

Abstract
A series of metalloprotein complexes embedded in a mitochondrial or bacterial membrane utilize electron transfer reactions to pump protons across the membrane and create an electrochemical potential (ΔμH+). Current understanding of the principles of electron-driven proton transfer is discussed, mainly with respect to the wealth of knowledge available from studies of cytochrome c oxidase. Structural, experimental, and theoretical evidence supports the model of long-distance proton transfer via hydrogen-bonded water chains in proteins as well as the basic concept that proton uptake and release in a redox-driven pump are driven by charge changes at the membrane-embedded centers. Key elements in the pumping mechanism may include bound water, carboxylates, and the heme propionates, arginines, and associated water above the hemes. There is evidence for an important role of subunit III and proton backflow, but the number and nature of gating mechanisms remain elusive, as does the mechanism of physiological control of efficiency.
Keywords: cytochrome c oxidase, hydrogen-bonded water
INTRODUCTION
Aerobic organisms have evolved the ability to obtain energy from their environment by extracting electrons from food stuffs and converting the electrical potential energy into a useful chemical form, ATP. Reduced substrates, such as NADH or succinate, donate electrons to a respiratory chain composed of metalloprotein complexes embedded in a mitochondrial or bacterial membrane (Figure 1). Three of these complexes have the ability to pump protons across the membrane and create a transmembrane electrochemical potential (ΔμH+) using a series of redox cofactors that achieve a step-wise drop in potential energy. The electrochemical gradient produced is then used for the synthesis of ATP by ATP synthase, also a membrane-inserted complex. In most systems, the ultimate acceptor of electrons in the respiratory chain is oxygen, which is reduced to water, providing a large redox potential drop and maximizing the free energy available for ATP synthesis.
Figure 1.
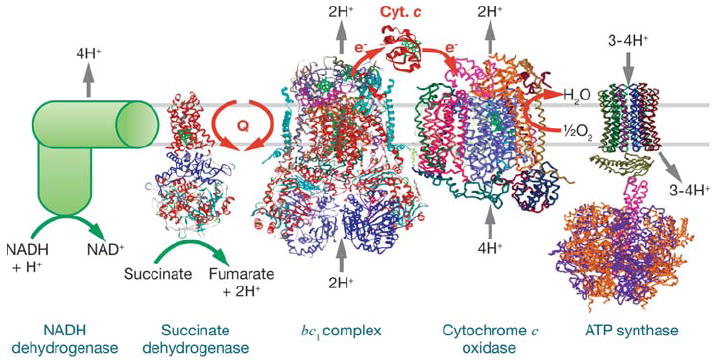
Complexes of the respiratory chain. These include Escherichia coli NADH dehydrogenase (145), succinate dehydrogenase 1NEN, bc1 complex 1PP9, cytochrome c oxidase 1V54, and cytochrome c 1HRC.
The respiratory system can be “uncoupled”; that is, the electron transfer process can proceed without resulting in net proton pumping or ATP synthesis, either because the pumped protons are leaked back across the membrane by protonophores or by uncoupling proteins that dissipate ΔμH+, leading to the production of heat. This mode of operation is of physiological importance in both plants and animals when there is the need to make heat as well as ATP. There is evidence that the respiratory complex cytochrome c oxidase (CcO) has the ability to control its own pumping efficiency as well by “intrinsic uncoupling” via proton backflow through the protein in response to a buildup of the membrane potential (1) or to signals such as phosphorylation (2). Proton backflow may be one mechanism to prevent the buildup of a high ΔμH+, which can inhibit electron transfer and allow intermediates, such as semiquinones, to persist (3). Semiquinone is known to reduce O2 directly, ultimately leading to the production of reactive O2 species (ROS) such as the hydroxyl radical. Mitochondria have sophisticated systems to scavenge ROS, including proteins such as superoxide dismutase, but even low levels of ROS can be damaging. One explanation for the complexity of the mammalian respiratory complexes, in terms of a larger number of subunits compared to the bacterial forms, is that the extra subunits provide protection and stability in the presence of toxic compounds. The far fewer subunits of bacterial complexes, along with a highly homologous catalytic core, make the prokaryotic enzymes excellent model systems for mechanistic studies of energy transduction, especially because they are more amenable to site-directed mutagenesis.
In this review, we discuss the current understanding of the energy transduction mechanism that allows electron transfer energy to be converted to a membrane potential gradient by means of a series of proton pumps. A diversity of chemistries is used by the three proton-pumping complexes, which work in series, each using different segments of the redox potential gradient. In spite of the diversity, the movement of protons through these proteins has many features in common. The principles of electron-driven proton transfer are illustrated in this review mainly by discussion of the wealth of information available from studies of CcO. This complex encompasses a variety of chemistries, including long-range proton transfer, vectorial charge separation, and redox-driven proton pumping. Analogies to the other well-characterized proton-motive system, the bc1 complex, are also explored.
LONG-RANGE PROTON MOVEMENT THROUGH PROTEINS
Pathways for proton transfer have been identified in the structures of the photosynthetic reaction center, bacteriorhodopsin (4), the bc1 complex (5), and in CcO structures (6). Within the protein, evidence supports the model that protons move rapidly through hydrogen-bonded chains of water and amino acid residues, presumably via a hopping mechanism (7). One mechanism proposes a sequential, isoenergetic exchange of hydrogen bonds and covalent bonds, which allows a proton to be added to one end of a chain of waters while another proton is released from the other end. Proton transfer through proteins may be rapid because a single file of hydrogen-bonded waters, as shown in model channel studies, is capable of moving a proton 40 times faster than proton transfer through bulk water (8). Proton pathways may begin or end with an exchangeable donor or acceptor, such as quinol, quinone, or O2. At the protein surface, residues that begin or terminate pathways are generally carboxylate or histidine residues and sometimes arginine. The flow of protons, from a donor at the protein surface through a hydrogen-bonded water chain to an acceptor, may be interrupted by a protonatable group, such as a carboxylate residue, that can store a proton and then release it as dictated by the demands of the next acceptor. The waters of the pathways are generally coordinated by polar side chains, but this is not an absolute requirement because molecular dynamics (MD) simulations of cytochrome oxidase and artificial channels (8, 9) predict water chains forming in hydrophobic regions. Not all of the waters that are predicted to play a role in proton transfer are seen in crystal structures—even at high resolution (10, 11), but unseen waters can be modeled into spaces in structures (12). In addition, when many waters are seen in a structure, the proton pathways are not necessarily obvious, e.g., the proton exit pathway of CcO. This may relate to the ability of proton pathways to form and dissipate during the catalytic cycle (4). For example, the proton uptake pathway used to reprotonate the Schiff base in bacteriorhodopsin was not observed in the earliest crystal structures, but it was found in the structure of a mutant that mimics a later intermediate of the photocycle (10). Transient proton transfer pathways may be used by proteins to “gate” the flow of protons in order to provide directionality or to control the timing of proton delivery.
CYTOCHROME c OXIDASE: AN ELECTRON TRANSFER-DRIVEN PUMP
Cytochrome c oxidase is the terminal electron acceptor in the electron transfer chain, carrying out the critical tasks of reducing oxygen to water and pumping protons across the membrane. Although it differs significantly in structure, prosthetic groups, and catalytic function from the other members of the chain, the concerted movement of protons and electrons is common to all of the energy-transducing complexes.
Proton Pathways and the Study of Proton Movements
Structural information now exists for two of the three energy-transducing electron transfer complexes of oxidative phosphorylation (Figure 1), with crystal structure resolutions often below 3 Å. However, the largest amount of structural, mutational, and kinetic information concerning proton transfer is available for CcO. The bovine CcO structure has been resolved to 1.8 Å (13) along with a considerable amount of well-defined water within the protein. Additionally, there are good crystal structures of the bacterial aa3-type oxidases from Paracoccus denitrificans (14, 15) and Rhodobacter sphaeroides (16), with a more recent isotropic crystal (2.35 Å) of the two subunit R. sphaeroides enzyme wherein the positions of bound lipids, detergents, and Cd2+, as well as water can be seen clearly (17). Both of these bacterial oxidases are amenable to genetic manipulation leading to the production of numerous mutant forms.
CcO has been well characterized spectroscopically with respect to its multiple metal centers: hemes a and a3, a dinuclear copper center (CuA), a type II copper (CuB), along with a non-redox active Mg and a Ca/Na-binding site (Figure 2) (18, 19). Time-resolved spectroscopic studies have shown that electrons from soluble cytochrome c are delivered first to CuA in subunit II, then to six-coordinate heme a in subunit I, and then laterally to the heme a3/CuB active site, where O2 is reduced to water (19).
Figure 2.
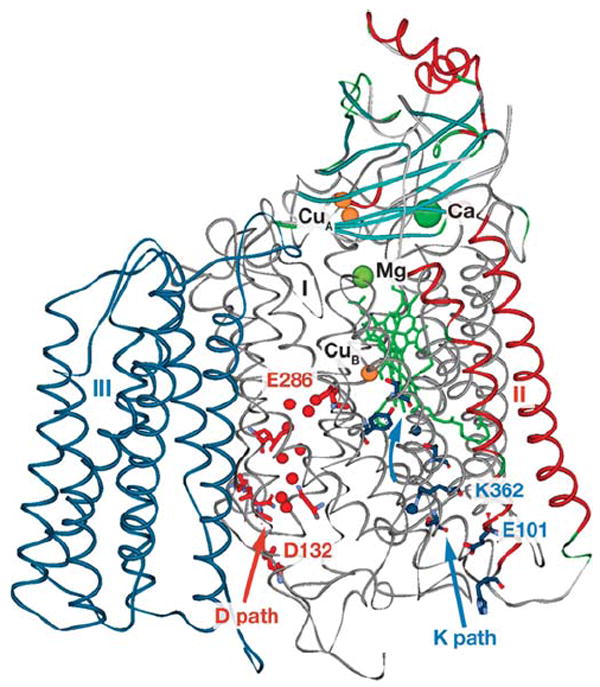
CcO (R. sphaeroides numbering in structure 1M56) showing the D path (red) and K path (blue) with D-path waters (red spheres) and K-path waters (blue spheres). Heme a and a3 (green) are stick structures with Ca and Mg metals (green spheres) and Cu metal (orange spheres).
Two proton uptake pathways lead from the inner surface of cytochrome oxidase (the negative side of the membrane) toward the buried heme a3-CuB active site where O2 binds to Fe of heme a3 (Figure 2). As yet, clear identification of an exit pathway for pump protons is lacking. Both proton uptake pathways were identified in the oxidase structures with the help of knowledge gleaned from prior mutagenesis experiments (20-22). The D pathway consists of a series of hydrogen-bonded waters anchored by D132 on the surface and E286 in the interior between hemes a and a3, approximately 26 Å above D132 (16) (R. sphaeroides CcO numbering is used throughout). Substrate protons (those destined to make water) flow from E286 to CuB, a distance of 10–12 Å, through a short series of waters that are not resolved in the oxidase structures but can be modeled into a hydrophobic cavity between E286 and the active site (23-26). The role of E286 in proton transfer has been extensively examined (27-29). In the native oxidase, E286 appears usually to be in its protonated form because of a high pKa and rapid reprotonation from the bulk solvent through D132 (30).
