Murray EJ, Booth RFG, Newby AC. Divergent regulation by growth factors and cytokines of 95 kDa and 72 kDa gelatinases and tissue inhibitors of metalloproteinases-1-2 and -3 in rabbit aortic smooth muscle cells. Biochem. J. 1996;315:335–342. doi: 10.1042/bj3150335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Faller DV. Endothelial cell responses to hypoxic stress. Clin. Exp. Pharmacol. Physiol. 1999;26:74–84. doi: 10.1046/j.1440-1681.1999.02992.x. [DOI] [PubMed] [Google Scholar]
- Farb A, Lee SJ, Min DH, et al. Vascular smooth muscle cell cytotoxicity and sustained inhibition of neointimal formation by fibroblast growth factor 2-saporin fusion protein. Circ. Res. 1997;80:542–550. doi: 10.1161/01.res.80.4.542. [DOI] [PubMed] [Google Scholar]
- Faxon DP, Weber VJ, Haudenschild C, Gottsman SB, McGovern WA, Ryan TJ. Acute effects of transluminal angioplasty in three experimental models of arterosclerosis. Arteriosclerosis. 1982;2:125–133. doi: 10.1161/01.atv.2.2.125. [DOI] [PubMed] [Google Scholar]
- Faxon DP, Sanborn TA, Haudenschild CC, Ryan TJ. Effect of antiplatelet therapy on restenosis after experimental angioplasty. Am. J. Cardiol. 1984;53:72C–72C. doi: 10.1016/0002-9149(84)90751-3. [DOI] [PubMed] [Google Scholar]
- Feldman LJ, Tahlil O, Steg PG. Adenovirus-mediated arterial gene therapy for restenosis: problems and perspectives. Sem. Intervent. Cardiol. 1996;1:203–208. [PubMed] [Google Scholar]
- Ferns G, Reidy M, Ross R. Vascular effects of cyclosporine A in vivo and in vitro. Am. J. Pathol. 1990;137:403–413. [PMC free article] [PubMed] [Google Scholar]
- Ferns GA, Motani AS, Anggαrd EE. The insulin-like growth factors: their putative role in atherogenesis. Artery. 1991a;18:197–225. [PubMed] [Google Scholar]
- Ferns GA, Raines EW, Sprugel KH, Motani AS, Reidy MA, Ross R. Inhibition of neointimal smooth muscle accumulation after angioplasty by an antibody to PDGF. Science. 1991b;253:1129–1132. doi: 10.1126/science.1653454. [DOI] [PubMed] [Google Scholar]
- Ferns GA, Reidy MA, Ross R. Balloon catheter de-endothelialization of the nude rat carotid. Response to injury in the absence of functional T lymphocytes. Am. J. Pathol. 1991c;138:1045–1057. [PMC free article] [PubMed] [Google Scholar]
- Ferns GA, Forster L, Stewart-Lee A, Konneh M, Nourooz-Zadeh J, Anggαrd EE. Probucol inhibits neointimal thickening and macrophage accumulation after balloon injury in the cholesterol-fed rabbit. Proc. Natl. Acad. Sci. USA. 1992a;89:11312–11316. doi: 10.1073/pnas.89.23.11312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferns GA, Stewart-Lee AL, Anggαrd EE. Arterial response to mechanical injury: balloon catheter de-endothelialization. Atheroscler. 1992b;92:89–104. doi: 10.1016/0021-9150(92)90268-l. [DOI] [PubMed] [Google Scholar]
- Ferns GA, Forster L, Stewart-Lee A, Nourooz-Zadeh J, Anggαrd EE. Probucol inhibits mononuclear cell adhesion to vascular endothelium in the cholesterol-fed rabbit. Atheroscler. 1993a;100:171–181. doi: 10.1016/0021-9150(93)90203-7. [DOI] [PubMed] [Google Scholar]
- Ferns GA, Konneh M, Anggαrd EE. Vitamin E: the evidence for an anti-atherogenic role. Artery. 1993b;20:61–94. [PubMed] [Google Scholar]
- Ferns GA, Konneh M, Rutherford C, Woolaghan E, Anggard EE. Hyaluronan (HYAL-BV 5200) inhibits neo-intimal macrophage influx after balloon-catheter induced injury in the cholesterol-fed rabbit. Atheroscler. 1995;114:157–164. doi: 10.1016/0021-9150(94)05479-3. [DOI] [PubMed] [Google Scholar]
- Fingerle J, Au YPT, Clowes AW, Reidy MA. intimal lesion formation in rat carotid arteries after endothelial denudation in absence on medial injury. Arteriosclerosis. 1990;10:1082–1087. doi: 10.1161/01.atv.10.6.1082. [DOI] [PubMed] [Google Scholar]
- Firulli AB, Miano JM, Bi W, et al. Myocyte enhancer binding factor-2 expression and activity in vascular smooth muscle cells: association with the activated phenotype. Circ. Res. 1996;78:196–204. doi: 10.1161/01.res.78.2.196. [DOI] [PubMed] [Google Scholar]
- Fischell TA, Nellessen U, Johnson DE, Ginsburg R. Endothelium-dependent arterial vasoconstriction after balloon angioplasty. Circulation. 1989;79:899–910. doi: 10.1161/01.cir.79.4.899. [DOI] [PubMed] [Google Scholar]
- Frid MG, Dempsey EC, Durmowicz AG, Stenmark KR. Smooth muscle cell heterogeneity in pulmonary and systemic vessels. Importance in vascular disease. Arterioscler. Thromb. Vasc. Biol. 1997;17:1203–1209. doi: 10.1161/01.atv.17.7.1203. [DOI] [PubMed] [Google Scholar]
- Fuster V, Dyken ML, Vokonas PS, Hennekens C. Aspirin as a therapeutic agent in cardiovascular disease. Special Writing Group. Circulation. 1993;87:659–675. doi: 10.1161/01.cir.87.2.659. [DOI] [PubMed] [Google Scholar]
- Galis ZS, Kranzhofer R, Fenton JW, Libby P. Thrombin promotes activation of matrix metalloproteinase-2 produced by cultured vascular smooth muscle cells. Arterioscler. Thromb. Vasc. Biol. 1997;17:483–489. doi: 10.1161/01.atv.17.3.483. [DOI] [PubMed] [Google Scholar]
- Geary RL, Williams JK, Golden D, Brown DG, Benjamin ME, Adams MR. Time course of cellular proliferation, intimal hyperplasia, and remodeling following angioplasty in monkeys with established atherosclerosis. A nonhuman primate model of restenosis. Arterioscler. Thromb. Vasc. Biol. 1996;16:34–43. doi: 10.1161/01.atv.16.1.34. [DOI] [PubMed] [Google Scholar]
- Geary RL, Nikkari ST, Wagner WD, Williams JK, Adams MR, Dean RH. Wound healing: a paradigm for lumen narrowing after arterial reconstruction. J. Vasc. Surg. 1998;27:96–106. doi: 10.1016/s0741-5214(98)70296-4. [DOI] [PubMed] [Google Scholar]
- Gellman J, Ezekowitz MD, Sarembock IJ, et al. Effect of lovastatin on intimal hyperplasia after balloon angioplasty: a study in an atherosclerotic hypercholesterolemic rabbit. J. Am. Coll. Cardiol. 1991;17:251–259. doi: 10.1016/0735-1097(91)90735-r. [DOI] [PubMed] [Google Scholar]
- George SJ, Zaltsman AB, Newby AC. Surgical preparative injury and neointima formation increase MMP-9 expression and MMP-2 activation in human saphenous vein. Cardiovasc. Res. 1997;33:447–459. doi: 10.1016/s0008-6363(96)00211-8. [DOI] [PubMed] [Google Scholar]
- George SJ, Baker AH, Angelini GD, Newby AC. Gene transfer of tissue inhibitor of metalloproteinase-2 inhibits metalloproteinase activity and neointima formation in human saphenous veins. Gene Ther. 1998;5:1552–1560. doi: 10.1038/sj.gt.3300764. [DOI] [PubMed] [Google Scholar]
- Gertz SD, Gimple LW, Banai S, et al. Geometric remodeling is not the principal pathogenetic process in restenosis after balloon angioplasty. Evidence from correlative angiographic-histomorphometric studies of atherosclerotic arteries in rabbits. Circulation. 1994;90:3001–3008. doi: 10.1161/01.cir.90.6.3001. [DOI] [PubMed] [Google Scholar]
- Gertz SD, Fallon JT, Gallo R, et al. Hirudin reduces tissue factor expression in neointima after balloon injury in rabbit femoral and porcine coronary arteries. Circulation. 1998;98:580–587. doi: 10.1161/01.cir.98.6.580. [DOI] [PubMed] [Google Scholar]
