

Abstract
Protein dynamics is currently an area of intense research because of its importance as complementary information to the huge quantity of available data relating protein structure and function. Because it is usually the influence of dynamics on function that is studied, the physical determinants of the distribution of flexibility in proteins have not been explored as thoroughly. In the present NMR study, an expanded suite of five 2H relaxation experiments was used to characterize the picosecond-nanosecond side-chain dynamics of chymotrypsin inhibitor 2 (CI2) and five hydrophobic core mutants, some of which are members of the folding nucleus. Because CI2 is a homologue of the serine protease inhibitor eglin c, which has already been extensively characterized in terms of its dynamics, it was possible to compare not only side-chain dynamics, but also the responses of these dynamics to analogous mutations. Remarkably, each of the five core mutations in CI2 led to similar and reproducible increases in side-chain flexibility throughout the entire structure. Although the expanded suite of 2H relaxation experiments does not affect model selection for the vast majority of residues, it did enable the detection of increasing levels of nanosecond-scale motions in CI2’s reactive site binding loop as the L68 side chain was progressively shortened by mutation. Collectively, we observed that the CI2 mutants are more dynamically similar to each other than to the more rigid wild-type CI2, from which we conclude that wild-type CI2 has been optimized to a specific level of rigidity which may aid in its function as a serine protease inhibitor. We also observed that the pattern of side-chain dynamics of CI2 is quantitatively similar to eglin c, but that this similarity is lost upon mutating both proteins at an equivalent position. Finally, 15N relaxation was used to characterize the backbone dynamics of wild-type and mutant CI2. Interestingly, mutation at folding nucleus positions led to widespread increases in backbone flexibility, whereas non-folding-nucleus positions led to increases in flexibility in the C-terminal half of the protein only.
Keywords: NMR, CI2, hydrophobic cores, side-chain dynamics, 2H relaxation, 15N relaxation, homologous proteins
The realization that internal dynamics across multiple time scales can impact both protein thermodynamics and function to a significant degree has changed the conceptual framework for phenomena such as catalysis and signal transduction. Motions in the picosecond-nanosecond (ps-ns) time regime impact a protein’s thermodynamics via contributions to its conformational entropy and thus total free energy (1-10), while slower motions taking place on the microsecond-to-millisecond (μs-ms) time scale have typically been linked to allostery (11, 12) and enzymatic catalysis (13, 14). Less well understood, however, are the physical determinants of protein internal motions, especially side-chain dynamics. Efforts to predict internal dynamics based on structure have shown promise (15, 16), but the moderate correlations observed in such attempts indicate that more than one or a few different factors must come into play.
In this study, we employ a range of sophisticated NMR methodologies to characterize the dynamical consequences of five hydrophobic core mutations in chymotrypsin inhibitor 2 (CI2), a 7.3 kDa serine protease inhibitor from barley seeds (17, 18). The five core mutants studied (L68V, L68A, I76V, V28A, and V66A) were selected to increase understanding of several specific issues concerning internal dynamics in hydrophobic cores (Figure 1). Residues L68 and I76 are known members of the CI2 folding nucleus, as determined by both experimental Φ-value analysis (19) and computational approaches (20, 21). We sought to determine whether these residues, which make important contacts that stabilize the transition state during folding, are also important for modulating CI2’s internal dynamics in the native state. Furthermore, computational studies of CI2 have suggested that L68 is one of two key energetic “linchpins” connecting CI2’s hydrophobic core to its reactive loop (22). For this reason, we have selected two L68 mutants in which the length of the side chain is progressively reduced (L→V→A) in order to determine whether the dynamical character of the L68 side chain has a role in mediating this connection. V28 is not part of the canonical CI2 folding nucleus but is located near both L68 and I76 in the hydrophobic core, and thus was chosen to reveal whether residues proximal to canonical folding nuclei show similar dynamical behavior. V66, on the other hand, is neither part of the folding nucleus nor pointed into the same cluster of core residues formed by L68, I76, and V28. Analyzing the dynamical consequences of mutating this position adds perspective for interpreting the results of mutation to the canonical core residues. As a whole, the selected core mutations differ in location, the chemical identity of the WT residue, and the degree of severity of the mutation (although all are generally conservative), and differences between the dynamics of these mutants and WT CI2 should yield information on the relative contribution of each position to modulating the observed dynamics of the WT protein. Furthermore, the acquisition of the dynamics data for WT CI2 and hydrophobic core mutants will also allow us to compare the internal dynamics of CI2 to data available for the homologue eglin c.
Figure 1.
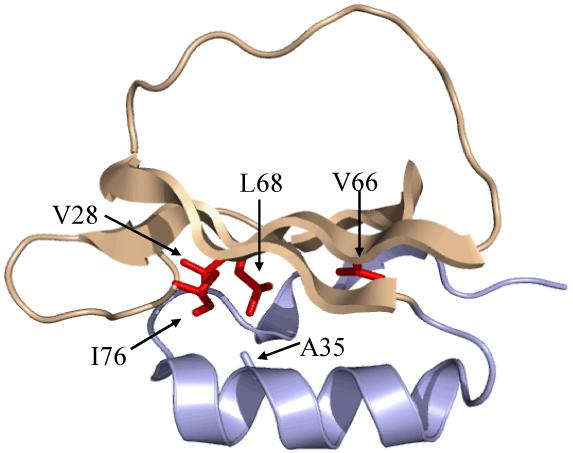
The structure of chymotrypsin inhibitor 2 (PDB code 2CI2), with the sites of mutation examined in this study shown in red. The third canonical member of the CI2 folding nucleus, A35, has not been examined in this work and is shown in gray. The backbone coloring corresponds to the two distinct regions of dynamical perturbation (N-terminal region, light blue; C-terminal region, beige) revealed by 15N relaxation experiments (see text).
Experimental Procedures
Protein Expression and Purification
Crystallographic evidence indicates that the first 19 amino acids of CI2 are unstructured (17). Consequently, experimental and computational studies of CI2 are routinely performed on a Δ1-19 truncation with residue 20 replaced by methionine, as is the case in the present work. WT CI2 DNA was generated from synthetic oligonucleotides using PCR and placed into the pET28a plasmid (Novagen). CI2 mutants were created by application of site-directed mutagenic PCR to the pET28a vector containing the WT sequence. Proteins were expressed in E. coli BL21 (DE3) cells (Invitrogen) grown at 37 °C to an optical density at 600 nm of 0.6-0.8 in 1 L of M9 minimal medium supplemented with 50 μg/mL kanamycin and containing 99%-15NH4Cl, U-13C-glucose, and up to 60% 2H2O (v/v) as required for particular NMR samples. Expression was induced by addition of IPTG to a final concentration of 1 mM and allowed to proceed for 4-4.5 hours at 37 °C in a rotary shaker. The cells were harvested by centrifugation at 6,000 g for 30 min, and the cell pellet was resuspended in 40 mL of Buffer A (25 mM Tris·HCl, pH 8) and stored at -20 °C until needed. Frozen cell suspensions were subjected to 3 rounds of freeze-thawing in a dry ice/ethanol bath, and cell lysis was completed by sonicating the sample 4 × 4 minutes. Cell debris from sonication was cleared by centrifugation at 20,000 g for 30 min, contaminant DNA was precipitated from the solution by the addition of polyethyleneimine to a final concentration of 0.2% (w/v), and the entire solution was allowed to stir on ice for 30 min. DNA was cleared from solution by centrifugation for 30 min at 20,000 g. The protein solution was then dialyzed overnight against 4 L of Buffer A. Chromatographic purification of CI2 was achieved by passing the dialyzed protein solution over an anion exchange column packed with Q-Sepharose FF resin (GE Healthcare) equilibrated in Buffer A and eluted by a linear gradient of Buffer B (2 M NaCl, 25 mM Tris·HCl, pH 8). Fractions containing CI2 (judged by the absorbance at 280 nm) were pooled, concentrated, and passed through a size-exclusion column consisting of Sephadex G-50 Superfine beads (GE Healthcare) equilibrated in NMR Buffer (20 mM KPO4, 50 mM KCl, 0.02% NaN3, pH 7). Pure CI2-containing fractions were again pooled, concentrated, and stored at -20 °C until needed. Typically, a 1 L culture yielded approximately 60 mg of pure protein.
NMR Resonance Assignments
Resonance assignment experiments were carried out on an 11.7 T Varian INOVA spectrometer equipped with a triple-resonance probe and z-axis pulsed field gradients using uniformly 13C,15N-enriched CI2 samples in NMR Buffer (10% D2O) at pH 7. Protein concentrations were 1-2 mM. Resonance assignments for the Cα, Cβ, N, and HN atoms of the non-proline residues of each CI2 variant were obtained using gradient-enhanced 3D HNCACB and CBCA(CO)NH experiments (23). The chemical shifts of side-chain methyl carbons (for A, V, L, I, and T) were assigned using the (H)CCH3-TOCSY experiment (24), while methionine methyl carbons were assigned using a 1H-13C HMBC experiment in conjunction with the other assignment experiments. The prochirality of each side-chain methyl carbon was determined using a 10% 13C-labeled sample (25) in conjunction with standard constant-time 1H-13C HSQCs. All spectra were processed using NMRPipe/NMRDraw (26) and analyzed using NMRView (27).