The K pathway appears to begin at E101 of subunit II and to sequentially involve S299, K362, T359, the hydroxyl farnesyl of heme a3 and Y288 of subunit I (Figure 2). Computational studies have suggested positions for K pathway waters (31) because only two are resolved in the structures. Side-chain movements of K362 (32), T359 (31), and Y288 (25) have been proposed.
The roles of the D and K pathways are best introduced in the context of the O2 reduction mechanism (Figure 3). Oxygen binds to heme a3 only after heme a3 and CuB are reduced. This splits the catalytic cycle of CcO into two parts. In the metal reduction phase, electrons flow one at a time from two cytochromes c through CuA and heme a to heme a3/CuB. Studies indicate the uptake of one proton with each of the two electrons introduced into the active site (33-36); the uptake is argued as a requirement to maintain electrostatic neutrality as the electrons move into the hydrophobic environment (34, 37). Both of the protons of the metal reduction phase may be taken up by the K pathway (35, 38) or the first by the K pathway and the second by the D pathway (39, 40). The initial steps of the subsequent O2 reduction phase are very rapid, beginning with O2 binding, moving through O2 bond scission, with the formation of the P oxoferryl state (Fe4+=O CuB2+−OH−) and likely a tyrosine radical. These steps can proceed rapidly because the four electrons (two from Fea3 one from CuB, and one from Tyr288) and one proton to be consumed are already available in the active site. However, the O2 reduction phase ends with two slower steps that are rate limited by the transfer of substrate protons from the D proton pathway into the active site (27). One proton is required for the P to F transition, where F is postulated to be a protonated form of P, and one or two for the F to O transition (this number depends upon whether the single-turnover reaction or the steady-state cycle is being described). The input of two more electrons (one to Fea34+ and one to Y288) returns the binuclear center to its oxidized form O (Figure 3).
Figure 3.
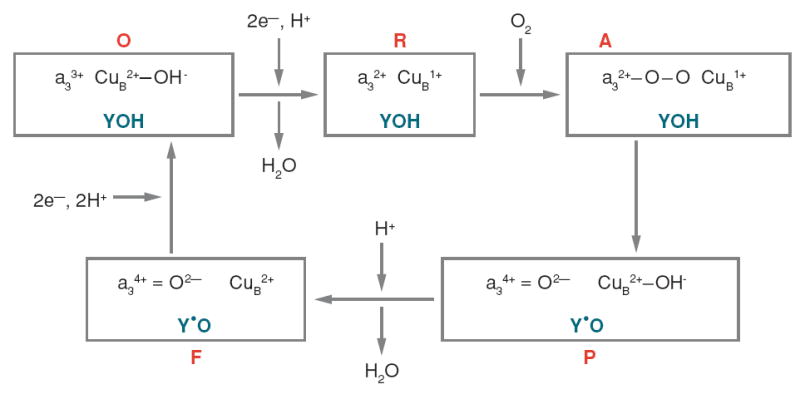
Proposed oxygen reduction reactions at the active site of CcO during steady-state turnover. Abbreviations used are as follows: Y, Y288 (R. sphaeroides numbering); R, reduced; A, oxy; P, F, and O are as described in the text. Only substrate protons are indicated. The two phases of the catalytic cycle merge to a certain extent, but the metal reduction phase is essentially O to R, whereas the O2 reduction phase is A to F.
Although mutagenesis studies indicate that alteration of the D pathway eliminates proton pumping (41, 42), mutagenesis of the K pathway does not (21); all four of the pump protons appear to be taken up by the D pathway. There is an as yet unidentified path beyond E286 that must take protons to the proton-pumping element(s) and then to a proton exit pathway, leading to the outer surface of the oxidase complex (the positive side of the membrane).
A variety of techniques initially developed to study O2 reduction and internal electron transfer has been adapted to study proton transfer through CcO. The flow-flash technique (43) has provided information on proton uptake (44, 45) and release (46, 47) during the P to F and F to O transitions in single-turnover experiments. Laser-induced electron injection has also been used to observe proton release (48) during the F to O transition. The proton uptake and release events are monitored with pH-sensitive dyes. Electrometric measurements that follow the movement of charge in the protein have been used to deduce proton transfer during the metal reduction phase (49-52) and during the F to O transition (53, 54). The Mg at the subunit I/II interface can be replaced in vivo with Mn (55) creating a sensitive paramagnetic probe for water and proton movement in the area above the hemes (56-58). The acquisition of high-resolution crystal structures of CcO has allowed the prediction of proton movements by computational methods (23, 59-61). Fourier transform infrared (FTIR) spectroscopy analysis, which has been used to great benefit in studies of bacteriorhodopsin, is beginning to yield information on the more complicated CcO enzyme (58, 62-64).
Postulated Mechanisms of Proton Pumping
Protons are known to move rapidly through ordered chains of water, but how the rate and direction of movement is controlled and how it is coordinated to electron transfer is controversial. Mechanistic questions also arise from the fact that protons are moved against an electrochemical gradient, requiring a gating process to prevent the backflow.
Theoretical considerations
Since the discovery of the proton-pumping function of cytochrome oxidase by Wikstrom (65), the theoretical requirements for redox-driven proton pumps have been discussed in detail (66-68). At a minimium, the protein must contain at least one group capable of existing as either a protonated or deprotonated species; this is termed the “pumping element.” The pumping element may be a protein residue, a heme propionate, a water, or some combination of these. The pumping element must at one point be accessible to protons from the inner surface of the oxidase (the negative surface of the membrane) and then at a later point be accessible to the outer surface (the positive surface of the membrane) to release the proton. The pumping element may change its pKa (proton affinity) during the pumping cycle, but this is not absolutely required. There are two absolute requirements. One is a mechanism to couple the free energy of O2 reduction to the proton pump. The other is for one or more “gating” mechanisms, which may be physical, chemical, or kinetic, that provide directionality to the flow of protons and inhibit a pump proton from dropping back through the electrical gradient to the inner surface of the protein. For full details of the coupling and gating mechanisms proposed for the pumping schemes, discussed below, the reader is directed to the references.
Coupling to electron transfer through CuA or heme a
On the basis of a great deal of work on the structure and chemistry of the CcO redox centers, two well-formulated pumping schemes were proposed in the 1980s involving CuA and heme a, the centers that transfer electrons to the heme a3-CuB active site. Because the mechanism of pumping is likely to be the same for each of the four pump protons, one attraction for focusing on these metal centers was that they should undergo the same redox chemistry with each transfer of an electron from cytochrome c to O2. In contrast, the active site is in different conformations at different points in the catalytic cycle. Babcock & Callahan (69) proposed that the alternating strength of a hydrogen bond to the formyl constituent of heme a, during redox cycling of this center, provided the energy for a geometric change of the hydrogen donor. Weakening of the hydrogen bond between the donor side chain and the formyl group allowed transfer of a proton from an input proton pathway to an output proton pathway.
Another mechanism, proposed by Chan and colleagues (70), involved a ligand-switching scheme driven by changes in the coordination geometry of CuA upon reduction and oxidation (71). Upon reduction, movement of CuA is posited to induce a nearby tyrosine to deprotonate and ligate the metal. The released proton is transferred to a cysteine ligand as the cysteine is displaced from the CuA. Relaxation to the oxidized coordination geometry reverses these events. Although ingenious, these schemes lost support with the discovery that the heme-Cu oxidase of Escherichia coli, cytochrome bo3, pumps protons without the benefit of CuA or a formyl group on its low-spin heme (72).
A more recent proton-pumping mechanism involving heme a has been proposed by Yoshikawa and colleagues; this is based on their deductions from high-resolution structures of bovine CcO (73) and recent analyses of mutant forms in HeLa cells (13). Comparison of the reduced and oxidized forms shows an alteration in the position of an aspartate residue near the outer surface of subunit I. In the oxidized enzyme, the aspartate is buried in the hydrophobic environment of the protein, its affinity for protons is high, and it is connected to a channel involving water clusters and the formyl group of heme a. In the reduced form, the aspartate is exposed to the outer (positive) surface, its affinity for protons is low, and it is disconnected from its proton source. Proton movement to the aspartate is proposed as coupled to the reduction and oxidation of heme a rather than directly to the chemistry at the active site. However, the critical aspartate and other important residues are not present in CcO of bacteria or plants, implying that the proton-pumping mechanism of CcO is not conserved throughout evolution.
Coupling to electron transfer events at the active site
In the 1990s, the paradigm that CcO activity is primarily regulated by the rate of proton uptake began to emerge from a number of studies. For example, the steps of O2 reduction seemed to be slowed by the rate of proton delivery (74), and an association was found between proton uptake and the reduction of heme a3 and CuB prior to O2 binding (75). The intrinsic rates of electron transfer between CuA, heme a, and heme a3/CuB were found to be much faster than that of maximum turnover (76, 77), indicating that the electron transfer reactions themselves are not rate determining. With this knowledge, plus comparative analyses of conserved residues in sequences of more than 30 CcO forms, pumping mechanisms emerged, linking the uptake of pump protons to O2 chemistry at the active site.
In a series of publications, Wikstrom and colleagues (78-80) concluded that proton pumping was restricted to the O2 reduction phase of the catalytic cycle. In terms of energetics, this was an attractive concept because the free energy available during the O2 reduction phase appears far greater than that of the metal reduction phase owing to the large redox drop from cytochrome c to O2 (ΔE ~ 580 mV). In essence, the electron transfer reactions can be viewed as using their energy to create O2 reduction intermediates with extremely high affinities for protons (high pKa). The original “histidine cycle” of Wikstrom’s group (81) posits that negatively charged O2 intermediates at the active site provide the driving force for pump proton uptake from the inner surface of the oxidase to the pumping element, a histidine ligand (H334) of CuB. The subsequent protonation of the O2 reduction intermediates by substrate protons provides the electrostatic repulsion that triggers the release of the pump protons from the histidine to the positive surface of the membrane. The histidine cycle mechanism requires that the pump protons are taken up prior to substrate protons in order to ensure the coupling of O2 reduction to proton pumping. In order to pump two protons in both the P to F and the F to O transitions, the model postulates that H334 is bound to CuB in its imidazolate form (Im−) and then dissociates when protonated to the imidazolium form (ImHH+). A critical gating feature is that the pump protons must not be allowed to combine with the O2 reduction intermediates at the active site, even though it is the electrostatic interaction between the pump protons and the active site charge that drives pump proton uptake.
Shortly following the proposal of the histidine cycle, the acquisition of crystal structures of CcO provided a wealth of information applicable to proton pumping, along with some disappointments. The D and K pathways leading from the inner surface of the oxidase to the active site (Figure 2) could be identified with the help of data from prior mutagenesis experiments. However, the plethora of hydrophilic residues and waters above the active site did not reveal an obvious proton exit pathway that would have helped to locate the site of pumping. Further, the D pathway appeared to terminate at an internal glutamate, E286, located between hemes a and a3, some 10–12 Å from the active site. The water chain that must provide the pathway for substrate protons between E286 and the active site was not resolved in any of the structures.