- Giachelli CM, Liaw L, Murry CE, Schwartz SM, Almeida M. Osteopontin expression in cardiovascular diseases. Ann. N.Y. Acad. Sci. 1995;760:109–126. doi: 10.1111/j.1749-6632.1995.tb44624.x. [DOI] [PubMed] [Google Scholar]
- Giddings JC, Banning AP, Ralis H, Lewis MJ. Redistribution of von Willebrand factor in porcine carotid arteries after balloon angioplasty. Arterioscler. Thromb. Vasc. Biol. 1997;17:1872–1878. doi: 10.1161/01.atv.17.10.1872. [DOI] [PubMed] [Google Scholar]
- Giese N, Marijianowski MMH, McCook O, et al. The role of alpha and beta platelet-derived growth factor receptor in the vascular response to injury in nonhuman primates. Arterioscler. Thromb. Vasc. Biol. 1999;19:900–909. doi: 10.1161/01.atv.19.4.900. [DOI] [PubMed] [Google Scholar]
- Glazebrook H, Hatch T, Brindle NP. Regulation of insulin-like growth factor-1 expression in vascular endothelial cells by the inflammatory cytokine interleukin-1. J. Vasc. Res. 1998;35:143–149. doi: 10.1159/000025577. [DOI] [PubMed] [Google Scholar]
- Glazier JJ, Varricchione TR, Ryan TJ, Ruocco NA, Jacobs AK, Faxon DP. Outcome in patients with recurrent restenosis after percutaneous transluminal balloon angioplasty. Br. Heart J. 1989;61:485–488. doi: 10.1136/hrt.61.6.485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Golino P, Ragni M, Cirillo P, et al. Effects of tissue factor induced by oxygen free radicals on coronary flow during reperfusion. Nature Med. 1996;2:35–40. doi: 10.1038/nm0196-35. [DOI] [PubMed] [Google Scholar]
- Gonzalez-Fernandez F, Lopez-Farre A, Rodriguez-Feo JA, et al. Expression of inducible nitric oxide synthase after endothelial denudation of the rat carotid artery: role of platelets. Circ. Res. 1998;83:1080–1087. doi: 10.1161/01.res.83.11.1080. [DOI] [PubMed] [Google Scholar]
- Greenlees C, Wadsworth RM, Martorana PA, Wainwright CL. The effects of L-arginine on neointimal formation and vascular function following balloon injury in heritable hyperlipidaemic rabbits. Cardiovasc. Res. 1997;35:351–359. doi: 10.1016/s0008-6363(97)00122-3. [DOI] [PubMed] [Google Scholar]
- Groves HM, Kinlough-Rathbone RL, Richardson M, Moore S, Mustard JF. Platelet interaction with damaged rabbit aorta. Lab. Invest. 1979;40:194–200. [PubMed] [Google Scholar]
- Groves PH, Lewis MJ, Cheadle HA, Penny WJ. SIN-1 reduces platelet adhesion and platelet thrombus formation in a porcine model of balloon angioplasty. Circulation. 1993;87:590–597. doi: 10.1161/01.cir.87.2.590. [DOI] [PubMed] [Google Scholar]
- Groves PH, Banning AP, Penny WJ, Lewis MJ, Cheadle HA, Newby AC. Kinetics of smooth muscle cell proliferation and intimal thickening in a pig carotid model of balloon injury. Atheroscler. 1995;117:83–96. doi: 10.1016/0021-9150(95)05562-b. [DOI] [PubMed] [Google Scholar]
- Gruentzig AR. Transluminal dilation of coronary artery stenoses. Lancet. 1978;i:263. [Google Scholar]
- Grunwald J, Fingerle J, Hammerle H, Betz E, Haudenschild CC. Cytocontractile structures and proteins of smooth muscle cells during the formation of experimental lesions. Exp. Mol. Pathol. 1987;46:78–88. doi: 10.1016/0014-4800(87)90032-3. [DOI] [PubMed] [Google Scholar]
- Gunn J, Holt CM, Francis SE, et al. The effect of oligonucleotides to c-myb on vascular smooth muscle cell proliferation and neointima formation after porcine coronary angioplasty. Circ. Res. 1997;80:520–531. doi: 10.1161/01.res.80.4.520. [DOI] [PubMed] [Google Scholar]
- Guth BD, Seewaldtbecker E, Himmelsbach F, Weisenberger H, Muller TH. Antagonism of the GPIIb/IIIa receptor with the nonpeptidic molecule BIBU52: Inhibition of platelet aggregation in vitro and antithrombotic efficacy in vivo. J. Cardiovasc. Pharmacol. 1997;30:261–272. doi: 10.1097/00005344-199708000-00017. [DOI] [PubMed] [Google Scholar]
- Guzman LA, Forudi F, Villa AE, Topol EJ. Role of leukocytes in neointimal formation after balloon angioplasty in the rabbit atherosclerotic model. Coron. Art. Dis. 1995;6:693–701. [PubMed] [Google Scholar]
- Hajjar DP, Falcone DJ, Fowler S, Minick CR. Endothelium modifies the altered metabolism of injured aortic wall. Am. J. Pathol. 1981;102:28–39. [PMC free article] [PubMed] [Google Scholar]
- Hamdan AD, Quist WC, Gagne JB, Feener EP. Angiotensin-converting enzyme inhibition suppresses plasminogen activator inhibitor-1 expression in the neointima of balloon-injured rat aorta. Circulation. 1996;93:1073–1078. doi: 10.1161/01.cir.93.6.1073. [DOI] [PubMed] [Google Scholar]
- Hancock WW, Adams DH, Wyner LR, Sayegh MH, Karnovsky MJ. CD4+ mononuclear cells induce cytokine expression, vascular smooth muscle proliferation, and arterial occlusion after endothelial injury. Am. J. Pathol. 1994;145:1008–1014. [PMC free article] [PubMed] [Google Scholar]
- Hansson GK, Jonasson L, Holm J, Clowes MM, Clowes AW. Gamma-interferon regulates vascular smooth muscle proliferation and Ia antigen expression in vivo and in vitro. Circ. Res. 1988;63:712–719. doi: 10.1161/01.res.63.4.712. [DOI] [PubMed] [Google Scholar]
- Hansson GK. Immunological control mechanisms in plaque formation. Basic Res. Cardiol. 1994;89(Suppl.):41–46. doi: 10.1007/978-3-642-85660-0_4. [DOI] [PubMed] [Google Scholar]
- Hatton MW, Moar SL, Richardson M. Behaviour of plasminogen at the luminal surface of normal and de-endothelialized rabbit aorta in vivo and in vitro. Blood. 1988;71:1260–1267. [PubMed] [Google Scholar]
- Haudenschild CC, Grunwald J. Proliferative heterogenecity of vascular smooth muscle cells and it alteration by injury. Exp. Cell. Res. 1985;157:364–370. doi: 10.1016/0014-4827(85)90121-1. [DOI] [PubMed] [Google Scholar]
- Henderson HE, Kastelein JJ, Zwinderman AH, et al. Lipoprotein lipase activity is decreased in a large cohort of patients with coronary artery disease and is associated with changes in lipids and lipoproteins. J. Lipid Research. 1999;40:735–743. [PubMed] [Google Scholar]
- Hu Y, Cheng L, Hochleitner B, Xu Q. Activation of mitogen-activated protein kinases (ERK/JNK) and AP-1 transcription factor in rat carotid arteries after balloon injury. Arterioscler. Thromb. Vasc. Biol. 1997;17:2808–2816. doi: 10.1161/01.atv.17.11.2808. [DOI] [PubMed] [Google Scholar]
- Huber KC, Schwartz RS, Edwards WD, et al. Effects of angiotensin converting enzyme inhibition on neointimal proliferation in a porcine coronary injury model. Am. Heart J. 1993;125:695–701. doi: 10.1016/0002-8703(93)90160-b. [DOI] [PubMed] [Google Scholar]
- Huckle WR, Drag MD, Acker WR, et al. Effects of subtype-selective and balanced angiotensin II receptor antagonists in a porcine coronary artery model of vascular restenosis. Circulation. 1996;93:1009–1019. doi: 10.1161/01.cir.93.5.1009. [DOI] [PubMed] [Google Scholar]
- Hultgardh-Nilsson A, Cercek B, Wang JW, et al. Regulated expression of the ets-1 transcription factor in vascular smooth muscle cells in vivo and in vitro. Circ. Res. 1996;78:589–595. doi: 10.1161/01.res.78.4.589. [DOI] [PubMed] [Google Scholar]