NMR Relaxation Experiments
The backbone 15N T1 and T2 relaxation time constants and the 1H-15N heteronuclear NOE were measured at 25 °C with standard experiments (28) on 11.7 T and 14.4 T Varian INOVA spectrometers equipped with triple resonance probes and z-axis pulsed field gradients. The experimental samples consisted of 2 mM uniformly 15N-labeled CI2 at pH 7 in 10% D2O NMR Buffer. For WT and L68A, the T1, T2, and NOE experiments were performed twice at each field strength on samples originating from independent protein expressions in order to verify the reproducibility of the experimental data. For each relaxation experiment, peak intensities as a function of relaxation time were fitted to a monoexponential decay to yield the T1 and T2 time constants. Error estimates for the T1 and T2 data were made using three duplicate relaxation delay times in each experiment, whereas the error in the calculated heteronuclear NOEs was conservatively estimated as twice the RMSD of the spectral noise. After using the backbone relaxation data to determine the global tumbling time (τm) of each variant and to check for the existence of rotational anisotropy, Lipari-Szabo “model free” dynamics parameters (29) for each residue were extracted using the program relxn2.2 (30) by fitting the experimental 15N data to one of five common forms of the spectral density function (31). The appropriate form of the spectra density function was selected on a per-residue basis by calculating both Akaike’s information criterion (AIC) and the Bayesian information criterion (BIC) (32). For the very few cases in which the AIC and BIC recommended different forms of the spectral density, the simpler form was selected.
The ps-ns dynamics of the methyl-bearing side chains of WT and mutant CI2 were characterized at 25 °C by monitoring the relaxation as a function of time of the following five types of 2H coherence (33, 34): transverse in-phase (D+), longitudinal (Dz), transverse antiphase (AP), double quantum (DQ), and quadrupolar order (QO). 2H relaxation experiments for each CI2 variant were conducted using samples of 2 mM CI2 uniformly labeled with 15N and 13C and 60% randomly labeled with 2H in 10% D2O NMR Buffer (pH 7) on the same spectrometers used for the 15N relaxation measurements. Only signals from -CH2D isotopomers could be detected in these experiments. The transverse and longitudinal relaxation time constants were measured at both 11.7 and 14.4 T, while the AP, DQ, and QO time constants were measured only at 11.7 T. As for the 15N data, the error in each 2H time constant measurement was estimated using three duplicate relaxation delays. To further verify the consistency of the data, measurements of the in-phase transverse and longitudinal time constants were repeated on separately prepared samples at both field strengths for both WT CI2 and L68A. As for the 15N data, peak intensities as a function of time for each 2H relaxation experiment were fitted to a monoexponential decay to extract the associated relaxation time constant. Raw DQ time constants were then corrected according to the method of Millet et al. (34) to yield estimates of the pure DQ time constants. The seven individual 2H relaxation measurements for each residue and the overall global tumbling time τm derived from the 15N relaxation measurements were then used to calculate model free dynamics parameters for each CI2 variant using the in-house program relxD (based on relxn2.2). RelxD generates “model free” parameters by fitting the data for each methyl group to two-parameter and three-parameter forms of the model free spectral density function. The statistical F-test was then used to determine whether the three-parameter model more accurately describes the experimental data, with α = 0.05.
CI2 Structural Characterization
To elucidate the mutations’ effects on CI2 backbone structure, N-HN and Cα-Hα residual dipolar couplings (RDCs) were measured using stretched 6% polyacrylamide gels (35) as the macromolecular alignment medium. A 2D IPAP-based experiment (36) was used to measure N-HN crosspeak splittings in the absence and presence of macroscopic alignment, with the RDC value being the difference between the aligned and isotropic splitting values. Cα-Hα RDCs were measured analogously using a 3D HNCA-based spectrum (37). RDC measurements were carried out at 25 °C on an 11.7 T Varian INOVA spectrometer using 1-2 mM CI2 samples in 10% D2O NMR Buffer at pH 7. To verify the quality of each variant’s measured RDCs, the program REDCAT (38) was used to compare the experimental values to values back-calculated from a high resolution WT CI2 crystal structure (PDB 2CI2).
To probe changes in side-chain structure, NMR measurements of the three-bond scalar couplings (39, 40) between γ-methyl groups and their respective backbone amide nitrogens and carbonyl carbons (3JNC and 3JCC’) were used to calculate χ1 rotamer populations (41, 42) for V, I, and T residues in the CI2 variants under study. Errors in determining the 3J values were estimated using the RMSD of the experimental noise. The experiments were carried out at 25 °C on samples containing 2 mM uniformly 13C,15N-labeled CI2 in 10% D2O NMR buffer at pH 7 using an 11.7 T Varian INOVA spectrometer equipped with a cryogenic probe and z-axis pulsed field gradients.
Results and Discussion
Mutation of Folding Nuclei Affects Backbone Dynamics More Universally than Non-Folding-Nucleus Mutations
In addition to WT CI2, we studied the backbone dynamics of five CI2 mutants. Mutants L68V, L68A, and I76V were chosen because they belong to the canonical CI2 folding nucleus. V28A and V66A are not members of the folding nucleus, but are located in proximity to it and were selected to provide context for interpreting the results of mutating folding nucleus residues. As a reference, the average T1 values at 500 MHz for non-reactive-loop residues in WT, L68V, L68A, I76V, V28A, and V66A were, respectively, 0.393±0.010, 0.387±0.003, 0.389±0.003, 0.396±0.003, 0.388±0.004, and 0.395±0.002 seconds. For T2, the averages of the 500 MHz data excluding reactive loop residues were 0.175±0.002, 0.170±0.001, 0.170±0.001, 0.165±0.003, 0.166±0.002, and 0.166±0.001 seconds. The respective 500 MHz NOE averages were 0.693±0.005, 0.698±0.004, 0.696±0.008, 0.699±0.007, 0.691±0.012, and 0.694±0.010. Based on the 15N T1, T2, and heteronuclear NOE measurements of rigid backbone amides at two static magnetic field strengths, the globally-optimized tumbling times (τm) for WT CI2, L68V, L68A, I76V, V28A, and V66A, were respectively calculated to be 4.05 ns, 4.13 ns, 4.10 ns, 4.29 ns, 4.07 ns, and 4.25 ns. Because the results of the Lipari-Szabo dynamics analysis are influenced by the type of rotational diffusion a macromolecule undergoes (43), we used the 15N data for WT CI2 and L68A to check for the presence of rotational diffusion anisotropy. The ratio D∥/D⊥ for WT CI2 and L68A, calculated using the local diffusion constant approach (44), was 1.09 and 1.12, respectively, suggesting that the use of an isotropic rotational diffusion tensor is appropriate; fitting the WT data to an axially symmetric diffusion model resulted in no significant changes in extracted dynamics parameters (data not shown). Furthermore, since the mutations studied are generally isolated from solvent and thus not expected to exert a significant influence on the mutants’ hydrodynamic properties, the 15N relaxation data for all CI2 variants were analyzed using the isotropic rotational diffusion model.
Fitting the 15N T1, T2, and heteronuclear NOE measurements for WT CI2 and each mutant to the Lipari-Szabo “model free” formalism yields on a per-residue basis the parameters S2 and τe, which, respectively, provide information on the amplitude and correlation time on the ps-ns time scale of the motion of a particular internuclear vector, in this case the vector between the amide nitrogen and the amide proton. The parameter S2 ranges from 0 to 1, with a value of 1 representing a completely rigid vector and a value of 0 indicating a bond vector undergoing isotropic reorientation, while the value of τe is ideally greater than one order of magnitude faster than overall rotational diffusion. The S2 data for each mutant were compared to the WT by further calculating for each residue; we consider significant changes to be those for which the magnitude of ΔS2 is at least twice that of the propagated error. These data can be seen in Figure 2. For all mutants, one overall trend is immediately obvious, namely that most of the ΔS2 values are negative, indicating that the backbones of the mutant proteins are slightly more flexible than the WT main chain. This effect, however, is small, as most of the changes are less than -0.05 and very few reach -0.1.
Figure 2.

Changes in the amplitude of backbone motions (ΔS2) for the CI2 hydrophobic core mutants based on Lipari-Szabo model free analysis of 15N relaxation data acquired at 25 °C. Plots in the left column are for folding nucleus mutants, whereas the right column shows data for core mutants that are not part of the canonical folding nucleus. In all plots, the site of mutation is indicated by a red bar, and residues exhibiting significant changes in dynamics are colored cyan.