With the possibility that E286 serves as a branch point that alternately directs protons to the pump or to the active site, it became possible to envision a pumping element that was not a metal ligand at the active site. Rich and colleagues (82) used the structural information to postulate the “glutamate trap,” in which the pumping element was proposed as being above E286, with E286 preventing backflow. A more detailed mechanism, based upon structural data indicating conformational changes of E286 depending upon its protonation state, was postulated by Brzezinski & Larsson (83, 84). In this scheme, E286 first donates a substrate proton to either the P or F oxygen reduction intermediates. Anionic E286 shifts upward toward a cluster of heme a and a3 propionate groups, closely associated with R481 and R482 (Figure 4). The proximity of anionic E286 induces a substantial increase in the pKa (proton affinity) of this cluster that drives the transfer of a proton from the D pathway (presumably through waters bypassing E286) to the cluster. Reprotonation of E286 from the inner surface of the oxidase allows it to relax to its original position, the pKa of the accepting cluster drops, and the proton is ejected to the positive surface of the membrane. The movement of E286 is supported by FTIR spectroscopy evidence (28, 64) and computational analyses (23, 59, 85) in addition to the structural data (16).
Figure 4.
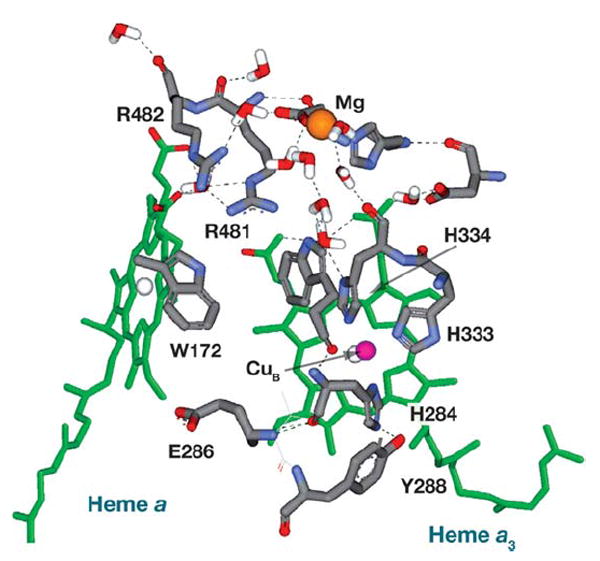
The area surrounding heme a and a3, including the nonredox Mg, is shown in the R. sphaeroides CcO (1M56) (16). The arginine pair interacts closely with the heme propionates. The nitrogen on W172 is close to the heme propionate and away from E286.
Another pumping mechanism at a distance from the active site is suggested by recent MD simulations that show movement of a conserved loop that brings W172 within hydrogen-bonding distance of E286 (86). If this movement is reversible, it could form the central element of a proton pump that moves each pump proton from E286 to a heme a3 propionate via W172.
In 1999, use of the electrometric technique led to the somewhat surprising conclusion that two protons are pumped during the metal reduction phase of the catalytic cycle, but only when this phase immediately follows the O2 reduction phase (49). Later experiments support this (51, 52). The energy required for pumping in the metal reduction phase may be conserved from the O2 reduction phase by the retention of hydroxyl ligands with a high pKa or by virtue of there being significantly more energy available in the metal reduction phase because of a high redox potential for heme a3/CuB during steady-state turnover (50, 52). In a modified form of the histidine cycle (39), E286 alternately directs protons to a dissociated histidine ligand of CuB (which binds a single proton in this model) or to O2 reduction intermediates on heme a3 and CuB. As before, the arrival of a substrate proton expels the pump protons from a heme propionate/water cluster to the outer surface of the oxidase. An evolved form of this model proposes specific pathways for the pump and substrate protons deduced from the positions of waters (25) conserved in all of the current CcO structures. Key conserved waters are proposed to be important pumping elements, which account for the number of CcO forms, including engineered mutants (29, 87), that can pump without a carboxyl residue at position 286. This model also invokes a novel gating mechanism, involving the dissociation of the H284-Y288 cross-linked pair from CuB in response to the overall charge at the heme a3-CuB center. A hydroxyl ligand on CuB provides the driving force for the uptake of pump protons at various stages of the catalytic cycle.
The bacterial protein bacteriorhodopsin is a light-driven proton pump in which light absorption induces isomerization of a bound retinal. Subsequent thermal reisomerization of the retinal drives the protein through a conformational series that pumps a proton across the bacterial membrane. Nine crystal structures, corresponding to different steps in the pumping cycle, have been analyzed to elucidate the mechanism of proton pumping (10) along with extensive FTIR spectroscopy analysis (88, 89). One of the findings from this extraordinary work is that the waters required to form connections in the input proton transfer pathway of bacteriorhodopsin are not all in position throughout the catalytic cycle. Rather, the proton pathway dissipates and reforms at various steps. By doing so, directionality is conferred on the pumping process. Transient water chains also provide the required gating function in a new scheme from the Wikstrom group (26). The model invokes the formation and dissipation of short chains of hydrogen-bonded water extending from E286 to either the propionate/arginine/water cluster above E286 or to the heme a3-CuB water center. As in other schemes, the arrival of a substrate proton provides the electrostatic repulsion that expels the pump proton from the propionate/arginine/water cluster to the outside.
CcO converts redox energy to a potential gradient by two mechanisms: a proton pump and vectorial redox chemistry. For the latter, electrons come from outside the membrane, and protons are drawn from inside the membrane to accomplish the reduction of O2 to water at the buried heme a3-CuB center. Here, the charges annihilate as O2 is reduced to water, resulting in the loss of one negative charge from the outside and one positive charge from the inside, equivalent to the transfer of one proton across the membrane. The addition of the electron transfer-driven proton pump translocates another proton for each electron consumed. Since the introduction of Wikstrom’s first histidine cycle, a common theme for proton pumping has emerged, although there is no consensus regarding the details of the process. The system is designed such that the introduction of negative charge (electrons) into the buried heme a/heme a3/CuB centers is the driving force for all of the protons taken up from the inner surface of the oxidase. Pump protons are directed to a site where they cannot protonate the O2 intermediates. Substrate protons both protonate the O2 intermediates and provide the charge repulsion that expels the pump protons to the outside.
This generalized mechanism provides a framework for efforts to identify the key pumping elements and to understand how rate and efficiency are controlled. Many questions remain. Where is the exit pathway? How is proton pumping gated to inhibit the movement of protons back down the electrical gradient, and how many gates are involved? In what form is free energy provided for pumping during the metal reduction phase? Work on all of these questions, and more, continues.
Experimental Insights
The mitochondrial CcO has 13 subunits, but the 3 largest core subunits are highly homologous to those of bacterial CcOs, which are usually made up of only four peptides. The simpler bacterial CcOs are experimentally more accessible because they can be easily grown and genetically altered. Removal of subunits and site-directed mutagenesis of protein residues have been powerful tools to study the mechanism of proton pumping and metal inhibition.
The effect of subunit III on proton transfer
Subunit III of CcO is a member of the highly conserved, three-subunit “catalytic core” of the enzyme. The subunit is closely associated with subunit I in the membrane (Figure 2), and recent work shows that subunit III has a profound effect on the transfer of protons through subunit I (90). The role of this entire subunit can be assessed because subunit III can be removed from CcO without strongly inhibiting the initial activity of the enzyme (90-92). Of the two pathways leading from the inner surface of CcO, only the D pathway is close to subunit III (Figure 2). In fact, the crystal structures of CcO show that D132 is located at a junction of subunits I and III such that half of the residues surrounding D132 come from subunit III. In the absence of subunit III, D132 is considerably more exposed to solvent. Single-turnover experiments show large differences in the rate of D pathway proton uptake in the presence and absence of subunit III, from >10,000 to ~350 s-1, respectively, at pH 8 (92). Rapid proton uptake is restored to subunit III-depleted CcO at low pH (<6). With steady-state turnover, the overall activity of subunit III-depleted CcO is limited by the rate of proton uptake through the D pathway at pH 7 and above. This situation can be exploited for the analysis of D pathway mutants.
Slow proton uptake in the absence of subunit III may result from changes in the pKa of D132 and/or the loss of proton-collecting groups on the protein surface (92-94). Such a “proton antenna” has been proposed to aid proton uptake in bacteriorhodopsin (95, 96), the photosynthetic reactions center, as well as in CcO (6). Surface groups, e.g., carboxylate residues and histidines, are argued to trap protons from the bulk solvent and transfer them along the surface of the protein by a process of release and recapture, effectively increasing the proton concentration near the initial acceptor of a pathway. In CcO, single-turnover experiments show that the rate at which E286 is reprotonated from the bulk solvent, via D132 and the D pathway water chain, may be 1000-fold greater than the bimolecular rate constant (4 · 1010 M−1s−1) for the diffusion-limited transfer of a buffer proton to D132 (97). This is interpreted as evidence for a proton antenna for the D pathway. By using the rate of O2 reduction by the wild-type and subunit III-depleted oxidases at different pH values (90) to estimate bimolecular rate constants of proton uptake, evidence for the continuous operation of a proton antenna during steady-state turnover can be derived (Figure 5). In the absence of subunit III, however, the estimated bimolecular rate constants for proton uptake do not significantly exceed the diffusion-limited value (Figure 5); the effect of the postulated proton antenna is clearly diminished. Without subunit III, the number of antenna residues on the inner surface of CcO may fall below the threshold required for its operation (98).
Figure 5.
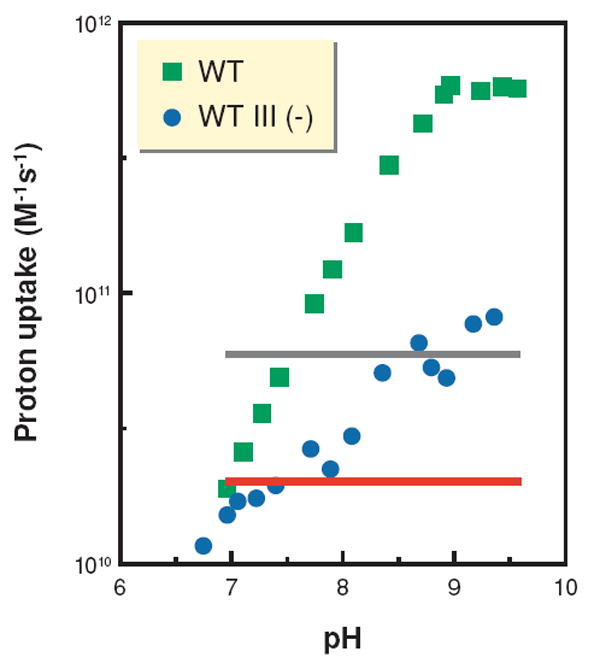
The absence of subunit III decreases the rate of proton uptake into the D pathway to the diffusion limit. Estimated bimolecular rate constants for the uptake of protons into the D pathway during steady-state turnover were calculated from the rates of steady-state activity of wild-type [WT, and without subunit III, WT III (−)] CcO. The red and gray lines represent diffusion-limited rate constants of 2–6 · 1010 M−1s−1, i.e., the range of rate constants expected if the diffusion of buffer to D132 through bulk solvent determines the rate of proton uptake. Rate constants above these limits are suggestive of the function of a proton antenna that increases the local concentration of protons near D132 of the D pathway.
