

Abstract
While the proteome defines the expressed gene products, the metabolome results from reactions controlled by such gene products. Plasma represents an accessible “window” to the metabolome both in regard of availability and content. The wide range of the plasma metabolome, in terms of molecular diversity and abundance, makes its comprehensive analysis challenging. Here we demonstrate an analytical method designed to target one region of the metabolome i.e. oxysterols. Since the discovery of their biological activity as ligands to nuclear receptors there has been a reawakening of interest in oxysterols and their analysis. In addition, the oxysterols, 24S- and 27-hydroxycholesterol, are currently under investigation as potential biomarkers associated with neurodegenerative disorders such as Alzheimer's disease and multiple sclerosis; widespread analysis of these lipids in clinical studies will require the development of robust, sensitive and rapid analytical techniques. In this communication we present results of an investigation of the oxysterols content of human plasma using a newly developed high-performance liquid chromatography – mass spectrometry (HPLC-MS) method incorporating charge-tagging and high-resolution MS. The method has allowed the identification in plasma of monohydroxylated cholesterol molecules, 7α-, 24S- and 27-hydroxycholesterol; the cholestenetriol 7α,27-dihydroxycholesterol; and 3β-hydroxycholest-5-en-27-oic acid and its metabolite and 3β,7α-dihydroxycholest-5-en-27-oic acid. The methodology described is also applicable for the analysis of other sterols in plasma i.e. cholesterol, 7-dehydrocholesterol, and desmosterol, as well as cholesterol 5,6-seco-sterols and steroid hormones. Although involving derivatisation, sample preparation is straight forward and chromatographic analysis rapid (17 min), while the MS method offers high sensitivity (ng/mL of sterol in plasma, or pg on-column) and specificity. The methodology is suitable for targeted metabolomic analysis of sterols, oxysterols and steroid hormones opening a “window” to view this region of the metabolome.
Keywords: metabolomics, lipidomics, steoidomics, sterols, steroids, high-performance liquid chromatography – mass spectrometry, tandem mass spectrometry, electrospray, ion-trap, derivatisation
Introduction
As the twenty first century progresses there is an ever increasing interest in “omic” science and its integration into systems biology. While the first draft of the human genome is complete1,2, and remarkable progress has been made in the definition of the human proteome3,4, the third arm of the system i.e. the metabolome, is still largely ill defined5. This is not surprising when one considers that the genome is made up from only four building blocks (or bases), the proteome from essentially 20 amino acids, while the metabolome shows variability at the atomic level. The complexity of the metabolome is such that its analysis in a single experimental format cf. shotgun proteomics6, is essentially impossible, simply on account of the widely differing physicochemical properties of metabolites e.g. amino acids and triglycerides. Despite this constraint major efforts are in progress to explore the variability of the metabolome and discover how it changes as a result of pharmacological intervention or disease7-9. Essentially, two analytical technologies are employed namely nuclear magnetic resonance (NMR) spectrometry and mass spectrometry (MS). NMR offers advantages for the complete structural determination of biomolecules including their stereochemistry, but is still comparatively insensitive, and its combination with on-line separation techniques is still in the development phase8,10,11. Mass spectrometry, in its various guises, is far more compatible with on-line chromatography and offers enhanced sensitivity, however, complete structural determination still requires comparison with synthetic standards5,12. The use of MS in metabolomics introduces a further complication as analytes must be converted into gas-phase ions for analysis. While volatile and thermally stable molecules are suitable for characterisation by gas-chromatography (GC)-MS, polar thermally labile molecules are more suited to investigation by atmospheric pressure ionisation (API)-MS in its various forms13. GC-MS has found a major role in plant metabolomics14, but with respect to animal systems electrospay ionisation (ESI)-MS is the favoured technique5,15.
In terms of mammalian and human metabolomics, urine, blood (serum or plasma) and cerebrospinal fluid (CSF) are popular fluids to profile. Urine on account of its ease of collection and representation of catabolomic/metabolomic end products, CSF as it reflects brain metabolism, and blood as it mirrors the organisms anabolic, metabolic and catabolic processes. Each medium has its advantages and disadvantages for metabolomic analysis. In regard of global metabolomics, where the goal is to reproducibly measure the greatest number of metabolites as possible and investigate how their pattern changes as a result of disease or pharmacological intervention, Siuzdak's group in California have been leading proponents in serum analysis5,15-17, however, as noted by this group the sensitivity of API-MS varies greatly with analyte physicochemical properties. For example, in the positive-ion ESI mode phospholipids containing the charged choline group give abundant ions, while phosphatidic acids or neutral molecules such as cholesterol (C5-3β-ol) are much more difficult to ionise18, and it is the metabolites of cholesterol which have attracted our interest in recent years19.
In a “standard” metabolomic investigation of human plasma/serum using positive-ion ESI-MS e.g. following cold methanol extraction, cholesterol, although present at a level of ∼2 mg/mL, is barely detectable15, and then only as the [M+H-H2O]+ ion at m/z 369 (C27H45+, Exact Mass: 369.3516 Da), by either high-resolution ESI-MS or by LC-ESI-MS (see http://metlin.scripps.edu/ftms_control.php?start=&end=&fraction=PlasmaO&formula=C27H45%2B%2C+ and supplementary Figures S1 & S2). As cholesterol is the most abundant sterol in plasma, the prospect of analysing cholesterol metabolites present at levels three orders of magnitude below that of cholesterol is daunting. However, cholesterol and its metabolites can be analysed against a metabolomic background by enhancing their ionisation properties. Such strategies have been used for many years20, dating back to the introduction of fast atom bombardment (FAB) as a soft ionisation method21. Furthermore, cholesterol metabolites can be separated from the parent molecule itself prior to analysis by group separation strategies, thereby reducing the dynamic range of detection required in the analysis. Again such strategies are not novel and have been used for many years in connection with GC-MS analysis of lipids19.
In this communication we present a targeted approach to plasma metabolomics designed with the specific objective of sterol analysis, but which also allows for the analysis of other lipids with similar polarities. In brief (Scheme 1), oxysterol are separated from cholesterol (and other lipids of lower polarity/hydropholicity) by reversed phase solid phase extraction (RP-SPE) giving two fractions i.e. SPE1-Fr-1 (oxysterol containing) and SPE1-Fr-2 (cholesterol containing). The sterols in each fraction are oxidised with cholesterol oxidase to convert 3β-hydroxy-5-ene- and 3β-hydroxy-5α(H) sterols to their 3-oxo-4-ene and 3-oxo-5α(H) analogues. The oxo groups are subsequently derivatised with the Girard P (GP) reagent, so as to introduce a quaternary nitrogen to the molecule22-25. Such charged molecules give very abundant gas-phase ions in the ESI process, improving the sensitivity of their analysis by two to three orders of magnitude23-25. LC-ESI-MS is then performed providing isomer separation and deconvolution of the still complex mixture. MS analysis is performed in an ion-trap instrument utilising tandem mass spectrometry (MS/MS or MS2) in the “data directed analysis” (DDA) mode, but with the incorporation of an include list to pre-programme the fragmentation of specified ions of interest. In a final step following MS2, if a specified neutral loss of 79 Da is observed, characteristic of the GP derivative, MS3 is performed on the [M-79]+ fragment-ion to give structural information and molecular characterisation.
Scheme 1.