More interesting than the magnitude of changes is the pattern of backbone dynamics changes. The two non-folding-nucleus mutants (V66A, V28A) show virtually no backbone perturbations in their N-terminal half (approximately to residue 46). In contrast, the folding nucleus mutants (L68V, L68A, I76V) show increases in backbone flexibility, many of them significant, in this same region. N-terminal changes caused by the L68 mutants cluster mainly to residues forming CI2’s α-helix, with the L68V mutant perturbing the N-terminal half of the helix (residues 30-36) and the L68A mutant perturbing nearly the whole helix. The I76V mutant, in contrast, perturbs the loop immediately prior to the start of the α-helix. The non-folding-nucleus mutants cause no perturbation of dynamics in these regions of the protein.
The C-terminal half of CI2 (approximately after residue 46) is perturbed by all mutants and generally becomes more flexible. It is also in this region where the greatest increases in flexibility (ΔS2 ≈ -0.1) are to be found. In fact, many of the residues showing changes near -0.1 are the same across the various mutants. These residues include 49, 56, 60, 67, 69, 72, and 79, and are scattered throughout the protein’s structure. The larger values of ΔS2 for the residues are in almost all cases attributable to the selection of model 5 (S2fast, S2slow, τe,slow) during data fitting, which indicates that the mutations all induce more complex backbone dynamics at these residues that cannot be described by simpler forms of the spectral density that contain fewer parameters.
Collectively, the 15N relaxation results indicate that CI2’s backbone becomes slightly more flexible upon larger-to-smaller mutation of aliphatic side chains in the hydrophobic core. What distinguishes the various mutants from each other is their ability to perturb the N-terminal half of the protein main chain. Only the folding nucleus mutants (L68V, L68A, I76V) were able to affect the N-terminal half of the protein, with the effects localized to CI2’s α-helix and the turn leading to it. The non-folding-nucleus core mutants were only able to perturb the C-terminal half of the protein, approximately after residue 46. Additionally, for all mutants, certain residues in the C-terminal half appear to undergo more complex dynamics, characterized by contributions from faster and slower ps-ns motions, than in the WT. We also note that initial CPMG-based 15N relaxation-dispersion experiments (45) did not reveal the presence of any motions taking place on the μs-ms time scale for either WT CI2 or CI2 L68A (data not shown). While all core mutations were able to increase backbone flexibility on the ps-ns time scale, they apparently have no effect on the backbone in the μs-ms time regime. Information about CI2 backbone flexibility over very slow time scales (∼ seconds) is available via hydrogen/deuterium exchange (HX) experiments, and HX data have been used as restraints in computational models of the CI2 native state ensemble (46). From these simulations, it was determined that individual RMS residue fluctuations averaged over all members of the native ensemble are mostly constant over the entire primary sequence. The profile of the largest observed fluctuations per residue, however, distinctly reveals that CI2’s α-helix is the most fluctuating unit of the whole structure. Furthermore, the largest-amplitude fluctuations in the native state ensemble were observed at residue A35 and immediately before residue I76, which are two of the three folding nucleus residues. Our study indicates that only the folding nucleus mutations were able to affect ps-ns flexibility throughout the whole protein. This is consistent with the notion that folding nucleus residues play an important role in the native state dynamics.
2H-Based Side-Chain Experiments Reveal that CI2’s Hydrophobic Core is Uniquely Rigid
15N relaxation is an excellent method for determining the flexibility of protein backbones, but the behavior of the backbone is often distinct from the side chains (47). It is interesting to study side-chain dynamics, because side chains are involved in key protein functions such as catalysis and molecular recognition. Studying side-chain dynamics is thus a way to gain further insight into those phenomena. 2H relaxation experiments applied to -CH2D methyl isotopomers are excellent for obtaining information about the motional amplitudes and time scales of the methyl-bearing side chains in proteins. CI2 is particularly suited to analysis by these types of experiments because of the large number of methyl-containing side chains distributed throughout its structure. Thus, CI2 contains enough -CH2D probes to reveal how core mutations affect ps-ns side-chain dynamics in all regions of the protein. Analogously to the 15N relaxation studies, 2H dynamics experiments lead to the calculation of and τe parameters for each methyl group. represents the amplitude of motion of the methyl group symmetry axis, and τe values as determined by 2H relaxation are motional correlation times that contain contributions from rotation about and reorientation of the methyl symmetry axis, although the values are dominated by the former (30).
Historically, 2H dynamics studies on methyl-bearing side chains have been conducted by measuring the relaxation of 2H longitudinal (Dz) and transverse (D+) magnetization as a function of time (33). One strength of the present study is that it expands our previous perturbation-response analyses of serine protease inhibitors (eglin c and CI2) by making use of three newer 2H relaxation experiments in addition to the two original experiments. The major advantage of applying all five 2H relaxation experiments (Dz, D+, AP, QO, DQ) is that it enables robust fitting of the relaxation data to a three-parameter form of the spectral density function, which in turn allows for identification of local nanosecond-scale dynamic processes (48).
Seven total 2H relaxation measurements (Dz and D+ at two fields; AP, DQ, QO at one field) were made for WT, L68V, L68A, I76V, and V66A. For V28A, only D+ and Dz data at two fields are available, which precludes robust fitting to the three-parameter spectral density function. One complete and independent set of data was acquired for each of two separate samples of WT CI2, and the extracted and τe values were compared to one another. Figure 3 demonstrates that the results of the two independent analyses of WT CI2 side-chain dynamics show the expected lack of change, a control that increases confidence in the methodology before analyzing the dynamical perturbations in CI2 mutants. Changes in and τe between mutant and WT were calculated by subtracting the WT values from the mutant values. The ΔS2axis data can be found in Figure 4. Most striking is the overall pattern of changes when comparing each mutant to WT CI2. For nearly every methyl group, ΔS2axis is negative and falls in the range of -0.05 to -0.1, with an average value of approximately -0.06. This indicates that, for all mutants, the vast majority of methyl-containing side chains have become more flexible on the ps-ns time scale as a result of mutation, without regard either to the specific chemical nature of the mutation or its particular location within the hydrophobic core. This result mirrors the backbone dynamics results, but the significant increases in flexibility are more numerous and globally distributed for the side chains. Such globally-distributed, uniform changes in the amplitude of motion of nearly every detectable methyl group (Figure 5) were not observed either in similar studies of eglin c (49-51) or in mutants of the Fyn SH3 domain (52). Additionally, while the amplitudes of side-chain motion are all perturbed to a similar degree seemingly irrespective of the position of mutation, the time scales of motion (reflected in the parameter τe) are not affected to any great degree (representative data in Figure 4). It thus appears that the internal side-chain dynamics of WT CI2 are constrained at or modulated to a unique level of rigidity, such that mutations in the hydrophobic core result in a small but general perturbation to the physical determinants of side-chain dynamics, with the observable result being a global increase in side-chain flexibility. This global coupling of dynamics to the mutation of core residues is a novel experimental manifestation of cooperativity. CI2 is a classically non-allosteric protein, but if these changes in side-chain dynamics, particularly at the reactive loop, were to lead to a change in functional competency, this would by definition be an allosteric effect, and appropriate functional assays will be carried out in future studies.
Figure 3.
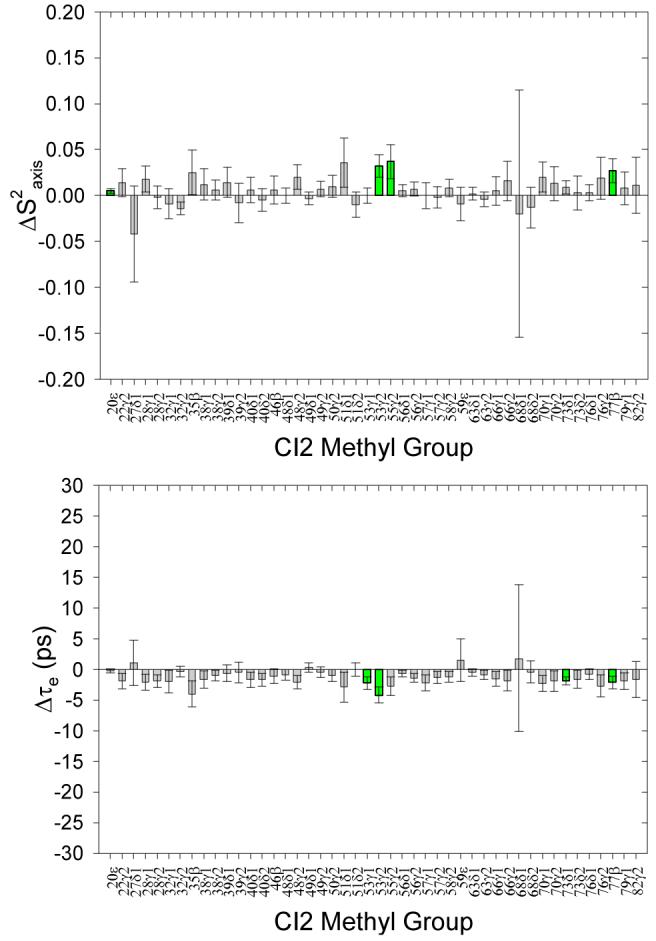
Control analysis of side-chain model free dynamics parameters extracted from independent replicate 2H relaxation measurements of different WT CI2 samples. For nearly every methyl group, the differences between the extracted S2axis and τe parameters for the two replicate sets of data are zero, within error. The very few small differences deemed significant are consistent with our 2σ standard and are colored green.