In experiments with CcO reconstituted into phospholipid vesicles in the absence of uncouplers or ionophores, the rate of O2 reduction is slow. In large part, this is due to the inhibition of proton uptake into the D and K pathways by the membrane potential. Nevertheless, a slow rate (~1/10 Vmax) of “controlled” turnover is observed, apparently supported by proton flow to the active site from the outer surface of the enzyme (99). The removal of subunit III inhibits this backflow of protons, as evidenced by a much slower rate of controlled turnover (100). The effect of removing subunit III is similar, but additive, to inhibiting the backflow pathway with zinc (see the section Metal inhibition of proton uptake, below). Assuming that the backflow proton pathway is actually the normal proton exit pathway operating in reverse, it appears that subunit III facilitates both the uptake and exit of pump protons. This is consistent with the long-known observation that the removal of subunit III decreases the efficiency of proton pumping to approximately half that of CcO containing subunit III (101).
Approximately a decade ago, it was noted that turnover of subunit III-depleted CcO resulted in the irreversible loss of activity (102). Later characterization showed that this “suicide inactivation” process decreased the catalytic life span of CcO (i.e., its total number of turnovers, independent of time) owing to the increased probability that an inactivating structural alteration of the CuB center occurs during the catalytic cycle (90, 91). The catalytic life span of subunit III-depleted CcO can be less than 0.1% that of the native enzyme. Recent experiments show that suicide inactivation and proton transfer are tightly linked (103). Mutations that inhibit the D pathway, but not the K pathway, induce suicide inactivation. Likewise, inhibition of proton uptake by a membrane potential increases suicide inactivation. Because the D pathway transfers substrate protons after O2 binds, it is argued that slow proton uptake increases the lifetime of reactive O2 reduction intermediates (such as heme a3 oxoferryl forms or a tyrosine radical), which in turn initiate the chemistry of inactivation. Simultaneous inhibition of the proton backflow pathway from the outer surface and the D pathway from the inner surface (using the D132A-R481K mutant described below) greatly increases the probability of suicide inactivation, even in the presence of subunit III. This result suggests a physiological role for proton backflow: Under conditions of a high membrane potential, where proton uptake by the D pathway is strongly inhibited, proton flow to the active site via the backflow pathway helps prevent inactivation of CcO. Measurements of suicide inactivation offer another experimental tool for the analysis of mutants affecting the D and backflow pathways because the catalytic lifetime of CcO is proportional to the rate of proton transfer to the active site.
Metal inhibition of proton uptake
Metal inhibition of voltage-gated proton channels has been well documented (104, 105), and more recently it has been found that proton uptake by the photosynthetic reaction center, the bc1 complex, and CcO are inhibited by micromolar concentrations of certain metals, including zinc, nickel, and cadmium (1, 106-109). In each case where the metal site has been identified, the coordinating groups include at least one histidine, one or more carboxylate residues, and waters. The sites of metal binding are difficult to determine as it appears that often two of the ligands must be removed in order to prevent metal binding and inhibition (110). In addition, metal binding is pH dependent because protons compete with the metal for binding (1, 104).
Low concentrations of zinc, cadmium, or nickel inhibit CcO catalytic turnover (KI ≤ 5 μM), and inhibition is specific for these three metals (1). The sum of current results suggests the presence of at least three inhibitory binding sites for metal on R. sphaeroides CcO because evidence exists for metal inhibition of proton uptake into the D and K pathways from the inner surface of the complex as well as inhibition of proton backflow from the outer surface.
Single-turnover experiments have shown that zinc slows the uptake of protons about 30-fold from the bulk solvent into the D pathway (109). However, the fact that zinc does not induce suicide inactivation, which is highly sensitive to the rate of proton uptake via the D pathway, indicates that zinc has little effect on D pathway activity during steady-state turnover (103). These two results need not be contradictory if zinc binds more slowly than protons at the D pathway-binding site. In the single-turnover experiment, preincubation with zinc gives it ample time to equilibrate into the proton site for binding at the D pathway, whereas in steady-state turnover, protons may successfully compete with zinc for binding. If the D pathway is not inhibited by zinc during steady-state turnover, it follows that the inhibition of steady-state activity is likely due to inhibition of the K pathway. In support of this, a new crystal structure of R. sphaeroides CcO reveals cadmium binding to residues E101 and H96 of subunit II at the entrance of the K pathway (17).
The fact that low concentrations of zinc further inhibit the slow turnover of CcO in vesicles in the presence of a potential gradient provides strong evidence for a specific proton pathway leading to the active site from the outer surface (1). The proton backflow pathway is inhibited with cadmium or zinc, but not nickel, whereas nickel does inhibit proton uptake from the inner surface. This selectivity argues against inhibition of proton movement through the lipid bilayer by alteration of its permeability by zinc and also argues against metal transfer across the lipid bilayer in the timescale of the experiment. It seems likely that the backflow of protons occurs by reversal of the normal exit pathway for pump protons. Zinc does not inhibit the activity of CcO in phospholipid vesicles in the absence of a membrane potential, suggesting that protons moving out of CcO successfully compete with the metal for binding or that a membrane potential is required for a conformational change that creates the zinc-binding site. The site of metal binding on the outside remains elusive, but identifying the site should help determine the location of the exit pathway, as in the case of the photosynthetic reaction center proton uptake pathway (111), and help clarify the pumping mechanism.
Mutants affecting proton movement
The mutation of selected amino acid residues has been particularly fruitful in defining the proton uptake pathways. With the advantage of high-resolution structures that resolve waters as well as side chains, it is possible to visualize regions where waters are held and to predict and test the involvement of specific side chains.
D pathway
The ends of the hydrogen-bonded water chain of the D pathway are anchored by D132 on the inner surface of CcO and E286, in between hemes a and a3 (Figure 2). Site-directed mutants of D132 and E286 in the aa3-type oxidases that remove a carboxylate residue from either of these positions strongly inhibit activity and proton uptake (21, 41, 45, 82).
The phenomenon of proton backflow from the outer surface was initially identified in the D132A mutant (41). The O2 reduction activity of this mutant is accelerated by a membrane potential, presumably because proton backflow is driven by the potential gradient (negative inside) across the membrane. Proton backflow is confirmed by measurement of rapid alkalinization on the outside of D132A vesicles upon initiation of turnover (112, 113). Because the structure of subunit I in the region of the proton backflow/exit pathway is unperturbed in the D132A mutant, it seems likely that proton backflow is a normal phenomenon that occurs with a high membrane potential. This may explain why there is no observation of proton pumping in wild-type CcO reconstituted into vesicles under the conditions of a high proton gradient (ΔpH) and transmembrane voltage gradient (ΔΨ) across the membrane (114).
The O2 reduction activity of D132A is partially rescued by micromolar concentrations of arachidonic acid, presumably by supplying a carboxylate group in the vicinity of the D pathway (41). The removal of subunit III from D132A further restores activity (99) apparently by exposing an alternative initial acceptor. The addition of arachidonic acid to D132A III (−) restores even more activity (99), but none of these CcO forms are capable of proton pumping (90).
A number of alterations of two amides further into the D pathway, N121 and N139, slow proton transfer (115). However, the substitution of N139 with aspartic acid either retains or substantially enhances (42) O2 reduction activity, but proton pumping is eliminated. This additional protonatable site in the D path is concluded to increase the effective pKa of E286 from 9.4 to 11.0 (54). Brzezinski and colleagues (116) argue that the resulting decrease in the anionic form of E286 eliminates proton pumping by decreasing the driving force for transfer to the proton-accepting cluster above E286. An alternative explanation is that the extra carboxyl alters the kinetics of proton transfer in the pathway such that the reverse reaction is facilitated to an extent that it successfully competes with proton pumping.
K pathway
The early observation that mutation of K362 inactivated the enzyme and prevented the reduction of heme a3 (53, 117) strongly supports the concept that the uptake of at least one charge-neutralizing proton via the K pathway is required during the metal reduction phase. The presumed binding site for this proton is a hydroxyl ligand of CuB, produced in the previous O2 reduction phase. The addition of a high concentration of hydrogen peroxide restores reasonable activity to K362M, presumably by bypassing the need for a K pathway proton in the metal reduction phase by supplying both oxygen and protons to the active site (38, 112). The mutation of T359 to alanine slows but does not eliminate K pathway proton transfer or CcO activity. The fact that T359A retains efficient proton-pumping activity supports the conclusion that pump protons are not supplied by the K pathway. Controversy remains about the roles of two subunit II residues at the entrance to the K path, E101 and H96, because they appear to affect CcO activity to differing extents depending on the bacterial enzyme that is studied (118).
Exit/backflow pathway
A pair of highly conserved arginine residues located above the hemes, R481 and R482, interact with the D propionates of hemes a and a3 (Figure 4). Arg481 bridges the two hemes by forming a hydrogen bond/ion pair interaction with propionates from each. Alteration of R481 to a lysine residue releases the interaction with the heme a3 propionate, with the intriguing result of decreased proton backflow. This is seen as inhibition of controlled turnover (plus ΔΨ) but a normal rate of uncontrolled turnover (119). The closer interaction of R481K with the heme a propionate produces additional effects, including a 20–40 mV decrease in the redox potential of heme a. The double mutation D132A-R481K blocks proton uptake through both the backflow pathway and the D pathway, with the results that O2 reduction is extremely inhibited (0.25% of native) (119) and that suicide inactivation is rapid (103). The precise role of R481 remains to be determined. The arginine may transfer protons itself, it may influence the protonation state of the propionates of the hemes, or it may help organize a water chain leading from E286 to the Mg center above the hemes (86).
Computational Insights
With the availability of high-resolution crystal structures of proteins and increased computational power, even large proteins can now be simulated in the computer. The movement of protein residues and explicit individual waters can be followed over time in molecular dynamics simulations. Along with electrostatic calculations, these theoretical analyses give insight into possible residue motion and water chain formation, and they suggest new experimental approaches and mechanistic ideas.
Water and proton dynamics
Although crystallography can reveal the positions of side chains and some of the water in a protein, many waters are mobile and hence not observed. For example, in a recent crystal structure of the two-subunit R. sphaeroides CcO at 2.35 Å resolution, 110 waters were observed (17), which is less than the calculated content. Yet, the current understanding that we have of energy transduction in the complexes of the respiratory chain critically involves water as part of proton conduction pathways. Thus, the positions of waters and changes in these positions during the reaction cycle are important for understanding proton transfer and pumping mechanisms. Computational methods to simulate water location and mobility are increasingly valuable tools for developing mechanistic models. Programs such as DOWSER (12) or GRID (Molecular Discovery) can be used to fill potential water sites in a structure with no van der Waals overlaps. The number of waters that have been placed within the two-subunit structures of CcO varies from 130 to 755 depending upon selection criteria, such as interaction energies (85, 120, 121).
Using MD simulations, methods have been developed to monitor the formation, nature, and persistence of hydrogen-bonded water chains (23, 31). Simulations are carried out with varying restraints on protein movement; either the complete protein backbone or everything outside the area of interest may be constrained to minimize computational cost. In order to predict the movement of protons themselves through water chains, quantum mechanics calculations have been combined with MD (23, 60, 122).