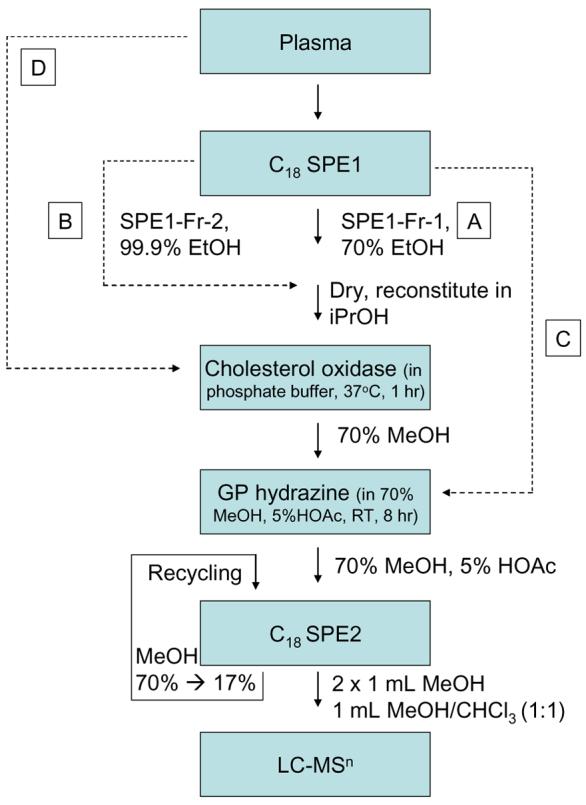
Illustration of the extraction, oxidation and derivatisation protocol applied to sterol analysis prior to LC-MS. Plasma (0.8 mL) in 15 mL of 70% ethanol (EtOH) is loaded on the first C18 column (SPE1, 0.75 – 1 g). Two fractions are eluted, SPE1-Fr-1 is the 70% EtOH flow-through (15 mL) plus a 20 mL 70% EtOH column-wash (route A), while SPE1-Fr-2 is 5 mL of 99.9% EtOH (route B). Oxysterols elute in SPE1-Fr-1, while cholesterol elutes in SPE1-Fr-2. Each fraction is reconstituted in isopropanol (iPrOH, 100 μL), and then added to a solution of cholesterol oxidase from Streptomyces sp in phosphate buffer (0.26 U in 1 mL) and incubated at 37°C for 1 hr. The reaction is quenched with 2 mL of methanol (MeOH), and 150 μL of glacial acetic acid (HOAc) and 150 mg of GP hydrazine added. The solution is incubated at room temperature for 8 hr. Derivatised sterols in the reaction mixture (70% MeOH, 5% HOAc, 3% iPrOH) are separated from un-reacted reagent by re-cycling solid phase extraction on a second C18 column (SPE2, 1 × 0.8 cm). The initial eluent is diluted with water to reduce its organic content by half, and re-cycled through the column and diluted again to eventually reach 17% MeOH which is finally re-cycled through the column. The flow-through and a column-wash (10 mL of 10% MeOH) are discarded and sterols eluted in 100% MeOH (2 × 1 mL) and MeOH/CHCl3 (1:1, v/v, 1 mL). In our study most attention has been paid to oxysterol analysis (SPE1-Fr-1), route A. Although cholesterol and similarly hydrophobic sterols can also be analysed (SPE1-Fr-2), route B. To distinguish between sterols which “naturally” posses an oxo group and those which are derivatised to contain one, route C can be followed in parallel to either or both of routes A and B. For the analysis of the most abundant sterols in blood or plasma, route D can be followed where the fluid, or a spot of blood applied to filter paper, can be treated directly with cholesterol oxidase.
Experimental
Materials
All solvents were of HPLC grade or above. GP reagent {1-(carboxymethyl)pyridinium chloride hydrazide} and cholesterol oxidase from Streptomyces sp were from Sigma-Aldrich Ltd (Poole, Dorset, UK). Sep-Pack tC18 and Sep-Pak C18 were from Waters (Elstree, UK). Reference sterols were from Steraloids Inc Ltd (London, UK), Sigma-Aldrich Ltd, or from previous studies in our laboratories24,26,27. Plasma samples were part of a GSK study, and were provided with institutional review board and ethical approval.
Methods
1. Extraction of plasma for analysis of sterols
Plasma (0.8 mL) was added dropwise to 10.5 mL of 99.9% ethanol in an ultrasonic bath. Note, 1 mL plasma is expected to contain ∼2 mg of cholesterol, 60 – 80 ng 24S-hydroxycholesterol (C5-3β,24S-diol), 120 - 180 ng 27-hydroxycholesterol (C5-3β,27-diol) and 70 −120 ng 3β-hydroxycholest-5-en-27-oic acid (CA5-3β-ol) [see supplementary Table S1, Wang and Griffiths28 and references therein]. This solution was diluted to 70% ethanol by the addition of 3.7 mL of water, ultrasonicated for 2 min, and centrifuged at 50,000 g at 4°C for ten minutes.
A bed of Sep-Pak tC18 (SPE1, 0.75 g or 1 g) was prepared in 70% ethanol by gravity flow in a glass column and washed with ∼ 20 mL of 70% ethanol. Plasma in 70% EtOH (15 mL) was applied to the column, and allowed to flow at a rate of ∼0.25 mL/min. The flow-through and four column washes of 5 mL of 70% ethanol were collected (SPE1-Fr-1, Scheme 1). By testing the method with a solution of 30,000 cpm [4-14C]cholesterol and [26,27-3H2]25-hydroxycholesterol (C5-3β,25-diol) in 70% ethanol, cholesterol was found to be retained on the column even after 4 column washes, while 25-hydroxycholesterol elutes in the flow-through and first two column washes (>87% recovery, data not shown). Cholesterol was eluted from the Sep-Pack column with 5 mL of 99.9% ethanol, as confirmed by [4-14C]cholesterol (SPE1-Fr-2, Scheme 1). The column can be further stripped with three additional 5 mL aliquots of 99.9% ethanol to elute more hydrophobic sterols. Each fraction was dried under reduced pressure using a rotor-evaporator.
2. Oxidation with cholesterol oxidase from streptomyces sp
The sterol fractions from above were reconstituted in 100 μL of isopropanol, and a solution of 1 mL 50 mM phosphate buffer (KH2PO4, pH 7) containing 3.0 μL of cholesterol oxidase (2 mg/mL in H2O, 44 units/mg of protein) added. The mixture was incubated at 37°C for 1 hr, and then quenched with 2 mL of methanol (Scheme 1).
3. Derivatisation with the Girard P reagent
Glacial acetic acid (150 μL) was added to the oxidation mixture above, now in ∼70% methanol, followed by 150 mg GP reagent. The mixture was thoroughly vortexed and incubated at room temperature overnight in the dark.
4. SPE extraction of oxidised/derivatised sterols
Even when derivatised with GP reagents, sterols may be difficult to solubilise (or retain in solution) when using a highly aqueous mixture of methanol and water. This can make their extraction using RP-SPE challenging. To circumvent this problem a re-cycling procedure is used24.
A bed of Sep-Pak C18 (SPE2, 1 × 0.8 cm) was prepared in a glass column and washed with 10 mL of chloroform/methanol, 50:50 (v/v), followed by 10 mL 100% of methanol, and 10 mL of 10% methanol prior to conditioning with 5 mL of 70% methanol. The derivatisation mixture from above (∼ 3 mL 70% methanol, 5% acetic acid, 3% isopropanol, containing 150 mg of GP reagent and 6 μg of cholesterol oxidase) was applied to the column followed by 1 mL of 70% methanol and 1 mL of 35% methanol. The combined effluent (5 mL) was diluted with water (4 mL) to give 9 mL of ∼ 35% methanol. The resulting solution was again applied to the column followed by a wash of 1 mL of 17% methanol. To the combined effluent, 9 mL of water was added to give 19 mL of ∼ 17% methanol. This solution was again applied to the column followed by a wash with 10 mL of 10% methanol. At this point all the derivatised sterols are extracted by the column while excess derivatisation reagent is in the flow-through and wash. Derivatised sterols were then eluted in two 1 mL portions of 100% methanol followed by 1 mL of chloroform/methanol 1:1 (v/v). ESI-MS analysis revealed that the derivatised sterols were present predominantly in the second mL of methanol.
LC-MSn on the LTQ-Orbitrap XL
Chromatographic separation of oxidised/derivatised sterols was performed on a Finnigan Surveyor HPLC (Thermo Fisher, San Jose, CA) system utilising a Hypersil GOLD reversed-phase column (1.9 μm particles, 50 × 2.1 mm, Thermo Fisher, San Jose, CA). Mobile phase A consisted of 50% methanol containing 0.1% formic acid, and mobile phase B consisted of 95% methanol containing 0.1% formic acid. After 1 min at 20% B, the proportion of B was raised to 80% B over the next 7 min, and maintained at 80% B for a further 5 min, before returning to 20% B in 6 s and re-equilibration for a further 3 min 54 s, giving a total run time of 17 min. The flow-rate was maintained at 200 μL min and eluent directed to the API source of a LTQ-Orbitrap XL (Thermo Fisher, San Jose, CA) mass spectrometer29. This instrument is a hybrid linear ion-trap (LIT) – Orbitrap analyser. Ions from the API source, operated in the positive ESI mode, are initially stored in the LIT and analysed in either MS or MSn (multistage fragmentation) modes. Ions can be detected at the LIT detector, or ejected axially and trapped in an intermediate C-trap from which they are “squeezed” into the Orbitrap. Trapped ions in the Orbitrap assume circular trajectories around the central electrode and perform axial oscillation. The oscillating ions induce an image current into the two halves of the Orbitrap, which can be detected. The axial oscillation frequency of an ion (ω) is proportional to the square root of the inverse of m/z, {ω = (k/m/z)½}, and the frequencies of complex signals derived from many ions can be determined using a Fourier transformation (FT).