Figure 4.

2H relaxation analysis of changes in the amplitudes of methyl-containing side-chain motions (ΔS2axis) with respect to WT CI2 for the hydrophobic core mutants studied. Green bars represent significant changes (≥ 2σ). Red bars represent the site of mutation, if data are available. The asterisks denote methyl groups experiencing slower nanosecond-scale motions based on LS-3 model selection during fitting of the relaxation data.
Figure 5.
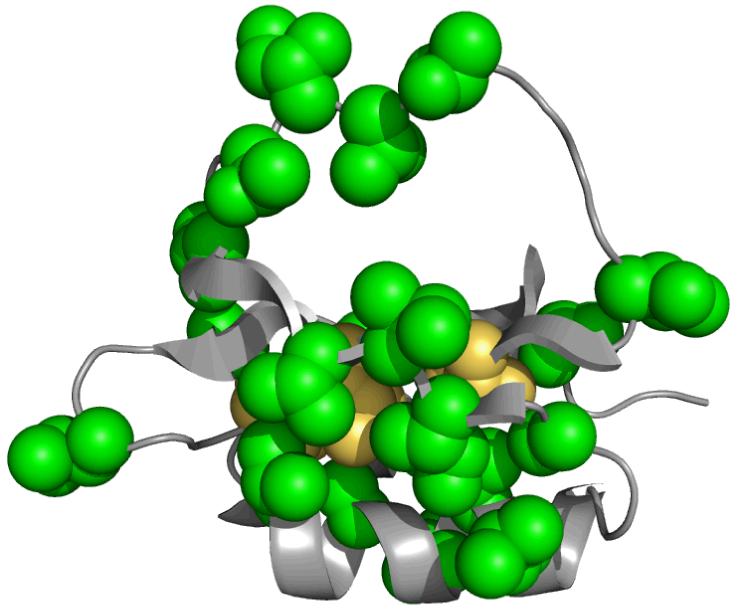
A graphical summary of the 2H side-chain dynamics results. In yellow are the sites of the individual single mutants (L68A, L68V, I76V, V66A, V28A). The green residues are those methyl-bearing side chains that have significant changes in S2axis in no fewer than 4 of the 5 single mutants investigated. Changes in the amplitude of side-chain motion radiate to all regions of the three dimensional structure and encompass nearly every methyl group which could be experimentally observed.
It is important to note that we do not claim that the side chains of CI2 are abnormally rigid in an absolute sense, but rather that WT CI2’s particular architecture and composition appear to support the dynamics of all the side chains such that any core perturbation “bumps” the side-chain dynamics away from the WT parameters to a nearly identical extent, as can be seen in Figure 6. These are each single mutants compared to WT CI2, but are double mutants compared to each other. Noticeably, there are many fewer significant differences in dynamics in this double mutant comparison than there are between either of the two single mutants and WT CI2, and many of the significant differences between the two single mutants cluster to the hydrophobic core. Furthermore, among the differences that do exist there are both increases and decreases in flexibility, which is in sharp contrast to the major behavior of the single mutants compared to the WT. The lack of many substantial differences among the side-chain dynamics of the various CI2 single mutants supports our conclusions that mutation seemingly anywhere in the hydrophobic core affects the global side-chain dynamics to a very similar degree.
Figure 6.
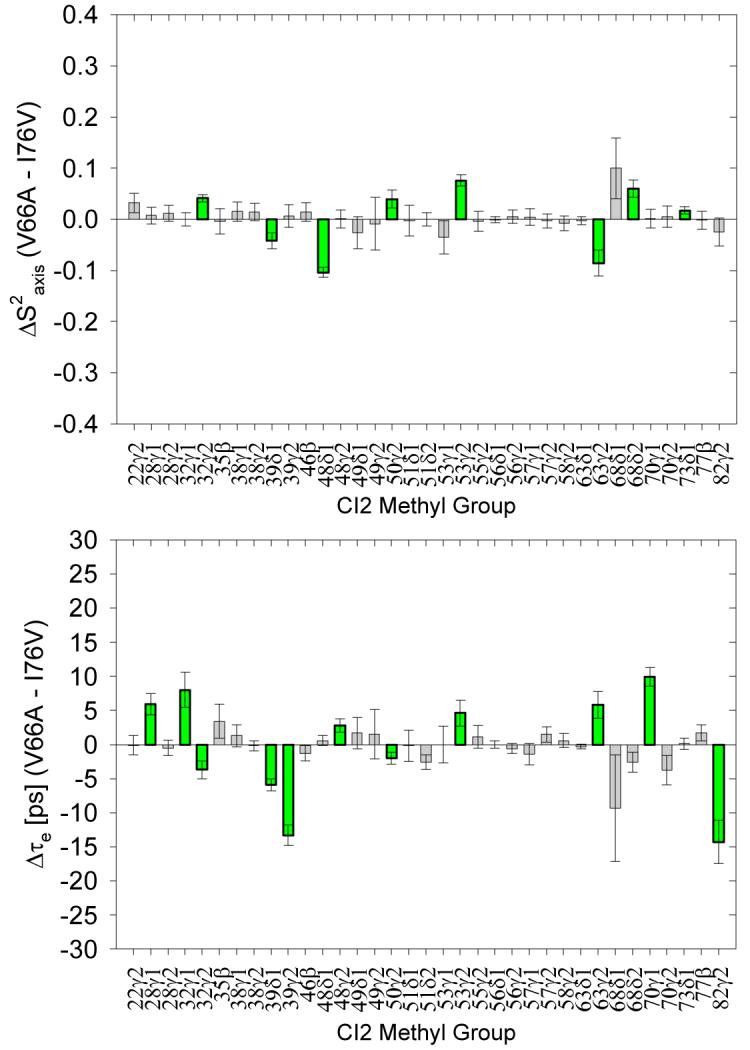
A comparison of the side-chain dynamics of a “double mutant” pair comprised of CI2 I76V and CI2 V66A. The dynamics of these two mutants are much more similar to each other than either is to WT CI2.
Mutation to Putative Energetic Linchpin L68 Results in Increased “Slower” Motions at the Reactive Loop
The chief benefit of the expansive set of side-chain 2H relaxation measurements obtained for each CI2 variant is the ability to detect the presence of “slower” nanosecond-scale motions that are not handled adequately by the standard two-parameter form of the spectral density. The data shown in Figure 4 resulted from model selection (two-parameter versus three-parameter spectral densities) for each methyl group in both WT CI2 and all mutants. Table 1 lists the methyl groups for which nanosecond-scale motions were detected in each variant; these methyl groups are marked with an asterisk in the panels of Figure 4. Except for one isoleucine, the side-chain dynamics of WT CI2 are adequately described by a two-parameter model in which the fitted motional correlation times are all on a time scale much faster than overall tumbling. This is also the case for mutants V66 and I76. The series of mutations at L68 (L→V→A) reveals clearly distinct behavior. As the length of the side chain at position 68 is decreased, more and more methyl groups appear to have a slower nanosecond-scale contribution to their dynamics. Especially for L68A, most of the residues which experience these nanosecond-scale motions in response to mutation are localized to the functional site of CI2, its reactive loop (residues 53-63). Whether these nanosecond-scale motions, which lead to increased rigidity (positive ΔS2axis), will impact any functional parameters such as binding affinity for or inhibitory ability towards serine proteases will be addressed in future studies. Taken together, the model-selected side-chain dynamics data suggest that L68 is unique among core residues, as suggested by previous work (22). Mutations at that position alone resulted in a noticeable presence of “slower” motions, and the number of methyl groups with such motions increased as the length of the side chain at position 68 decreased. It is conceivable that the changes in reactive loop dynamics reflect the loss of the “linchpin” connection between the loop and CI2’s hydrophobic core.
Table 1.
Selection of LS-3 model for CI2 mutants
| Variant | Methyl Groups with Detected Nanosecond-Scale Motions |
|---|---|
| WT | I49δ |
| V66A | I49γ2, I49δ |
| I76V | I49γ2, |
| L68V | I49δ1, I49γ2, M59ε, V68δ1, I76δ1 |
| L68A | L40δ1, I49δ1, I49δ2, V53γ1, V53γ2, T55γ2, I56δ1, T58γ2, M59ε, I63γ2, V79γ2 |
Effects of Mutations on CI2 Structure
We desired to determine the extent to which any structural perturbations caused by the mutations might be responsible for the observed widespread changes in dynamics. Structural perturbation to the backbone was assessed by measuring N-HN and Cα-Hα residual dipolar couplings (RDCs). RDCs are exquisitely sensitive to local environment, so any change in local structure should be readily detectable (except for pure translations in the frame of alignment). By comparing RDC data for mutants to WT data, we can assess the degree to which the backbones of the two variants are similar. Representative data are shown in Figure 7. The extremely high squared correlation coefficients for mutant/WT comparisons suggest that there are no gross structural perturbations caused by the mutations.