Simulations of the K pathway
In all CcO structures, the K path is found to be fairly “dry,” with only two conserved positions for water (25). However, water chains are formed during MD simulations of the two-subunit CcO of R. sphaeroides after the computational addition of waters (31). Interestingly, rotation of the amino acid side chain of T359 was required for completion of a hydrogen-bonded water chain that reached from S299 to the hydroxyl group of the heme a3 farnesyl chain. Another water chain was formed from the farnesyl hydroxyl to the active site via Tyr288 despite this being a hydrophobic region. These water chains were formed without any movement of the essential residue K362, which has been suggested to undergo a conformational change (123). However, this may not be inconsistent with the MD simulations, which were run for 2 ns, whereas a full turnover of CcO takes a millisecond, and hemes a, a3, and CuB were kept reduced for the duration of the analysis (31).
Simulations of the D pathway
Simulation of water chain formation in the D pathway shows a completely hydrogen-bonded chain from D132 to N139 (Figure 2) and then from N139 to E286. There is a break or discontinuity in the water chain around N139 (23). Proton movement through the D pathway varies with the protonation state of E286 at the top of the pathway. Deprotonated E286 causes a proton, introduced as a hydronium at the entrance to the channel, to move through the waters to protonate E286. However, when the simulation is started with protonated E286, the proton is held up near S200 and S201, about halfway along the D path, suggesting a proton trap at this site. Mutation of these serines to alanines inhibits CcO activity and slows proton movement in the simulations (23). These studies suggest that the D pathway could accommodate, or trap, a second (86) proton above N139.
Simulations of the proton exit
Simulation of wild-type CcO by MD reveals a possible segment of the exit pathway for pump protons as a continuous hydrogen-bonded water chain extending from E286 to the Mg center (23). Interestingly, the single-file nature of this series of waters is dictated by a narrow hydrophobic channel (Figure 6). It is observed that in the R481K mutant a movement of a loop containing the hydrophobic residues leads to the collapse of this water chain. This suggests that significant conformational changes may occur in response to highly conservative single mutations, which in this case could account for the observed phenotype of inhibited proton backflow in R481K (119).
Figure 6.
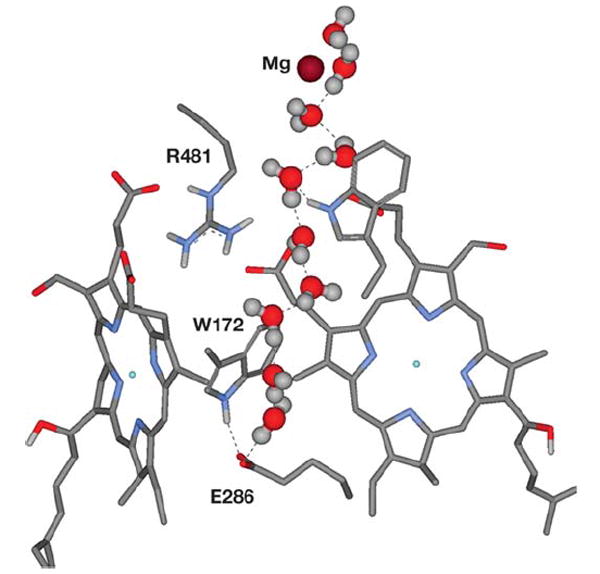
After over a nanosecond of an MD simulation of the R. sphaeroides CcO structure with added water (31, 86), a chain of hydrogen-bonded waters is clearly seen—stretching from E286 through a hydrophobic cavity to Mg. The glutamate has its carboxyl pointed up and is interacting with W172.
Protons move through CcO both for proton pumping across the membrane and for the reduction of O2 to water. The bifurcation of these two proton streams, at least in the O2 reduction phase of the catalytic cycle, has often been ascribed to E286, which appears to be capable of some rotational movement based on experimental, crystallographic, and computational methods (16, 26, 27, 59). An alternative to the movement of E286, however, is a conformational change of W172 (86). In MD simulations of wild-type CcO, W172 rotates to interact with E286, and such a movement could deliver a proton from E286 to the area above the hemes (Figure 4). In the R481K mutant, W172 remains hydrogen bonded to the D propionate of heme a3.
Electrostatic calculations
Electrostatic calculations are important for estimating the pKas of amino acid residues that may depend upon the redox states of hemes a, a3, and CuB, and thus aid in predictions of proton movement. Using a continuum dielectric model and Poisson-Boltzmann solution (124), a cluster of 18 residues in CcO were calculated to form an electrostatic network (125). The effect of the redox status of the metal centers in subunit I on the pKa values of these residues was examined. Although K362 in the K pathway did not alter its protonation state with redox state changes, E101 at the entrance to the K pathway did (by 2.6 pH units), even though it is 25 Å from the active site. The redox status of heme a was linked to changes in the arginine pair (R481/R482) above the propionates of hemes a and a3 and linked with Arg52, which closely interacts with the formyl group of heme a (125, 126). In the D pathway, E286 remained protonated in all redox states. However, later electrostatic analysis of P. denitrificans CcO suggested that the protonation of E286 was sensitive to redox changes (127).
Using comparative modeling, the caa3 oxidase of Rhodothermus marinus appears to substitute a tyrosine for E286, as does the ba3-type oxidase of Thermus thermophilus (127, 128). Calculations suggest that this tyrosine does not undergo a protonation change upon alteration of metal redox status (127). A water associated with the tyrosine at this site may be the protonatable and redox responsive element, opening the possibility that it is water that fufills this function even in the presence of E286 (25).
Olsson et al. (122) evaluated proton movements using a novel quantum mechanical analysis. From their results, they propose a movement of a proton from within the D pathway to E286 in concert with the movement of a proton from E286 to an acceptor toward the outer surface. Their analysis also supports pumping schemes discussed above (in the Coupling to electron transfer events at the active site section) in that protonation of the D propionate of heme a3 is followed by protonation of a hydroxyl bound at the heme a3/CuB center. Neutralization of this hydroxyl repulses the proton from the propionate to the outside and causes a high energy barrier for the backflow of protons.
A histidine ligand of CuB can be considered to be an unlikely site for protonation because of its nominally high pKa (122). However, other electrostatic calculations suggest that one or more of the CuB ligands can act as a proton loading site (61, 129), allowing for pumping mechanisms that include histidine protonation (see the Coupling to electron transfer events at the active site section, above).
Because of the salt bridges between the arginine pair (R481, R482) and the D propionates of hemes a and a3, the propionates were predicted to not be part of the proton pump (68). However, mutagenesis (119, 130), computation (86), and FTIR spectroscopy experiments (62, 131) implicate the arginine/ propionate cluster as part of the exit route for protons.
The variety of answers coming from different computational approaches suggests that systematic errors continue to be a challenge. Consensus among different analyses on different CcO structures could be the hallmark of valuable predictions.
COMPARISON OF THE PROTON MOTIVE COMPLEXES OF OXIDATIVE PHOSPHORYLATION
Little can be said about the pathways of proton transfer through NADH dehydrogenase given the current lack of high-resolution structures. One conundrum is that the metal centers of NADH dehydrogenase are largely or completely located in the extramembrane domain of the complex, although the proton-pumping mechanism must, by definition, be located in the transmembrane domain (132). How the redox reactions are coupled to the pump is a mystery (133).
In the case of the bc1 complex, a number of structures are available (108, 134-136) and interesting comparative features emerge. The bc1 complex moves protons from the negative to the positive surface of the membrane using a redox loop mechanism called the Q-cycle. The mechanism of the Q-cycle has been extensively reviewed elsewhere (137-140), but a brief summary follows. Reduced quinol, carrying two electrons and two protons, docks at the ubiquinone (Qo)-binding site toward the upper (positive) surface of the complex. A concerted two electron oxidation at Qo sends one electron to a high potential Fe/S center and the other to heme bL, and the two protons are released to the outer surface. The iron-sulfur protein transfers its electron to soluble cytochrome c, via cytochrome c1, whereas heme bL transfers its electron to heme bH toward the negative or inner surface of the complex. Near heme bH, there is a second quinone-binding site, ubiquinone (Qi), that binds oxidized quinone. This quinone is reduced by one electron from bH and one proton from the interior to produce a temporally stable semiquinone. A second round of quinol oxidation at Qo reduces another cytochrome c and reduces the semiquinone waiting at Qi. With the addition of another proton, neutral quinol is released from Qi into the membrane. When all of the charge movements are accounted for, it can be seen that the Q-cycle moves the equivalent of one proton from the negative to the positive surface of the membrane for each electron delivered to cytochrome c. This is precisely the same charge stoichiometry as in CcO, where only the electrons and protons used to reduce O2 to water are tallied.
A major difference between CcO and the bc1 complex is that CcO has evolved a proton pump that moves an additional proton per electron. The bc1 complex has not evolved a proton pump because there is only sufficient redox energy to drive the Q-cycle in the presence of a transmembrane voltage gradient. With the lack of a pump, there is no requirement for, or observation of, a proton transfer pathway, which extends all of the way through the bc1 complex. However, the Qo and Qi sites are each buried within the transmembrane domain such that proton transfer pathways are required for the exit and uptake of protons. Here the structure of quinones adds complication because the proton-binding carbonyls are separated by the length of the quinone ring. Thus, there exist two proton uptake pathways into Qi, one leading to each carbonyl. Likewise, there are two exit pathways for the protons released from Qo. The location of these pathways in the high-resolution yeast bc1 structure has been elegantly described (5).
The critical reaction in the bc1 mechanism is the initial oxidation of quinol at Qo, which is postulated to be a simultaneous reduction of the Fe/S protein and heme bL that sends the two electrons in two different directions (141, 142). The ability of Qo to carry out this concerted two-electron reaction is argued to be enhanced by the formation of hydrogen bonds between both quinol hydroxyl groups and two residues that subsequently accept the protons from the quinol (5). These residues are H181 of the Rieske Fe/S protein (yeast numbering) and E272 of cytochrome b. His181 of the Rieske protein is also a ligand of the Fe/S center, so that in accepting a quinol proton, the Fe/S center is acting as a one electron, one proton carrier. In the other proton exit pathway starting with E272, the glutamate rotates nearly 180° after it accepts the quinol proton in order to pass the proton to a heme propionate of the exit pathway via an intervening water (143). This movement is reminiscent of the postulated movement of deprotonated E286 of CcO up toward the propionates of hemes a and a3 (23, 83, 85). Interestingly, E272 of the bc1 complex is located in a conserved loop with the sequence PEWY, and E286 of CcO is located in a conserved sequence of PEVY. Possibly this structural motif is important in allowing glutamate movement.
As in CcO, zinc inhibits the activity of the bc1 complex at submicromolar concentration (106). A zinc-binding site involving a histidine and an aspartic acid has been observed in the structure of the chicken bc1 complex at the end of the Qo exit pathway, approximately 10 Å above E272 (108).