The Orbitrap was calibrated externally prior to each analytical session, and mass accuracy was in all cases better than 5 ppm. For LC-MS and LC-MSn analysis of reference compounds, sample (3 - 30 pg/μL in 60% methanol) was injected (10 μL) onto the reversed-phase column and eluted into the LTQ-Orbitrap at a flow-rate of 200 μL/min. Three scan events were performed, first a FTMS scan in the Orbitrap over the m/z range 400 – 650 at 30,000 resolution (full width at half maximum height, FWHM, definition) followed by data dependent MS2 and MS3 events performed in the LIT. For the MS2 and MS3 events the precursor-ion isolation width was set at 2 (to select the monoisotopic ion) and the normalised collision energy at 30 and 35 (instrument settings) respectively. A precursor-ion include list was defined according to the m/z of the [M]+ ions of expected sterols (see supplementary Table S2) so that MS2 was preferentially performed on these ions in the LIT if their intensity exceeded a pre-set minimum (500 counts). If a fragment-ion corresponding to a neutral loss of 79 Da from the precursor-ion was observed in the MS2 event and was above a minimal signal setting (200 counts), MS3 was performed on this fragment. To maximise efficiency, either the MS2 or MS3 event was performed at the same time as the next high-resolution mass spectrum was being recorded in the Orbitrap29.
For the analysis of oxidised/derivatised sterols from plasma, 10 μL of the methanol eluent (2 × 1 mL) from the SepPak C18 bed SPE2 (equivalent to ∼5 μL plasma, assuming all the oxysterols elute in the methanol fractions) was diluted with 90 μL of 60% methanol and 10 μL injected onto the LC column (equivalent to ∼0.5 μL plasma). MS, MS2 and MS3 spectra were recorded in a data dependent mode as described above.
LC-MSn on the LCQ
Sterol analysis was additionally performed by nano-ESI and capillary-LC-ESI-MSn utilising an LCQ ion-trap mass spectrometer (Finnigan, now Thermo Fisher Scientific, San Jose, CA, USA) fitted with a nano-ESI source. LC separation was performed using a Hypersil Gold C18 column (180 μm × 100 mm, 3 μm, Thermo Fisher) utilising a Ultimate 3000 Capillary HPLC system (Dionex, Surrey, UK). Further details are given in supplementary information.
Results
The above methodology was developed as part of a programme designed for the specific analysis of oxysterols in biological tissues and fluids23-,25,28. Oxysterols are present in both these matrices against a background of excess of cholesterol (103 – 105)28. This presents two major problems. The first is one of dynamic range, although the sensitivity of the final detection system may be sufficient, the low abundance of an oxysterol in a biological sample (e.g. ng/mL in plasma, see supplementary Table S1) may require sample loading which results in an overloading of the system (e.g. GC or LC column) with cholesterol (mg/mL in plasma) and the resultant degradation of performance and persistent carry-over. Secondly, and equally importantly, is the fact that cholesterol is prone to autoxidation30, with the formation of oxysterols which mirror those of biological origin. Thus, to avoid this problem it is necessary to separate, as far as possible, cholesterol from oxysterols in an initial step. Here we have achieved this by SPE of cholesterol on a Sep-Pak tC18 column (SPE1, 0.75 – 1 g). The method was optimised using radio-labelled [4-14C]cholesterol and [26,27-3H2]25-hydroxycholesterol so that when added in 70% ethanol, cholesterol is retained on the column while 25-hydroxycholesterol is in the flow-through and 70% ethanol wash. Thus, the oxysterol fraction (SPE1-Fr-1) will contain molecules with hydrophilicity of 25-hydroxycholesterol or greater (Scheme 1). The column was washed further with 70% ethanol to ensure high recovery of oxysterols (recovery of 25-hydroxycholesterol >87%, data not shown). Cholesterol and other less hydrophilic sterols were eluted with 99.9% ethanol (SPE1-Fr-2) as confirmed with radio-labelled cholesterol.
Thus, SPE1-Fr-1 will be enriched with oxysterols, but contain all molecules with similar or greater hydrophilicity i.e. those not retained by Sep-Pak tC18 in 70% ethanol. At this point direct analysis by API-MS or LC-MS is possible31, but the low ionisation efficiency of molecules without an acidic, basic or pre-charged group will make detection difficult32. Our preference is to incorporate a positively charged group onto the sterol structure thereby enhancing analysis by ESI-MS22-25,33. This is easily achieved by introduction of an oxo group (where necessary) by oxidation of the 3β-hydroxy group, and subsequent derivatisation with the GP reagent to “tag” a positively charged quaternary nitrogen to the molecule i.e. charge-tagging (Scheme 2). The resulting oxidised/derivatised sterols give a 102 – 103 fold improvement in sensitivity and importantly give characteristic MS2 spectra (loss of 79.04 Da) and structurally informative MS3 spectra24. Using this methodology the on-column detection limit for reference oxysterols is of the order of 1.5 pg (data not shown). At this level, MS2 and MS3 spectra are sufficiently informative to allow structure determination.
Scheme 2.

Oxidation of sterols with cholesterol oxidase and derivatsation with GP hydrazine i.e. charge-tagging. Oxidation/derivatisation of 24S-hydroxycholesterol is shown as an example. Note, some confusion can arise concerning the nomenclature used to describe 27-hydroxycholesterol, which was previously denoted as 26-hydroxycholesterol. According to rules of priority of numbering the correct description of 27-hydroxycholesterol is 25R,26-hydroxycholesterol, however, as the medical community uses the name 27-hydroxycholesterol, we will use this name in this article.
Previous studies using GC-MS and LC-MS have shown that 7α-hydroxycholesterol (C5-3β,7α-diol), 24S-hydroxycholesterol and 27-hydroxycholesterol34-38; 3β-hydroxycholest-5-en-27-oic acid, 3β,7α-dihydroxycholest-5-en-27-oic acid (CA5-3β,7α-diol) and their 3-oxo-4-ene analogues (CA4-3-one, CA4-7α-ol-3-one)35,39,40; and the cholesterol 5,6-seco-sterol 3β-hydroxy-5-oxo-5,6-secocholestan-6-al (5,6-seco-sterol) and its aldol, 3β,5β-dihydroxy-B-norcholestane-6β-carboxyaldehyde are present in plasma41 (see supplementary Figure S3 for sterol structures, and supplementary Table S1 for quantities). Thus, our LC-MS methodology was developed with this in mind. Each of the above metabolites contain a 3β-hydroxy group susceptible to oxidation (or a free oxo group) and to subsequent derivatisation. The m/z values of the [M]+ ions of the above oxidized/derivatised sterols were incorporated into an include list (see supplementary Table S2) so that MS2 would be preferentially performed on these ions. MS3 was subsequently performed on [M-79]+ ions if observed in the MS2 spectra.
Using this methodology ten different oxysterols were identified in plasma (see supplementary Table S1). For example, shown in Figure 1a is the reconstructed ion chromatogram (RIC) for m/z 534.4054 (± 5ppm) recorded at high-resolution on the LTQ-Orbitrap. Five peaks (A – E) are clearly discernable, each of which gave a [M-79]+ fragment-ion in its MS2 spectrum. Further fragmentation of the [M-79]+ ions gave structurally informative MS3 spectra which in combination with exact mass, MS2 spectra and retention time data, and spectra and chromatograms of reference standards, allowed the identification of the eluting compounds as oxidized/derivatised 24S-hydroxycholesterol (peaks A & B as syn and anti forms of the derivative), 27-hydroxycholesterol (peak C), 7α-hydroxycholesterol (peak D) and 6β-hydroxycholesterol (C5-3β,6β-diol) (peak E) (Figure 1b-e). Reference spectra of authentic standards are shown in supplementary data (Figure S4). Peak D was not always observed in biological replicates from different donors. The absence of 7β-hydroxycholesterol (C5-3β,7β-diol), the authentic standard of which is resolved from the 7α- isomer by about 40 s in the LC system, and is a tell tale sign of autoxidation, provides evidence for the gentleness of the methodology employed30. This was further confirmed by the absence of 7-oxocholesterol (C5-3β-ol-7-one) from the RIC for m/z 532.3898 (see supplementary Figure S5a and Karu et al24). Monohydroxycholesterols can be converted by hepatic and extrahepatic cytochrome P450 (CYP) enzymes to dihydroxycholesterols42-45, the oxidized/derivatised versions of which give a [M]+ ion which appears at m/z 550.4003. In the RIC of this m/z one major peak was observed which gave MS2 and MS3 spectra corresponding to 7α,27-dihydroxycholesterol (C5-3β,7α,27-triol) (Figure 2).
Figure 1.