Figure 7.
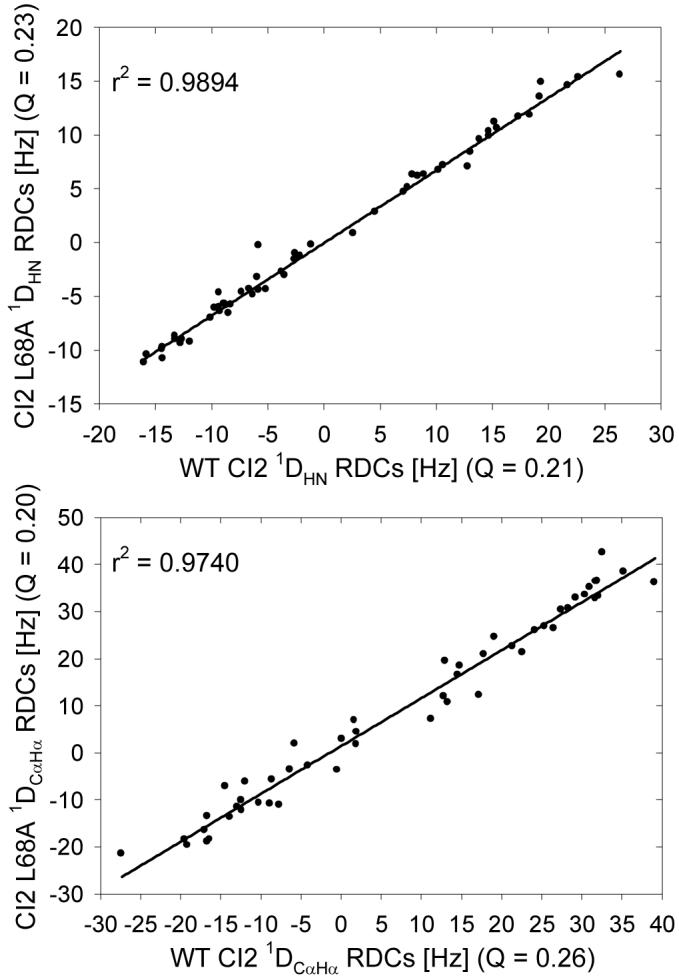
A comparison of residual dipolar couplings (RDCs) for CI2 L68A versus WT CI2. Mutant N-H (top panel) and Cα-Hα (bottom panel) RDCs correlate extremely well with WT values, indicating that no significant structural rearrangements occur upon mutation. Q-factors for all variants were calculated using the WT CI2 structure. The correlations observed here are highly representative of the mutant/WT RDC comparisons for all mutants tested.
Side-chain structure was investigated for WT CI2 and the L68V and L68A mutants via measurement of three-bond scalar couplings between the backbone and side-chain γ-methyls (Val, Ile, Thr). From those measurements, we calculated χ1 rotamer populations and compared mutant populations to those of the WT. Representative data are presented in Figure 8. Probing the populations of χ1 rotameric states using scalar couplings also revealed that side-chain structure is generally conserved as well, with one notable exception. The V32 side chain (green dot in the figure) shows an increasing preference for the +60° rotamer over the 180° rotamer as the side chain at position 68 is shortened from L to V to A. The amplitudes and time scales of the ps-ns dynamics of the V32 side chain are relatively constant between the L68V and L68A, but the rotamer populations of V32 in both mutants show signs of motional averaging, as all three rotamers are populated to some degree. Scalar couplings (and thus rotamer populations) are sensitive to motions across many time scales; since the ps-ns dynamics at V32 are relatively insensitive to mutation but the rotamer populations show increasing changes with mutation, it is likely that some other, slower dynamic process is occurring at this residue. Examples of that type of dynamics were also found in protein L (53).
Figure 8.
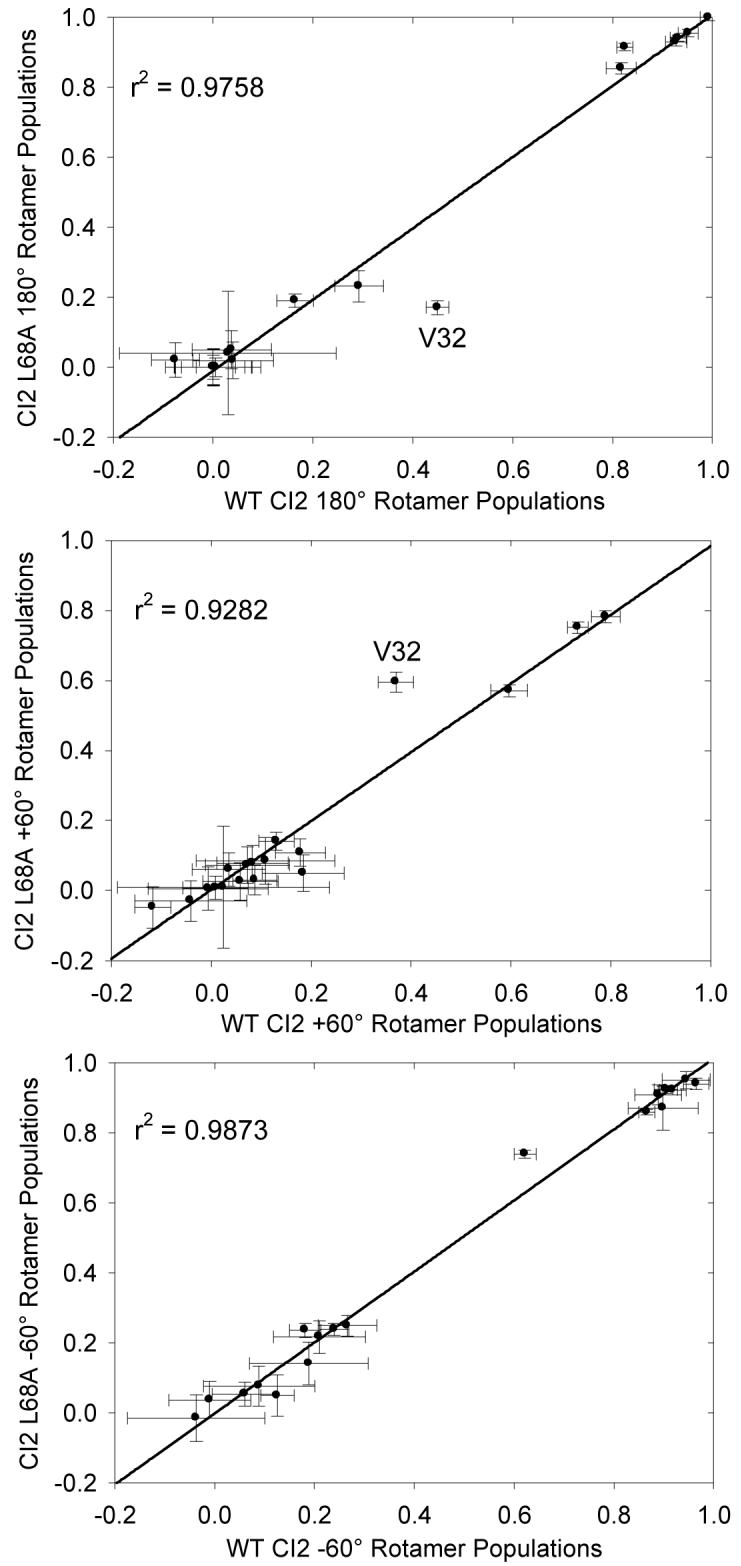
A comparison of the χ1 rotamer populations of WT CI2 and CI2 L68A calculated from three-bond scalar coupling measurements as a probe of side-chain structure. The populations of the three canonical rotameric states (+60°, -60°, 180°) are well conserved between WT CI2 and L68A, suggesting that the mutation does not grossly affect side-chain structure. The one significant exception is residue V32, which experiences a population shift from the major rotamer of 180° to the +60° rotamer as the side chain of residue 68 is progressively shortened.
In addition to ruling out gross alterations to side-chain structure, the aggregate rotamer population data suggest that the increases in mutant CI2 side-chain flexibility occur within the energy wells of individual rotameric states and are not attributable to significantly increased rotamer sampling. It is known that rotameric transitions result in decreased S2axis values measured by NMR (54, 55), but we do not observe significant changes in rotamer populations upon mutation, except for the single example discussed above. Thus, the global decrease in S2axis values observed for the CI2 mutants results not from increased transitioning among rotameric states, but rather from increased dynamics within individual rotamer energy wells.