Two proton uptake pathways consisting of hydrogen-bonded water chains coordinated by polar groups lead to the quinone bound at Qi (144). One of these pathways begins with a glutamate residue, leading to an arginine residue and then onto the quinone via another series of waters. The structure suggests that upon reduction of quinone at Qi, the arginine is deprotonated and then reprotonated by the water chain leading from the glutamate on the inner surface of the complex. This chemistry is similar to that postulated for the D pathway of CcO, where E286 transfers a proton via a water chain to the O2 reduction intermediates and is then reprotonated by the water chain leading from D132 on the inner surface. In yeast, a cardiolipin molecule is well positioned to be the initial proton donor for the second proton uptake pathway leading to Qi. The cardiolipin could transfer protons via water to a lysine and then to the quinone carbonyl via a series of waters. Proton uptake by both the bc1 complex and CcO may be assisted by structural lipids (5, 90).
If the bc1 complex keeps pace with the activity of CcO, where Vmax ≥ 2000 e−/sec, calculations indicate that a proton antenna would be required at pH values above 7. The cardiolipin at the entrance to one of the proton uptake pathways has been suggested as part of a putative proton-collecting antenna.
In general, it appears that proton movements in CcO and the bc1 complex have many similarities and that obtaining a high-resolution structure of both has been an important factor in reaching a clearer understanding of the mechanism of coupling to the electron transfer processes.
SUMMARY POINTS.
Comprehensive knowledge of CcO structure, function, and kinetic/spectroscopic properties create a valuable model for examining the function of an electron transfer-driven proton pump.
A central theme has emerged that charge interactions drive both the uptake and the release of pump protons.
A cluster of heme propionates, two arginines, and associated waters is likely to be an acceptor site for pump protons en route to the outer surface, regardless of the mechanism of the pump. Similar clusters are involved in the exit of protons from the bc1 complex.
Proton transfer in CcO involves hydrogen-bonded water chains, but experimental results and computational analyses reveal complexities, such as the likely control by side-chain movements, the existence of proton traps, apparent discontinuities in the water chains, and the formation of water chains through both hydrophilic and hydrophobic regions.
An increasing understanding of proton transfer mechanisms within CcO has provided the basis for rational models for mechanisms of coupling proton pumping to the transfer of electrons to oxygen.
Subunit III of the catalytic core plays a profound role in facilitating the transfer of pump protons through subunit I and may supply a major portion of a proton-collecting antenna, even though it contains no metal centers.
The existence of a specific exit pathway for pump protons is supported by evidence for (a) proton backflow from the outer surface of CcO, which is specifically inhibited by Zn/Cd in CcO vesicles; (b) membrane potential-stimulated activity of D-path mutants, which are inhibited in proton uptake from the inner surface; and (c) mutant forms of CcO that are inhibited in proton backflow.
FUTURE ISSUES TO BE RESOLVED.
Which residues are the critical elements of the electron transfer-driven proton pump of CcO?
Is there a defined, controlled exit pathway for pump protons? If so, where is it?
What controls the directionality of proton pumping? How many gates are involved? Are the gates physical barriers, e.g., a space through which a proton cannot hop; thermodynamic barriers, e.g., pKa changes; or kinetic barriers, e.g., dissipation of water chains?
Is there physiologically significant variability in the efficiency of proton pumping? If so, is this controlled by regulating the backflow of protons? Does the catalytic core of cytochrome oxidase have a mechanism to regulate its efficiency? Could this be a key function of subunit III or is this only achieved by the additional nuclear-encoded subunits in eukaryotes?
How important is the regulation of efficiency of proton pumping in the respiratory chain in the overall process of physiological energy balance? Do signaling systems in the cell, such as phosphorylation, disease states, and aging, play a role in regulating mitochondrial efficiency?
Acknowledgments
This work was supported by NIH GM 56802 (to J.H.) and NIH GM 26916 (to S. F.-M.). We thank Robert Cukier and Steve Seibold for their input on the computational section.
- CcO
cytochrome c oxidase
- Respiratory complexes
the membrane-embedded metallocomplexes that make up the respiratory chain used for energy transduction
- Energy transduction
change of form of energy such as the electrical potential energy of an electron into a membrane pH gradient
- MD
molecular dynamics
- FTIR
Fourier transform infrared
- ΔpH
pH gradient across the membrane
- ΔΨ
transmembrane voltage gradient
- Qo
ubiquinone-binding site toward the outer side of bc1
- Qi
ubiquinone-binding site toward the inner side of bc1
LITERATURE CITED
- 1.Mills DA, Schmidt B, Hiser C, Westley E, Ferguson-Miller S. J Biol Chem. 2002;277:14894–901. doi: 10.1074/jbc.M111922200. [DOI] [PubMed] [Google Scholar]
- 2.Kadenbach B. Biochim Biophys Acta. 2003;1604:77–94. doi: 10.1016/s0005-2728(03)00027-6. [DOI] [PubMed] [Google Scholar]
- 3.Brand MD, Buckingham JA, Esteves TC, Green K, Lambert AJ, et al. Biochem Soc Symp. 2004;71:203–13. doi: 10.1042/bss0710203. [DOI] [PubMed] [Google Scholar]
- 4.Lanyi JK. Annu Rev Physiol. 2004;66:665–88. doi: 10.1146/annurev.physiol.66.032102.150049. [DOI] [PubMed] [Google Scholar]
- 5.Hunte C, Palsdottir H, Trumpower BL. FEBS Lett. 2003;545:39–46. doi: 10.1016/s0014-5793(03)00391-0. [DOI] [PubMed] [Google Scholar]
- 6.Adelroth P, Brzezinski P. Biochim Biophys Acta. 2004;1655:102–15. doi: 10.1016/j.bbabio.2003.10.018. [DOI] [PubMed] [Google Scholar]
- 7.Nagle JF, Tristram-Nagle S. J Membr Biol. 1983;74:1–14. doi: 10.1007/BF01870590. [DOI] [PubMed] [Google Scholar]
- 8.Dellago C, Naor MM, Hummer G. Phys Rev Lett. 2003;90:105902. doi: 10.1103/PhysRevLett.90.105902. [DOI] [PubMed] [Google Scholar]
- 9.Wu YJ, Voth GA. Biophys J. 2003;85:864–75. doi: 10.1016/S0006-3495(03)74526-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Schobert B, Brown LS, Lanyi JK. J Mol Biol. 2003;330:553–70. doi: 10.1016/s0022-2836(03)00576-x. [DOI] [PubMed] [Google Scholar]
- 11.Luecke H, Schobert B, Richter H-T, Cartailler J-P, Lanyi JK. J Mol Biol. 1999;291:899–911. doi: 10.1006/jmbi.1999.3027. [DOI] [PubMed] [Google Scholar]
- 12.Zhang L, Hermans J. Proteins. 1996;24:433–38. doi: 10.1002/(SICI)1097-0134(199604)24:4<433::AID-PROT3>3.0.CO;2-F. [DOI] [PubMed] [Google Scholar]
- 13.Tsukihara T, Shimokata K, Katayama Y, Shimada H, Muramoto K, et al. Proc Natl Acad Sci USA. 2003;100:15304–9. doi: 10.1073/pnas.2635097100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Iwata S, Ostermeier C, Ludwig B, Michel H. Nature. 1995;376:660–69. doi: 10.1038/376660a0. [DOI] [PubMed] [Google Scholar]
- 15.Ostermeier C, Harrenga A, Ermler U, Michel H. Proc Natl Acad Sci USA. 1997;94:10547–53. doi: 10.1073/pnas.94.20.10547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Svensson-Ek M, Abramson J, Larsson G, Tornroth S, Brzezinski P, Iwata S. J Mol Biol. 2002;321:329–39. doi: 10.1016/s0022-2836(02)00619-8. [DOI] [PubMed] [Google Scholar]
- 17.Qin L. PhD thesis. Mich. State Univ.; East Lansing: 2005. X-ray crystallographic studies of cytochrome c oxidase from Rhodobacter sphaeroides; p. 205. [Google Scholar]
- 18.Gennis R, Ferguson-Miller S. Science. 1995;269:1063–64. doi: 10.1126/science.7652553. [DOI] [PubMed] [Google Scholar]
- 19.Ferguson-Miller S, Babcock G. Chem Rev. 1996;96:2889–907. doi: 10.1021/cr950051s. [DOI] [PubMed] [Google Scholar]
- 20.Hosler JP, Ferguson-Miller S, Calhoun MW, Thomas JW, Hill J, et al. J Bioenerg Biomembr. 1993;25:121–36. doi: 10.1007/BF00762854. [DOI] [PubMed] [Google Scholar]
- 21.Fetter JR, Qian J, Shapleigh J, Thomas JW, García-Horsman JA, et al. Proc Natl Acad Sci USA. 1995;92:1604–8. doi: 10.1073/pnas.92.5.1604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Gennis RB. Science. 1998;280:1712–13. doi: 10.1126/science.280.5370.1712. [DOI] [PubMed] [Google Scholar]
- 23.Cukier RI. Biochim Biophys Acta. 2004;1656:189–202. doi: 10.1016/j.bbabio.2004.03.006. [DOI] [PubMed] [Google Scholar]
- 24.Tashiro M, Stuchebrukhov AA. J Phys Chem B. 2005;109:1015–22. doi: 10.1021/jp0462456. [DOI] [PubMed] [Google Scholar]