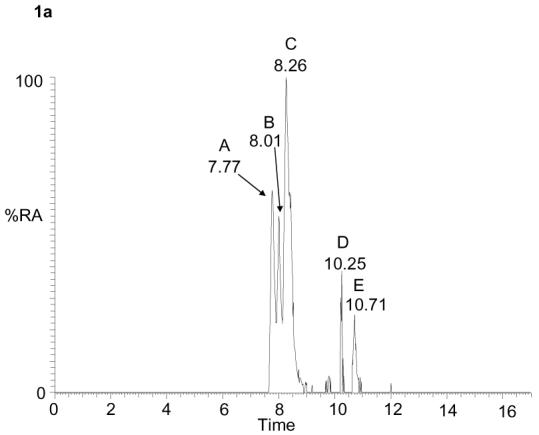

RIC for m/z 534.4054 (± 5 ppm) corresponding to the [M]+ ion of oxidised/derivatised monohydroxycholesterols in plasma (a). MS2 (534→; upper panels) and MS3 (534→455→; lower panels) spectra of the oxidised/derivatised monohydroxycholesterols eluting at 7.77 (b), 8.26 (c), 10.25 (d) and 10.71 min (e). These spectra are consistent with those of oxidised/derivatised 24S-hydroxycholesterol (b), 27-hydroxycholesterol (c), 7α-hydroxycholesterol (d), and 6β-hydroxycholesterol (e), respectively. The RIC was plotted from mass spectra recorded in the Orbitrap analyser, the MS2 and MS3 spectra were recorded by the LIT. Chromatographic conditions are described in the Experimental. Fragmentation nomenclature is illustrated in supplementary Figure S6.
Figure 2.
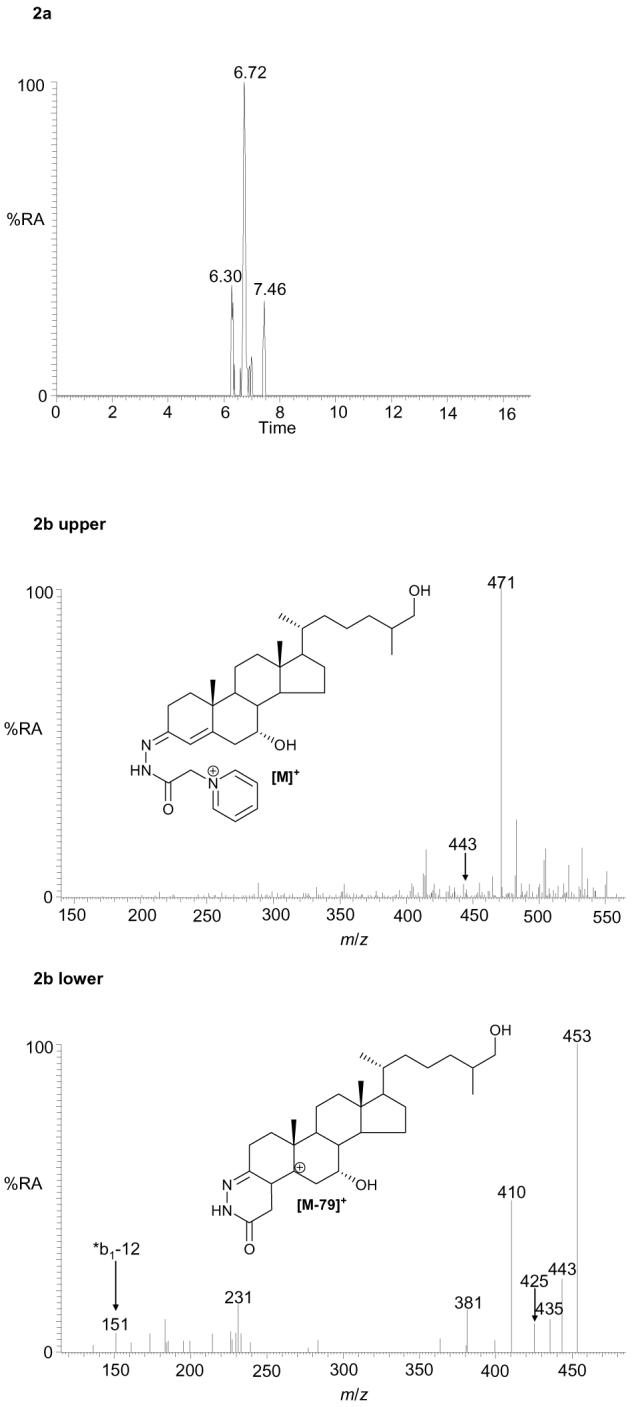
RIC for m/z 550.4003 (± 5 ppm) corresponding to the [M]+ ion of oxidised/derivatised dihydroxycholesterols in plasma (a). MS2 (550→; upper panel) and MS3 (550→471→; lower panel) spectra of the oxidised/derivatised dihydroxycholesterol eluting at 6.72 min (b). These spectra are consistent with those of oxidised/derivatised 7α,27-dihydroxycholesterol. The peaks eluting at 6.30 and 7.46 min were of insufficient intensity to allow interpretation from MSn spectra. The RIC was plotted from mass spectra recorded in the Orbitrap analyser, the MS2 and MS3 spectra were recorded by the LIT. Chromatographic conditions are described in the Experimental section.
Our extraction procedure is suitable not only for hydroxycholesterols but also for cholestenoic acids which are intermediates in the acidic pathway of bile acid biosynthesis44. Thus, we plotted RICs for m/z 548.3847 and 564.3796 corresponding to the [M]+ ions of oxidised/derivatised 3β-hydroxycholest-5-en-27-oic acid and 3β,7α-dihydroxycholest-5-en-27-oic acid, respectively (Figure 3a). Exact mass, MS2, MS3 and retention time data confirmed the presence of these acids in plasma (Figure 3, see also supplementary Figure S7 for a comparison with spectra of authentic samples). It should be noted that in this preliminary study we chose not to discriminate between these acids and their 3-oxo-4-ene forms generated endogenously by ubiquitous 3β-hydroxy-Δ5-C27-steroid dehydrogenase. If this is required the methodology can be replicated in the absence of bacterial cholesterol oxidase (see route C in Scheme 1).
Figure 3.

RICs for m/z 548.3847 (upper panel) and 564.3796 (lower panel), both at a mass tolerance of 5 ppm, corresponding to the [M]+ ions of oxidised/derivatised hydroxychoestenoic and dihydroxycholestenoic acids in plasma (a). MS2 (548→; upper panel) and MS3 (548→469→; lower panel) spectra of the oxidised/derivatised hydroxychoestenoic acid eluting at 8.08 min (b), and the MS2 (564→; upper panel) and MS3 (564→485→; lower panel) spectra of the oxidised/derivatised dihydroxycholestenoic acid eluting at 6.55 min (c). These spectra are consistent with those of oxidised/derivatised 3β-hydroxychoest-5-en-27-oic acid (b), and 3β,7α-dihydroxychoest-5-en-27-oic acid (c). The second peak in the RIC of m/z 564.3796 at 7.20 min is the syn/anti isomer of that eluting at 6.55 min. The RICs were plotted from mass spectra recorded in the Orbitrap analyser, the MS2 and MS3 spectra were recorded by the LIT. Chromatographic conditions are described in the Experimental.
Recently, it has been proposed that cholesterol 5,6-seco-sterol and its aldol have proatherogenic properties, are present in atherosclerotic plaque material, and that the aldol is present in plasma at significantly elevated levels in humans warranting carotid endartectomy, i.e. 29 – 706 ng/mL compared to control values of less than 4 ng/mL41. Both the 5,6-seco-sterol and its aldol possess oxo group(s) available for GP derivatisation and form GP derivatives24 (see supplementary Figure S3 for structures). The RIC for m/z 552.4160 corresponding to their [M]+ ions revealed three peaks (see supplementary Figure S5a), the second of which (10.24 min) eluted at a similar retention time to that of the aldol authentic standard (supplementary Table S1). The MS2 spectra recorded were similar but not identical to those of the authentic standard presumably on account of co-eluting contaminants. Unfortunately, there was insufficient ion-current to record an MS3 spectrum (see supplementary Figure S8). In the current study the equivalent of 0.5 μL of plasma was injected on-column. As the levels of oxysterols and cholestenoic acids in plasma range from ∼200 pg/μL to <5 pg/μL this represents the lower limit of plasma required for the structure determination of the low abundance compounds.
Not only oxysterols and cholestenoic acids, but also steroid hormones can be analysed by our methodology. Although we did not optimised the chromatography for steroid hormones, which eluted soon after the solvent front, a combination of accurate mass, MS2 and MS3 allowed the identification of an androst-5-ene-3-ol-17-one (A5-3-ol-17-one) 3-sulphate, almost certainly dehydroepiandrosterone (A5-3β-ol-17-one, DHEA) sulphate the most abundant hormonal steroid sulphate in human plasma46-48, and isomeric forms of androstan-3-ol-17-one (A-3-ol-17-one) 3-sulphate, and a 3-glucuronide (GlcA) also known to occur in human plasma49,50 (see supplementary Table 1, Figures S5b, S9 & S10 and supplementary Results). If analysis of hormonal steroids was to be perused further it would be necessary to start the chromatographic separation with a lower proportion of organic solvent in the mobile phase, this could then allow further isomer separation.