- Indolfi C, Esposito G, Di Lorenzo E, et al. Smooth muscle cell proliferation is proportional to the degree of balloon injury in a rat model of angioplasty. Circulation. 1995;92:1230–1235. doi: 10.1161/01.cir.92.5.1230. [DOI] [PubMed] [Google Scholar]
- Ismail NA, Wasty F, Moore S, Alavi MZ. Injury-induced alterations in newly synthesized sulphated proteoglycans from rabbit arterial neointima covered by regenerated endothelium. Int. J. Exp. Path. 1997;78:71–79. doi: 10.1046/j.1365-2613.1997.d01-242.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iwai N, Izumi M, Inagami T, Kinoshita M. Induction of renin in medial smooth muscle cells by balloon injury. Hypertension. 1997;29:1044–1050. doi: 10.1161/01.hyp.29.4.1044. [DOI] [PubMed] [Google Scholar]
- Iwata A, Masago A, Yamada K. Angiotensin-converting enzyme inhibitor cilazapril suppresses expression of basic fibroblast growth factor messenger ribonucleic acid and protein in endothelial and intimal smooth muscle cells in a vascular injury model of spontaneous hypertensive rats. Neurologia Med.-Chirurgica. 1998;38:257–264. doi: 10.2176/nmc.38.257. [DOI] [PubMed] [Google Scholar]
- Jackson CL, Reidy MA. Basic fibroblast growth factor: its role in the control of smooth muscle cell migration. Am. J. Pathol. 1993;143:1024–1031. [PMC free article] [PubMed] [Google Scholar]
- Jandeleit-Dahm K, Burrell LM, Johnston CI, Koch KM. Elevated vascular angiotensin converting enzyme mediates increased neointima formation after balloon injury in spontaneously hypertensive rats. J. Hypertension. 1997;15:643–650. doi: 10.1097/00004872-199715060-00011. [DOI] [PubMed] [Google Scholar]
- Jang Y, Guzman LA, Lincoff AM, et al. Influence of blockade at specific levels of the coagulation cascade on restenosis in a rabbit atherosclerotic femoral artery injury model. Circulation. 1995;92:3041–3050. doi: 10.1161/01.cir.92.10.3041. [DOI] [PubMed] [Google Scholar]
- Janssens S, Flaherty D, Nong Z, et al. Human endothelial nitric oxide synthase gene transfer inhibits vascular smooth muscle cell proliferation and neointima formation after balloon injury in rats. Circulation. 1998;97:1274–1281. doi: 10.1161/01.cir.97.13.1274. [DOI] [PubMed] [Google Scholar]
- Jawien A, Bowen-Pope DF, Lindner V, Schwartz SM. Platelet-derived growth factor promotes smooth muscle migration and intimal thickening in a rat model of balloon angioplasty. J. Clin. Invest. 1992;89:507–511. doi: 10.1172/JCI115613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jenkins GM, Crow MT, Bilato C, et al. Increased expression of membrane-type matrix metalloproteinase and preferential localization of matrix metalloproteinase-2 to the neointima of balloon-injured rat carotid arteries. Circulation. 1998;97:82–90. doi: 10.1161/01.cir.97.1.82. [DOI] [PubMed] [Google Scholar]
- Jonasson L, Holm J, Hansson GK. Cyclosporin A inhibits smooth muscle proliferation in the vascular response to injury. Proc. Natl. Acad. Sci. USA. 1988;85:2303–2306. doi: 10.1073/pnas.85.7.2303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kakuta T, Currier JW, Haudenschild CC, Ryan TJ, Faxon DP. Differences in compensatory vessel enlargement not intimal formation account for restenosis after angioplasty in the hypercholesterolemic rabbit model. Circulation. 1994;89:2809–2815. doi: 10.1161/01.cir.89.6.2809. [DOI] [PubMed] [Google Scholar]
- Kanamasa K, Ishida N, Kato H, et al. Recombinant tissue plasminogen activator prevents intimal hyperplasia after balloon angioplasty in hypercholesterolemic rabbits. Jap. Circulation J. 1996;60:889–894. doi: 10.1253/jcj.60.889. [DOI] [PubMed] [Google Scholar]
- Karas SP, Gravanis MB, Santoian EC, Robinson KA, Erberg KA, King SB. Coronary intimal proliferation after balloon injury and stenting in swine: an animal model of restenosis. J. Am. Coll. Cardiol. 1992;20:467–474. doi: 10.1016/0735-1097(92)90119-8. [DOI] [PubMed] [Google Scholar]
- Kenagy RD, Hart CE, Stetler-Stevenson WG, Clowes AW. Primate smooth muscle cell migration from aortic explants is mediated by endogenous platelet-derived growth factor and basic fibroblast growth factor acting through matrix metalloproteinases 2 and 9. Circulation. 1997;96:3555–3560. doi: 10.1161/01.cir.96.10.3555. [DOI] [PubMed] [Google Scholar]
- Kim S, Iwao H. Involvement of angiotensin II in cardiovascular and renal injury: effects of an AT1-receptor antagonist on gene expression and the cellular phenotype. J. Hypertension. 1997;15:S3–S7. [PubMed] [Google Scholar]
- Kinlough-Rathbone RL, Packham MA, Mustard JF. Vessel injury, platelet adherence and platelet survival. Arteriosclerosis. 1983;3:529–546. doi: 10.1161/01.atv.3.6.529. [DOI] [PubMed] [Google Scholar]
- Kocher O, Skalli O, Bloom WS, Gabbiani G. Cytoskeleton of rat aortic smooth muscle cells. Normal conditions and experimental intimal thickening. Lab. Invest. 1984;50:645–652. [PubMed] [Google Scholar]
- Kollum M, Kaiser S, Kinscherf R, Metz J, Kuebler W, Hehrlein C. Apoptosis after stent implantation compared with balloon angioplasty in rabbits. Role of macrophages. Arterioscler. Thromb. Vasc. Biol. 1997;17:2383–2388. doi: 10.1161/01.atv.17.11.2383. [DOI] [PubMed] [Google Scholar]
- Konneh MK, Rutherford C, Li SR, Anggαrd EE, Ferns GA. Vitamin E inhibits the intimal response to balloon catheter injury in the carotid artery of the cholesterol-fed rat. Atheroscler. 1995;113:29–39. doi: 10.1016/0021-9150(94)05423-g. [DOI] [PubMed] [Google Scholar]
- Koyama N, Kinsella MG, Wight TN, Hedin U, Clowes AW. Heparan sulfate proteoglycans mediate a potent inhibitory signal for migration of vascular smooth muscle cells. Circ. Res. 1998;83:305–313. doi: 10.1161/01.res.83.3.305. [DOI] [PubMed] [Google Scholar]
- Kranzhofer A, Baker AH, George SJ, Newby AC. Expression of tissue inhibitor of metalloproteinase-1-2, and -3 during neointima formation in organ cultures of human saphenous vein. Arterioscler. Thromb. Vasc. Biol. 1999;19:255–265. doi: 10.1161/01.atv.19.2.255. [DOI] [PubMed] [Google Scholar]
- Krasinski K, Spyridopoulos I, Asahara T, Van Der Zee R, Isner JM, Losordo DW. Estradiol accelerates functional endothelial recovery after arterial injury. Circulation. 1997;95:1768–1772. doi: 10.1161/01.cir.95.7.1768. [DOI] [PubMed] [Google Scholar]
- Kumar A, Lindner V. Remodeling with neointima formation in the mouse carotid artery after cessation of blood flow. Arterioscler. Thromb. Vasc. Biol. 1997;17:2238–2244. doi: 10.1161/01.atv.17.10.2238. [DOI] [PubMed] [Google Scholar]
- La Veau PJ, Sarembock IJ, Sigal SL, Yang TL, Ezekowitz MD. Vascular reactivity after balloon angioplasty in an atherosclerotic rabbit. Circulation. 1990;82:1790–1801. doi: 10.1161/01.cir.82.5.1790. [DOI] [PubMed] [Google Scholar]
- Lafont A, Guzman LA, Whitlow PL, Goormastic M, Cornhill JF, Chisolm GM. Restenosis after experimental angioplasty. Intimal medial, and adventitial changes associated with constrictive remodeling. Circ. Res. 1995;76:996–1002. doi: 10.1161/01.res.76.6.996. [DOI] [PubMed] [Google Scholar]