- 25.Sharpe MA, Qin L, Ferguson-Miller S. In: Biophysical and Structural Aspects of Bioenergetics. Wikstrom M, editor. Cambridge: R. Soc. Chem.; 2005. pp. 26–54. [Google Scholar]
- 26.Wikstrom M, Verkhovsky MI, Hummer G. Biochim Biophys Acta. 2003;1604:61–65. doi: 10.1016/s0005-2728(03)00041-0. [DOI] [PubMed] [Google Scholar]
- 27.Adelroth P, Karpefors M, Gilderson G, Tomson FL, Gennis RB, Brzezinski P. Biochim Biophys Acta. 2000;1459:533–39. doi: 10.1016/s0005-2728(00)00194-8. [DOI] [PubMed] [Google Scholar]
- 28.Lubben M, Prutsch A, Mamat B, Gerwert K. Biochemistry. 1999;38:2048–56. doi: 10.1021/bi981859k. [DOI] [PubMed] [Google Scholar]
- 29.Aagaard A, Gilderson G, Mills DA, Ferguson-Miller S, Brzezinski P. Biochemistry. 2000;39:15847–50. doi: 10.1021/bi0012641. [DOI] [PubMed] [Google Scholar]
- 30.Namslauer A, Brzezinski P. FEBS Lett. 2004;567:103–10. doi: 10.1016/j.febslet.2004.04.027. [DOI] [PubMed] [Google Scholar]
- 31.Cukier RI. Biochim Biophys Acta. 2005;1706:134–46. doi: 10.1016/j.bbabio.2004.10.004. [DOI] [PubMed] [Google Scholar]
- 32.Brändén M, Sigurdson H, Namslauer A, Gennis RB, Adelroth P, Brzezinski P. Proc Natl Acad Sci USA. 2001;98:5013–18. doi: 10.1073/pnas.081088398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Rich PR. Aust J Plant Physiol. 1995;22:479–84. [Google Scholar]
- 34.Rich P, Meunier B, Mitchell R, Moody R. Biochim Biophys Acta. 1996;1275:91–95. [Google Scholar]
- 35.Forte E, Scandurra F, Richter O-MH, D’Itri E, Sarti P, et al. Biochemistry. 2004;43:2957–63. doi: 10.1021/bi035863u. [DOI] [PubMed] [Google Scholar]
- 36.Parul D, Palmer G, Fabian M. Biochemistry. 2005;44:4562–71. doi: 10.1021/bi048435c. [DOI] [PubMed] [Google Scholar]
- 37.Artzatbanov VY, Konstantinov A, Skulachev VP. FEBS Lett. 1978;87:180–85. doi: 10.1016/0014-5793(78)80327-5. [DOI] [PubMed] [Google Scholar]
- 38.Vygodina TV, Pecoraro C, Mitchell D, Gennis R, Konstantinov AA. Biochemistry. 1998;37:3053–61. doi: 10.1021/bi971876u. [DOI] [PubMed] [Google Scholar]
- 39.Wikstrom M, Jasaitis A, Backgren C, Puustinen A, Verkhovsky MI. Biochim Biophys Acta. 2000;1459:514–20. doi: 10.1016/s0005-2728(00)00191-2. [DOI] [PubMed] [Google Scholar]
- 40.Ruitenberg M, Kannt A, Bamberg E, Ludwig B, Michel H, Fendler K. Proc Natl Acad Sci USA. 2000;97:4632–36. doi: 10.1073/pnas.080079097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Fetter JR, Sharpe M, Qian J, Mills D, Ferguson-Miller S, Nicholls P. FEBS Lett. 1996;393:155–60. doi: 10.1016/0014-5793(96)00874-5. [DOI] [PubMed] [Google Scholar]
- 42.Pawate AS, Morgan J, Namslauer A, Mills D, Brzezinski P, et al. Biochemistry. 2002;41:13417–23. doi: 10.1021/bi026582+. [DOI] [PubMed] [Google Scholar]
- 43.Gibson Q, Greenwood C. Biochem J. 1963;86:541–54. doi: 10.1042/bj0860541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Oliveberg M, Hallén S, Nilsson T. Biochemistry. 1991;30:436–40. doi: 10.1021/bi00216a019. [DOI] [PubMed] [Google Scholar]
- 45.Adelroth P, Ek MS, Mitchell DM, Gennis RB, Brzezinski P. Biochemistry. 1997;36:13824–29. doi: 10.1021/bi9629079. [DOI] [PubMed] [Google Scholar]
- 46.Nilsson T, Hallén S, Oliveberg M. FEBS Lett. 1990;260:45–47. [Google Scholar]
- 47.Flaxen K, Gilderson G, Adelroth P, Brzezinski P. Nature. 2005;437:286–89. doi: 10.1038/nature03921. [DOI] [PubMed] [Google Scholar]
- 48.Zaslavsky D, Sadoski RC, Rajagukguk S, Geren L, Millett F, et al. Proc Natl Acad Sci USA. 2004;101:10544–47. doi: 10.1073/pnas.0401521101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Verkhovsky MI, Jasaitis A, Verkhovskaya ML, Morgan JE, Wikstrom M. Nature. 1999;400:480–83. doi: 10.1038/22813. [DOI] [PubMed] [Google Scholar]
- 50.Wikstrom M, Verkhovsky MI. Biochim Biophys Acta. 2002;1555:128–32. doi: 10.1016/s0005-2728(02)00267-0. [DOI] [PubMed] [Google Scholar]
- 51.Ruitenberg M, Kannt A, Bamberg E, Fendler K, Michel H. Nature. 2002;417:99–102. doi: 10.1038/417099a. [DOI] [PubMed] [Google Scholar]
- 52.Bloch D, Belevich I, Jasaitis A, Ribacka C, Puustinen A, et al. Proc Natl Acad Sci USA. 2004;101:529–33. doi: 10.1073/pnas.0306036101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Konstantinov AA, Siletsky S, Mitchell D, Kaulen A, Gennis RB. Proc Natl Acad Sci USA. 1997;94:9085–90. doi: 10.1073/pnas.94.17.9085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Siletsky SA, Pawate AS, Weiss K, Gennis RB, Konstantinov AA. J Biol Chem. 2004;279:52558–65. doi: 10.1074/jbc.M407549200. [DOI] [PubMed] [Google Scholar]
- 55.Hosler JP, Espe MP, Zhen Y, Babcock GT, Ferguson-Miller S. Biochemistry. 1995;34:7586–92. doi: 10.1021/bi00023a004. [DOI] [PubMed] [Google Scholar]
- 56.Florens L, Schmidt B, McCracken J, Ferguson-Miller S. Biochemistry. 2001;40:7491–97. doi: 10.1021/bi0101188. [DOI] [PubMed] [Google Scholar]
- 57.Schmidt B, McCracken J, Ferguson-Miller S. Proc Natl Acad Sci USA. 2003;100:15539–42. doi: 10.1073/pnas.2633243100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Schmidt B, Hillier W, McCracken J, Ferguson-Miller S. Biochim Biophys Acta. 2004;1655:248–55. doi: 10.1016/j.bbabio.2003.11.008. [DOI] [PubMed] [Google Scholar]
- 59.Pomes R, Hummer G, Wikstrom M. Biochim Biophys Acta. 1998;1365:255–60. [Google Scholar]
- 60.Xu JC, Voth GA. Proc Natl Acad Sci USA. 2005;102:6795–800. doi: 10.1073/pnas.0408117102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Popovic DM, Stuchebrukhov AA. J Phys Chem B. 2005;109:1999–2006. doi: 10.1021/jp0464371. [DOI] [PubMed] [Google Scholar]
- 62.Behr J, Hellwig P, Mantele W, Michel H. Biochemistry. 1998;37:7400–6. doi: 10.1021/bi9731697. [DOI] [PubMed] [Google Scholar]
- 63.Iwaki M, Puustinen A, Wikstrom M, Rich PR. Biochemistry. 2003;42:8809–17. doi: 10.1021/bi034522d. [DOI] [PubMed] [Google Scholar]
- 64.Nyquist RM, Heitbrink D, Bolwien C, Gennis RB, Heberle J. Proc Natl Acad Sci USA. 2003;100:8715–20. doi: 10.1073/pnas.1530408100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Wikstrom MK. Nature. 1977;266:271–73. doi: 10.1038/266271a0. [DOI] [PubMed] [Google Scholar]
- 66.Wikstrom M, Krab K, Saraste M. Annu Rev Biochem. 1981;50:623–55. doi: 10.1146/annurev.bi.50.070181.003203. [DOI] [PubMed] [Google Scholar]
- 67.Malmstrom BG. Biochim Biophys Acta. 1985;811:1–12. [PubMed] [Google Scholar]
- 68.Popovic DM, Stuchebrukhov AA. FEBS Lett. 2004;566:126–30. doi: 10.1016/j.febslet.2004.04.016. [DOI] [PubMed] [Google Scholar]
- 69.Babcock GT, Callahan PM. Biochemistry. 1983;22:2314–19. doi: 10.1021/bi00279a002. [DOI] [PubMed] [Google Scholar]
- 70.Gelles J, Blair DF, Chan SI. Biochim Biophys Acta. 1986;853:205–36. doi: 10.1016/0304-4173(87)90002-4. [DOI] [PubMed] [Google Scholar]
- 71.Chan SI, Li PM. Biochemistry. 1990;29:1–12. doi: 10.1021/bi00453a001. [DOI] [PubMed] [Google Scholar]
- 72.Puustinen A, Finel M, Virkki M, Wikström M. FEBS Lett. 1989;249:163–67. doi: 10.1016/0014-5793(89)80616-7. [DOI] [PubMed] [Google Scholar]
- 73.Tsukihara T, Aoyama H, Yamashita E, Tomizaki T, Yamaguchi H, et al. Science. 1995;269:1069–74. doi: 10.1126/science.7652554. [DOI] [PubMed] [Google Scholar]
- 74.Verkhovsky MI, Morgan JE, Wikstrom M. Biochemistry. 1995;34:7483–91. doi: 10.1021/bi00022a023. [DOI] [PubMed] [Google Scholar]
- 75.Mitchell R, Mitchell P, Rich PR. Biochim Biophys Acta. 1992;1101:188–91. [PubMed] [Google Scholar]
- 76.Winkler JR, Malmstrom BG, Gray HB. Biophys Chem. 1995;54:199–209. doi: 10.1016/0301-4622(94)00156-e. [DOI] [PubMed] [Google Scholar]
- 77.Verkhovsky MI, Morgan JE, Wikstrom M. Biochemistry. 1992;31:11860–63. doi: 10.1021/bi00162a026. [DOI] [PubMed] [Google Scholar]
- 78.Wikstrom M. Nature. 1989;338:776–78. doi: 10.1038/338776a0. [DOI] [PubMed] [Google Scholar]
- 79.Babcock GT, Wikstrom M. Nature. 1992;356:301–9. doi: 10.1038/356301a0. [DOI] [PubMed] [Google Scholar]
- 80.Wikstrom M. Biochim Biophys Acta. 2004;1655:241–47. doi: 10.1016/j.bbabio.2003.07.013. [DOI] [PubMed] [Google Scholar]
- 81.Wikström M, Bogachev A, Finel M, Morgan JE, Puustinen A, et al. Biochim Biophys Acta. 1994;1187:106–11. doi: 10.1016/0005-2728(94)90093-0. [DOI] [PubMed] [Google Scholar]
- 82.Rich PR, Junemann S, Meunier B. J Bioenerg Biomembr. 1998;30:131–38. doi: 10.1023/a:1020524014920. [DOI] [PubMed] [Google Scholar]
- 83.Brzezinski P, Larsson G. Biochim Biophys Acta. 2003;1605:1–13. doi: 10.1016/s0005-2728(03)00079-3. [DOI] [PubMed] [Google Scholar]
- 84.Brzezinski P. Trends Biochem Sci. 2004;29:380–87. doi: 10.1016/j.tibs.2004.05.008. [DOI] [PubMed] [Google Scholar]
- 85.Hofacker I, Schulten K. Proteins. 1998;30:100–7. [PubMed] [Google Scholar]
- 86.Seibold SA, Mills DA, Ferguson-Miller S, Cukier RI. Biochemistry. 2005;44:10475–85. doi: 10.1021/bi0502902. [DOI] [PubMed] [Google Scholar]