The current study has been one of sterol/steroid discovery and as such we have not attempted to perform absolute quantification by isotope dilution mass spectrometry. However, our data is semi-quantitative in that once derivatised to the GP hydrazone, sterols give an approximately equal response upon ESI24. Thus, it is possible to determine the approximate relative abundance of different oxysterols in plasma. Our data indicates that the ratio of 7α-hydroxycholesterol to 24S-hydroxycholesterol to 27-hydroxycholesterol is about 05:75:100, this compares to literature values for these ratios of 28:42:100 (43/64/154 in ng/mL)34, and 28:52:100 (44/83/159 in ng/mL)35. Other studies give the ratio of 24S-hydroxycholesterol to 27-hydroxycholesterol in plasma as 53:100 (64/120 in ng/mL)36, 60:10051 and 31:100 (57/181 in ng/mL) in males and 45:100 (63/139 in ng/mL) in females37 (see supplementary Table S1).
Although the methodology is designed for the analysis of oxysterols, the second fraction from the initial SPE (SPE1-Fr-2) contains cholesterol and related sterols with similar polarity/hydrophobicity. If required this fraction can also be taken through the analytical system and analysed after oxidation/derivatisation by LC-MSn (route B in Scheme 1). In the case of abundant sterols, the initial SPE step can be omitted, and plasma (or blood) added directly to cholesterol oxidase in phosphate buffer (route D in Scheme 1). This can potentially allow the diagnosis of inborn errors of cholesterol metabolism e.g. Smith-Lemli-Opitz syndrome (SLOS) by direct infusion or nano-ESI-MSn (see supplementary information, Experimental and Results).
By using an initial RP-SPE (SPE1) for depletion of cholesterol and similarly hydrophobic lipids and a second RP-SPE (SPE2) for removal of the more hydrophilic molecules (i.e. those not retained by the column in 10% methanol) a “window” is opened on the plasma metabolome allowing the view of oxysterols, cholestenoic acids and steroid hormones. Other parts of the picture include phospholipids particularly lysophospholipids. Those partially identified by accurate mass and MS2 are listed in Table S3 of supplementary data.
Discussion
The methodology described herein is suitable for the analysis of a wide range of sterols/steroids which posses either an oxo group or a 3β-hydroxy group that can be oxidised to a 3-oxo group. At one end of the spectrum are the hydrophobic monohydroxycholesterols (or cholesterol and dehydrocholesterols if route B or D is followed), while at the other end of the spectrum are molecules as hydrophilic as DHEA-sulphate. The methodology provides specificity based on MS3 fragmentation of [M-79]+ product-ions explicitly generated by the neutral loss of pyridine (C5H5N) from the GP derivative, but is not exclusive in that other biomolecules that fall within the hydrophilicity window selected can also be “viewed”. To our knowledge no other methodology is suitable for the high-sensitivity (pg) analysis of such a broad range of molecules from a biological matrix in a single experiment. GC-MS is well documented for the analysis of hydroxycholesterols34,35 and cholestenoic acids39, but is not suitable for the analysis of steroid sulphates or glucuronides without prior solvolysis or hydrolysis52. Additionally, polyhydroxylated sterols even after appropriate derivatisation for GC-MS tend to fragment in the ion-source providing only a very weak molecular ion, or one none at all52. Alternatively, atmospheric pressure chemical ionisation (APCI)-MS can be used for oxysterol36, cholestenoic acid38, and steroid sulphate/glucuronide analysis53,54, but is comparatively insensitive requiring multiple reaction monitoring (MRM) scans and often analysis relies on detection of the [M+H-H2O]+ a [M+H-A]+ ion (where A is the conjugating group), thereby discarding valuable structural information. In contrast the LC-ESI-MSn methodology described herein offers high sensitivity over a wide-range of derivatised sterols/steroids and structurally informative MSn spectra, making this methodology more compatible with discovery metabolomics, or more specifically steroidomics.
The current study has been driven by our interest in the correlation between neurodegenerative disease and oxysterols28,55. 24S-Hydroxycholesterol is an oxysterol formed exclusively in brain and can cross the blood brain barrier and enter the circulation, hence its level in blood in comparison to other oxysterols e.g. 27-hydroxycholesterol, may provide a biomarker, or indication, of the progression of neurodegenerative disease51. In addition, the ratio of these two lipid species may serve as useful surrogate indicator of neuronal function in response to therapeutic intervention. The development of the robust and rapid methodology presented in this study is an important step in allowing these analytes to be routinely monitored, and their relevance assessed, both in longitudinal disease studies and clinical studies.
As monohydroxycholesterols, following oxidation/derivatisation, give similar response factors in LC-ESI-MS the ratio of 24S- to and 27-hydroxycholesterol can easily be measured. The ratio observed for the two samples reported here is 74-81:100 which is in good agreement with the range of values found in the literature e.g. 42:10034, 52:10035, 53:10036, 60:10051, 31:100 in males and 45:100 in females37 (see supplementary Table S1). Future studies will involve the collection of additional plasma samples to allow a statistical appreciation of data.
3β-Hydroxycholest-5-en-27-oic acid, its metabolite 3β,7α-dihydroxycholest-5-en-27-oic acid and their 3-oxo-4-ene analogues are known constituents of blood39. These molecules represent intermediates in the acidic pathway of bile acid biosynthesis following mitochondrial hydroxylation of cholesterol by CYP27A144. In the current study we did not attempt to distinguish between 3β-hydroxy-5-ene sterols and their 3-oxo-4-ene metabolites formed endogenously by ubiquitous 3β-hydroxy-Δ5-C27-steroid dehydrogenase, and for simplicity herein we classify them together as the oxidised/derivatised acids. CYP27A1 is expressed hepatically and extrahepatically in a wide range of tissues44,56, while the microsomal enzyme CYP7A1 responsible for 7α-hydroxylation of cholesterol in the first step of the classic pathway of bile acid biosynthesis is liver specific44. In our study we were able to identify both 27-hydroxycholesterol and 7α-hydroxycholesterol, along with 7α,27-dihydroxycholesterol, a further intermediated in the acidic pathway of bile acid biosynthesis, notoriously difficult to identify in plasma40. The enzyme responsible for 7α-hydroxylation of 27-hydroxycholesterol and 3β-hydroxycholest-5-en-27-oic acid (i.e. CYP7B1) is different from that responsible for 7α-hydroxylation of cholesterol (i.e. CYP7A1)42,57-59. CYP7B1 is expressed in many tissues including brain Zhang27,60,61. Thus, the sequence of 27-hydroxylation (CYP27A1 catalysed conversion of cholesterol to 27-hydroxycholesterol and further to C27 acids), 7α-hydroxylation (CYP7B1) and finally 3-dehydrogenation (by ubiquitous 3β-hydroxysteroid-Δ5-C27-steroid dehydrogenase) may represent a pathway for extrahepatic cholesterol metabolism in humans (Scheme 3)19. 7α-Hydroxy-3-oxocholest-4-en-27-oic acid can be formed by rat fetal astrocytes and neurons from 27-hydroxycholesterol via 3β-hydroxycholest-5-en-27-oic acid or from 3β,7α-dihydroxycholesterol via 3β,7α-dihydroxycholest-5-en-27-oic acid27, and is abundant in chronic subdural hematoma62. It has recently been shown that 7α-hydroxy-3-oxocholest-4-en-27-oic acid can cross an in vitro model of the blood brain barrier40 and is a net export product from brain. Given the emerging connection between cholesterol and neurodegeneration, measurement of components of the extrahepatic pathway for bile acid biosynthesis may be of importance in respect to development of these conditions.
Scheme 3.
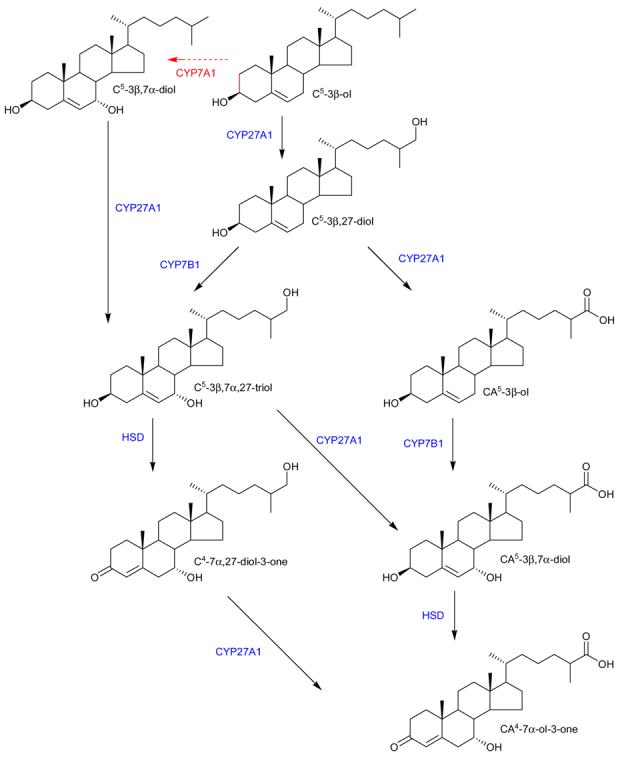
Extrahepatic metabolism of cholesterol. The liver specific 7α-hydroxylation by CYP7A1 is shown by the broken red arrow. Extra hepatic enzymes are shown in blue.