- Lai K, Wang H, Lee WS, Jain MK, Lee ME, Haber E. Mitogen-activated protein kinase phosphatase-1 in rat arterial smooth muscle cell proliferation. J. Clin. Invest. 1996;98:1560–1567. doi: 10.1172/JCI118949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lam J, Chesebro JH, Steele PM, Badimon L, Fuster V. Is vasospasm related to platelet deposition? Relationship in a porcine preparation of arterial injury in vivo. Circulation. 1987;75:243–248. doi: 10.1161/01.cir.75.1.243. [DOI] [PubMed] [Google Scholar]
- Lam J, Badimon JJ, Ellefson RD, Fuster V, Chesebro JH. Cod liver oil alters platelet-arterial wall response to injury in pigs. Circ. Res. 1992;71:769–775. doi: 10.1161/01.res.71.4.769. [DOI] [PubMed] [Google Scholar]
- Lamawansa MD, Wysocki SJ, House AK, Norman PE. Morphometric changes seen in balloon-injured porcine iliac arteries: the influence of sympathectomy on intimal hyperplasia and remodeling. Eur. J. Vascul. Endovascul. Surg. 1997;13:43–47. doi: 10.1016/s1078-5884(97)80049-4. [DOI] [PubMed] [Google Scholar]
- Landry DB, Couper LL, Bryant SR, Lindner V. Activation of the NF-kappa B and I kappa B system in smooth muscle cells after rat arterial injury. Induction of vascular cell adhesion molecule-1 and monocyte chemoattractant protein-1. Am. J. Pathol. 1997;151:1085–1095. [PMC free article] [PubMed] [Google Scholar]
- Law MM, Colburn MD, Hajjar KA, Gelabert HA, Quinones-Baldrich W, Moore WS. Suppression of intimal hyperplasia in a rabbit model of arterial balloon injury by enalaprilat but not dimethyl sulfoxide. Ann. Vasc. Surg. 1994;8:158–165. doi: 10.1007/BF02018864. [DOI] [PubMed] [Google Scholar]
- Lawrence M, Watson L, Pettijohn T, Doyle T, Tasca SI, Stoica G. Immunohistochemical localization of proliferating cells basic fibroblast growth factor and Fos protein following iliac artery balloon angioplasty in hypercholesterolemic rabbits. In Vivo. 1995;9:27–34. [PubMed] [Google Scholar]
- Le Tourneau T, Van Belle E, Corseaux D, et al. Role of nitric oxide in restenosis after experimental balloon angioplasty in the hypercholesterolemic rabbit: effects on neointimal hyperplasia and vascular remodeling. J. Am. Coll. Cardiol. 1999;33:876–882. doi: 10.1016/s0735-1097(98)00621-4. [DOI] [PubMed] [Google Scholar]
- Leppanen P, Luoma JS, Hofker MH, Havekes LM, Yla-Herttuala S. Characterization of atherosclerotic lesions in apo E3-leiden transgenic mice. Atheroscler. 1998;136:147–152. doi: 10.1016/s0021-9150(97)00196-2. [DOI] [PubMed] [Google Scholar]
- Liao L, Starzyk RM, Granger DN. Molecular determinants of oxidized low-density lipoprotein-induced leukocyte adhesion and microvascular dysfunction. Arterioscler. Thromb. Vasc. Biol. 1997;17:437–444. doi: 10.1161/01.atv.17.3.437. [DOI] [PubMed] [Google Scholar]
- Liao X, Escobedo JA, Williams LT. Viability of transgenic mice expressing a platelet derived growth factor (PDGF) antagonist in plasma. J. Invest. Med. 1996;44:139–143. [PubMed] [Google Scholar]
- Lincoff AM, Tcheng JE, Califf RM, et al. Sustained suppression of ischemic complications of coronary intervention by platelet GP IIb/IIIa blockade with abciximab: one-year outcome in the EPILOG trial. Evaluation in PTCA to Improve Long-term Outcome with abciximab GP IIb/IIIa blockade. Circulation. 1999;99:1951–1958. doi: 10.1161/01.cir.99.15.1951. [DOI] [PubMed] [Google Scholar]
- Lindner V, Reidy MA. Proliferation of smooth muscle cells after vascular injury is inhibited by an antibody against basic fibroblast growth factor. Proc. Natl. Acad. Sci. USA. 1991;88:3739–3743. doi: 10.1073/pnas.88.9.3739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lindner V, Olson NE, Clowes AW, Reidy MA. Inhibition of smooth muscle cell proliferation in injured rat arteries. Interaction of heparin with basic fibroblast growth factor. J. Clin. Invest. 1992;90:2044–2049. doi: 10.1172/JCI116085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lindner V, Fingerle J, Reidy MA. Mouse model of arterial injury. Circ. Res. 1993;73:792–796. doi: 10.1161/01.res.73.5.792. [DOI] [PubMed] [Google Scholar]
- Lindner V, Giachelli CM, Schwartz SM, Reidy MA. A subpopulation of smooth muscle cells in injured rat arteries expresses platelet-derived growth factor-B chain mRNA. Circ. Res. 1995;76:951–957. doi: 10.1161/01.res.76.6.951. [DOI] [PubMed] [Google Scholar]
- Liu MW, Roubin GS, Robinson KA, et al. Trapidil in preventing restenosis after balloon angioplasty in the atherosclerotic rabbit. Circulation. 1990;81:1089–1093. doi: 10.1161/01.cir.81.3.1089. [DOI] [PubMed] [Google Scholar]
- Luoma PV. Gene activation, apolipoprotein A-I/high density lipoprotein, atherosclerosis prevention and longevity. Pharmacol. Toxicol. 1997;81:57–64. doi: 10.1111/j.1600-0773.1997.tb00032.x. [DOI] [PubMed] [Google Scholar]
- Maillard L, Walsh K. Growth-arrest homeobox gene Gax: a molecular strategy to prevent arterial restenosis. Schweizer. Med. Wochensch. J. Suisse Med. 1996;126:1721–1726. [PubMed] [Google Scholar]
- Maillard L, Van Belle E, Smith RC, et al. Percutaneous delivery of the gax gene inhibits vessel stenosis in a rabbit model of balloon angioplasty. Cardiovasc. Res. 1997;35:536–546. doi: 10.1016/s0008-6363(97)00147-8. [DOI] [PubMed] [Google Scholar]
- Majesky MW, Reidy MA, Bowen-Pope DF, Hart CE, Wilcox JN, Schwartz SM. PDGF ligand and receptor gene expression during repair of arterial injury. J. Cell Biol. 1990;111:2149–2158. doi: 10.1083/jcb.111.5.2149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Majesky MW, Lindner V, Twardzik DR, Schwartz SM, Reidy MA. Production of transforming growth factor beta 1 during repair of arterial injury. J. Clin. Invest. 1991;88:904–910. doi: 10.1172/JCI115393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manderson JA, Mosse PRL, Safstrom JA, Young SB, Cambell GR. Balloon catheter injury to rabbit carotid artery. I.Changes in smooth muscle cell phenotype. Arteriosclerosis. 1989;9:289–298. doi: 10.1161/01.atv.9.3.289. [DOI] [PubMed] [Google Scholar]
- Mattsson E, Brunkwall J, Falt K, Bergqvist D. Vessel repair after balloon angioplasty: morphological appearance and prostacyclin synthesising capacity. Eur. J. Vasc. Surg. 1992;6:585–592. doi: 10.1016/s0950-821x(05)80832-1. [DOI] [PubMed] [Google Scholar]
- Mattsson E, Jansen I, Edvinsson L, Bergqvist D. Endothelin does not contribute to the vasospasm after balloon angioplasty in vivo. J. Surg. Res. 1993a;55:298–303. doi: 10.1006/jsre.1993.1144. [DOI] [PubMed] [Google Scholar]
- Mattsson E, Brunkwall J, Falt K, Bergqvist D. Prostanoid release immediately after balloon angioplasty in three models of atheroscelrosis in rabbits. Eur. J. Vasc. Surg. 1993b;159:15–21. [PubMed] [Google Scholar]
- McCormick ML, Gaut JP, Lin TS, Britigan BE, Buettner GR, Heinecke JW. Electron paramagnetic resonance detection of free tyrosyl radical generated by myeloperoxidase lactoperoxidase and horseradish peroxidase. J. Biol. Chem. 1998;273:32030–32037. doi: 10.1074/jbc.273.48.32030. [DOI] [PubMed] [Google Scholar]