- 87.Gilderson G, Aagaard A, Gomes CM, Adelroth P, Teixeira M, Brzezinski P. Biochim Biophys Acta. 2001;1503:261–70. doi: 10.1016/s0005-2728(00)00195-x. [DOI] [PubMed] [Google Scholar]
- 88.Braiman MS, Bousché O, Rothschild KJ. Proc Natl Acad Sci USA. 1991;88:2388–92. doi: 10.1073/pnas.88.6.2388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Garczarek F, Brown LS, Lanyi JK, Gerwert K. Proc Natl Acad Sci USA. 2005;102:3633–38. doi: 10.1073/pnas.0500421102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Hosler JP. Biochim Biophys Acta. 2004;1655:332–39. doi: 10.1016/j.bbabio.2003.06.009. [DOI] [PubMed] [Google Scholar]
- 91.Bratton M, Pressler M, Hosler J. Biochemistry. 1999;38:16236–45. doi: 10.1021/bi9914107. [DOI] [PubMed] [Google Scholar]
- 92.Gilderson G, Salomonsson L, Aagaard A, Gray J, Brzezinski P, Hosler J. Biochemistry. 2003;42:7400–9. doi: 10.1021/bi0341298. [DOI] [PubMed] [Google Scholar]
- 93.Marantz Y, Nachliel E, Aagaard A, Brzezinski P, Gutman M. Proc Natl Acad Sci USA. 1998;95:8590–95. doi: 10.1073/pnas.95.15.8590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Marantz Y, Einarsdottir OO, Nachliel E, Gutman M. Biochemistry. 2001;40:15086–97. doi: 10.1021/bi010453w. [DOI] [PubMed] [Google Scholar]
- 95.Gutman M, Nachliel E. Biochem Biophys Acta. 1990;1015:391–414. [Google Scholar]
- 96.Checover S, Marantz Y, Nachliel E, Gutman M, Pfeiffer M, et al. Biochemistry. 2001;40:4281–92. doi: 10.1021/bi002574m. [DOI] [PubMed] [Google Scholar]
- 97.Namslauer A, Aagaard A, Katsonouri A, Brzezinski P. Biochemistry. 2003;42:1488–98. doi: 10.1021/bi026524o. [DOI] [PubMed] [Google Scholar]
- 98.Georgievskii Y, Medvedev ES, Stuchebrukhov AA. Biophys J. 2002;82:2833–46. doi: 10.1016/S0006-3495(02)75626-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Mills DA, Tan Z, Ferguson-Miller S, Hosler J. Biochemistry. 2003;42:7410–17. doi: 10.1021/bi0341307. [DOI] [PubMed] [Google Scholar]
- 100.Mills DA, Ferguson-Miller S. FEBS Lett. 2003;545:47–51. doi: 10.1016/s0014-5793(03)00392-2. [DOI] [PubMed] [Google Scholar]
- 101.Wilson KS, Prochaska LJ. Arch Biochem Biophys. 1990;282:413–20. doi: 10.1016/0003-9861(90)90137-n. [DOI] [PubMed] [Google Scholar]
- 102.Haltia T, Saraste M, Wikstrom M. EMBO J. 1991;10:2015–21. doi: 10.1002/j.1460-2075.1991.tb07731.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Mills DA, Hosler JP. Biochemistry. 2005;44:4656–66. doi: 10.1021/bi0475774. [DOI] [PubMed] [Google Scholar]
- 104.Cherny V, DeCoursey T. J Gen Physiol. 1999;114:819–38. doi: 10.1085/jgp.114.6.819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.DeCoursey T. Physiol Rev. 2003;83:475–579. doi: 10.1152/physrev.00028.2002. [DOI] [PubMed] [Google Scholar]
- 106.Link T, von Jagow G. J Biol Chem. 1995;270:25001–6. doi: 10.1074/jbc.270.42.25001. [DOI] [PubMed] [Google Scholar]
- 107.Axelrod H, Abresch E, Paddock M, Okamura M, Feher G. Proc Natl Acad Sci USA. 2000;97:1542–47. doi: 10.1073/pnas.97.4.1542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Berry E, Zhang Z, Bellamy H, Huang L. Biochim Biophys Acta. 2000;1459:440–48. doi: 10.1016/s0005-2728(00)00182-1. [DOI] [PubMed] [Google Scholar]
- 109.Aagaard A, Namslauer A, Brzezinski P. Biochim Biophys Acta. 2002;1555:133–39. doi: 10.1016/s0005-2728(02)00268-2. [DOI] [PubMed] [Google Scholar]
- 110.Paddock ML, Adelroth P, Chang C, Abresch EC, Feher G, Okamura MY. Biochemistry. 2001;40:6893–902. doi: 10.1021/bi010280a. [DOI] [PubMed] [Google Scholar]
- 111.Paddock M, Graige M, Feher G, Okamura M. Proc Natl Acad Sci USA. 1999;96:6183–88. doi: 10.1073/pnas.96.11.6183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Mills DA, Ferguson-Miller S. Biochim Biophys Acta. 1998;1365:46–52. doi: 10.1016/s0005-2728(98)00040-1. [DOI] [PubMed] [Google Scholar]
- 113.Zhen Y, Mills D, Hoganson CW, Lucas RL, Shi W, et al. In: Frontiers of Cellular Bioenergetics: Molecular Biology, Biochemistry and Physiopathology. Papa S, Guerrieri F, Tager JM, editors. New York: Kluwer Acad./Plenum; 1999. pp. 157–78. [Google Scholar]
- 114.Proteau G, Wrigglesworth JM, Nicholls P. Biochem J. 1983;210:199–205. doi: 10.1042/bj2100199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Pfitzner U, Hoffmeier K, Harrenga A, Kannt A, Michel H, et al. Biochemistry. 2000;39:6756–62. doi: 10.1021/bi992235x. [DOI] [PubMed] [Google Scholar]
- 116.Namslauer A, Pawate AS, Gennis RB, Brzezinski P. Proc Natl Acad Sci USA. 2003;100:15543–47. doi: 10.1073/pnas.2432106100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Hosler JP, Shapleigh JP, Mitchell DM, Kim Y, Pressler M, et al. Biochemistry. 1996;35:10776–83. doi: 10.1021/bi9606511. [DOI] [PubMed] [Google Scholar]
- 118.Richter O-MH, Durr K, Kaant A, Ludwig B, Scandurra F, et al. FEBS J. 2005;272:404–12. doi: 10.1111/j.1742-4658.2004.04480.x. [DOI] [PubMed] [Google Scholar]
- 119.Mills DA, Geren L, Hiser C, Schmidt B, Durham B, et al. Biochemistry. 2005;44:10457–65. doi: 10.1021/bi050283d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Olkhova E, Hutter MC, Lill MA, Helms V, Michel H. Biophys J. 2004;86:1873–89. doi: 10.1016/S0006-3495(04)74254-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Zheng XH, Medvedev DM, Swanson J, Stuchebrukhov AA. Biochim Biophys Acta. 2003;1557:99–107. doi: 10.1016/s0005-2728(03)00002-1. [DOI] [PubMed] [Google Scholar]
- 122.Olsson MH, Sharma PK, Warshel A. FEBS Lett. 2005;579:2026–34. doi: 10.1016/j.febslet.2005.02.051. [DOI] [PubMed] [Google Scholar]
- 123.Adelroth P, Gennis RB, Brzezinski P. Biochemistry. 1998;37:2470–76. doi: 10.1021/bi971813b. [DOI] [PubMed] [Google Scholar]
- 124.Tiede DM, Vashishta A-C, Gunner MR. Biochemistry. 1993;32:4515–31. doi: 10.1021/bi00068a006. [DOI] [PubMed] [Google Scholar]
- 125.Kannt A, Roy C, Lancaster D, Michel H. Biophys J. 1998;74:708–21. doi: 10.1016/S0006-3495(98)73996-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Lancaster CRD. FEBS Lett. 2003;545:52–60. doi: 10.1016/s0014-5793(03)00393-4. [DOI] [PubMed] [Google Scholar]
- 127.Soares CM, Baptista AM, Pereira MM, Teixeira M. J Biol Inorg Chem. 2004;9:124–34. doi: 10.1007/s00775-003-0509-9. [DOI] [PubMed] [Google Scholar]
- 128.Chen Y, Hunsicker-Wang L, Pacoma RL, Luna E, Fee JA. Protein Expr Purif. 2005;40:299–318. doi: 10.1016/j.pep.2004.11.014. [DOI] [PubMed] [Google Scholar]
- 129.Kannt A, Lancaster CR, Michel H. Biophys J. 1998;74:708–21. doi: 10.1016/S0006-3495(98)73996-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Brändén G, Brändé M, Schmidt B, Mills DA, Ferguson-Miller S, Brzezinski P. Biochemistry. 2005;44:10466–74. doi: 10.1021/bi0502745. [DOI] [PubMed] [Google Scholar]
- 131.Behr J, Michel H, Mantele W, Hellwig P. Biochemistry. 2000;39:1356–63. doi: 10.1021/bi991504g. [DOI] [PubMed] [Google Scholar]
- 132.Friedrich T, Abelmann A, Brors B, Guenebaut V, Kintscher L, et al. Biochem Biophys Acta. 1998;1365:215–19. doi: 10.1016/s0005-2728(98)00070-x. [DOI] [PubMed] [Google Scholar]
- 133.Brandt U, Kerscher S, Drose S, Zwicker K, Zickermann V. FEBS Lett. 2003;545:9–17. doi: 10.1016/s0014-5793(03)00387-9. [DOI] [PubMed] [Google Scholar]
- 134.Xia D, Yu C-A, Kim H, Xia J-Z, Kachurin AM, et al. Science. 1997;277:60–66. doi: 10.1126/science.277.5322.60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Iwata S, Lee JW, Okada K, Lee JK, Iwata M, et al. Science. 1998;281:64–71. doi: 10.1126/science.281.5373.64. [DOI] [PubMed] [Google Scholar]
- 136.Lange C, Hunte C. Proc Natl Acad Sci USA. 2002;99:2800–5. doi: 10.1073/pnas.052704699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Crofts AR. Annu Rev Physiol. 2004;66:689–733. doi: 10.1146/annurev.physiol.66.032102.150251. [DOI] [PubMed] [Google Scholar]
- 138.Crofts AR, Hacker B, Barquera B, Yun CH, Gennis RB. Biochim Biophys Acta. 1992;1101:162–65. [PubMed] [Google Scholar]
- 139.Trumpower BL. Biochim Biophys Acta. 2002;1555:166–73. doi: 10.1016/s0005-2728(02)00273-6. [DOI] [PubMed] [Google Scholar]
- 140.Osyczka A, Moser CC, Dutton PL. Trends Biochem Sci. 2005;30:176–82. doi: 10.1016/j.tibs.2005.02.001. [DOI] [PubMed] [Google Scholar]
- 141.Junemann S, Heathcote P, Rich PR. J Biol Chem. 1998;273:21603–7. doi: 10.1074/jbc.273.34.21603. [DOI] [PubMed] [Google Scholar]
- 142.Darrouzet E, Moser CC, Dutton PL, Daldal F. Trends Biochem Sci. 2001;26:445–51. doi: 10.1016/s0968-0004(01)01897-7. [DOI] [PubMed] [Google Scholar]
- 143.Crofts AR, Barquera B, Gennis RB, Kuras R, Guergova-Kuras M, Berry EA. Biochemistry. 1999;38:15807–26. doi: 10.1021/bi990962m. [DOI] [PubMed] [Google Scholar]
- 144.Hunte C, Koepke J, Lange C, Rossmanith T, Michel H. Struct Fold Des. 2000;8:669–84. doi: 10.1016/s0969-2126(00)00152-0. [DOI] [PubMed] [Google Scholar]
- 145.Holt P, Morgan DJ, Sazanov LA. J Biol Chem. 2003;278:43114–20. doi: 10.1074/jbc.M308247200. [DOI] [PubMed] [Google Scholar]