Of the conjugated steroids identified in our study, the most abundant, at least in terms of signal intensity, was unsurprisingly DHEA sulphate, this molecule is present in high levels in plasma i.e, 1000 - 3300 ng/mL46-48. Sulphated androsterone and also glucuronidated androsterone were also observed.
In summary, LC-MS analysis of plasma can provide a view of the metabolome. The exact window through which the metabolome is viewed will depend on the sample preparation method employed. In this study we have opened a widow to view a “high definition” picture of sterols.
Supplementary Material
Acknowledgement
This work was supported by the UK Biotechnology and Biological Sciences Research Council (BBSRC grant no. BB/C515771/1, & BB/C511356/1), Swansea University and GSK. The authors wish to acknowledge Kersti Karu and Emmanuel Samuel for technical assistance, and Drs Cedric Shackleton and Forbes Porter for provision of the SLOS bloodspot. We are grateful to Dr Gary Siuzdak and his group at the Scripps Institute for permission to include data from the Metlin database.
Footnotes
Supporting Information Available
Supporting information available. This material is available free at http://pubs.acs.org
References
- 1.Shoemaker DD, Schadt EE, Armour CD, He YD, Garrett-Engele P, McDonagh PD, Loerch PM, Leonardson A, Lum PY, Cavet G, Wu LF, Altschuler SJ, Edwards S, King J, Tsang JS, Schimmack G, Schelter JM, Koch J, Ziman M, Marton MJ, Li B, Cundiff P, Ward T, Castle J, Krolewski M, Meyer MR, Mao M, Burchard J, Kidd MJ, Dai H, Phillips JW, Linsley PS, Stoughton R, Scherer S, Boguski MS. Experimental annotation of the human genome using microarray technology. Nature. 2001;409(6822):922–7. doi: 10.1038/35057141. [DOI] [PubMed] [Google Scholar]
- 2.Venter JC, Adams MD, Myers EW, Li PW, Mural RJ, Sutton GG, Smith HO, Yandell M, Evans CA, Holt RA, Gocayne JD, Amanatides P, Ballew RM, Huson DH, Wortman JR, Zhang Q, Kodira CD, Zheng XH, Chen L, Skupski M, Subramanian G, Thomas PD, Zhang J, Gabor Miklos GL, Nelson C, Broder S, Clark AG, Nadeau J, McKusick VA, Zinder N, Levine AJ, Roberts RJ, Simon M, Slayman C, Hunkapiller M, Bolanos R, Delcher A, Dew I, Fasulo D, Flanigan M, Florea L, Halpern A, Hannenhalli S, Kravitz S, Levy S, Mobarry C, Reinert K, Remington K, Abu-Threideh J, Beasley E, Biddick K, Bonazzi V, Brandon R, Cargill M, Chandramouliswaran I, Charlab R, Chaturvedi K, Deng Z, Di Francesco V, Dunn P, Eilbeck K, Evangelista C, Gabrielian AE, Gan W, Ge W, Gong F, Gu Z, Guan P, Heiman TJ, Higgins ME, Ji RR, Ke Z, Ketchum KA, Lai Z, Lei Y, Li Z, Li J, Liang Y, Lin X, Lu F, Merkulov GV, Milshina N, Moore HM, Naik AK, Narayan VA, Neelam B, Nusskern D, Rusch DB, Salzberg S, Shao W, Shue B, Sun J, Wang Z, Wang A, Wang X, Wang J, Wei M, Wides R, Xiao C, Yan C, Yao A, Ye J, Zhan M, Zhang W, Zhang H, Zhao Q, Zheng L, Zhong F, Zhong W, Zhu S, Zhao S, Gilbert D, Baumhueter S, Spier G, Carter C, Cravchik A, Woodage T, Ali F, An H, Awe A, Baldwin D, Baden H, Barnstead M, Barrow I, Beeson K, Busam D, Carver A, Center A, Cheng ML, Curry L, Danaher S, Davenport L, Desilets R, Dietz S, Dodson K, Doup L, Ferriera S, Garg N, Gluecksmann A, Hart B, Haynes J, Haynes C, Heiner C, Hladun S, Hostin D, Houck J, Howland T, Ibegwam C, Johnson J, Kalush F, Kline L, Koduru S, Love A, Mann F, May D, McCawley S, McIntosh T, McMullen I, Moy M, Moy L, Murphy B, Nelson K, Pfannkoch C, Pratts E, Puri V, Qureshi H, Reardon M, Rodriguez R, Rogers YH, Romblad D, Ruhfel B, Scott R, Sitter C, Smallwood M, Stewart E, Strong R, Suh E, Thomas R, Tint NN, Tse S, Vech C, Wang G, Wetter J, Williams S, Williams M, Windsor S, Winn-Deen E, Wolfe K, Zaveri J, Zaveri K, Abril JF, Guigó R, Campbell MJ, Sjolander KV, Karlak B, Kejariwal A, Mi H, Lazareva B, Hatton T, Narechania A, Diemer K, Muruganujan A, Guo N, Sato S, Bafna V, Istrail S, Lippert R, Schwartz R, Walenz B, Yooseph S, Allen D, Basu A, Baxendale J, Blick L, Caminha M, Carnes-Stine J, Caulk P, Chiang YH, Coyne M, Dahlke C, Mays A, Dombroski M, Donnelly M, Ely D, Esparham S, Fosler C, Gire H, Glanowski S, Glasser K, Glodek A, Gorokhov M, Graham K, Gropman B, Harris M, Heil J, Henderson S, Hoover J, Jennings D, Jordan C, Jordan J, Kasha J, Kagan L, Kraft C, Levitsky A, Lewis M, Liu X, Lopez J, Ma D, Majoros W, McDaniel J, Murphy S, Newman M, Nguyen T, Nguyen N, Nodell M, Pan S, Peck J, Peterson M, Rowe W, Sanders R, Scott J, Simpson M, Smith T, Sprague A, Stockwell T, Turner R, Venter E, Wang M, Wen M, Wu D, Wu M, Xia A, Zandieh A, Zhu X. The sequence of the human genome. Science. 2000;291(5507):1304–51. doi: 10.1126/science.1058040. Erratum in: Science2001, 292(5523),1838. [DOI] [PubMed] [Google Scholar]
- 3.Uhlen M, Ponten F. Antibody-based proteomics for human tissue profiling. Mol Cell Proteomics. 2005;4(4):384–93. doi: 10.1074/mcp.R500009-MCP200. [DOI] [PubMed] [Google Scholar]
- 4.Desiere F, Deutsch EW, King NL, Nesvizhskii AI, Mallick P, Eng J, Chen S, Eddes J, Loevenich SN, Aebersold R. The PeptideAtlas project. Nucleic Acids Res. 2006;34(Database issue):D655–8. doi: 10.1093/nar/gkj040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Want EJ, Nordström A, Morita H, Siuzdak G. From exogenous to endogenous: the inevitable imprint of mass spectrometry in metabolomics. J Proteome Res. 2007;6(2):459–68. doi: 10.1021/pr060505+. [DOI] [PubMed] [Google Scholar]
- 6.Wolters DA, Washburn MP, Yates JR., 3rd An automated multidimensional protein identification technology for shotgun proteomics. Anal Chem. 2001;73(23):5683–90. doi: 10.1021/ac010617e. [DOI] [PubMed] [Google Scholar]
- 7.Wilson ID, Nicholson JK. Metabonomics and Global Systems Biology. In: Griffiths WJ, editor. Metabolomics, Metabonomics and Metabolite Profiling. RSC Biomolecular Sciences Series; Cambridge, UK: 2008. [Google Scholar]
- 8.Rantalainen M, Cloarec O, Beckonert O, Wilson ID, Jackson D, Tonge R, Rowlinson R, Rayner S, Nickson J, Wilkinson RW, Mills JD, Trygg J, Nicholson JK, Holmes E. Statistically integrated metabonomic-proteomic studies on a human prostate cancer xenograft model in mice. J Proteome Res. 2006;5(10):2642–55. doi: 10.1021/pr060124w. [DOI] [PubMed] [Google Scholar]
- 9.Wikoff WR, Gangoiti JA, Barshop BA, Siuzdak G. Metabolomics Identifies Perturbations in Human Disorders of Propionate Metabolism. Clin Chem. 2007 Oct 19; doi: 10.1373/clinchem.2007.089011. [Epub ahead of print] [DOI] [PubMed] [Google Scholar]
- 10.Viant MR, Ludwig C, Günther UL. 1D and 2D NMR Spectroscopy: from Metabolic Fingerprinting to Profiling. In: Griffiths WJ, editor. Metabolomics, Metabonomics and Metabolite Profiling. RSC Biomolecular Sciences Series; Cambridge, UK: 2008. [Google Scholar]