- McNamara DB, Bedi B, Aurora H, et al. L-arginine inhibits balloon catheter-induced intimal hyperplasia. Biochem. Biophys. Res. Commun. 1993;193:291–296. doi: 10.1006/bbrc.1993.1622. [DOI] [PubMed] [Google Scholar]
- Meier B. Prevention of restenosis after coronary angioplasty: a pharmacological approach. Eur. Heart J. 1989;10(Suppl.):64–68. doi: 10.1093/eurheartj/10.suppl_g.64. [DOI] [PubMed] [Google Scholar]
- Merhi Y, Lacoste LL, Lam JY. Neutrophil implications in platelet deposition and vasoconstriction after deep arterial injury by angioplasty in pigs. Circulation. 1994;90:997–1002. doi: 10.1161/01.cir.90.2.997. [DOI] [PubMed] [Google Scholar]
- Miles LA, Fless GM, Levin EG, Scanu AM, Plow EF. A potential basis for the thrombotic risks associated with lipoprotein (a) Nature. 1989;339:301–303. doi: 10.1038/339301a0. [DOI] [PubMed] [Google Scholar]
- Minamino T, Kurihara H, Takahashi M, et al. Endothelin-converting enzyme expression in the rat vascular injury model and human coronary atherosclerosis. Circulation. 1997;95:221–230. doi: 10.1161/01.cir.95.1.221. [DOI] [PubMed] [Google Scholar]
- Miyauchi K, Kawai S, Okada R, Yamaguchi H. Limitations of angiotensin-converting enzyme inhibitor in restenosis of a deep arterial injury model. Jap. Circulation J. 1998;62:53–60. doi: 10.1253/jcj.62.53. [DOI] [PubMed] [Google Scholar]
- Miyazawa N, Watanabe S, Matsuda A, et al. Role of histamine H1 and H2 receptor antagonists in the prevention of intimal thickening. Eur. J. Pharmacol. 1998;362:53–59. doi: 10.1016/s0014-2999(98)00716-x. [DOI] [PubMed] [Google Scholar]
- More RS, Rutty G, Underwood MJ, Gershlick AH. Assessment of myointimal cellular kinetics in a model of angioplasty by means of proliferating cell nuclear antigen expression. Am. Heart J. 1994;128:681–686. doi: 10.1016/0002-8703(94)90264-x. [DOI] [PubMed] [Google Scholar]
- More RS, Underwood MJ, Brack MJ, De Bono DP, Gershlick AH. Changes in vessel wall plasminogen activator activity and smooth muscle cell proliferation and activation after arterial injury. Cardiovasc. Res. 1995;29:22–26. [PubMed] [Google Scholar]
- Morishita R, Gibbons GH, Kaneda Y, Ogihara T, Dzau VJ. Pharmacokinetics of antisense oligodeoxyribonucleotides (cyclin B1 and CDC 2 kinase) in the vessel wall in vivo: enhanced therapeutic utility for restenosis by HVJ-liposome delivery. Gene. 1994;149:13–19. doi: 10.1016/0378-1119(94)90406-5. [DOI] [PubMed] [Google Scholar]
- Motani A, Rutherford C, Anggard EE, Ferns GA. Insulin-like growth factor binding protein-1 inhibits arterial smooth muscle cell proliferation in vitro but does not reduce the neointimal response to balloon catheter injury. Atheroscler. 1995;118:57–66. doi: 10.1016/0021-9150(95)05593-l. [DOI] [PubMed] [Google Scholar]
- Muller DW. The role of proto-oncogenes in coronary restenosis. Prog. Cardiovasc. Dis. 1997;40:117–128. doi: 10.1016/s0033-0620(97)80004-7. [DOI] [PubMed] [Google Scholar]
- Nakamura Y, Nakamura K, Matsukura T. Vascular angiotensin converting enzyme activity in spontaneously hypertensive rats and its inhibition with cilazapril. J. Hypertension. 1988;6:105–110. [PubMed] [Google Scholar]
- Nathan CF. Secretory products of macrophages. J. Clin. Invest. 1987;79:319–326. doi: 10.1172/JCI112815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neschis DG, Safford SD, Hanna AK, Fox JC, Golden MA. Antisense basic fibroblast growth factor gene transfer reduces early intimal thickening in a rabbit femoral artery balloon injury model. J. Vasc. Surg. 1998;27:126–134. doi: 10.1016/s0741-5214(98)70299-x. [DOI] [PubMed] [Google Scholar]
- Nielsen LB, Stender S, Kjeldsen K, Nordestgaard BG. Specific accumulation of lipoprotein (a) in balloon-injured rabbit aorta in vivo. Circ. Res. 1996;78:615–626. doi: 10.1161/01.res.78.4.615. [DOI] [PubMed] [Google Scholar]
- Nielsen LB, Juul K, Nordestgaard BG. Increased degradation of lipoprotein (a) in atherosclerotic compared with nonlesioned aortic intima-inner media of rabbits: in vivo evidence that lipoprotein (a) may contribute to foam cell formation. Arterioscler. Thromb. Vasc. Biol. 1998;18:641–649. doi: 10.1161/01.atv.18.4.641. [DOI] [PubMed] [Google Scholar]
- Nishida K, Abiko T, Ishihara M, Tomikawa M. Arterial injury induced smooth muscle cell proliferation in rats is accompanied by increase in polyamine synthesis and level. Atheroscler. 1990;83:119–125. doi: 10.1016/0021-9150(90)90157-e. [DOI] [PubMed] [Google Scholar]
- Nobuyoshi M, Tanaka M, Fujii M, Asanuma A, Takimoto M. Thrombus formation on the aorta injured by angioplasty and its prevention with dilazep in atherosclerotic rabbits. Thromb. Res. 1989;54:561–572. doi: 10.1016/0049-3848(89)90122-9. [DOI] [PubMed] [Google Scholar]
- Oberhoff M, Novak S, Herdeg C, et al. Local and systemic delivery of low molecular weight heparin stimulates the re-endothelialization after balloon angioplasty. Cardiovasc. Res. 1998;38:751–762. doi: 10.1016/s0008-6363(98)00049-2. [DOI] [PubMed] [Google Scholar]
- Oparil S, Levine RL, Chen SJ, Durand J, Chen YF. Sexually dimorphic response of the balloon-injured rat carotid artery to hormone treatment. Circulation. 1997;95:1301–1307. doi: 10.1161/01.cir.95.5.1301. [DOI] [PubMed] [Google Scholar]
- Panek RL, Dahring TK, Olszewski BJ, Keiser JA. PDGF receptor protein tyrosine kinase expression in the balloon-injured rat carotid artery. Arterioscler. Thromb. Vasc. Biol. 1997;17:1283–1288. doi: 10.1161/01.atv.17.7.1283. [DOI] [PubMed] [Google Scholar]
- Parlavecchia M, Skalli O, Gabbiani G. LDL accumulation in cultured rat aortic smooth muscle cells with different cytoskeletal phenotypes. J. Vasc. Med. Biol. 1989;1:308. [Google Scholar]
- Pasterkamp G, Mali WP, Borst C. Application of intravascular ultrasound in remodeling studies. Sem. Intervent. Cardiol. 1997;2:11–18. [PubMed] [Google Scholar]
- Perlman H, Maillard L, Krasinski K, Walsh K. Evidence for the rapid onset of apoptosis in medial smooth muscle cells after balloon injury. Circulation. 1997;95:981–987. doi: 10.1161/01.cir.95.4.981. [DOI] [PubMed] [Google Scholar]
- Pocock SJ, Henderson HE, Rickards A, et al. Meta-analysis of randomised trials comparing coronary angioplasty with bypass surgery. Lancet. 1995;346:1184–1189. doi: 10.1016/s0140-6736(95)92897-9. [DOI] [PubMed] [Google Scholar]
- Pocock SJ, Henderson RA, Treasure T, Hampton JR. Quality of life employment status, and anginal symptoms after coronary angioplasty or bypass surgery. 3-year follow-up in the Randomized Intervention Treatment of Angina (RITA) Trial. Circulation. 1996;94:135–142. doi: 10.1161/01.cir.94.2.135. [DOI] [PubMed] [Google Scholar]
- Pollman MJ, Hall JL, Gibbons GH. Determinants of vascular smooth muscle cell apoptosis after balloon angioplasty injury. Influence of redox state and cell phenotype. Circ. Res. 1999;84:113–121. doi: 10.1161/01.res.84.1.113. [DOI] [PubMed] [Google Scholar]