Comparing CI2 Side-Chain Dynamics to the Homologue Eglin c
One particularly interesting open question in the field of protein dynamics is whether a given protein fold encodes the dynamics of its constituent amino acids and thus whether homologous proteins should have similar dynamics regardless of their sequence dissimilarity. By analyzing the CI2 dynamics data obtained in this study, we can contribute to the discussion of whether the dynamical character of proteins is encoded in the three-dimensional fold or whether local, sequence-specific considerations are more important. A large body of structure and dynamics information from NMR exists for the serine protease inhibitor eglin c (49, 50), a homologue of CI2. Similar dynamical responses upon mutating the two proteins at equivalent positions argues that the overall fold is a major determinant of internal dynamics, whereas different consequences upon mutating equivalent positions argue that sequence-specific local effects are more important. Despite the host of NMR dynamics studies published in the literature, examples of direct comparisons of the side-chain dynamics of homologous proteins are comparatively lacking. Best et al. compared the side-chain dynamics of homologous fnIII domains from human fibronectin and human tenascin and found the side chains in the core of the tenascin fnIII domain to be much more flexible than those in the fibronectin domain; the increased flexibility of the tenascin fnIII side chains was attributed in part to both the amino acid composition and excess packing volume of the hydrophobic core (56). In contrast, other analyses of pooled side-chain dynamics data from numerous proteins suggest that, in general, there is little correlation between side-chain dynamics and structural properties such as depth of burial, solvent accessible surface area, packing density, or crystallographic B-factors (47).
Thus, we turned our attention to whether the side-chain dynamics of CI2 are similar to those of the homologue eglin c. Because the two proteins have different amino acids at structurally equivalent positions, the S2axis values were normalized to yield S2norm according to the method of Mittermaier et al. (57) in order to facilitate direct comparison. By calculating ΔS2norm for structurally equivalent positions in CI2 and eglin c, we can estimate the similarity of the side-chain dynamics at those positions. In addition to WT CI2 and WT eglin c, S2axis (and thus S2norm) data at pH 7 and 25 °C exist for the mutant pair comprised of CI2 I76V and eglin c V63A. ΔS2norm calculations for the WT/WT and mutant/mutant pairs are presented in Figure 9.
Figure 9.
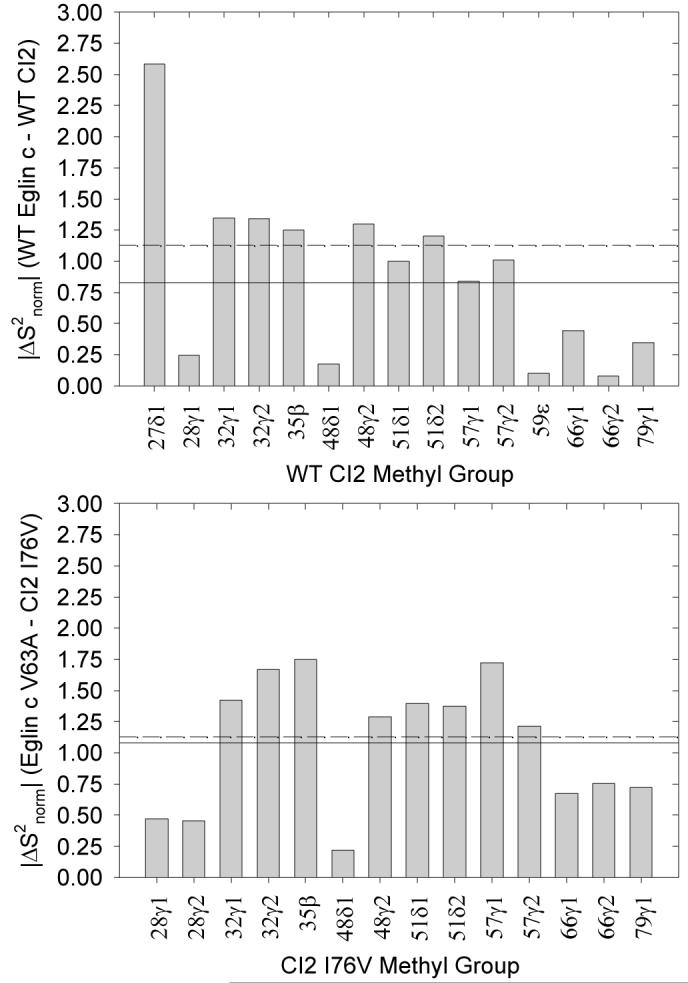
Comparisons of CI2 side-chain dynamics with respect to the homologue eglin c. Plotted are |ΔS2norm| values for WT CI2 vs. WT eglin c (top) and the homologous mutant pair CI2 I76V and eglin c V63A (bottom). The dashed line in both panels is |ΔS2norm| = 1.13, the pairwise average value expected if the two proteins shared no more than random dynamical similarity. The actual pairwise averages (solid lines) are |ΔS2norm| = 0.83 (top) and 1.08 (bottom).
In order to interpret these data in terms of similarity of dynamics, it is important to appreciate what value of ΔS2norm might be expected for two proteins of no significant dynamical similarity. We approached this question by generating a Gaussian distribution of S2norm values centered at zero and having a standard deviation of one. We then selected 100,000 random pairs of S2norm values based on this Gaussian distribution, calculated the absolute value of ΔS2norm for each pair, and averaged over all the pairs to determine that two proteins of only random dynamical similarity can be expected to have a pairwise average |ΔS2norm| of approximately 1.13. This value is marked with a dashed line in Figure 9.
Our ability to compare CI2 and eglin c is limited by the small number of positions in the two proteins that are structurally equivalent and both occupied by amino acids with methyl-bearing side chains. While CI2 has a large number of such side chains, eglin c contains a larger degree of aromatic side chains, and thus the number of positions available for direct comparison is limited. Based on the comparisons available, however, the average |ΔS2norm| for WT CI2 versus WT eglin c is approximately 0.83, while the average value for CI2 I76V versus eglin c V63A is 1.08. This suggests semi-quantitatively that there is greater similarity between the side-chain dynamics of WT CI2 and WT eglin c than would be expected at random and would seem to support the possibility that the protein fold, rather than the local environment, encodes the dynamics of its constituent amino acids. In fact, Figure 9 shows that most of the |ΔS2norm| values for WT CI2 and WT eglin are at worst near and at best significantly below the calculated average value for random similarity; were it not for the peculiar behavior at residue 27, the |ΔS2norm| average across all comparable methyl groups would be smaller than 0.83 and thus indicate an even greater degree of similarity between the side-chain dynamics of the two WT proteins. The CI2 I76V and eglin c V63A mutants, however, are hardly more than randomly dynamically related to one another. This supports our proposition that native CI2 side-chain dynamics, whatever their specific physical determinants, are regulated or constrained to a unique global level and that any core mutation perturbs the side-chain dynamics away from their norm. In essence, the CI2 core mutations rob the mutants of their characteristic dynamical “CI2-ness” such that the CI2 I76V mutant shares little more than random dynamical similarity with the homologous eglin c mutant. This also indicates that the determinants of side-chain dynamics act with some subtlety, since structural characterizations revealed that there are no gross backbone or side-chain structural perturbations upon mutating CI2 core residues. These conclusions are tentative on account of the lack of available dynamics data collected under the same experimental conditions for structurally homologous positions in CI2 and eglin c.
Conclusions
This paper presents the results of an extensive NMR study of the effects of hydrophobic core mutations in chymotrypsin inhibitor 2 on the internal dynamics of both the backbone and methyl-containing side-chains. The core mutations generally resulted in increased flexibility on the ps-ns time scale, but while the particular backbone results were mutant-dependent, the side chains showed the same slight but global increase in flexibility regardless of the position or chemical nature of the mutation. We suggest that the internal dynamics of WT CI2 are modulated in such a way as to be at some maximum of possible internal rigidity and that any small mutational perturbation in the core knocks the cohesive whole off balance, with the particular result being a small global increase in backbone and side-chain flexibility. Additionally, a progressive shortening of the L68 side chain leads to the increasing appearance of slower nanosecond-scale dynamics, primarily localized to the reactive loop. If it is indeed the case that L68 serves as a connection between the reactive loop and the hydrophobic core, as evidence in the literature suggests, then abolishing this link by shortening the side chain might decouple the behavior of the loop from the behavior of the hydrophobic core in the ps-ns time regime.
We have also performed an initial comparison of the degree of similarity in the side-chain dynamics of two homologues, CI2 and eglin c, for both the pair of WT proteins and a pair of structurally homologous mutants. The available data suggest that the ps-ns side-chain dynamics of WT CI2 and WT eglin c exhibit more than random similarity, while the dynamics of the mutant pair are no more similar than two random proteins would be. We interpret this idea in the context of alteration to the physical determinants of side-chain dynamics upon mutation, with the CI2 I76V mutation altering these determinants in such a way that the resultant side-chain dynamics are no longer characteristic of the WT protein. Thus the similarity in the side-chain dynamics of WT CI2 and WT eglin c is abolished upon mutating two proteins at the equivalent position. The dynamical similarity of the two WT proteins suggests the importance of a protein’s fold in determining internal dynamics; however, the reduced degree of similarity in the side-chain dynamics of the homologous mutants suggests that some local, sequence-dependent effect(s) from the mutations must also be contributing to the observed dynamics. Further investigation of the effects of mutation on the dynamics of homologous proteins is needed in order to clarify the relative importance of global and local influences in regulating the observed dynamics.