- 11.Sýkora J, Bernásek P, Zarevúcká M, Kurfürst M, Sovová H, Schraml J. High-performance liquid chromatography with nuclear magnetic resonance detection. A method for quantification of alpha- and gamma-linolenic acids in their mixtures with free fatty acids. J Chromatogr A. 2007;1139(1):152–5. doi: 10.1016/j.chroma.2006.11.041. [DOI] [PubMed] [Google Scholar]
- 12.McLafferty FW, Turecek F. Interpretation of Mass Spectra. Fourth edition University Science Books; Mill Valley, California: 1993. [Google Scholar]
- 13.Griffiths WJ, Karu K, Hornshaw M, Woffendin G, Wang Y. Metabolomics and metabolite profiling: past heroes and future developments. Eur J Mass Spectrom (Chichester, Eng) 2007;13(1):45–50. doi: 10.1255/ejms.850. [DOI] [PubMed] [Google Scholar]
- 14.Jonsson P, Johansson AI, Gullberg J, Trygg J, A J, Grung B, Marklund S, Sjöström M, Antti H, Moritz T. High-throughput data analysis for detecting and identifying differences between samples in GC/MS-based metabolomic analyses. Anal Chem. 2005;77(17):5635–42. doi: 10.1021/ac050601e. [DOI] [PubMed] [Google Scholar]
- 15.Nordström A, Want E, Northen T, Lehtiö J, Siuzdak G. Multiple Ionization Mass Spectrometry Strategy Used To Reveal the Complexity of Metabolomics. Anal Chem. 2008;80(2):421–429. doi: 10.1021/ac701982e. [DOI] [PubMed] [Google Scholar]
- 16.Want EJ, O'Maille G, Smith CA, Brandon TR, Uritboonthai W, Qin C, Trauger SA, Siuzdak G. Solvent-dependent metabolite distribution, clustering, and protein extraction for serum profiling with mass spectrometry. Anal Chem. 2006;78(3):743–52. doi: 10.1021/ac051312t. [DOI] [PubMed] [Google Scholar]
- 17.Smith CA, Want EJ, O'Maille G, Abagyan R, Siuzdak G. XCMS: processing mass spectrometry data for metabolite profiling using nonlinear peak alignment, matching, and identification. Anal Chem. 2006;78(3):779–87. doi: 10.1021/ac051437y. [DOI] [PubMed] [Google Scholar]
- 18.Han X, Gross RW. Shotgun lipidomics: electrospray ionization mass spectrometric analysis and quantitation of cellular lipidomes directly from crude extracts of biological samples. Mass Spectrom Rev. 2005;24(3):367–412. doi: 10.1002/mas.20023. [DOI] [PubMed] [Google Scholar]
- 19.Sjövall J. Fifty years with bile acids and steroids in health and disease. Lipids. 2004;39(8):703–22. doi: 10.1007/s11745-004-1288-1. [DOI] [PubMed] [Google Scholar]
- 20.Chatman K, Hollenbeck T, Hagey L, Vallee M, Purdy R, Weiss F, Siuzdak G. Nanoelectrospray mass spectrometry and precursor ion monitoring for quantitative steroid analysis and attomole sensitivity. Anal Chem. 1999;71(13):2358–63. doi: 10.1021/ac9806411. [DOI] [PubMed] [Google Scholar]
- 21.Nakamura T, Takazawa T, Maruyama-Ohki Y, Nagaki H, Kinoshita T. Location of double bonds in unsaturated fatty alcohols by microderivatization and liquid secondary ion tandem mass spectrometry. Anal Chem. 1993;65(6):837–840. [Google Scholar]
- 22.Shackleton CH, Chuang H, Kim J, de la Torre X, Segura J. Electrospray mass spectrometry of testosterone esters: potential for use in doping control. Steroids. 1997;62(7):523–9. doi: 10.1016/s0039-128x(97)00004-4. [DOI] [PubMed] [Google Scholar]
- 23.Griffiths WJ, Wang Y, Alvelius G, Liu S, Bodin K, Sjövall J. Analysis of oxysterols by electrospray tandem mass spectrometry. J Am Soc Mass Spectrom. 2006;17(3):341–62. doi: 10.1016/j.jasms.2005.10.012. [DOI] [PubMed] [Google Scholar]
- 24.Karu K, Hornshaw M, Woffendin G, Bodin K, Hamberg M, Alvelius G, Sjövall J, Turton J, Wang Y, Griffiths WJ. Liquid chromatography-mass spectrometry utilizing multi-stage fragmentation for the identification of oxysterols. J Lipid Res. 2007;48(4):976–87. doi: 10.1194/jlr.M600497-JLR200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Griffiths WJ, Liu S, Alvelius G, Sjövall J. Derivatisation for the characterisation of neutral oxosteroids by electrospray and matrix-assisted laser desorption/ionisation tandem mass spectrometry: the Girard P derivative. Rapid Commun Mass Spectrom. 2003;17(9):924–35. doi: 10.1002/rcm.1002. [DOI] [PubMed] [Google Scholar]
- 26.Liu S, Sjövall J, Griffiths WJ. Neurosteroids in rat brain: extraction, isolation, and analysis by nanoscale liquid chromatography-electrospray mass spectrometry. Anal Chem. 2003;75(21):5835–46. doi: 10.1021/ac0346297. [DOI] [PubMed] [Google Scholar]
- 27.Zhang J, Akwa Y, el-Etr M, Baulieu EE, Sjövall J. Metabolism of 27-, 25- and 24-hydroxycholesterol in rat glial cells and neurons. Biochem J. 1997;322(Pt 1):175–84. doi: 10.1042/bj3220175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wang Y, Griffiths WJ. Steroids, Sterols and the Nervous System. In: Griffiths WJ, editor. Metabolomics, Metabonomics and Metabolite Profiling. RSC Biomolecular Sciences Series; Cambridge, UK: 2008. [Google Scholar]
- 29.Makarov A, Denisov E, Kholomeev A, Balschun W, Lange O, Strupat K, Horning S. Performance evaluation of a hybrid linear ion trap/orbitrap mass spectrometer. Anal Chem. 2006;78(7):2113–20. doi: 10.1021/ac0518811. [DOI] [PubMed] [Google Scholar]
- 30.Schroepfer GJ., Jr Oxysterols: modulators of cholesterol metabolism and other processes. Physiol Rev. 2000;80(1):361–554. doi: 10.1152/physrev.2000.80.1.361. [DOI] [PubMed] [Google Scholar]
- 31.McDonald JG, Thompson BM, McCrum EC, Russell DW. Extraction and analysis of sterols in biological matrices by high performance liquid chromatography electrospray ionization mass spectrometry. Methods Enzymol. 2007;432:145–70. doi: 10.1016/S0076-6879(07)32006-5. [DOI] [PubMed] [Google Scholar]
- 32.Cech NB, Enke CG. Practical implications of some recent studies in electrospray ionization fundamentals. Mass Spectrom Rev. 2001;20(6):362–87. doi: 10.1002/mas.10008. [DOI] [PubMed] [Google Scholar]
- 33.Kirk JM, Tarbin J, Keely BJ. Analysis of androgenic steroid Girard P hydrazones using multistage tandem mass spectrometry. Rapid Commun Mass Spectrom. 2006;20(8):1247–52. doi: 10.1002/rcm.2442. [DOI] [PubMed] [Google Scholar]
- 34.Dzeletovic S, Breuer O, Lund E, Diczfalusy U. Determination of cholesterol oxidation products in human plasma by isotope dilution-mass spectrometry. Anal Biochem. 1995;225(1):73–80. doi: 10.1006/abio.1995.1110. [DOI] [PubMed] [Google Scholar]
- 35.Babiker A, Diczfalusy U. Transport of side-chain oxidized oxysterols in the human circulation. Biochim Biophys Acta. 1998;1392(2-3):333–9. doi: 10.1016/s0005-2760(98)00047-2. [DOI] [PubMed] [Google Scholar]
- 36.Burkard I, Rentsch KM, von Eckardstein A. Determination of 24S- and 27-hydroxycholesterol in plasma by high-performance liquid chromatography-mass spectrometry. J Lipid Res. 2004;45(4):776–81. doi: 10.1194/jlr.D300036-JLR200. [DOI] [PubMed] [Google Scholar]
- 37.Burkard I, von Eckardstein A, Waeber G, Vollenweider P, Rentsch KM. Lipoprotein distribution and biological variation of 24S- and 27-hydroxycholesterol in healthy volunteers. Atherosclerosis. 2007;194(1):71–8. doi: 10.1016/j.atherosclerosis.2006.09.026. [DOI] [PubMed] [Google Scholar]