- Post MJ, Borst C, Kuntz RE. The relative importance of arterial remodeling compared with intimal hyperplasia in lumen renarrowing after balloon angioplasty. A study in the normal rabbit and the hypercholesterolemic Yucatan micropig. Circulation. 1994;89:2816–2821. doi: 10.1161/01.cir.89.6.2816. [DOI] [PubMed] [Google Scholar]
- Post MJ, Borst C, Pasterkamp G, Haudenschild, Christian C. Arterial remodeling in atherosclerosis and restenosis: a vague concept of a distinct phenomenon. Atheroscler. 1995;118:S115–S123. [PubMed] [Google Scholar]
- Powell JS, Muller RK, Baumgartner HR. Suppression of the vascular response to injury: the role of angiotensin-converting enzyme inhibitors. J. Am. Coll. Cardiol. 1991;17:137B–137B. doi: 10.1016/0735-1097(91)90950-e. [DOI] [PubMed] [Google Scholar]
- Provost P, Tremblay J, Merhi Y. The antiadhesive and antithrombotic effects of the nitric oxide donor SIN-1 are combined with a decreased vasoconstriction in a porcine model of balloon angioplasty. Arterioscler. Thromb. Vasc. Biol. 1997;17:1806–1812. doi: 10.1161/01.atv.17.9.1806. [DOI] [PubMed] [Google Scholar]
- Rade JJ, Schulick AH, Virmani R, Dichek DA. Local adenoviral-mediated expression of recombinant hirudin reduces neointima formation after arterial injury. Nature Med. 1996;2:293–298. doi: 10.1038/nm0396-293. [DOI] [PubMed] [Google Scholar]
- Ragosta M, Barry WL, Gimple LW, et al. Effect of thrombin inhibition with desulfatohirudin on early kinetics of cellular proliferation after balloon angioplasty in atherosclerotic rabbits. Circulation. 1996;93:1194–1200. doi: 10.1161/01.cir.93.6.1194. [DOI] [PubMed] [Google Scholar]
- Raines EW, Dower SK, Ross R. Interleukin-1 mitogenic activity for fibroblasts and smooth muscle cells is due to PDGF-AA. Science. 1989;243:393–396. doi: 10.1126/science.2783498. [DOI] [PubMed] [Google Scholar]
- Reidy MA. Biology of disease. A reassessment of endothelial injury and arterial lesion formation. Lab. Invest. 1985;53:513–520. [PubMed] [Google Scholar]
- Reidy MA, Silver M. Endothelail regeneration VII. Lack of intimal proliferation after defined injury in the rat aorta. Am. J. Pathol. 1985;118:173–177. [PMC free article] [PubMed] [Google Scholar]
- Reidy MA, Clowes AW, Schwartz SM. Endothelial regeneration. V. Inhibition of endothelial regrowth in arteries of rat and rabbit. Lab. Invest. 1983;49:569–575. [PubMed] [Google Scholar]
- Reidy MA, Irvin C, Lindner V. Migration of arterial wall cells. Expression of plasminogen activators and inhibitors in injured rat arteries. Circ. Res. 1996;78:405–414. doi: 10.1161/01.res.78.3.405. [DOI] [PubMed] [Google Scholar]
- Reilly MP, Delanty N, Roy L, et al. Increased formation of the isoprostanes IPF2alpha-I and 8-epi-prostaglandin F2alpha in acute coronary angioplasty: evidence for oxidant stress during coronary reperfusion in humans. Circulation. 1997;96:3314–3320. doi: 10.1161/01.cir.96.10.3314. [DOI] [PubMed] [Google Scholar]
- Richardson M, Kinlough-Rathbone RL, Groves HM, Jorgensen L, Mustard JF, Moore S. Ultrastructural changes in re-endothelialized and non-endothelialized rabbit aortic neo-intima following re-injury with a balloon catheter. Br. J. Exp. Pathol. 1984;65:597–611. [PMC free article] [PubMed] [Google Scholar]
- Rogers C, Edelman ER, Simon DI. A mAb to the beta2-leukocyte integrin Mac-1 (CD11b/CD18) reduces intimal thickening after angioplasty or stent implantation in rabbits. Proc. Natl. Acad. Sci. USA. 1998;95:10134–10139. doi: 10.1073/pnas.95.17.10134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenfeld ME, Ross R. Macrophage and smooth muscle cell proliferation in atherosclerotic lesions of WHHL and comparably hypercholesterolemic fat-fed rabbits. Arteriosclerosis. 1990;10:680–687. doi: 10.1161/01.atv.10.5.680. [DOI] [PubMed] [Google Scholar]
- Ross R, Glomset JA. The pathogenesis of atherosclerosis. N. Engl. J. Med. 1976;295:369–377. doi: 10.1056/NEJM197608122950707. [DOI] [PubMed] [Google Scholar]
- Rutherford C, Martin W, Salame M, Carrier M, Anggard E, Ferns G. Substantial inhibition of neo-intimal response to balloon injury in the rat carotid artery using a combination of antibodies to platelet-derived growth factor-BB and basic fibroblast growth factor. Atheroscler. 1997;130:45–51. doi: 10.1016/s0021-9150(96)06042-x. [DOI] [PubMed] [Google Scholar]
- Ryan MJ, Emig LL, Hicks GW, et al. Localization of lipoprotein (a) in a monkey model of rapid neointimal growth. Arterioscler. Thromb. Vasc. Biol. 1997;17:181–187. doi: 10.1161/01.atv.17.1.181. [DOI] [PubMed] [Google Scholar]
- Ryan MJ, Emig LL, Hicks GW, et al. Influence of lipoprotein (a) plasma concentration on neointimal growth in a monkey model of vascular injury. Atheroscler. 1998;139:137–145. doi: 10.1016/s0021-9150(98)00072-0. [DOI] [PubMed] [Google Scholar]
- Sarkar R, Webb RC. Does nitric oxide regulate smooth muscle cell proliferation? A critical appraisal. J. Vasc. Res. 1998;35:135–142. doi: 10.1159/000025576. [DOI] [PubMed] [Google Scholar]
- Sata M, Perlman H, Muruve DA, et al. Fas ligand gene transfer to the vessel wall inhibits neointima formation and overrides the adenovirus-mediated T cell response. Proc. Natl. Acad. Sci. USA. 1998;95:1213–1217. doi: 10.1073/pnas.95.3.1213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwartz RS, Murphy JG, Edwards WD, Camrud AR, Vliestra RE, Holmes DR. Restenosis after balloon angioplasty. A practical proliferative model in porcine coronary arteries. Circulation. 1990;82:2190–2200. doi: 10.1161/01.cir.82.6.2190. [DOI] [PubMed] [Google Scholar]
- Schwarzacher SP, Lim TT, Wang B, et al. Local intramural delivery of L-arginine enhances nitric oxide generation and inhibits lesion formation after balloon angioplasty. Circulation. 1997;95:1863–1869. doi: 10.1161/01.cir.95.7.1863. [DOI] [PubMed] [Google Scholar]
- Schwenke DC, Zilversmit DB. Enhanced accumulation and turnover of esterified cholestrol in injured rabbit aorta. Arteriosclerosis. 1987;7:367–377. doi: 10.1161/01.atv.7.4.367. [DOI] [PubMed] [Google Scholar]
- Seddon AM, Woolf N, La Ville A, et al. Hereditary hyperlipidemia and atherosclerosis in the rabbit due to overproduction of lipoproteins. II. Preliminary report of arterial pathology. Arterioscler. 1987;7:113–124. doi: 10.1161/01.atv.7.2.113. [DOI] [PubMed] [Google Scholar]
- Seki J, Nishio M, Kato Y, Motoyama Y, Yoshida K. FK409, a new nitric-oxide donor, suppresses smooth muscle proliferation in the rat model of balloon angioplasty. Atheroscler. 1995;117:97–106. doi: 10.1016/0021-9150(95)05563-c. [DOI] [PubMed] [Google Scholar]
- Shanahan CM, Cary NR, Osbourn JK, Weissberg PL. Identification of osteoglycin as a component of the vascular matrix. Differential expression by vascular smooth muscle cells during neointima formation and in atherosclerotic plaques. Arterioscler. Thromb. Vasc. Biol. 1997;17:2437–2447. doi: 10.1161/01.atv.17.11.2437. [DOI] [PubMed] [Google Scholar]