Acknowledgements
The authors thank Dr. Karl Koshlap of the UNC School of Pharmacy NMR Facility and Dr. Greg Young of the UNC Biomolecular NMR Laboratory for technical assistance. We also thank Anthony Law for helpful discussions and acknowledge Steve Gilmore for generating the CI2 expression construct.
This work was supported in part by NIH Grant GM066009 to A.L.L. and an NSF Graduate Research Fellowship to M.J.W.
Abbreviations
- AIC
Akaike’s information criterion
- AP
deuterium antiphase magnetization
- BIC
Bayesian information criterion
- CI2
chymotrypsin inhibitor 2
- Dz
deuterium longitudinal magnetization
- D+
deuterium in-phase transverse magnetization
- DQ
deuterium double quantum coherence
- μs-ms
microsecond-to-millisecond
- NMR
nuclear magnetic resonance
- NOE
nuclear Overhauser enhancement
- PCR
polymerase chain reaction
- ps-ns
picosecond-to-nanosecond
- QO
deuterium quadrupolar order
- RDCs
residual dipolar couplings
- S2
generalized order parameter characterizing amplitude of N-H bond motions
- S2axis
order parameter characterizing amplitude of methyl symmetry axis motions
- τe
correlation time for internal motions
- τm
a protein’s global tumbling time
- WT
wild-type
References
- 1.D’Aquino JA, Gomez J, Hilser VJ, Lee KH, Amzel LM, Freire E. The magnitude of the backbone conformational entropy change in protein folding. Proteins. 1996;25:143–156. doi: 10.1002/(SICI)1097-0134(199606)25:2<143::AID-PROT1>3.0.CO;2-J. [DOI] [PubMed] [Google Scholar]
- 2.Yang D, Kay LE. Contributions to conformational entropy arising from bond vector fluctuations measured from NMR-derived order parameters: application to protein folding. J. Mol. Biol. 1996;263:369–382. doi: 10.1006/jmbi.1996.0581. [DOI] [PubMed] [Google Scholar]
- 3.Zidek L, Novotny MV, Stone MJ. Increased protein backbone conformational entropy upon hydrophobic ligand binding. Nat. Struct. Biol. 1999;6:1118–1121. doi: 10.1038/70057. [DOI] [PubMed] [Google Scholar]
- 4.Wrabl JO, Shortle D, Woolf TB. Correlation between changes in nuclear magnetic resonance order parameters and conformational entropy: molecular dynamics simulations of native and denatured staphylococcal nuclease. Proteins. 2000;38:123–133. doi: 10.1002/(sici)1097-0134(20000201)38:2<123::aid-prot2>3.0.co;2-h. [DOI] [PubMed] [Google Scholar]
- 5.Stone MJ. NMR relaxation studies of the role of conformational entropy in protein stability and ligand binding. Acc. Chem. Res. 2001;34:379–388. doi: 10.1021/ar000079c. [DOI] [PubMed] [Google Scholar]
- 6.Wand AJ. Dynamic activation of protein function: a view emerging from NMR spectroscopy. Nat. Struct. Biol. 2001;8:926–931. doi: 10.1038/nsb1101-926. [DOI] [PubMed] [Google Scholar]
- 7.Bocharov EV, Korzhnev DM, Blommers MJ, Arvinte T, Orekhov VY, Billeter M, Arseniev AS. Dynamics-modulated biological activity of transforming growth factor beta3. J. Biol. Chem. 2002;277:46273–46279. doi: 10.1074/jbc.M206274200. [DOI] [PubMed] [Google Scholar]
- 8.Johnson E, Chazin WJ, Rance M. Effects of calcium binding on the side-chain methyl dynamics of calbindin D9k: a 2H NMR relaxation study. J. Mol. Biol. 2006;357:1237–1252. doi: 10.1016/j.jmb.2006.01.031. [DOI] [PubMed] [Google Scholar]
- 9.Frederick KK, Marlow MS, Valentine KG, Wand AJ. Conformational entropy in molecular recognition by proteins. Nature. 2007;448:325–329. doi: 10.1038/nature05959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Cooper A, Dryden DTF. Allostery without conformational change: a plausible model. Eur. Biophys. J. 1984;11:103–109. doi: 10.1007/BF00276625. [DOI] [PubMed] [Google Scholar]
- 11.Li Z, Lukasik SM, Liu Y, Grembecka J, Bielnicka I, Bushweller JH, Speck NA. A mutation in the S-switch region of the Runt domain alters the dynamics of an allosteric network responsible for CBFbeta regulation. J. Mol. Biol. 2006;364:1073–1083. doi: 10.1016/j.jmb.2006.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Popovych N, Sun S, Ebright RH, Kalodimos CG. Dynamically driven protein allostery. Nat. Struct. Mol. Biol. 2006;13:831–838. doi: 10.1038/nsmb1132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Eisenmesser EZ, Millet O, Labeikovsky W, Korzhnev DM, Wolf-Watz M, Bosco DA, Skalicky JJ, Kay LE, Kern D. Intrinsic dynamics of an enzyme underlies catalysis. Nature. 2005;438:117–121. doi: 10.1038/nature04105. [DOI] [PubMed] [Google Scholar]
- 14.Watt ED, Shimada H, Kovrigin EL, Loria JP. The mechanism of rate-limiting motions in enzyme function. Proc. Natl. Acad. Sci. USA. 2007;104:11981–11986. doi: 10.1073/pnas.0702551104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ming D, Bruschweiler R. Prediction of methyl-side chain dynamics in proteins. J. Biomol. NMR. 2004;29:363–368. doi: 10.1023/B:JNMR.0000032612.70767.35. [DOI] [PubMed] [Google Scholar]
- 16.Zhang F, Bruschweiler R. Contact model for the prediction of NMR N-H order parameters in globular proteins. J. Am. Chem. Soc. 2002;124:12654–12655. doi: 10.1021/ja027847a. [DOI] [PubMed] [Google Scholar]
- 17.McPhalen CA, James MN. Crystal and molecular structure of the serine proteinase inhibitor CI-2 from barley seeds. Biochemistry. 1987;26:261–269. doi: 10.1021/bi00375a036. [DOI] [PubMed] [Google Scholar]
- 18.Svendsen I, Jonassen I, Hejgaard J, Boisen S. Amino acid sequence homology between a serine protease inhibitor from barley and potato I inhibitor. Carlsberg Res. Commun. 1980;45:389–395. [Google Scholar]
- 19.Itzhaki LS, Otzen DE, Fersht AR. The structure of the transition state for folding of chymotrypsin inhibitor 2 analysed by protein engineering methods: evidence for a nucleation-condensation mechanism for protein folding. J. Mol. Biol. 1995;254:260–288. doi: 10.1006/jmbi.1995.0616. [DOI] [PubMed] [Google Scholar]
- 20.Shakhnovich E, Abkevich V, Ptitsyn O. Conserved residues and the mechanism of protein folding. Nature. 1996;379:96–98. doi: 10.1038/379096a0. [DOI] [PubMed] [Google Scholar]
- 21.Dokholyan NV, Li L, Ding F, Shakhnovich EI. Topological determinants of protein folding. Proc. Natl. Acad. Sci. USA. 2002;99:8637–8641. doi: 10.1073/pnas.122076099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hilser VJ, Dowdy D, Oas TG, Freire E. The structural distribution of cooperative interactions in proteins: analysis of the native state ensemble. Proc. Natl. Acad. Sci. USA. 1998;95:9903–9908. doi: 10.1073/pnas.95.17.9903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Muhandiram DR, Kay LE. Gradient-enhanced triple-resonance three-dimensional NMR experiments with improved sensitivity. J. Magn. Reson. Ser. A. 1994;103:203–216. [Google Scholar]
- 24.Uhrín D, Uhrínová S, Leadbeater C, Nairn J, Price NC, Barlow PN. 3D HCCH3-TOCSY for resonance assignment of methyl-containing side chains in 13C-labeled proteins. J. Magn. Reson. 2000;142:288–293. doi: 10.1006/jmre.1999.1951. [DOI] [PubMed] [Google Scholar]
- 25.Neri D, Szyperski T, Otting G, Senn H, Wüthrich K. Stereospecific nuclear magnetic resonance assignments of the methyl groups of valine and leucine in the DNA-binding domain of the 434 repressor by biosynthetically directed fractional 13C labeling. Biochemistry. 1989;28:7510–7516. doi: 10.1021/bi00445a003. [DOI] [PubMed] [Google Scholar]
- 26.Delaglio F, Grzesiek S, Vuister GW, Zhu G, Pfeifer J, Bax A. NMRPipe: a multidimensional spectral processing system based on UNIX pipes. J. Biomol. NMR. 1995;6:277–293. doi: 10.1007/BF00197809. [DOI] [PubMed] [Google Scholar]