- 38.Lövgren-Sandblom A, Heverin M, Larsson H, Lundström E, Wahren J, Diczfalusy U, Björkhem I. Novel LC-MS/MS method for assay of 7alpha-hydroxy-4-cholesten-3-one in human plasma. Evidence for a significant extrahepatic metabolism. J Chromatogr B Analyt Technol Biomed Life Sci. 2007;856(1-2):15–9. doi: 10.1016/j.jchromb.2007.05.019. [DOI] [PubMed] [Google Scholar]
- 39.Axelson M, Mörk B, Sjövall J. Occurrence of 3 b eta-hydroxy-5-cholestenoic acid, 3 beta,7 alpha-dihydroxy-5-cholestenoic acid, and 7 alpha-hydroxy-3-oxo-4-cholestenoic acid as normal constituents in human blood. J Lipid Res. 1988;29(5):629–41. [PubMed] [Google Scholar]
- 40.Meaney S, Heverin M, Panzenboeck U, Ekström L, Axelsson M, Andersson U, Diczfalusy U, Pikuleva I, Wahren J, Sattler W, Björkhem I. Novel route for elimination of brain oxysterols across the blood-brain barrier: conversion into 7alpha-hydroxy-3-oxo-4-cholestenoic acid. J Lipid Res. 2007;48(4):944–51. doi: 10.1194/jlr.M600529-JLR200. [DOI] [PubMed] [Google Scholar]
- 41.Wentworth P, Jr, Nieva J, Takeuchi C, Galve R, Wentworth AD, Dilley RB, DeLaria GA, Saven A, Babior BM, Janda KD, Eschenmoser A, Lerner RA. Evidence for ozone formation in human atherosclerotic arteries. Science. 2003;302(5647):1053–6. doi: 10.1126/science.1089525. [DOI] [PubMed] [Google Scholar]
- 42.Toll A, Shoda J, Axelson M, Sjövall J, Wikvall K. 7 alpha-hydroxylation of 26-hydroxycholesterol, 3 beta-hydroxy-5-cholestenoic acid and 3 beta-hydroxy-5-cholenoic acid by cytochrome P-450 in pig liver microsomes. FEBS Lett. 1992;296(1):73–6. doi: 10.1016/0014-5793(92)80406-7. [DOI] [PubMed] [Google Scholar]
- 43.Shoda J, Toll A, Axelson M, Pieper F, Wikvall K, Sjövall J. Formation of 7 alpha- and 7 beta-hydroxylated bile acid precursors from 27-hydroxycholesterol in human liver microsomes and mitochondria. Hepatology. 1993;17(3):395–403. [PubMed] [Google Scholar]
- 44.Russell DW. The enzymes, regulation, and genetics of bile acid synthesis. Annu Rev Biochem. 2003;72:137–74. doi: 10.1146/annurev.biochem.72.121801.161712. [DOI] [PubMed] [Google Scholar]
- 45.Pikuleva IA. Cytochrome P450s and cholesterol homeostasis. Pharmacol Ther. 2006;112(3):761–73. doi: 10.1016/j.pharmthera.2006.05.014. [DOI] [PubMed] [Google Scholar]
- 46.Liu S, Griffiths WJ, Sjövall J. Capillary liqui d chromatography/electrospray mass spectrometry for analysis of steroid sulfates in biological samples. Anal Chem. 2003;75(4):791–7. doi: 10.1021/ac0262154. [DOI] [PubMed] [Google Scholar]
- 47.Tagawa N, Tamanaka J, Fujinami A, Kobayashi Y, Takano T, Fukata S, Kuma K, Tada H, Amino N. Serum dehydroepiandrosterone, dehydroepiandrosterone sulfate, and pregnenolone sulfate concentrations in patients with hyperthyroidism and hypothyroidism. Clin Chem. 2000;46(4):523–8. [PubMed] [Google Scholar]
- 48.Shackleton CH, Kletke C, Wudy S, Pratt JH. Dehydroepiandrosterone sulfate quantification in serum using high-performance liquid chromatography/mass spectrometry and a deuterated internal standard: a technique suitable for routine use or as a reference method. Steroids. 1990;55(10):472–8. doi: 10.1016/0039-128x(90)90016-5. [DOI] [PubMed] [Google Scholar]
- 49.Peng SH, Segura J, Farré M, de la Torre X. Oral testosterone administration detected by testosterone glucuronidation measured in blood spots dried on filter paper. Clin Chem. 2000;46(4):515–22. [PubMed] [Google Scholar]
- 50.Sjövall J, Vihko R. Aalysis of solvolyzable ste roids in human plasma by combined gas chromatography - mass spectrometry. Acta Endocrinol (Copenh) 1968;57(2):247–60. doi: 10.1530/acta.0.0570247. [DOI] [PubMed] [Google Scholar]
- 51.Kölsch H, Heun R, Kerksiek A, Bergmann KV, Maier W, Lütjohann D. Altered levels of plasma 24S- and 27-hydroxycholesterol in demented patients. Neurosci Lett. 2004;368(3):303–8. doi: 10.1016/j.neulet.2004.07.031. [DOI] [PubMed] [Google Scholar]
- 52.Griffiths WJ, Shackleton C, Sjövall J. Steroid analysis. In: Caprioli R, editor. Encyclopedia of mass spectrometry volume 3. Elsevier; Oxford: 2005. [Google Scholar]
- 53.Nakajima M, Yamato S, Shimada K. Determination of dehydroepiandrosterone sulphate in biological samples by liquid chromatography/atmospheric pressure chemical ionization-mass spectrometry using [7,7,16,16-2H4]-dehydroepiandrosterone sulphate as an internal standard. Biomed Chromatogr. 1998;12(4):211–6. doi: 10.1002/(SICI)1099-0801(199807/08)12:4<211::AID-BMC737>3.0.CO;2-N. [DOI] [PubMed] [Google Scholar]
- 54.Kuuranne T, Vahermo M, Leinonen A, Kostianen R. Electrospray and atmospheric pressure chemical ionization tandem mass spectrometric behaviour of eight anabolic steroid glucuronides. J Am Soc Mass Spectrom. 2000;11(8):722–30. doi: 10.1016/s1044-0305(00)00135-5. [DOI] [PubMed] [Google Scholar]
- 55.Björkhem I, Heverin M, Leoni V, Meaney S, Diczfalusy U. Oxysterols and Alzheimer's disease. Acta Neurol Scand Suppl. 2006;185:43–9. doi: 10.1111/j.1600-0404.2006.00684.x. [DOI] [PubMed] [Google Scholar]
- 56.Lund E, Andersson O, Zhang J, Babiker A, Ahlborg G, Diczfalusy U, Einarsson K, Sjövall J, Björkhem I. Importance of a novel oxidative mechanism for elimination of intracellular cholesterol in humans. Arterioscler Thromb Vasc Biol. 1996;16(2):208–12. doi: 10.1161/01.atv.16.2.208. [DOI] [PubMed] [Google Scholar]
- 57.Axelson M, Shoda J, Sjövall J, Toll A, Wikvall K. Cholesterol is converted to 7 alpha-hydroxy-3-oxo-4-cholestenoic acid in liver mitochondria. Evidence for a mitochondrial sterol 7 alpha-hydroxylase. J Biol Chem. 1992;267(3):1701–4. [PubMed] [Google Scholar]
- 58.Martin KO, Budai K, Javitt NB. Cholesterol and 27-hydroxycholesterol 7 alpha-hydroxylation: evidence for two different enzymes. J Lipid Res. 1993;34(4):581–8. [PubMed] [Google Scholar]
- 59.Schwarz M, Lund EG, Russell DW. Two 7 alpha-hydroxylase enzymes in bile acid biosynthesis. Curr Opin Lipidol. 1998;9(2):113–8. doi: 10.1097/00041433-199804000-00006. [DOI] [PubMed] [Google Scholar]
- 60.Stapleton G, Steel M, Richardson M, Mason JO, Rose KA, Morris RG, Lathe R. A novel cytochrome P450 expressed primarily in brain. J Biol Chem. 1995;270(50):29739–45. doi: 10.1074/jbc.270.50.29739. [DOI] [PubMed] [Google Scholar]
- 61.Rose K, Allan A, Gauldie S, Stapleton G, Dobbie L, Dott K, Martin C, Wang L, Hedlund E, Seckl JR, Gustafsson JA, Lathe R. Neurosteroid hydroxylase CYP7B: vivid reporter activity in dentate gyrus of gene-targeted mice and abolition of a widespread pathway of steroid and oxysterol hydroxylation. J Biol Chem. 2001;276(26):23937–44. doi: 10.1074/jbc.M011564200. [DOI] [PubMed] [Google Scholar]
- 62.Nagata K, Takakura K, Asano T, Seyama Y, Hirota H, Shigematsu N, Shima I, Kasama T, Shimizu T. Identification of 7 alpha-hydroxy-3-oxo-4-cholestenoic acid in chronic subdural hematoma. Biochim Biophys Acta. 1992;1126(2):229–36. doi: 10.1016/0005-2760(92)90295-7. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.