- Shanley CJ, Gharaee-Kermani M, Sarkar R, et al. Transforming growth factor-beta 1 increases lysyl oxidase enzyme activity and mRNA in rat aortic smooth muscle cells. J. Vasc. Surg. 1997;25:446–452. doi: 10.1016/s0741-5214(97)70254-4. [DOI] [PubMed] [Google Scholar]
- Shi Y, O'brien JE, Fard A, Zalewski A. Transforming growth factor-beta-1 expression and myofibroblast formation during arterial repair. Arterioscler. Thromb. Vasc. Biol. 1996;16:1298–1305. doi: 10.1161/01.atv.16.10.1298. [DOI] [PubMed] [Google Scholar]
- Shi Y, O'brien JEJ, Ala-Kokko L, Chung W, Mannion JD, Zalewski A. Origin of extracellular matrix synthesis during coronary repair. Circulation. 1997;95:997–1006. doi: 10.1161/01.cir.95.4.997. [DOI] [PubMed] [Google Scholar]
- Sibinga NE, Foster LC, Hsieh CM, et al. Collagen VIII is expressed by vascular smooth muscle cells in response to vascular injury. Circ. Res. 1997;80:532–541. doi: 10.1161/01.res.80.4.532. [DOI] [PubMed] [Google Scholar]
- Smith RC, Wills KN, Antelman D, et al. Adenoviral constructs encoding phosphorylation-competent full-length and truncated forms of the human retinoblastoma protein inhibit myocyte proliferation and neointima formation. Circulation. 1997;96:1899–1905. doi: 10.1161/01.cir.96.6.1899. [DOI] [PubMed] [Google Scholar]
- Snow AD, Bolender RP, Wight TN, Clowes AW. Heparin modulates the composition of the extracellular matrix domain surrounding arterial smooth muscle cells. Am. J. Pathol. 1990;137:313–330. [PMC free article] [PubMed] [Google Scholar]
- Softeland E, Framstad T, Thorsen T, Holmsen H. Porcine platelets in vitro and in vivo studies: relevance to human thrombosis research. Eur. J. Haematol. 1992;49:161–173. doi: 10.1111/j.1600-0609.1992.tb00043.x. [DOI] [PubMed] [Google Scholar]
- Srivatsa SS, Fitzpatrick LA, Tsao PW, et al. Selective alpha v beta3 integrin blockade potently limits neointimal hyperplasia and lumen stenosis following deep coronary arterial stent injury: Evidence for the functional importance of integrin αVβ3 and osteopontin expression during neointima formation. Cardiovasc. Res. 1997;36:408–428. doi: 10.1016/s0008-6363(97)00184-3. [DOI] [PubMed] [Google Scholar]
- Stadius ML, Gown AM, Kernoff R, Collins C. Cell proliferation after balloon injury of iliac arteries in the cholesterol-fed New Zealand White rabbit. Arterioscler. Thromb. 1994;14:727–733. doi: 10.1161/01.atv.14.5.727. [DOI] [PubMed] [Google Scholar]
- Stadius ML, Gown AM, Kernoff R, Schwartz SM. Does sequential balloon injury of an artery lead to a different outcome than a single injury? An experimental study of angioplasty. Coron. Art. Dis. 1996;7:247–255. [PubMed] [Google Scholar]
- Steele PM, Chesebro JH, Stanson AW, et al. Balloon angioplasty. Natural history of the pathophysiological response to injury in a pig model. Circ. Res. 1985;57:105–112. doi: 10.1161/01.res.57.1.105. [DOI] [PubMed] [Google Scholar]
- Stemerman MB, Spaet TH, Pitlick F, Cintron J, Lejnieks I, Tiell ML. Intimal healing. the pattern of re-endothelialization and intimal thickening. Am. J.Pathol. 1977;87:125–142. [PMC free article] [PubMed] [Google Scholar]
- Stemerman MB. Effects of moderate hypercholesterolaemia on rabbit endothelium. Arteriosclerosis. 1981;1:25–32. doi: 10.1161/01.atv.1.1.25. [DOI] [PubMed] [Google Scholar]
- Strauss BH, Chisholm RJ, Keeley FW, Gotlieb AI, Logan RA, Armstrong PW. Extracellular matrix remodeling after balloon angioplasty injury in a rabbit model of restenosis. Circ. Res. 1994;75:650–658. doi: 10.1161/01.res.75.4.650. [DOI] [PubMed] [Google Scholar]
- Strauss BH, Robinson R, Batchelor WB, et al. In vivo collagen turnover following experimental balloon angioplasty injury and the role of matrix metalloproteinases. Circ. Res. 1996;79:541–550. doi: 10.1161/01.res.79.3.541. [DOI] [PubMed] [Google Scholar]
- Sun YP, Zhu BQ, Sievers RE, Isenberg WM, Parmley WW. Aspirin inhibits platelet activity but does not attenuate experimental atherosclerosis. Am. Heart J. 1993;125:79–86. doi: 10.1016/0002-8703(93)90059-i. [DOI] [PubMed] [Google Scholar]
- Takagi M, Ueda M, Becker AE, Takeuchi K, Takeda T. The Watanabe heritable hyperlipidemic rabbit is a suitable experimental model to study differences in tissue response between intimal and medical injury after balloon angioplasty. Arterioscler. Thromb. Vasc. Biol. 1997;17:3611–3619. doi: 10.1161/01.atv.17.12.3611. [DOI] [PubMed] [Google Scholar]
- Tanaka H, Sukhova GK, Swanson SJ, et al. Sustained activation of vascular cells and leukocytes in the rabbit aorta after balloon injury. Circulation. 1993;88:1788–1803. doi: 10.1161/01.cir.88.4.1788. [DOI] [PubMed] [Google Scholar]
- Tanaka H, Sukhova G, Schwartz D, Libby P. Proliferating arterial smooth muscle cells after balloon injury express TNF-alpha but not interleukin-1 or basic fibroblast growth factor. Arterioscler. Thromb. Vasc. Biol. 1996;16:12–18. doi: 10.1161/01.atv.16.1.12. [DOI] [PubMed] [Google Scholar]
- Tardif JC, Coto G, Lesperance J, et al. Probucol and multivitamins in the prevention of restenosis after coronary angioplasty. Multivitamins and Probucol Study Group. N. Engl. J. Med. 1997;337:365–372. doi: 10.1056/NEJM199708073370601. [DOI] [PubMed] [Google Scholar]
- Tashiro J, Takahashi K, Yokote K, et al. Administration of a small amount of lard enhances intimal thickening in the balloon catheter injury model without affecting serum lipids. Scand. J. Clin. Lab. Invest. 1998;58:149–154. doi: 10.1080/00365519850186733. [DOI] [PubMed] [Google Scholar]
- Thorlacius H, Lindbom L, Raud J. Cytokine-induced leukocyte rolling in mouse cremaster muscle arterioles in P-selectin dependent. Am. J. Physiol. 1997;272:H1725–H1729. doi: 10.1152/ajpheart.1997.272.4.H1725. [DOI] [PubMed] [Google Scholar]
- Thyberg J, Fredholm BB. Induction of ornithine decarboxylase activity and putrescine synthesis in arterial smooth muscle cells stimulated with platelet derived growth factor. Exp. Cell. Res. 1987;170:160–169. doi: 10.1016/0014-4827(87)90125-x. [DOI] [PubMed] [Google Scholar]
- Tsujino M, Hirata Y, Eguchi S, Watanabe T, Chatani F, Marumo F. Nonselective ETA/ETB receptor anatgonist blocks proliferation of rat vascular smooth muscle cells after balloon angioplasty. Life Sci. 1995;56:PL449–PL454. doi: 10.1016/0024-3205(95)00219-v. [DOI] [PubMed] [Google Scholar]
- Uchida K, Sasahara M, Morigami N, Hazama F, Kinoshita M. Expression of platelet-derived growth factor B-chain in neointimal smooth muscle cells of balloon injured rabbit femoral arteries. Atheroscler. 1996;124:9–23. doi: 10.1016/0021-9150(95)05742-0. [DOI] [PubMed] [Google Scholar]
- Ueno H, Yamamoto H, Ito S, Li JJ, Takeshita A. Adenovirus-mediated transfer of a dominant-negative H-ras suppresses neointimal formation in balloon-injured arteries in vivo. Arterioscler. Thromb. Vasc. Biol. 1997;17:898–904. doi: 10.1161/01.atv.17.5.898. [DOI] [PubMed] [Google Scholar]
- Van Belle E, Bauters C, Wernert N, et al. Neointimal thickening after balloon denudation is enhanced by aldosterone and inhibited by spironolactone and aldosterone antagonist. Cardiovasc. Res. 1995;29:27–32. [PubMed] [Google Scholar]