- 27.Johnson BA, Blevins RA. NMRView: a computer program for the visualization and analysis of NMR data. J. Biomol. NMR. 1994;4:603–614. doi: 10.1007/BF00404272. [DOI] [PubMed] [Google Scholar]
- 28.Farrow NA, Muhandiram R, Singer AU, Pascal SM, Kay CM, Gish G, Shoelson SE, Pawson T, Forman-Kay JD, Kay LE. Backbone dynamics of a free and phosphopeptide-complexed Src homology 2 domain studied by 15N NMR relaxation. Biochemistry. 1994;33:5984–6003. doi: 10.1021/bi00185a040. [DOI] [PubMed] [Google Scholar]
- 29.Lipari G, Szabo A. Model-free approach to the interpretation of nuclear magnetic resonance relaxation in macromolecules. 1. Theory and range of validity. J. Am. Chem. Soc. 1982;104:4546–4559. [Google Scholar]
- 30.Lee AL, Flynn PF, Wand AJ. Comparison of 2H and 13C NMR Relaxation Techniques for the Study of Protein Methyl Group Dynamics in Solution. J. Am. Chem. Soc. 1999;121:2891–2902. [Google Scholar]
- 31.Jarymowycz VA, Stone MJ. Fast time scale dynamics of protein backbones: NMR relaxation methods, applications, and functional consequences. Chem. Rev. 2006;106:1624–1671. doi: 10.1021/cr040421p. [DOI] [PubMed] [Google Scholar]
- 32.Chen J, Brooks CL, III, Wright PE. Model-free analysis of protein dynamics: assessment of accuracy and model selection protocols based on molecular dynamics simulation. J. Biomol. NMR. 2004;29:243–257. doi: 10.1023/B:JNMR.0000032504.70912.58. [DOI] [PubMed] [Google Scholar]
- 33.Muhandiram DR, Yamazaki T, Sykes BD, Kay LE. Measurement of 2H T1 and T1ρ Relaxation Times in Uniformly 13C-Labeled and Fractionally 2H-Labeled Proteins in Solution. J. Am. Chem. Soc. 1995;117:11536–11544. [Google Scholar]
- 34.Millet O, Muhandiram DR, Skrynnikov NR, Kay LE. Deuterium spin probes of side-chain dynamics in proteins. 1. Measurement of five relaxation rates per deuteron in 13C-labeled and fractionally 2H-enriched proteins in solution. J. Am. Chem. Soc. 2002;124:6439–6448. doi: 10.1021/ja012497y. [DOI] [PubMed] [Google Scholar]
- 35.Chou JJ, Gaemers S, Howder B, Louis JM, Bax A. A simple apparatus for generating stretched polyacrylamide gels, yielding uniform alignment of proteins and detergent micelles. J. Biomol. NMR. 2001;21:377–382. doi: 10.1023/a:1013336502594. [DOI] [PubMed] [Google Scholar]
- 36.Ottiger M, Delaglio F, Bax A. Measurement of J and dipolar couplings from simplified two-dimensional NMR spectra. J. Magn. Reson. 1998;131:373–378. doi: 10.1006/jmre.1998.1361. [DOI] [PubMed] [Google Scholar]
- 37.Permi P. Measurement of residual dipolar couplings from 1Hα to 13Cα and 15N using a simple HNCA-based experiment. J. Biomol. NMR. 2003;27:341–349. doi: 10.1023/a:1025866606252. [DOI] [PubMed] [Google Scholar]
- 38.Valafar H, Prestegard JH. REDCAT: A Residual Dipolar Coupling Analysis Tool. J. Magn. Reson. 2004;167:228–241. doi: 10.1016/j.jmr.2003.12.012. [DOI] [PubMed] [Google Scholar]
- 39.Grzesiek S, Vuister GW, Bax A. A simple and sensitive experiment for measurement of JCC couplings between backbone carbonyl and methyl carbons in isotopically enriched proteins. J. Biomol. NMR. 1993;3:487–493. doi: 10.1007/BF00176014. [DOI] [PubMed] [Google Scholar]
- 40.Vuister GW, Wang AC, Bax A. Measurement of three-bond nitrogen-carbon J-couplings in proteins uniformly enriched in 15N and 13C. J. Am. Chem. Soc. 1993;115:5334–5335. [Google Scholar]
- 41.Hennig M, Bermel W, Spencer A, Dobson CM, Smith LJ, Schwalbe H. Side-chain conformations in an unfolded protein: χ1 distributions in denatured hen lysozyme determined by heteronuclear 13C, 15N NMR spectroscopy. J. Mol. Biol. 1999;288:705–723. doi: 10.1006/jmbi.1999.2722. [DOI] [PubMed] [Google Scholar]
- 42.Schnell JR, Dyson HJ, Wright PE. Effect of cofactor binding and loop conformation on side chain methyl dynamics in dihydrofolate reductase. Biochemistry. 2004;43:374–383. doi: 10.1021/bi035464z. [DOI] [PubMed] [Google Scholar]
- 43.Luginbuhl P, Pervushin KV, Iwai H, Wuthrich K. Anisotropic molecular rotational diffusion in 15N spin relaxation studies of protein mobility. Biochemistry. 1997;36:7305–7312. doi: 10.1021/bi963161h. [DOI] [PubMed] [Google Scholar]
- 44.Lee LK, Rance M, Chazin WJ, Palmer AG., 3rd. Rotational diffusion anisotropy of proteins from simultaneous analysis of 15N and 13C alpha nuclear spin relaxation. J. Biomol. NMR. 1997;9:287–298. doi: 10.1023/a:1018631009583. [DOI] [PubMed] [Google Scholar]
- 45.Loria JP, Rance M, Palmer AG. A Relaxation-Compensated Carr-Purcell-Meiboom-Gill Sequence for Characterizing Chemical Exchange by NMR Spectroscopy. J. Am. Chem. Soc. 1999;121:2331–2332. [Google Scholar]
- 46.Best RB, Vendruscolo M. Structural interpretation of hydrogen exchange protection factors in proteins: characterization of the native state fluctuations of CI2. Structure. 2006;14:97–106. doi: 10.1016/j.str.2005.09.012. [DOI] [PubMed] [Google Scholar]
- 47.Igumenova TI, Frederick KK, Wand AJ. Characterization of the fast dynamics of protein amino acid side chains using NMR relaxation in solution. Chem. Rev. 2006;106:1672–1699. doi: 10.1021/cr040422h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Skrynnikov NR, Millet O, Kay LE. Deuterium spin probes of side-chain dynamics in proteins. 2. Spectral density mapping and identification of nanosecond time-scale side-chain motions. J. Am. Chem. Soc. 2002;124:6449–6460. doi: 10.1021/ja012498q. [DOI] [PubMed] [Google Scholar]
- 49.Clarkson MW, Gilmore SA, Edgell MH, Lee AL. Dynamic coupling and allosteric behavior in a nonallosteric protein. Biochemistry. 2006;45:7693–7699. doi: 10.1021/bi060652l. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Clarkson MW, Lee AL. Long-range dynamic effects of point mutations propagate through side chains in the serine protease inhibitor eglin c. Biochemistry. 2004;43:12448–12458. doi: 10.1021/bi0494424. [DOI] [PubMed] [Google Scholar]
- 51.Boyer JA, Lee AL. Monitoring Aromatic Picosecond to Nanosecond Dynamics in Proteins via 13C Relaxation: Expanding Perturbation Mapping of the Rigidifying Core Mutation, V54A, in Eglin c. Biochemistry. 2008 doi: 10.1021/bi702330t. In press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Mittermaier A, Kay LE. The response of internal dynamics to hydrophobic core mutations in the SH3 domain from the Fyn tyrosine kinase. Protein Sci. 2004;13:1088–1099. doi: 10.1110/ps.03502504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Millet O, Mittermaier A, Baker D, Kay LE. The effects of mutations on motions of side-chains in protein L studied by 2H NMR dynamics and scalar couplings. J. Mol. Biol. 2003;329:551–563. doi: 10.1016/s0022-2836(03)00471-6. [DOI] [PubMed] [Google Scholar]
- 54.Chou JJ, Case DA, Bax A. Insights into the Mobility of Methyl-Bearing Side Chains in Proteins from 3JCC and 3JCN Couplings. J. Am. Chem. Soc. 2003;125:8959–8966. doi: 10.1021/ja029972s. [DOI] [PubMed] [Google Scholar]
- 55.Hu H, Hermans J, Lee AL. Relating side-chain mobility in proteins to rotameric transitions: insights from molecular dynamics simulations and NMR. J. Biomol. NMR. 2005;32:151–162. doi: 10.1007/s10858-005-5366-0. [DOI] [PubMed] [Google Scholar]
- 56.Best RB, Rutherford TJ, Freund SM, Clarke J. Hydrophobic core fluidity of homologous protein domains: relation of side-chain dynamics to core composition and packing. Biochemistry. 2004;43:1145–1155. doi: 10.1021/bi035658e. [DOI] [PubMed] [Google Scholar]
- 57.Mittermaier A, Davidson AR, Kay LE. Correlation between 2H NMR side-chain order parameters and sequence conservation in globular proteins. J. Am. Chem. Soc. 2003;125:9004–9005. doi: 10.1021/ja034856q. [DOI] [PubMed] [Google Scholar]