- Van DV, Carpenter KL, Taylor SE, McDonald JA, Mitchinson MJ. Factors affecting events during oxidation of low density lipoprotein: correlation of multiple parameters of oxidation. Free Rad. Res. 1997;27:459–476. doi: 10.3109/10715769709065786. [DOI] [PubMed] [Google Scholar]
- Verheyen AK, Vlaminckx EM, Lauwers FM, Saint-Guillain ML, Borgers MJ. Identification of macrophages in intimal thickening of rat carotid arteries by cytochemical localisation of purine nucleoside phosphorylase. Arterioscler. 1988;8:759–767. doi: 10.1161/01.atv.8.6.759. [DOI] [PubMed] [Google Scholar]
- Villa AE, Guzman LA, Poptic EJ, et al. Effects of antisense c-myb oligonucleotides on vascular smooth muscle cell proliferation and response to vessel wall injury. Circ. Res. 1995;76:505–513. doi: 10.1161/01.res.76.4.505. [DOI] [PubMed] [Google Scholar]
- Viswanathan M, De Oliveira AM, Johren O, Saavedra JM. Increased endothelin ET (A) receptor expression in rat carotid arteries after balloon injury. Peptides. 1997;18:247–255. doi: 10.1016/s0196-9781(96)00285-9. [DOI] [PubMed] [Google Scholar]
- Walker LN, Ramsay MM, Bowyer DE. Endothelial healing following defined injury to rabbit aorta. Depth of injury and mode of repair. Atheroscler. 1983;47:123–130. doi: 10.1016/0021-9150(83)90149-1. [DOI] [PubMed] [Google Scholar]
- Walker LN, Bowyer DE. Endothelial healing in the rabbit aorta and the effect of risk factors for atherosclerosis: Hypercholesterolemia. Arterioscler. 1984;4:479–488. doi: 10.1161/01.atv.4.5.479. [DOI] [PubMed] [Google Scholar]
- Waller BF. Crackers, stretches, drillers, scrapers, shavers, burners, welders, melter. The future treatment of coronary artery disease? A clinic-morphologic assessment. J. Am. Coll. Cardiol. 1989;13:969–987. doi: 10.1016/0735-1097(89)90248-9. [DOI] [PubMed] [Google Scholar]
- Waller BF, Pinkerton CA, Orr CM, Slack JD, Van Tassel J, Peters T. Morphological observations late (greater than 30 days) after clinically successful balloon coronary angioplasty. Circulation. 1991;83(Suppl.):28–41. [PubMed] [Google Scholar]
- Wang H, Moore S, Alavi MZ. Synthesis of tissue inhibitor of metalloproteinase-1 (TIMP-1) in rabbit aortic neointima after selective de-endothelialization. Atheroscler. 1996a;126:95–104. doi: 10.1016/0021-9150(96)05898-4. [DOI] [PubMed] [Google Scholar]
- Wang H, Keiser JA. Expression of membrane-type matrix metalloproteinase in rabbit neointimal tissue and its correlation with matrix-metalloproteinase-2 activation. J. Vasc. Res. 1998;35:45–54. doi: 10.1159/000025564. [DOI] [PubMed] [Google Scholar]
- Wang H, Li Z, Moore S, Alavi MZ. Collagen biosynthesis by neointimal smooth muscle cells cultured from rabbit aortic explants 15 weeks after de-endothelialization. Int. J. Exp. Path. 1998;79:47–53. doi: 10.1046/j.1365-2613.1998.00048.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X, Feuerstein GZ, Clark RK, Yue TL. Enhanced leucocyte adhesion to interleukin-1 beta stimulated vascular smooth muscle cells is mainly through intercellular adhesion molecule-1. Cardiovasc. Res. 1994;28:1808–1814. doi: 10.1093/cvr/28.12.1808. [DOI] [PubMed] [Google Scholar]
- Wang X, Louden C, Ohlstein EH, Stadel JM, Gu JL, Yue TL. Osteopontin expression in platelet-derived growth factor-stimulated vascular smooth muscle cells and carotid artery after balloon angioplasty. Arterioscler. Thromb. Vasc. Biol. 1996b;16:1365–1372. doi: 10.1161/01.atv.16.11.1365. [DOI] [PubMed] [Google Scholar]
- Wang X, Douglas SA, Louden C, et al. Expression of endothelin-1, endothelin-3, endothelin-converting enzyme-1, and endothelin-A and endothelin-B receptor mRNA after angioplasty-induced neointimal formation in the rat. Circ. Res. 1996c;78:322–328. doi: 10.1161/01.res.78.2.322. [DOI] [PubMed] [Google Scholar]
- Ward MR, Agrotis A, Kanellakis P, Dilley R, Jennings G, Bobik A. Inhibition of protein tyrosine kinases attenuates increases in expression of transforming growth factor-beta isoforms and their receptors following arterial injury. Arterioscler. Thromb. Vasc. Biol. 1997;17:2461–2470. doi: 10.1161/01.atv.17.11.2461. [DOI] [PubMed] [Google Scholar]
- Ward MR, Sasahara T, Agrotis A, Dilley RJ, Jennings GL, Bobik A. Inhibitory effects of tranilast on expression of transforming growth factor-beta isoforms and receptors in injured arteries. Atheroscler. 1998;137:267–275. doi: 10.1016/s0021-9150(97)00275-x. [DOI] [PubMed] [Google Scholar]
- Weidinger FF, McLenachan JM, Cybulsky MI, et al. Persistent dysfunction of regenerated endothelium after balloon angioplasty of rabbit iliac artery. Circulation. 1990;81:1667–1679. doi: 10.1161/01.cir.81.5.1667. [DOI] [PubMed] [Google Scholar]
- Weir L, Chen D, Pastore C, Isner JM, Walsh K. Expression of gax, a growth arrest homeobox gene is rapidly down-regulated in the rat carotid artery during the proliferative response to balloon injury. J. Biol. Chem. 1995;270:5457–5461. doi: 10.1074/jbc.270.10.5457. [DOI] [PubMed] [Google Scholar]
- Wempe F, Lindner V, Augustin HG. Basic fibroblast growth factor (bFGF) regulates the expression of the CC chemokine monocyte chemoattractant protein-1 (MCP-1) in autocrine-activated endothelial cells. Arterioscler. Thromb. Vasc. Biol. 1997;17:2471–2478. doi: 10.1161/01.atv.17.11.2471. [DOI] [PubMed] [Google Scholar]
- West JL, Hubbell JA. Separation of the arterial wall from blood contact using hydrogel barriers reduces intimal thickening after balloon injury in the rat: the roles of medial and luminal factors in arterial healing. Proc. Natl. Acad. Sci. USA. 1996;93:13188–13193. doi: 10.1073/pnas.93.23.13188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wight TN. The extracellular matrix and atherosclerosis. Curr. Opin. Lipidol. 1995;6:326–334. doi: 10.1097/00041433-199510000-00013. [DOI] [PubMed] [Google Scholar]
- Winocour PD, Kinlough-Rathbone RL, Richardson M, Mustard JF. Platelet accumulation and turnover on de-endothelialized aortae in rats. Br. J.Exp. Pathol. 1989;70:337–348. [PMC free article] [PubMed] [Google Scholar]
- Witztum JL, Berliner JA. Oxidized phospholipids and isoprostanes in atherosclerosis. Curr. Opin. Lipidol. 1998;9:441–448. doi: 10.1097/00041433-199810000-00008. [DOI] [PubMed] [Google Scholar]
- Yoshida Y, Mitsumata M, Ling G, Jiang J, Shu Q. Migration of medial smooth muscle cells to the intima after balloon injury. Ann. N.Y. Acad. Sci. 1997;811:459–470. doi: 10.1111/j.1749-6632.1997.tb52027.x. [DOI] [PubMed] [Google Scholar]
- Yumi K, Fagin JA, Yamashita M, et al. Direct effects of somatostatin analog octreotide on insulin-like growth factor-I in the arterial wall. Lab. Invest. 1997;76:329–338. [PubMed] [Google Scholar]
- Zahger D, Fishbein MC, Garfinkel LI, et al. VCL, an antagonist of platelet GP1B receptor, markedly inhibits platelet adhesion and intimal thickening after balloon injury in the rat. Circulation. 1995;92:1269–1273. doi: 10.1161/01.cir.92.5.1269. [DOI] [PubMed] [Google Scholar]